Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(2):842-859. doi:10.7150/ijbs.101937 This issue Cite
Research Paper
BMSCs-derived small extracellular vesicles antagonize cerebral endothelial Caveolin-1 driven autophagic degradation of tight-junction proteins to protect blood-brain barrier post-stroke
Yiyang Li1, Xingping Quan1, Jiacheng Hu1, Yan Han1, Jinfen Chen1, Manfei Zhou1, Fan Zhang1, Yayue Yang1,3, Mingchun Liao1,3, Bin Wang3, Yonghua Zhao1,2, ![]()
1. Institute of Chinese Medical Sciences, State Key Laboratory of Quality Research in Chinese Medicine, University of Macau, Taipa, Macau SAR, China.
2. Department of Pharmaceutical Sciences, Faculty of Health Sciences, University of Macau, Taipa, Macau SAR, China.
3. Guangdong Institute of Intelligence Science and Technology, Zhuhai, Guangdong, China.
Received 2024-8-5; Accepted 2024-12-22; Published 2025-1-1
Abstract

Bone marrow mesenchymal stem cells (BMSCs) -derived extracellular vesicles (EVs), especially small EVs (sEVs), were vastly reported to enable multiple restorative effects on ischemic stroke, yet the protective mechanism of blood-brain barrier (BBB) has not been fully illustrated. In the present study, we investigated the therapeutic effects and mechanism of BMSCs-derived sEVs on BBB injury after ischemic stroke. In-vivo, administering sEVs to transient middle cerebral artery occlusion (tMCAo) mice mitigated the brain infarct volume, BBB permeability and neural apoptosis, and improved the cerebral blood flow perfusion and neurological function. Simultaneously, cerebral vascular endothelial overexpressed Caveolin-1 (Cav-1) together with its strong co-localization with autophagosome protein LC3B were suppressed, and ZO-1 and Occludin expressions were enhanced, whose results were consistent with those of oxygen-glucose-deprivation/reperfusion (OGD/R)-insulted brain endothelial cells (BECs) in vitro. Furthermore, by employing Cav-1 siRNA and pcDNA3.1 transfection, Co-immunoprecipitation, cycloheximide assay, and molecular docking, it proved that brain endothelial Cav-1 was an essential upstream of autophagy activation, contributing to tight-junction proteins delegation via the autophagy-lysosomal pathway. Altogether, our study demonstrates the novel mechanism of Cav-1-dependent tight-junction proteins autophagic disruption on BBB integrity after ischemic stroke, and BMSC-sEVs treatment can reverse such hazard cascades.
Keywords: Autophagy, Blood-brain barrier, Caveolin-1, Ischemic stroke, Small extracellular vesicles
Introduction
Ischemic stroke has become a secondary death and disability cause worldwide, and over 13 million cases of stroke onset globally, accompanied by sorrowful stress to patients' families and a great financial burden to society [1]. However, unsatisfied therapeutic interventions for ischemic stroke remain to be an obstacle for medical measures to advance. Till nowadays, although many novel thrombolysis drugs have emerged for ischemic stroke treatment (e.g. Tenecteplase), recombinant tissue plasminogen activator (r-tPA) treatment within a narrow therapeutic time window (within 4.5 hours from stroke onset) is still the only FDA-approved thrombolysis agent for ischemic stroke patients [2, 3]. Moreover, though previous studies revealed that eligible patients identified by advanced imaging can receive intravenous thrombolysis with a longer time window (4.5-9 hours) [4, 5], most of the patients who do not meet the criteria fail to receive due interventions on time, and the potential risk such as intracerebral hemorrhage (ICH) cannot be fully avoided. All the dark sides mentioned above make an extremely low percentage of patients who can benefit from r-tPA administration [1, 6], and the intravenous r-tPA administration strategy has a relatively low recanalization rate (20-50%, dependent on the occluded vessel sites. [7, 8]. Another strategy is mechanical thrombectomy within 6 to 24 hours after stroke onset in patients with large vessel occlusion (LVO), however, the criteria for the selection of eligible patients and endovascular risk should also be considered [9]. In addition, dual antiplatelet therapy (DAPT), is only suitable for minor stroke patients based on a recent clinical trial [10]. Hence, developing novel therapeutics for ischemic stroke is urgent and necessary.
Extracellular vesicles (EVs) secreted by stem cells are widely reported to show robust potential for ischemic stroke treatment, especially those smaller-sized extracellular vesicles (sEVs) which can cross the blood-brain barrier (BBB), maintain BBB integrity, mediate neurovascular remodeling and neuroinflammatory response [11, 12] in central nervous system diseases including ischemic stroke. Compared with their parent stem cells therapy, EVs have the capability to reach similar efficacy with more low immunogenicity, tumor formation concern and ethical issues [13, 14]. The possible mechanisms of EVs therapy on ischemic stroke may be attributed to their contained non-coding RNAs and proteome [15, 16], nonetheless, the detailed mechanism of how EVs improve stroke recovery is still ambiguous.
Autophagy is a crucial self-degradation biological process that delicately controls the disruption of proteins and substances via the autophagosome-lysosome pathway, dynamically regulating cells survival and maintaining cellular homeostasis [17]. In the acute phase of ischemic stroke, autophagy is a double-edged sword, on the one hand recycling the damaged cellular compartments and toxic substances, while on the other hand aggravating cellular death of neurons, endothelial, and glia [18]. In the acute cerebral ischemia phase, autophagy also participates in the maintenance of BBB integrity, which is essential for stroke prognosis. Previous studies have identified that autophagy is responsible for tight junction proteins degradation, inducing BBB disruption [19-21], and notably, a scaffolding protein, Caveolin-1 (Cav-1), was proven to be increased and involved in the sub-cellular redistribution of tight junction proteins [21-25]. Apart from the broad bio-regulation process (such as anti-apoptosis, inflammation, and promoting neuro-angiogenesis) of Cav-1 on acute ischemic recovery as widely documented [26, 27], interestingly, recent studies revealed that Cav-1 also directly regulated autophagy in the endothelium, affecting endothelial barrier permeability and function in diverse diseases [28-31], yet, the exact role of Cav-1 affecting BBB integrity by brain endothelial autophagy still awaits to be demonstrated. To better bridge this gap and understand the mechanism of stem cell-derived EVs treatment after ischemic stroke, in this study, we investigated the therapeutic effects of BMSC-derived small EVs (sEVs) on BBB injury in cerebral ischemia/reperfusion model, and firstly illustrated the mechanism was related to Cav-1 mediated autophagic degradation of tight junction protein ZO-1 and Occludin.
Materials and Methods
sEVs isolation from bone marrow mesenchymal stem cells (BMSCs)
sEVs were isolated and purified from BMSCs by using ultracentrifugation. Briefly, rat BMSCs (Purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd) were cultured in 75 cm2 flasks with Dulbecco's modified Eagle's medium (DMEM) with 1% penicillin/streptomycin (PS) and 10% fetal bovine serum (FBS) in a density of 2.5 ⅹ106 per flask in an incubator with 37 ℃ and 5% CO2. 24 hours later, the culture medium was replaced by EVs-free medium containing EVs-free FBS produced by ultracentrifuged under 100,000 g for 18 h at 4 ℃ as a previous report [32]. Cells were allowed for growth for another 72 hours, then, the cells' supernatant was centrifuged under 2000 rpm for 10 minutes and passed through a 0.22 μm filter. Afterward, the supernatant was ultracentrifuged under 110,000 g for 120 mins at 4 ℃ by an ultracentrifuge (XPN-100, Beckman Coulter). The pellet was collected and reconstituted in PBS, and re-ultracentrifuged under 110,000 g for 120 mins at 4 ℃. The final sEVs pellet was collected and diluted in PBS for further experiments. For each time of isolation, 8 flasks of BMSCs (about 2 ⅹ107 in the original seeded cells) were used for isolation and diluted in 1.6 ml PBS. Each 100 μl of the sEVs solution was packed in a 1.5 ml microtube and stored in -80 ℃ till use.
sEVs nano-size, dosage, phagocytosis, distribution and morphology determination
The size and concentration of sEVs were evaluated by a nanoparticle tracking analysis (NTA) device (Nanosight-NS500, Malvern Panalytical Ltd), and BCA assay by a BCA kit (Biosharp Lifescience, China) was used for sEVs protein concentration evaluation. For sEVs phagocytosis experiment, 5 μM DiI dye (Invitrogen) was incubated with sEVs, and then washed by PBS with ultracentrifugation at 110,000 g for 120 mins. The isolated DiI-labeled sEVs were co-cultured with b. End3 cells under 37 ℃ for 120 mins. The cells were then fixed by 4% paraformaldehyde (PFA), and permeabilized by 0.5% Triton X-100. DAPI (Beyotime Biotechnology, China) was stained to visualize the nucleus, and then samples were observed by a confocal microscope (SP8, Leica). For in-vivo DiI-sEVs distribution, mice were subjected to tMCAo surgery, and 200 μg DiI-sEVs were injected via tail vein, and 6, 12, and 24 hours after injection, mice were perfused, and sacrificed for brain sampling. After slicing into 20 μm frozen slices, the brains were stained with vessel marker Lectin, and neuron marker Tuj-1 before imaging. Furthermore, sEVs morphology was detected by transmission electron microscope (TEM, Hitachi) (Service provided by Servicebio Inc., Wuhan, China).
OGD/R-insulted brain endothelial cells and neurons and administrations
Mice brain cells line b. End 3 (Purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd) was cultured with DMEM with 1% PS and 10% FBS, and human neuroblastoma cells line SH-SY5Y were cultured with DMEM/F12 medium with 1% PS and 10% FBS (Gibco). All cells were kept in an incubator with 37 ℃ and 5% CO2. For the in-vitro brain endothelial and neuronal oxygen-glucose-deprivation/reperfusion (OGD/R) model, the two kinds of cells medium was replaced by DMEM without glucose (Gibco), and cells cultures were placed under the hypoxia chamber (MIC-101, Billups-Rothenberg Inc., United States) charged with 95% N2, and 5% CO2 in 37 ℃. 2.5 hours later, the cells were transferred to the normoxia culture condition and replaced the medium with the normal culture medium for 24 hours of re-oxygen. For the sEVs treatment, low (2×106 sEVs/ml, 0.1 μg/ml), medium (2×107 sEVs/ml, 1 μg/ml), and high (2×108 sEVs/ml, 10 μg/ml) dosages of sEVs were added to the cells medium after the OGD/R treatment. For brain endothelial cells autophagy regulation, autophagy inhibitor 3-methyladenine (3-MA, 10 mM, MedChemExpress, United States), and lysosomal inhibitor bafilomycin A1 (bafA1, 100 nM, MedChemExpress, United States) were administered for 1 hour before OGD/R insult, whereas autophagy inducer rapamycin (100 nmol/L, MedChemExpress, United States) was employed 24 hours before OGD/R insult. For cycloheximide (CHX) chase assay, CHX (10 μM, MedChemExpress, United States) was applied in OGD/R insulted b. End 3 cells for 3-12 hours to inhibit proteasome pathway for the degradation of tight junction proteins.
