Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(3):1032-1046. doi:10.7150/ijbs.105513 This issue Cite
Research Paper
Exacerbation by knocking-out metallothionein gene of obesity-induced cardiac remodeling is associated with the activation of CARD9 signaling
Haina Zhang1,2, Wenqian Zhou1,3, Xiang Wang1,3, Hongbo Men1,3, Jiqun Wang1,3, Jianxiang Xu1, Shanshan Zhou3, Quan Liu3, Lu Cai1,4 ![]()
1. Pediatric Research Institute, Department of Pediatrics, University of Louisville, Louisville, KY, 40202, USA.
2. Department of Cardiology, The Second Hospital of Jilin University, Jilin University, Changchun, 130041, China.
3. Department of Cardiovascular Diseases, The First Hospital of Jilin University, Jilin University, Changchun, 130021, China.
4. Departments of Radiation Oncology, Pharmacology and Toxicology, University of Louisville, Louisville, KY, 40202, USA.
Received 2024-10-20; Accepted 2024-12-28; Published 2025-1-13
Abstract

Obesity increases the risk of metabolic syndrome including insulin resistance, dyslipidemia, and cardiovascular disease. We demonstrated insulin resistance, cardiac hypertrophy, and cardiac inflammation in an obese mouse model induced by a high-fat diet (HFD). Caspase recruitment domain-containing protein 9 (CARD9) and B-cell lymphoma/leukemia 10 (BCL10) were upregulated, and p38 MAPK was activated in these mice. Zinc supplementation prevented these changes with upregulation of metallothionein (MT). Deletion of MT exacerbated palmitate-triggered expression of BCL10 and p38 MAPK activation and eliminated the protective benefits of zinc in palmitate-treated cardiomyocytes. Here we further investigated the mechanisms by which endogenous MT expression affects HFD-induced cardiac remodeling and the CARD9/BCL10/p38 MAPK pathway. Male MT knockout and 129S wild-type mice were assigned to receive either a normal diet or a HFD from 8-week-age for 18 weeks. MT knockout (KO) aggravated HFD-induced obesity and systemic metabolic disorder, reflected by increased body weight, perirenal white adipose tissue, and plasma cholesterol, and cardiac hypertrophy and fibrosis. Obese MT-KO mice had abundant cardiac macrophages, upregulated cardiac proinflammatory cytokines, chemokines, adhesion molecules, CARD9, and BCL10 and activated NF-κB. MT-KO exacerbated HFD-induced trace metal dyshomeostasis and oxidative stress. MT-KO combined with HFD-induced obesity synergistically promotes cardiac remodeling, possibly via trace metal dyshomeostasis-induced oxidative stress to trigger CARD9/BCL10-mediated NF-κB activation.
Keywords: Obesity cardiomyopathy, Cardiac remodeling, Metallothionein, CARD9, Inflammation
Introduction
Obesity is the most common chronic medical condition worldwide. It affects over 1 billion people globally, based on the latest published data [1]. Metabolically abnormal obesity (MAO) is a disorder affecting blood pressure, glucose, and lipid metabolism. This disorder contributes to various cardiovascular diseases (CVDs) such as coronary artery disease (CAD), hypertension, and cardiac dysfunction or even heart failure (HF), called obesity cardiomyopathy [2]. Obesity cardiomyopathy is characterized by cardiac metabolic, structural, and functional abnormalities mediated by obesity alone without other CVDs such as hypertension and CAD [3]. These abnormalities include cardiac pathological remodeling and impaired systolic and diastolic function. These dysfunctions then develop into HF, including those with preserved ejection fraction and HF with reduced ejection fraction [3,4]. Obesity cardiomyopathy is becoming a major global public health problem without efficacious therapeutic targets. It's a progressive process from metabolically healthy but obese to MAO and cardiac remodeling. Hence, early identification and prevention of MAO are vital for the prevention of obesity cardiomyopathy.
Obesity cardiomyopathy is associated with systemic and local metabolic disturbances, inflammation, and subcellular abnormalities such as oxidative stress, endoplasmic reticulum stress, mitochondrial dysfunction and autophagy [3,5]. Thus, local and systemic chronic inflammation may have pivotal roles in the occurrence and progression of obesity cardiomyopathy [5].
Expressed on myeloid cells, caspase recruitment domain-containing protein 9 (CARD9) contributes to the transduction of signals related to innate immunity and the inflammatory response, thereby protecting against pathogen invasion [6]. When activated, CARD9 forms the signaling complex CARD9/B-cell lymphoma/leukemia 10 (BCL10)/mucosa-associated lymphoid tissue 1 (MALT1) (CBM) with BCL10 and MALT1. Then MAPKs and/or NF-κB were activated to mediate proinflammatory cytokine and chemokine production [7]. CARD9 promotes inflammation in CVDs, including HF, myocardial infarction, viral myocarditis, cardiac arteritis, myocardial ischemia/reperfusion, hypertension, and obesity cardiomyopathy [6,8,9]. We previously reported the regulation of CARD9 and BCL10 expression in the hearts of mice with obesity and diabetic cardiomyopathy. In both cases, cytokine production and p38 MAPK activation were augmented [10,11]. Furthermore, CARD9 deletion in mice with metabolic disorders and obesity improved glucose intolerance, decreased cardiac fibrosis and cardiac dysfunction, reduced cardiac macrophage infiltration into the heart, lowered proinflammatory cytokine production, and attenuated MAPK activity [6,12,13].
Metallothionein (MT) is a protein characterized by a high cysteine content and ability to bind metals. Its roles include zinc (Zn), copper (Cu), and other metal homeostasis maintaining, free radical scavenging, oxidative stress and damage protecting, and heavy metal detoxifying [14]. Polymorphisms in MT1A gene have been linked to a heightened susceptibility to type 2 diabetes mellitus. Meanwhile, diabetics have higher levels of proinflammatory cytokines than healthy participants [15]. Moreover, we demonstrated that MT upregulation by Zn supplementation and cardiac-specific MT overexpression prevented diabetic cardiomyopathy (DCM) [16,17]. Conversely, MT deletion aggravated diabetes-induced oxidative damage, inflammation, and cardiac remodeling [18].
Owing to the common link of MT and CARD9 signaling with inflammation, we preliminarily explored these interactions in obesity cardiomyopathy. We observed that obesity induces cardiac hypertrophy by activating p38 MAPK through CARD9/BCL10 complex upregulation, which was assumed to be mediated by increased oxidative stress [10,19]. In vitro, MT activation by Zn supplementation ameliorated palmitate-induced oxidative stress, upregulated BCL10 expression, and p38 MAPK activation in wild type (WT) cardiomyocytes. However, MT deletion counteracted the beneficial effect of Zn on palmitate-induced BCL10 overexpression and p38 MAPK activation [10,19].
Here, we used MT knockout (KO) mice to explore whether MT protects against HFD-induced obesity cardiomyopathy via the CARD9/MAPKs pathway in vivo. We further investigated the mechanisms through which this protein activates CARD9 signaling. As NF-κB is a downstream component of CARD9 that is in parallel with p38 MAPK, we examined whether its activation is implicated in obesity cardiomyopathy.
Materials and Methods
Animals and experimental models
MT-KO mice were generated by interbreeding homozygous mutants (MT-/-, both MT1 and MT2 gene knockout) within a 129S1 WT background. All breeding pairs (Jackson Laboratory, Bar Harbor, ME, USA), were mated according to the vendor's instructions. Male WT 129S1 (MT+/+) mice were also procured from Jackson Laboratory. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Louisville (Louisville, KY, USA) which is certified by the American Association for the Accreditation of Laboratory Animal Care.
