Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. Origins of viral mimicry and...
2. The synergistic interaction...
3. Mechanisms of viral mimicry...
4. Potential and challenges of...
5. Conclusions and future...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(3):958-973. doi:10.7150/ijbs.103877 This issue Cite
Review
Exploring viral mimicry combined with epigenetics and tumor immunity: new perspectives in cancer therapy
Ruirui Wang1#, Xin Dong2#, Xiongjian Zhang1, Jinzhuang Liao1,3, Wei Cui2 ![]() , Wei Li1
, Wei Li1 ![]()
1. Department of Radiology, The Third Xiangya Hospital of Central South University. Tongzipo Road 138, Changsha, Hunan, People's Republic of China.
2. Department of Clinical Laboratory, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
3. Department of Interventional Therapy, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
# These authors contributed equally to this work.
Received 2024-9-19; Accepted 2024-12-20; Published 2025-1-6
Abstract

Viral mimicry refers to an active antiviral response triggered by the activation of endogenous retroviruses (ERVs), usually manifested by the formation of double-stranded RNA (dsRNA) and activation of the cellular interferon response, which activates the immune system and produces anti-tumor effects. Epigenetic studies have shown that epigenetic modifications (e.g. DNA methylation, histone modifications, etc.) play a crucial role in tumorigenesis, progression, and treatment resistance. Particularly, alterations in DNA methylation may be closely associated with the suppression of ERVs expression, and treatment by demethylation may restore ERVs activity and thus strengthen the tumor immune response. Therefore, we propose that viral mimicry can induce immune responses in the tumor microenvironment by activating the expression of ERVs, and that epigenetic alterations may play a key regulatory role in this process. In this paper, we review the intersection of viral mimicry, epigenetics and tumor immunotherapy, and explore the possible interactions and synergistic effects among the three, aiming to provide a new theoretical basis and potential strategies for cancer immunotherapy.
Keywords: Viral mimicry, Endogenous retroviruses, DNA methylation, Hypomethylating agents, Epigenetic therapy, Tumor treatment
Introduction
Viral mimicry
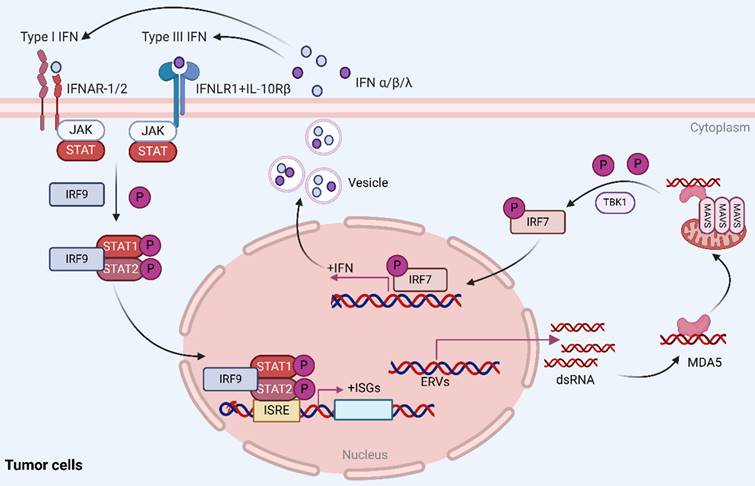
Viral mimicry is a cellular state of active antiviral response induced by endogenous stimuli rather than exogenous viral infection that affects tumor immunity by activating endogenous retroviruses (ERVs) that are epigenetically silenced and inducing an interferon response[1-3]. In this process, activation of ERVs leads to the formation of double-stranded RNA (dsRNA), which can be sensed by pattern recognition receptors (PRRs) such as the cytosolic melanoma differentiation-associated gene 5 (MDA5), which further activates the mitochondrial antiviral signaling protein (MAVS) pathway[4, 5]. In addition, TANK binding kinase 1 (TBK1), a central node protein involved in multiple intrinsic immune signaling pathways, activates both NF-κB and IRFs and is a critical protein kinase in the body's resistance to infection[6, 7]. In viral mimicry response, TBK1 promotes interferon regulatory factor 7 (IRF7) dimerization and translocation to the nucleus by phosphorylating IRF7 to form an active transcriptional complex, which in turn initiates type I and type III interferon responses, stimulates cytokine production, and enhances the body's antiviral, antimicrobial, antitumor, and immunomodulatory functions[8, 9] (Figure 1).
Reactivation of ERVs in tumor cells induces viral mimicry responses. Activation of ERVs leads to the formation of dsRNA, which is sensed by MDA5, further activating the MAVS pathway. Additionally, TBK1 phosphorylates IRF7, causing it to dimerize and ectopically translocate to the nucleus, where it forms an active transcriptional complex that induces a type I/III interferon response and activates the transcription of IFN-stimulated genes (ISGs). Reproduced with permission from BioRender publisher.
Epigenetics
Cancer has long been regarded as a hereditary disease, but with advances in epigenetic research, there is growing evidence of the important role of epigenetic alterations in tumorigenesis and progression[10, 11]. Thus, cancer can be regarded not only as a hereditary disease, but also as an epigenetic disease[12]. The central concept of epigenetics is that epigenetic modifications of chromosomes can lead to persistent changes in gene expression, although the DNA sequence itself is unaltered, and these changes can be transmitted to offspring through cell division. In addition, classical mechanisms of epigenetic inheritance include alterations in DNA methylation, histone modifications, chromatin remodeling, and non-coding RNA-mediated gene regulation[12, 13]. These epigenetic abnormalities play a crucial role in the stability of chromatin structure, the regulation of gene expression, and the maintenance of basic cellular physiological functions, especially in the process of tumorigenesis and progression, and disruptions in epigenetic mechanisms are closely related to tumor formation and treatment resistance[14]. Therefore, epigenetics is expected to be an attractive therapeutic target in cancer treatment. With the deepening of oncology research, researchers have found an increasing role for epigenetics in aspects such as diagnosis and prognosis of tumors[15]. The most widely used epigenetic therapies in cancer treatment are small molecule inhibitors (i.e., demethylating drugs) that use DNA methyltransferase (DNMT). Notably, previous reports have indicated that DNA methylation prevents the activation of retroviral progenitors in drug-resistant cells and that using hypomethylating drugs can reactivate ERVs and inhibit the growth of tumor cells[16, 17]. These studies suggest that viral mimicry could enhance the therapeutic effect on cancer by combining it with epigenetic therapies.
Tumor immunity
Tumor immunotherapy aims to harness the body's natural immune system to activate and enhance its ability to attack tumors. By activating specific immune cells, such as T-cells and natural killer cells, and by proliferating the antitumor immune response in the body, the therapy can direct the body's immune system to recognize and destroy tumor cells[18-20]. Its basic principle lies in breaking the evasion mechanism of the tumor cells against the immune system and reawakening the immune cells so that they can recognize and attack the tumor cells, thus clearing the tumor[21]. Tumor immunotherapy has specific therapeutic effects for cancer patients with few side effects and is listed as one of the four major tumor treatment techniques, together with surgery, radiotherapy and chemotherapy[22, 23]. The origins of tumor immunotherapy can be traced back to 1893, when Cloey discovered that sarcoma patients infected with Streptococcus pyogenes experienced tumor regression following surgery. Thereafter, he further explored the mechanism and, for the first time, used attenuated bacterial mixtures to stimulate the immune system to enhance the patient's resistance to disease, a discovery that laid the groundwork for the emergence of modern immunotherapy and provided important insights into subsequent therapeutic strategy exploration[24, 25]. Subsequently, with a large number of studies, the mechanisms of tumor immunity have been better understood, and tumor immunotherapy has been classified into four modalities: non-specific immune stimulation, immune checkpoint blockade, tumor vaccines, and overdose immune cell therapy[26]. In addition, recent studies have revealed that the expression of ERVs can trigger the activation of innate immune receptors, thereby initiating an immune response against viral, a process that may induce tumor cell death[2, 27, 28]. This discovery has led to new research directions in cancer treatment and may provide a theoretical basis for developing more effective immunotherapy strategies in the future.
1. Origins of viral mimicry and its progress in cancer therapy
1.1 The role of endogenous retroviruses
As remnants of ancient retroviral infections, ERVs have long been in equilibrium with the host[29]. Human endogenous retroviruses (HERVs) account for 8% of the human genome and are hardly expressed under normal conditions due to strict epigenetic regulation[30]. HERVs can be classified into 3 prominent families, which are the Class I family: gamma retroviral-like elements, including HERV-T, HERV-I, HERV-H, HERV-W, HERV-R, etc.; the Class II family: β-endotransposon-like elements (HERV-K superfamily); Class Ⅲ family: foamy viral-like elements, including HERV-L, HERV-S, etc.[31]. Previous studies have reported that HERVs play a role in human pathological processes (except cancer) such as type 1 diabetes (T1D)[32], autoimmune diseases such as amyotrophic lateral sclerosis (ALS), systemic lupus erythematosus (SLE), and Sjogren's syndrome (SS) are associated with HERVs[33-36]. For this review, we will focus on the mechanism of action of HERVs in tumors.
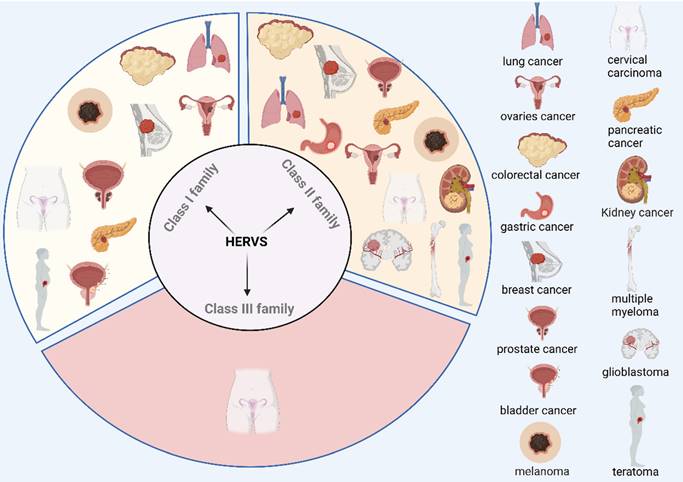
Increasing evidence indicates that the expression of the HERV family shows significant differences in various cancers, highlighting its potential role in tumorigenesis. Studies have shown that HERVs are associated with the development of several types of cancer, including colorectal cancer[37-41], gastric cancer[37], breast cancer[42-53], prostate cancer[54-59], melanoma[60-70], teratoma[71-74], ovarian cancer[75], lung cancer[76, 77], cervical cancer[78, 79], glioblastoma[80-82], pancreatic cancer[83, 84], multiple myeloma[85], kidney cancer[86-88], and bladder cancer[89] (Figure 2). For example, research has demonstrated that expression of ERVs correlates with melanoma development[70], and the use of antiretroviral drugs (doravirine, lamivudine and cabotegravir) inhibits cell viability, invasion and colony-forming ability of melanoma cells, while having no inhibitory effect on normal human epithelial melanocytes[74]. Tumor cells are specific for invasive metastasis, unlimited proliferation, and resistance to death[90], and a growing number of studies have shown that activation of ERVs correlates with the invasion of tumor cells[91]. Additionally, cancer stem cells (CSCs) are associated with tumorigenesis, invasion and metastasis, and resistance to radiotherapy. It has been suggested that the activation of ERVs may contribute to tumor progression by modulating the functions of CSCs[92]. For example, a study by DO-Ye Kim et al. found that knockdown of the HERV-K env gene significantly inhibited the induction and proliferation of CSCs in the SKOV3 cell line[93]. However, it is worth noting that the role of ERVs may exhibit duality in different biological and therapeutic contexts. Specifically, on the one hand, recent studies have shown that bifunctional inhibitors (J208) of DNA methyltransferases and histone deacetylases by epigenetic means induce ERVs expression, which in turn triggers a viral mimicry response that activates the immune system and exerts an anti-Triple Negative Breast Cancer (TNBC) effect[94]. In addition, Yang et al. found that a dual inhibitor (C02S) of DNA methyltransferases and histone deacetylases, not only upregulated ERVs and activated viral mimicry responses through the MDA5-MAVS signaling pathway in colorectal cancer (CRC) model, but also remodeled the tumor immune microenvironment (TME), enhanced immune cell infiltration, and significantly improved the efficacy of anti-PD-L1 therapy in CRC mouse model. These results suggest that activation of ERVs not only induces immune responses but also enhances the efficacy of immune checkpoint inhibitors (ICIs)[95]. On the other hand, recent studies have shown that ERVs expression is also closely associated with the malignant features of TNBC, and a genome-wide transcriptomic analysis of HERV sequences revealed that TROJAN, a primate long-stranded non-coding RNA, is highly expressed in TNBC and is strongly associated with a poor prognosis by promoting the proliferation and invasion of tumor cells[96]. This phenomenon reflects the complex and diverse roles of ERVs in the tumor microenvironment. Different epigenetic mechanisms, cellular environments, and immune responses may lead to distinct biological effects of ERVs in different contexts. In conclusion, the activation of ERVs and their induced viral mimicry responses have important regulatory roles in cancer development. Although the results suggest that ERVs may serve as biomarkers for cancer and have the potential to become new targets for cancer therapy, further in-depth studies on their mechanisms in different cancer types are needed. The challenge for the future is how to precisely target ERVs to maximize anti-tumor effects while avoiding side effects.