Transwell assay
To determine the in-vitro brain endothelial barrier permeability, a transwell assay was performed as our previous study [33]. After high dosages of sEVs were added to the upper chamber, the transwell cell culture plate was subjected to OGD treatment. Then, medium from upper and lower chambers was collected for detection by a microplate reader (SpectraMax M5) at excitation/ emission: 550 nm/ 572 nm. An equation was provided in Supplemental Method 1. was used for the permeability coefficient determination.
Autophagic flux measurement
To determine the autophagy flux of brain endothelial cells, pCMV-mCherry-GFP-LC3B (Beyotime Biotechnology, China) was transfected to b. End3 cells seeded in confocal dishes, and cultured for 48h, then after the treatment of sEVs or autophagy regulator, the cells were imaged under a confocal microscope (SP8, Leica).
Cell viability, live/dead cells staining, and ROS level evaluation
Cell viability was tested by using CCK-8 kit (Beyotime Biotechnology, China), 7000 to 10000 cells were seeded into 96 well plates per well, and after OGD/R and sEVs treatment, 10% CCK-8 solution was added to the wells, and cultured for 2 hours before the plates were read at 450 nm absorbance. Cell viability was determined as the fold of Ctrl groups. Moreover, the live and dead cells rates were evaluated by staining cells with HiPer Calcein AM /PI kit according to the protocol (Mei5 Biotechnology, Co., China), by which live cells were stained green and red for dead cells observed by fluorescent microscope (Lecia, DMi8). The percentage of live cells was calculated. To test the ROS level of cells, the Reactive Oxygen Species Assay Kit (Beyotime Biotechnology, China) with DCFH-DA solution was used, and the labeled ROS green fluorescence was observed and calculated.
Animal cerebral ischemia/reperfusion modeling and sEVs administration
The transient mice middle cerebral artery occlusion (tMCAo) model was established for ischemic stroke modeling. All animal experiment procedures were conducted following the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines and were approved by the Ethics Committee of the University of Macau (Ethics Protocol ID: UMARE-010-2023). Briefly, male C57BL/6 mice (8-10 weeks, 20-25g) were anesthetized by isoflurane (4% for surgical induction, and 1.5-2.5% for maintenance), and an incision was made in the neck of the mouse, and the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were exposed. Subsequently, a silicon-coated filament (with a head diameter of 0.19±0.01 mm, Changsha Meyue Bio Co., Ltd, China) was introduced from ECA and advanced to the middle cerebral artery (MCA) to temporarily block the blood flow. Reperfusion was established by withdrawing the filament after 60 minutes. The incision was sutured closed and disinfected. Mice in Sham group was only made the incision without filament insertion. Animals were all kept on a warming surgery pad with a set temperature of 37 ℃ after anesthetization and surgery. sEVs (4×109 particles in 200 μl volume, 200 μg) and the same volume of PBS were intravenously administrated via the tail vein after the reperfusion, respectively. For 14 days of long-term experiments, the same dosage of sEVs was once administrated every 3 days.
Isolation of brain cortex microvessels
Brain microvessels were isolated as previously reported [34]. Briefly, after mice were sacrificed, the brain cortexs were collected and homogenated in MCDB 131 medium (Gibco), and was centrifuged at 2000g under 4°C for 5 mins. The precipitation was resuspended in 15% dextran (MW ~ 70 kDa; Sigma-Aldrich), and centrifugated at 10,000 g for 15 min under 4 °C. Consequently, the red pellets enriched with brain cortex microvessels were obtained.
Intracerebroventricular (ICV) injection
Small interfering RNA was injected intracerebroventricularly to in-vivo regulate the cerebral expressed level of Cav-1. Briefly, after anesthetized by isoflurane, mice were fixed on a stereotaxic apparatus, and a hole was drilled 1mm right lateral from the midline, and 0.34 mm behind the bregma according to The Mouse Brain in Stereotaxic Coordinates (second edition, Copyright © 200 by ACADEMIC PRESS, United States). Hamilton syringe (Hamilton Co, Reno, NV) with 33G needle was inserted 2.5 mm from the surface of the drilled skull. The prepared 4 μl Cav-1 and negative ctrl siRNA solution was injected at the rate of 0.8 μl/min. After finishing the injection, the needle was kept for 10 minutes and then withdrawn slowly. The incision was closed and disinfected, and mice were placed on the warm pad for recovery. 48 hours later the mice were subjected to tMCAo surgery and further experimental treatments. The Cav-1 and negative ctrl siRNA solution preparation and siRNA sequences were illustrated in the Supplemental Method 2.
The evaluation of BBB permeability, infarct volume, and cerebral blood flow in tMACo mice
Evans Blue assay was employed to test BBB permeability, and TTC staining was for brain infarction evaluation as previous protocol [33]. Briefly, 2% Evans Blue (4 ml/kg) was intravenously injected and allowed to circulate for 1 hour. Then mice were sacrificed by intracardiac PBS perfusion under deep anesthesia. The brains were imaged by IVIS® Spectrum small animal image system (PerkinElmer) at excitation/emission wavelength of 620 nm/710 nm. For Evans blue quantification, brain ischemic hemispheres were homogenized by 50% trichloroacetic acid, and centrifuged at 15,000 g for 15 min under 4 °C. The collected supernatant was mixed with 4 times the volume of ethanol, and the fluorescence intensity was determined by a microplate reader (SpectraMax M5) at the excitation/emission wavelength of 620 nm/710 nm. The Evans Blue standard curve was created to evaluate the Evans Blue concentration. For the assessment of brain infarct volume and edema, brain samples were sliced into 2mm sections and stained by 2% 2,3,5-Triphenyl tetrazolium chloride (TTC, Macklin) for 15 minutes at 37 °C. Brain infarct volume and swelling were calculated by using the equation described in Supplemental Method 3. The cerebral blood flow was determined and analyzed by using a laser speckle PeriCam PSI System (Perimed, Sweden), and the ipsilateral blood flow signal was compared with the contralateral site.
Neurological function assessment
The modified neurological severity score (mNSS) was employed to evaluate the neurological function deficit of tMCAo mice, and the detailed scoring standard was provided in Supplemental Method 4. For the rotarod test, all mice were trained on the rotarod (Ugo Basile, Italy) three days before the testing, and the speed was set to 10 rpm on day 1, 20 rpm on day 2, and 40 rpm on day 3. All mice were trained for 10 minutes each day. On the testing day, the acceleration program was set from 0 to 45 rpm in 2 minutes. The latency to fall and the rpm at which the mouse fell were recorded. Grip strength was recorded by using a grip strength machine (Bioseb, United States). Mice were placed on a metal grid, and by pulling the tail, the max grip strength was recorded, and the data was corrected by body weight. 3 times of measurements were recorded, and the average was determined for the final result. The mice gait analysis was performed after 14 days of the stroke onset by using the DigiGait™ imaging system (Mouse Specifics, Inc., United States). Mice were trained by running on the treadmill belt (15 cm/s) for 3 days before the experiments, and during the experiment day, the running gait parameters of left forelimb, right forelimb, left hindlimb, and right hindlimb were recorded by image system camera (150-200 frames/s), and analyzed by DigiGait™ Analysis system.
Western blotting
Protein samples from cells, sEVs and mice brain tissues were collected, and the sub-cellular protein was isolated by ProteoExtract Subcellular Proteome Extraction Kit (539790, Calbiochem), and after protein concentration quantifying by BCA assay, the samples were mixed with 5 X loading buffer, and electrophoretic separated by using the 10-12% SDS-polyacrylamide gel. Afterward, the proteins were transferred to PVDF membrane and blocked by 5% BSA. The primary and secondary antibodies were incubated with the membrane, and the protein band was imaged by using the ChemiDoc™ imaging system (Bio-Rad, United States) after incubated with ECL super sensitive luminescent reagent (Biosharp Lifescience, China). The antibodies used were listed in Supplemental Method 5. The relative protein expression levels were calculated by ImagJ software (ImageJ 1.5, NIH, United States).
Co-immunoprecipitation
Protein G-Magnetic Beads (MedChemExpress, United States) were conjugated with Anti-Cav-1 antibody or Anti-Normal rabbit IgG antibody for 2 h at 4 ℃, after washed by PBS-T (0.5% Tween-20 in PBS, pH 7.4) for 4 times, the same protein mass of samples (300 μg) in each group was incubated overnight at 4 ℃ with the antibody-conjugated beads. After washing by PBS-T, samples were heated under 95 ℃ for 5 min with loading buffer and separated by electrophoresis as western blotting.
Immunofluorescence
Mice were sacrificed by intraventricular perfusion by cold PBS slowly under deep anesthesia. The brain samples were isolated and fixed, dehydrated, then sliced into 20 μm frozen slices by a cryostat (CryoStar NX70, Thermo Fisher Scientific, United of States). Cell samples were seeded into confocal dishes and subjected to further experiments. The brain slices and cell samples were fixed by 4% PFA and permeabilized by 0.1% Triton X-100, blocked by 2.5% BSA. Primary and secondary antibodies listed in Supplemental Method 5 were incubated with samples, and then DAPI was stained for the nucleus. All samples were sealed to be imaged by a fluorescent microscope (Lecia, DMi8). The relative fluorescent intensity of the targets was calculated by ImagJ software (ImageJ 1.5, NIH, United States).
Hematoxylin-Eosin (H&E) staining
Tissues were fixed in 4% PFA overnight and then prepared for paraffin slices. Hematoxylin and Eosin were stained in slices, after washing, the slices were imaged by a light microscope.