These mice were started on a normal diet (ND; 10% kcal from fat; No. D14020202, Research Diets, New Brunswick, NJ, USA) or a high-fat diet (HFD; 60% kcal from fat; No. D14020205, Research Diets) at 8 weeks and maintained on each diet for 18 weeks. Weekly body weight (BW) measurements were taken. The mice were anesthetized through an intraperitoneal avertin injection (250 mg/kg) after an 18-week dietary intervention, and blood samples were obtained from the inferior vena cava. The mice were then euthanized by exsanguination. Heart weight, tibia length, and peri-renal white adipose tissue weight were recorded, and the tissues were harvested.
Intraperitoneal glucose tolerance test (IPGTT)
The IPGTT was conducted at 26 weeks of age. Mice were subjected to a 6-h fast that began at 8 a.m. and ended at 2 p.m., followed by an intraperitoneal administration of glucose solution (2 g/kg BW). Blood glucose levels were measured at 0, 15, 30, 60, 90, and 120 minutes post-glucose administration, with a FreeStyle Lite glucometer (Abbott Diabetes Care, Alameda, CA, USA).
Echocardiography
At the end of the study, transthoracic echocardiography was conducted utilizing a high-resolution imaging system (Vevo 770; Visual Sonics, Toronto, ON, Canada) as previously described [20]. Left ventricular (LV) parameters, including LV internal dimension (LVID), LV posterior wall thickness (LVPW), and interventricular septum (IVS), were measured. LV mass and volume, ejection fraction (EF), and fractional shortening (FS) were calculated using Vevo 770 software.
Assessment of metal concentration in the heart tissue
Zn, iron (Fe), and Cu concentrations in the heart tissue were determined by inductively coupled plasma mass spectrometry (ICP-MS; X series II, Thermo Fisher Scientific, Waltham, MA, USA). Each sample underwent digestion with 0.6 mL of concentrated nitric acid (70%) under conditions of 85 °C for a duration of 4 hours. After cooling to room temperature, the samples experienced 1 minute of centrifugation at 5,000 rpm and dilution with 9 mL deionized Milli-Q water (EMD Millipore, Billerica, MA, USA) (constituting a 4.38% nitric acid solution), which was then vortexed and subjected to ICP-MS analysis.
Biochemical plasma analysis
Plasma triglyceride and cholesterol levels were measured utilizing Infinity™ triglyceride/cholesterol liquid stable reagents (Cat#TR22421, Cat#TR13421; Thermo Fisher Scientific), following the provided protocols. Briefly, the plasma samples underwent a 1:100 dilution using the aforementioned reagents, followed by thorough mixing and incubation at 37 ℃ for 5 minutes. Subsequently, a microplate reader measured the absorbance at 500 nm.
Histopathological analysis
The heart tissue was embedded in paraffin and sectioned as previously described [10]. The tissues were fixed in 10% buffered formalin, followed by paraffin embedding and sectioning at a thickness of 5 μm. To determine the cross-sectional areas of the cardiomyocytes and collagen deposition in cardiac tissues, we used fluorescein isothiocyanate (FITC)-conjugated wheat germ agglutinin (WGA; Alexa Fluor 488 conjugate; Molecular Probes/Invitrogen, Carlsbad, CA, USA) and Picro-Sirius Red staining. Immunohistochemical (IHC) staining with anti-CD68 (1:150 dilution; Abcam, Cambridge, MA, USA) was performed to examine cardiac macrophage infiltration. All stained sections were examined with an Olympus BX43 microscope (Olympus Life Science, Tokyo, Japan), and quantitative measurements were performed using ImageJ software.
Western blot analysis
Western blotting was performed as previously described [10]. Heart tissues were collected and lysed. The protein samples were separated by SDS-PAGE and then electrotransferred onto nitrocellulose membranes. The membranes were incubated with primary antibodies, followed by appropriate secondary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA). Images were acquired using a ChemiDoc Touch Imaging System (Bio-Rad Laboratories). Protein content was determined with Image Lab software (Bio-Rad Laboratories) and normalized against the respective controls. A modified western blotting protocol, as described before, was employed for the evaluation of MT expression [16].
Primary antibodies used included those against TGF-β1 (1:1000; ab92486; Abcam, Cambridge, MA, USA), CD68 (1:1000; ab125212), vascular cell adhesion molecule-1 (VCAM-1; 1: 1000; ab134047), intercellular adhesion molecule-1 (ICAM-1; 1:1000; ab179707), IL-6 (1:1000; ab208113), TNF-α (1:1000; ab6671), IL-1β (1:1000; ab9722), GAPDH (1:3000; ab37168), BCL10 (1 : 1000; sc-5611; Santa Cruz Biotechnology), collagen1A1 (COL1A1; 1:500; SC293182), CARD9 (1:1000; 12283s; Cell Signaling Technology(CST), Danvers, MA, USA), phosphorylation-NF-κB p65 (p-p65; 1:1000; 3033s; CST), p-p38 MAPK/p38 MAPK (p-p38/p38; 1:1000; 9211s/9212s; CST), p-JNK/JNK (1:1000; 9255s/9252s; CST), p-Erk1/2/Erk1/2 (1:1000; 4370s/4696s; CST), 3-NT (1:1000; ab5411; EMD Millipore, Billerica, MA, USA), 4-HNE (1:2000; Alpha Diagnostic International, San Antonio, TX, USA), MT (1:1000; M0639; Dako, Glostrup, Denmark).
Quantitative real-time polymerase chain reaction (qRT-PCR)
TRIzol reagent (Invitrogen) was used to extract total RNA from heart tissues, followed by assessment of RNA purity and concentration using a NanoDrop ND-1000 spectrophotometer. A reverse transcription kit (Promega, Madison, WI, USA) was then used to synthesize cDNA from 1 μg total RNA, following the provided instructions. The qRT-PCR was performed in a 10 μl solution including TagMan universal PCR master mix (Invitrogen) using a LightCycler 96 RT-PCR system (Roche Diagnostics, Indianapolis, IN, USA). Primers for GAPDH (Mm99999915_g1), collagen I (col1a1, Mm00801666_g), collagen III (col3a1, Mm00802300_m1), fibronectin, (Fn, Mm01256744_m1), TGF-β (Mm01178820_m1), monocyte chemoattractant protein-1 (MCP-1/CCL2, Mm00441242_m1), and CD68 (4331182 Mm03047343_m1) were acquired from Thermo Fisher Scientific.
Statistical analysis
The data were processed using GraphPad Prism v. 8 (San Diego, CA, USA). Data are presented as mean ± standard deviation (SD; n = 5). Between-group differences were determined by one-way analysis of variance (ANOVA) followed by Tukey's post-hoc test or two-way ANOVA. P < 0.05 indicated statistically significant difference.
Results
MT knockout exacerbated HFD-induced obesity and metabolic disorders
Fig. 1A-C shows that both WT and MT-KO mice fed a HFD for 18 weeks displayed a significant increase in BW and perirenal white adipose tissue weight in comparison to their corresponding ND-fed mice. Moreover, HFD-fed caused significantly different results between WT and MT-KO mice. In addition, MT-KO alone resulted in a significantly elevated BW relative to WT mice fed a ND (Fig. 1A, B). These outcomes propose that MT deletion increased BW and perirenal white adipose accumulation in response to HFD feeding, i.e., aggravated HFD-induced obesity.
