Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. The lysosome as an effector...
2. Dysregulation of lysosomal...
3. Lysosomal dysfunction in...
4. Lysosomal Stress Response...
5. TFEB as a target of...
6. Autophagy and lysosomal...
Conclusion and prospect
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(3):1014-1031. doi:10.7150/ijbs.103028 This issue Cite
Review
Regulatory Mechanisms and Therapeutic Implications of Lysosomal Dysfunction in Alzheimer's Disease
Yeji Kim1, Tae-Young Ha2,3, Myung-Shik Lee4,5, ![]() , Keun-A Chang1,2,3,
, Keun-A Chang1,2,3, ![]()
1. Department of Health Sciences and Technology, Gachon Advanced Institute for Health Sciences & Technology, Gachon University, Incheon 21999, Korea.
2. Department of Pharmacology, College of Medicine, Gachon University, Incheon 21999, Korea.
3. Neuroscience Research Institute, Gachon University, Incheon 21565, Korea.
4. Soonchunhyang Institute of Medi-bio Science & Division of Endocrinology, Department of Internal Medicine & Immunology, Soonchunhyang University College of Medicine, Cheonan 31151, Korea.
5. Chief Scientific Officer, LysoTech, Inc., Seoul 03766, Korea.
Received 2024-8-30; Accepted 2024-12-26; Published 2025-1-13
Abstract

Alzheimer's disease (AD) is characterized by the accumulation of amyloid-beta (Aβ) plaques, neurofibrillary tangles (NFTs) formed from hyperphosphorylated Tau, and widespread neuronal loss. The autophagy-lysosomal pathway plays a crucial role in maintaining cellular homeostasis by degrading and recycling of damaged organelles and aggregate amyloid proteins implicated in AD. Lysosomes are key effectors of autophagic process, responsible for the breakdown of a variety of damaged organelles and aggregate or dysfunctional proteins. This review examines the role of lysosomal dysfunction in AD pathophysiology, focusing on genetic factors, acidification abnormalities, and other contributing factors. We also explore the involvement of lysosomal dysfunction of microglia in AD pathology, and cover the role of lysosomal stress response (LSR) in cellular response to neuronal injury associated with AD. Furthermore, we discuss potential therapeutic strategies targeting lysosomal proteolysis pathway and addressing lysosomal dysfunction for AD treatment, including the pharmacologically activating lysosomal activity, regulating TFEB, and considering other emerging approaches.
Keywords: Alzheimer's disease, Autophagy-lysosomal pathway, Lysosomal dysfunction, Lysosomal stress response
Introduction
Alzheimer's disease (AD), which is characterized by amyloid deposition in the brain, memory loss, and cognitive dysfunction, is the most common cause of dementia worldwide. The pathological hallmarks of AD include the deposition of amyloid-beta (Aβ) aggregates, commonly known as amyloid plaques, and accumulation of hyperphosphorylated Tau (p-Tau) protein, resulting in neurofibrillary tangles (NFTs), along with widespread neuronal degeneration [1] (Fig. 1). Despite extensive characterization of the pathological features of AD, the precise mechanisms underlying AD pathogenesis remain incompletely understood.
Pathological hallmarks of Alzheimer's disease. Alzheimer's disease (AD) is characterized by several key pathological features. (A) The deposition of amyloid-beta (Aβ) plaques. (B) Increased production of hyperphosphorylated Tau (p-Tau) leading to the formation and accumulation of neurofibrillary tangles (NFTs). (C) Activation or dysfunction of microglia and astrocyte induced by Aβ plaques or NFTs, leading to neuroinflammation and neuronal damage. These pathological features contribute to lysosomal and mitochondria stress, resulting in neuronal degeneration, further exacerbating AD pathology.
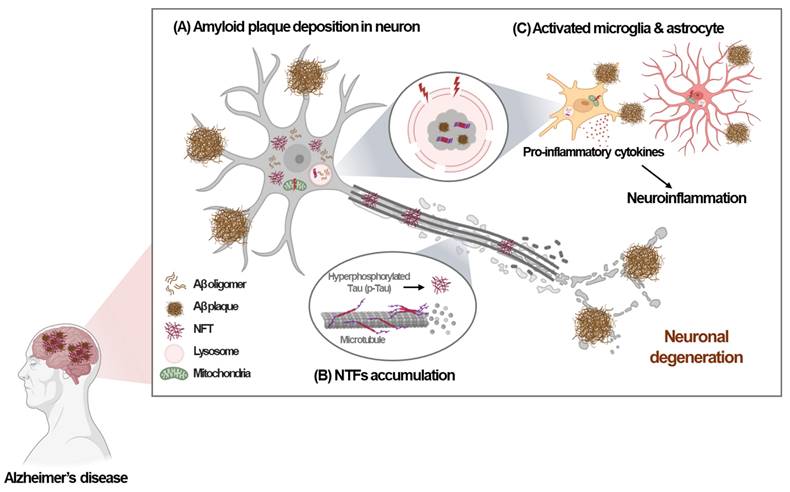
Recent studies have shown the crucial role of the autophagy-lysosomal pathway in the clearance of Aβ and Tau aggregates, suggesting that dysfunction in this pathway could significantly contribute to AD development [2]. Autophagy is a conserved cellular process responsible for degrading and recycling cytoplasmic components, including damaged organelles and protein aggregates via autophagosome formation followed by lysosomal degradation [3]. Emerging evidence indicates that dysfunction of lysosome, effector organelle of autophagy, is causally associated with the accumulation of toxic protein aggregates and the progression of neurodegenerative diseases, including AD [2].
Previous studies focused on lysosomal dysfunction in AD have primarily examined lysosomal function in neurons. However, recent research has revealed the importance of the microglial lysosomal system in the pathogenesis of AD. Microglia are critical immune cells that play an important role in maintaining brain homeostasis. In AD, microglia may exert protective effects by clearing Aβ through phagocytosis and lysosomal degradation and by preventing the accumulation of toxic aggregates [4]. However, if microglial reactions become chronic or dysfunctional due to genetic risk factors, they may contribute to neuroinflammation and neuronal damage, exacerbating AD pathology [4] (Fig. 1).
This review provides a comprehensive updated overview of the mechanisms of the lysosomal dysfunction implicated in AD pathogenesis not only in neurons but also in glial cells, and its effects on neurological deficit characterizing AD. Thus, our review might provide additional cutting-edge information regarding the role of lysosomal dysfunction in the pathogenesis of AD to the current reviews [5, 6]. We additionally discuss the mechanism of lysosomal adaptation to the lysosomal dysfunction or stress associated with AD, and finally address pharmacological strategies that target lysosome for a potential therapeutic intervention. Modulating the autophagic flux and restoring lysosomal function may provide promising avenues for AD treatment.
1. The lysosome as an effector organelle in autophagy
Ubiquitin-proteasome system and autophagy-lysosome pathway are the two primary mechanisms responsible for protein degradation within cells. Ubiquitin-proteasome system primarily targets short-lived, soluble, or unfolded proteins [7], while autophagy-lysosome pathway preferentially degrades long-lived, amyloidogenic, or aggregated proteins [8]. Autophagy-lysosome pathway is also essential for maintaining cellular homeostasis and nutrient balance [3].
Lysosomes are membrane-bound organelles enclosed by a lipid bilayer, containing more than 60 types of hydrolases that break down macromolecules within an acidic environment [9]. This degradation process occurs in autophagy and phagocytosis, helping to ensure cellular homeostasis and support immune responses [9]. Beyond their role in degradation, lysosomes are also critical for nutrient sensing and cellular signaling [9].
Transcription Factor EB (TFEB) serves as a master regulator of autophagy-lysosome pathway, responding to cellular stress by translocating to the nucleus and activating the transcription of autophagy and lysosomal genes [3, 10]. Under nutrient-rich conditions, TFEB is phosphorylated and retained in the cytosol. However, under stress conditions, TFEB is dephosphorylated by calcineurin or other phosphatases, enabling its movement to the nucleus. Once in the nucleus, TFEB binds to coordinated lysosomal expression and regulatory (CLEAR) elements, promoting the expression of genes involved in autophagy and lysosomal biogenesis [11].
2. Dysregulation of lysosomal function in AD
Numerous studies have reported abnormal autophagic activity in AD, which may contribute to the failure to clear amyloidogenic or aggregate-prone proteins that are pathogenically linked to AD development [12, 13]. Interestingly, an increased number of autophagosomes has been observed in the brain tissues of AD patients [14]. This accumulation may result from diminished lysosomal function or impaired autophagosome-lysosomal fusion [15]. Consistent with this, defective lysosomal function has been detected even before the formation of amyloid plaque [16], suggesting that lysosomal dysfunction may play a fundamental role in the accumulation of amyloid plaques and in the pathogenesis of AD.
2.1 Genes or proteins related to lysosomal function in AD
As strong evidence suggesting that lysosomal dysfunction plays a key role in the development of AD, several genes associated with early-onset AD have been linked to lysosomal dysfunction. Furthermore, a significant association has been observed between single nucleotide polymorphisms (SNPs) of various lysosomal genes and the development of sporadic AD.
One notable gene related to autophagy and lysosomal function in AD is the mammalian target of rapamycin (mTOR), a serine threonine kinase that negatively regulates autophagy. mTOR inhibits autophagy by phosphorylating key residues of TFEB and suppressing the unc-51-like autophagy-activating kinase (ULK1) complex [17]. In AD patients, increased expression and phosphorylation of mTOR have been observed in the hippocampus, together with elevated levels of its downstream targets, such as S6 kinase 1 (S6K1) and regulatory-associated protein of TOR (RAPTOR). These changes were correlated with cognitive impairment, linking mTOR to autophagy-lysosomal dysfunction in AD [18, 19]. In 3xTg-AD mouse model, increased mTOR activity is observed in the hippocampus and cortex. This heightened activity is evident at both 6 and 12 months of age, corresponding with the presence of Aβ and Tau pathologies. The increase in mTOR signaling is marked by elevated levels of phosphorylated p70S6K, a downstream target of mTOR, in these brain regions [20] (Table 1).