TUNEL, Fluoro-Jade® C (FJC), Nissl and silver glycine staining
Ischemic cerebral and cultured neuronal apoptotic cells were stained by using the One Step TUNEL Apoptosis Assay Kit (Beyotime Biotechnology, China) according to the manufacturer's protocol, and the apoptotic cells were stained in green fluorescence. The relative number of TUNEL+ cells was calculated. For degradative neuron staining, the FJC staining kit (Solarbio Science & Technology Co., Ltd, China) was employed according to the manufacturer's protocol, and the degradative neurons were stained in green light. The fluorescent intensity of FJC was calculated. To determine the neuronal morphology after cerebral ischemia, brain paraffin slices were stained by Nissl staining, and silver glycine reagents (Servicebio Inc., China) respectively, and the slices were imaged by a light microscope.
siRNA and pcDNA3.1 transfection
Caveolin-1 and negative Ctrl siRNA or pcDNA3.1 plasmid were transfected to cells to manipulate the mRNA expression of target gene, and lipofectamine 3000 (Invitrogen, United States) was used for the transfection experiments according to the manufacturer's protocol. Detailed siRNA sequences and pcDNA3.1 plasmid were provided in Supplemental Method 2.
Immunoelectron microscopy
Brain samples were collected on ice and fixed by 2.5% glutaraldehyde in 4 °C for 24 h, the immunoelectron microscopy was carried out with a TEM imaging system (Hitachi) by (Service provided by Servicebio Inc., China).
Protein-protein docking
Cav-1 and LC3B interaction was predicted by using an online HDOCK server (http://hdock.phys.hust.edu.cn/) [35]. Cav-1 (PDB code: 7SC0) and LC3 (PDB code: 1UGM) crystal structure files were downloaded from Protein Data Bank (PDB, NIH, United States), the top 10 docking models were recognized and listed. The protein docking images were visualized by using PyMOL 2.5.8 software.
Statistical analysis
All data was processed by GraphPad Prism 8 Software and presented as mean ± SD. Data distribution was assessed by using Shapiro-Wilk normality test. Two-tailed independent Student's t-test was employed for comparisons between two groups, and one-way analysis of variance (ANOVA) followed by the Tukey's post-hoc-test was performed for multiple groups comparison. P value of <0.05 was determined statistically significant.
Results
BMSC-sEVs isolation and characteristics
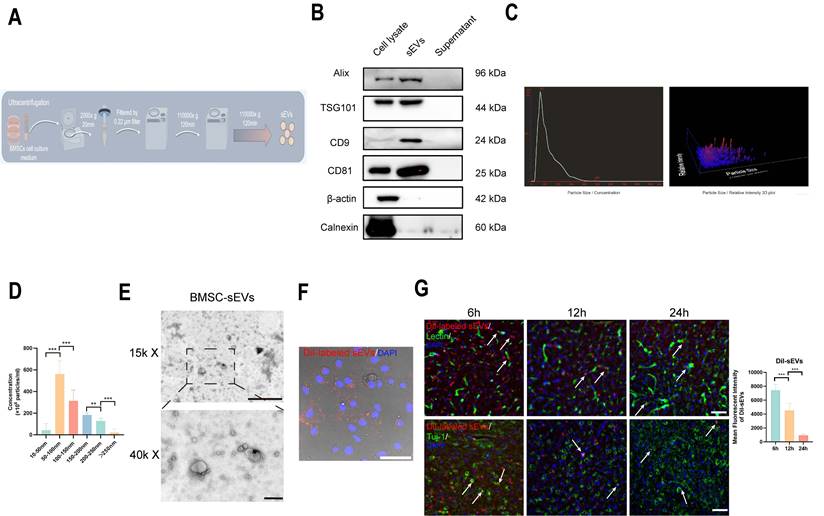
BMSC-sEVs were isolated and purified by ultracentrifugation method, the brief experimental procedure was shown in (Fig. 1A), and the sEVs typical marker Alix, TSG101, CD9, and CD81 was highly expressed in BMSC-sEVs, and their parent BMSC cells, and was absent in the supernatant of BMSC culture medium after the sEVs isolation, whereas cell cytoskeletal protein β-actin, and sEVs negative marker Calnexin were mainly expressed in BMSCs (Fig. 1B), and the NTA experiment revealed the isolated sEVs have a high concentrated peak around 50-100 nm (Fig. 1C-D), which was a typical sEVs size range [36]. Furthermore, the TEM image of sEVs displayed that the isolated sEVs had a typical cup shape (Fig. 1E), and the DiI-labeled sEVs were phagocytosed by brain endothelial cells (Fig. 1F). Moreover, we administrated DiI-labeled sEVs to tMCAo mice, and the DiI-labeled sEVs could both be reserved in Lectin- labeled brain vessels, and in brain parenchyma beyond vessels from 6 to 24h after sEVs injection, and gradually vanished over time (Fig. 1G). Additionally, DiI-labeled sEVs can retained within 24h in Tuj-1-labeled neurons (Fig. 1G). The above results indicated the isolated sEVs kept a nice phenotype and function.
The isolation and identification of BMSC-sEVs. (A) The scheme of sEVs isolation and purification (By Figdraw). (B) Western blotting of sEVs markers in original cells, sEVs, and supernatant after sEVs isolation samples. (C) The NTA analysis of BMSC-sEVs. (D) The size distribution of BMSC-sEVs (n=3). (E) The TEM image of BMSC-sEVs scale bar above: 1 μm, scale bar below: 200 nm. (F) DiI-labeled sEVs phagocytosis by b. End3 cells. Scale bar: 50 μm. (G) DiI-labeled sEVs retention in tMCAo mice brain parenchyma and neurons from 6 to 24h after administration. (Indicated by white arrow) ***P < 0.001, **P < 0.01.
BMSC-sEVs treatment alleviated brain infarct volume and neurological function deficit after cerebral ischemia
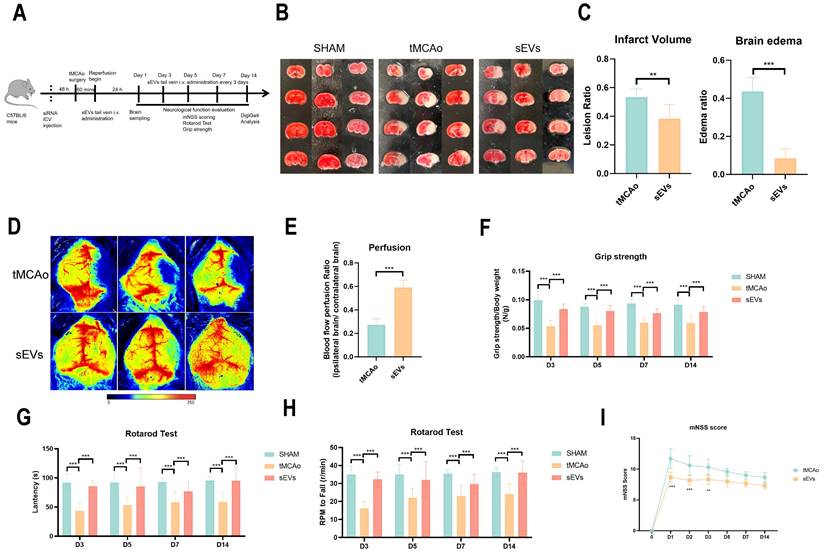
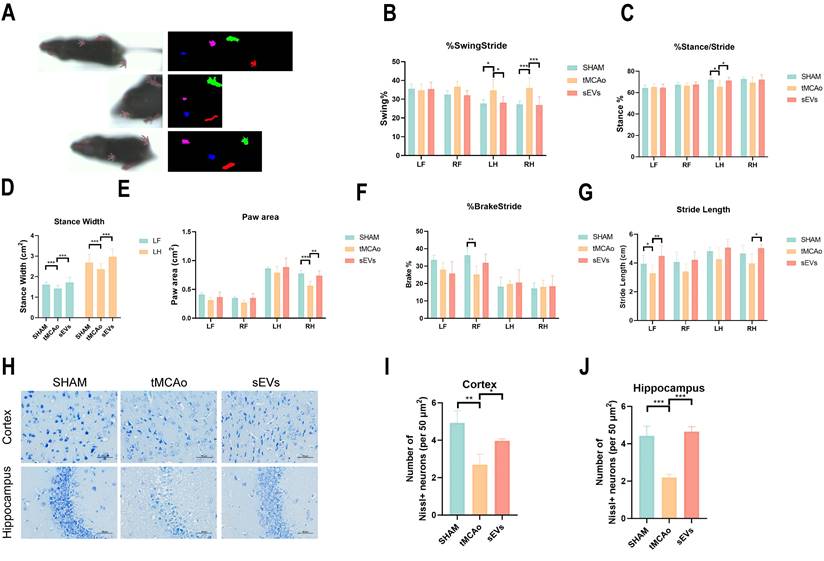
To illustrate BMSC-sEVs therapeutic efficacies, we administrated BMSC-sEVs to tMCAo mice for 14 days treatment after cerebral ischemia onset (Fig. 2A). The results indicated sEVs treatment significantly decreased the brain infarct volume and swelling after 24 hours tMCAo (Fig. 2B-C). Meanwhile, the cerebral blood flow determined by laser speckle imaging was highly recovered in sEVs-treated tMCAo mice (Fig. 2D-E). Inspiringly, in the evaluation of the long-term neurological function in tMCAo mice, BMSC-sEVs treatment obviously enhanced the grip strength, and the performance in the rotarod test, together with the better mNSS scoring during 14 days of cerebral ischemia (Fig. 2F-I). To systematically assess the motor nerve function of tMCAo mice by sEVs treatment, we utilized the DigiGait™ imaging system to analyze the gait abnormality of mice at day 14. The tMCAo mice displayed abnormal gait when compared with the sham group, and sEVs treatment recovered the overall mice gait performance (Fig. 3A). In detail, the abnormality of the left hind limb and right hind limb swing percentage, left hind limb stance percentage, left fore and hind limb stance width, right hind limb paw area, and left fore and right hind limb stride length in tMCAo mice were significantly recovered in sEVs group, however, there is no significant recovery effect by sEVs treatment in brake percentage parament in all limbs (Fig. 3B-G). We further applied nissl staining in mice brain slices to characterize neurons in the cortex and hippocampus, and the number of nissl body-positive neurons was deficient, whereas partly restored in the sEVs treatment group (Fig. 3H-J). Similarly, the silver glycine staining results showed consistency with those of nissl staining (Supplemental Fig. 1). Additionally, we also utilized H&E staining for the hearts, livers, spleens, lungs, and kidneys after sEVs administration in tMCAo mice after 14 days, and it showed there was no obvious occurrence of pathological damages (Supplemental Fig. 2).