Correlation of the HERV family with multiple tumorigenesis. Correlations between different families of HERVs and the occurrence and development of various tumors have been reported in the literature. These findings not only reveal a potentially important role in the mechanism of tumorigenesis but also provide an academic foundation for further investigation into the role of HERVs in tumor development. Reproduced with permission from BioRender publisher.
1.2 Potential application of viral mimicry in tumor immunotherapy
Viral mimicry is an endogenous cellular state that affects tumorigenesis and progression by activating normally epigenetically silenced ERVs, further inducing type I/Ⅲ interferon responses. Type I interferons (e.g., IFN-α and IFN-β) promote activating natural killer cells and CD8+ T-cells, augmenting their tumor cell-killing effects. Meanwhile, type Ⅲ interferons (IFN-λ) can modulate immune responses and enhance antitumor immunity in the tumor microenvironment[97-99].
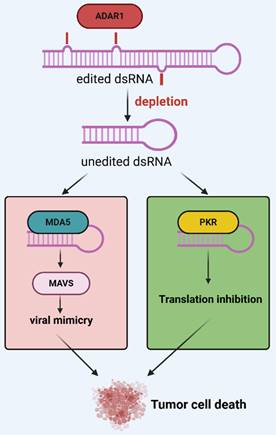
In viral mimicry models, it is more interesting to note that a certain amount of ERV dsRNA is recognized as nonself by pattern recognition receptors to trigger an immune response. However, an adenosine deaminase that acts on RNA (ADAR) prevents MDA5 from sensing endogenous dsRNA as nonself by catalyzing adenosine-to-inosine (A-to-I) editing of dsRNA[100]. In detail, ADAR is a group of enzymes that bind dsRNA to homodimers and catalyze the hydrolytic deamination of adenosine nucleotides to form inosine[101, 102]. Humans have three ADAR proteins: ADAR1, ADAR2, and ADAR3[103-105]. Of these, ADAR1 is universally expressed in almost all tissues and contains both nuclear p110 and interferon-inducible p150 isoforms and expression of ADAR1p150 is thought to be associated with interferon response[106-110]. Further studies have shown that interferon-inducible p150 is predominantly found in the nucleus and cytoplasm, that cytoplasmic p150 isoforms specifically regulate MDA5-MAVS-IFN signaling, and that A-to-I editing reduces the ability of MDA5 to carry out its function, making edited dsRNA less efficient at binding to MDA5[111]. In addition, ADARp150 was shown to inhibit another dsRNA sensor, protein kinase R (PKR)[112, 113]. These RNA sensors are part of the innate immunity against viral infections[114]. Researchers have recently identified ADAR1 as a potential therapeutic target for various cancers. For example, Kyle A and his team proposed in 2021 that ADAR1 is highly expressed in TNBC and that knockdown of ADAR1 attenuates the proliferation of tumor cells[115]; a report by Kyle A and his team in April 2024 showed that targeting ADAR1 and DHX9 could exert anti-tumor effects by inducing viral mimicry, suggesting that both could serve as effective tools for breast and other cancers[116]. Similarly, Hyeongjwa et al. proposed targeting DEAD-box RNA helicase 3X (DDX3X) and ADAR1 triggers antitumor immunity through dsRNA-mediated endogenous tumor type I interferon response[117]. Therefore, we propose that ADAR1 acts as an interferon-stimulated gene (ISG) that labels dsRNA as itself and inhibits interferon responses, providing negative feedback regulation of viral mimicry responses (Figure 3).
Negative feedback regulation of ADAR1 in the viral mimicry response. After ADAR1 depletion, unedited dsRNA triggers pattern recognition receptors MDA5, PKR, etc., ultimately inducing a viral mimicry response and activating a translation-stopping antiviral mechanism. Reproduced with permission from BioRender publisher.
The most classic studies on the mechanisms of viral mimicry in cancer therapy are two articles reported in 2015: Roulois et al. proposed that the use of low-dose DNA methyltransferase inhibitors (DNMTis), such as 5-aza-2-deoxycytidine (5-AZA-CdR), could induce a viral mimicry response to target colorectal cancer-initiating cells (CICs), resulting in an anti-tumor effect. However, by disrupting the viral mimicry pathway (e.g., knockdown of MDA5, MAVS, or IRF7) the targeting of CICs by 5-AZA-CdR can be inhibited and its long-term growth effect significantly reduced[118]. Furthermore, Chiappinelli et al. proposed that in ovarian cancer (OC), DNMTis activates the type I interferon response and induces apoptosis by triggering dsRNA perception. Knockdown of the dsRNA sensors TLR3 and MAVS significantly inhibited this immune response, and blockade of IFN-β or its receptor also inhibited the response[16]. These two studies reveal in detail the mechanism of action of viral mimicry in cancer therapy for the first time, validating the direct association between DNMTis, ERVs and anti-tumor immunity. This mechanism provides a new theoretical basis for the combination of epigenetic therapy and tumor immunotherapy, and lays preliminary evidence for future combined treatment strategies. These findings broaden the idea of cancer treatment and provide potential directions for developing new immunotherapies and improving the effectiveness of existing treatments.
In recent studies, RNA deconjugating enzyme DHX9 was found to be a repressor of dsDNA sensing, and further studies have shown that deletion of DHX9 induces activation of the dsRNA sensing pathway and viral mimicry responses and suggested that DHX9 would be a potential target for enhancing antitumor immunity[116, 119]. In pancreatic cancer, trametinib, as an MEK1/2 inhibitor, induces activation of ERVs and IFN responses, increasing the potential for tumor immunogenicity[120]. Plant homeodomain finger protein 8 (PHF8), a histone lysine demethylase with cancer-restricted antitumor immune function, and in colorectal cancer, deletion of PHF8 activates the antiviral response and significantly improves the therapeutic efficacy of immune checkpoint blockade (ICB)[121]. In addition, studies have also reported that activation of viral mimicry response can increase the sensitivity of tumor cells to radiotherapy. For example, treatment of cervical cancer (CC) with low-dose decitabine (DAC) activated the viral mimicry response, thereby enhancing the sensitivity of CC to chemotherapy[122]. The deletion of histone methyltransferase SETDB1 has been shown to significantly promote the activation of ERVs and induce a type I interferon response, which promotes the sensitivity of cancer cells to radiation therapy[123]. These findings suggest that viral mimicry responses are prevalent in a wide range of cancer types and that their activation not only enhances anti-tumor immune responses, but may also improve tumor sensitivity to radiotherapy and chemotherapy (Table 1). Therefore, therapeutic strategies targeting viral mimicry responses are not only expected to enhance the efficacy of cancer immunotherapy, but may also be an important adjuvant therapy to improve the clinical efficacy of existing treatments. These findings provide an important theoretical basis and potential clinical applications for developing new anti-cancer therapeutic strategies in the future.
Mechanisms that trigger viral mimicry in malignant tumors.
| Cancer | Mechanisms for triggering viral mimicry | Reference |
|---|---|---|
| Breast cancer | Combined knockdown of adenosine deaminase acting on RNA 1 (ADAR1) and RNA helicase DHX9 leads to the activation of multiple dsRNA-sensing pathways to induce viral mimicry; Bifunctional inhibitors of HDAC and DNMT are effective in inducing expression of ERVs and viral mimicry; The use of the chemical probe BAY-299 targets TATA-box binding protein-associated factor 1 (TAF1) and induces ERVs and dsRNA formation; Type I protein arginine methyltransferase (PRMT) inhibitors have anti-tumor biological activity in TNBC and induce viral mimicry; Spliceosome-targeted therapy (STT) leads to accumulation of misspliced mRNAs in cytoplasm, which can further lead to dsRNA formation; HDAC and DNMT bifunctional inhibitors can effectively induce expression of ERVs and viral mimicry; (STT) leads to the accumulation of mis-spliced mRNAs in the cytoplasm, which can further form dsRNA, triggering antiviral signaling and exogenous apoptosis; Transcriptomics results show an atypical viral mimicry response in all-trans-retinoic acid (ATRA)-treated breast cancer cells, which leads to an increase in expression of interferon-responsive factor 1 (IRF1) transcription factors and downstream effector expression of Deltex-E3-ubiquitin ligase-3L (DTX3L); Cell cycle protein-dependent kinases are also known to induce viral mimicry in TNBC; The use of inhibitors of cell cycle protein-dependent kinases 4 and 6 (CDK4/6) inhibits tumor cell cycle arrest and activates the expression of ERVs, which in turn induces viral mimicry and enhances tumor antigen presentation. | [94, 116, 124-129] |
| Pancreatic cancer | Targeting epigenetic factors (e.g. DNMTs, LSD1, KDM5B, SETDB1, SUV39H1, G9A, EZH2) induces a viral mimicry response and triggers interferon signaling to sensitize non-immunogenic tumor cells to immune checkpoint inhibitors; Inhibition of MEK1/2 by trametinib leads to increased dsRNA production and INF gene expression. | [1, 120] |
| Colorectal cancer | Low-dose 5-AZA-CdR targets colorectal cancer-initiating cells by inducing viral mimicry; Deletion of histone demethylase PHF8 inhibits tumor cell growth and induces viral mimicry to enhance the sensitivity of mouse models of colorectal cancer to ICB treatment; The combination of DNMTi and EZH2i activates viral mimicry to exert anti-tumor effects; Exposure of colorectal cancer cells to low-dose RRx-001 induces viral mimicry to enhance the anticancer activity of RRx-001. | [118, 121, 130, 131] |
| Small cell lung cancer | DExD/H-box deconjugase 9 (DHX9) is a deconjugating enzyme that plays an important role in small-cell lung cancer and effectively inhibits dsRNA. Its absence leads to the accumulation of dsRNA in the cytoplasm and triggers an innate immune response within the tumor. | [119, 132] |
| Glioblastoma | Treatment with the corresponding compounds increased macroH2A2 levels and strongly activated the viral mimicry response in Glioblastoma cells, and this effect was attenuated upon macroH2A2 knockdown; Combination therapy targeting HERV-mediated viral mimetics and immunotherapy improves GBM treatment. | [133, 134] |
| Hepatocellular carcinoma | Chromatin assembly factor 1 (CAF-1) knockdown leads to the enrichment of autosomal H3.3, which may activate viral mimicry, i.e., targeting CAF-1 may enhance anti-tumor immune responses. | [135] |
| Prostate cancer | Inhibition of CDK9 leads to dsRNA production, which in turn induces viral mimicry to enhance anti-tumor immunity; methyltestosterone (MeT) is an androgenic and anabolic compound, and sustained MeT treatment induces viral mimicry responses; targeting FBXO44 leads to DNA replication stress and induces viral mimicry, thereby improving anti-tumor immunotherapy effects. | [136-139] |
| Melanoma/Human non-small cell carcinoma | Inhibition of SETDB1 significantly enhances the anti-tumor effect of radiotherapy by promoting radiation-induced viral mimicry up-regulation of type I interferon. | [123] |
| Clear cell renal cell carcinoma | HERV may activate the interferon (IFN) signaling pathway by means of viral mimicry, thereby enhancing the effect of tumor immunotherapy; RNA splicing errors can elicit a viral mimicry response, and further studies have found that SETD2-deficient renal cancers are more prone to splicing errors and that DAC treatment further exacerbates this effect, which in turn facilitates viral mimicry and enhances anti-tumor effects. | [140, 141] |
| Thyroid cancer | Inhibition of the coatomer protein complex zeta 1 (COPZ1) leads to apoptosis, reduced cell viability, activation of the type I interferon response signaling pathway and induction of viral mimicry. | [142] |
| Ovaries cancer | DNA and histone methyltransferase inhibition increases viral mimicry. | [16, 143] |
2. The synergistic interaction between epigenetics and viral mimicry in cancer therapy
2.1 Epigenetic modifications and their integration in cancer therapy: mechanisms and applications of DNA methylation and histone acetylation inhibitors
Epigenetic-targeting drugs have increasingly been used to treat malignant tumors, with common types including DNA methyltransferase inhibitors and histone deacetylase inhibitors[144]. This review focuses on the mechanisms and therapeutic applications of DNA methylation and histone acetylation inhibitors in cancer treatment and further explores the mechanisms of combining epigenetic therapy and viral mimicry against tumors. DNA methylation is the transfer of methyl provided by S adenosine methionine (SAM) to the carbon atom at the 5-position of cytosine, catalyzed by DNA methyltransferases (DNMT1, DNMT3A, DNMT3B), ultimately resulting in the formation of 5'methylcytosine[145]. The link between DNA methylation and cancer has been the subject of numerous research personnel. It is one of the most common and well-studied epigenetic modifications in mammals, and DNA methylation analysis has been initially used as a complementary diagnostic tool for various tumors[146-148]. Therefore, aberrant DNA methylation is associated with tumorigenesis. Tumors such as colorectal cancer[149, 150], breast cancer[151, 152], glioblastoma[153, 154], hepatocellular carcinoma[155, 156], and renal cell carcinoma[157, 158] have been reported to be associated with DNA methylation abnormalities are associated. In addition, it has been recently reported that the detection of methylation differences in circulating free DNA can be used to sensitively monitor the treatment effect of CRC and detect early pancreatic cancer. As a non-invasive biomarker, it has the advantage of being less cost-effective[159-162].