Altered genes or proteins related to lysosomal dysfunction in AD.
| Gene name | Functions in autophagy-lysosome | Changes in AD progression | References |
|---|---|---|---|
| mTOR | mTOR is a negative regulator of autophagy by phosphorylating and suppressing the ULK1 complex and TFEB. | The expression levels of mTOR/p-mTOR and its downstream targets S6K1/p-S6K1 and RAPTOR/p-RAPTOR were significantly increased in the hippocampus of AD patients' brain in the late stage. In the 3xTg-AD mouse model, increased mTOR activity, indicated by elevated phosphorylated p70S6K levels, is observed in the cortex and hippocampus at 6 and 12 months of age, correlating with Aβ and Tau pathologies. | [18] [19] [20] |
| NRBF2 | NRBF2, component of the class III PIK3C3 complex, directly interacts with ATG14L, thereby boosting the activity of ATG14L-associated VPS34 kinase, initiating autophagy. | In the AD human brain, NRBF2-related kinase complexes, BECN1-PIK3C3 was significantly downregulated specifically in the parahippocampal gyrus. In the 5xFAD AD mouse model, reduced expression levels of NRBF2 may linked to AD pathology, as overexpressing NRBF2 decreases APP-CTFs and Aβ levels. | [22] [23] |
| CTSD | CTSD is a lysosomal protease belonging to the aspartic proteas and crucial for degrading AD-associated proteins such as APP and Tau. | Mutations in the CTSD are associated with an increased risk of AD, in men but not in women. CTSD deletion in APP transgenic mice increases intracellular Aβ aggregates and induces severe early onset tauopathy with hyperphosphorylated tau accumulation, independent of human tau overexpression. | [25] [26] |
| CTSE | CTSE is a lysosomal protease belonging to the aspartic proteas and crucial for degrading AD-associated proteins such as APP and Tau. | Increased protein levels CTSE were observed in the cortex of AD patients and in the hippocampus of 6-month-old APP KI mice, suggesting its potential relevance in the pathology of AD. | [27] |
| CTSE has emerged as a novel biomarker of FAP. | [28] | ||
| PSEN1 | Mutations of PSEN1 disrupt lysosome acidification, leading to markedly impaired autophagy and causing early onset of familial AD. | Defect in lysosomal acidification occurred in PSEN1 knockdown, KO, and PSEN1 mutant cells. | [29] [30] |
| PGRN | Progranulin regulates cathepsin maturation, BMP levels, and lysosomal acidity. | Progranulin mutation is a risk factor for AD progression. Progranulin level in CSF or serum is a diagnostic/prognostic factor in AD. | [32] [34] [35] [36] |
| SORL1 | SORL1 helps in the sorting and trafficking of APP between the endosome, Golgi complex, and lysosome. | SORL1 protects APP from amyloidogenic processing and guides Aβ to lysosome for degradation. | [37] |
| BIN1 | BIN1 transports BACE1 to lysosome for degradation to inhibit Aβ generation. BIN1 interacts with Tau. | BIN1 mutants increase amyloid burden and attenuates GFAP induction in glial cells. BIN1 decreases Tau load in the cortex. | [38] [40] |
| PICALM | PICALM controls clathrin-coated endocytic vesicle formation, clathrin-mediated endocytosis (CME), and autophagy through endocytosis of VAMP proteins. | A PICALM variant is associated with cortical thickness and CSF Aβ42 level. Endothelial PICALM expression is reduced in the brains of AD patients. Endothelial PICALM deficiency impairs clearance and enhances Aβ pathology. | [41] [42] [43] |
Another gene of interest is Nuclear Receptor Binding Factor 2 (NRBF2), which modulates autophagy by interacting with autophagy-related protein 14-like protein (Atg14L) and enhancing the kinase activity of the BECN1-PIK3C3 complex (VPS34) [21]. In AD patients, NRBF2 activity was found to be reduced in the parahippocampal gyrus and hippocampus [22]. In the hippocampus of 5xFAD AD mice, the expression levels of NRBF2 are reduced, indicating a potential link between decreased NRBF2 levels and AD pathology. Overexpression of NRBF2 in human mutant APP-overexpressing cells promotes the degradation of APP-CTFs and reduces levels of Aβ. Conversely, when NRBF2 is knocked out, there is an increase in APP-CTFs and Aβ, suggesting that lower NRBF2 levels may exacerbate AD-related processes [23]. Additionally, overexpression of NRBF2 in 5xFAD mouse model led to a reduction in Aβ deposition and improvements in memory [22], suggesting that NRBF2 plays a role in regulating autophagy and lysosomal function related to Aβ clearance and neuronal health (Table 1).
A classic example of a lysosomal protein related to AD is Cathepsin D (CTSD), a lysosomal protease involved in degrading amyloid precursor protein (APP) and Tau. Mutations and polymorphisms in the CTSD gene, such as SNP rs17571, have been linked to an increased risk of AD, which is likely to be associated with its effect on Tau processing [24]. Notably, the SNP rs17571 was associated with a higher risk of AD in male carriers, with no significant effect observed in women, suggesting a potential gender-specific effect [25]. The deletion of CTSD in APP transgenic mice resulted in significant increases in both soluble and insoluble forms of Aβ, which forms intense intracellular aggregates, while extracellular Aβ deposition is not affected by CTSD deletion [26]. Interestingly, CTSD KO mice develop significant tauopathy by about three weeks of age, characterized by the accumulation of hyperphosphorylated tau, which is more severe than in other mouse models of tauopathy, such as JNPL3, without the need for overexpression of human tau with disease-associated mutations [26] (Table 1).
Another lysosomal protease, Cathepsin E (CTSE), is also implicated in AD pathogenesis. Elevated levels of CTSE have been observed in the cortex of AD patients and in the hippocampus of 6-month-old APP knockin (KI) mice [27]. Inhibition of CTSE improved memory function and reduced Aβ accumulation and neuroinflammation in APP KI mice [27], positioning CTSE as a potential therapeutic target for treatment of AD. CTSE has also been suggested to be a biomarker for familial amyloidotic polyneuropathy (FAP) [28] (Table 1).
Preseniln-1 (PS1), a component of the γ-secretase complex that catalyzes the intramembrane proteolysis of type I membrane proteins such as APP and Notch, is another key molecule linking lysosomal dysfunction and the development of AD [29]. Mutations in the PSEN1 gene, which encodes PS1, are the most common causes of familial AD [29]. Recent studies have shown that PS1 regulates lysosomal function and autophagy by modulating vacuolar ATPase (v-ATPase) activity, lysosomal acidity, and lysosomal Ca2+ content [29, 30], as discussed in the following chapter (Table 1).
Another example of protein which could be associated with the development of AD and also regulates lysosomal function is progranulin. Mutations of PGRN encoding neuroprotective glycoprotein, progranulin, is linked to the development of neurodegenerative diseases such as frontotemporal dementia (FTD) or amyotrophic lateral sclerosis (ALS) [31]. Progranulin regulates lysosomal function such as maturation of cathepsins and maintenance of the level of bis(monoacylglycerol)phosphate (BMP), a late endosome/lysosome-specific phospholipid [32], and PGRN knockout (KO) is associated with severe lysosomal dysfunction such as defective lysosomal acidification. Progranulin binding to Sortilin leads to endocytosis of progranulin and after delivery of progranulin to lysosome, Sortilin is cycled back to the plasma membrane [33]. In addition to FTP and ALS, progranulin mutation can be a risk factor of AD [34]. Furthermore, it has been reported that progranulin level in the cerebrospinal fluid (CSF) or serum could be a diagnostic or prognostic factor in AD [35], and PRGN gene delivery could ameliorate signs of AD in the Tg2576 mouse model of AD [36] (Table 1).
Sortilin-related receptor 1 (SORL1) variant is also a risk gene for both rare genetic AD and common sporadic one. SORL1 plays a role in sorting and trafficking of APP between endosome, Golgi complex and lysosome. SORL1 binds and protects APP from amyloidogenic processing. SORL1 can also bind newly-formed Aβ and guide to the lysosome for degradation. Without SORL1, APP moves to late endosome where APP can be cleaved by secretase forming Aβ [37], which could explain the mechanism of AD associated with SORL1 variants (Table 1).
BIN1 encoding amphiphysin 2 has been identified as a locus of genetic risk to AD, and the second most significant susceptibility locus of sporadic AD after apolipoprotein E (APOE). BIN1 functions in membrane trafficking and endocytosis. In the absence of BIN1, BACE1 is no longer transported to the lysosome and degraded but instead accumulates in early endosomes, promoting Aβ generation [38]. BIN1 K358R mice expressing a risk allele showed increased amyloid burden when crossed to 5xFAD mice which was accompanied by attenuated response of glial cells such as GFAP induction but not of microglial cells [39]. BIN1 has been shown to interact with Tau. Tau PS19 BIN1 -KO mice exhibited more severe neurological deficit compared to PS19 control mice with increased Tau load in the somatosensory cortex but decreased Tau load in the hippocampus, suggesting region-specific effects of BIN1 [40].
PICALM encoding phosphatidylinositol-binding clathrin assembly protein (PICALM) is a risk gene for sporadic AD identified in GWAS studies. PICALM senses membrane curvature and controls size, maturation and completion of clathrin-coated endocytic vesicles. PICALM plays an important role in clathrin-mediated endocytosis (CME) and also in autophagy through endocytosis of vesicle-associated membrane proteins (VAMPS) proteins and regulation of lysosomal function such as cathepsin processing [41, 42]. PICALM has also been reported to rescue endocytic uptake defect mediated by APOE4 [43]. PICALM complexed with AP2 has been reported to crosslink LC3 with APP C-terminal fragment (APP-CTF) for autophagic degradation [44]. The association of the PICALM rs3851179 allele with cortical thickness, as well as CSF levels of Aβ42 and p-Tau, has been reported in elderly subjects [45]. Reduced endothelial PICALM was observed in the brains of AD patients, and this deficiency is associated with delayed Aβ clearance, exacerbating Aβ pathology [46].
2.2 Faulty acidification as a causative factor of lysosomal dysfunction in AD
Recent studies have highlighted the crucial role of impaired lysosomal acidifications in the pathogenesis of AD. Proper lysosomal function depends on the acidic pH of 4.5-5, which is regulated by the v-ATPase that pumps protons into lysosomes via ATP hydrolysis [9]. Dysfunction in v-ATPase disrupts lysosomal acidification, leading to impaired clearance of aggregate proteins, which contributes to AD. Notably, lysosomal acidification impairment has been observed early in the disease process, before neurodegeneration and the appearance of overt pathological changes [16]. In Tg2576 AD mouse models, a significant decline in autolysosome acidification occurs prior to extracellular amyloid deposition, which is linked to reduced v-ATPase activity and the accumulation of Aβ and β-C-terminal fragments (CTFs) in enlarged, de-acidified autolysosomes [16]. While Aβ fibril and Tau aggregates would accumulate in dysfunctional lysosome with impaired proteolytic activity, both Aβ and Tau been identified as key inducers of lysosomal dysfunction in AD, culminating in the feed-forward mechanism between lysosomal dysfunction and Aβ/ Tau accumulation. For instance, the luminal subunit Atp6v0c of v-ATPase binds to internalized Aβ, while the cytosolic subunit Atp6v1b2 interacts with p-Tau [47]. These interactions disrupt v-ATPase function, leading to endolysosomal dysfunction, disrupted endolysosomal integrity, and neurotoxicity or neurodegeneration [47] (Fig. 2A).
Faulty lysosomal acidification in AD. (A) Luminal and cytosolic subunits of the v-ATPase complex interact with internalized Aβ and cytosolic p-tau, impairing v-ATPase activity in neurons. (B) Deletion of PSEN1 results in abnormal Ca2+ efflux via TRPML1, thereby increasing cytosolic Ca2+ levels in neurons. (C) Mutant PSEN1 disturbs the lysosomal pH by enhancing Ca2+ release via TPC2, resulting in lysosomal acidification defect in neurons.
