Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Methods
3. INPP5E expression in...
4. Regulation of INPP5E during...
5. Role of INPP5E in cellular...
6. INPP5E regulation in...
7. INPP5E mutations in human...
8. Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(2):579-594. doi:10.7150/ijbs.99010 This issue Cite
Review
Regulation of INPP5E in Ciliogenesis, Development, and Disease
Abdulaziz Hakeem1,2, Shuying Yang1,3,4 ![]()
1. Department of Basic & Translational Sciences, School of Dental Medicine, University of Pennsylvania, USA.
2. Department of Basic and Translation Science, School of Dentistry, Umm Al Qura University, Saudi Arabia.
3. The Penn Center for Musculoskeletal Disorders, Perelman School of Medicine, University of Pennsylvania, Philadelphia, USA.
4. Center for Innovation & Precision Dentistry, Penn Dental Medicine and School of Engineering and Applied Sciences, University of Pennsylvania, Philadelphia, USA.
Received 2024-5-29; Accepted 2024-12-3; Published 2025-1-1
Abstract

Inositol polyphosphate-5-phosphatase E (INPP5E) is a 5-phosphatase critically involved in diverse physiological processes, including embryonic development, neurological function, immune regulation, hemopoietic cell dynamics, and macrophage proliferation, differentiation, and phagocytosis. Mutations in INPP5E cause Joubert and Meckel-Gruber syndromes in humans; these are characterized by brain malformations, microphthalmia, situs inversus, skeletal abnormalities, and polydactyly. Recent studies have demonstrated the key role of INPP5E in governing intracellular processes like endocytosis, exocytosis, vesicular trafficking, and membrane dynamics. Moreover, it regulates cellular signaling pathways by dephosphorylating the 5-phosphate of phosphatidylinositol-3,4,5-trisphosphate, phosphatidylinositol 4,5-bisphosphate, and phosphatidylinositol 3,5-bisphosphate. Despite recent advances, knowledge gaps persist regarding the function and molecular mechanism of INPP5E in various cells and species. This review integrates recent findings on the role of INPP5E in regulating cellular function, development, and the pathogenesis of various human disorders, emphasizing the molecular mechanism by which INPP5E regulates primary cilia assembly and function and critical signaling pathways. Identifying the importance of INPP5E in healthy and diseased states can advance our understanding of cellular processes and disease pathogenesis and provide a foundation for developing targeted therapeutic interventions.
Keywords: INPP5E, inositol polyphosphate-5-phosphatase E, disease, cell behavior, primary cilia
1. Introduction
Inositol polyphosphate 5-phosphatases (INPP5s) represent a superfamily of phosphatases that remove the fifth-position phosphate from phosphatidylinositol-3,4,5-trisphosphate [PI(3,4,5)P3], phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], and phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2] to form phosphatidylinositol 3,4-bisphosphate [PI(3,4)P2], phosphatidylinositol 4-phosphate [PI(4)P], and phosphatidylinositol 3-phosphate [PI(3)P], respectively [1]. INPP5s are involved in multiple cellular processes, including ion channel control, membrane trafficking, synaptic vesicle formation, hematopoietic cell proliferation, and signal transmission [2] by regulating inositol polyphosphate and phosphatidylinositol signaling [3, 4]. The INPP5 family includes INPP5A (also called proline-rich INPP5 or PIPP), INPP5B, INPP5C, INPP5D (also known as SH-2 containing inositol 5′ polyphosphatase 1 or SHIP1), INPP5E (also called PIPP1), INPP5F (also called SAC2), INPP5J (also referred to as PIPP2), and INPP5K (SKIP) members [5]. These proteins have non-redundant roles in regulating cell functions and important signaling pathways. The crucial roles of these enzymes in biological processes have been summarized elsewhere [6-11].
Phosphatidylinositol-4,5-bisphosphate 5-phosphatase E (INPP5E), also known as pharbin, is a 72-kDa protein comprising 644 amino acids in humans. It is encoded by INPP5E on chromosome 9. INPP5E was first identified by Kisseleva et al. in 2000 [10]. Subsequently, increasing evidence has shown that INPP5E regulates various cellular activities, including ion channel function, membrane trafficking, synaptic vesicle formation, hematopoietic cell proliferation, and signal transmission [10, 11]. INPP5E mutations are associated with various pathologies, including Joubert syndrome, human syndromic ciliopathies, mental retardation, truncal obesity, retinal dystrophy, and micropenis (MORM) syndrome [11-14]. These findings highlight the importance of the spatial localization and enzymatic activity of INPP5E within cilia [15, 16]. However, gaps remain regarding how INPP5E functions in healthy cells and how its inactivation leads to diseases. In this review, we comprehensively summarize the role of INPP5E in regulating cellular proliferation, differentiation, and function and highlight the importance of INPP5E in ciliogenesis. We also discuss the contribution of INPP5E to organ development and disease pathogenesis, including ciliopathies, Joubert syndrome, polycystic kidney disease (PKD), sonic hedgehog (Shh) medulloblastoma, cystic renal dysplasia, hepatic fibrosis, colorectal carcinoma, inherited retinal degeneration (IRD), MORM syndrome, and inflammation. Finally, we discuss the signaling pathways regulated by INPP5E under physiological and pathophysiological conditions.
2. Methods
A literature search was performed using “PubMed,” “Embase,” and “Scopus” with the following search terms: “Inositol Polyphosphate-5-Phosphatase E” and “INPP5E gene.” The search yielded 360 articles. The inclusion criteria included studies published up to 2024 that contained specific keywords, such as “INPP5E” and “inositol polyphosphate-5-phosphatase E.” Additionally, only studies published in English and those specifically focused on INPP5E were considered. Studies that mentioned INPP5E but did not study or investigate it directly were excluded. After review, 49 articles describing specific studies on INPP5E were included. The other articles served as sources of background information.
3. INPP5E expression in organelles and tissues
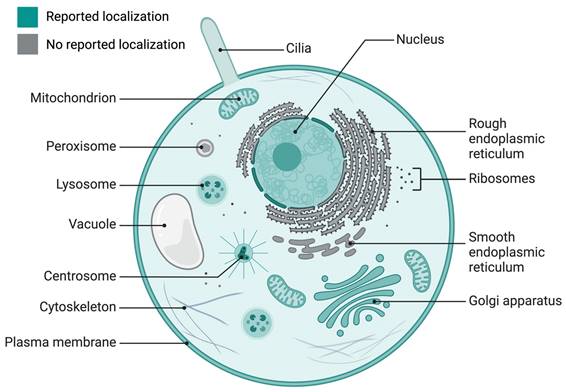
INPP5E expression has been reported in different cellular organelles, including the Golgi apparatus [17, 18], lysosomes [19, 20], membranes [21, 22], nucleus [23], and centrosomes [23] (Fig. 1). Additionally, INPP5E localizes in many cells and tissues, such as epithelial cells [4, 22, 24-26], Kupffer's vesicles, pronephric ducts [4], retinal and renal tissue [24], kidneys [22], pronephric ducts, otic vesicles [25], colorectum [26], and neuroepithelium in the cerebellum [27]. Furthermore, it is expressed in immune cells, including macrophages [21] and T-cells [28], in addition to photoreceptor cells in the inner segment tissue of the retina [29], mesenchymal cells in the hindlimbs and ribcage [30], and hepatocytes in the liver [31]. INPP5E is also reportedly expressed in different neural cells [24, 30, 32-35].
INPP5E localization in the cell. The literature-reported localization of INPP5E in a cell is presented in green (cilia, Golgi apparatus, mitochondria, lysosome, centrosome, nucleus, and plasma membrane); the unreported localization appears in gray/white (rough and smooth endoplasmic reticulum, ribosome, cytoskeleton, and peroxisome). Created in BioRender. Hakeem, A. (2024) BioRender.com/h58k985.
INPP5E protein is found in different locations within cilia. In mouse embryonic fibroblasts, INPP5E has been reported to partially localizes to the transition zone (TZ) near specific lipid molecules (PI(4,5)P2 and PI(3,4,5)P3) during Hedgehog (Hh) pathway activation [30]. INPP5E is localized to the apical membrane and basal bodies, not cilia, in the pronephric epithelium of zebrafish to break down PI(3,4,5)P3 [25]. INPP5E is also present in the ciliary membrane, directly interacting with ARL13B [36]. In IMCD3 cells, INPP5E primarily localizes to the ciliary axoneme and, to a lesser extent, the basal end of cilia [37], mediated by the intraflagellar transport (IFT) system [38].
4. Regulation of INPP5E during ciliogenesis
4.1. Structure and function of primary cilia
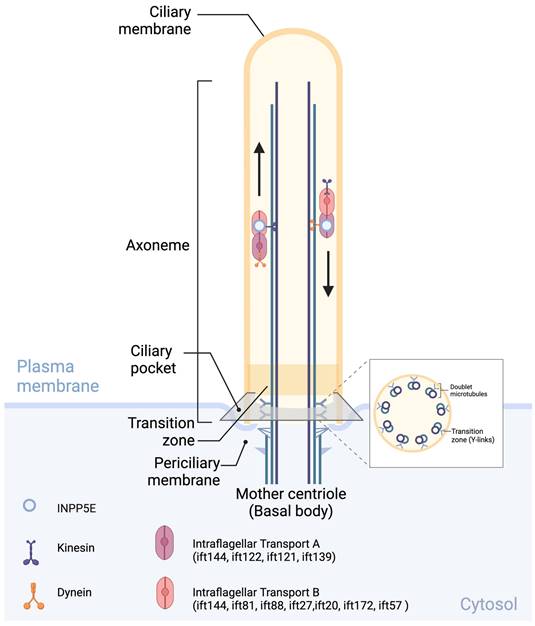
The primary cilium is a dynamic and sensory subcellular organelle that extends from the cell membrane surface (Fig. 2) [35, 39]. Primary cilia comprise four essential components [39] including basal body, axoneme, transition zone (TZ) and ciliary membrane. The basal body develops from the mother centriole and serves as an anchor for the ciliary membrane. The axoneme consists of a ring of nine outer microtubule doublets and provides a scaffold for multiple protein complexes such as Intraflagellar Transport B (IFT-B) and Intraflagellar Transport A (IFT-A), crucial components of the IFT machinery and their associated motor proteins dynein and kinesin. The IFT-B complex, including ift144, ift81, ift88, ift27, ift20, ift172, and ift57, moves cargo from the base of the cilium to the tip using kinesin motor proteins, whereas the IFT-A complex, including ift144, ift122, ift121, and ift139, moves cargo from the tip of the cilium back to the base using dynein motor proteins. The ciliary sheath covers axoneme and it is a continuous with the plasma membrane and the TZ (the region between the basal body and axoneme) separate them functionally [29, 39]. The TZ in primary cilia, also known as the ciliary gate, is a critical region located at the base of the cilium and is characterized by Y-shaped linkers, a ciliary necklace, and transition fibers. This region functions as a selective gate that controls transport, acts as a diffusion barrier, and regulates signaling pathways within the cilium [39]. Finally, the ciliary membrane covers the microtubule-based axoneme [24-29].