DNMTis were developed and approved well before the complexity of methylation patterns had been discerned[163, 164]. Studies have shown that low doses of DNA methyltransferase inhibitors cause inactivation of DNMT1, which in turn causes DNA demethylation[165]. Commonly used DNMTis include azacitidine (AZA) and DAC[166]. These two drugs have remained the mainstay of treatment for elderly AML and MDS patients since their first approval for use to date[167, 168]. In addition, in some clinical trials, using these two drugs has improved overall survival and quality of life in elderly patients who are not candidates for intense chemotherapy and has also shown that epigenetic therapies are efficacious[169, 170]. More importantly, it has been reported that DNMTis combined with cytostatic agents promotes apoptosis in CRC cells[171, 172]; in both in vivo and ex vivo models, the use of DNMTis significantly inhibited the growth of smooth muscle sarcoma cells[173]; and in the treatment of breast and ovarian cancers, the combination of PARP inhibitor (PARPi) and DNMTis in combination will restore the sensitivity of breast and ovarian cancer to PARPi treatment[174]; in a mouse ovarian cancer model, the results showed that DNMTis could activate the type I interferon response, reduce the percentage of macrophages in the tumor microenvironment, and in combination with α-difluoromethylornithine (DFMO) the therapeutic effect was more significant[175]. These results suggest that DNMTis plays an active role in cancer therapy and may improve the therapeutic outcome of a wide range of tumors.
Histones are highly conserved proteins consisting of five types of core proteins, H1, H3, H2A, H2B, and H4, and histone modifications include phosphorylation, acetylation, methylation, ubiquitination, glycosylation, and other modification processes[176]. Histone acetylation is a dynamic modification, and histone acetylation and histone deacetylation work together to maintain normal gene transcription[177], and the balance between them is tightly regulated by histone acetyltransferase (HAT) and histone deacetylase (HDAC)[178]. Moreover, it was shown that HDAC could be one of the potential targets in cancer therapy[179]. Histone deacetylase inhibitors (HDACis) are new antitumor agents that exert their antitumor effects by regulating gene expression[180-182]. In recent years, reports have indicated that the combination of HDACis and DNMTis at low doses significantly improved the antitumor effects in non-small cell lung cancer (NSCLC)[183] and multiple myeloma (MM)[184], as well as the decrease of cell viability in oral squamous carcinoma (OSCC) after the combination treatment[185]. In addition, in the treatment of breast cancer, HDACis and DNMTis, in combination with conventional chemotherapeutic agents, can exert a positive antitumor mechanism of action by inhibiting the proliferation of breast cancer cells as well as promoting apoptosis[186, 187]. These results all suggest that targeting epigenetics is an extremely promising cancer treatment.
2.2 Synergistic anti-tumor potential of epigenetic therapies and viral mimicry
As mentioned earlier, DNMTis and HDACis are being extensively investigated as epigenetic regulatory drugs for cancer therapy[17]. These two classes of drugs are currently being used in several clinical trials, alone or in combination with other therapeutic agents, to evaluate their therapeutic effects on a wide range of cancers (Table 2). Furthermore, many basic studies have shown that using these two classes of drugs enhances immune signaling, including promoting an interferon response, which induces a viral mimicry response in the body and enhances antitumor effects[188-190]. For example, it has been shown that epigenetic inhibitor therapy may trigger the expression of multiple epigenetically silenced genes in gastrointestinal mesenchymal stromal tumor (GIST) cells as well as the activation of the interferon signaling pathway, resulting in antitumor effects[191]. In addition to DNMTis and HDACis, other epigenetic drugs can exert antitumor effects by inducing viral mimicry responses. For example, the zeste enhancer homolog 2 (EZH2) gene, a human homolog of the drosophila zeste gene enhancer, belongs to a key member of the Polycomb group (PcG) family, and possesses histone methylase activity, which catalyzes the methylation of the lysine residue 27 (H3K27) of histone H3 to regulate the expression of oncogenes[192, 193]. It has been found that EZH2 is highly expressed in a variety of tumors and correlates with tumor prognosis[194, 195]. Therefore, developing inhibitors targeting EZH2 has become an important research direction in cancer therapy. Recent studies have shown that EZH2 inhibitors (EZH2is) can trigger viral mimicry via RNA and DNA sensing pathways, effectively targeting atypical teratoid rhabdomyosarcomas (ATRTs)[196]. More interestingly, a study found that epigenetic alterations in drug-resistant TNBC, particularly DNA demethylation and modulation of H3K27me3 markers, could evade chemotherapy-induced viral mimicry responses, thereby promoting tumor progression. However, the altered epigenetic state of tumor cells “sensitizes” them to EZH2is after chemoresistance, thereby reversing resistance and restoring the immune system's anti-tumor response[197]. Thus, epigenetic therapies, through multiple mechanisms (e.g., cell cycle regulation and apoptosis induction), can trigger viral mimicry responses and enhance anti-tumor effects, suggesting that epigenetic status modulation may provide new strategies for tumor immunotherapy. These combined effects make the combination of epigenetic therapies and viral mimicry responses a promising strategy for tumor treatment and show broad promise in clinical care.
Epigenetic Drugs DNMTis and HDACis in Cancer Clinical Trials.
| Cancer | Register Trial Code | Drugs | Phase | Clinical Trial Effects |
|---|---|---|---|---|
| Colorectal cancer | NCT01105377[198] | Entinostat+Azacitidine | II | These findings indicate that epigenetic drugs commonly upregulate immune genes in a wide range of solid tumor types, suggesting a strong immunomodulatory role for these drugs in cancer |
| Ovarian cancer | NCT02901899[199] | Guadecitabine+Pembrolizumab | II | These results suggest that epigenetic therapies enhance the immune response and benefit patients |
| Breast cancer | NCT01105312[200] | Letrozole+Panobinostat | I/II | These results suggest a broad immunostimulatory role for epigenetic therapies in a variety of cancers, including breast cancer, and that patients have improved progression-free survival (PFS) and overall survival (OS) |
| NCT01349959[198] | Azacitidine+Entinostat | II | ||
| NCT04296942[201] | BN-Brachyury+Entinostat+Adotrastuzumab Emtansine+M7824 | I | ||
| NCT02623751[202] | KHK2375+Exemestane | I | ||
| NCT02115282[203, 204] | Entinostat | III | ||
| NCT02833155[205] | Entinostat+Exemestane | I | ||
| NCT02632071[206] | ACY-1215+Nab-paclitaxel | I | ||
| Lymphoma | NCT01742988[207, 208] | CUDC-907 | I | These studies show that the combination of epigenetic drugs and clinical chemotherapeutic agents demonstrates favourable safety and efficacy, supporting further clinical trials |
| NCT00691210[209] | Vorinostat+Niacinamide+Romidepsin | I | ||
| NCT03770000[210] | Tenalisib+Romidepsin | I/II | ||
| Melanoma | NCT02836548[211] | Vorinostat | I/II | These studies have shown that drug combination therapy eliminates cells carrying these secondary mutations that lead to resistance in the short term, enhances the immune response, and has good response rates, but with high levels of toxicity |
| NCT03565406[212] | Mocetinostat+Ipilimumab+Nivolumab | I | ||
| Gastrointestinal stromal tumor | NCT03165721[213] | Guadecitabine | II | These findings indicate that Guadecitabine was tolerated in patients with succinate dehydrogenase (dSDH) tumors with manageable toxicity |
| Thyroid cancer | NCT00134043[214] | Vorinostat | II | Previous studies have shown that Vorinostat induces cell death and sensitises thyroid cancer cells to chemotherapy, but this phase II study suggests that it is not an effective treatment for advanced thyroid cancer |
| Renal cell cancer | NCT01582009[215] | LBH-589+Everolimus | I/II | This study demonstrated the safety of combination therapy, but it did not improve clinical outcomes in the group of patients with advanced RCC |
| Tumors of the thymus | NCT01100944[216, 217] | Belinostat | I/II | These results suggest that Belinostat has modest antitumor activity in thymic malignancies |
| Brain tumors | NCT02282917[218, 219] | AR-42 | Early I | These studies suggest that AR-42 may be a well-tolerated and effective epigenetic drug that needs to be evaluated in further clinical trials |
| Prostatic cancer | NCT01075308[220] | SB939 | II | This study demonstrated that SB939 was tolerable at the given dose/regimen and showed a decrease in circulating tumor cells in the majority of evaluable patients, but it did not show sufficient activity according to the PSA RR. Therefore, further clinical trials are required to evaluate |
| Non-small cell lung cancer | NCT02805660[221] | Mocetinostat+Durvalumab | I/II | This study demonstrated that combination therapy is usually well tolerated. In addition, clinical activity was observed in NSCLC patients who had not responded to prior anti-PD-(L) 1 therapy |
| Chondrosarcoma | NCT04340843[222] | Belinostat+Guadecitabine/ASTX727 | II | These results suggest that epigenetic therapies are effective in inhibiting preclinical activity in chondrosarcoma, and clinical trials are ongoing |
| Multiple myeloma | NCT02569320[218] | AR-42+Pomalidomide | I | These studies suggest that epigenetic therapies are effective in treating patients with multiple myeloma |
| NCT00773838[223] | Vorinostat+Bortezomib | II | ||
| NCT01583283[224] | ACY-1215+Lenalidomide+Dexamethssone | I | ||
| Hodgkin lymphoma | NCT01460940[225] | Panobinostat+Lenalidomide | II | This study suggests that combination therapy appears to be safe in patients with relapsed/refractory HL, but does not have better clinical outcomes. Therefore, further evaluation of this combination therapy in HL is not supported |
3. Mechanisms of viral mimicry in the tumor immune response
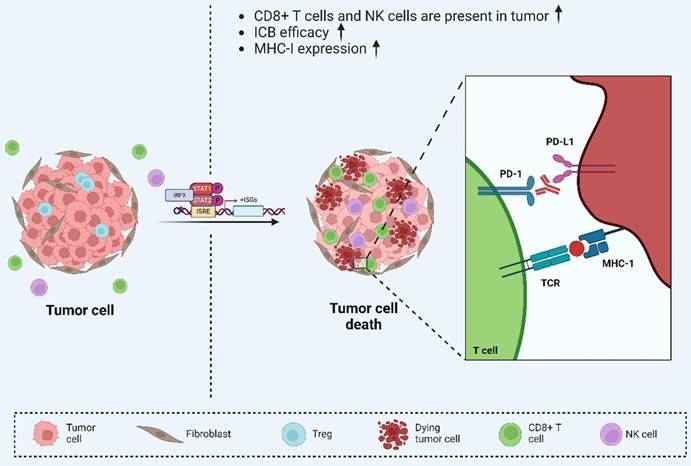
Several recent studies have reported the impact of viral mimicry phenomena on tumor immunotherapy sensitivity[226-228]. These studies have found that viral mimicry can enhance tumor sensitivity to immunotherapy through multiple mechanisms (Figure 4). Specifically, viral mimicry has been reported that viral mimicry can promote the immunogenicity of tumor cells, making them easier to recognize and attack by the immune system[94]. Additionally, it can modulate the epigenetic modifications of tumor cells and change their gene expression patterns, thus enhancing the efficacy of immunotherapy[16, 118]. Furthermore, viral mimicry can improve the therapeutic effect of immune checkpoint blockade (ICB) and promote synergistic inhibition of tumor progression through viral mimicry and tumor immunity[1, 229]. Collectively, these findings indicate that viral mimicry can potentiate the body's antitumor immune response at multiple levels, providing a strong theoretical and experimental foundation for advancing more effective tumor immunotherapies[230-233].
Viral mimicry enhances anti-tumor immunity. Viral mimicry-mediated upregulation of ISGs increased tumor immunogenicity and further enhanced the efficacy of ICB therapy, suggesting that viral mimicry enhances anti-tumor immunity and is may serve as a new target for tumor immunotherapy. Reproduced with permission from BioRender publisher.
Recent studies have demonstrated that DAC, as one of the DNMTis, activates viral mimicry responses in renal cell carcinoma. Following DAC treatment, ERVs show increased binding to RIG-I and MDA5, which modulates T-cell activity and induces antitumor immunity[234]. Similarly, treatment with an aurora kinase inhibitor (AURKi) in colorectal cancer has been shown to activate the type I IFN response, which is dependent on MAVS and RIG-I expression[235]. In the treatment of prostate cancer, where metastasis and hormone therapy resistance are major factors in treatment failure, Charles Spruck and his team have proposed a new therapeutic strategy based on viral mimicry. The mechanism of action of this treatment, which is entirely different from traditional treatment, is to target FBXO44 to induce viral mimicry and thus enhance the antitumor immune response, effectively reducing drug resistance. Subsequently, Charles Spruck's team further developed drugs that can induce viral mimicry responses in prostate cancer, which have not yet entered clinical trials due to drug potency and specificity[138, 139]. These findings suggest an interaction between viral mimicry responses and the tumor immune system, leading to new therapeutic strategies in cancer treatment. This represents a significant step forward in developing next-generation cancer immunotherapies and presents promising prospects for future therapeutic strategies.