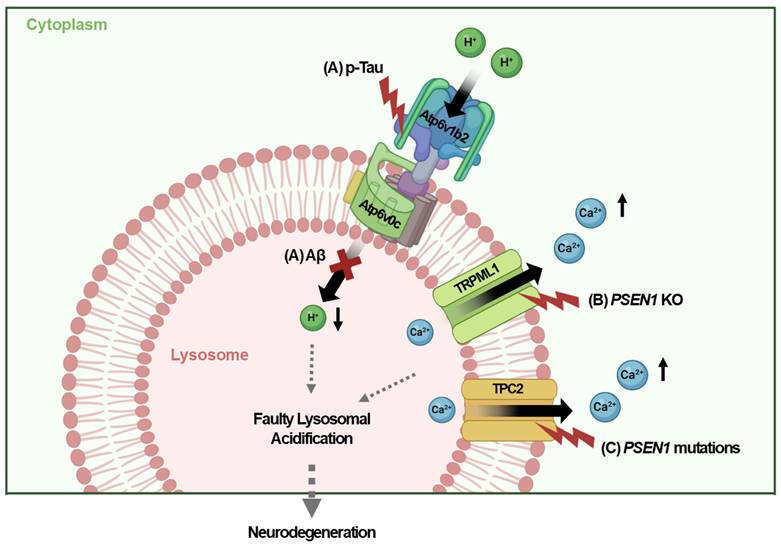
Genetic mutations associated with AD also impact lysosomal dysfunction and acidification. For example, the genetic deletion or mutation of PSEN1, a gene implicated in AD, has been reported to disrupt lysosomal acidification and proteolysis due to defective v-ATPase maturation, which impairs autophagy [29, 30]. Elevated lysosomal pH in PSEN1 KO neuronal cells affects transient receptor potential mucolipin 1 (TRPML1) activity, leading to abnormal Ca2+ efflux from lysosomes into the cytosol [30] (Fig. 2B). In contrast to these results suggesting the primary role of disturbed v-ATPase maturation accompanied by subsequent alteration of Ca2+ efflux, primary role of abnormal Ca2+ efflux in PSEN1 mutations has also been reported [47]. Recent research has also suggested the role of lysosomal Ca2+ channels other than TRPML1 in the context of PSEN1 mutations [48]. Mutant PSEN1 has been shown to increase Ca2+ release through two-pore channel 2 (TPC2), resulting in lysosomal alkalization and impaired lysosome function. Inhibition of TPC2 can restore Ca2+ homeostasis in lysosomes, normalizing both pH and function [49] (Fig. 2C). Thus, the regulation of lysosomal Ca2+ is crucial not only for the expression of autophagy genes and lysosomal biogenesis via TFEB activation but also for maintaining lysosomal acidity, which is essential for neuronal function and the pathogenesis of AD [50, 51].
Additionally, dysregulation of other organelles that interact with lysosomes can contribute to defective lysosomal function and acidification. For instance, abnormal Ca2+ flux through the endoplasmic reticulum (ER)-resident ryanodine receptor (RyR) has been shown to impair lysosomal acidification by reducing the expression of v-ATPase subunits, such as V0a1. This leads to lysosome deacidification and disrupted proteolytic activity in 3xTg AD mouse models and human neurons derived from AD patients [52]. Restoring ER Ca2+ balance can reverse lysosomal dysfunction in 3xTg AD mouse models [52]. Therefore, ER-lysosome Ca2+ coupling is crucial for lysosomal acidification, vesicular trafficking, nutrient sensing, and the processes of autophagy and mitophagy [53, 54]. Lysosomal acidification and Ca²⁺ regulation are key aspects of cell physiology in the pathogenesis of AD. However, further research is necessary to develop therapeutics that target this pathway.
2.3 Other mechanisms of lysosomal dysfunction in AD
Lysosomal dysfunction in AD involves several mechanisms beyond faulty lysosomal acidification, including defective autophagy-lysosome fusion, release of lysosomal hydrolases, and disruptions associated with APOE4. These mechanisms highlight the broad roles of lysosomal dysfunction in AD pathogenesis.
2.3.1 Defective autophagosome-lysosome fusion
Fusion between autophagosome and lysosomes is an essential event in autophagy [55]. Defective fusion between them or premature fusion of unsealed autophagosomes can impair the degradation of cargos or autophagosome inner membrane [56]. The autophagy-related protein 8 (ATG8) family such as gamma-aminobutyric acid receptor-associated protein (GABARAP) plays a vital role in regulating autophagosome-lysosome fusion by recruiting pleckstrin homology domain containing protein family member 1 (PLEKHM1) to autophagosomes. This process is facilitated by soluble N-ethylmaleimide-sensitive factor attachment protein receptor SNARE proteins, such as STX17, SNAP29, and lysosome-localized VAMP8 or VAMP7, with YKT6 acting independently of STX17 via SNAP29 [57]. Other regulators, like RAB GTPases (RAB7A, RAB33B, and RAB2A), and effectors such as PLEKHM1, are also involved in this fusion process [58].
Tripartite motif-containing 22 (TRIM22) further aids in autophagosome-lysosome fusion by associating GABARAP family proteins with PLEKHM1. TRIM22 interacts with GABARAP proteins rather than LC3 family proteins, indicating its specialized role in the fusion process rather than in autophagosome formation [15, 58]. The TRIM22 R321K variant disrupts the association between PLEKHM1 and GABARAPs, potentially hindering their fusion and subsequent autophagic clearance [15]. This variant is associated with early-onset familial AD, highlighting the critical importance of TRIM22-mediated autophagosome-lysosomal fusion in the clearance of amyloid or protein aggregates and presenting potential therapeutic targets for addressing autophagic or lysosomal dysfunction in AD [15] (Fig. 3A).
Autophagy lysosomal dysfunction in AD. (A) The TRIM22 R321K variant inhibits the association between GABARAPs and PLEKHM1, thereby impeding autophagosome-lysosome fusion and interfering with intracellular autophagic clearance. (B) Release of intra-lysosomal hydrolases such as cathepsin B, C, H, E into the cytoplasm caused by lysosome membrane permeabilization impairs intra-lysosomal enzyme activity and promotes neuroinflammation, leading to neuronal degeneration. (C) APOE4 is transported to the lysosome by LAMP2A from post-Golgi compartment and accumulates in lysosome. Delayed dissociation of APOE4 from its receptors in the endosome prevents endolysosomal trafficking. Accumulated APOE4 and resultant cholesterol build-up in lysosome disturb autophagy flux and inhibit mitophagy. APOE4 also impairs TFEB-mediated lysosomal biogenesis and autophagy.
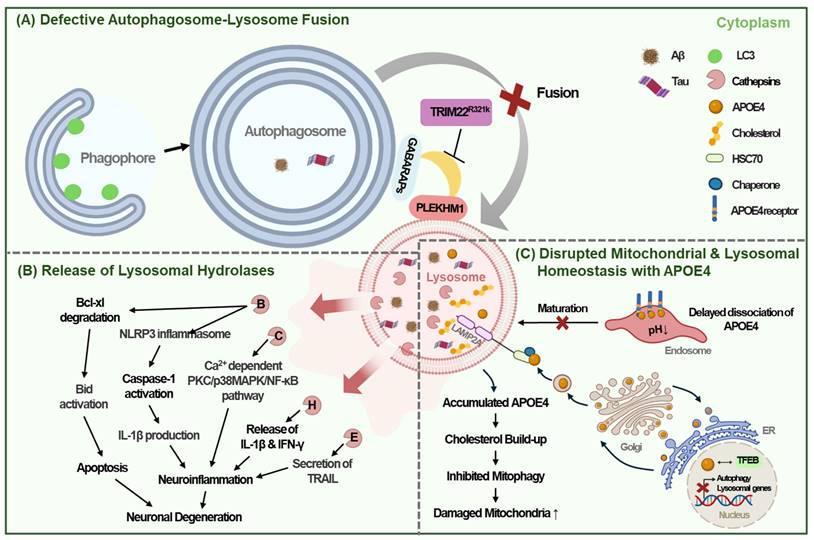
2.3.2 Release of lysosomal hydrolases
The lysosomal system comprises over 80 hydrolases, including proteases, nucleases, phosphatases, sulfatases, lipases, and glycosidases, all essential for maintaining proper lysosomal function. Lysosomal cathepsins are the most abundant lysosomal proteases and include cysteine (cathepsins B, C, F, H, K, L, O, S, V, W, and X), aspartic (CTSD and CTSE), and serine (cathepsin G) proteases [59]. To prevent leakage of lysosomal enzymes and damage to cytosolic constituents and also to protect lysosomal acidity, maintenance of lysosomal membrane integrity is vital. Saposins (SAPs) are lysosome-restricted proteases lysosome activator/lysosomal restricted proteins that are involved in cathepsin maturation and progranulin transport. Elevated levels of SAPs and LAMP1 were observed around amyloid plaques in the brains of APP KI, 5xFAD and APP/PS1 mice in the early stages of the disease, potentially contributing to lysosomal membrane leakage and the subsequent hydrolase loss [60].
Regarding the consequences of lysosomal membrane leakage, lysosome membrane permeabilization can release intracellular components, such as cathepsins, into the cytoplasm, leading to lysosomal-dependent cell death, which may contribute to neuronal loss in AD [61]. For instance, cytosolic cathepsin B (CTSB) initiates a proteolytic cascade that results in apoptosis by degrading the apoptosis-preventing protein Bcl-xL and activating the apoptosis-inducing protein Bid [62]. Moreover, the release of cathepsins can trigger neuroinflammation by inducing the release of inflammatory cytokines from microglia. Cathepsin C, for example, exacerbates neuroinflammation by inducing the M1 polarization of microglia through activation of the Ca2+ dependent PKC/p38MAPK/NF-κB pathway [63]. Similarly, cathepsin H induces neuroinflammation by releasing interleukin-1β (IL-1β) and interferon-γ (IFN-γ), leading to neuronal death [64]. Furthermore, lysosomal CTSB leakage promotes the assembly of the NLR family pyrin domain containing 3 (NLRP3) inflammasome, which triggers IL-1β production and caspase-1 activation in macrophages [65]. Microglial CTSE also contributes to neuroinflammation by enhancing the secretion of TNF-related apoptosis-inducing ligand (TRAIL), which further increased both NF-κB-dependent microglial neuroinflammation and BACE-mediated neuronal Aβ production [27] (Fig. 3B).
2.3.3 Disrupted mitochondrial and lysosomal homeostasis associated with APOE4
APOE4 is a major genetic risk factor for AD and associated with lysosomal dysfunction and abnormal nutrient metabolism. Carriers of the APOE4 allele exhibit increased aerobic glycolysis and impaired oxygen consumption in the brain [66, 67], which is linked to altered glucose metabolism, dysregulated lipid homeostasis, and lysosomal/autophagosomal impairments in neurons and astrocytes [68].
Individuals carrying the APOE4 allele have APOE4 transported from the post-Golgi compartment to the endolysosome via lysosome-associated membrane glycoprotein 2A (LAMP2A)-mediated; chaperon-mediated autophagy; consequently, APOE4 accumulates in the lysosomes [69].
Dissociation of APOE4 from its receptors in the endosome has been reported to be delayed due to high isoelectric point, which may impair endolysosomal trafficking through progressive acidification [43, 70]. In the lysosomes of glial cells or human induced pluripotent stem cells (iPSCs) with APOE4 variants, there is excessive cholesterol accumulation due to upregulated de novo synthesis, despite elevated intracellular cholesterol due to sequestration in lysosomes [71, 72]. This excessive lysosomal cholesterol can inhibit autophagic flux and mitophagy, leading to mitochondrial dysfunction [73]. Reducing lysosomal cholesterol accumulation has been shown to restore both autophagy and mitochondrial homeostasis, highlighting a critical role of elevated lysosomal cholesterol in impaired lysosomal and mitochondrial function [73] (Fig. 3C).
Consistent with these findings, astrocytes from AD patients exhibit reduced mitochondrial respiration, increased mitochondrial reactive oxygen species (ROS), circular mitochondria, impaired mitophagy, and the accumulation of dysfunctional mitochondria [73]. Additionally, APOE4 has been shown to inhibit autophagy gene expression by directly binding to the CLEAR sites in the promoters of TFEB target genes [74] (Fig. 3C). Importance of microglial endolysosomal system in APOE4-mediated aggravation of AD has also been reported. A recent paper reported that APOE, particularly APOE4, aggregates primarily in endolysosome of microglia which was aggravated by lysosomal dysfunction and could initiate amyloid seeding [75]. Intriguingly, enhanced binding of APOE4 with low-density lipoprotein receptor (LDLR) was also reported to contribute to the increased endolysosomal delivery of LDLR-associated cholesterol esterified with polyunsaturated fatty acids inducing lipofuscin accumulation [76]. Furthermore, an APOE R136S mutation (APOE Christchurch, APOEch) which has protective effect against AD development could downregulate APOE binding to LDLR reducing lipid burden which might explain the mechanism of clinical benefits of APOEch mutation [76]. These findings were observed not only in neurons but also in astrocytes and microglial cells, supporting effect of APOE in a wide variety of cells in the CNS participating in the pathogenesis of AD [76].