The effect of MT knockout on HFD-induced obesity and metabolic disorders. A: Body weights during 26 weeks of ages and 18 weeks of HFD feeding (started at 8 weeks old). B-C: Weight of body and perirenal adipose tissue weight at the end of 18-week feeding. D: The changes of blood glucose levels over time after IPGTT in 26-week-old mice (Da, i.e., at the end of 18-wks HFD feeding) and the IPGTT results via calculation of the integrated area under the curves (AUC, Db). E-F: Plasma cholesterol and triglyceride. Data are presented as Mean ± SD (n=5). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, HFD vs. ND in WT and MT-KO groups; #P<0.05, ##P<0.01, ####P<0.0001, MT-KO vs. WT in ND and HFD groups.
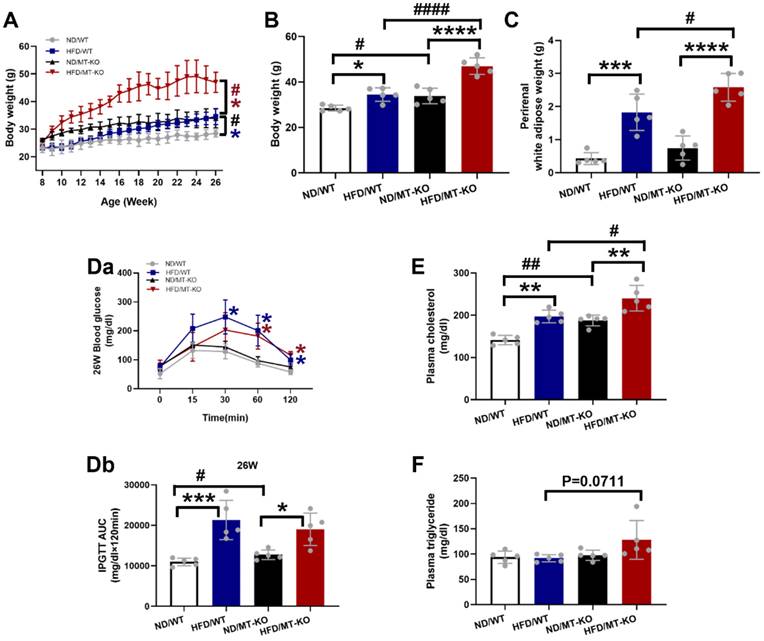
As shown in Fig. 1D, MT-KO or obese mice showed significantly reduced glucose tolerance, as reflected by a larger area under the curve at 26 weeks of age (i.e. ,18 weeks of HFD feeding) in both WT and MT-KO strains. Combined MT-KO and HFD did not significantly worse glucose tolerance compared to either HFD/WT or MT-KO alone group (P > 0.05).
The obese mice displayed augmented plasma cholesterol in the WT and MT-KO groups compared to their corresponding ND-fed mice. Moreover, the MT-KO obese mice exhibited higher plasma cholesterol levels than the WT mice fed with HFD (Fig. 1E). As for plasma triglyceride, neither MT deletion nor HFD has effect, but obese MT-KO mice showed slightly increased plasma triglyceride levels compared to other groups (Fig. 1F).
MT gene knockout promoted HFD-induced cardiac remodeling
Echocardiographic analysis revealed no significant impact on cardiac dysfunction after 18 weeks of exposure to a HFD, as indicated by functional parameters EF% and FS%, in WT and MT-KO mice (Table 1). As cardiac structural parameters, IVS, LV mass, and LVPW (Table 1) were significantly increased in HFD/MT-KO group than in HFD/WT and ND/MT-KO groups. This indicated that MT gene KO combined with HFD-induced obesity caused cardiac structure remodeling.
Cardiac structure and function parameters measured by echocardiography.
| ND/WT | HFD/WT | ND/ MT-KO | HFD/ MT-KO | |
|---|---|---|---|---|
| IVS;d (mm) | 0.58±0.04 | 0.59±0.01 | 0.56±0.02 | 0.71±0.005#& |
| IVS;s (mm) | 0.90±0.05 | 0.89±0.04 | 0.83±0.11 | 1.01±0.12& |
| LVID;d (mm) | 3.84±0.11 | 3.6±0.09* | 4.00±0.06* | 3.93±0.05# |
| LVID;s (mm) | 2.43±0.45 | 2.05±0.08 | 2.41±0.18 | 2.41±0.23 |
| LVPW;d (mm) | 0.73±0.04 | 0.73±0.02 | 0.72±0.03 | 0.84±0.01#& |
| LVPW;s (mm) | 1.02±0.04 | 1.02±0.04 | 1.05±0.05 | 1.19±0.03#& |
| LV Vol;d (mm) | 63.68±4.56 | 54.61±3.33* | 69.93±2.50* | 67.02±2.25# |
| LV Vol;s (mm) | 22.05±11.32 | 13.66±1.41 | 20.48±3.47 | 20.65±5.08 |
| EF (%) | 66.38±13.93 | 75.03±1.66 | 70.84±4.24 | 69.36±6.59 |
| FS (%) | 37.01±9.38 | 43.02±1.45 | 39.86±3.70 | 38.74±5.08 |
| LV mass (mg) | 83.99±4.71 | 76.55±4.85 | 87.57±4.91 | 108.83±2.66#& |
IVS: d: end-diastolic interventricular septum; IVS: s: end-systolic interventricular septum; LVID: d: left ventricular (LV) end-diastolic diameter; LVID: s: LV end-systolic diameter; LVPW: d: LV end-diastolic posterior wall; LVPW: s: LV end-systolic posterior wall; LV Vol: d: LV end-diastolic volume; LV Vol: s: LV end-systolic volume; EF: ejection fraction; FS: fractional shortening; LV mass: left ventricular mass. Data are presented as Mean ± SD (n=5). *P < 0.05 vs. ND/WT; #P<0.05 vs. HFD/WT: &P< 0.05 vs. ND/MT-KO.
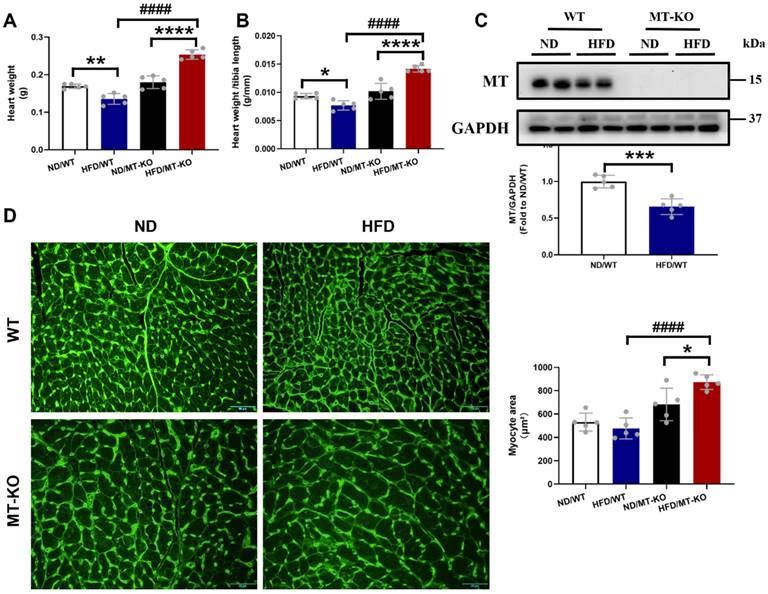
Consistent with cardiac hypertrophy, estimated by echocardiographic LV mass, MT-KO obese mice showed significantly higher heart weight (HW, Fig. 2A) and HW/tibia length ratio (Fig. 2B) than those in two corresponding control groups. We confirmed the absence of the cardiac MT abundance in MT-KO mice with Western blotting analysis since MT expression was undetectable in the hearts of MT-KO mice, whereas it was strongly evident in the WT mice (Fig. 2C). Figure 2C also shows that HFD exposure for 18 weeks significantly downregulated MT in the WT mice. Finally, we used WGA staining to confirm that the cardiomyocytes in HFD/MT-KO group were enlarged compared to those in the other two control groups (Fig. 2D).