4. Potential and challenges of epigenetic and viral mimicry combination immunotherapy
4.1 Potential applications of epigenetic regulation in immunotherapy
At the onset of tumorigenesis, epigenetic abnormalities disrupt critical cellular processes such as the cell cycle, DNA repair, and apoptosis[236, 237]. Epigenetic therapies have been shown to stimulate antitumor immune responses in both tumor and host cells[238, 239]. Previous reports have indicated that epigenetic mechanisms have critical regulatory roles in CD4+ T cells[240], CD8+ T cells[241], and NK cells[242]. By using immune checkpoint blockade (ICB) and chimeric antigen receptor T cells are the most important therapeutic tools for the tumor immune system[243]. Recent studies have highlighted the potential of novel epigenetic modulators, such as CN133, a novel HDACis, which has been reported to sensitize prostate cancer (PCa) to immunotherapy by remodeling the TME in combination with anti-PD-L1 therapy[244]. Additionally, combination therapies involving DAC, PD-1 blockade, and conventional treatments have demonstrated enhanced tumor cell sensitivity to paclitaxel (PTX) alongside a therapeutic effect on reversing T-cell depletion and improving ICB efficacy in TNBC[245]. Recent reports on PCa have shown that combining HDACi with anti-PD-1 antibody and CTAL-4 antibody can enhance antitumor immunity in ICB-resistant PCa cells[246]. Furthermore, epigenetic abnormalities impact tumor responses to reactive oxygen species (ROS)-based therapies. In colorectal cancer, regulation of ubiquitination and phosphorylation pathways within the epigenome has been identified as a key mechanism in overcoming ROS resistance in the TME, thus enhancing the efficacy of ROS-targeted treatments[247]. Neurogliomas have a low immune response and high drug resistance, which makes them much more challenging to treat. Recent studies have shown that DAC combined with anti-PD-1 immunotherapy can effectively inhibit disease progression and improve antitumor efficacy in neurogliomas[248]. Ovarian cancer is also refractory to treatment, and recent studies have shown that low-dose DAC administration increased NK cell and CD8+ T cell recruitment and prolonged mouse survival in a murine transplantation tumor model of ovarian cancer, while the combination of DAC enhanced the therapeutic efficacy of anti-CTAL-4 treatment, and further studies have found that the combination of DNMTis can enhance the cytotoxic T cell response[249]. These studies suggest that the combination of epigenetic therapy and immunotherapy holds great promise and may become a critical strategy in the future of cancer treatment.
Epigenetic therapy targets abnormal epigenetic markers in cancer cells by modulating epigenetic modifying enzymes, aiming to restore normal cellular function or enhance immune system recognition of tumor cells. This therapeutic strategy differs from traditional radiotherapy, chemotherapy and immunotherapy as it focuses on gene regulatory mechanisms[250, 251]. Compared to conventional treatments that directly kill cancer cells or prevent their proliferation, epigenetic therapies can maximize the destruction of cancer cells by modulating the epigenetic state of tumor cells, often accompanied by fewer side effects[252, 253]. Thus, epigenetic drugs show important therapeutic potential as stand-alone therapies or in combination with other treatments[254]. In particular, clinical studies in recent years have highlighted the promise of combining epigenetic drugs with ICIs, especially in tumors that are resistant or refractory to ICIs[255]. For example, a phase II clinical study evaluated the use of the hypomethylating drug Guadecitabine in combination with the anti-PD-1 antibody pembrolizumab in patients with recurrent platinum-resistant ovarian cancer. Of 35 evaluable patients, three experienced partial remission and eight had stable disease, with an overall clinical benefit rate of 31.4%. The median duration of clinical benefit was 6.8 months. Following treatment, patients' peripheral blood mononuclear cells (PBMCs) showed hypomethylation of the Long-interspersed element 1 (LINE1) gene, and tumor biopsies and genomic analyses revealed activation of the tumor immune response. This study demonstrated that epigenetic initiation of combined immune checkpoint inhibitor therapy with hypomethylating agents is feasible and resulted in durable clinical benefit in selected patients with recurrent ovarian cancer[199]. Additionally, in a phase I/Ib study, the combination of pembrolizumab and vorinostat, a histone deacetylase inhibitor, was evaluated in 24 patients with ICI-resistant metastatic NSCLC. The results showed partial remission in one patient and stable disease in eight patients[256]. These findings suggest that the combination of epigenetic drugs and ICIs can effectively overcome drug resistance in conventional immunotherapy, and that combination therapy is promising in the clinic, providing new directions and possibilities for tumor therapy.
4.2 Synergy between epigenetic therapy and immunotherapy: the potential of viral mimicry responses in tumor therapy
Immunotherapy and epigenetic therapy are very promising therapies for treating tumors, and they have great potential and research value in antitumor mechanisms. However, tumor cells have the specificity to evade the immune response[257, 258], and more profound research has proposed that tumor cells can also evade immune cells in this way through epigenetic silencing mechanisms[259]. This finding reveals that epigenetic mechanisms can modulate the immune response of tumor cells by inducing viral mimicry, which promotes sensitivity to immunotherapy and improves therapeutic efficacy in tumor patients[183, 260]. More importantly, Roulois et al. proposed that DNA demethylating agents can activate the interferon response by inducing dsRNA, thereby allowing the organism to mimic viral infection[261]. This study suggests the possibility that epigenetic therapies can improve cancer immunotherapy through viral mimicry responses. This possibility has also been confirmed by recent studies, such as DNMTis in combination with conventional compounds for the treatment of advanced breast cancer, which improves the therapeutic efficacy[262], and in colorectal cancer, where it has been found that the histone demethylase PHF8 can act as an essential mediator of immune evasion and its absence can stimulate a viral mimicry response. A recent study has shown that using a PHF8-specific small molecule inhibitor iPHF8 can effectively regulate colorectal cancer cell growth and ETC gene transcription[263, 264]. In ovarian cancer, DNMTis induced a viral mimicry response that triggered a type I interferon response and promoted apoptosis, and a concurrent study found that the combination of CTAL-4 and DAC was more effective against CTAL-4 than when used alone in a melanoma mouse model[16]. In clear renal cell carcinoma (ccRCC), DNMTis, which induces the expression of ERVs and other transposable elements, also enhances T-cell activation, promoting antitumor immune mechanisms of action[234]. In addition, RRx-001, a novel immunomodulatory anticancer agent, can increase immunomodulatory effects directly or indirectly by modulating tumor-associated macrophages and T lymphocytes. The report also indicated that low-dose RRx-001 transient treatment of colorectal cancer cells induced a viral mimicry response, which increased the pharmacological efficacy and therapeutic potential of immunomodulatory RRx-001[131]. These studies suggest that viral mimicry is an intermediate mediator in linking epigenetic therapy and immunotherapy, providing new ideas for developing antitumor drugs and studying antitumor mechanisms[228, 265].
5. Conclusions and future perspectives
Viral mimicry therapy, epigenetic therapy and tumor immunotherapy complement each other to build a comprehensive treatment strategy, which brings new therapeutic prospects for cancer patients. Viral mimicry activates the immune system, epigenetic therapy enhances the therapeutic effect, and tumor immunotherapy improves the body's immune response. Combining the three can effectively inhibit tumor growth and metastasis, which is one of the critical directions for future cancer treatment. However, at the same time, it also faces many challenges, such as the selection of suitable inducers in viral mimicry therapy and in-depth study of the therapeutic mechanism, the regulation of epigenetic drug dosage and the development of new drugs, as well as how to effectively circumvent the adverse events that may be induced by immunotherapy. Furthermore, while epigenetic therapy and immunotherapy both show great potential, their safety and tolerability profiles need further exploration in large-scale trials, particularly regarding the cumulative toxicities from prolonged use. Additionally, the inherent heterogeneity of tumors complicates the treatment's effectiveness, as not all tumor cells may respond equally to the three therapies. Identifying biomarkers for selecting patients most likely to benefit from this integrated strategy will be crucial for improving outcomes. More importantly, the interactions between viral mimetic therapy, epigenetic therapy, and tumor immunotherapy have not been fully elucidated, and further research is needed to reveal the links between them. The synergistic effect of these therapies is essential to improve the clinical outcome of cancer patients. Therefore, more basic research and clinical trials are needed to refine these therapeutic strategies to provide more effective treatment options for most cancer patients and maximize their survival and quality of life.
Abbreviations
ERVs: endogenous retroviruses; HERVs: human endogenous retroviruses; dsRNAs: double-stranded RNAs; PRRs: pattern recognition receptors; MDA5: melanoma differentiation-associated gene 5; MAVS: mitochondrial antiviral signaling; TBK1: TANK binding kinase 1; IRF7: interferon regulatory factor 7; DNMT: DNA methyltransferase; DNMTis: DNA methyltransferase inhibitors; AZA: azacitidine; DAC: decitabine; HDAC: histone deacetylase; HDACis: Histone deacetylase inhibitors; ADAR: adenosine deaminase that acts on RNA; ISG: interferon-stimulated gene; 5-AZA-CdR: 5-aza-2-deoxycytidine; EZH2is: EZH2 inhibitors; ICB: immune checkpoint blockade; ICIs: immune checkpoint inhibitors; TME: tumor microenvironment.
Acknowledgements
We want to express our gratitude for the drawing materials provided by BioRender.
Funding
This work was supported by the Hunan Natural Science Foundation Outstanding Youth Fund [2023JJ10091], the Wisdom Accumulation and Talent Cultivation Project of the Third Xiangya Hospital of Central South University [BJ202203], the Beijing Natural Science Foundation [L244031], the National Natural Science Foundation of China [82302625].