While both mutations of presenilin and APOE4 can affect lysosomal function in association with the development of AD, their interaction is far from clear. PSEN1/2 mutations and APOE4 are associated with different types of AD in that PSEN1/2 mutations are the most prevalent causes of genetic AD, and APOE4 is the strongest risk factor for sporadic AD. However, they can interact with each other in either genetic or sporadic AD. For instance, APOE4 has been reported to inhibit γ-secretase activity of presenilin through direct binding. APOE4 KO was associated with increased Aβ40 level and γ-secretase activity. In contrast, APOE4 KI resulted in lower Aβ40 level and γ-secretase activity but increased Aβ42/Aβ40 ratio, which might be related to the increased Aβ42/Aβ40 ratio in the brain of AD [77]. An intriguing finding related to the interaction between APOE and PSEN mutations is amelioration of AD phenotypes in patients with E280A PSEN1 mutation by APOE3 R136S mutation (APOE3ch). APOE3ch has been found to impair APOE binding to heparan sulfate proteoglycan (HSPG) and LDLR-related protein 1 (LRP1) involved in neuronal Tau uptake and spreading, which can explain reduced Tau accumulation and spreading in patients with homozygous APOE3ch mutation [78]. Recent papers reported enhanced microglial lysosomal activity and increased microglial phagocytosis of Tau fibrils followed through degradation by APOE3ch mutation [79], supporting the effect of APOE3 on lysosomal function. Furthermore, APOE3ch mutation restored lysosomal enlargement observed in astrocytes of a mouse model expressing a mutant Tau and APOE4 [80], supporting the effect of APOE4 on lysosomal function and its reversal by APOE3 R136S.
Despite these results suggesting the relationship between APOE4 and lysosomal dysfunction associated with AD, the detailed mechanism of endolysosomal dysfunction by APOE4 and differential effect of APOE3 vs. APOE4 on the pathogenesis of AD are still far from clear and warrants further studies [69, 81].
While these observations suggest a link between APOE4 and lysosomal dysfunction associated with AD, the precise mechanisms of endolysosomal dysfunction caused by APOE4 remain unclear and require further investigation.
3. Lysosomal dysfunction in microglia associated with AD
Autophagy and lysosomal function are crucial not only in neuronal cells but also in glial cells, particularly in microglia, which are key components of the innate immune system in the central nervous system (CNS). Microglia are responsible for engulfing and degrading brain-derived products, including apoptotic cells, synapses, myelin, and Aβ aggregates, thereby maintaining physiological function and cellular homeostasis in the CNS [82]. Under the pathological conditions, defective lysosomal acidification in microglia contributes to neuroinflammation and neurodegeneration, exacerbating inflammatory cytokine production through mechanisms such as altered polarization and lysosomal membrane permeabilization [82].
In AD, microglia surrounding amyloid plaques exhibit disrupted lysosomal function caused by the mislocalization of the CIC-7 transporter. This mislocalization is linked to low levels of osteoclastogenesis-associated transmembrane protein 1 in quiescent microglia, impairing the degradation of Aβ. Without proper CIC-7 function, lysosomal acidification is impaired and negatively affects Aβ degradation [4]. Additionally, the dysregulation of TFEB has been observed in microglia associated with AD. Microglia with PSEN1 S367A mutations exhibit decreased TFEB mRNA levels, leading to impaired autophagy and inability to degrade Aβ [83]. Impaired lysosomal function in microglia reduces both phagocytic and autophagic capabilities, preventing the clearance of damaged organelles, such as mitochondria, myelin debris, and toxic protein aggregates. As a result, it promotes neuroinflammation, neuronal death, and neurodegeneration in the CNS [84-86]. Lysosomal dysfunction in microglia can lead to the accumulation of Aβ due to defective clearance, thereby exacerbating AD pathology [86]. The importance of microglial lysosomal function in the pathogenesis of AD is strongly supported by the localization of AD-risk gene variants, such as BIN1, APOE and PICALM, in microglia-specific enhancer-promoter regions [87]. In particular, the effects of APOE4 on the endolysosomal system of microglia through aggregation facilitating Aβ seeding [84] or increased lipid burden leading to aggravated inflammation [76] suggest the critical role of endolysosomal function in microglia as an important target of APOE4 [75], as mentioned earlier.
Aβ deposition in microglia can also trigger neuroinflammatory responses, through mechanisms such as the activation of the NLRP3-dependent inflammasome due to extracellular Aβ. The relevance of microglial inflammation or inflammasome activation in AD is further supported by studies showing that switching microglia to an anti-inflammatory state through NLRP3 inflammasome KO prevents memory loss in APP/PS1 AD mice [88]. Furthermore, microglial-specific deletion of Autophagy-related 7 (ATG7), a key gene for autophagosome biogenesis, results in microglial transition to a pro-inflammatory state and inflammasome activation in vitro, which in turn enhances intraneuronal Tau pathology and its spread [89]. Aβ-mediated inflammasome activation could be mediated by protein kinase AMP-activated catalytic subunit alpha 1 (PRKAA1) pathway [88], potentially contributing to the disease process through cell-to-cell propagation. For example, apoptosis-associated speck-like protein containing a CARD specks, a key component of inflammasome complex, released from microglia can bind to Aβ, forming oligomers and aggregates that promote cross-seeding [90]. Similarly, Tau proteins or aggregates activate inflammasomes [91]. Tau phosphorylation can be induced by IL-1β-dependent Tau kinases, forming a feedback loop between inflammasome activation and Tau pathology [92]. Accentuated NLRP3 inflammasome activation in microglia with autophagy insufficiency or lysosomal dysfunction could arise from the accumulation of Aβ or Tau, as well as from the impaired clearance of the NLRP3 inflammasome complex itself by microglial autophagy [93, 94].
These findings underscore the critical role of the microglial autophagy-lysosomal system in modulating Aβ clearance, Aβ-dependent inflammatory responses, and the overall development of AD. As a corollary, enhancing microglial lysosomal function may have the potential to ameliorate AD-related neuroinflammation (Kim et al., unpublished data).
4. Lysosomal Stress Response (LSR) in AD
The lysosomal stress response (LSR) is crucial for counteracting lysosomal damage and maintaining cellular integrity in AD. Cells have evolved several mechanisms to address lysosomal stress, including repair, lysophagy, replacement, and autophagic lysosomal reformation (ALR), making these pathways promising therapeutic targets for AD [95].
4.1 Lysosomal Repair
Minor disruptions in lysosomal membrane integrity activate repair mechanisms involving the endosomal sorting complex required for transport (ESCRT) [96, 97] and heat shock protein 70 (HSP70) [98]. Proteins such as ESCRT-I, ESCRT-III, and ALG-2-interacting protein X (ALIX) are rapidly recruited to the ruptured lysosomal membranes to facilitate repair [99]. Essential ESCRT components, such as Hrs and Tsg101, are required for lysosomal targeting of APP, and their reduction results in increased intracellular Aβ and decreased Aβ secretion [100]. The knockdown of ESCRT components, including charged multivesicular body proteins (CHMP6, CHMP2A, and CHMP2B), promotes Tau aggregation [101]. Furthermore, defects in the ESCRT machinery are associated with both sporadic and familial AD [99], suggesting the role of lysosomal recovery via the ESCRT pathway in protecting against Aβ- or Tau-mediated damage in the development of AD. Severe lysosomal damage triggers lysophagy, a selective autophagic process that degrade damaged lysosomes [98, 102] (Fig. 4A).
Lysosomal Stress Response (LSR) in AD. (A) Small disruptions in lysosomal membrane integrity can trigger lysosomal repair mechanisms that facilitate membrane sealing via the recruitment of ESCRT machinery and ALIX proteins, resulting in their rapid localization to the lysosome membrane when the lysosomal membrane is ruptured. (B) If this disruption is too severe to repair, lysosomes are targeted for self-degradation via lysophagy, a selective autophagy process. TRIM16 overexpression can enhance lysophagy by recruiting LC3, p62, OPTN, and TAX1BP1 to damaged lysosomes, thus inhibiting the high-glucose-induced accumulation of Aβ and tau. (C) Lysosome replacement involves the creation of new lysosomes to replace those that are damaged or lost, thereby ensuring the cell can continue to break down and recycle materials effectively. Lysosomal replacement is triggered by lysosome stress, and TFEB is a key regulator in lysosome regeneration by promoting lysosomal function. (D) ALR might also help restore impaired lysosomal function by lysosomal damage. During ALR, LIMP2 binds to ATG8 and then recruits TBC1D15, providing scaffold for assembly of ALR machinery. TBC1D15 hydrolyzes Rab7-GTP for segregation of damaged lysosome and Dynamin2-dependent scission of lysosomal tubule.
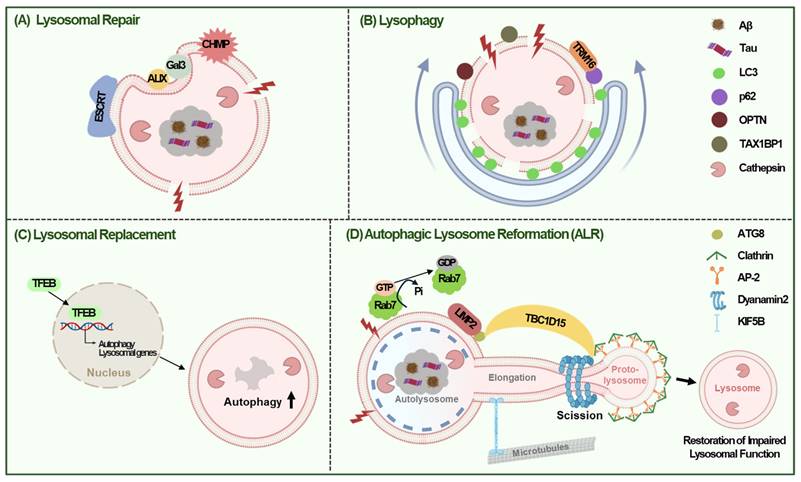
4.2 Lysophagy
When lysosomal repair through ESCRT pathway fails, lysosomes are tagged with ubiquitin and selective autophagy of lysosome called 'lysophagy' is initiated (66). During lysophagy, glycosylated proteins on damaged lysosomes are recognized by galectins, which recruit autophagy receptors like p62, Optineurin (OPTN), or Tax1 binding protein 1 (TAX1BP1) after ubiquitination [103]. These receptors facilitate the engulfment of ubiquitinated lysosomes into autophagosomes, which then fuse with intact lysosomes for degradation. For example, galectin 3 recruits tripartite motif-containing 16 (TRIM16), while galectin 8 binds to nuclear dot protein 52 (NDP52), signaling lysosomal damage to mTORC1 and AMPK, thereby inducing autophagy [93, 104]. Overexpression of TRIM16 enhances lysophagy by recruiting LC3, p62, and ubiquitin to damaged lysosomes, effectively reducing Aβ and p-Tau accumulation [105] (Fig. 4B).
4.3 Lysosomal Replacement
Lysosomal replacement involves creation of new lysosomes to replace damaged ones. This process requires production and endocytosis of new lysosomal proteins, which are triggered by lysosomal stress [3, 10]. TFEB plays a critical role in promoting function and regeneration of lysosome by interacting with the CLEAR elements in lysosomal gene promoters, especially when mTOR is inhibited [106] (Fig. 4C).