Primary cilia structure: Intraflagellar transport B (IFT-B), such as IFT144, IFT81, IFT88, IFT27, IFT20, IFT172, and IFT57, as well as Intraflagellar transport A (IFT-A), including IFT144, IFT122, IFT121, and IFT13, contribute to INPP5E transport; their components and machinery are essential for the assembly, maintenance, and function of primary cilia. Created in BioRender. Hakeem, A. (2024) BioRender.com/d83l169.
Primary cilia are essential for vertebrate embryonic development as they coordinate multiple signaling cascades [35]. Primary cilia dynamics are closely linked to cell cycle progression, assembling and disassembling when cells exit and enter the cell cycle, respectively [39]. In non-dividing cells, primary cilia maintain the balance of phosphoinositide, which serves as a second messenger in signal transduction [24, 25]. As a cellular antenna and environmental sensor, primary cilia can sense changes in external mechanical (physical) stimuli such as mucus, fluid flow, and chemical signals (such as hormones and nutrients) and transmit the signals between cells, which can lead to cellular responses [39]. The sensory function of primary cilia is essential for embryonic development, sensory perception, and tissue balance. Moreover, primary cilia help regulate the balance of various organs and tissues in vertebrates [29, 35].
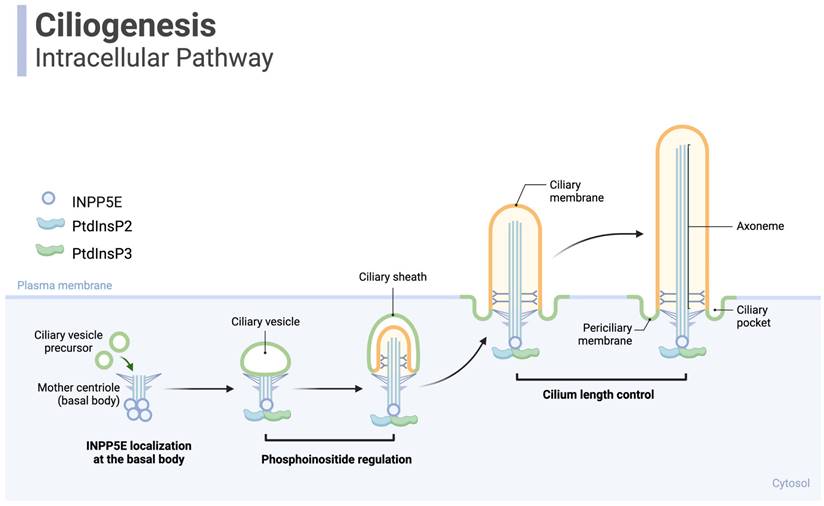
INPP5E is critically involved in multiple cellular processes, particularly ciliogenesis and the microtubule assembly of tactile organelles in most mammalian cells (Fig. 3) [35]. INPP5E is essential for cilia formation and function across various tissues in myriad physiological processes, including regulating lipid levels [39]. INPP5E controls the lipid and protein connections at the ciliary base and participates in hydrolyzing two key phospholipids—PI(3,4,5)P3 and PI(4,5)P2 into PI(3,4)P2 and PI(4)P, respectively [39]. These products are vital for preserving the structure and length of primary cilia, especially in dormant cells [39].
Ciliogenesis: INPP5E is a crucial protein in developing primary cilia. Primary cilia are microtubule-based organelles extending from the surfaces of most mammalian cells and are involved in various cellular signaling pathways. Inositol polyphosphate-5-phosphatase E (INPP5E) is localized to the base of primary cilia, acting as a phosphoinositide phosphatase to explicitly target the lipid molecules phosphatidylinositol 4,5-bisphosphate (PIP2) and phosphatidylinositol 3,4,5-bisphosphate (PIP3). It dephosphorylates PIP2 and PIP3, converting them to PI4P. Proper levels of PIP2 are crucial for localizing various proteins involved in cilia assembly, including intraflagellar transport (IFT) machinery, which facilitates the trafficking of components required for cilium elongation and maintenance. INPP5E also helps control cilium elongation. Regulating PIP2 levels influences the activation of various downstream signaling pathways that control protein trafficking into and out of the cilium. This regulation of protein trafficking contributes to the modulation of cilium length. Created in BioRender. Hakeem, A. (2024) BioRender.com/g24c237.
4.2. Role of INPP5E in ciliogenesis in different animal models
4.2.1. Inpp5e regulates ciliogenesis during zebrafish development
Roles for inpp5e in zebrafish ciliogenesis have been defined. Luo et al. [4] first revealed that inpp5e is required for ciliogenesis in zebrafish kidneys. They found that suppressing inpp5e expression with morpholinos results in shorter and fewer cilia in the Kupffer's vesicle of young larvae and the pronephric ducts of the kidneys, as well as impaired epinephrine-induced melanosome retraction [4]. Zebrafish with an inpp5e mutation exhibit abnormal ocular size, kidney function, heart development, eye development, and body symmetry [4]. Additionally, Xu et al. [25] reported that zebrafish inpp5e promotes ciliogenesis in renal epithelium by breaking down PI(3,4,5)P3 and stabilizing apical PI(4,5)P2, helping recruit Ezrin, F-actin, and basal bodies to the apical side of the renal epithelium. Meanwhile, downregulating inpp5e in zebrafish causes abnormalities in cilia formation and leads to cystic kidney development [25]. Moreover, inpp5e is crucial in suppressing PI(3,4,5)P3 expression in the apical membrane of the pronephric epithelia in zebrafish embryos [25]. When inpp5e is absent or depleted, PI(3,4,5)P3 is localized at the apical membrane, causing PI3K to relocate via a positive feedback mechanism to convert PI(4,5)P2 to PI(3,4,5)P3, decreasing PI(4,5)P2 levels [25]. These findings provide evidence that inpp5e is essential for ciliogenesis and organ development and function in zebrafish.
4.2.2. Inpp5e regulates ciliogenesis during mouse development
Consistent with the findings in zebrafish, Inpp5e is also required for ciliogenesis in mouse organs (i.e., brain and retina) and cells (i.e., inner medullary collecting duct cells, NIH3T3 cells, fibroblasts, and rod outer segments) [21-25]. Xu et al. [24] conditionally deleted INPP5E from human renal cortical tubular epithelial cells and 293 cells, causing shortened cilia and ciliary disassembly in inner medullary collecting duct cells. They showed that phosphatidylinositol-4-phosphate 5-kinase type I (PIPKI) and INPP5E regulate PI(4)P lipid levels in centrosomes, which develop into primary cilia [24]. PIPKI and INPP5E also localize tau tubulin kinase 2 (TTBK2) to the cilial basal body [24]. Furthermore, for functional cilia to develop, centriolar coiled-coil protein 110, which blocks axoneme formation in cilia, must be absent, and TTBK2 must be present [24]. The ciliary assembly protein centrosomal protein 164 (CEP164) binds to PI(4)P, preventing its interaction with TTBK2 [24]. Therefore, PIPKI and INPP5E regulate PI(4)P-dependent TTBK2 recruitment to initiate ciliogenesis, and INPP5E improves the coordination of ciliogenesis in mouse inner medullary collecting duct and renal cortical tubular epithelial cells [24].
Similar findings have shown that INPP5E is necessary for ciliogenesis during mouse embryonic neurodevelopment [35]. Organoids harboring an Inpp5e mutation exhibit fewer cilia and extra ciliary smoothened (SMO) proteins [40]. This alters the expression of several cilium-related genes, including Kras, Ift80, Pkhd1, Prkca, Mkks, and Smo [41]. Most importantly, radial glial cells exhibit abnormal sonic hedgehog (Shh) signaling [35]. During neural tube closure, INPP5E expression is markedly decreased in mice with abnormal neurodevelopment and/or neural tube defects compared to normal control mice [35]. This finding suggests that decreased expression of Inpp5e is closely associated with neural tube deformities, and INPP5E is essential for ciliogenesis and brain development [35].
INPP5E is also strongly associated with the development and function of other organs. Sharif et al. [29] recently reported that Inpp5e deletion in the retina results in a regular-length connecting cilium; however, this does not extend axonemes into the outer segment (OS) or form disks [29]. Moreover, the deletion of Inpp5e in neural stem cells impairs cilia formation and results in the accumulation of PI(4,5)P2, tubby-like protein 3 (TULP3), and IFT particles, inhibiting OS growth [29]. Furthermore, Dewees et al. [18] reported that the transportation of IFT140 and INPP5E to the cilia requires ADP-ribosylation factor-like GTPase (ARL) 16, whereas INPP5E relies solely on retinal rod rhodopsin-sensitive cGMP 3′,5′-cyclic phosphodiesterase subunit delta (PDE6D). Consequently, the prenylated INPP5E cargo binds to the PDE6D shuttle [18]. The removal of Arl16 from mouse embryonic fibroblasts (MEFs) inhibits ciliogenesis and alters ciliary protein content, resulting in a loss of ARL13B, ARL3, INPP5E, and the IFT-A core component IFT140 [18]. Hence, INPP5E is critical in organ development, regulating cilia formation and function [18].