Author contributions
C.W., L.W., D.X. and W.R. designed the review, D.X., W.R., and L.J. undertook the literature research, manuscript writing, and drafting, C.W. and L.W. further checked and revised the manuscript. W.R., Z.X.J and L.J.Z. participated in the revision. All authors have read and corrected the article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Micevic G, Bosenberg MW, Yan Q. The Crossroads of Cancer Epigenetics and Immune Checkpoint Therapy. Clin Cancer Res. 2023;29:1173-82
2. Lindholm HT, Chen R, De Carvalho DD. Endogenous retroelements as alarms for disruptions to cellular homeostasis. Trends Cancer. 2023;9:55-68
3. Petrizzo A, Ragone C, Cavalluzzo B, Mauriello A, Manolio C, Tagliamonte M. et al. Human Endogenous Retrovirus Reactivation: Implications for Cancer Immunotherapy. Cancers (Basel). 2021;13:1999
4. Jansz N, Faulkner GJ. Endogenous retroviruses in the origins and treatment of cancer. Genome Biol. 2021;22:147
5. Jiang Y, Zhang H, Wang J, Chen J, Guo Z, Liu Y. et al. Exploiting RIG-I-like receptor pathway for cancer immunotherapy. J Hematol Oncol. 2023;16:8
6. Perry AK, Chen G, Zheng D, Tang H, Cheng G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005;15:407-22
7. Yu T, Yi YS, Yang Y, Oh J, Jeong D, Cho JY. The pivotal role of TBK1 in inflammatory responses mediated by macrophages. Mediators Inflamm. 2012;2012:979105
8. Cui J, Chen Y, Wang HY, Wang RF. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum Vaccin Immunother. 2014;10:3270-85
9. Hu YW, Zhang J, Wu XM, Cao L, Nie P, Chang MX. TANK-Binding Kinase 1 (TBK1) Isoforms Negatively Regulate Type I Interferon Induction by Inhibiting TBK1-IRF3 Interaction and IRF3 Phosphorylation. Front Immunol. 2018;9:84
10. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380
11. Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284-99
12. Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol. 2016;8:a019505
13. Ge T, Gu X, Jia R, Ge S, Chai P, Zhuang A. et al. Crosstalk between metabolic reprogramming and epigenetics in cancer: updates on mechanisms and therapeutic opportunities. Cancer Commun (Lond). 2022;42:1049-82
14. Biswas S, Rao CM. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol Ther. 2017;173:118-34
15. Janin M, Esteller M. Epigenetic Awakening of Viral Mimicry in Cancer. Cancer Discov. 2020;10:1258-60
16. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B. et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015;162:974-86
17. Daskalakis M, Brocks D, Sheng YH, Islam MS, Ressnerova A, Assenov Y. et al. Reactivation of endogenous retroviral elements via treatment with DNMT- and HDAC-inhibitors. Cell Cycle. 2018;17:811-22
18. Benmelech S, Le T, McKay M, Nam J, Subramaniam K, Tellez D. et al. Biophysical and biochemical aspects of immune cell-tumor microenvironment interactions. APL Bioeng. 2024;8:021502
19. Guo Q, Qian ZM. Macrophage based drug delivery: Key challenges and strategies. Bioact Mater. 2024;38:55-72
20. Oliveira G, Wu CJ. Dynamics and specificities of T cells in cancer immunotherapy. Nat Rev Cancer. 2023;23:295-316
21. Yu Q, Ding J, Li S, Li Y. Autophagy in cancer immunotherapy: Perspective on immune evasion and cell death interactions. Cancer Lett. 2024;590:216856
22. Alizadeh D, Larmonier N. Chemotherapeutic targeting of cancer-induced immunosuppressive cells. Cancer Res. 2014;74:2663-8
23. Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature. 2019;574:45-56
24. Evdokimova V, Gassmann H, Radvanyi L, Burdach SEG. Current State of Immunotherapy and Mechanisms of Immune Evasion in Ewing Sarcoma and Osteosarcoma. Cancers (Basel). 2022;15:272
25. Karbach J, Neumann A, Brand K, Wahle C, Siegel E, Maeurer M. et al. Phase I clinical trial of mixed bacterial vaccine (Coley's toxins) in patients with NY-ESO-1 expressing cancers: immunological effects and clinical activity. Clin Cancer Res. 2012;18:5449-59
26. Rui R, Zhou L, He S. Cancer immunotherapies: advances and bottlenecks. Front Immunol. 2023;14:1212476
27. Zhong F, Lin Y, Zhao L, Yang C, Ye Y, Shen Z. Reshaping the tumour immune microenvironment in solid tumours via tumour cell and immune cell DNA methylation: from mechanisms to therapeutics. Br J Cancer. 2023;129:24-37
28. Wolff F, Leisch M, Greil R, Risch A, Pleyer L. The double-edged sword of (re)expression of genes by hypomethylating agents: from viral mimicry to exploitation as priming agents for targeted immune checkpoint modulation. Cell Commun Signal. 2017;15:13
29. Heij HA, Obertop H, van Blankenstein M, ten Kate FW, Westbroek DL. Relationship between functional and histological changes in chronic pancreatitis. Dig Dis Sci. 1986;31:1009-13
30. Fang Y, Zhang MC, He Y, Li C, Fang H, Xu PP. et al. Human endogenous retroviruses as epigenetic therapeutic targets in TP53-mutated diffuse large B-cell lymphoma. Signal Transduct Target Ther. 2023;8:381
31. Suntsova M, Garazha A, Ivanova A, Kaminsky D, Zhavoronkov A, Buzdin A. Molecular functions of human endogenous retroviruses in health and disease. Cell Mol Life Sci. 2015;72:3653-75
32. Levet S, Medina J, Joanou J, Demolder A, Queruel N, Réant K. et al. An ancestral retroviral protein identified as a therapeutic target in type-1 diabetes. JCI Insight. 2017;2:e94387
33. Halcrow PW, Quansah DNK, Kumar N, Steiner JP, Nath A, Geiger JD. HERV-K (HML-2) Envelope Protein Induces Mitochondrial Depolarization and Neurotoxicity via Endolysosome Iron Dyshomostasis. J Neurosci. 2024;44:e0826232024
34. Talal N, Flescher E, Dang H. Are endogenous retroviruses involved in human autoimmune disease? J Autoimmun. 1992 5 Suppl A: 61-6
35. Herrmann M, Hagenhofer M, Kalden JR. Retroviruses and systemic lupus erythematosus. Immunol Rev. 1996;152:145-56
36. Nakagawa K, Harrison LC. The potential roles of endogenous retroviruses in autoimmunity. Immunol Rev. 1996;152:193-236
37. Tavakolian S, Iranshahi M, Faghihloo E. The Evaluation of HERV-K np9, rec, gag Expression in Isolated Human Peripheral Blood Mononuclear Cell (PBMC) of Gastric and Colon Cancer. Adv Biomed Res. 2023;12:131
38. Peng B, Reeves KKL, Lee SWY, Chung THY, Hui HWL, Leung AHL. et al. Physical, psychological, and behavioral problems among children and adolescents in countries with different economic statuses during the COVID-19 pandemic: a systematic review and meta-analysis. Front Pediatr. 2023;11:1181186
39. Ko EJ, Ock MS, Choi YH, Iovanna JL, Mun S, Han K. et al. Human Endogenous Retrovirus (HERV)-K env Gene Knockout Affects Tumorigenic Characteristics of nupr1 Gene in DLD-1 Colorectal Cancer Cells. Int J Mol Sci. 2021;22:3941
40. Dolci M, Favero C, Toumi W, Favi E, Tarantini L, Signorini L. et al. Human Endogenous Retroviruses Long Terminal Repeat Methylation, Transcription, and Protein Expression in Human Colon Cancer. Front Oncol. 2020;10:569015
41. Liang Q, Xu Z, Xu R, Wu L, Zheng S. Expression patterns of non-coding spliced transcripts from human endogenous retrovirus HERV-H elements in colon cancer. PLoS One. 2012;7:e29950
42. Zhou F, Li M, Wei Y, Lin K, Lu Y, Shen J. et al. Activation of HERV-K Env protein is essential for tumorigenesis and metastasis of breast cancer cells. Oncotarget. 2016;7:84093-117
43. Wei Y, Wei H, Wei Y, Tan A, Chen X, Liao X. et al. Screening and Identification of Human Endogenous Retrovirus-K mRNAs for Breast Cancer Through Integrative Analysis of Multiple Datasets. Front Oncol. 2022;12:820883
44. Wang-Johanning F, Frost AR, Jian B, Epp L, Lu DW, Johanning GL. Quantitation of HERV-K env gene expression and splicing in human breast cancer. Oncogene. 2003;22:1528-35
45. Zhou F, Krishnamurthy J, Wei Y, Li M, Hunt K, Johanning GL. et al. Chimeric antigen receptor T cells targeting HERV-K inhibit breast cancer and its metastasis through downregulation of Ras. Oncoimmunology. 2015;4:e1047582
46. Tavakolian S, Goudarzi H, Faghihloo E. Evaluating the expression level of HERV-K env, np9, rec and gag in breast tissue. Infect Agent Cancer. 2019;14:42
47. Golan M, Hizi A, Resau JH, Yaal-Hahoshen N, Reichman H, Keydar I. et al. Human endogenous retrovirus (HERV-K) reverse transcriptase as a breast cancer prognostic marker. Neoplasia. 2008;10:521-33
48. Rinkoff S. Letter to the editor: How well is the NHS set up for issues surrounding gender identity? Int J Surg. 2019;68:91
49. Wang-Johanning F, Li M, Esteva FJ, Hess KR, Yin B, Rycaj K. et al. Human endogenous retrovirus type K antibodies and mRNA as serum biomarkers of early-stage breast cancer. Int J Cancer. 2014;134:587-95
50. Wang-Johanning F, Radvanyi L, Rycaj K, Plummer JB, Yan P, Sastry KJ. et al. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. 2008;68:5869-77
51. Wang-Johanning F, Frost AR, Johanning GL, Khazaeli MB, LoBuglio AF, Shaw DR. et al. Expression of human endogenous retrovirus k envelope transcripts in human breast cancer. Clin Cancer Res. 2001;7:1553-60
52. Rhyu DW, Kang YJ, Ock MS, Eo JW, Choi YH, Kim WJ. et al. Expression of human endogenous retrovirus env genes in the blood of breast cancer patients. Int J Mol Sci. 2014;15:9173-83
53. Lemaître C, Tsang J, Bireau C, Heidmann T, Dewannieux M. A human endogenous retrovirus-derived gene that can contribute to oncogenesis by activating the ERK pathway and inducing migration and invasion. PLoS Pathog. 2017;13:e1006451
54. Manca MA, Solinas T, Simula ER, Noli M, Ruberto S, Madonia M. et al. HERV-K and HERV-H Env Proteins Induce a Humoral Response in Prostate Cancer Patients. Pathogens. 2022;11:95
55. Rezaei SD, Hayward JA, Norden S, Pedersen J, Mills J, Hearps AC. et al. HERV-K Gag RNA and Protein Levels Are Elevated in Malignant Regions of the Prostate in Males with Prostate Cancer. Viruses. 2021;13:449
56. Wallace TA, Downey RF, Seufert CJ, Schetter A, Dorsey TH, Johnson CA. et al. Elevated HERV-K mRNA expression in PBMC is associated with a prostate cancer diagnosis particularly in older men and smokers. Carcinogenesis. 2014;35:2074-83
57. Schulz WA. Does HERV-K represent a potential therapeutic target for prostate cancer? Expert Opin Ther Targets. 2017;21:921-4
58. Agoni L, Guha C, Lenz J. Detection of Human Endogenous Retrovirus K (HERV-K) Transcripts in Human Prostate Cancer Cell Lines. Front Oncol. 2013;3:180
59. Reis BS, Jungbluth AA, Frosina D, Holz M, Ritter E, Nakayama E. et al. Prostate cancer progression correlates with increased humoral immune response to a human endogenous retrovirus GAG protein. Clin Cancer Res. 2013;19:6112-25
60. Cardelli M, Doorn RV, Larcher L, Donato MD, Piacenza F, Pierpaoli E. et al. Association of HERV-K and LINE-1 hypomethylation with reduced disease-free survival in melanoma patients. Epigenomics. 2020;12:1689-706
61. Argaw-Denboba A, Balestrieri E, Serafino A, Cipriani C, Bucci I, Sorrentino R. et al. HERV-K activation is strictly required to sustain CD133+ melanoma cells with stemness features. J Exp Clin Cancer Res. 2017;36:20
62. Krishnamurthy J, Rabinovich BA, Mi T, Switzer KC, Olivares S, Maiti SN. et al. Genetic Engineering of T Cells to Target HERV-K, an Ancient Retrovirus on Melanoma. Clin Cancer Res. 2015;21:3241-51
63. Schmitt K, Reichrath J, Roesch A, Meese E, Mayer J. Transcriptional profiling of human endogenous retrovirus group HERV-K(HML-2) loci in melanoma. Genome Biol Evol. 2013;5:307-28
64. Büscher K, Trefzer U, Hofmann M, Sterry W, Kurth R, Denner J. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005;65:4172-80
65. Serafino A, Balestrieri E, Pierimarchi P, Matteucci C, Moroni G, Oricchio E. et al. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp Cell Res. 2009;315:849-62
66. Singh M, Cai H, Bunse M, Feschotte C, Izsvák Z. Human Endogenous Retrovirus K Rec forms a Regulatory Loop with MITF that Opposes the Progression of Melanoma to an Invasive Stage. Viruses. 2020;12:1303
67. Singh S, Kaye S, Francis N, Peston D, Gore M, McClure M. et al. Human endogenous retrovirus K (HERV-K) rec mRNA is expressed in primary melanoma but not in benign naevi or normal skin. Pigment Cell Melanoma Res. 2013;26:426-8