BMSC-sEVs treatment reduced tMCAo insulted mice brain infarct volume and BBB leakage together with neurological function performance. (A) The experimental scheme of animal experiments (B-C) TTC staining and quantification of brain infarct volume and edema of SHAM and tMCAo mice treated with sEVs (n=6). (D-E) Blood flow perfusion of tMCAo mice treated with sEVs by laser speckle (n=6). (F) Grip strength tests of SHAM and tMCAo mice treated with sEVs from 3 to 14 days after surgery (n=6-12). (G-H) Rotarod tests of SHAM and tMCAo mice treated with sEVs from 3 to 14 days after surgery (n=6). (I) mNSS scoring of tMCAo mice treated with sEVs from 1 to 14 days after surgery (n=6-13). ***P < 0.001, **P < 0.01.
BMSC-sEVs treatment improved the long-term neurological function of tMCAo mice. (A) DigiGait recorded paw print characters of mice in different groups. (B-G) Gait parameters analyzed by the DigiGait system on the 14-day post-surgery. LF: Left forelimb, RF: Right forelimb, LH: Left hindlimb, RH: Right hindlimb. (H-J) Nissl staining and quantification in brain cortex and hippocampus of tMCAo mice treated with sEVs (n=3). Scale bar: 50 μm. ***P < 0.001, **P < 0.01, *P < 0.05.
BMSC-sEVs treatment attenuated neuronal injury
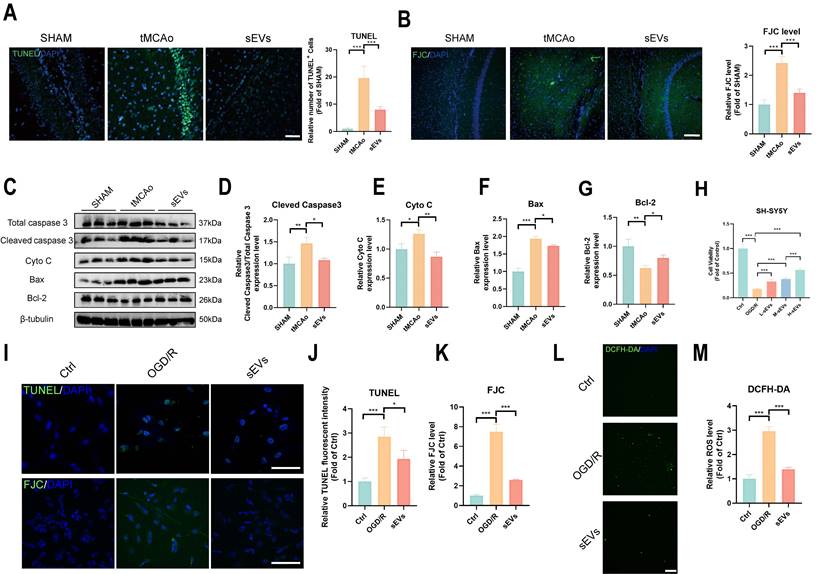
Furthermore, we investigated the neural protection by BMSC-sEVs treatment. The increased TUNEL-positive apoptotic cells and FJC-labeled degradative neurons were both inhibited by sEVs treatment in tMCAo mice (Fig. 4A-B). The pro-apoptosis protein cleaved caspase 3, Bax, and cytochrome c were downregulated, and the anti-apoptosis protein Bcl-2 was upregulated after sEVs treatment (Fig. 4C-G). Moreover, we treated OGD/R insulted neuronal cell lines SH-SY5Y with low, medium, and high dosages of sEVs, and figured out that the sEVs treatment significantly recovered the neuron viability in a dose-dependent manner (Fig. 4H). The high dosage of sEVs treatment obviously decreased the number of apoptotic neurons marked by TUNEL staining and the neuron degeneration by FJC staining in OGD/R-insulted neurons (Fig.4I-K). At the same time, the high ROS production was also suppressed by sEVs therapeutic (Fig. 4L-M).
BMSC-sEVs treatment attenuated neuronal apoptosis after cerebral ischemia. (A) TUNEL staining and quantification of mice brain slices in different groups (n=3). Scale bar: 50 μm. (B) FJC staining and quantification of mice brain slices in different groups (n=3) Scale bar: 100 μm. (C-G) Western blotting of Caspase3, CytoC, Bax and Bcl-2 expression in tMCAo mice brain samples and quantification (n=3). (H) Cell viability of SH-SHY5Y cells after sEVs treatments by CCK-8 assay (n=6). (I-K) TUNEL and FJC staining and quantification of SH-SY5Y cells after OGD/R and sEVs treatment (n=4) Scale bar: 100 μm. (L-M) DCFH-DA labeled ROS activity and quantification in SH-SY5Y cells after OGD/R and sEVs treatment (n=4). Scale bar: 200 μm. ***P < 0.001, **P < 0.01, *P < 0.05.
BMSC-sEVs treatment decreased BBB permeability and upregulated tight-junction proteins expressions
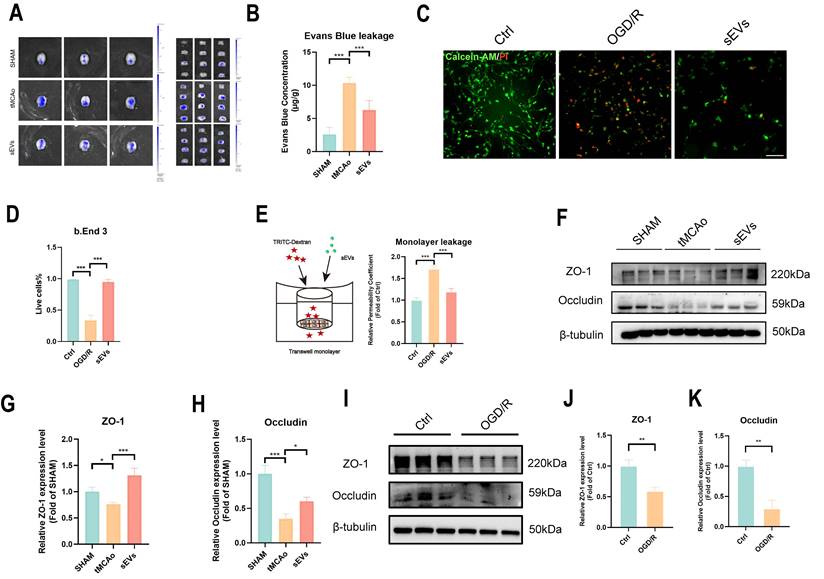
In tMCAo mice, the drastic rise of Evans Blue across-BBB leakage was hindered after sEVs treatment (Fig. 5A-B). In addition, we imaged and examined the viability of brain endothelial cells subjected to OGD/R, and the result suggested that the treatment of sEVs enhanced the live cells percentage (Fig. 5C-D). The barrier permeability of the brain endothelial cells monolayer after OGD/R was evaluated by using transwell assay, and the sEVs treatment significantly restrained the upregulated monolayer barrier leakage (Fig.5E). In addition, we evaluated the tight junction protein ZO-1 and Occudin expression in brain microvessels isolated from tMCAo mice, and it showed the expressions of ZO-1 and Occludin were both downregulated, which were reversed in sEVs administrated group 24 hours after modeling (Fig 5F-H). In vitro, it suggested that tight junction proteins ZO-1 and Occludin expressions were decreased after OGD/R insult (Fig. 5I-K). and the treatment of sEVs also uplifted the expression levels of these two tight junction proteins, dose-dependently in OGD/R insulted brain endothelial cells (Fig. 6A-C). The immunofluorescent staining was in compliance with the western blotting results (Fig. 6D-G).
BMSC-sEVs treatment harnessed BBB leakage and recovered tight junction proteins in tMCAo mice. (A-B) Evans blue leakage of SHAM and tMCAo mice treated with sEVs (n=6). (C-D) Live/dead cell staining of b. End3 cells and quantification (n=5) Scale bar: 50 μm. (E) Transwell assay of brain endothelial cells monolayer permeability analysis (n=4). (F-H) Western blotting of ZO-1 and Occludin expressions in tMCAo mice brain microvessel and quantification (n=3). (I-K) Western blotting of ZO-1 and Occludin expression in b. End3 cells after OGD/R and quantification (n=3). ***P < 0.001, **P < 0.01, *P < 0.05.
BMSC-sEVs treatment improved tight junction proteins expression in OGD/R insulted b. End3 cells. (A-C) Western blotting of ZO-1 and Occludin expression in b. End3 cells after OGD/R and sEVs treatment and quantification (n=3). (D-G) Immunofluorescence and quantification of ZO-1 and Occludin in brain endothelial cells after OGD/R and sEVs treatment (n=4) Scale bar: 50 μm; 2.5 μm in zoomed image. ***P < 0.001, **P < 0.01, *P < 0.05.