5. Role of INPP5E in cellular function
INPP5E plays crucial roles in regulating cellular functions, such as inflammation, immune cell responses, phagocytosis, autophagy, the functionality of olfactory sensory neurons, and the cell cycle, highlighting its importance in maintaining cellular homeostasis.
5.1. Role of INPP5E in inflammation and immune cells
The inositol family has well-defined effects on immune cell function. Specifically, the role of inositol 1,4,5-triphosphate hydrolysis and Ca2+ release in immune cells, specifically macrophages, was first described in 1986 by Kukita et al. [42]. The study examined the role of Ca2+ in the hydrolysis of PI(4,5)P2 in isolated guinea pig macrophages stimulated with fMet-Leu-Phe. This potent chemotactic factor attracts immune cells, such as macrophages, to infection or inflammation sites and participates in degranulation, superoxide production, and intracellular calcium mobilization. fMet-Leu-Phe causes a rapid decrease in PIP2 levels, accompanied by phosphatidic acid and IP3 accumulation [42].
INPP5E is crucial for the formation and function of immune synapses, specialized interfaces between T lymphocytes and antigen-presenting cells [28]. INPP5E is enriched in cilia and regulates the localization of phosphoinositides [28]. It is highly concentrated at the immune synapse in Jurkat T-cells in antibody-mediated crosslinking of T-cell receptor (TCR) complexes or superantigen-mediated conjugation [28] and regulates immune synapses by interacting with CD3ζ, ZAP-70, and Lck. Repressing INPP5E in T-cells disrupts CD3ζ polarized distribution at the immune synapse and causes failed PI(4,5)P2 clearance at the center of the synapse [28]. Additionally, silencing INPP5E reduces proximal TCR signaling and decreases interleukin (IL)-2 secretion [28]. These findings highlight the significant role of INPP5E in manipulating phosphoinositides at the synapse, especially in controlling the TCR signaling cascade.
INPP5E regulates hematopoietic lineage by regulating centrosome interactions with immune synapses in lymphocytes [28, 43]. Chiu et al. reported that INPP5E is crucial in T-cell activation, regulating phosphoinositide distribution at the immune synapse [28]. This helps in the polarized distribution of CD3ζ, a vital component of the TCR complex. Moreover, INPP5E actively participates in the formation of CD3ζ, ZAP-70, and Lck complexes at the immune synapse to properly activate the TCR signaling cascade. Given its role as a primary activating signal for T-cells, CD3ζ is used as the driver of signal transmission by all currently approved chimeric antigen receptor T-cells targeting CD19 or B-cell maturation antigens. These findings highlight the crucial role of the ciliary-enriched phosphatase INPP5E in immune synapses and suggest that chimeric antigen receptor T-cell therapy in relation to INPP5E is a promising strategy for immunotherapy [28, 43].
INPP5E also regulates viral infections and their mediated immune responses. Several viruses trigger PI3K signaling pathways to elevate PI(3,4,5)P3 levels, activating the protein kinase B (AKT)-mammalian target of rapamycin (mTOR) signaling axis and impacting infection efficiency [44]. PI(4,5)P2 and PI(3,4,5)P3 are INPP5E substrates and well-defined mediators of actin remodeling [44]. Hoang et al. revealed that the removal of INPP5E enhances herpes simplex virus type 1 and vesicular stomatitis virus D51 infection in 4T1 cells [44] by altering the actin cytoskeleton and membrane ruffling, improving viral attachment and enhancing binding of the viruses to the cell surface [44]. Despite INPP5E's localization to the cilium, viral attachment does not show separation as expected if enhanced attachment occurs at or close to ciliary structures. Thus, INPP5E's antiviral outcomes may not be connected to its ciliary localization. Centrosomal PIs can also be influenced by INPP5E loss, causing spindle microtubule deterioration, potentially through a discrepancy in PI(4,5)P2 expression. Whether these immune mechanisms mediate enhanced virus binding and infection is subject to future investigation.
5.2. Role of INPP5E in phagosome function
Studies have shown that INPP5E influences macrophage phagocytosis and T-cell synapse by regulating the TCR signaling cascade [21, 28]. Decreased INPP5E levels (shInpp5e cells) can result in reduced RAS-related protein RAB20 levels in phagosomes, accelerating phagosome acidification [21]. Moreover, PI(3,4,5)P3 and PI(3,4)P2 levels become altered during phagocytic cup formation in murine macrophages (RAW 264.7) deficient in INPP5E [21]. Furthermore, the expression of a constitutively active form of RAB5B in these cells rescues PI(3)P accumulation [21]. Therefore, INPP5E plays a crucial role in RAB5 activation by functionally interacting with RAB20 in phagosomes, leading to increased PI(3)P levels and delayed phagosome acidification [21].
5.3. Role of INPP5E in autophagy
Autophagy is a natural cellular process in which cells break down and recycle their components. It plays a crucial role in maintaining cellular health and homeostasis by removing cellular debris, recycling damaged components, and providing energy and building blocks for cellular renewal and repair [19, 20]. Autophagy is also involved in various physiological processes, such as development, growth, immunity, and adaptation to nutrient availability and stress conditions [19, 20]. During autophagy, autophagosomes engulf damaged organelles, misfolded proteins, and other cellular waste [20]; they then fuse with lysosomes to break down the engulfed materials into their basic building blocks [20]. INPP5E plays a crucial role in the fusion of autophagosomes and lysosomes in neuronal cells [20]. Lysosomes require actin filaments on their surface for fusion with autophagosomes, and activated cortactin stabilizes these filaments [20]. INPP5E catalyzes PI(3,5)P2—a phosphatase substrate that counteracts cortactin-mediated actin filament stabilization—to PI(3)P in lysosomes, decreasing PI(3,5)P2 levels and promoting cortactin activation [20]. Cortactin binds to actin filaments, stabilizing them on lysosomes and ensuring membrane anchoring and autophagosome-lysosome fusion [20]. Consequently, INPP5E knockdown impairs autophagosome-lysosome fusion, markedly inhibiting autophagy [19, 20].
5.4. Role of INPP5E in olfactory sensory neurons
The lipid composition of the primary ciliary membrane is a crucial factor regulating cilium formation, maintenance, and function [34]. In mammals, odors are recognized through the conversion of chemicals into neural signal cues by olfactory sensory neurons [34]. When odor molecules activate olfactory cilia, PI(3,4,5)P3 levels are decreased by INPP5E, which stops Ca2+ influx and redistributes PI(3,5)P2 [34]. This redistribution of PI(3,5)P2 enhances the sensitivity of the neurons to odorants by increasing the exposure of receptors to the extracellular environment [34]. Conditional knockout mice with Inpp5e deletion in olfactory sensory neurons (OSNs) exhibit [34] impaired odor adaptation and reduced recovery from the short-lived excitation. Deletion of Inpp5e in mouse OSNs decreased cilia numbers and length, increased PI(3,4,5)P3 levels, altered PI(3,5)P2 distribution, and resulted in AKT signaling pathway activation and tremendous intraciliary Ca2+ elevation [34]. These findings demonstrate that INPP5E is involved in the modulation of odorant receptor trafficking and signaling via the regulation of PI(3,5)P2 levels to trigger the redistribution of odorant receptors within the plasma membrane of OSNs [34].
5.5. Role of INPP5E in mitosis
The life cycle of a primary cilium, which begins at the quiescence stage and ends before mitosis, has been extensively studied [23, 45, 46]. Sierra Potchanant et al. [23] reported that INPP5E is essential for maintaining cellular homeostasis and development via cell division regulation [23]. INPP5E silencing or knockout in murine/human cells weakens spindle aggregation checkpoint, centrosome formation, spindle function, and chromosomal integrity. Additionally, INPP5E expression is cell cycle-dependent, heightening during mitotic entry [23]. INPP5E protein localizes to chromosomes, centrosomes and kinetochores during early mitosis and transports to the midzone spindle at the mitotic exit [23]. Upon nuclear envelope breakdown, INPP5E diffuses from the nucleus through the cell, with a portion accumulating around chromosomes; its abundance increases during the prophase [23]. INPP5E translocates to the nucleus during telophase after nuclear envelope reformation [23]. Moreover, INPP5E prevents aneuploidy. INPP5E knockdown causes chromosomal instability in primary human fibroblasts, a hallmark of carcinogenesis or cellular dysfunction. However, INPP5E transcription is reportedly upregulated in cervical cancers, uterine leiomyomas, and lymphomas and downregulated in gastric carcinomas and metastatic adenocarcinomas [23]. Further studies are needed to better understand the true impact of INPP5E's role in the chromosome [23]. Nevertheless, these findings highlight the crucial role of INPP5E in mitosis and aneuploidy prevention.
INPP5E also regulates the cilia life cycle and cell cycle through cilia decapitation to induce cell mitogenic signaling [45]. Deletion or mutation of INPP5E causes PI(4,5)P2 accumulation at the distal cilia, leading to cilia decapitation; that is, the excision of cilia tips from cilia [45]. Moreover, Aurora kinase A (AURKA) functions downstream of INPP5E and HDAC6 in complementary pathways to regulate cilia decapitation. AURKA phosphorylates INPP5E to modulate its 5-phosphatase activity and governs the re-localization of INPP5E, suggesting that AURKA may have a broader role in coordinating ciliary dynamics and signaling [45].
Beside regulating cell cycle, INPP5E also regulates extracellular vesicles, including exosomes and micro-vesicles, that participate in intercellular communication within a single organism or between different organisms, species, and kingdoms [46]. Distal cilia tips are shed or “decapitated” when there is growth stimulation or high levels of ciliary PI(4,5)P2. During growth induction, PI(4,5)P2 accumulates at the distal ends of cilia and displaces the ciliary protein INPP5E [45, 46]. This displacement triggers actin (F-actin) polymerization, which enters the primary cilia, leading to decapitation [45, 46]. Although cilia disassembly is traditionally thought to occur through resorption, the acute loss of IFT-B protein components (ARL13B, IFT88, and IFT81) due to cilia decapitation precedes resorption [45]. In summary, the exit of INPP5E from the cilium leads to F-actin accumulation at the sites of ciliary decapitation [45, 46]. Concurrently, the shedding of extracellular vesicles from decapacitated cilia precedes cilia disassembly and influences the cell cycle [46].