4.4 Autophagic Lysosomal Reformation (ALR)
Besides the three mechanisms discussed above, ALR might also contribute to the lysosomal regeneration in response to lysosomal damage [107]. ALR is characterized by extrusion of proto-lysosome from autolysosome and its maturation into functional lysosome, which can help restore impaired lysosomal function by lysosomal injury [107]. In ALR following lysosomal injury, LIMP2, a lysosomal integral membrane protein-2 was found to bind Atg8 and then recruit TBC1 domain family member 15 (TBC1D15), which provides scaffold for assembly of ALR machinery. This process comprises hydrolysis of Rab7-GTP for segregation of damaged lysosome and Dynamin2-dependent but TFEB-independent scission of lysosomal tubule [107] (Fig. 4D).
These LSR mechanisms are essential for maintaining cellular homeostasis against lysosomal stress and offer potential therapeutic strategies for mitigating lysosomal dysfunction in AD.
5. TFEB as a target of therapeutic approaches enhancing autophagy-lysosome pathway
Several studies have reported that TFEB dysregulation is associated with AD, consistent with the role of lysosomal dysfunction in AD pathogenesis. For example, reduced nuclear TFEB levels have been observed in the brains of AD patients or aged experimental animals [109, 110]. In contrast, increased expression of TFEB downstream genes has been noted in the brains of patients with AD. Although TFEB mRNA expression remained unchanged in cortical neurons, it is increased in the hippocampal region, likely due to its expression in glial cells. In contrast, the mRNA expression of TFE3, another member of the MiT family of bHLH-leucine zipper transcription factors alongside TFEB, has been shown to increase in both cortical and hippocampal regions in the brains of AD patients [18]. This discrepancy may reflect variations in the disease stages studied, as TFEB-mediated adaptive changes to lysosomal dysfunction or stress in AD may fluctuate over the course of disease progression. However, these adaptive changes may not be sufficient to fully counteract the lysosomal dysfunction or reverse the disease course. Reinforcing these adaptive changes may provide an opportunity to halt or even reverse AD progression, making TFEB a promising target for these approaches.
6. Autophagy and lysosomal enhancers targeting TFEB in AD
Given the central role of dysregulated autophagy and lysosomal function in AD pathogenesis, various pharmacological strategies have been developed to target this pathway [5]. These strategies focus on enhancing autophagy initiation, promoting lysosomal biogenesis, and activating TFEB-mediated transcriptional programs [110]. Below are several pharmacological agents that have shown promise in preclinical models and are being evaluated for their potential therapeutic benefits in AD (Table 2).
Autophagy/lysosomal enhancers in AD.
| Drugs or Chemicals | Mechanism and effects | References |
|---|---|---|
| Atractylenolide III, Ferulic acid and Paeoniflorin | Activation of the AMPK/ULK1/TFEB autophagic signaling pathway. | [111] [112] |
| Inhibition of autophagy and neuroinflammation in BV2 microglial cells. | ||
| Caudatin | Activates PPARα. | [113] |
| Improves cognitive dysfunction & enhances lysosomal degradation of Aβ and p-tau aggregates in 3xTg-AD model mice. | ||
| Celastrol | mTORC1 inhibition. | [114] [115] |
| Promotes the degradation of p-Tau aggregates in P301S AD model mice. | ||
| Corynoxine | Activating TFEB/TFE3 by inhibiting AKT/mTOR signalling and stimulating lysosomal Ca2+ release via TRPML1 | [116] |
| Improves learning and memory function in 5xFAD AD mice. | ||
| Crocetin | Activating the STK11/LKB1-mediated AMPK pathway. | [117] |
| Reducing Aβ levels and neuroinflammation, improving memory function in 5xFAD AD mice. | ||
| Curcumin-analog compound C1 | Direct binding to and activation of TFEB. | [118] |
| Improves synaptic and cognitive function and reduced Aβ and tau aggregates in P301S, 5xFAD and 3xTg AD mice models. | ||
| Hederagenin | Enhanced expressions of TFEB, LC3II/LC3I, LAMP2A. | [119] |
| Improves cognitive impairment and pathological changes by promoting autophagy in APP/PS1 AD mice. | ||
| Ibudilast | Inhibition of mTORC1 and activation of TFEB. | [120] |
| Improves spatial learning and memory, and increases clearance of Aβ plaque and tau protein aggregates in Aβ/Tau in 344-AD rats. | ||
| Latrepirdine | Stimulates mTOR- and ATG5-dependent autophagy | [121] |
| Reducing Aβ deposition, and prevents further behavioral decline in TgCRND8 mice expressing a double mutant form of APP 695 (KM670/671NL+V717F) | ||
| LH2-051 | Promotion of TFEB activation and lysosome biogenesis via inhibition of DAT. | [122] |
| Improves learning, memory, and cognitive function in APP/PS1 AD mice. | ||
| ML246 | Activating BECN1. | [123] |
| Aβ removal and memory in 5xFAD AD mice. | ||
| ML-SA1 | Activation of TRPML1 endolysosomal Ca2+ signaling | [124] |
| Rescued enlargement and perinuclear clustering of endolysosomes, autophagic vesicle accumulation, and early endosomal enlargement in primary neurons. | ||
| Oleoylethanolamide, KDS-5104 | Promotion of TFEB-lysosomal function in a PPARα -dependent but mTORC1-independent manner. | [125] |
| Reduces reactive gliosis and Aβ pathology, and rescues cognitive impairment in 5xFAD AD mice. | ||
| Spermidine | Promotion of eIF5A hypusination | [126] [127] |
| Reduction in Aβ deposition and neuroinflammation in APP/PS1 AD mouse model. | ||
| Thonningianin A | Activation of the AMPK/ULK1 and Raf/MEK/ERK pathways | [128] |
| Improving cognitive function, reducing Aβ expression, and decreasing neuronal apoptosis in microglial cells treated with Aβ. | ||
| Trehalose | Activation of TFEB through mTOR-independent, FOXO1-dependent manner | [129] |
| Reduction in Aβ deposition in the hippocampus of APP/PS1 mice | ||
| Uric Acid | Activation of microglia and upregulation of the autophagy-related proteins such as LC3II/I ratio, Beclin-1, and LAMP1 | [130] |
| Improves cognitive function in APP/PS1 mouse model of AD. |
Atractylenolide III, Ferulic acid and Paeoniflorin, either alone or in combination, counteract lipopolysaccharide (LPS)-induced autophagy inhibition and neuroinflammation. They enhance the expression of autophagy-related proteins including AMP-activated protein kinase (AMPK), ULK1, and TFEB in BV2 microglial cells, leading to improved autophagy and reduced neuroinflammation [111, 112]. Caudatin activates peroxisome proliferator-activated receptor α (PPARα), which enhances lysosomal degradation of Aβ and p-Tau aggregates. This reduces the onset of AD and improves cognitive dysfunction in 3xTg-AD model mice [113]. Celastrol promotes autophagy and lysosomal biogenesis by activating TFEB through mTORC1 inhibition. It ameliorates Tau pathology in P301S AD model mice [114, 115]. Corynoxine, a natural autophagy enhancer, activates TFEB/TFE3 via inhibitions of the AKT/mTOR pathway and TRPML1-mediated Ca2+ release. This results in increased neuronal autophagy and lysosomal biogenesis, reduced APP-CTF levels, and improved cognitive function in 5xFAD AD mice [116]. Crocetin stimulates autophagy by activating the serine/threonine kinase 11 (STK11/LKB1)-mediated AMPK pathway. It effectively crosses the blood-brain barrier, induces autophagy specifically in the hippocampus, reduces Aβ levels and neuroinflammation, and enhances memory function in 5xFAD AD mice [117]. Curcumin-analog C1 activates TFEB, leading to enhanced autophagy and lysosomal activity. It reduces levels of APP, CTF-β/α, Aβ, and Tau aggregates, and improves cognitive function in P301S, 5xFAD and 3xTg AD mice models [118]. Hederagenin upregulates TFEB and promotes the expression of autophagy-related proteins like LC3II/LC3I and LAMP2. This enhancement of autophagy and Aβ degradation alleviates cognitive impairment in APP/PS1 AD mice [119]. Ibudilast inhibits mTORC1 activity and enhances TFEB-mediated lysosomal biogenesis and autophagy. This results in improved cognitive function and clearance of Aβ/Tau in 344-AD rats [120]. Latrepirdine decreased mTOR phosphorylation and stimulated Atg5-dependent autophagy, reducing intracellular levels of Aβ. In TgCRND8 mice expressing a double mutant form of APP 695 (KM670/671NL+V717F) under the control of the PrP gene promoter, latrepirdine enhances autophagy, reduces Aβ deposition, and prevents further decline in behavioral abnormalities [121]. LH2-051 enhances TFEB activation and lysosome biogenesis by inhibiting dopamine transporters (DAT). This improves cognitive function and Aβ clearance in APP/PS1 AD mice [122]. ML246 is a small-molecule autophagy inducer that exerts BECN1-dependent protective effects. It enhances Aβ removal and memory in 5xFAD AD mice [123]. ML-SA1, an agonist of the essential endosomal-lysosomal Ca2+ channel TRPML1, has been shown to reverse several key abnormalities in the endosomal-autophagic-lysosomal pathway in primary neurons. This effect is similar to those observed in AD-affected neurons after PIKfyve inhibition [124]. Oleoylethanolamide enhances microglial Aβ clearance and improves cognitive function in 5xFAD AD mice by promoting TFEB lysosomal function in a PPARα-dependent manner, independent of mTORC1 [125]. Spermidine activates autophagy by promoting the hypusination of eukaryotic translation initiation factor 5A-1 (eIF5A), which upregulates TFEB translation [126]. Treatment with spermidine in APP/PS1 AD mouse model activates autophagy, particularly in microglia, leading to reduced Aβ and neuroinflammation [127]. Thonningianin A triggers autophagy via the AMPK/ULK1 and Raf/MEK/ERK pathways and promotes the degradation of NLRP3 inflammasome in microglial cells treated with Aβ [128]. In APP/PS1 AD mice, it improves cognitive function, reduces Aβ accumulation, and decreases neuronal apoptosis [128]. Trehalose, a natural disaccharide, activates the transcription of autophagy genes and TFEB. This activation occurs through low-grade lysosomal stress in an mTOR-independent, forkhead box protein O1 (FOXO1)-dependent manner, leading to a reduction in amyloid-β deposition in the hippocampus of APP/PS1 AD mice [129]. Uric acid activates microglial autophagy and increases levels of autophagy-related proteins, including the LC3II/I ratio, Beclin-1, and LAMP1. It also helps alleviate memory decline and promotes Aβ degradation in APP/PS1 mouse model of AD [130] (Table 2).
Conclusion and prospect
AD is a devastating neurodegenerative disorder marked by the accumulation of Aβ plaques and Tau tangles, leading to progressive cognitive decline and memory loss. Recent studies on the autophagy-lysosome pathway have revealed promising therapeutic strategies by targeting lysosomal dysfunction, a critical factor in AD pathogenesis.
Lysosomal dysfunction, especially due to faulty acidification, contributes significantly to the accumulation of Aβ and Tau, key factors in AD. Reduced lysosomal acidification, often caused by impaired v-ATPase activity, leads to the buildup of these toxic proteins, exacerbating the disease. Genetic mutations, such as those in PSEN1, further exacerbate lysosomal dysfunction by affecting v-ATPase maturation and Ca2+ homeostasis. Additionally, defective autophagy-lysosome fusion and the release of lysosomal hydrolases contribute to neurodegeneration and neuroinflammation. The APOE4 allele, a major genetic risk factor for AD, is associated with abnormal cholesterol accumulation within lysosomes, disrupting both autophagic and mitochondrial functions and accelerating disease progression.