The effect of MT knockout on HFD-induced cardiac hypertrophy. A-B, Heart weight and heart weight to tibia length ratio. C, Cardiac MT protein expression evaluated by Western blot. D, WGA staining of cardiac tissue sections and the quantification of myocyte cross-sectional areas (scale bar = 50 μm). Data are presented as Mean ± SD (n=5). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, HFD vs. ND in WT and MT-KO groups, ####P<0.0001, MT-KO vs. WT.
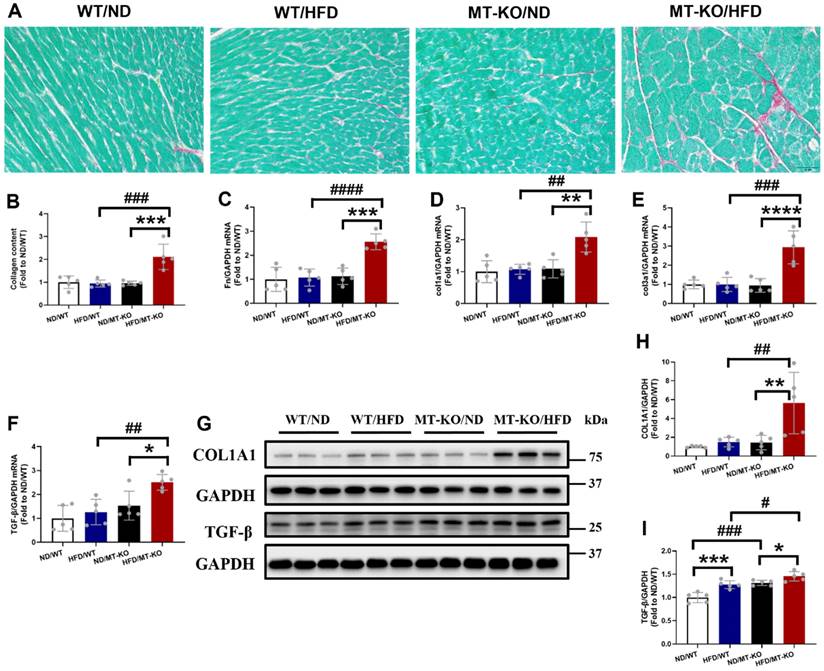
Picro-Sirius Red staining, a general fibrotic index, showed increased collagen accumulation in the hearts of HFD/MT-KO group compared to HFD/WT and ND/MT-KO groups (Fig. 3A, B). Meanwhile, the MT-KO obese group exhibited markedly elevated mRNA expression levels of FN, Col1a1, Col3a1, and TGF-β (Fig. 3C-F) compared to the HFD-fed WT and ND-fed MT-KO groups. Furthermore, the protein abundance of the profibrotic mediators collagen1A1 (Fig 3G, H) and TGF-β1 (Fig. 3G, I) was consistent with their mRNA expression levels (Fig.3 E, F). Thus, MT-KO promoted cardiac hypertrophy and fibrosis in HFD-induced obesity in the mouse model.
The effect of MT knockout on HFD-induced cardiac fibrosis. A, Picro-Sirius Red staining results of heart tissue sections. B, Quantitative analysis of Picro-Sirius Red staining for collagen accumulation. C-F: Cardiac pro-fibrotic mRNA expression of Fn, col1a1, col3a1, and TGF-β evaluated by qRT-PCR. G-I, Protein expression of COL1A1 and TGF-β1 detected by Western blot. Data are presented as Mean ± SD (n=5). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, HFD vs. ND in WT and MT-KO groups, #P<0.05, ##P<0.01, ###P<0.001, ####P<0.0001, MT-KO vs. WT in ND and HFD groups.
Potential mechanisms by which MT knockout exacerbated HFD-induced obesity cardiomyopathy
MT gene knockout increased HFD-induced macrophage infiltration and inflammation in mouse cardiac tissue
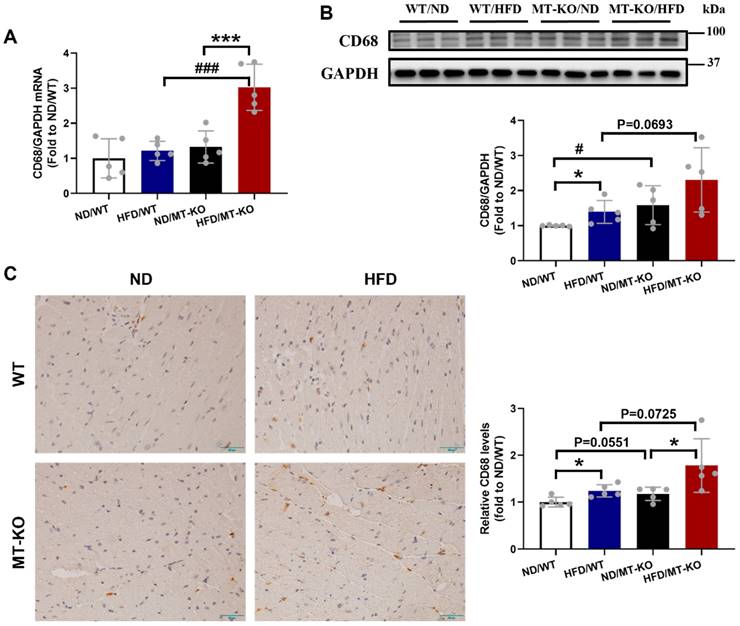
Obesity cardiomyopathy, characterized by macrophage infiltration in both adipose and cardiac tissues, is associated with low-grade inflammation [5,13]. Therefore, we assessed mRNA and protein expression of CD68, a macrophage marker, in the heart following the 18-week HFD. Compared to the ND/WT group, HFD/WT and ND/MT-KO mice only trended increases in the transcriptional expression of CD68 (Fig. 4A) but significantly increased protein expression of CD68 (Fig. 4B). In contrast, HFD/MT-KO mice showed a synergistic increase in CD68 mRNA expression, whereas its protein expression did not significantly increase (Fig. 4A, B). Finally, IHC staining revealed similar mRNA and protein expression profiles for CD68 (Fig. 4C).
The effect of MT knockout on HFD-induced macrophage infiltration into the heart. A, mRNA expression of CD68 detected by qRT-PCR.B, Cardiac protein expression of CD68 evaluated by Western blot. C, Immunohistochemistry staining of CD68 results in the heart and quantitative analysis of IHC staining (brown considered positive staining; scale bar = 50 μm). Data are presented as Mean ± SD (n=5). *P<0.05, ***P<0.001, HFD vs. ND in WT and MT-KO groups, #P<0.05, ###P<0.001, MT-KO vs. WT in ND and HFD groups.