6. INPP5E regulation in signaling pathways and other mechanistic analyses
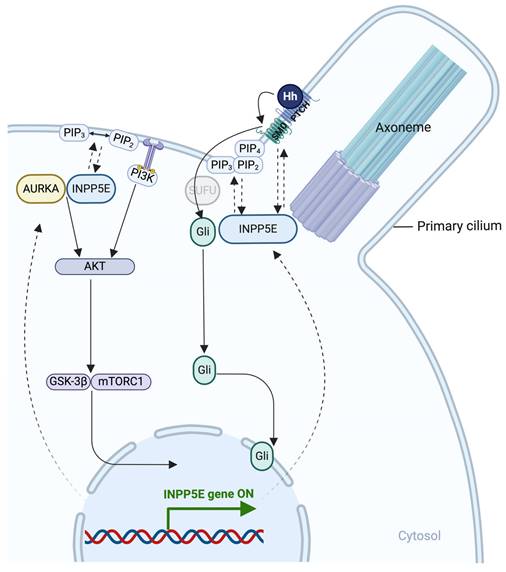
The interactions, associations, and regulatory roles of INPP5E make it essential for maintaining cellular functions and processes, particularly in ciliated cells. In addition to the Hh pathway, INPP5E functions through the GSK-3β, Wingless-related integration site, PI3K, and nuclear factor kappa B (NF-κB) pathways, with particular importance in the PdtIns(3,4,5)P3/PI3K/AKT and Hh signaling pathways (Fig. 4). The primary function of INPP5E in cilia is to produce a PI(4)P-rich membrane environment by converting PdtIns(4,5)P2 to PI(4)P, which is crucial for controlling the localization of receptors involved in the Hh signaling pathway. Although further studies are needed to elucidate the mechanisms underlying the roles of INPP5E and develop restorative interventions for ciliopathies, some important findings have been published, as presented herein.
The main pathways involved in INPP5E activation. Shh signaling pathway. In the unstimulated state, patched (PTCH) localizes in the cilium membrane, inhibiting SMO entry. GLI family zinc finger (GLI) proteins are suppressed by SuFu at the ciliary tip. Upon SHH binding to PTCH, SMO repression is released, allowing its entry while PTCH remains in the cilium. SMO then represses SuFu, enabling GLI activation. Activated GLI (GLIA) translocates to the nucleus, driving target gene expression. During Hh pathway activation, phosphatidylinositol-4,5-bisphosphate 5-phosphatase E (INPP5E) helps regulate SMO and GLI2/GLI1 translocation. Moreover, INPP5E regulates GLI3 processing, possibly due to increased PI(3,4,5)P3. PIP2 and PIP3 activate the SHH signaling pathway to control downstream target gene expression. INPP5E may also influence GLI3 processing by impacting the transition zone (TZ). TKR signaling pathway: INPP5E can mediate tyrosine kinase receptor (TKR) pathway activation by regulating Aurora kinase A (AURKA) via phosphatidylinositol 4,5-bisphosphate (PIP2) control and PIP3 conversion, activating signaling cascades, including protein kinase B (AKT), glycogen synthase kinase-3 (GSK-3)β, and mammalian target of rapamycin (mTOR)C1. Subsequently, INPP5E expression is upregulated. Created in BioRender. Hakeem, A. (2024) BioRender.com/y60b182.
6.1. PI(3,4,5)P3/PI3K/AKT pathway
INPP5E regulates PI(3,4,5)P3 hydrolysis, which is required for appropriate AKT activation [24, 37]. At the primary cilia, the hydrolysis of PI(3,4,5)P3 by INPP5E changes the different phosphoinositide levels in species and facilitates the functional interaction between INPP5E and AURKA, activating AURKA through auto-phosphorylation [23, 37]. When this pathway is activated, INPP5E hydrolysis of PI(3,4,5)P3 facilitates the binding of AURKA to INPP5E and its phosphorylation, thus increasing INPP5E's phosphatase activity [23, 44]. This process subsequently reduces AURKA transcription downstream of AKT [23, 44]. AURKA phosphorylates INPP5E, enhancing its activity and suppressing the PI3K/AKT pathway [23, 37]. Ciliary INPP5E is required for PI(4,5)P2 hydrolysis in cilia and subsequent activation of the Shh pathway [24, 30-33, 35]. In the absence of INPP5E, upregulated PI(3,4,5)P3 levels increase AKT phosphorylation, suggesting that INPP5E also regulates PI(3,4,5)P3 outside cilia [23, 24, 37].
6.2. Hh signaling pathway
The Hh pathway is a primary cellular formation and development pathway [30, 35]. In the absence of Hh, patched (PTCH) is localized on the cilia and cell membrane, preventing high expression of SMO, a G protein-coupled receptor, trafficking to cilia. Hh binds to PTCH to release the inhibition in SMO expression and translocation to cilia. Hh activates SMO and converts the signal cascade to INPP5E activation[30, 35]. SMO accumulation is increased in wild-type mice, promoting Inpp5e activation, i.e., positive regulation [30, 35]. In contrast, reduced Inpp5e expression in neural and NIH3T3 cells in mice causes primary cilia malformation and neural maldevelopment [35]. Furthermore, Inpp5e deletion suppresses Hh signaling in mouse embryos despite an average number of cilia and low SMO levels [35]. This low SMO level in the cilia is due to TZ diffusion failure as a gatekeeper [30, 35]. SMO translocates into cilia to activate Hh signaling; in contrast, reduced ciliary localization of SMO reduces its ability to activate Hh signaling [30, 35]. At the TZ, INPP5E is important in regulating PI(4,5)P2 and PI(3,4,5)P3, which influence SMO accumulation and Hh signal activation [30]. INPP5E deletion negatively affects the accumulation of SMO, GLI family zinc finger 2 (GLI2), IFT, and other proteins at the ciliary base in human neural cells [40]. However, as a compensatory response, SHH signaling accumulates at the site of INPP5E deletion, leading to the formation of more neurons and progenitors [40]. Moreover, the development of certain diseases may be attributed to abnormalities in the SHH signaling pathway, potentially due to ciliary dysfunction [40]. Further research is needed to determine the effects of INPP5E dysfunction, such as its impact on inositol phosphate and other signaling pathways.
Additionally, it is vital to understand how the various cellular defects associated with the loss of INPP5E function are related to human phenotypes [47]. In cells cultured from mice harboring an inactivated Inpp5e allele, MEFs exhibit no Shh transcriptional response, whereas neural cells exhibit an enhanced Shh transcriptional response [33]. Inpp5e weakens SHH signaling in developing mouse neural cells and enhances it in MEF cells [33]. SMO, PTCH, and SuFu keeps the pathway in an “off” state while the ligand is absent; therefore, their loss completes the pathway activation besides +GliA/-GliR production [33].
Further studies should address how Inpp5e helps Hh ligand binds to its receptor PTCH. We think that PTCH saturation on the ciliary membrane, causing its removal from the cilium, permitting the GPCR SMO entry [48]. The current understanding is that PTCH remains localized on the cilia and cell membrane without SHH stimulation. In contrast, with SHH, PTCH disassociates from the cilia, and SMO translocates into the cilia.
Moreover, the need to understand the ciliary expression of SUFU and GLI in relation to Inpp5e protein during Hh signaling is essential. We think that the activation of Hh signaling leads GLI and SUFU entering the cilium and gathering at the ciliary tip. In activation, SHH binds to PTCH to release the inhibition in SMO; then, SMO, SUFU, GLI proteins accumulate at the ciliary tip [49]. There, the GLI proteins phosphorylates to transcriptional activators that move to the nucleus and incite the target genes transcription of SHH pathway [49].
The deletion [27, 30] (germline deletion, precursors granule cell neurons deletion) of INPP5E in mice reduces the ciliary localization of GLI2/GLI1 and SMO during Hh pathway activation. Moreover, in this pathway, the Hh ligand binds to its receptor PTCH in the ciliary membrane [18] to trigger the removal of PTCH from the cilium, enabling SMO, a GPCR, to translocate into cilia. This action results in the cleavage and initiation of GLI transcription factors, which then move to the nucleus and initiate the target genes transcription [18]. Inpp5e is also involved in controlling direct and indirect neurogenesis through GLI3 processing [50]. In Inpp5e mutant embryos, the GLI3R level and GLI3R/GLI3FL ratio decrease, increasing the prevalence of direct neurogenesis [50]. The loss of INPP5E results in structural abnormalities in cilia, possibly due to increased PI(3,4,5)P3. INPP5E may also influence GLI3 processing by impacting the TZ. These findings suggest that INPP5E is crucial in regulating cilia formation and function and GLI3R formation [50].
Taken together, Inpp5e regulation of the Hh response involves a more complicated mechanism than previously appreciated, and it differs from one cell type to another [24, 30-33, 35, 50].
6.3. Interactions between INPP5E and other proteins
INPP5E exhibits complex action mechanisms in the cilia and interacts with many proteins (ARL13B, ATG16L1, CEP164, IFT20, PDE6D, retinitis pigmentosa GTPase regulator (RPGR), and TULP3) to achieve ciliary homeostasis [38, 51, 52]. Dynamic equilibrium between these proteins and INPP5E is essential to maintain normal function within the cilia and promote cell survival [38, 51, 52]. Accordingly, the interactions between INPP5E and different proteins and the effects of these interactions on cell function have garnered substantial research interest [50, 51].