In AD, lysosomal dysfunction is further aggravated by Aβ or Tau oligomer accumulation, which impairs essential lysosomal functions. Aging, a major risk factor for sporadic AD, exacerbates lysosomal dysfunction by reducing autophagic activity, leading to a buildup of toxic aggregates that overwhelm the lysosomal system. This cascade creates a vicious cycle that is further complicated by oxidative damage, altered enzymatic activity, and compromised lysosomal membrane integrity, ultimately resulting in neuronal toxicity and cell death. In AD, there is disrupted lysosomal acidification and Ca²⁺ regulation, leading to impaired autophagic flux. Impaired autophagy and lysosomal function also contribute to neuronal cell death through necroptosis [131], with autophagy deficits causing the accumulation of proteins such as receptor-interacting protein kinase-1 (RIPK1) and receptor-interacting protein kinase-3 (RIPK3) that are typically degraded by lysosomes [132]. Elevated levels of RIPK1 and RIPK3 can trigger necroptosis, a programmed cell death pathway, and increased tumor necrosis factor receptor-1 (TNFR1) signaling in AD further promotes necroptosis through the RIPK1/RIPK3/MLKL pathway [131].
To counteract lysosomal dysfunction, the LSR plays a crucial role in maintaining cellular integrity by addressing lysosomal damage via mechanisms such as repair, lysophagy, replacement, and ALR. Repair processes, including those facilitated by the ESCRT machinery, are associated with intracellular Aβ and Tau aggregation when defective. Lysophagy, which is mediated by proteins such as galectins and autophagy receptors, selectively degrades damaged lysosomes. Lysosomal replacement regulated by TFEB promotes the generation of new lysosomes, essential for restoring lysosomal function. While ALR contributes to lysosome regeneration through membrane remodeling, independently of TFEB. These pathways are vital for maintaining cellular homeostasis and may present potential therapeutic targets for AD.
Given the crucial role of the dysregulated autophagy-lysosome pathway system, particularly, lysosomal dysfunction in AD development, targeting the autophagy-lysosome pathway and modulating lysosomal stress offers significant therapeutic potential through addressing the fundamental mechanisms underlying AD, enhancing the clearance of pathological proteins, and reducing neuroinflammation. TFEB has emerged as a promising therapeutic target for treating neurodegenerative disorders. However, there are several challenges and safety concerns associated with the development of TFEB-targeting drugs. Due to its influence on multiple cellular pathways, non-specific activation may lead to excessive lysosomal activity, disrupt cellular balance, and cause unintended side effects. Persistent TFEB activation could overdrive autophagy, potentially causing damage to healthy proteins and organelles, and this can also impair immune function, increasing the risk of autoimmune diseases [132]. Additionally, TFEB-targeting drugs often struggle to cross the blood-brain barrier, potentially limiting their effectiveness in treating brain-related diseases. Therefore, further research is essential to optimize drug delivery systems and formulations for stability and targeted action, particularly for brain-specific therapies.
Interestingly, natural compounds such as berberine [133], rifampicin [134], curcumin [118], genistein [135], and kaempferol [136] have shown potential as TFEB activators, which aligns with our findings on TFEB modulation as a therapeutic approach [137, 138]. These compounds may offer alternative or complementary strategies for enhancing TFEB activity with potentially fewer side effects due to their natural origin and inherent bioactivities. However, similar challenges with bioavailability and blood-brain barrier penetration remain.
To address these challenges, it is crucial to develop drugs that finely modulate TFEB activation. Nanotechnology-based approaches for targeted tissue delivery and ligand-based strategies to activate drugs within specific cells or tissues are required [139, 140]. Incorporating natural TFEB activators into advanced delivery systems, such as nanoparticles, may further enhance their therapeutic potential by improving stability, targeting efficiency, and brain-specific delivery. Futures studies should investigate the integration of these natural compounds into TFEB-targeting strategies to complement synthetic approaches and overcome existing barriers.
In summary, this review highlights that 1) lysosomal dysfunction caused by genetic causes (PSEN1/2 mutations, APOE4 and variants of BIN1, PICALM or SOLR1), aging, or other factors such as accumulated Aβ or Tau fibril plays a critical role in initiating or amplifying the AD disease process. This dysfunction is associated with faulty acidification, impaired autophagy, and release of lysosomal hydrolases, and 2) therapeutic strategies focused on improving lysosomal stress, lysosomal repair, replacement, reformation, or lysophagy through pharmacologic agents or genetic measure that activate TFEB or related genes may provide new approaches to retard, halt, or reverse disease progression and to treat AD. Future research should focus on translating these findings into safe and effective clinical interventions that provide hope to patients with AD.
Abbreviations
AD: Alzheimer's disease; ALIX: alg-2-interacting protein x; ALR: autophagic lysosomal reformation; ALS: amyotrophic lateral sclerosis; AMPK: amp-activated protein kinase; APOE4: apolipoprotein e4; APP: amyloid-β precursor protein; ASC: apoptosis-associated speck-like protein containing a card; ATG14L: autophagy-related protein 14-like protein; ATG7: autophagy-related 7; ATG8: autophagy-related protein; Aβ: amyloid β; BMP: bis(monoacylglycerol)phosphate; CHMP2A: charged multivesicular body protein 2a; CHMP2B: charged multivesicular body protein 2b; CHMP6: charged multivesicular body protein 6; CLEAR: coordinated lysosomal expression and regulatory; CME: clathrin-mediated endocytosis; CNS: central nervous system; CSF: cerebrospinal fluid; CTF: c-terminal fragment; CTSB: cathepsin b; CTSD: cathepsin d; CTSE: cathepsin e; DAT: dopamine transporters; eIF5A: eukaryotic translation initiation factor 5a-1; ER: endoplasmic reticulum; ESCRT: endosomal sorting complex required for transport; FAP: familial amyloidotic polyneuropathy; FOXO1: forkhead box protein o1; FTD: frontotemporal dementia; GABARAP: gamma-aminobutyric acid receptor-associated protein; Hsp70: heat shock protein 70; HSPG: heparan sulfate proteoglycan; IFN-γ: interferon-γ; IL-1β: interleukin-1β; iPSCs: induced pluripotent stem cells; KI: knockin; KO: knockout; LAMP2: lysosome-associated membrane glycoprotein 2; LPS: lipopolysaccharide; LSR: lysosomal stress response; LRP1: low density lipoprotein receptor-related protein 1; mTOR: mammalian target of rapamycin; NDP52: nuclear dot protein 52; NFTs: neurofibrillary tangles; NLRP3: NLR family pyrin domain containing 3; NRBF2: nuclear receptor binding factor 2; OPTN: optineurin; OSTM1: osteoclastogenesis associated transmembrane protein1; PICALM: phosphatidylinositol-binding clathrin assembly protein; PLEKHM1: pleckstrin homology domain containing protein family member 1; PPARα: peroxisome proliferator-activated receptor α; PRKAA1: protein kinase amp-activated catalytic subunit alpha 1; PS1: presenilin-1; p-Tau: phosphorylated tau; RAPTOR: regulatory-associated protein of tor; ROS: reactive oxygen species; RIPK1: receptor-interacting protein kinase-1; RyR: resident ryanodine receptor; S6K1: S6 kinase 1; SAPs: saposins; SNARE: soluble N-ethylmaleimide-sensitive factor attachment protein receptors; SNP: single nucleotide polymorphism; SORL1: sortilin-related receptor 1; RIPK3: receptor-interacting protein kinase-3; STK11/LKB1: serine/threonine kinase 11; TAX1BP1: tax1 binding protein 1; TBC1D15: tbc1 domain family member 15; TFEB: transcription factor eb; TNFR1: tumor necrosis factor receptor-1; TPC2: two-pore channel 2; TRAIL: TNF-related apoptosis-inducing ligand; TRIM16: tripartite motif-containing 16; TRIM22: tripartite motif-containing 22; TRPML1: transient receptor potential mucolipin 1; ULK1: unc-51-like autophagy-activating kinase; VAMPS: vesicle-associated membrane proteins; v-ATPase: vacuolar ATPase.
Acknowledgements
Figures were created through BioRender.com.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2022R1A2C1092597), a grant of the Korea Dementia Research Project through the Korea Dementia Research Center (KDRC), funded by the Ministry of Health & Welfare and Ministry of Science and ICT, Republic of Korea (RS-2024-00338662), and the Gachon University research fund of 2022 (GCU202209230001). The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.
Author contributions
YJK, T-YH, and K-AC conceived and designed the review. YJK, T-YH, and K-AC reviewed the literature and drafted the manuscript. M-SL and K-AC revised the manuscript. All authors read and approved the final manuscript.