To elucidate the mechanisms by which macrophages infiltrate the cardiac tissues of MT-KO obese group, we evaluated the levels of macrophage-associated chemokine CCL2, along with ICAM-1 and VCAM-1. CCL2 signaling modulates macrophage recruitment and polarization. Additionally, ICAM-1 and VCAM-1 facilitate monocyte adherence to endothelial cells by regulating macrophage cellular chemotaxis and adhesion during inflammation (21-23). The qRT-PCR analysis for CCL2 mRNA showed that the combination of HFD and MT-KO significantly upregulated CCL2 transcription whereas either treatment alone did not (Fig. 5A). We then used western blotting to measure VCAM-1 and ICAM-1 protein expression. HFD or MT gene KO alone slightly or non-significantly upregulated VCAM-1 (Fig. 5B, C) and ICAM-1 expression (Fig. 5B, D). However, their combination substantially increased the expression levels of both adhesion molecules.
The effect of MT knockout on HFD-induced secretion of adhesion molecules, proinflammation cytokines, and chemokines. A, mRNA expression of chemokines including CCL2 evaluated by qRCT-PCR. B-G, Cardiac protein expression of VCAM-1(C), ICAM-1(D), TNF-α(E), IL-6(F), IL-1β(G) detected by Western blot. Data are presented as Mean ± SD (n=5). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, HFD vs. ND in WT and MT-KO groups, #P<0.05, ##P<0.01, ###P<0.001, ####P<0.0001, MT-KO vs. WT in ND and HFD groups.
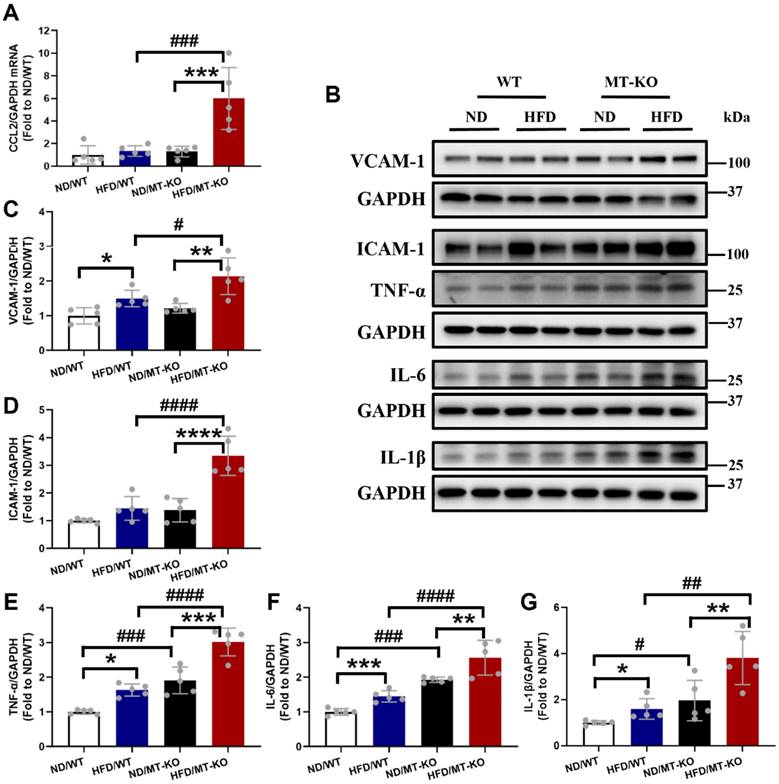
We then measured the expression of cardiac TNF-α, IL-6, and IL-1β. As shown in Fig. 5B, E-G, HFD and MT gene KO significantly upregulated these three inflammatory cytokines expression compared with the cytokine levels observed in WT mice receiving a ND. Moreover, the HFD-fed MT-KO group had higher cardiac TNF-α, IL-6, and IL-1β expression than its two corresponding control groups. Thus, both obesity and MT-KO increased cardiac macrophage infiltration and proinflammatory cytokine production, and the combination of MT-KO and obesity synergistically enhanced these responses.
The exacerbation of HFD-induced cardiac macrophage infiltration and inflammation by MT knockout may be associated with CARD9 activation-mediated signalling pathway
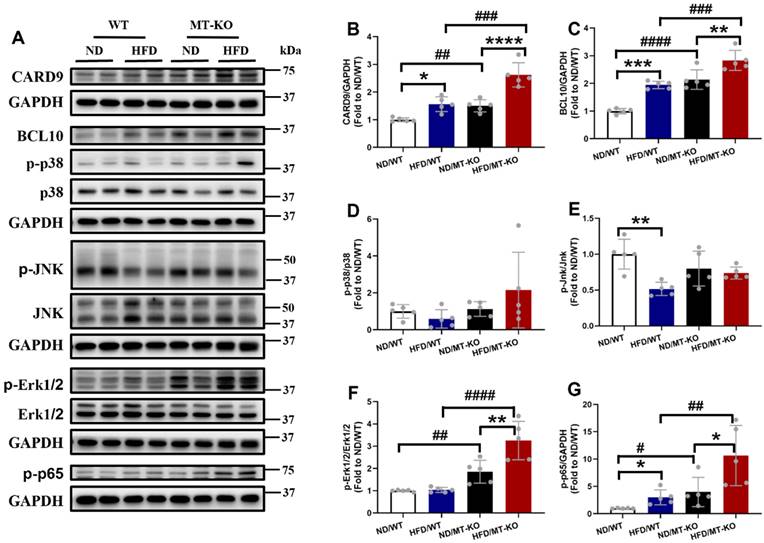
In the myeloid lineage, including macrophages, dendritic cells, and neutrophils, CARD9 forms a CBM complex with BCL10 during innate immune responses, activates NF-κB and/or MAPKs, and stimulates proinflammatory cytokine production [7]. Our previous in vitro study indicated that MT deletion excessively upregulated BCL10 expression and activated p38 MAPK in palmitate-treated cardiomyocytes compared to those in untreated WT controls [19]. Therefore, we hypothesized that hyperactivation of the CARD9 signaling pathway in response to MT gene knockout would contribute to adhesion molecule and proinflammatory cytokine overproduction in MT-KO obese mice. Our results indicated that the CARD9 and BCL10 expression (Fig. 6A, B, C), in parallel to the phosphorylation level of NF-κB (Fig. 6A, G), but not those of p38 MAPK (Fig. 6A, D), JNK (Fig. 6A, E), or Erk1/2 (Fig. 6A, F), were higher in the HFD/WT and ND/MT-KO groups than in the ND/WT group. The extent of these increments was markedly greater in the HFD/MT-KO group.
The effect of MT knockout on HFD-induced activation of inflammation pathway. A-G, Cardiac protein expression of CARD9 (B), BCL10 (C), phosphorylation and total p38 MAPK (D), phosphorylation and total JNK (E), phosphorylation and total ERK1/2 (F), and phospho-NF-κB p65 (G) were detected by Western blot, with GAPDH used as the loading control. Data are presented as Mean ± SD (n=5). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, HFD vs. ND in WT and MT-KO groups, #P<0.05, ##P<0.01, ###P<0.001, ####P<0.0001, MT-KO vs. WT in ND and HFD groups.