PDE6δ is important in ciliary sorting and localization mechanisms. PDE6δ sorts farnesylated INPP5E into cilia through high-affinity binding and release by the ADP-ribosylation factor (ARF)-like protein ARL3·GTP1 [38]. PDE6δ-free INPP5E can be specifically retained in the cilia [50]. INPP5E is transported to the ciliary base upon binding to PDE6δ [51]. After diffusing into the cilium, INPP5E is released from PDE6δ by ARL3·GTP, and the farnesyl moiety connects it to the ciliary membrane [51]. INPP5E is then carried by the IFT system and retained inside the cilium [38, 50]. ATG16L1 partially affects the primary ciliary phosphoinositide equilibrium [38, 51]. However, it directly regulates the distribution of ciliary phosphoinositides and INPP5E trafficking, which is associated with primary cilia membrane regulation [17]. Humbert et al. [53] investigated an ARL13B/PDE6D/CEP164 complex to track the ciliary location of INPP5E. ARL13B and PDE6D are ciliary proteins that connect to INPP5E, directing them toward the target area inside cilia [54].
Other specific carrier proteins also facilitate INPP5E transportation to cilia [38, 54]. ARL13B—a small GTPase—also contributes to the localization of INPP5E, although the precise mechanism is not fully understood [38, 54]. Once inside the cilia, the inner ciliary transport of INPP5E is regulated solely by the IFT system, independent of PDE6δ activity and INPP5E farnesylation [38]. Therefore, INPP5E depends on these carrier proteins and transport systems for its localization and movement within the cilia [38, 54]. In particular, TULP3 plays a crucial role in the localization of phosphoinositide phosphatase INPP5E in the cilia [54]. It interacts with the intraflagellar IFT-A, which is responsible for the ciliary localization of transmembrane proteins [54]. TULP3 is an adapter for transporting various integral membrane cargo to the cilia [54]. The Tubby domain of TULP3 binds to PI(4,5)P2—a phosphoinositide—essential for retrograde ciliary protein trafficking mediated by IFT-A and TULP3 [54]. In the absence of ciliary INPP5E, particularly under PI(4,5)P2-rich conditions, this trafficking is impaired [38, 54]. Furthermore, TULP3 regulates the association of INPP5E with other proteins, such as ARL13B. This complex interplay ensures the correct localization of INPP5E in the cilia. Hence, TULP3 acts as a carrier of INPP5E to the cilia, playing a critical role in its ciliary localization and function [54].
6.4. Mechanisms of INPP5E ciliary targeting and localization
INPP5E functions as a basic controller and effector in many cells, especially those with cilia [36, 37, 55]. On ciliary cells, such as murine embryonic fibroblasts, human embryonic kidney cells, and retinal pigment epithelial cells, INPP5E targets an intricate interaction of proteins, such as ARL13B, PDE6D, and CEP164, ensuring its localization to the targeted site [36, 37, 55]. Deletion of INPP5E causes cilia loss and alters the total level of AKT, upregulating AURKA transcription, a centrosomal kinase that regulates mitosis and ciliary disassembly, resulting in its accumulation in the remaining cilia [37]. In response to physiological stimuli, an increased pool of AURKA at this site drives an abnormal cilia disassembly phenotype [37]. Meanwhile, AURKA inhibition may rescue the defects caused by INPP5E loss [37]. The interaction between INPP5E and AURKA at the primary cilia may occur as follows [37]. PI(3,4,5)P3 hydrolysis by INPP5E alters the levels of different phosphoinositide species, mediating the functional interaction between INPP5E and AURKA and activating AURKA via auto-phosphorylation. Activated AURKA attaches to and phosphorylates INPP5E, rising its phosphatase levels and decreasing AURKA transcription downstream of AKT [37]. In the case of INPP5E deletion, elevated AKT signaling increases AURKA transcription, causing in a general AURKA buildup at the cilia. Responding to physiological stimuli, the heightened AURKA accumulation may drive the abnormal cilia disassembly phenotype seen in cells depriving INPP5E [44]. Moreover, Fujisawa et al. [36] highlighted the association between ARL3 and ARL13B GTPases and the different forms of INPP5E [36]. Their findings underscore the multifaceted nature of the INPP5E restriction cycle [36]. The association of INPP5E with ARL13B is fundamental in maintaining ciliogenesis and supporting ciliary homeostasis [36]. RPGR isoforms play a crucial role in regulating binding partners in the primary cilia, potentially affecting the functional properties of INPP5E and RPGR-interacting protein 1-like (RPGRIP1) [55]. RPGR may act as a molecular link that fine-tunes the functions of these binding partners in the primary cilium [55].
7. INPP5E mutations in human diseases
A ciliopathy is a genetic disorder that affects the cilia structures (basal bodies) and/or function [31, 56-66]. These disorders are characterized primarily by several unrelated genes, such as INPP5E, NPHP1, AHI1, CEP290, CSPP1, KIF7, TCTN1, OFD1, and ARL13B, that affect the ciliary structure or function [31, 56-66]. INPP5E mutations are predominantly homozygous; the following mutations have been reported in the literature: R378C, G286R, V303M, R345S, T426N, R512W, R435Q, W474R, R515W, Y534D, R563H, K580E, R585C, Y588C, R621, C641R, Q309X, Y543X, and Q627X [56]. Diseases linked to INPP5E mutations include Joubert syndrome, Leber congenital amaurosis, and MORM syndrome. However, many other diseases, including PKD, SHH medulloblastoma, cystic renal dysplasia, hepatic fibrosis, colorectal carcinoma, IRD, monogenic obesity syndrome, and Senior-Loken syndrome, are associated with INPP5E mutations [31, 56-66].
7.1. Joubert syndrome
Joubert syndrome is a rare, autosomal recessive, X-linked disorder of neurological impairment characterized by mid- and hindbrain malformation, hypo-dysplastic cerebellar vermis, and neuro-dialogical (structural) or molar tooth sign [16, 67]. This disorder is accompanied by dysregulated breathing patterns, ataxia, and delayed development [16, 67]. Typical neurological signs in infants include dysregulated breathing patterns, ataxia, hypotonia, apnea, oculomotor dysfunction/ocular anomalies (reported in all cases with the INPP5E mutation), delayed psychomotor function, and multi-organ anomalies involving the digits, eyes, liver, and kidneys [16, 67]. Joubert syndrome is characterized by clinical and genetic heterogeneity and is associated with mutations in the following genes: INPP5E, NPHP1, AHI1, CEP290, CSPP1, TCTN3, MKS11, MKS10, MKS9, TCTN2, NPHP3, CC2D2A, RPGRIP1L, MKS4, TMEM67, TMEM216, MKS1, CEP120, C2CD3, B9D2, B9D1, MKS1, PDE6D, IFT172, TCTN3, C5ORF42, TMEM231, TMEM138, CEP41, TMEM237, KIF7, TCTN1, OFD1, and ARL13B [68].
Mutations in INPP5E may lead to the formation of an unstable protein, resulting in reduced levels of INPP5E in the fibroblasts and impaired ciliary development [33]. Biochemical data from patients with Joubert syndrome has shown a missense mutation in INPP5E, which is associated with a reduced number of cilia and shorter ciliary length, indicating that INPP5E is essential for cilia formation and function [33]. INPP5E coordinates with the centrosomal enzyme Ptdlns(4)P 5′-kinase or PIPKly to initiate cilia formation [33]. INPP5E deletion suppresses cilia formation, resulting in TULP3 and G protein-coupled receptor 161 accumulation, which represses the SHH signaling pathway and downregulates phosphoinositide production [33]. ARL13B, CEP164, and PDE6D are involved in the ciliary localization of INPP5E; mutations in INPP5E lead to the pathological alterations underlying Joubert syndrome, resulting from differences in cilia length and localization patterns [47].
Mutations in INPP5E are associated with a limited genotype-phenotype correlation but pronounced phenotypic variation in Joubert syndrome [69]. For example, because INPP5E is necessary for ciliary signaling pathway regulation, mutations in INPP5E result in cilium dysfunction and dysregulated brain development, specifically of the mid- and hindbrain, which are crucial for body coordination, ocular and renal function (preventing cystic kidneys), breathing pattern maintenance, and intellectual ability [69]. An unusual homozygous mutation in INPP5E, c.1303C>T, leads to the replacement of arginine by tryptophan at residue 435 (p.Arg435Trp), affecting protein functionality and altering brain function [68, 70].
INPP5E regulates many of proteins localized in the TZ, which is located in the proximal region of the cilia [66,68]. This zone begins at the distal end of the ciliary basal and links to the plasma membrane and axoneme which allows the movement of proteins “in-and-out” of the cilium [71].
Joubert syndrome and related anomalies represent a group of genetically heterogeneous, cilium-related pathologies characterized by a cerebellar deformity known as the molar tooth sign on brain MRI [72]. Furthermore, INPP5E mutations are related to dysfunctional cilia formation or ciliopathy, dysregulated cilia signaling pathways, structural abnormalities in the brain, heterogeneity in genes leading to Joubert syndrome onset, and the related clinical signs and symptoms shown by Joubert syndrome patients [72]. Gene profiling of patients with Joubert syndrome has revealed interactions between different genes with INPP5E in modulating ciliary function; mutations in INPP5E downregulate phosphoinositol metabolism [73]. Exome and next-generation sequencing have been used to analyze INPP5E mutations related to Joubert syndrome and associated syndromes [73]. This has led to the development of a novel diagnostic technique based on the identified heterozygous INPP5E variations [73], emphasizing the importance of molecular and genetic testing for achieving a precise diagnosis of this complex and rare neurociliopathy.
7.2. MORM syndrome
MORM syndrome is a rare condition in men that is characterized by obesity, micropenis, intellectual disability, and retinal dystrophy [56, 74]. The gene affected by this autosomal recessive disorder is located on chromosome 9q34.3 [56, 74]. As INPP5E is located at this same chromosomal position, the condition has been linked to INPP5E [56, 74]. Although the exact mechanistic role of INPP5E in the disease has not been defined, it has been hypothesized that it may involve defective primary cilia [56, 74].