68. Schiavetti F, Thonnard J, Colau D, Boon T, Coulie PG. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res. 2002;62:5510-6
69. Schanab O, Humer J, Gleiss A, Mikula M, Sturlan S, Grunt S. et al. Expression of human endogenous retrovirus K is stimulated by ultraviolet radiation in melanoma. Pigment Cell Melanoma Res. 2011;24:656-65
70. Muster T, Waltenberger A, Grassauer A, Hirschl S, Caucig P, Romirer I. et al. An endogenous retrovirus derived from human melanoma cells. Cancer Res. 2003;63:8735-41
71. Chan SM, Sapir T, Park SS, Rual JF, Contreras-Galindo R, Reiner O. et al. The HERV-K accessory protein Np9 controls viability and migration of teratocarcinoma cells. PLoS One. 2019;14:e0212970
72. Morozov VA, Morozov AV. A Comprehensive Analysis of Human Endogenous Retroviruses HERV-K (HML.2) from Teratocarcinoma Cell Lines and Detection of Viral Cargo in Microvesicles. Int J Mol Sci. 2021;22:12398
73. Götzinger N, Sauter M, Roemer K, Mueller-Lantzsch N. Regulation of human endogenous retrovirus-K Gag expression in teratocarcinoma cell lines and human tumours. J Gen Virol. 1996;77( Pt 12):2983-90
74. Zanrè V, Bellinato F, Cardile A, Passarini C, Monticelli J, Di Bella S. et al. Lamivudine, Doravirine, and Cabotegravir Downregulate the Expression of Human Endogenous Retroviruses (HERVs), Inhibit Cell Growth, and Reduce Invasive Capability in Melanoma Cell Lines. Int J Mol Sci. 2024;25:1615
75. Iramaneerat K, Rattanatunyong P, Khemapech N, Triratanachat S, Mutirangura A. HERV-K hypomethylation in ovarian clear cell carcinoma is associated with a poor prognosis and platinum resistance. Int J Gynecol Cancer. 2011;21:51-7
76. Yang C, Guo X, Li J, Han J, Jia L, Wen HL. et al. Significant Upregulation of HERV-K (HML-2) Transcription Levels in Human Lung Cancer and Cancer Cells. Front Microbiol. 2022;13:850444
77. Zare M, Mostafaei S, Ahmadi A, Azimzadeh Jamalkandi S, Abedini A, Esfahani-Monfared Z. et al. Human endogenous retrovirus env genes: Potential blood biomarkers in lung cancer. Microb Pathog. 2018;115:189-93
78. Curty G, Menezes AN, Brant AC, de Mulder Rougvie M, Moreira MÂ M, Soares MA. Expression of Retroelements in Cervical Cancer and Their Interplay with HPV Infection and Host Gene Expression. Cancers (Basel). 2021;13:3513
79. Soleimani-Jelodar R, Arashkia A, Shoja Z, Akhavan S, Yarandi F, Sharifian K. et al. The expression analysis of human endogenous retrovirus-K Env, Np9, and Rec transcripts in cervical cancer. J Med Virol. 2024;96:e29501
80. Shah AH, Rivas SR, Doucet-O'Hare TT, Govindarajan V, DeMarino C, Wang T. et al. Human endogenous retrovirus K contributes to a stem cell niche in glioblastoma. J Clin Invest. 2023;133:e167929
81. Shah AH, Govindarajan V, Doucet-O'Hare TT, Rivas S, Ampie L, DeMarino C. et al. Differential expression of an endogenous retroviral element [HERV-K(HML-6)] is associated with reduced survival in glioblastoma patients. Sci Rep. 2022;12:6902
82. Hothi P, Cobbs C. The potential role of human endogenous retrovirus K in glioblastoma. J Clin Invest. 2023;133:e170885
83. Li M, Radvanyi L, Yin B, Rycaj K, Li J, Chivukula R. et al. Downregulation of Human Endogenous Retrovirus Type K (HERV-K) Viral env RNA in Pancreatic Cancer Cells Decreases Cell Proliferation and Tumor Growth. Clin Cancer Res. 2017;23:5892-911
84. Li M, Radvanyi L, Yin B, Rycaj K, Li J, Chivukula R. et al. Correction: Downregulation of Human Endogenous Retrovirus Type K (HERV-K) Viral env RNA in Pancreatic Cancer Cells Decreases Cell Proliferation and Tumor Growth. Clin Cancer Res. 2019;25:2936
85. Masuda Y, Ishihara R, Murakami Y, Watanabe S, Asao Y, Gotoh N. et al. Clinical significance of human endogenous retrovirus K (HERV-K) in multiple myeloma progression. Int J Hematol. 2023;117:563-77
86. Cherkasova E, Scrivani C, Doh S, Weisman Q, Takahashi Y, Harashima N. et al. Detection of an Immunogenic HERV-E Envelope with Selective Expression in Clear Cell Kidney Cancer. Cancer Res. 2016;76:2177-85
87. Takahashi Y, Harashima N, Kajigaya S, Yokoyama H, Cherkasova E, McCoy JP. et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J Clin Invest. 2008;118:1099-109
88. Cherkasova E, Malinzak E, Rao S, Takahashi Y, Senchenko VN, Kudryavtseva AV. et al. Inactivation of the von Hippel-Lindau tumor suppressor leads to selective expression of a human endogenous retrovirus in kidney cancer. Oncogene. 2011;30:4697-706
89. Park EG, Lee DH, Kim WR, Lee YJ, Bae WH, Kim JM. et al. Human Endogenous Retrovirus-H-Derived miR-4454 Inhibits the Expression of DNAJB4 and SASH1 in Non-Muscle-Invasive Bladder Cancer. Genes (Basel). 2023;14:1410
90. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
91. Cherkasova EA, Chen L, Childs RW. Mechanistic regulation of HERV activation in tumors and implications for translational research in oncology. Front Cell Infect Microbiol. 2024;14:1358470
92. Matteucci C, Balestrieri E, Argaw-Denboba A, Sinibaldi-Vallebona P. Human endogenous retroviruses role in cancer cell stemness. Semin Cancer Biol. 2018;53:17-30
93. Kim DY, Kim H, Ko EJ, Koh SB, Kim H, Lee JY. et al. Correlation analysis of cancer stem cell marker CD133 and human endogenous retrovirus (HERV)-K env in SKOV3 ovarian cancer cells. Genes Genomics. 2024;46:511-8
94. Fan W, Li W, Li L, Qin M, Mao C, Yuan Z. et al. Bifunctional HDAC and DNMT inhibitor induces viral mimicry activates the innate immune response in triple-negative breast cancer. Eur J Pharm Sci. 2024;197:106767
95. Yang Z, Chu B, Tu Y, Li L, Chen D, Huang S. et al. Dual inhibitors of DNMT and HDAC remodels the immune microenvironment of colorectal cancer and enhances the efficacy of anti-PD-L1 therapy. Pharmacol Res. 2024;206:107271
96. Jin X, Xu XE, Jiang YZ, Liu YR, Sun W, Guo YJ. et al. The endogenous retrovirus-derived long noncoding RNA TROJAN promotes triple-negative breast cancer progression via ZMYND8 degradation. Sci Adv. 2019;5:eaat9820
97. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16:131-44
98. Kelly JM, Darcy PK, Markby JL, Godfrey DI, Takeda K, Yagita H. et al. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat Immunol. 2002;3:83-90
99. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK. et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69-77
100. Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC. et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349:1115-20
101. Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R. et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24:365-76
102. Wagner RW, Smith JE, Cooperman BS, Nishikura K. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc Natl Acad Sci U S A. 1989;86:2647-51
103. Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc Natl Acad Sci U S A. 1994;91:11457-61
104. Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996;379:460-4
105. Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. Rna. 2000;6:755-67
106. George CX, Samuel CE. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci U S A. 1999;96:4621-6
107. Desterro JM, Keegan LP, Lafarga M, Berciano MT, O'Connell M, Carmo-Fonseca M. Dynamic association of RNA-editing enzymes with the nucleolus. J Cell Sci. 2003;116:1805-18
108. Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;15:5376-88
109. Shiromoto Y, Sakurai M, Minakuchi M, Ariyoshi K, Nishikura K. ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat Commun. 2021;12:1654
110. Baker AR, Slack FJ. ADAR1 and its implications in cancer development and treatment. Trends Genet. 2022;38:821-30
111. Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity. 2015;43:933-44
112. Koval L, Kalashnyk O, Lykhmus O, Skok M. α7 nicotinic acetylcholine receptors are involved in suppression of the antibody immune response. J Neuroimmunol. 2018;318:8-14
113. Song B, Shiromoto Y, Minakuchi M, Nishikura K. The role of RNA editing enzyme ADAR1 in human disease. Wiley Interdiscip Rev RNA. 2022;13:e1665
114. Paget M, Cadena C, Ahmad S, Wang HT, Jordan TX, Kim E. et al. Stress granules are shock absorbers that prevent excessive innate immune responses to dsRNA. Mol Cell. 2023;83:1180-96.e8
115. Kung CP, Cottrell KA, Ryu S, Bramel ER, Kladney RD, Bao EA. et al. Evaluating the therapeutic potential of ADAR1 inhibition for triple-negative breast cancer. Oncogene. 2021;40:189-202
116. Cottrell KA, Ryu S, Pierce JR, Soto Torres L, Bohlin HE, Schab AM. et al. Induction of Viral Mimicry Upon Loss of DHX9 and ADAR1 in Breast Cancer Cells. Cancer Res Commun. 2024;4:986-1003
117. Choi H, Kwon J, Cho MS, Sun Y, Zheng X, Wang J. et al. Targeting DDX3X Triggers Antitumor Immunity via a dsRNA-Mediated Tumor-Intrinsic Type I Interferon Response. Cancer Res. 2021;81:3607-20
118. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY. et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell. 2015;162:961-73
119. Chiappinelli KB. Targeting the DHX9 RNA Helicase to Induce Antitumor Immunity in Small-Cell Lung Cancer. Cancer Discov. 2024;14:389-91
120. Cortesi A, Gandolfi F, Arco F, Di Chiaro P, Valli E, Polletti S. et al. Activation of endogenous retroviruses and induction of viral mimicry by MEK1/2 inhibition in pancreatic cancer. Sci Adv. 2024;10:eadk5386
121. Liu Y, Hu L, Wu Z, Yuan K, Hong G, Lian Z. et al. Loss of PHF8 induces a viral mimicry response by activating endogenous retrotransposons. Nat Commun. 2023;14:4225
122. Alexandraki A, Strati K. Decitabine Treatment Induces a Viral Mimicry Response in Cervical Cancer Cells and Further Sensitizes Cells to Chemotherapy. Int J Mol Sci. 2022;23:14042
123. Pan D, Bao X, Hu M, Jiao M, Li F, Li CY. SETDB1 Restrains Endogenous Retrovirus Expression and Antitumor Immunity during Radiotherapy. Cancer Res. 2022;82:2748-60
124. Huang W, Zhu Q, Shi Z, Tu Y, Li Q, Zheng W. et al. Dual inhibitors of DNMT and HDAC induce viral mimicry to induce antitumour immunity in breast cancer. Cell Death Discov. 2024;10:143
125. Zhang S, Liu X, Chen W, Zhang K, Wu Q, Wei Y. Targeting TAF1 with BAY-299 induces antitumor immunity in triple-negative breast cancer. Biochem Biophys Res Commun. 2023;665:55-63
126. Wu Q, Nie DY, Ba-Alawi W, Ji Y, Zhang Z, Cruickshank J. et al. PRMT inhibition induces a viral mimicry response in triple-negative breast cancer. Nat Chem Biol. 2022;18:821-30
127. Bowling EA, Wang JH, Gong F, Wu W, Neill NJ, Kim IS. et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell. 2021;184:384-403.e21
128. Bolis M, Paroni G, Fratelli M, Vallerga A, Guarrera L, Zanetti A. et al. All-Trans Retinoic Acid Stimulates Viral Mimicry, Interferon Responses and Antigen Presentation in Breast-Cancer Cells. Cancers (Basel). 2020;12:1169
129. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB. et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548:471-5
130. Chomiak AA, Tiedemann RL, Liu Y, Kong X, Cui Y, Wiseman AK. et al. Select EZH2 inhibitors enhance viral mimicry effects of DNMT inhibition through a mechanism involving NFAT:AP-1 signaling. Sci Adv. 2024;10:eadk4423
131. Zhao H, Ning S, Nolley R, Scicinski J, Oronsky B, Knox SJ. et al. The immunomodulatory anticancer agent, RRx-001, induces an interferon response through epigenetic induction of viral mimicry. Clin Epigenetics. 2017;9:4
132. Murayama T, Nakayama J, Jiang X, Miyata K, Morris AD, Cai KQ. et al. Targeting DHX9 Triggers Tumor-Intrinsic Interferon Response and Replication Stress in Small Cell Lung Cancer. Cancer Discov. 2024;14:468-91
133. Nikolic A, Maule F, Bobyn A, Ellestad K, Paik S, Marhon SA. et al. macroH2A2 antagonizes epigenetic programs of stemness in glioblastoma. Nat Commun. 2023;14:3062
134. Ramsoomair CK, Ceccarelli M, Heiss JD, Shah AH. The epitranscriptome of high-grade gliomas: a promising therapeutic target with implications from the tumor microenvironment to endogenous retroviruses. J Transl Med. 2023;21:893
135. Chan FF, Yuen VW, Shen J, Chin DW, Law CT, Wong BP. et al. Inhibition of CAF-1 histone chaperone complex triggers cytosolic DNA and dsRNA sensing pathways and induces intrinsic immunity of hepatocellular carcinoma. Hepatology. 2024;80:295-311