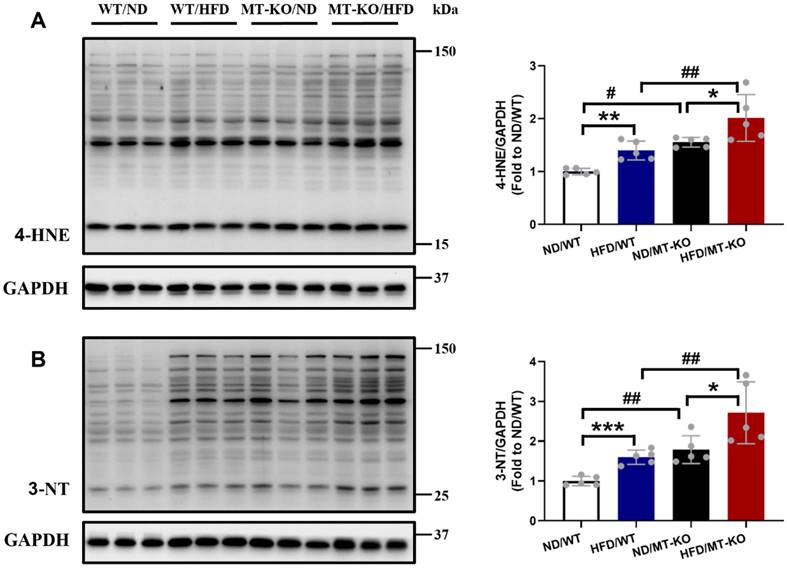
To explore the reason for excessive inflammation, we examined cardiac oxidative stress damage by measuring the lipid peroxidation marker 4-HNE (Fig. 7A) and the protein oxidation marker 3-NT (Fig. 7B) using immunoblotting. The HFD-fed WT group exhibited upregulated levels of the two molecules compared to the ND-fed WT group (Fig. 7A, B). MT-KO mice with ND also had significantly increased expression of these two molecules in contrast with the ND-fed WT group (Fig. 7A, B). Meanwhile, upregulated levels of the two molecules were observed in the MT-KO group subjected to HFD, surpassing those in its two corresponding control groups (Fig. 7A, B). Thus, MT-KO aggravated obesity-induced oxidative stress and damage in the heart.
The effect of MT knockout on HFD-induced oxidative stress damage in hearts. Cardiac expression of 4-HNE (A) and 3-NT (B), as markers of oxidative stress damage, was detected by Western blot, with GAPDH used as loading control. Data are presented as Mean ± SD (n=5). *P<0.05, **P<0.01, ***P<0.001, HFD vs. ND in WT and MT-KO groups, #P<0.05, ##P<0.01, MT-KO vs. WT in ND and HFD groups.
The exacerbation of HFD-induced cardiac macrophage infiltration and inflammation by MT knockout may be associated with trace element dyshomeostasis caused by MT knockout
Trace element dyshomeostasis is involved in the inflammatory and oxidative stress damage in certain diseases [24]. We measured Zn (Fig. 8A), Fe (Fig. 8B), and Cu (Fig. 8C) levels in the mouse hearts to assess how obesity and MT-KO affect trace element homeostasis. Obesity slightly reduced Zn levels in both WT and MT-KO groups compared to their corresponding ND controls (p > 0.05). However, no significant variations were detected between two groups receiving HFD. Fe levels were slightly decreased in MT-KO mice with HFD in contrast to HFD/WT and ND/MT-KO mice. Conversely, Cu levels were slightly elevated in ND/MT-KO and HFD/MT-KO groups compared to their WT controls, respectively (p > 0.05).
The effect of MT knockout on HFD-induced changes of the essential metal level in the heart. Zinc (Zn) (A), Iron (Fe) (B), Copper (Cu) (C) levels in the heart were measured in WT and MT-KO mice. The ratios of Fe/Zn (D), Cu/Zn (E), and Cu/Fe (F) in the heart were then calculated. Data are presented as Mean ± SD (n=5). *P<0.05, HFD vs. ND, #P<0.05, MT-KO vs. WT in ND and HFD groups.
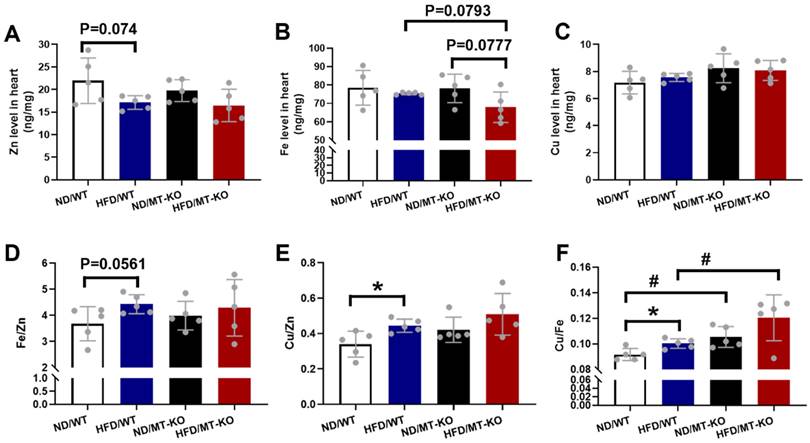
We then calculated Fe/Zn (Fig. 8D), Cu/Zn (Fig. 8E), and Cu/Fe (Fig. 8F). The HFD-fed WT group exhibited a minor increase in Fe/Zn compared to the ND-fed WT group. A HFD increased Cu/Zn and Cu/Fe in the WT group, whereas the MT-KO obesity mice presented with higher Cu/Fe than the HFD-fed WT group. These results suggest that MT-KO exacerbated obesity-mediated trace element dysregulation.
Discussion
In the present study, 18 weeks HFD feeding and MT gene knockout, respectively, caused obesity with glucose intolerance, hypercholesterolemia, essential trace element dyshomeostasis, oxidative stress, and inflammation without any significant changes in cardiac structure. The combination of obesity and MT deletion led to more severe chronic, low-grade inflammation and oxidative stress than either factor alone. In addition, this combination contributed to cardiac remodeling in the form of hypertrophy and fibrosis. These findings indicate that the endogenous stress protein MT protects the heart against HFD-induced oxidative stress and inflammation and consequent cardiac remodeling. Moreover, MT may prevent HFD-induced cardiac remodeling by inhibiting CARD9/BCL10-mediated NF-κB (but not MAPK) activation. Finally, HFD and/or MT gene knockout caused cardiac trace element dyshomeostasis which may trigger cardiac oxidative stress and activation of the CARD9 signaling pathway (Fig. 9).
An illustration outlining the potential protective mechanisms of MT against obesity-induced adverse impacts on the heart that we studied.
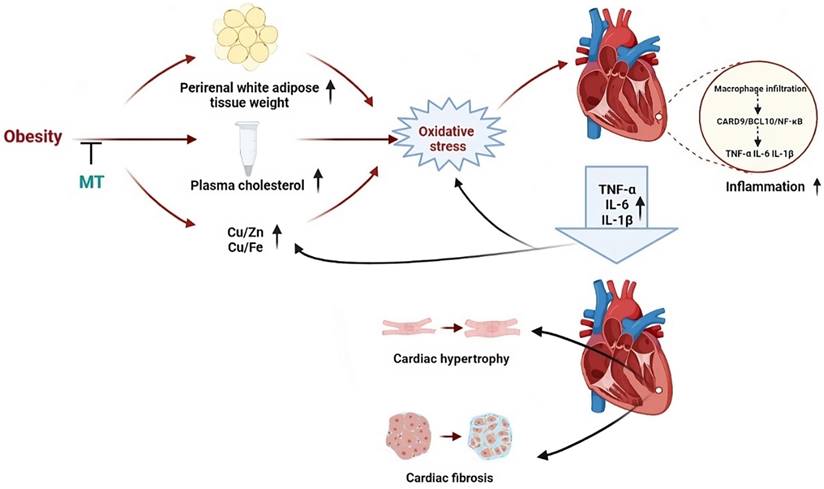
An 18-week HFD (60% kcal fat) in WT mice led to obesity as shown by the augmentation of BW and perirenal white adipose tissue, as well as systemic metabolism disorder including impaired glucose tolerance and increased blood total cholesterol. These adverse metabolic changes induced by obesity were evident in previous studies [25,26]. Moreover, MT-KO with ND feeding resulted in obesity accompanied by the lipid and glucose metabolism abnormalities as WT obese mice. This suggests that MT may help prevent obesity, as indicated by an earlier study [27]. Here, the combination of HFD and MT-KO induced more severe obesity and higher blood cholesterol levels than either factor alone. This indicates that MT could also prevent HFD-induced obesity, as previously reported [28].