Consent for publication
All authors have consented for this review paper to be published.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT. et al. Alzheimer disease. Nature Reviews Disease Primers. 2021;7:33
2. Martini-Stoica H, Xu Y, Ballabio A, Zheng H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends in Neurosciences. 2016;39:221-34
3. Gu Z, Cao H, Zuo C, Huang Y, Miao J, Song Y. et al. TFEB in Alzheimer's disease: From molecular mechanisms to therapeutic implications. Neurobiol Dis. 2022;173:105855
4. Van Acker ZP, Perdok A, Bretou M, Annaert W. The microglial lysosomal system in Alzheimer's disease: Guardian against proteinopathy. Ageing Research Reviews. 2021;71:101444
5. Zhang W, Xu C, Sun J, Shen HM, Wang J, Yang C. Impairment of the autophagy-lysosomal pathway in Alzheimer's diseases: Pathogenic mechanisms and therapeutic potential. Acta Pharm Sin B. 2022;12:1019-40
6. Xue W, Zhang J, Li Y. Enhancement of lysosome biogenesis as a potential therapeutic approach for neurodegenerative diseases. Neural regeneration research. 2023;18:2370-6
7. Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895-9
8. Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931-7
9. Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nature Cell Biology. 2019;21:133-42
10. Settembre C, Di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S. et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science. 2011;332:1429-33
11. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo j. 2012;31:1095-108
12. Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nature Reviews Neuroscience. 2015;16:345-57
13. Orr ME, Oddo S. Autophagic/lysosomal dysfunction in Alzheimer's disease. Alzheimers Res Ther. 2013;5:53
14. Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. Journal of Neuropathology & Experimental Neurology. 2005;64:113-22
15. Heo H, Park H, Lee MS, Kim J, Kim J, Jung S-Y. et al. TRIM22 facilitates autophagosome-lysosome fusion by mediating the association of GABARAPs and PLEKHM1. Autophagy. 2024;20:1098-1113
16. Lee J-H, Yang D-S, Goulbourne CN, Im E, Stavrides P, Pensalfini A. et al. Faulty autolysosome acidification in Alzheimer's disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nature Neuroscience. 2022;25:688-701
17. Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW. et al. Galectins Control mTOR in Response to Endomembrane Damage. Mol Cell. 2018;70:120-35.e8
18. Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S. et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12:2467-83
19. Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R. et al. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem. 2015;133:739-49
20. Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107-20
21. Cao Y, Wang Y, Abi Saab WF, Yang F, Pessin JE, Backer JM. NRBF2 regulates macroautophagy as a component of Vps34 Complex I. Biochem J. 2014;461:315-22
22. Lachance V, Wang Q, Sweet E, Choi I, Cai CZ, Zhuang XX. et al. Autophagy protein NRBF2 has reduced expression in Alzheimer's brains and modulates memory and amyloid-beta homeostasis in mice. Mol Neurodegener. 2019;14:43
23. Yang C, Cai CZ, Song JX, Tan JQ, Durairajan SSK, Iyaswamy A. et al. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy. 2017;13:2028-40
24. Riemenschneider M, Blennow K, Wagenpfeil S, Andreasen N, Prince JA, Laws SM. et al. The cathepsin D rs17571 polymorphism: effects on CSF tau concentrations in Alzheimer disease. Hum Mutat. 2006;27:532-7
25. Albayrak Ö, Tirniceriu A, Riemenschneider M, Kurz A, Scherag A, Egensperger R. The Cathepsin D (224C/T) Polymorphism Confers an Increased Risk to Develop Alzheimer's Disease in Men. The Journals of Gerontology: Series A. 2010;65A:219-24
26. Terron HM, Parikh SJ, Abdul-Hay SO, Sahara T, Kang D, Dickson DW. et al. Prominent tauopathy and intracellular β-amyloid accumulation triggered by genetic deletion of cathepsin D: implications for Alzheimer disease pathogenesis. Alzheimer's Research & Therapy. 2024;16:70
27. Xie Z, Meng J, Kong W, Wu Z, Lan F, Narengaowa. et al. Microglial cathepsin E plays a role in neuroinflammation and amyloid β production in Alzheimer's disease. Aging Cell. 2022;21:e13565
28. Gonçalves NP, Moreira J, Martins D, Vieira P, Obici L, Merlini G. et al. Differential expression of Cathepsin E in transthyretin amyloidosis: from neuropathology to the immune system. Journal of Neuroinflammation. 2017;14:115
29. Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM. et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146-58
30. Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y. et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015;12:1430-44
31. Chitramuthu BP, Bennett HPJ, Bateman A. Progranulin: a new avenue towards the understanding and treatment of neurodegenerative disease. Brain. 2017;140:3081-104
32. Logan T, Simon MJ, Rana A, Cherf GM, Srivastava A, Davis SS. et al. Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell. 2021;184:4651-68.e25
33. Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR. et al. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654-67
34. Zhu M, Wang X, Hjorth E, Colas RA, Schroeder L, Granholm AC. et al. Pro-Resolving Lipid Mediators Improve Neuronal Survival and Increase Aβ42 Phagocytosis. Mol Neurobiol. 2016;53:2733-49
35. Suárez-Calvet M, Capell A, Araque Caballero M, Morenas-Rodríguez E, Fellerer K, Franzmeier N. et al. CSF progranulin increases in the course of Alzheimer's disease and is associated with sTREM2, neurodegeneration and cognitive decline. EMBO Mol Med. 2018;10:e9712
36. Van Kampen JM, Kay DG. Progranulin gene delivery reduces plaque burden and synaptic atrophy in a mouse model of Alzheimer's disease. PLOS ONE. 2017;12:e0182896
37. Barthelson K, Newman M, Lardelli M. Sorting Out the Role of the Sortilin-Related Receptor 1 in Alzheimer's Disease. J Alzheimers Dis Rep. 2020;4:123-40
38. Zhang R, Witkowska K, Afonso Guerra-Assunção J, Ren M, Ng FL, Mauro C. et al. A blood pressure-associated variant of the SLC39A8 gene influences cellular cadmium accumulation and toxicity. Hum Mol Genet. 2016;25:4117-26
39. Garcia-Agudo LF, Shi Z, Smith IF, Kramár EA, Tran K, Kawauchi S. et al. BIN1(K358R) suppresses glial response to plaques in mouse model of Alzheimer's disease. Alzheimers Dement. 2024;20:2922-42
40. Ponnusamy M, Wang S, Yuksel M, Hansen MT, Blazier DM, McMillan JD. et al. Loss of forebrain BIN1 attenuates hippocampal pathology and neuroinflammation in a tauopathy model. Brain. 2023;146:1561-79
41. Maninger JK, Nowak K, Goberdhan S, O'Donoghue R, Connor-Robson N. Cell type-specific functions of Alzheimer's disease endocytic risk genes. Philos Trans R Soc Lond B Biol Sci. 2024;379:20220378
42. Hattersley KJ, Carosi JM, Hein LK, Bensalem J, Sargeant TJ. PICALM regulates cathepsin D processing and lysosomal function. Biochem Biophys Res Commun. 2021;570:103-9
43. Narayan P, Sienski G, Bonner JM, Lin YT, Seo J, Baru V. et al. PICALM Rescues Endocytic Defects Caused by the Alzheimer's Disease Risk Factor APOE4. Cell Rep. 2020;33:108224
44. Tian Y, Chang JC, Fan EY, Flajolet M, Greengard P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer's APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci U S A. 2013;110:17071-6
45. Wu Z, Yang Y, Song Z, Ma M, Feng M, Liu Y. et al. PICALM rs3851179 Variants Modulate Left Postcentral Cortex Thickness, CSF Amyloid β42, and Phosphorylated Tau in the Elderly. Brain Sciences. 2022;12:1681
46. Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K. et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat Neurosci. 2015;18:978-87
47. Kim SH, Cho YS, Kim Y, Park J, Yoo SM, Gwak J. et al. Endolysosomal impairment by binding of amyloid beta or MAPT/Tau to V-ATPase and rescue via the HYAL-CD44 axis in Alzheimer disease. Autophagy. 2023;19:2318-37
48. Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W. et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol. 2012;198:23-35
49. Tong BC, Wu AJ, Huang AS, Dong R, Malampati S, Iyaswamy A. et al. Lysosomal TPCN (two pore segment channel) inhibition ameliorates beta-amyloid pathology and mitigates memory impairment in Alzheimer disease. Autophagy. 2022;18:624-42
50. Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R. et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288-99
51. Zhang X, Yu L, Xu H. Lysosome calcium in ROS regulation of autophagy. Autophagy. 2016;12:1954-5
52. Mustaly-Kalimi S, Gallegos W, Marr RA, Gilman-Sachs A, Peterson DA, Sekler I, Stutzmann GE. Protein mishandling and impaired lysosomal proteolysis generated through calcium dysregulation in Alzheimer's disease. Proceedings of the National Academy of Sciences. 2022;119:e2211999119
53. Oulès B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J. et al. Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci. 2012;32:11820-34
54. Lacampagne A, Liu X, Reiken S, Bussiere R, Meli AC, Lauritzen I. et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer's disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017;134:749-67
55. Lőrincz P, Juhász G. Autophagosome-Lysosome Fusion. Journal of Molecular Biology. 2020;432:2462-82
56. Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036-41
57. Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy. 2021;17:2680-8
58. McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D. et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015;57:39-54
59. Di Domenico F, Tramutola A, Perluigi M. Cathepsin D as a therapeutic target in Alzheimer's disease. Expert Opin Ther Targets. 2016;20:1393-5
60. Sharoar MG, Palko S, Ge Y, Saido TC, Yan R. Accumulation of saposin in dystrophic neurites is linked to impaired lysosomal functions in Alzheimer's disease brains. Molecular Neurodegeneration. 2021;16:45
61. Wang F, Gómez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19:918-31
62. de Castro MAG, Bunt G, Wouters FS. Cathepsin B launches an apoptotic exit effort upon cell death-associated disruption of lysosomes. Cell Death Discovery. 2016;2:16012
63. Liu Q, Zhang Y, Liu S, Liu Y, Yang X, Liu G. et al. Cathepsin C promotes microglia M1 polarization and aggravates neuroinflammation via activation of Ca(2+)-dependent PKC/p38MAPK/NF-κB pathway. J Neuroinflammation. 2019;16:10
64. Nakanishi H. Cathepsin regulation on microglial function. Biochim Biophys Acta Proteins Proteom. 2020;1868:140465
65. Chevriaux A, Pilot T, Derangère V, Simonin H, Martine P, Chalmin F. et al. Cathepsin B Is Required for NLRP3 Inflammasome Activation in Macrophages, Through NLRP3 Interaction. Frontiers in Cell and Developmental Biology. 2020;8:167
66. Cho S, Lee H, Seo J. Impact of Genetic Risk Factors for Alzheimer's Disease on Brain Glucose Metabolism. Mol Neurobiol. 2021;58:2608-19
67. Farmer BC, Williams HC, Devanney NA, Piron MA, Nation GK, Carter DJ. et al. APOΕ4 lowers energy expenditure in females and impairs glucose oxidation by increasing flux through aerobic glycolysis. Mol Neurodegener. 2021;16:62
68. Qi G, Mi Y, Shi X, Gu H, Brinton RD, Yin F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021;34:108572
69. Fote GM, Steffan JS. APOE4 dysregulates autophagy in cultured cells. Autophagy Rep. 2022;1:29-33
70. Pohlkamp T, Xian X, Wong CH, Durakoglugil MS, Werthmann GC, Saido TC. et al. NHE6 depletion corrects ApoE4-mediated synaptic impairments and reduces amyloid plaque load. eLife. 2021;10:e72034
71. Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J. et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer's Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018;98:1141-54.e7
72. Tcw J, Qian L, Pipalia NH, Chao MJ, Liang SA, Shi Y. et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell. 2022;185:2213-33.e25
73. Lee H, Cho S, Kim MJ, Park YJ, Cho E, Jo YS. et al. ApoE4-dependent lysosomal cholesterol accumulation impairs mitochondrial homeostasis and oxidative phosphorylation in human astrocytes. Cell Rep. 2023;42:113183
74. Parcon PA, Balasubramaniam M, Ayyadevara S, Jones RA, Liu L, Shmookler Reis RJ. et al. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement. 2018;14:230-42
75. Kaji S, Berghoff SA, Spieth L, Schlaphoff L, Sasmita AO, Vitale S. et al. Apolipoprotein E aggregation in microglia initiates Alzheimer's disease pathology by seeding β-amyloidosis. Immunity. 2024;57:2651-2668.e12
76. Guo JL, Braun D, Fitzgerald GA, Hsieh Y-T, Rougé L, Litvinchuk A. et al. Decreased lipidated ApoE-receptor interactions confer protection against pathogenicity of ApoE and its lipid cargoes in lysosomes. Cell. 2024: S0092-8674(24)01209-1.