136. Yalala S, Gondane A, Poulose N, Liang J, Mills IG, Itkonen HM. CDK9 inhibition activates innate immune response through viral mimicry. Faseb j. 2024;38:e23628
137. Alizadeh-Ghodsi M, Owen KL, Townley SL, Zanker D, Rollin SPG, Hanson AR. et al. Potent Stimulation of the Androgen Receptor Instigates a Viral Mimicry Response in Prostate Cancer. Cancer Res Commun. 2022;2:706-24
138. Shen JZ, Spruck C. Targeting FBXO44/SUV39H1 elicits tumor cell-specific DNA replication stress and viral mimicry. Cell Stress. 2021;5:37-9
139. Shen JZ, Qiu Z, Wu Q, Finlay D, Garcia G, Sun D. et al. FBXO44 promotes DNA replication-coupled repetitive element silencing in cancer cells. Cell. 2021;184:352-69.e23
140. Cao W, Kang R, Xiang Y, Hong J. Human Endogenous Retroviruses in Clear Cell Renal Cell Carcinoma: Biological Functions and Clinical Values. Onco Targets Ther. 2020;13:7877-85
141. Li HT, Jang HJ, Rohena-Rivera K, Liu M, Gujar H, Kulchycki J. et al. RNA mis-splicing drives viral mimicry response after DNMTi therapy in SETD2-mutant kidney cancer. Cell Rep. 2023;42:112016
142. Di Marco T, Bianchi F, Sfondrini L, Todoerti K, Bongarzone I, Maffioli EM. et al. COPZ1 depletion in thyroid tumor cells triggers type I IFN response and immunogenic cell death. Cancer Lett. 2020;476:106-19
143. Liu M, Thomas SL, DeWitt AK, Zhou W, Madaj ZB, Ohtani H. et al. Dual Inhibition of DNA and Histone Methyltransferases Increases Viral Mimicry in Ovarian Cancer Cells. Cancer Res. 2018;78:5754-66
144. Bates SE. Epigenetic Therapies for Cancer. N Engl J Med. 2020;383:650-63
145. Nishiyama A, Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021;37:1012-27
146. Jain S, Wojdacz TK, Su YH. Challenges for the application of DNA methylation biomarkers in molecular diagnostic testing for cancer. Expert Rev Mol Diagn. 2013;13:283-94
147. Roy D, Tiirikainen M. Diagnostic Power of DNA Methylation Classifiers for Early Detection of Cancer. Trends Cancer. 2020;6:78-81
148. Papanicolau-Sengos A, Aldape K. DNA Methylation Profiling: An Emerging Paradigm for Cancer Diagnosis. Annu Rev Pathol. 2022;17:295-321
149. Karlstrom L, Kelly KA. Roux-Y gastrectomy for chronic gastric atony. Am J Surg. 1989;157:44-9
150. Chouhan H, Ferrandon S, DeVecchio J, Kalady MF, Church JM. A Changing Spectrum of Colorectal Cancer Biology With Age: Implications for the Young Patient. Dis Colon Rectum. 2019;62:21-6
151. Li SY, Wu HC, Mai HF, Zhen JX, Li GS, Chen SJ. Microarray-based analysis of whole-genome DNA methylation profiling in early detection of breast cancer. J Cell Biochem. 2019;120:658-70
152. Williams KE, Jawale RM, Schneider SS, Otis CN, Pentecost BT, Arcaro KF. DNA methylation in breast cancers: Differences based on estrogen receptor status and recurrence. J Cell Biochem. 2019;120:738-55
153. Park AK, Kim P, Ballester LY, Esquenazi Y, Zhao Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro Oncol. 2019;21:59-70
154. Feng J, Zhang Y, She X, Sun Y, Fan L, Ren X. et al. Hypermethylated gene ANKDD1A is a candidate tumor suppressor that interacts with FIH1 and decreases HIF1α stability to inhibit cell autophagy in the glioblastoma multiforme hypoxia microenvironment. Oncogene. 2019;38:103-19
155. Liu J, Jiang J, Mo J, Liu D, Cao D, Wang H. et al. Global DNA 5-Hydroxymethylcytosine and 5-Formylcytosine Contents Are Decreased in the Early Stage of Hepatocellular Carcinoma. Hepatology. 2019;69:196-208
156. Nakamura M, Chiba T, Kanayama K, Kanzaki H, Saito T, Kusakabe Y. et al. Epigenetic dysregulation in hepatocellular carcinoma: an up-to-date review. Hepatol Res. 2019;49:3-13
157. Wang L, Fan Y, Zhang L, Li L, Kuang G, Luo C. et al. Classic SRY-box protein SOX7 functions as a tumor suppressor regulating WNT signaling and is methylated in renal cell carcinoma. Faseb j. 2019;33:254-63
158. Grube WA, Liming KW. ATTACHMENT AND BIOBEHAVIORAL CATCH-UP: A SYSTEMATIC REVIEW. Infant Ment Health J. 2018;39:656-73
159. Mariella E, Grasso G, Miotto M, Buzo K, Reilly NM, Andrei P. et al. Transcriptome-wide gene expression outlier analysis pinpoints therapeutic vulnerabilities in colorectal cancer. Mol Oncol. 2024;18:1460-1485
160. Ajithkumar P, Vasantharajan SS, Pattison S, McCall JL, Rodger EJ, Chatterjee A. Exploring Potential Epigenetic Biomarkers for Colorectal Cancer Metastasis. Int J Mol Sci. 2024;25:874
161. Yasui K, Toshima T, Inada R, Umeda Y, Yano S, Tanioka H. et al. Circulating cell-free DNA methylation patterns as non-invasive biomarkers to monitor colorectal cancer treatment efficacy without referencing primary site mutation profiles. Mol Cancer. 2024;23:1
162. Ben-Ami R, Wang QL, Zhang J, Supplee JG, Fahrmann JF, Lehmann-Werman R. et al. Protein biomarkers and alternatively methylated cell-free DNA detect early stage pancreatic cancer. Gut. 2024;73:639-48
163. Sorm F, Vesely J. THE ACTIVITY OF A NEW ANTIMETABOLITE, 5-AZACYTIDINE, AGAINST LYMPHOID LEUKAEMIA IN AK MICE. Neoplasma. 1964;11:123-30
164. Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y. et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001;97:1172-9
165. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8-13
166. Kurahashi Y, Watanabe T, Yamamoto Y, Ureshino H, Kamachi K, Yoshida-Sakai N. et al. Dual targeting of aberrant DNA and histone methylation synergistically suppresses tumor cell growth in ATL. Blood Adv. 2023;7:1545-59
167. Derissen EJ, Beijnen JH, Schellens JH. Concise drug review: azacitidine and decitabine. Oncologist. 2013;18:619-24
168. Šimoničová K, Janotka Ľ, Kavcová H, Sulová Z, Breier A, Messingerova L. Different mechanisms of drug resistance to hypomethylating agents in the treatment of myelodysplastic syndromes and acute myeloid leukemia. Drug Resist Updat. 2022;61:100805
169. Navada SC, Steinmann J, Lübbert M, Silverman LR. Clinical development of demethylating agents in hematology. J Clin Invest. 2014;124:40-6
170. Lübbert M, Suciu S, Baila L, Rüter BH, Platzbecker U, Giagounidis A. et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol. 2011;29:1987-96
171. Flis S, Gnyszka A, Misiewicz-Krzemińska I, Spławiński J. Decytabine enhances cytotoxicity induced by oxaliplatin and 5-fluorouracil in the colorectal cancer cell line Colo-205. Cancer Cell Int. 2009;9:10
172. Flis S, Gnyszka A, Flis K. DNA methyltransferase inhibitors improve the effect of chemotherapeutic agents in SW48 and HT-29 colorectal cancer cells. PLoS One. 2014;9:e92305
173. De Carvalho Fischer C, Hu Y, Morreale M, Lin WY, Wali A, Thakar M. et al. Treatment with epigenetic agents profoundly inhibits tumor growth in leiomyosarcoma. Oncotarget. 2018;9:19379-95
174. Pulliam N, Fang F, Ozes AR, Tang J, Adewuyi A, Keer H. et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin Cancer Res. 2018;24:3163-75
175. Travers M, Brown SM, Dunworth M, Holbert CE, Wiehagen KR, Bachman KE. et al. DFMO and 5-Azacytidine Increase M1 Macrophages in the Tumor Microenvironment of Murine Ovarian Cancer. Cancer Res. 2019;79:3445-54
176. Allfrey VG, Mirsky AE. Structural Modifications of Histones and their Possible Role in the Regulation of RNA Synthesis. Science. 1964;144:559
177. Kelly RDW, Stengel KR, Chandru A, Johnson LC, Hiebert SW, Cowley SM. Histone deacetylases maintain expression of the pluripotent gene network via recruitment of RNA polymerase II to coding and noncoding loci. Genome Res. 2024;34:34-46
178. Xu L, Yan X, Wang J, Zhao Y, Liu Q, Fu J. et al. The Roles of Histone Deacetylases in the Regulation of Ovarian Cancer Metastasis. Int J Mol Sci. 2023;24:15066
179. Cheshmazar N, Hamzeh-Mivehroud M, Nozad Charoudeh H, Hemmati S, Melesina J, Dastmalchi S. Current trends in development of HDAC-based chemotherapeutics. Life Sci. 2022;308:120946
180. Mercurio C, Minucci S, Pelicci PG. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol Res. 2010;62:18-34
181. Grant PA. A tale of histone modifications. Genome Biol. 2001;2:Reviews0003
182. Biersack B, Nitzsche B, Höpfner M. Immunomodulatory properties of HDAC6 inhibitors in cancer diseases: New chances for sophisticated drug design and treatment optimization. Semin Cell Dev Biol. 2024;154:286-94
183. Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC. et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell. 2017;171:1284-300.e21
184. Bruyer A, Maes K, Herviou L, Kassambara A, Seckinger A, Cartron G. et al. DNMTi/HDACi combined epigenetic targeted treatment induces reprogramming of myeloma cells in the direction of normal plasma cells. Br J Cancer. 2018;118:1062-73
185. Ushio R, Hiroi M, Matsumoto A, Mori K, Yamamoto N, Ohmori Y. Enhanced Cytotoxic Effects in Human Oral Squamous Cell Carcinoma Cells Treated with Combined Methyltransferase Inhibitors and Histone Deacetylase Inhibitors. Biomedicines. 2022;10:763
186. Salahuddin A, Ghanem H, Omran GA, Helmy MW. Epigenetic restoration and activation of ERβ: an inspiring approach for treatment of triple-negative breast cancer. Med Oncol. 2022;39:150
187. Szczepanek J, Skorupa M, Jarkiewicz-Tretyn J, Cybulski C, Tretyn A. Harnessing Epigenetics for Breast Cancer Therapy: The Role of DNA Methylation, Histone Modifications, and MicroRNA. Int J Mol Sci. 2023;24:7235
188. Hong H, Sui C, Qian T, Xu X, Zhu X, Fei Q. et al. Long noncoding RNA LINC00460 conduces to tumor growth and metastasis of hepatocellular carcinoma through miR-342-3p-dependent AGR2 up-regulation. Aging (Albany NY). 2020;12:10544-55
189. Fresquet V, Garcia-Barchino MJ, Larrayoz M, Celay J, Vicente C, Fernandez-Galilea M. et al. Endogenous Retroelement Activation by Epigenetic Therapy Reverses the Warburg Effect and Elicits Mitochondrial-Mediated Cancer Cell Death. Cancer Discov. 2021;11:1268-85
190. Lee AV, Nestler KA, Chiappinelli KB. Therapeutic targeting of DNA methylation alterations in cancer. Pharmacol Ther. 2024;258:108640
191. Niinuma T, Kitajima H, Yamamoto E, Maruyama R, Aoki H, Harada T. et al. An Integrated Epigenome and Transcriptome Analysis to Clarify the Effect of Epigenetic Inhibitors on GIST. Anticancer Res. 2021;41:2817-28
192. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624-9
193. Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM. et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107:20980-5
194. Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA. et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606-11
195. Poirier JT, Gardner EE, Connis N, Moreira AL, de Stanchina E, Hann CL. et al. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene. 2015;34:5869-78
196. Feng S, Marhon SA, Sokolowski DJ, D'Costa A, Soares F, Mehdipour P. et al. Inhibiting EZH2 targets atypical teratoid rhabdoid tumor by triggering viral mimicry via both RNA and DNA sensing pathways. Nat Commun. 2024;15:9321
197. Deblois G, Tonekaboni SAM, Grillo G, Martinez C, Kao YI, Tai F. et al. Epigenetic Switch-Induced Viral Mimicry Evasion in Chemotherapy-Resistant Breast Cancer. Cancer Discov. 2020;10:1312-29
198. Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R. et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587-98
199. Chen S, Xie P, Cowan M, Huang H, Cardenas H, Keathley R. et al. Epigenetic priming enhances antitumor immunity in platinum-resistant ovarian cancer. J Clin Invest. 2022;132:e158800
200. Tan WW, Allred JB, Moreno-Aspitia A, Northfelt DW, Ingle JN, Goetz MP. et al. Phase I Study of Panobinostat (LBH589) and Letrozole in Postmenopausal Metastatic Breast Cancer Patients. Clin Breast Cancer. 2016;16:82-6
201. Gatti-Mays ME, Gameiro SR, Ozawa Y, Knudson KM, Hicks KC, Palena C. et al. Improving the Odds in Advanced Breast Cancer With Combination Immunotherapy: Stepwise Addition of Vaccine, Immune Checkpoint Inhibitor, Chemotherapy, and HDAC Inhibitor in Advanced Stage Breast Cancer. Front Oncol. 2020;10:581801
202. Masuda N, Tamura K, Yasojima H, Shimomura A, Sawaki M, Lee MJ. et al. Phase 1 trial of entinostat as monotherapy and combined with exemestane in Japanese patients with hormone receptor-positive advanced breast cancer. BMC Cancer. 2021;21:1269