7.3. Other ciliopathies
Disorders characterized by primary ciliary dysfunction caused by genetic mutations are termed ciliopathies [75]. Ciliopathies can include brain anomalies, kidney cysts, retinal degeneration, obesity, polydactyly, and infertility [50, 76, 77]. Control of ciliary phosphoinositide metabolism is crucial for mammalian development and is essential for maintaining tissue integrity in adulthood, as lack of control due to INPP5E mutations can lead to ciliopathies [75]. Mutations in INPP5E affect various cell types that rely on ciliary signaling, such as neural stem cells [50], ciliated epithelial cells [15], photoreceptor cells [76], and kidney cells [77]. In the primary cilia, INPP5E modifies the functions of the PI3K/AKT, Hh, and GSK-3β signaling pathways, markedly affecting cell development, viability, and specialization.
7.3.1. Neural maldevelopment
The role of INPP5E in ciliopathy has been investigated using various animal and human models. In mice [50], Inpp5e deletion leads to increased cortical generation of basal neuronal progenitors. INPP5E influences the balance between indirect and direct neurogenesis, and its absence causes cortical thinning and reduced brain size [50]. Furthermore, INPP5E regulates the ciliary localization of GLI3, a transcription factor that mediates SHH signaling [50]. This process is necessary for the morphogenesis and patterning of different tissues. Hasenpusch-Theil et al. [50] highlighted the short-term function of INPP5E and its association with GLI3 in regulating direct and indirect neurogenesis during cortical development. Hoang et al. [44] reported an alternative 5′ leader of Inpp5e mRNA that enhances its translation under stress conditions and demonstrated its impact on cellular resistance to infection by oncolytic viruses. Meanwhile, Khan et al. [77] identified two new INPP5E variants in families with ciliopathic illnesses in Pakistan and expanded the genetic and clinical ranges of Bardet-Biedl syndrome and Joubert syndrome. Rao et al. [76] studied the relationship among RPGR, PDE6, and INPP5E, as well as their roles in the homeostasis and composition of ciliary proteins in neural maldevelopment. They detected RPGR in cilia; its localization in the cilia is vital for its functions. PDE6δ interacts with RPGR, which is essential for RPGR's localization in cilia. INPP5E interacts with RPGR and PDE6δ, and the transportation of INPP5E to cilia depends on RPGR's ciliary localization and interaction with PDE6δ. Moreover, Yinsheng et al. [78] studied the role of transmembrane protein 67 in gating ARL13B and INPP5E proteins across the primary cilia membranes. ARL13B interacts with and helps target INPP5E to cilia. Disruption of this interaction by ARL13B missense mutations leads to impaired neural development. These proteins form a functional network crucial for ciliary function and neural development [50, 76-78].
7.3.2. Polycystic kidney disease (PKD)
PKD causes progressive dilation of renal tubules and kidney enlargement, eventually leading to renal failure [57]. The symptoms include high blood pressure, side or back pain, and a swollen abdomen [57]. It is linked primarily to PKD1 and PKD2; however, germline Inpp5e deletion may also be linked to PKD [22, 58]. The loss of INPP5E in the kidney epithelium leads to aggressive PKD development. Meanwhile, AKT hyperactivation activates the mammalian target of rapamycin (mTOR) complex 1 signaling in renal epithelial cells [57, 58]. Additionally, INPP5E is a key inhibitor of PI3K/AKT and acts downstream in mTORC1 signaling in the kidney epithelium in vivo [57, 58]. Thus, dual PI3K/mTORC1 inhibition may be effective for identifying kidney-related pathologies and therapies [22]. Treatment includes tolvaptan, an agonist for cAMP generation, and mTOR inhibitors [57].
7.3.3. SHH medulloblastoma
Medulloblastoma is a highly malignant brain tumor typically affecting children [59]. It originates from embryonal cells in the cerebellum and can arise from various stem cell or progenitor cell populations in early development [59]. Although it can occur during infancy or adulthood, the typical age of diagnosis is 6-8 years [59]. The SHH medulloblastoma subgroup is well-defined genetically, with most patients carrying somatic or germline mutations and copy-number alterations in critical genes of the SHH signaling pathway [27, 59]. The best survival rates have been achieved through maximal surgical resection and adjuvant chemotherapy [59]. Meanwhile, INPP5E is a crucial regulator of primary ciliary dynamics that promotes the progression of SHH medulloblastoma [27]. INPP5E expression was downregulated in a subset of patients with SHH medulloblastomas and correlated with enhanced overall survival [27]. INPP5E confines to the primary cilia, regulating a compartmentalized PI3K/AKT/GSK-3β signaling axis in medulloblastoma and maintain the primary cilia [27]. The loss of INPP5E causes elevated PI(3,4,5)P3, AKT, and GSK-3β levels in cilia [27]. A decreased number of ciliated cells leads to reduced SHH signaling and a subsequent switch from a proliferative to a differentiated phenotype, resulting in slower tumor progression [27].
7.3.4. Cystic renal dysplasia and hepatic fibrosis
Neonatal mortality in Norwich terriers is caused by renal cystic dysplasia and congenital hepatic fibrosis [31]. INPP5E c.1572+5G>A is responsible for hepatorenal fibrocystic disorder in Norwich terriers [31]. The affected dogs and carriers are part of an isolated Finnish family with a common ancestor tracing back 15 generations [31]. This genetically defined syndromic ciliopathy leads to neonatal mortality in Norwich terriers [31].
7.3.5. Inherited retinal diseases (IRDs)
IRDs are a group of disorders that affect the eyes and other parts of the body, including the central nervous system, ear, skeleton, kidney, and cardiovascular system [61, 62]. In some cases, they cause inborn errors of metabolism and ciliopathies [61, 62]. These diseases are caused by various genetic mutations [61, 62]. Currently, 200 genes, including INPP5E, have been identified as associated with IRDs [61, 62]. Many of these genes are rare and inherited in a recessive manner [61]. Defects in INPP5E can lead to early-onset, non-syndromic IRD [61, 62]. Sangermano et al. reported 16 non-syndromic patients with IRD from ten families and two mildly syndromic Joubert syndrome (JBTS) cases, all with rare INPP5E gene variants [62]. They identified 14 INPP5E variants, of which 12 were novel, mainly missense changes in conserved amino acid residues within the phosphatase catalytic domain, suggesting a broader genetic diversity in INPP5E-associated diseases [62]. Although INPP5E variants are linked to syndromic and non-syndromic cases, no clear correlation between specific genetic changes and disease severity was observed, indicating that additional genetic factors may modify the clinical outcomes [62]. Meanwhile, in zebrafish [4], inpp5e knockdown causes defects in membrane left-right asymmetry, eye development, and pronephric duct formation. In humans, INPP5E mutations impair phosphatase activity and ciliary stability, disrupting ciliary function and development [61, 62]. These findings demonstrate the broad phenotypic spectrum of INPP5E-related diseases [61, 62].
7.3.6. Leber congenital amaurosis (LCA)
LCA is a severe IRD and a genetic disorder causing vision loss in children [63]. Over 38 genes have been identified as the cause of LCA, including INPP5E and RPE65, resulting in a wide range of clinical symptoms [63]. Clinical assessment, fundoscopic images, optical coherence tomography examination, and electroretinogram tests can help establish the correct diagnosis [63]. Moreover, gene therapy holds promise for LCA, with various clinical trials focusing on other LCA-related genes to treat the condition [63].
7.3.7. Senior-Loken syndrome
A recent study presented a case of Senior-Loken syndrome attributed to the deletion of a critical TZ protein that ultimately leads to renal failure and retinal degeneration [66]. This report offered critical insights into primary ciliary morphological aberrations in a renal biopsy, which resulted in impaired function [66]. Moreover, the study highlighted altered ciliary localization of INPP5E in a patient with nephrocystin 1 (NPHP1) deletion [66]. Abnormal expression of NPHP4 was also detected in human NPHP1-deficient tissues, underscoring the crucial role of NPHP1 in TZ formation, where NPHP4 functions normally [66]. This helps regulate INPP5E transition and its ability to influence cilia stability, providing crucial insights into the pathophysiology of Senior-Loken syndrome [66].
7.3.8. Monogenic obesity syndrome
A recent case study reported that a Caucasian girl diagnosed with monogenic obesity syndrome was attributed to a novel INPP5E truncating variant [64]. The study identified a cone-rod-type progressive retinal dystrophy that emerged during the first decade of the patient's life [64]. In addition, severe obesity was observed within the first year of life, leading to the early onset of prediabetes, arterial hypertension and, dyslipidemia [64]. Notably, a delay in acquiring language skills was observed [64]. These findings are important for prognosis and informed treatment decision-making [64].
Another study demonstrated the involvement of INPP5E in insulin signal transduction in muscle and adipose tissue. Reduced INPP5E levels enhance insulin signaling by regulating PI3K/AKT and FOXO1 signal transduction [65]. As a glimpse of the potential application of INPP5E in therapy, Bertelli et al. [65] found that reducing Inpp5e expression through the PI3K pathway in mice and rats resulted in reduced weight gain and improved insulin production to manage blood sugar in obese animals. This finding opens numerous possibilities for treating obesity and diabetes.
7.3.9. Colorectal carcinoma (CRC)
CRC is a common cancer with risk factors including age, chronic disease history, lifestyle, and gut microbiota [26, 60]. CRC is triggered by mutations in oncogenes, tumor suppressor genes, and mechanistic error in DNA repair genes [26, 60]. In CRC, chromosomal changes, common mutations, and translocations have been reported to influence major pathways (such as WNT and PI3K), and mutations, such as PIK3CA, can be used to predict prognosis [60]. INPP5E is linked to CRC through miR-598, which is frequently upregulated in CRC. miR-598 overexpression promotes cell proliferation and cell cycle progression in CRC by silencing INPP5E [26]. This provides a better understanding of the molecular pathways underlying CRC pathogenesis [26].
8. Conclusion
In this review, we have summarized the reported cellular pathogenesis pathways associated with INPP5E. However, the specific mechanisms for different cell types and tissues warrant further investigation. Moreover, whether INPP5E can move independently to the cilia and the carrier molecules responsible for its transport require evaluation. This may provide further insights into the levels of PI(4,5)P2 and INPP5E proteins in cilia, ultimately characterizing the specific mechanism(s) underlying cilia formation.