77. Sun Y, Islam S, Gao Y, Nakamura T, Zou K, Michikawa M. Apolipoprotein E4 inhibits γ-secretase activity via binding to the γ-secretase complex. J Neurochem. 2023;164:858-74
78. Zalocusky KA, Nelson MR, Huang Y. An Alzheimer's-disease-protective APOE mutation. Nat Med. 2019;25:1648-9
79. Chen Y, Song S, Parhizkar S, Lord J, Zhu Y, Strickland MR. et al. APOE3ch alters microglial response and suppresses Aβ-induced tau seeding and spread. Cell. 2024;187:428-45.e20
80. Nelson MR, Liu P, Agrawal A, Yip O, Blumenfeld J, Traglia M. et al. The APOE-R136S mutation protects against APOE4-driven Tau pathology, neurodegeneration and neuroinflammation. Nat Neurosci. 2023;26:2104-21
81. Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W. et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523-7
82. Quick JD, Silva C, Wong JH, Lim KL, Reynolds R, Barron AM. et al. Lysosomal acidification dysfunction in microglia: an emerging pathogenic mechanism of neuroinflammation and neurodegeneration. Journal of Neuroinflammation. 2023;20:185
83. Ledo JH, Liebmann T, Zhang R, Chang JC, Azevedo EP, Wong E. et al. Presenilin 1 phosphorylation regulates amyloid-β degradation by microglia. Mol Psychiatry. 2021;26:5620-35
84. Lin M, Yu H, Xie Q, Xu Z, Shang P. Role of microglia autophagy and mitophagy in age-related neurodegenerative diseases. Frontiers in Aging Neuroscience. 2023;14:1100133
85. Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S. et al. Age-related myelin degradation burdens the clearance function of microglia during aging. Nature Neuroscience. 2016;19:995-8
86. Guo X, Tang P, Chen L, Liu P, Hou C, Zhang X. et al. Amyloid β-Induced Redistribution of Transcriptional Factor EB and Lysosomal Dysfunction in Primary Microglial Cells. Front Aging Neurosci. 2017;9:228
87. Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R. et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 2019;366:1134-9
88. Cho MH, Cho K, Kang HJ, Jeon EY, Kim HS, Kwon HJ. et al. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10:1761-75
89. Xu Y, Propson NE, Du S, Xiong W, Zheng H. Autophagy deficiency modulates microglial lipid homeostasis and aggravates tau pathology and spreading. Proceedings of the National Academy of Sciences. 2021;118:e2023418118
90. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D. et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer's disease. Nature. 2017;552:355-61
91. Stancu IC, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S. et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019;137:599-617
92. Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A. et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669-73
93. Jiang S, Bhaskar K. Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front Mol Neurosci. 2020;13:586731
94. Zhang D, Zhang Y, Pan J, Cao J, Sun X, Li X. et al. Degradation of NLRP3 by p62-dependent-autophagy improves cognitive function in Alzheimer's disease by maintaining the phagocytic function of microglia. CNS Neurosci Ther. 2023;29:2826-42
95. Saftig P, Puertollano R. How Lysosomes Sense, Integrate, and Cope with Stress. Trends Biochem Sci. 2021;46:97-112
96. Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360:eaar5078
97. Radulovic M, Schink KO, Wenzel EM, Nähse V, Bongiovanni A, Lafont F, Stenmark H. ESCRT‐mediated lysosome repair precedes lysophagy and promotes cell survival. The EMBO Journal. 2018;37:e99753
98. Papadopoulos C, Kravic B, Meyer H. Repair or Lysophagy: Dealing with Damaged Lysosomes. Journal of Molecular Biology. 2020;432:231-9
99. Chou CC, Vest R, Prado MA, Wilson-Grady J, Paulo JA, Shibuya Y. et al. Proteostasis and lysosomal quality control deficits in Alzheimer's disease neurons. bioRxiv. 2023
100. Edgar JR, Willén K, Gouras GK, Futter CE. ESCRTs regulate amyloid precursor protein sorting in multivesicular bodies and intracellular amyloid-β accumulation. Journal of Cell Science. 2015;128:2520-8
101. Chen JJ, Nathaniel DL, Raghavan P, Nelson M, Tian R, Tse E. et al. Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J Biol Chem. 2019;294:18952-66
102. Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T. et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev Cell. 2016;39:13-27
103. Eapen VV, Swarup S, Hoyer MJ, Paulo JA, Harper JW. Quantitative proteomics reveals the selectivity of ubiquitin-binding autophagy receptors in the turnover of damaged lysosomes by lysophagy. eLife. 2021;10:e72328
104. Thurston TLM, Wandel MP, von Muhlinen N, Foeglein Á, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414-8
105. Chae CW, Yoon JH, Lim JR, Park JY, Cho JH, Jung YH. et al. TRIM16-mediated lysophagy suppresses high-glucose-accumulated neuronal Aβ. Autophagy. 2023;19:2752-68
106. Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903-14
107. Bhattacharya A, Mukherjee R, Kuncha SK, Brunstein ME, Rathore R, Junek S. et al. A lysosome membrane regeneration pathway depends on TBC1D15 and autophagic lysosomal reformation proteins. Nat Cell Biol. 2023;25:685-98
108. Wang H, Wang R, Xu S, Lakshmana MK. Transcription Factor EB Is Selectively Reduced in the Nuclear Fractions of Alzheimer's and Amyotrophic Lateral Sclerosis Brains. Neurosci J. 2016;2016:4732837
109. Wang B, Huang M, Shang D, Yan X, Zhao B, Zhang X. Mitochondrial Behavior in Axon Degeneration and Regeneration. Front Aging Neurosci. 2021;13:650038
110. Chen M, Dai Y, Liu S, Fan Y, Ding Z, Li D. TFEB Biology and Agonists at a Glance. Cells. 2021;10:333
111. Zhou X, Chen X, Cheng X, Lin L, Quan S, Li S. et al. Paeoniflorin, ferulic acid, and atractylenolide III improved LPS-induced neuroinflammation of BV2 microglia cells by enhancing autophagy. J Pharmacol Sci. 2023;152:151-61
112. Mahaman YAR, Huang F, Salissou MTM, Yacouba MBM, Wang JZ, Liu R. et al. Ferulic Acid Improves Synaptic Plasticity and Cognitive Impairments by Alleviating the PP2B/DARPP-32/PP1 Axis-Mediated STEP Increase and Aβ Burden in Alzheimer's Disease. Neurotherapeutics. 2023;20:1081-108
113. Krishnamoorthi S, Iyaswamy A, Sreenivasmurthy SG, Thakur A, Vasudevan K, Kumar G. et al. PPARɑ Ligand Caudatin Improves Cognitive Functions and Mitigates Alzheimer's Disease Defects By Inducing Autophagy in Mice Models. J Neuroimmune Pharmacol. 2023;18:509-28
114. Zhang W, Wang J, Yang C. Celastrol, a TFEB (transcription factor EB) agonist, is a promising drug candidate for Alzheimer disease. Autophagy. 2022;18:1740-2
115. Yang C, Su C, Iyaswamy A, Krishnamoorthi SK, Zhu Z, Yang S. et al. Celastrol enhances transcription factor EB (TFEB)-mediated autophagy and mitigates Tau pathology: Implications for Alzheimer's disease therapy. Acta Pharm Sin B. 2022;12:1707-22
116. Guan XJ, Deng ZQ, Liu J, Su CF, Tong BC, Zhu Z. et al. Corynoxine promotes TFEB/TFE3-mediated autophagy and alleviates Aβ pathology in Alzheimer's disease models. Acta Pharmacol Sin. 2024;45:900-13
117. Wani A, Al Rihani SB, Sharma A, Weadick B, Govindarajan R, Khan SU. et al. Crocetin promotes clearance of amyloid-β by inducing autophagy via the STK11/LKB1-mediated AMPK pathway. Autophagy. 2021;17:3813-32
118. Song JX, Malampati S, Zeng Y, Durairajan SSK, Yang CB, Tong BC. et al. A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer's disease models. Aging Cell. 2020;19:e13069
119. Xie ZS, Zhao JP, Wu LM, Chu S, Cui ZH, Sun YR. et al. Hederagenin improves Alzheimer's disease through PPARα/TFEB-mediated autophagy. Phytomedicine. 2023;112:154711
120. Oliveros G, Wallace CH, Chaudry O, Liu Q, Qiu Y, Xie L. et al. Repurposing ibudilast to mitigate Alzheimer's disease by targeting inflammation. Brain. 2023;146:898-911
121. Steele JW, Gandy S. Latrepirdine (Dimebon®), a potential Alzheimer therapeutic, regulates autophagy and neuropathology in an Alzheimer mouse model. Autophagy. 2013;9:617-8
122. Yin L, Zhou Y, Liu H, Li Y. LYECs: lysosome-enhancing compounds as potential therapeutic approaches for Alzheimer disease. Autophagy. 2023;19:1863-4
123. Rocchi A, Yamamoto S, Ting T, Fan Y, Sadleir K, Wang Y. et al. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer's disease. PLoS Genet. 2017;13:e1006962
124. Somogyi A, Kirkham ED, Lloyd-Evans E, Winston J, Allen ND, Mackrill JJ. et al. The synthetic TRPML1 agonist ML-SA1 rescues Alzheimer-related alterations of the endosomal-autophagic-lysosomal system. J Cell Sci. 2023;136:jcs259875
125. Comerota MM, Gedam M, Xiong W, Jin F, Deng L, Wang MC. et al. Oleoylethanolamide facilitates PPARα and TFEB signaling and attenuates Aβ pathology in a mouse model of Alzheimer's disease. Mol Neurodegener. 2023;18:56
126. Metur SP, Klionsky DJ. The curious case of polyamines: spermidine drives reversal of B cell senescence. Autophagy. 2020;16:389-90
127. Freitag K, Sterczyk N, Wendlinger S, Obermayer B, Schulz J, Farztdinov V. et al. Spermidine reduces neuroinflammation and soluble amyloid beta in an Alzheimer's disease mouse model. Journal of Neuroinflammation. 2022;19:172
128. Zhou XG, Qiu WQ, Yu L, Pan R, Teng JF, Sang ZP. et al. Targeting microglial autophagic degradation of the NLRP3 inflammasome for identification of thonningianin A in Alzheimer's disease. Inflamm Regen. 2022;42:25
129. Portbury SD, Hare DJ, Sgambelloni C, Perronnes K, Portbury AJ, Finkelstein DI, Adlard PA. Trehalose Improves Cognition in the Transgenic Tg2576 Mouse Model of Alzheimer's Disease. J Alzheimers Dis. 2017;60:549-60
130. Xiao Q, Wang J, Tian Q, Tian N, Tian Q, He X. et al. Uric Acid Mitigates Cognitive Deficits via TFEB-Mediated Microglial Autophagy in Mice Models of Alzheimer's Disease. Mol Neurobiol. 2024;61:3678-96
131. Asimakidou E, Reynolds R, Barron AM, Lo CH. Autolysosomal acidification impairment as a mediator for TNFR1 induced neuronal necroptosis in Alzheimer's disease. Neural Regen Res. 2024;19:1869-70
132. Gros F, Muller S. The role of lysosomes in metabolic and autoimmune diseases. Nature Reviews Nephrology. 2023;19:366-83
133. Zheng Y, Kou J, Wang P, Ye T, Wang Z, Gao Z. et al. Berberine-induced TFEB deacetylation by SIRT1 promotes autophagy in peritoneal macrophages. Aging (Albany NY). 2021;13:7096-119
134. Umeda T, Ono K, Sakai A, Yamashita M, Mizuguchi M, Klein WL. et al. Rifampicin is a candidate preventive medicine against amyloid-β and tau oligomers. Brain. 2016;139:1568-86
135. Argüello G, Balboa E, Tapia PJ, Castro J, Yañez MJ, Mattar P. et al. Genistein Activates Transcription Factor EB and Corrects Niemann-Pick C Phenotype. Int J Mol Sci. 2021;22:4220
136. Xu J, Zhang XQ, Zhang Z. Transcription factor EB agonists from natural products for treating human diseases with impaired autophagy-lysosome pathway. Chin Med. 2020;15:123
137. Islam F, Khadija JF, Harun-Or-Rashid M, Rahaman MS, Nafady MH, Islam MR. et al. Bioactive Compounds and Their Derivatives: An Insight into Prospective Phytotherapeutic Approach against Alzheimer's Disease. Oxid Med Cell Longev. 2022;2022:5100904
138. Kabir MT, Uddin MS, Jeandet P, Emran TB, Mitra S, Albadrani GM. et al. Anti-Alzheimer's Molecules Derived from Marine Life: Understanding Molecular Mechanisms and Therapeutic Potential. Mar Drugs. 2021;19:251
139. Asimakidou E, Tan JKS, Zeng J, Lo CH. Blood-Brain Barrier-Targeting Nanoparticles: Biomaterial Properties and Biomedical Applications in Translational Neuroscience. Pharmaceuticals. 2024;17:612
140. Lo CH, Zeng J. Defective lysosomal acidification: a new prognostic marker and therapeutic target for neurodegenerative diseases. Transl Neurodegener. 2023;12:29
Author contact
![]() Corresponding authors: Keun-A Chang (keuna705ac.kr); Myung-Shik Lee (mslee0923ac.kr).
Corresponding authors: Keun-A Chang (keuna705ac.kr); Myung-Shik Lee (mslee0923ac.kr).