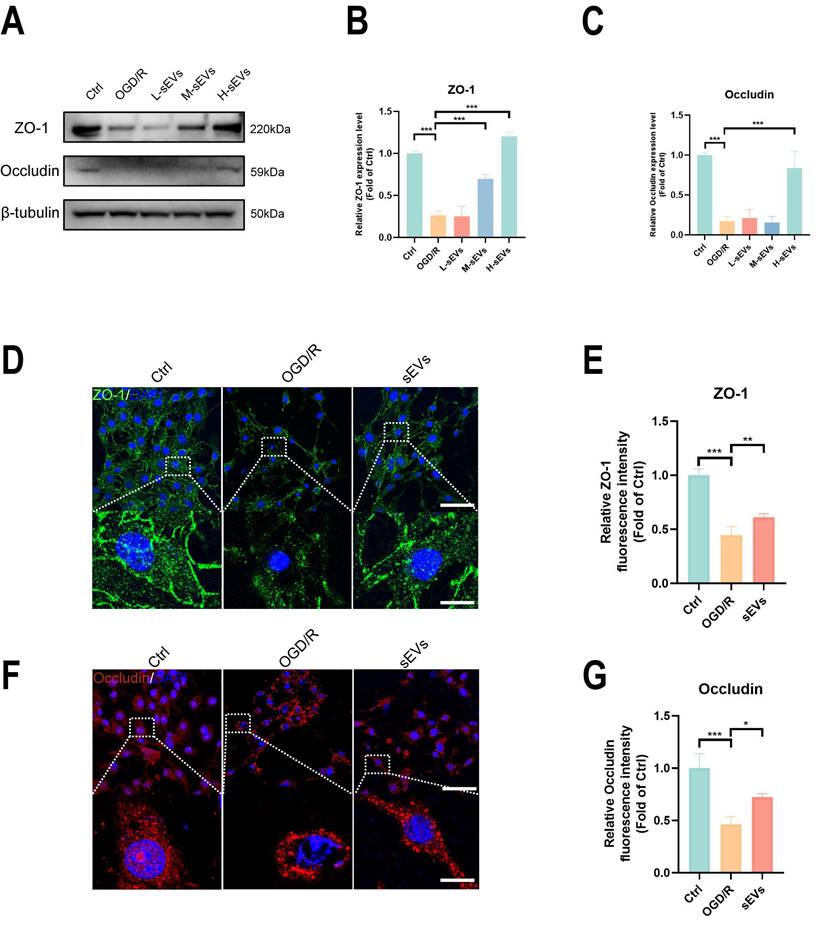
BMSC-sEVs treatment suppressed the autophagy of brain endothelial cells against the degradation of ZO-1 and Occludin
Autophagy is an important pathway for protein clearance and has been proven to damage tight junctions after cerebral ischemia [20, 37]. To illustrate the influence of brain endothelial cells autophagy on BBB integrity and verify whether sEVs therapeutic regulated this pathological abnormality, we examined the autophagosome protein LC3B and autophagic degradation substrates p62 in vivo and in vitro. In tMCAo modeled mice brain microvessels. LC3B and p62 expressions indicated the activation of microvascular autophagy, and all these trends were reversed in sEVs administrated group after 24 hours modeling (Fig 7A-C). In vitro, the western blotting results revealed that LC3B II expression was upregulated after OGD/R, and in contrast, p62 was downregulated in brain endothelial cells, and interestingly, these trends of enhanced autophagy can be reversed by sEVs treatment (Fig. 7D-F). The high dosage of sEVs significantly reduced the LC3B II expression, whereas low, medium and high dosage sEVs all intensified the p62 expression (Fig. 7D-F).
BMSC-sEVs inhibited brain endothelial cells' autophagy flux in-vitro. (A-C) Western blotting of LC3B, and p62 expressions in tMCAo mice brain microvessel and quantification (n=3). (D-F) Western blotting of LC3B and p62 expression in b. End3 cells after OGD/R and quantification (n=3). (G-H) Western blotting of LC3B, p62, and ZO-1/Occludin expression in b. End3 cells after OGD/R co-treated with bafA1 and quantification (n=3). (I-J) Autophagy flux visualized by GFP-RFP-LC3B transfection in b. End3 cells after OGD/R treated with sEVs and rapamycin (n=6) Scale bar: 20 μm. ***P < 0.001, **P < 0.01, *P < 0.05.
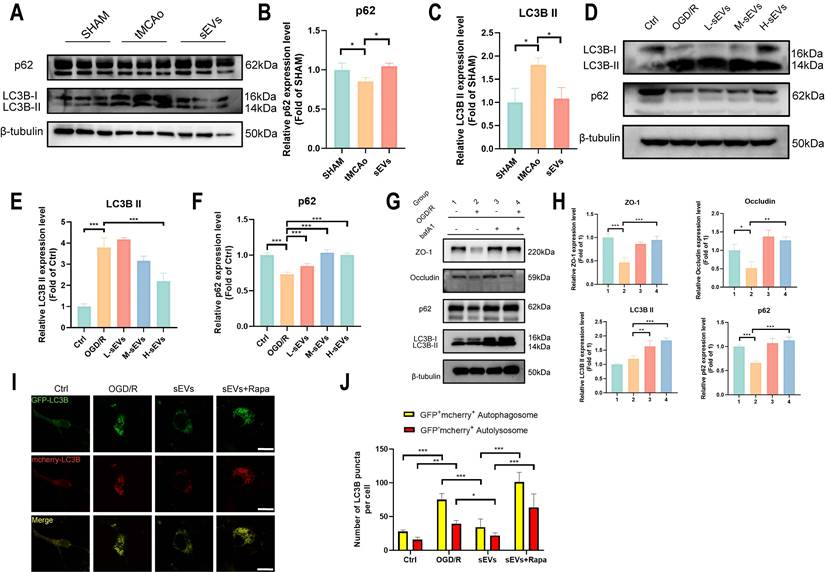
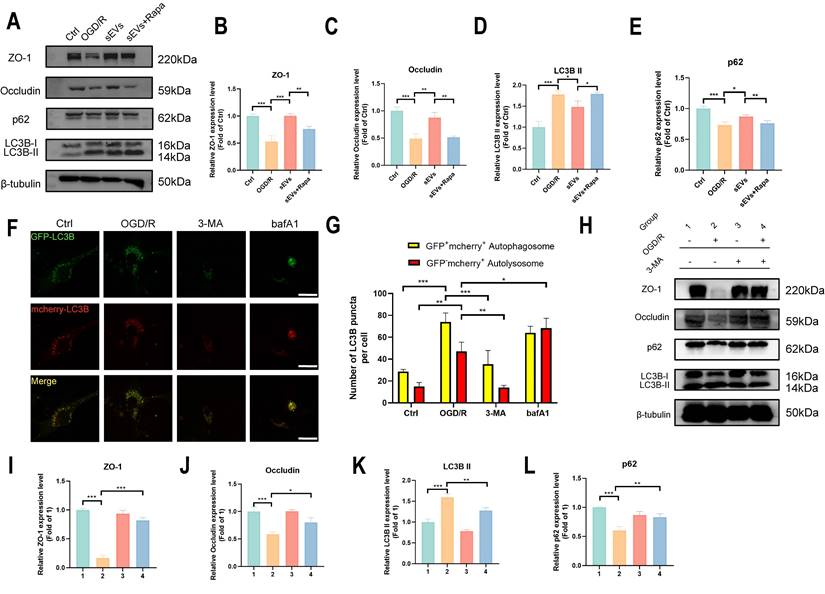
Subsequently, we investigated if the degradation of ZO-1 and Occludin was autophagy-lysosomal pathway dependent. We applied an autolysosome blocker bafA1, and tested the ZO-1 and Occludin, together with autophagy indicator LC3B and p62 expressions after OGD/R in brain endothelial cells. A restorative trend of ZO-1 and Occludin was observed in bafA1 treating group after OGD/R, together with an upregulation of LC3B II and p62, indicating inhibiting autolysosome pathway rescued the tight junction proteins degradation after OGD/R injury (Fig. 7G-H). Furthermore, the enhanced autophagy flux after OGD/R observed by using GFP-mcherry-LC3B plasmid transfection was remarkably reversed by sEVs treatment, whereas the therapeutic effect was abolished by autophagy enhancer rapamycin administration (Fig. 7I-J). Similarly, the immunofluorescence of lysosome marker LAMP-1 was observed after OGD/R and reduced by sEVs treatment (Supplemental Fig. 3). The western blotting results also indicated the enhanced efficacy of sEVs treatment on OGD/R insulted endothelial tight junction proteins expression was compromised after the cells were administered rapamycin, additionally, and the rise of LC3B II and the decline of p62 simultaneously occurred (Fig. 8A-E). To extensively illustrate the role of early-phase autophagy on ZO-1 and Occludin, we utilized an early-phase autophagy inhibitor 3-MA and lysosome inhibitor bafA1 to OGD/R induced brain endothelial cells, and the autophagy flux test displayed a sharp decrease of both red and yellow puncta number after 3-MA intervention, whereas an upregulation of both fluorescence puncta was observed after bafA1 treatment (Fig. 8F-G). The results indicated the successful manipulation of autophagy by two different autophagy inhibitors. Interestingly, the 3-MA treatment inhibited the LC3B II expression, restored the p62 expression, and at the same time significantly enhanced the ZO-1 and Occludin expressions in OGD/R insulted brain endothelial cells (Fig. 8H-L). The autophagy inhibitors demonstrated similar therapeutic effects on tight junction proteins and autophagy compared to sEVs treatment. Collectively, the results demonstrated the sEVs treatment intervened with the autophagy-lysosome dependent ZO-1 and Occludin degradation.
Autophagy is essential for ZO-1 and Occludin degradation after cerebral ischemia. (A-E) Western blotting of ZO-1/Occludin, LC3B and p62 expression in b. End3 cells treated with sEVs and rapamycin after OGD/R and quantification (n=3). (F-G) Autophagy flux visualized by GFP-RFP-LC3B transfection in b. End3 cells after OGD/R treated with 3-MA and bafA1 (n=6) Scale bar: 20 μm. (H-L) Western blotting of ZO-1/Occludin, LC3B and p62 expression in b. End3 cells treated with 3-MA after OGD/R and quantification (n=3). ***P < 0.001, **P < 0.01, *P < 0.05.