203. Connolly RM, Zhao F, Miller KD, Lee MJ, Piekarz RL, Smith KL. et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor-Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J Clin Oncol. 2021;39:3171-81
204. Connolly RM, Rudek MA, Piekarz R. Entinostat: a promising treatment option for patients with advanced breast cancer. Future Oncol. 2017;13:1137-48
205. Wang J, Zhang Q, Li Q, Mu Y, Jing J, Li H. et al. Phase I Study and Pilot Efficacy Analysis of Entinostat, a Novel Histone Deacetylase Inhibitor, in Chinese Postmenopausal Women with Hormone Receptor-Positive Metastatic Breast Cancer. Target Oncol. 2021;16:591-9
206. Zeleke TZ, Pan Q, Chiuzan C, Onishi M, Li Y, Tan H. et al. Network-based assessment of HDAC6 activity predicts preclinical and clinical responses to the HDAC6 inhibitor ricolinostat in breast cancer. Nat Cancer. 2023;4:257-75
207. Oki Y, Kelly KR, Flinn I, Patel MR, Gharavi R, Ma A. et al. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: results from an expanded phase I trial. Haematologica. 2017;102:1923-30
208. Younes A, Berdeja JG, Patel MR, Flinn I, Gerecitano JF, Neelapu SS. et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016;17:622-31
209. Amengual JE, Clark-Garvey S, Kalac M, Scotto L, Marchi E, Neylon E. et al. Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood. 2013;122:2104-13
210. Iyer SP, Huen A, Ai WZ, Jagadeesh D, Lechowicz MJ, Okada C. et al. Safety and efficacy of tenalisib in combination with romidepsin in patients with relapsed/refractory T-cell lymphoma: results from a phase I/II open-label multicenter study. Haematologica. 2024;109:209-19
211. Huijberts S, Wang L, de Oliveira RL, Rosing H, Nuijen B, Beijnen J. et al. Vorinostat in patients with resistant BRAF(V600E) mutated advanced melanoma: a proof of concept study. Future Oncol. 2020;16:619-29
212. Weber JS, Levinson BA, Laino AS, Pavlick AC, Woods DM. Clinical and immune correlate results from a phase 1b study of the histone deacetylase inhibitor mocetinostat with ipilimumab and nivolumab in unresectable stage III/IV melanoma. Melanoma Res. 2022;32:324-33
213. Ligon JA, Sundby RT, Wedekind MF, Arnaldez FI, Del Rivero J, Wiener L. et al. A Phase II Trial of Guadecitabine in Children and Adults with SDH-Deficient GIST, Pheochromocytoma, Paraganglioma, and HLRCC-Associated Renal Cell Carcinoma. Clin Cancer Res. 2023;29:341-8
214. Woyach JA, Kloos RT, Ringel MD, Arbogast D, Collamore M, Zwiebel JA. et al. Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J Clin Endocrinol Metab. 2009;94:164-70
215. Wood A, George S, Adra N, Chintala S, Damayanti N, Pili R. Phase I study of the mTOR inhibitor everolimus in combination with the histone deacetylase inhibitor panobinostat in patients with advanced clear cell renal cell carcinoma. Invest New Drugs. 2020;38:1108-16
216. Giaccone G, Rajan A, Berman A, Kelly RJ, Szabo E, Lopez-Chavez A. et al. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J Clin Oncol. 2011;29:2052-9
217. Gharwan H, Tomita Y, Lee MJ, Thomas A, Berman A, Giaccone G. et al. Alterations of immune cell subsets in relapsed, thymoma-associated minimal change disease: A case report. Oncol Lett. 2015;10:1155-8
218. Cheng H, Xie Z, Jones WP, Wei XT, Liu Z, Wang D. et al. Preclinical Pharmacokinetics Study of R- and S-Enantiomers of the Histone Deacetylase Inhibitor, AR-42 (NSC 731438), in Rodents. Aaps j. 2016;18:737-45
219. Welling DB, Collier KA, Burns SS, Oblinger JL, Shu E, Miles-Markley BA. et al. Early phase clinical studies of AR-42, a histone deacetylase inhibitor, for neurofibromatosis type 2-associated vestibular schwannomas and meningiomas. Laryngoscope Investig Otolaryngol. 2021;6:1008-19
220. Eigl BJ, North S, Winquist E, Finch D, Wood L, Sridhar SS. et al. A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Invest New Drugs. 2015;33:969-76
221. Johnson ML, Strauss J, Patel MR, Garon EB, Eaton KD, Neskorik T. et al. Mocetinostat in Combination With Durvalumab for Patients With Advanced NSCLC: Results From a Phase I/II Study. Clin Lung Cancer. 2023;24:218-27
222. Sheikh TN, Chen X, Xu X, McGuire JT, Ingham M, Lu C. et al. Growth Inhibition and Induction of Innate Immune Signaling of Chondrosarcomas with Epigenetic Inhibitors. Mol Cancer Ther. 2021;20:2362-71
223. Siegel DS, Dimopoulos M, Jagannath S, Goldschmidt H, Durrant S, Kaufman JL. et al. VANTAGE 095: An International, Multicenter, Open-Label Study of Vorinostat (MK-0683) in Combination With Bortezomib in Patients With Relapsed and Refractory Multiple Myeloma. Clin Lymphoma Myeloma Leuk. 2016;16:329-34.e1
224. Yee AJ, Bensinger WI, Supko JG, Voorhees PM, Berdeja JG, Richardson PG. et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol. 2016;17:1569-78
225. Maly JJ, Christian BA, Zhu X, Wei L, Sexton JL, Jaglowski SM. et al. A Phase I/II Trial of Panobinostat in Combination With Lenalidomide in Patients With Relapsed or Refractory Hodgkin Lymphoma. Clin Lymphoma Myeloma Leuk. 2017;17:347-53
226. Zhou X, Singh M, Sanz Santos G, Guerlavais V, Carvajal LA, Aivado M. et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021;11:3090-105
227. Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer. 2019;19:151-61
228. Chen R, Ishak CA, De Carvalho DD. Endogenous Retroelements and the Viral Mimicry Response in Cancer Therapy and Cellular Homeostasis. Cancer Discov. 2021;11:2707-25
229. Tao H, Jin C, Zhou L, Deng Z, Li X, Dang W. et al. PRMT1 Inhibition Activates the Interferon Pathway to Potentiate Antitumor Immunity and Enhance Checkpoint Blockade Efficacy in Melanoma. Cancer Res. 2024;84:419-33
230. Xu B, He Y, Wu X, Luo C, Liu A, Zhang J. Exploration of the correlations between interferon-γ in patient serum and HEPACAM in bladder transitional cell carcinoma, and the interferon-γ mechanism inhibiting BIU-87 proliferation. J Urol. 2012;188:1346-53
231. Yoon N, Park MS, Shigemoto T, Peltier G, Lee RH. Activated human mesenchymal stem/stromal cells suppress metastatic features of MDA-MB-231 cells by secreting IFN-β. Cell Death Dis. 2016;7:e2191
232. Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD. et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6:722-9
233. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557-69
234. de Cubas AA, Dunker W, Zaninovich A, Hongo RA, Bhatia A, Panda A. et al. DNA hypomethylation promotes transposable element expression and activation of immune signaling in renal cell cancer. JCI Insight. 2020;5:e137569
235. Choy L, Norris S, Wu X, Kolumam G, Firestone A, Settleman J. et al. Inhibition of Aurora Kinase Induces Endogenous Retroelements to Induce a Type I/III IFN Response via RIG-I. Cancer Res Commun. 2024;4:540-55
236. Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene. 2002;21:5462-82
237. Lee JH, Park SJ, Abraham SC, Seo JS, Nam JH, Choi C. et al. Frequent CpG island methylation in precursor lesions and early gastric adenocarcinomas. Oncogene. 2004;23:4646-54
238. Hu A, Sun L, Lin H, Liao Y, Yang H, Mao Y. Harnessing innate immune pathways for therapeutic advancement in cancer. Signal Transduct Target Ther. 2024;9:68
239. Liang Y, Wang L, Ma P, Ju D, Zhao M, Shi Y. Enhancing anti-tumor immune responses through combination therapies: epigenetic drugs and immune checkpoint inhibitors. Front Immunol. 2023;14:1308264
240. Cribbs AP, Terlecki-Zaniewicz S, Philpott M, Baardman J, Ahern D, Lindow M. et al. Histone H3K27me3 demethylases regulate human Th17 cell development and effector functions by impacting on metabolism. Proc Natl Acad Sci U S A. 2020;117:6056-66
241. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol. 2018;18:340-56
242. Shreeve N, Depierreux D, Hawkes D, Traherne JA, Sovio U, Huhn O. et al. The CD94/NKG2A inhibitory receptor educates uterine NK cells to optimize pregnancy outcomes in humans and mice. Immunity. 2021;54:1231-44.e4
243. Avella Patino DM, Radhakrishnan V, Suvilesh KN, Manjunath Y, Li G, Kimchi ET. et al. Epigenetic Regulation of Cancer Immune Cells. Semin Cancer Biol. 2022;83:377-83
244. Chen Z, Yang X, Chen Z, Li M, Wang W, Yang R. et al. A new histone deacetylase inhibitor remodels the tumor microenvironment by deletion of polymorphonuclear myeloid-derived suppressor cells and sensitizes prostate cancer to immunotherapy. BMC Med. 2023;21:402
245. Gao T, Sang X, Huang X, Gu P, Liu J, Liu Y. et al. Macrophage-camouflaged epigenetic nanoinducers enhance chemoimmunotherapy in triple negative breast cancer. Acta Pharm Sin B. 2023;13:4305-17
246. Murphy S, Rahmy S, Gan D, Liu G, Zhu Y, Manyak M. et al. Ketogenic diet alters the epigenetic and immune landscape of prostate cancer to overcome resistance to immune checkpoint blockade therapy. Cancer Res. 2024;84:1597-1612
247. Bu Z, Yang J, Zhang Y, Luo T, Fang C, Liang X. et al. Sequential Ubiquitination and Phosphorylation Epigenetics Reshaping by MG132-Loaded Fe-MOF Disarms Treatment Resistance to Repulse Metastatic Colorectal Cancer. Adv Sci (Weinh). 2023;10:e2301638
248. Long S, Huang G, Ouyang M, Xiao K, Zhou H, Hou A. et al. Epigenetically modified AP-2α by DNA methyltransferase facilitates glioma immune evasion by upregulating PD-L1 expression. Cell Death Dis. 2023;14:365
249. Wang L, Amoozgar Z, Huang J, Saleh MH, Xing D, Orsulic S. et al. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer Immunol Res. 2015;3:1030-41
250. Pang L, Zhou F, Liu Y, Ali H, Khan F, Heimberger AB. et al. Epigenetic regulation of tumor immunity. J Clin Invest. 2024;134:e178540
251. Mabe NW, Perry JA, Malone CF, Stegmaier K. Pharmacological targeting of the cancer epigenome. Nat Cancer. 2024;5:844-65
252. Qin S, Xie B, Wang Q, Yang R, Sun J, Hu C. et al. New insights into immune cells in cancer immunotherapy: from epigenetic modification, metabolic modulation to cell communication. MedComm (2020). 2024;5:e551
253. Tang F, Choy E, Tu C, Hornicek F, Duan Z. Therapeutic applications of histone deacetylase inhibitors in sarcoma. Cancer Treat Rev. 2017;59:33-45
254. Dai W, Qiao X, Fang Y, Guo R, Bai P, Liu S. et al. Epigenetics-targeted drugs: current paradigms and future challenges. Signal Transduct Target Ther. 2024;9:332
255. Morel D, Jeffery D, Aspeslagh S, Almouzni G, Postel-Vinay S. Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat Rev Clin Oncol. 2020;17:91-107
256. Gray JE, Saltos A, Tanvetyanon T, Haura EB, Creelan B, Antonia SJ. et al. Phase I/Ib Study of Pembrolizumab Plus Vorinostat in Advanced/Metastatic Non-Small Cell Lung Cancer. Clin Cancer Res. 2019;25:6623-32
257. DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405-9
258. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-8
259. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235-71
260. Beyer K, Partecke LI, Roetz F, Fluhr H, Weiss FU, Heidecke CD. et al. LPS promotes resistance to TRAIL-induced apoptosis in pancreatic cancer. Infect Agent Cancer. 2017;12:30
261. Roulois D, Yau HL, De Carvalho DD. Pharmacological DNA demethylation: Implications for cancer immunotherapy. Oncoimmunology. 2016;5:e1090077
262. Connolly RM, Li H, Jankowitz RC, Zhang Z, Rudek MA, Jeter SC. et al. Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand Up to Cancer Study. Clin Cancer Res. 2017;23:2691-701
263. Markus A, Krekel D, Lingens F. Purification and some properties of component A of the 4-chlorophenylacetate 3,4-dioxygenase from Pseudomonas species strain CBS. J Biol Chem. 1986;261:12883-8
264. Li N, Ma T, Deng X. Analysis of the coupling degree between regional logistics efficiency and economic development coordination. PLoS One. 2024;19:e0293175
265. Dumetier B, Sauter C, Hajmirza A, Pernon B, Aucagne R, Fournier C. et al. Repeat Element Activation-Driven Inflammation: Role of NFκB and Implications in Normal Development and Cancer? Biomedicines. 2022;10:3101
Author contact
![]() Corresponding authors: Wei Cui, Ph.D., E-mail: wendycuiweicn; Wei Li, Ph.D., E-mail: weililxedu.cn.
Corresponding authors: Wei Cui, Ph.D., E-mail: wendycuiweicn; Wei Li, Ph.D., E-mail: weililxedu.cn.