Although the role of inositol deficiency in ciliogenesis is known, the contributions of INPP5E-specific mechanisms remain to be elucidated. To this end, the specific mechanisms under deficient conditions among different cells must be defined. Moreover, INPP5E is essential for ciliogenesis initiation and coordination. However, the precise mechanism(s) and role(s) of INPP5E in cilia formation are highly complex, particularly among cell types and tissues. A better understanding of the molecular and cellular mechanisms of INPP5E in regulating development and diseases could inform the development of novel therapeutic approaches.
Furthermore, promising results have demonstrated the potential role of INPP5E in lowering weight and improving insulin production to manage blood sugar in obese animals [64]. Nonetheless, further studies are required to gain a complete understanding of the therapeutic role of INPP5E in animal and human models using various cells, tissues, and treatment conditions. Finally, proteomic studies and pathway analysis may help elucidate the regulatory cross-talk between INPP5E and other proteins involved in cellular signaling pathways, clarifying how these interactions influence ciliary dynamics.
Abbreviations
AKT: protein kinase B; ARL: ADP-ribosylation factor-like GTPase; AURKA: Aurora kinase A; CC: connecting cilium; CEP164: centrosomal protein 164; GLI3: GLI family zinc finger 3; GSK-3β: glycogen synthase kinase 3β; Hh: Hedgehog; IFT: intraflagellar transport; INPP5: inositol polyphosphate 5-phosphatase; IRD: inherited retinal degeneration; MEF: mouse embryonic fibroblast; MORM: mental retardation, truncal obesity, retinal dystrophy, and micropenis; mTORC1: mammalian target of rapamycin complex 1; NPHP1: nephrocystin 1; OS: outer segment; P10: photoreceptor 10; PDE: phosphodiesterase; PI3K: phosphoinositide 3-kinase; PIPP: proline-rich inositol polyphosphate 5-phosphatase; PIPKI: phosphatidylinositol-4-phosphate 5-kinase type I; PIPKly: Ptdlns(4)P 5′-kinase; PKD: polycystic kidney disorder; PI(3,4)P2: phosphatidylinositol 3,4-bisphosphate; PI(3,5)P2: phosphatidylinositol 3,5-bisphosphate; PI(3,4,5)P3: phosphatidylinositol (3,4,5)-trisphosphate; PI(4)P: phosphatidylinositol 4-phosphate; PI(4,5)P2: phosphatidylinositol 4,5-bisphosphate; RPGR: retinitis pigmentosa GTPase regulator; Shh: Sonic Hedgehog; SMO: smoothened; TTBK2: tau tubulin kinase 2; TULP3: tubby-like protein 3; TZ: transition zone.
Acknowledgements
This work was supported by the U.S. Department of Defense grants PR201467 and NIH Grants R01AR084491 to Dr. Shuying Yang and R01AR080895 to Drs. Shuying Yang and Dana Graves. Figures were created with BioRender.com.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ramos AR, Elong Edimo W, Erneux C. Phosphoinositide 5-phosphatase activities control cell motility in glioblastoma: Two phosphoinositides PI(4,5)P2 and PI(3,4)P2 are involved. Adv Biol Regul. 2018;67:40-8
2. Downes CP, Mussat MC, Michell RH. The inositol trisphosphate phosphomonoesterase of the human erythrocyte membrane. Biochem J. 1982;203:169-77
3. Horan KA, Watanabe K, Kong AM, Bailey CG, Rasko JE, Sasaki T. et al. Regulation of FcgammaR-stimulated phagocytosis by the 72-kDa inositol polyphosphate 5-phosphatase: SHIP1, but not the 72-kDa 5-phosphatase, regulates complement receptor 3 mediated phagocytosis by differential recruitment of these 5-phosphatases to the phagocytic cup. Blood. 2007;110:4480-91
4. Luo N, Lu J, Sun Y. Evidence of a role of inositol polyphosphate 5-phosphatase INPP5E in cilia formation in zebrafish. Vision Res. 2012;75:98-107
5. Dyson JM, Fedele CG, Davies EM, Becanovic J, Mitchell CA. Phosphoinositide phosphatases: just as important as the kinases. Subcell Biochem. 2012;58:215-79
6. Berridge MJ. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol Rev. 2016;96:1261-96
7. Droubi A, Wallis C, Anderson KE, Rahman S, de Sa A, Rahman T. et al. The inositol 5-phosphatase INPP5B regulates B cell receptor clustering and signaling. J Cell Biol. 2022;221:e202112018
8. Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J. et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat. 2013;34:1208-15
9. Pauls SD, Marshall AJ. Regulation of immune cell signaling by SHIP1: A phosphatase, scaffold protein, and potential therapeutic target. Eur J Immunol. 2017;47:932-45
10. Kisseleva MV, Wilson MP, Majerus PW. The isolation and characterization of a cDNA encoding phospholipid-specific inositol polyphosphate 5-phosphatase. J Biol Chem. 2000;275:20110-6
11. Ooms LM, Horan KA, Rahman P, Seaton G, Gurung R, Kethesparan DS. et al. The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem J. 2009;419:29-49
12. Hakim S, Bertucci MC, Conduit SE, Vuong DL, Mitchell CA. Inositol polyphosphate phosphatases in human disease. Curr Top Microbiol Immunol. 2012;362:247-314
13. Barkovich AJ, Millen KJ, Dobyns WB. A developmental and genetic classification for midbrain-hindbrain malformations. Brain. 2009;132:3199-230
14. Cincinnati P, Neri ME, Valentini A. Dandy-Walker anomaly in Meckel-Gruber syndrome. Clin Dysmorphol. 2000;9:35-8
15. Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L. et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41:1032-6
16. Hardee I, Soldatos A, Davids M, Vilboux T, Toro C, David KL. et al. Defective ciliogenesis in INPP5E-related Joubert syndrome. Am J Med Genet A. 2017;173:3231-7
17. Boukhalfa A, Roccio F, Dupont N, Codogno P, Morel E. The autophagy protein ATG16L1 cooperates with IFT20 and INPP5E to regulate the turnover of phosphoinositides at the primary cilium. Cell Rep. 2021;35:109045
18. Dewees SI, Vargova R, Hardin KR, Turn RE, Devi S, Linnert J. et al. Phylogenetic profiling and cellular analyses of ARL16 reveal roles in traffic of IFT140 and INPP5E. Mol Biol Cell. 2022;33:ar33
19. Nakamura S, Hasegawa J, Yoshimori T. Regulation of lysosomal phosphoinositide balance by INPP5E is essential for autophagosome-lysosome fusion. Autophagy. 2016;12:2500-1
20. Hasegawa J, Iwamoto R, Otomo T, Nezu A, Hamasaki M, Yoshimori T. Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. EMBO J. 2016;35:1853-67
21. Segawa T, Hazeki K, Nigorikawa K, Morioka S, Guo Y, Takasuga S. et al. Inpp5e increases the Rab5 association and phosphatidylinositol 3-phosphate accumulation at the phagosome through an interaction with Rab20. Biochem J. 2014;464:365-75
22. Hakim S, Dyson JM, Feeney SJ, Davies EM, Sriratana A, Koenig MN. et al. Inpp5e suppresses polycystic kidney disease via inhibition of PI3K/Akt-dependent mTORC1 signaling. Hum Mol Genet. 2016;25:2295-313
23. Sierra Potchanant EA, Cerabona D, Sater ZA, He Y, Sun Z, Gehlhausen J. et al. INPP5E Preserves Genomic Stability through Regulation of Mitosis. Mol Cell Biol. 2017;37:e00500-16
24. Xu Q, Zhang Y, Wei Q, Huang Y, Hu J, Ling K. Phosphatidylinositol phosphate kinase PIPKIgamma and phosphatase INPP5E coordinate initiation of ciliogenesis. Nat Commun. 2016;7:10777
25. Xu W, Jin M, Hu R, Wang H, Zhang F, Yuan S. et al. The Joubert Syndrome Protein Inpp5e Controls Ciliogenesis by Regulating Phosphoinositides at the Apical Membrane. J Am Soc Nephrol. 2017;28:118-29
26. Li KP, Fang YP, Liao JQ, Duan JD, Feng LG, Luo XZ. et al. Upregulation of miR-598 promotes cell proliferation and cell cycle progression in human colorectal carcinoma by suppressing INPP5E expression. Mol Med Rep. 2018;17:2991-7
27. Conduit SE, Ramaswamy V, Remke M, Watkins DN, Wainwright BJ, Taylor MD. et al. A compartmentalized phosphoinositide signaling axis at cilia is regulated by INPP5E to maintain cilia and promote Sonic Hedgehog medulloblastoma. Oncogene. 2017;36:5969-84
28. Chiu TY, Lo CH, Lin YH, Lai YD, Lin SS, Fang YT. et al. INPP5E regulates CD3zeta enrichment at the immune synapse by phosphoinositide distribution control. Commun Biol. 2023;6:911
29. Sharif AS, Gerstner CD, Cady MA, Arshavsky VY, Mitchell C, Ying G. et al. Deletion of the phosphatase INPP5E in the murine retina impairs photoreceptor axoneme formation and prevents disc morphogenesis. J Biol Chem. 2021;296:100529
30. Dyson JM, Conduit SE, Feeney SJ, Hakim S, DiTommaso T, Fulcher AJ. et al. INPP5E regulates phosphoinositide-dependent cilia transition zone function. J Cell Biol. 2017;216:247-63
31. Dillard KJ, Hytonen MK, Fischer D, Tanhuanpaa K, Lehti MS, Vainio-Siukola K. et al. A splice site variant in INPP5E causes diffuse cystic renal dysplasia and hepatic fibrosis in dogs. PLoS One. 2018;13:e0204073
32. Yue H, Zhu X, Li S, Wang F, Wang X, Guan Z. et al. Relationship Between INPP5E Gene Expression and Embryonic Neural Development in a Mouse Model of Neural Tube Defect. Med Sci Monit. 2018;24:2053-9