BMSC-sEVs treatment regulated Cav-1-dependent autophagic degradation of ZO-1 and Occludin
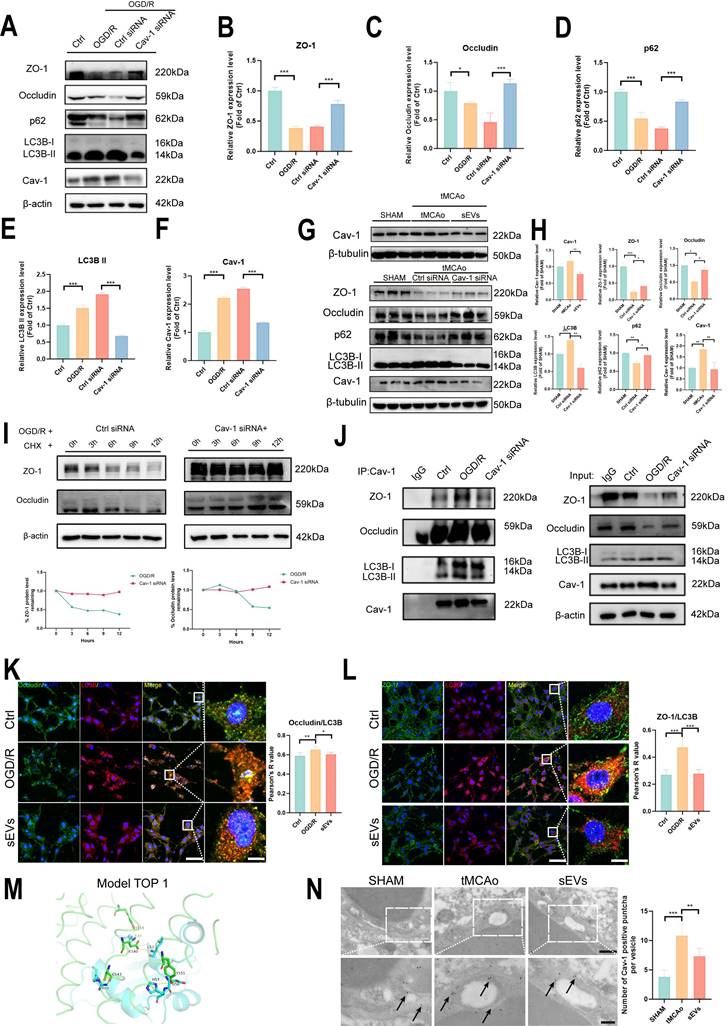
Lastly, we questioned in which pathway BMSC-sEVs mediated autophagy-dependent ZO-1 and Occludin degradation and focused on Cav-1 and its relationship with autophagy. We employed Cav-1 siRNA to knock down the Cav-1 expression in-vitro. The effectiveness of our designed Cav-1 siRNA sequences was presented as the Cav-1 significant knockdown by western blotting, and the siRNA FAM label was positive in brain endothelial after 48 h after transfection (Supplemental Fig. 4). In accordance with the knockdown of Cav-1 in OGD/R-subjected brain endothelial cells, the ZO-1 and Occludin were both recovered, and significant inhibition of autophagy was observed. The result indicated that Cav-1 itself regulated the autophagy process and affected the expression of tight junction proteins (Fig. 9A-F). Subsequently, in-vivo experiment, the increased Cav-1 expression was verified in tMCAo mice brain microvessels and downregulated by sEVs treatment (Fig. 9G-H), similarly, in-vitro OGD induced expression upregulation of Cav-1 in b. End3 cells were decreased dose-dependently after sEVs treatment, especially by the high dosage sEVs (Supplemental Fig. 5). Furthermore, we injected the cholesterol-modified Cav-1 siRNA by ICV approach and examined the tight junction and autophagy proteins expression in ischemic mice brain microvessels after 24 h cerebral ischemia. The increased Cav-1 expression showed a decline in the Cav-1 siRNA administrated tMCAo mice group, when compared with the Ctrl siRNA group, accompanied by a significant restoration of ZO-1 and Occludin, and inhibition of autophagy (Fig. 9G-H). To confirm the degradation of ZO-1 and Occludin was by Cav-1 dependent manner, we applied cycloheximide (CHX) chase assay, and the knockdown of Cav-1 in OGD/R insulted brain endothelial cells. The expressions of ZO-1 and Occludin were gradually decreased over time after CHX treatment, whereas Cav-1 siRNA transfection abolished the declining trend of these two tight junction proteins expression (Fig. 9I). The result indicated that Cav-1 was responsible for ZO-1 and Occludin degradation via the non-proteasomal pathway. Consequently, in order to verify whether sEVs treatment intervened the crosstalk among Cav-1, LC3B, and tight junction proteins, Co-ip experiment validated that the interaction between Cav-1 with ZO-1/Occludin and LC3B was enhanced after OGD/R, whereas downregulated by Cav-1 knockdown (Fig. 9J), and the immunofluorescence results indicated the co-localization between ZO-1/Occludin and LC3B was dramatically upregulated after OGD/R in brain endothelial cells, whereas partly abolished by sEVs treatment (Fig. 9K-L). As we proved the strong autophagy regulation by Cav-1, we next demonstrated the crosstalk between Cav-1 and LC3B. The subcellular protein distribution experiment presented that, after OGD/R, the co-distributions of ZO-1/Occludin, Cav-1, and LC3B were obviously decreased in cytoskeleton fraction, however, accumulated in membrane and cytoplasm fractions, interestingly, this abnormal trend was reversed by sEVs treatment in brain endothelial cells (Supplemental Fig. 6). The in-vitro co-localization of Cav-1 and LC3B was also observed after OGD/R and rescued by sEVs (Supplemental Fig. 7). Next, the binding of Cav-1 and LC3B was predicted by using HDOCK, and the Top 1 binding model was presented (Fig. 9M). All the Top 10 binding models, the docking confidence, and scores were all listed in (Supplemental Fig. 8). In addition, we overexpressed Cav-1 in b.End3 cells after OGD/R and sEVs treatment. The Cav-1 artificial upregulation partly abolished the sEVs' recovery on ZO-1 and Occludin (Supplemental Fig. 9). Furthermore, the number of Cav-1 labeled puncta located in brain endothelial Caveolae vesicles imaged by immunoelectron microscopy was significantly upregulated in tMCAo mice, and the density of the puncta was relatively lower in sEVs administrated mice group (Fig. 9N). All our results suggested that BMSC-sEVs could regulate Cav-1-mediated autophagic degradation of ZO-1 and Occludin after cerebral ischemia.
BMSC-sEVs treatment regulated Cav-1 mediated ZO-1 and Occludin autophagic degradation after cerebral ischemia. (A-F) Western blotting and quantification of ZO-1/Occludin, LC3B, p62, and Cav-1 expression in b. End3 cells after OGD/R and Cav-1 siRNA transfection (n=3). (G-H) Western blotting of Cav-1 and ZO-1/Occludin, LC3B, and p62 expression in brain microvessels of tMCAo mice treated by sEVs or ICV injected Cav-1 siRNA (n=3). (I) CHX assay for ZO-1 and Occludin degradation after Cav-1 siRNA transfection in OGD/R insulted b. End3 cells. (J) Co-ip for verifying the interaction between Cav-1 and ZO-1/Occludin or LC3B. (K) Immunofluorescence and Pearson's R co-localization analysis of Occludin and LC3B in b. End3 cells after OGD/R and sEVs treatment (n=4). Scale bar: 50 μm, and 25 μm in zoomed images. (L) Immunofluorescence and Pearson's R co-localization analysis of ZO-1 and LC3B in b. End3 cells after OGD/R and sEVs treatment (n=4). Scale bar: 50 μm, and 25 μm in zoomed images. (M) Protein-protein docking top 1 model of LC3B (Blue) and Cav-1 (Green). (N) Immunoelectron microscopy image of Cav-1 in mice brain microvessels parts, and the quantification of the Cac-1 positive puncta (black arrow indicated) (n=6) Scale bar: 500 nm and 200 nm in zoomed images. ***P < 0.001, **P < 0.01, *P < 0.05.
Discussion
In the current study, we utilized in-vitro OGD insulted brain endothelial cells and neurons together with an in-vivo tMCAo mice model to demonstrate that BMSC-sEVs treatment effectively restored tight junction protein ZO-1 and Occludin expression levels together with protecting the injured neurons after cerebral ischemia, which contributed to the long-term neurological function amelioration in tMCAo mice. By employing different phase autophagy regulators and Cav-1 siRNA and pcDNA3.1 plasmid, we verified that the therapeutic efficacy of BMSC-sEVs on BBB integrity was Caveolin-1-dependent autophagic degradation of ZO-1 and Occludin after ischemic stroke.
Autophagy is a comprehensive biological cell self-eating progress, and it has multiple roles after ischemic stroke. After cerebral ischemia, autophagy can be an adaptive process for deficient organelles and protein clearance, thus maintaining homeostasis, and facilitating recovery, while on the other hand, uncontrolled excessive autophagy contributes to no benefit but, the activation of endoplasmic reticulum (ER) stress, accumulation of ROS, and mitochondrial deficit can lag restoration and damage of cerebral cells [38, 39]. Not only in neurons, but autophagy in brain endothelial cells is also important for their viability, especially affecting BBB permeability [38]. The over-activated autophagy degrades tight junction proteins, directly intensifies the BBB permeability after ischemic stroke, and worsens the prognosis [19]. Developing effective therapeutic interventions to regulate the over-activated autophagy after ischemic stroke can be a novel and promising way for ischemic stroke treatment.
sEVs have been extensively documented to treat ischemic stroke via regulating excessive neuronal autophagy after ischemic stroke. For instance, human-induced pluripotent stem cells derived sEVs inhibited autophagy via STAT3 activation in MCAo-induced rats, improving rats' cerebral angiogenesis and long-term neurological function after cerebral ischemia [40]. Similarly, adipose-derived stem cells inhibited neuronal autophagy in cerebral ischemia mice via miR-25 dependent pathway, which exerted neuroprotection [41]. For brain endothelial cells' autophagy regulation, healthy rat serum-originated sEVs downregulated brain endothelial autophagy in MCAo rat, and OGD modeled b. End3 cells, further mediated BBB tight junction protection after ischemic stroke [42]. Apart from autophagy regulation, sEVs treatment can directly compose BBB protection, apoptosis inhibition and inflammation suppressing, and promoting angio-neurogenesis after ischemic stroke [15, 43]. Compared with traditional stem cell transplantation stroke therapeutics and neuroactive agents, sEVs can cross BBB, and showed low immunogenicity and a relatively low risk of thrombosis [43, 44] which provided a novel solution for stroke treatment. However, the detailed mechanism of how sEVs affect tight junction disruption via autophagy is unclear.
Physiologically, tight junction proteins remain in the plasma membrane to maintain endothelial barrier integrity, and under stimulus, the internalized tight junction proteins were subjected to complex endosomal sorting pathways for recycling or degradation [45], Caveolae-mediated internalization of tight junction proteins were previously reported as one of the predominant endocytosis pathway in brain endothelial cells [25, 45, 46], and the Cav-1 phosphorylation mediated the generation of caveolae, and driven the endocytosis [47, 48]. The endocytosed tight junction proteins were then subjected to caveosomes or endosome-dependent sorting and degraded by lysosome or recycled to the membrane [45, 49]. Cav-1 was previously reported to translocate ZO-1 and Occludin, intensify the BBB permeability [20, 21, 25], and our previous study also illustrated Cav-1 regulated ZO-1 and Claudin-5 by endocytosis after ischemic stroke [23]. The degradation pathways of tight junction proteins are mainly divided into two parts, one is ubiquitin-dependent pathway, after tight junction proteins ubiquitination, they can be degraded by proteasome or lysosome, and Claudin-5 was reported to be degraded via ubiquitin-proteasome routine [45, 50]. Another important routine is the autophagy-lysosome pathway, the autophagosome wraps endocytosed tight junction proteins, and fuses with the lysosome to accomplish the degradation, for example, Claudin-2, Occludin and ZO-1 were reported to be degraded via autophagy in intestinal epithelial barrier [51-53]. And in BBB damage after ischemic stroke, ZO-1 and Claudin-5 were both documented to be degraded via autophagy-lysosome [21, 22], which were in accord with our finding to find ZO-1 and Occludin were damaged via autophagy-lysosome pathway after ischemic stroke.