Eighteen weeks of HFD-induced obesity combined with MT deletion led to cardiac hypertrophy and fibrosis, whereas HFD-induced obesity only increased oxidative stress and inflammation but did not cause significant cardiac remodeling in the WT group. Thus, the basal MT level may be crucial for alleviating obesity-induced adverse cardiac effects, consistent with our previous in vitro study [19]. Currently, there have been no prior in vivo studies exploring the protective effects of MT on HFD-induced obesity cardiomyopathy in MT-KO mice. However, we previously showed that MT alleviated diabetic cardiomyopathy and nephropathy [18,29]. Here, obesity or MT-KO alone did not cause significant cardiac remodeling or dysfunction. Similarly, a previous study also did not find MT-KO showing cardiac remodeling or dysfunction even in the long term [18]. However, three months of HFD feeding contributed to cardiac hypertrophy in our previous study [10]. This finding differs from the results of the present study. This discrepancy may be attributed to variations in mouse strains and the ages at which a HFD was initiated across studies. In our previous study, we used the obesity-prone C57BL/6J strain [10], whereas we used the obesity-resistant 129S1 strain in the present study [30,31]. Furthermore, HFD was initiated at 4 weeks of age in the previous investigation [10] but at 8 weeks in the present study.
Obesity-induced systemic metabolic disturbances includes impaired lipid and glucose metabolism and contributes to chronic low-grade cardiac inflammation [3]. IL-6, TNF-α, IL-1β, and TGF-β, as downstream proinflammatory cytokines of NF-κB, produced and secreted by immune cells infiltrating the heart muscle, can contribute to cardiac remodeling [6,32]. In addition, the activation of NF-κB can lead to the upregulated expression of chemokines and adhesion molecules, which contribute to immune cells infiltration in the heart [33]. The inflammatory changes observed herein suggest that obesity and MT-KO synergistically affected macrophage infiltration in the heart, proinflammatory cytokine production, CARD9 signaling pathway activation, and, by extension, cardiac hypertrophy and fibrosis.
More specifically, in this process, we first observed macrophages infiltration, along with the expression of CCL2 and ICAM-1, VCAM-1, which are essential in recruiting macrophages and mediating macrophages adhesion to endothelial cells, showed an increasing trend in obese WT mice and MT-KO mice with ND, and a significant increase in MT-KO obese mice. Meanwhile, the expression of IL-6, TNF-α, IL-1β, and TGF-β had the same changes in parallel with macrophage infiltration. Consistent with previous studies, HFD-induced obesity caused macrophage infiltration into the heart and increased secretion of these proinflammatory cytokines [10,13], which contributed to cardiac remodeling. Moreover, diabetic MT-KO mice showed increased cardiac remodeling, mRNA levels of CCL2, and expression of TGF-β and IL-6 compared with diabetic WT mice [18]. These findings indicate that MT prevented cardiac macrophage infiltration by inhibiting chemokine and adhesion molecule expression. Furthermore, the resulting limited inflammation could further restrain the progress of cardiac remodeling.
In addition, we discovered that CARD9 and BCL10 expression and NF-κB activation were markedly elevated in MT-KO obese mice than in WT obese or ND-fed MT-KO mice. However, MAPK (p38 MAPK, JNK, and Erk1/2) activation was not in parallel with the upregulated expression of CARD9 or BCL10. Our previous study reported that HFD-induced obesity led to cardiac remodeling, increased proinflammatory cytokine production, upregulation of CARD9 expression and phospho-p38 MAPK in the heart [19], whereas these obesity-induced adverse effects and p38 MAPK activation were blunted following CARD9 knockout [13]. Moreover, deletion of CARD9 and BCL10 decreased hypertension-induced cardiac fibrosis and electrical remodeling, and reduced NF-κB activity [34,35]. These studies suggest CARD9 mediates its downstream inflammatory effect by triggering either MAPK or NF-κB signaling pathways under different conditions. Here our in vivo data suggest that CARD9/BCL10 may activate NF-κB, but not MAPKs, to mediate obesity- or MT-induced cardiac inflammation. Furthermore, CARD9/BCL10 may be implicated in the synergistic effects of MT-KO and obesity on cardiac remodeling.
Oxidative stress leads to protein modification, lipid peroxidation, and DNA damage [24], and is closely linked to inflammation in chronic disorders, including obesity, diabetes, atherosclerosis, and CVDs [36]. Our earlier work showed that HFD-induced obesity significantly increases cardiac oxidative stress, which leads to cardiac hypertrophy [10]. In line with these studies, our data indicate that obesity led to the downregulated expression of MT, along with increased protein oxidation (indexed by 3-NT) and lipid peroxidation (indexed by 4-HNE) in WT mice. Metallothionein decreases oxidative stress, thereby protecting the heart against metabolic disorders [18]. MT prevents DCM, intermittent hypoxia-induced cardiomyopathy in vivo, and obesity-induced cardiac injury in vitro through its potent antioxidant effects against oxidative stress damage [18,19,37]. In our study, MT-KO obese mice exhibited more severe oxidative stress-mediated heart damage than WT obese mice or MT-KO mice fed a ND. This finding suggests that the downregulation and deletion of MT contribute to decreased antioxidant capacity, leading to and exacerbating cardiac damage under obesity condition. Lipid peroxidation and reactive oxygen species (ROS) upregulate the expression of chemokines and adhesion molecules, thereby facilitating the recruitment of circulating inflammatory cells in the heart [38,39]. Proinflammatory cytokines, in turn, increase ROS production-induced oxidative stress damage [40]. Thus, our findings suggest that MT exerts its beneficial role in HFD by eliminating oxidative stress damage and further limits macrophage infiltration and proinflammatory cytokines-mediated cardiac remodeling.
Obesity causes essential trace elements metabolism disorders, including serum Zn deficiency [41], Fe deficiency [42], and Cu excess [43]. The present study showed these trends in the myocardial concentrations of Zn, Fe, and Cu in WT obese mice in comparison to WT controls. As shown in our previous study, Zn deficiency increased oxidative stress and inflammation, which in turn contributed to cardiac remodeling and dysfunction [19]. As a metal-binding protein, MT mostly binds Zn (and a little Cu) under physiological conditions, playing an important role in storing and donating Zn to other Zn-containing enzymes and transcriptional factors [44,45]. Therefore, MT is crucial for maintaining metal homeostasis [45]. However, the effects of MT on obesity cardiomyopathy and the cardiac essential metal remain largely understudied. We initially observed a decreasing trend in Zn levels in MT-KO obese mice compared to their counterparts on a ND, although this difference was not statistically significant. Moreover, cardiac Fe levels were slightly lower in MT-KO obese mice than in WT obese and ND-fed MT-KO mice.