33. Constable S, Long AB, Floyd KA, Schurmans S, Caspary T. The ciliary phosphatidylinositol phosphatase Inpp5e plays positive and negative regulatory roles in Shh signaling. Development. 2020;147:dev183301
34. Ukhanov K, Uytingco C, Green W, Zhang L, Schurmans S, Martens JR. INPP5E controls ciliary localization of phospholipids and the odor response in olfactory sensory neurons. J Cell Sci. 2022;135:jcs258364
35. Yue H, Li S, Qin J, Gao T, Lyu J, Liu Y. et al. Down-Regulation of Inpp5e Associated With Abnormal Ciliogenesis During Embryonic Neurodevelopment Under Inositol Deficiency. Front Neurol. 2021;12:579998
36. Fujisawa S, Qiu H, Nozaki S, Chiba S, Katoh Y, Nakayama K. ARL3 and ARL13B GTPases participate in distinct steps of INPP5E targeting to the ciliary membrane. Biol Open. 2021;10:bio058843
37. Plotnikova OV, Seo S, Cottle DL, Conduit S, Hakim S, Dyson JM. et al. INPP5E interacts with AURKA, linking phosphoinositide signaling to primary cilium stability. J Cell Sci. 2015;128:364-72
38. Kosling SK, Fansa EK, Maffini S, Wittinghofer A. Mechanism and dynamics of INPP5E transport into and inside the ciliary compartment. Biol Chem. 2018;399:277-92
39. Zhang R, Tang J, Li T, Zhou J, Pan W. INPP5E and Coordination of Signaling Networks in Cilia. Front Mol Biosci. 2022;9:885592
40. Schembs L, Willems A, Hasenpusch-Theil K, Cooper JD, Whiting K, Burr K. et al. The ciliary gene INPP5E confers dorsal telencephalic identity to human cortical organoids by negatively regulating Sonic hedgehog signaling. Cell Rep. 2022;39:110811
41. Wang X, Yu J, Yue H, Li S, Yang A, Zhu Z. et al. Inpp5e Regulated the Cilium-Related Genes Contributing to the Neural Tube Defects Under 5-Fluorouracil Exposure. Mol Neurobiol. 2024;61:6189-99
42. Kukita M, Hirata M, Koga T. Requirement of Ca2+ for the production and degradation of inositol 1,4,5-trisphosphate in macrophages. Biochim Biophys Acta. 1986;885:121-8
43. Stinchcombe JC, Griffiths GM. Communication, the centrosome and the immunological synapse. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130463
44. Hoang HD, Graber TE, Jia JJ, Vaidya N, Gilchrist VH, Xiang X. et al. Induction of an Alternative mRNA 5' Leader Enhances Translation of the Ciliopathy Gene Inpp5e and Resistance to Oncolytic Virus Infection. Cell Rep. 2019;29:4010-23 e5
45. Phua SC, Chiba S, Suzuki M, Su E, Roberson EC, Pusapati GV. et al. Dynamic Remodeling of Membrane Composition Drives Cell Cycle through Primary Cilia Excision. Cell. 2017;168:264-79 e15
46. Wang J, Barr MM. Cell-cell communication via ciliary extracellular vesicles: clues from model systems. Essays Biochem. 2018;62:205-13
47. Slaats GG, Isabella CR, Kroes HY, Dempsey JC, Gremmels H, Monroe GR. et al. MKS1 regulates ciliary INPP5E levels in Joubert syndrome. J Med Genet. 2016;53:62-72
48. Goetz SC, Ocbina PJ, Anderson KV. The primary cilium as a Hedgehog signal transduction machine. Methods Cell Biol. 2009;94:199-222
49. Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53
50. Hasenpusch-Theil K, Laclef C, Colligan M, Fitzgerald E, Howe K, Carroll E. et al. A transient role of the ciliary gene Inpp5e in controlling direct versus indirect neurogenesis in cortical development. Elife. 2020;9:e58162
51. Fansa EK, Kosling SK, Zent E, Wittinghofer A, Ismail S. PDE6delta-mediated sorting of INPP5E into the cilium is determined by cargo-carrier affinity. Nat Commun. 2016;7:11366
52. Cilleros-Rodriguez D, Martin-Morales R, Barbeito P, Deb Roy A, Loukil A, Sierra-Rodero B. et al. Multiple ciliary localization signals control INPP5E ciliary targeting. Elife. 2022;11:e78383
53. Humbert MC, Weihbrecht K, Searby CC, Li Y, Pope RM, Sheffield VC. et al. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci U S A. 2012;109:19691-6
54. Qiu H, Fujisawa S, Nozaki S, Katoh Y, Nakayama K. Interaction of INPP5E with ARL13B is essential for its ciliary membrane retention but dispensable for its ciliary entry. Biol Open. 2021;10:bio057653
55. Vossing C, Atigbire P, Eilers J, Markus F, Stieger K, Song F. et al. The Major Ciliary Isoforms of RPGR Build Different Interaction Complexes with INPP5E and RPGRIP1L. Int J Mol Sci. 2021;22:3583
56. de Goede C, Yue WW, Yan G, Ariyaratnam S, Chandler KE, Downes L. et al. Role of reverse phenotyping in interpretation of next generation sequencing data and a review of INPP5E related disorders. Eur J Paediatr Neurol. 2016;20:286-95
57. Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers. 2018;4:50
58. Tham MS, Cottle DL, Zylberberg AK, Short KM, Jones LK, Chan P. et al. Deletion of Aurora kinase A prevents the development of polycystic kidney disease in mice. Nat Commun. 2024;15:371
59. Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC. et al. Medulloblastoma. Nat Rev Dis Primers. 2019;5:11
60. Marmol I, Sanchez-de-Diego C, Pradilla Dieste A, Cerrada E, Rodriguez Yoldi MJ. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int J Mol Sci. 2017;18:197
61. Tatour Y, Ben-Yosef T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics (Basel). 2020;10:779
62. Sangermano R, Deitch I, Peter VG, Ba-Abbad R, Place EM, Zampaglione E. et al. Broadening INPP5E phenotypic spectrum: detection of rare variants in syndromic and non-syndromic IRD. NPJ Genom Med. 2021;6:53
63. Huang CH, Yang CM, Yang CH, Hou YC, Chen TC. Leber's Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes (Basel). 2021;12:1261
64. Drole Torkar A, Avbelj Stefanija M, Bertok S, Trebusak Podkrajsek K, Debeljak M, Stirn Kranjc B. et al. Novel Insights Into Monogenic Obesity Syndrome Due to INPP5E Gene Variant: A Case Report of a Female Patient. Front Endocrinol (Lausanne). 2021;12:581134
65. Bertelli DF, Coope A, Caricilli AM, Prada PO, Saad MJ, Velloso LA. et al. Inhibition of 72 kDa inositol polyphosphate 5-phosphatase E improves insulin signal transduction in diet-induced obesity. J Endocrinol. 2013;217:131-40
66. Ning K, Song E, Sendayen BE, Prosseda PP, Chang KC, Ghaffarieh A. et al. Defective INPP5E distribution in NPHP1-related Senior-Loken syndrome. Mol Genet Genomic Med. 2021;9:e1566
67. Travaglini L, Brancati F, Silhavy J, Iannicelli M, Nickerson E, Elkhartoufi N. et al. Phenotypic spectrum and prevalence of INPP5E mutations in Joubert syndrome and related disorders. Eur J Hum Genet. 2013;21:1074-8
68. Shetty M, Ramdas N, Sahni S, Mullapudi N, Hegde S. A Homozygous Missense Variant in INPP5E Associated with Joubert Syndrome and Related Disorders. Mol Syndromol. 2017;8:313-7
69. Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, Abhyankar A. et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat. 2014;35:137-46
70. Tsurusaki Y, Kobayashi Y, Hisano M, Ito S, Doi H, Nakashima M. et al. The diagnostic utility of exome sequencing in Joubert syndrome and related disorders. J Hum Genet. 2013;58:113-5
71. Garcia G 3rd, Raleigh DR, Reiter JF. How the Ciliary Membrane Is Organized Inside-Out to Communicate Outside-In. Curr Biol. 2018;28:R421-R34
72. Nozaki S, Katoh Y, Terada M, Michisaka S, Funabashi T, Takahashi S. et al. Regulation of ciliary retrograde protein trafficking by the Joubert syndrome proteins ARL13B and INPP5E. J Cell Sci. 2017;130:563-76
73. Kumar N, Nomakuchi T, Vossough A, Leonard JMM, Dubbs H, Agarwal S. A Case of INPP5E-Related Joubert Syndrome: Connecting Evolving Phenotype With Novel Genotype. Pediatr Neurol. 2023;145:112-4
74. Hampshire DJ, Ayub M, Springell K, Roberts E, Jafri H, Rashid Y. et al. MORM syndrome (mental retardation, truncal obesity, retinal dystrophy and micropenis), a new autosomal recessive disorder, links to 9q34. Eur J Hum Genet. 2006;14:543-8
75. Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M. et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41:1027-31
76. Rao KN, Zhang W, Li L, Anand M, Khanna H. Prenylated retinal ciliopathy protein RPGR interacts with PDE6delta and regulates ciliary localization of Joubert syndrome-associated protein INPP5E. Hum Mol Genet. 2016;25:4533-45
77. Khan S, Lin S, Harlalka GV, Ullah A, Shah K, Khalid S. et al. BBS5 and INPP5E mutations associated with ciliopathy disorders in families from Pakistan. Ann Hum Genet. 2019;83:477-82
78. Yinsheng Z, Miyoshi K, Qin Y, Fujiwara Y, Yoshimura T, Katayama T. TMEM67 is required for the gating function of the transition zone that controls entry of membrane-associated proteins ARL13B and INPP5E into primary cilia. Biochem Biophys Res Commun. 2022;636:162-9
Author contact
![]() Corresponding author: Dr. Shuying Yang; Email: shuyingyedu; Tel.: 2158982685.
Corresponding author: Dr. Shuying Yang; Email: shuyingyedu; Tel.: 2158982685.