On the other hand, Cav-1 involves multiple pathways related to BBB permeability, neuron survival, apoptosis, and autophagy after ischemic stroke [26, 54, 55], and we also revealed that the Cav-1/CD147/VEGFR2/MMP pathway was responsible for BBB disruption and rat neurological function deficit after cerebral ischemia [33]. The phosphorylation of Cav-1 bound with BECN1 and thus activated autophagy after cerebral ischemia [56]. However, the effect of Cav-1 on autophagy is controversial, in other non-cerebral diseases, Cav-1 was also reported to inhibit autophagy [29, 30, 57]. Based on previous work, we endeavored to explore whether Cav-1 not only internalized ZO-1 and Occludin on membrane of brain endothelial cells, but also affected their downstream autolysosome degradation.
Our results firstly revealed that BMSC-derived sEVs inhibited the degradation of tight junction protein ZO-1 and Occludin in brain endothelial cells through the autophagy-lysosomal pathway. We applied 3-MA, a PI3K inhibitor to inhibit early-phased autophagy, and bafA1, a lysosome inhibitor to suppress late-phased autolysosome. The two kinds of inhibitors protected ZO-1 and Occludin in OGD/R insulted brain endothelial cells with the two different autophagy inhibition characters. The Cav-1 knockdown applied in vitro and in vivo significantly inhibited autophagy and restored tight junction proteins. Using CHX assay, co-ip, co-localization analysis, HDOCK protein-protein docking, and Immunoelectron microscopy, we extensively proved ZO-1 and Occludin closely interact with Cav-1 and deteriorated through the autophagy pathway. Contrary to our findings, other previous studies reported that Cav-1 mediated tight junction protein Claudin-5 endocytosis and then degradation via autophagy, notably prevented the accumulation of damaged Claudin-5 in the cytoplasm and benefitted cerebral ischemia recovery [37], and in intestinal epithelial barrier inflammatory model, autophagy is necessary to maintain barrier function via inhibiting the loss of tight junction protein Occludin [52]. Different tight junction proteins and their functions, and different experimental models can explain the possible reasons for these opposite conclusions. In addition, the tight junctions should be right-functioned in the cytoskeleton, their accumulation in the cytoplasm can be harmful to cell survival, and that's why autophagy-lysosome is essential to clear them out. In our study, we employed subcellular protein isolation assay and proved ZO-1 and Occludin detached from the cytoskeleton to membrane and cytoplasm after OGD/R treatment, however, the sEVs treatment reversed these trends. The sEVs treatment not only restored the ZO-1/Occludin expression level but also maintained their right subcellular location. Regarding the in-depth mechanism of how sEVs treatment can regulate the autophagy pathway and the degradation of tight junction proteins, we supposed non-coding RNAs especially microRNAs (miRNA) or circularRNAs (circRNA) are vital for their discovered therapeutic effects. Non-coding RNAs are essential for transcriptional and post-transcriptional gene expression regulation [58], consequently regulating various biological processes and diseases. In cerebral ischemia, sEVs-derived non-coding RNAs have been broadly documented to mediate acute ischemic stroke protection and long-term function recovery [59, 60]. Particularly, sEVs-derived non-coding RNAs can regulate autophagy and intervene in the progression of central neural system injury diseases, including ischemic stroke [61]. Kuang et, al. reported that adipose-derived mesenchymal stem cells can transfer miR-25-3p to inhibit neuron autophagy to activate neuron production in OGD-induced primary neurons and MCAo-induced mice model [41]. Additionally, miR-133a-3p from BMSCs can reduce rat cerebral ischemia injury by intervening in neuron apoptosis and autophagy [62]. Similarly, neuron-derived miR-21-5p enriched exosomes can promote neuroplasticity in traumatic brain injury mice model [63]. Based on the current knowledge of sEVs treatment on autophagy after acute cerebral ischemia, we supposed that the autophagy inhibition on brain endothelial cells after stroke by sEVs is mediated by dozens of sEVs derived non-coding RNAs. Additional studies should be conducted to define the non-coding RNA or proteomic in BMSC-sEVs and bridge the better understanding of BMSC-sEVs therapeutic on BBB and tight junction proteins. Moreover, it should be highlighted that we proved autophagy is responsible for BBB disruption in our experimental conditions, and further studies are required to clarify the role of autophagy in different pathological stages of ischemic stroke, and autophagy outcomes in different cerebral cells subjected to ischemia. Different stroke models also matter on the pathologic mechanism and therapeutic evaluation [64], further studies should test BMSC-sEVs therapeutic and the autophagy regulation pathways in more different stroke models such as permeant MCAo models or photothrombotic models. Besides, before sEVs treatment studies are widely applied to clinics, long-term safety information should be acquired, although we tested sEVs safety in 14 days by organs H&E staining during our study, a longer period of safety verification should be performed. Also, clinical samples from stroke patients should be tested in the study for the mechanism confirmation, which further benefits the translation of sEVs application in clinical practice.
In summary, our work firstly provides a novel BMSC-sEVs therapeutic on alleviating BBB disruption post-ischemic stroke and defines the mechanism is related to Cav-1 mediated autophagic degradation of tight junction protein ZO-1 and Occludin, contributing to the long-term neurological function amelioration.
Supplementary Material
Supplementary methods and figures.
Acknowledgements
We thank the technical and resource support from Bing Wang's group and Di Mu's group at the Guangdong Institute of Intelligent Science and Technology, and group members from Dr. Huanxing Su and Dr. Jiahong Lu's group at the University of Macau.
Funding
This work is supported by the National Natural Science Foundation of China (NSFC No. 82074051), Guangdong Natural Science Fund (General Programme, no. 2023A1515010034), and the University of Macau (MYRG2022-00221-ICMS).
Availability of data and materials
All datasets that support the study are available from the corresponding author on reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Saini V, Guada L, Yavagal DR. Global Epidemiology of Stroke and Access to Acute Ischemic Stroke Interventions. Neurology. 2021;97:S6-s16
2. Emberson J, Lees KR, Lyden P, Blackwell L, Albers G, Bluhmki E. et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet. 2014;384:1929-35
3. Tsivgoulis G, Katsanos AH, Sandset EC, Turc G, Nguyen TN, Bivard A. et al. Thrombolysis for acute ischaemic stroke: current status and future perspectives. Lancet Neurol. 2023;22:418-29
4. Berge E, Whiteley W, Audebert H, De Marchis GM, Fonseca AC, Padiglioni C. et al. European Stroke Organisation (ESO) guidelines on intravenous thrombolysis for acute ischaemic stroke. Eur Stroke J. 2021;6:I-lxii
5. Campbell BCV, Ma H, Ringleb PA, Parsons MW, Churilov L, Bendszus M. et al. Extending thrombolysis to 4·5-9 h and wake-up stroke using perfusion imaging: a systematic review and meta-analysis of individual patient data. Lancet. 2019;394:139-47
6. Kleindorfer D, de los Rios La Rosa F, Khatri P, Kissela B, Mackey J, Adeoye O. Temporal trends in acute stroke management. Stroke. 2013;44:S129-31
7. Mathews MS, Sharma J, Snyder KV, Natarajan SK, Siddiqui AH, Hopkins LN. et al. Safety, effectiveness, and practicality of endovascular therapy within the first 3 hours of acute ischemic stroke onset. Neurosurgery. 2009;65:860-5 discussion 5
8. Wolpert SM, Bruckmann H, Greenlee R, Wechsler L, Pessin MS, del Zoppo GJ. Neuroradiologic evaluation of patients with acute stroke treated with recombinant tissue plasminogen activator. The rt-PA Acute Stroke Study Group. AJNR Am J Neuroradiol. 1993;14:3-13
9. Turc G, Bhogal P, Fischer U, Khatri P, Lobotesis K, Mazighi M. et al. European Stroke Organisation (ESO) - European Society for Minimally Invasive Neurological Therapy (ESMINT) Guidelines on Mechanical Thrombectomy in Acute Ischemic Stroke. J Neurointerv Surg. 2023;15:e8
10. Chen HS, Cui Y, Zhou ZH, Zhang H, Wang LX, Wang WZ. et al. Dual Antiplatelet Therapy vs Alteplase for Patients With Minor Nondisabling Acute Ischemic Stroke: The ARAMIS Randomized Clinical Trial. JAMA. 2023;329:2135-44
11. Li Y, Cheng Q, Hu G, Deng T, Wang Q, Zhou J. et al. Extracellular vesicles in mesenchymal stromal cells: A novel therapeutic strategy for stroke. Exp Ther Med. 2018;15:4067-79
12. Rajendran L, Bali J, Barr MM, Court FA, Krämer-Albers EM, Picou F. et al. Emerging roles of extracellular vesicles in the nervous system. J Neurosci. 2014;34:15482-9
13. Boltze J, Arnold A, Walczak P, Jolkkonen J, Cui L, Wagner DC. The Dark Side of the Force - Constraints and Complications of Cell Therapies for Stroke. Front Neurol. 2015;6:155
14. Cai Y, Liu W, Lian L, Xu Y, Bai X, Xu S. et al. Stroke treatment: Is exosome therapy superior to stem cell therapy? Biochimie. 2020;179:190-204
15. Zhang ZG, Buller B, Chopp M. Exosomes - beyond stem cells for restorative therapy in stroke and neurological injury. Nat Rev Neurol. 2019;15:193-203
16. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367:eaau6977
17. Wollert T. Autophagy. Curr Biol. 2019;29:R671-r7
18. Wang P, Shao BZ, Deng Z, Chen S, Yue Z, Miao CY. Autophagy in ischemic stroke. Prog Neurobiol. 2018;163-164:98-117
19. Kim KA, Kim D, Kim JH, Shin YJ, Kim ES, Akram M. et al. Autophagy-mediated occludin degradation contributes to blood-brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS. 2020;17:21
20. Liu J, Weaver J, Jin X, Zhang Y, Xu J, Liu KJ. et al. Nitric Oxide Interacts with Caveolin-1 to Facilitate Autophagy-Lysosome-Mediated Claudin-5 Degradation in Oxygen-Glucose Deprivation-Treated Endothelial Cells. Mol Neurobiol. 2016;53:5935-47
21. Zhang S, An Q, Wang T, Gao S, Zhou G. Autophagy- and MMP-2/9-mediated Reduction and Redistribution of ZO-1 Contribute to Hyperglycemia-increased Blood-Brain Barrier Permeability During Early Reperfusion in Stroke. Neuroscience. 2018;377:126-37
22. Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. 2012;32:3044-57
23. Li Y, Liu B, Zhao T, Quan X, Han Y, Cheng Y. et al. Comparative study of extracellular vesicles derived from mesenchymal stem cells and brain endothelial cells attenuating blood-brain barrier permeability via regulating Caveolin-1-dependent ZO-1 and Claudin-5 endocytosis in acute ischemic stroke. J Nanobiotechnology. 2023;21:70
24. Nag S, Venugopalan R, Stewart DJ. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 2007;114:459-69
25. Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem. 2009;284:19053-66
26. Huang Q, Zhong W, Hu Z, Tang X. A review of the role of cav-1 in neuropathology and neural recovery after ischemic stroke. J Neuroinflammation. 2018;15:348
27. Chow BW, Nunez V, Kaplan L, Granger AJ, Bistrong K, Zucker HL. et al. Caveolae in CNS arterioles mediate neurovascular coupling. Nature. 2020;579:106-10
28. Bai X, Yang X, Jia X, Rong Y, Chen L, Zeng T. et al. CAV1-CAVIN1-LC3B-mediated autophagy regulates high glucose-stimulated LDL transcytosis. Autophagy. 2020;16:1111-29
29. Chen ZH, Cao JF, Zhou JS, Liu H, Che LQ, Mizumura K. et al. Interaction of caveolin-1 with ATG12-ATG5 system suppresses autophagy in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2014;306:L1016-25
30. Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ. et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107:18880-5
31. Zhang X, Ramírez CM, Aryal B, Madrigal-Matute J, Liu X, Diaz A. et al. Cav-1 (Caveolin-1) Deficiency Increases Autophagy in the Endothelium and Attenuates Vascular Inflammation and Atherosclerosis. Arterioscler Thromb Vasc Biol. 2020;40:1510-22
32. Tian Y, Gong M, Hu Y, Liu H, Zhang W, Zhang M. et al. Quality and efficiency assessment of six extracellular vesicle isolation methods by nano-flow cytometry. J Extracell Vesicles. 2020;9:1697028
33. Li Y, Chen J, Quan X, Chen Y, Han Y, Chen J. et al. Extracellular Vesicles Maintain Blood-Brain Barrier Integrity by the Suppression of Caveolin-1/CD147/VEGFR2/MMP Pathway After Ischemic Stroke. Int J Nanomedicine. 2024;19:1451-67
34. Lee YK, Uchida H, Smith H, Ito A, Sanchez T. The isolation and molecular characterization of cerebral microvessels. Nat Protoc. 2019;14:3059-81
35. Yan Y, Tao H, He J, Huang SY. The HDOCK server for integrated protein-protein docking. Nat Protoc. 2020;15:1829-52
36. Welsh JA, Goberdhan DCI, O'Driscoll L, Buzas EI, Blenkiron C, Bussolati B. et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J Extracell Vesicles. 2024;13:e12404
37. Yang Z, Lin P, Chen B, Zhang X, Xiao W, Wu S. et al. Autophagy alleviates hypoxia-induced blood-brain barrier injury via regulation of CLDN5 (claudin 5). Autophagy. 2021;17:3048-67
38. Ajoolabady A, Wang S, Kroemer G, Penninger JM, Uversky VN, Pratico D. et al. Targeting autophagy in ischemic stroke: From molecular mechanisms to clinical therapeutics. Pharmacol Ther. 2021;225:107848
39. Tuo QZ, Zhang ST, Lei P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev. 2022;42:259-305
40. Xia Y, Ling X, Hu G, Zhu Q, Zhang J, Li Q. et al. Small extracellular vesicles secreted by human iPSC-derived MSC enhance angiogenesis through inhibiting STAT3-dependent autophagy in ischemic stroke. Stem Cell Res Ther. 2020;11:313
41. Kuang Y, Zheng X, Zhang L, Ai X, Venkataramani V, Kilic E. et al. Adipose-derived mesenchymal stem cells reduce autophagy in stroke mice by extracellular vesicle transfer of miR-25. J Extracell Vesicles. 2020;10:e12024
42. Huang LY, Song JX, Cai H, Wang PP, Yin QL, Zhang YD. et al. Healthy Serum-Derived Exosomes Improve Neurological Outcomes and Protect Blood-Brain Barrier by Inhibiting Endothelial Cell Apoptosis and Reversing Autophagy-Mediated Tight Junction Protein Reduction in Rat Stroke Model. Front Cell Neurosci. 2022;16:841544
43. Lee EC, Ha TW, Lee DH, Hong DY, Park SW, Lee JY. et al. Utility of Exosomes in Ischemic and Hemorrhagic Stroke Diagnosis and Treatment. Int J Mol Sci. 2022;23:8367
44. Rahmani A, Saleki K, Javanmehr N, Khodaparast J, Saadat P, Nouri HR. Mesenchymal stem cell-derived extracellular vesicle-based therapies protect against coupled degeneration of the central nervous and vascular systems in stroke. Ageing Res Rev. 2020;62:101106
45. Stamatovic SM, Johnson AM, Sladojevic N, Keep RF, Andjelkovic AV. Endocytosis of tight junction proteins and the regulation of degradation and recycling. Ann N Y Acad Sci. 2017;1397:54-65
46. Li Z, Liang G, Ma T, Li J, Wang P, Liu L. et al. Blood-brain barrier permeability change and regulation mechanism after subarachnoid hemorrhage. Metab Brain Dis. 2015;30:597-603
47. Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857-902
48. Echarri A, Del Pozo MA. Caveolae. Curr Biol. 2012;22:R114-R6
49. Nabi IR, Le PU. Caveolae/raft-dependent endocytosis. J Cell Biol. 2003;161:673-7
50. Mandel I, Paperna T, Volkowich A, Merhav M, Glass-Marmor L, Miller A. The ubiquitin-proteasome pathway regulates claudin 5 degradation. J Cell Biochem. 2012;113:2415-23
51. Ganapathy AS, Saha K, Suchanec E, Singh V, Verma A, Yochum G. et al. AP2M1 mediates autophagy-induced CLDN2 (claudin 2) degradation through endocytosis and interaction with LC3 and reduces intestinal epithelial tight junction permeability. Autophagy. 2022;18:2086-103
52. Saha K, Ganapathy AS, Wang A, Morris NM, Suchanec E, Ding W. et al. Autophagy Reduces the Degradation and Promotes Membrane Localization of Occludin to Enhance the Intestinal Epithelial Tight Junction Barrier against Paracellular Macromolecule Flux. J Crohns Colitis. 2023;17:433-449
53. Huang L, Zhang D, Han W, Guo C. High-mobility group box-1 inhibition stabilizes intestinal permeability through tight junctions in experimental acute necrotizing pancreatitis. Inflamm Res. 2019;68:677-89
54. Xu L, Guo R, Xie Y, Ma M, Ye R, Liu X. Caveolae: molecular insights and therapeutic targets for stroke. Expert Opin Ther Targets. 2015;19:633-50
55. Chen HS, Chen X, Li WT, Shen JG. Targeting RNS/caveolin-1/MMP signaling cascades to protect against cerebral ischemia-reperfusion injuries: potential application for drug discovery. Acta Pharmacol Sin. 2018;39:669-82
56. Nah J, Yoo SM, Jung S, Jeong EI, Park M, Kaang BK. et al. Phosphorylated CAV1 activates autophagy through an interaction with BECN1 under oxidative stress. Cell Death Dis. 2017;8:e2822
57. Jiang Y, Krantz S, Qin X, Li S, Gunasekara H, Kim YM. et al. Caveolin-1 controls mitochondrial damage and ROS production by regulating fission - fusion dynamics and mitophagy. Redox Biol. 2022;52:102304
58. Collado A, Gan L, Tengbom J, Kontidou E, Pernow J, Zhou Z. Extracellular vesicles and their non-coding RNA cargos: Emerging players in cardiovascular disease. J Physiol. 2023;601:4989-5009
59. Fullerton JL, Cosgrove CC, Rooney RA, Work LM. Extracellular vesicles and their microRNA cargo in ischaemic stroke. J Physiol. 2023;601:4907-21
60. Hermann DM, Xin W, Bähr M, Giebel B, Doeppner TR. Emerging roles of extracellular vesicle-associated non-coding RNAs in hypoxia: Insights from cancer, myocardial infarction and ischemic stroke. Theranostics. 2022;12:5776-802
61. Xiaoqing S, Yinghua C, Xingxing Y. The autophagy in ischemic stroke: A regulatory role of non-coding-RNAs. Cell Signal. 2023;104:110586
62. Yang X, Xu J, Lan S, Tong Z, Chen K, Liu Z. et al. Exosomal miR-133a-3p Derived from BMSCs Alleviates Cerebral Ischemia-Reperfusion Injury via Targeting DAPK2. Int J Nanomedicine. 2023;18:65-78
63. Li D, Huang S, Zhu J, Hu T, Han Z, Zhang S. et al. Exosomes from MiR-21-5p-Increased Neurons Play a Role in Neuroprotection by Suppressing Rab11a-Mediated Neuronal Autophagy In Vitro After Traumatic Brain Injury. Med Sci Monit. 2019;25:1871-85
64. Barthels D, Das H. Current advances in ischemic stroke research and therapies. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165260
Author contact
![]() Corresponding author: Yonghua Zhao, M.D., Institute of Chinese Medical Sciences, State Key Laboratory of Quality Research in Chinese Medicine, University of Macau, Taipa, Macao SAR, China. Tel.: +853-88224877 Fax: +853-28841358 E-mail address: yonghuazhaoedu.mo.
Corresponding author: Yonghua Zhao, M.D., Institute of Chinese Medical Sciences, State Key Laboratory of Quality Research in Chinese Medicine, University of Macau, Taipa, Macao SAR, China. Tel.: +853-88224877 Fax: +853-28841358 E-mail address: yonghuazhaoedu.mo.