Based on the existing antagonistic effects between Cu and Fe and between Cu and Zn [46], the application of essential trace element ratios (Fe/Zn, Cu/Zn, and Cu/Fe) has been appreciated as an index for the etiology, diagnosis, treatment, and prognosis of various diseases (47-50). Here, the Cu/Fe ratio was higher in the WT obese and ND-fed MT-KO mice than the WT control. Furthermore, this ratio synergistically increased in the MT-KO obese mice. In addition, Cu/Zn ratio exhibited similar trends to those of the Cu/Fe ratio. These findings are in concordance with the study by Wang et al., wherein the Fe/Cu and Zn/Cu ratios were lowest in the hair of the morbidly obese group compared to the slim, normal, and overweight or obese groups [46]. Meanwhile, higher blood Cu concentrations, Cu/Zn ratio, and reduced Zn levels were found in patients with diabetes mellitus, or cardiomyopathy compared with normal controls [51,52]. Abnormally high serum Cu/Fe ratios can distinguish individuals with progressive mild cognitive impairment (MCI) from cognitively stable MCI subjects [50]. Moreover, the overproduction of free radicals/ROS induced by disruption of the essential metal homeostasis may lead to oxidative stress [24,53]. Therefore, we hypothesize that simultaneous obesity and MT deletion triggered the metal metabolism disorders in the heart and led to the onset of oxidative stress damage and inflammation, which were responsible for the corresponding cardiac remodeling. In addition, the Cu/Fe and Cu/Zn ratios may provide markers for the stage of obesity-related cardiac metabolism in the heart and potentially facilitate the early intervention of MAO.
In conclusion, MT may prevent obesity-induced oxidative stress, inflammation, and, by extension, cardiac remodeling. Abnormal trace element metabolism may trigger oxidative stress, and MT could prevent this pathological process by inhibiting cardiac macrophage infiltration, downregulating CARD9 and BCL10, and activating NF-κB, but not MAPKs (Fig. 9). The discovery that MT-mediated CARD9 signaling activation is implicated in cardiac remodeling may serve as a foundation for future studies investigating the regulation of cardiac inflammation. This finding could also facilitate the identification of potential therapeutic targets for cardiac remodeling and dysfunction. Finally, the Cu/Fe and Cu/Zn ratios may enable clinical monitoring of the metabolic state of obesity.
Abbreviations
MAO: metabolically abnormal obesity; CVDs: cardiovascular diseases; CAD: coronary artery disease; HF: heart failure; CARD9: caspase recruitment domain-containing protein 9; BCL10: B-cell lymphoma/leukemia 10; MALT1: mucosa-associated lymphoid tissue 1; CBM: CARD9/BCL10/MALT1; MT: metallothionein; Zn: zinc; Cu: copper; WT: wild type; KO: knockout; ND: normal diet; HFD: high-fat diet; BW: body weight; IPGTT: intraperitoneal glucose tolerance test; LV: left ventricular; LVID: LV internal dimension; LVPW: LV posterior wall thickness; IVS: interventricular septum; EF: ejection fraction; FS: fractional shortening; Fe: iron; ICP-MS: inductively coupled plasma mass spectrometry; WGA: wheat germ agglutinin; IHC: Immunohistochemistry; 3-NT: 3-nitrotyrosine; 4-HNE: 4-hydroxynonenal; VCAM-1: vascular cell adhesion molecule-1; ICAM-1: intercellular adhesion molecule-1; qRT-PCR: quantitative real-time polymerase chain reaction; COL1A1: collagen1A1; Fn: fibronectin; MCP-1/CCL2: monocyte chemoattractant protein-1; SD: standard deviation; HW: heart weight; ROS: reactive oxygen species.
Acknowledgements
We thank Editage (www.editage.cn) for the language editing of the manuscript.
Funding
This work was supported in part by the China-U.S. University of Louisville Pediatric Research Training Exchange Program (to LC 2014 - 2021, no salary support) and National Institute Environmental Health Science (P30ES030283 to LC). All personal expenses for HZ, WZ, XW, HM, and JW and partial research-related expenses when they worked in the Louisville during 2017 - 2021 were provided by the First Hospital of Jilin University, Changchun, China, under the agreement of the U.S.-China Pediatric Research Exchange Training Program.
Author contributions
Lu Cai: Conceptualization, Supervision, Reviewing and editing. Haina Zhang: Model building, Data curation, original draft writing. Wenqian Zhou, Xiang Wang, Hongbo Men, Jiqun Wang, Jianxiang Xu, Shanshan Zhou: Literature searching, Methodology, result interpretation. Quan Liu: Writing-reviewing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Collaboration (NCD-RisC)* NRF. Worldwide trends in underweight and obesity from 1990 to 2022: a pooled analysis of 3663 population-representative studies with 222 million children, adolescents, and adults. Lancet Lond Engl. 2024;403(10431):1027
2. Zhang Y, Ren J. Epigenetics and obesity cardiomyopathy: From pathophysiology to prevention and management. Pharmacol Ther. 2016;161:52-66
3. Ren J, Wu NN, Wang S. et al. Obesity cardiomyopathy: evidence, mechanisms, and therapeutic implications. Physiol Rev. 2021;101(4):1745-807
4. Lewis AJ, Neubauer S, Tyler DJ. et al. Pyruvate dehydrogenase as a therapeutic target for obesity cardiomyopathy. Expert Opin Ther Targets. 2016;20(6):755-66
5. Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res. 2017;113(4):389-98
6. Zhang H, Wang Y, Men H. et al. CARD9 Regulation and its Role in Cardiovascular Diseases. Int J Biol Sci. 2022;18(3):970-82
7. Ruland J, Hartjes L. CARD-BCL-10-MALT1 signalling in protective and pathological immunity. Nat Rev Immunol. 2019;19(2):118-34
8. Qian J, Wang Q, Xu J. et al. Macrophage OTUD1-CARD9 axis drives isoproterenol-induced inflammatory heart remodelling. Clin Transl Med. 2024;14(8):e1790
9. Liu Y, Shao Y, Zhang J. et al. Macrophage CARD9 mediates cardiac injury following myocardial infarction through regulation of lipocalin 2 expression. Signal Transduct Target Ther. 2023;8(1):394
10. Wang S, Luo M, Zhang Z. et al. Zinc deficiency exacerbates while zinc supplement attenuates cardiac hypertrophy in high-fat diet-induced obese mice through modulating p38 MAPK-dependent signaling. Toxicol Lett. 2016;258:134-46
11. Wang S, Wang B, Wang Y. et al. Zinc Prevents the Development of Diabetic Cardiomyopathy in db/db Mice. Int J Mol Sci. 2017;18(3):E580
12. Zeng X, Du X, Zhang J. et al. The essential function of CARD9 in diet-induced inflammation and metabolic disorders in mice. J Cell Mol Med. 2018;22(6):2993-3004
13. Cao L, Qin X, Peterson MR. et al. CARD9 knockout ameliorates myocardial dysfunction associated with high fat diet-induced obesity. J Mol Cell Cardiol. 2016;92:185-95
14. Shabb JB, Muhonen WW, Mehus AA. Quantitation of Human Metallothionein Isoforms in Cells, Tissues, and Cerebrospinal Fluid by Mass Spectrometry. Methods Enzymol. 2017;586:413-31
15. Yang L, Li H, Yu T. et al. Polymorphisms in metallothionein-1 and -2 genes associated with the risk of type 2 diabetes mellitus and its complications. Am J Physiol Endocrinol Metab. 2008;294(5):E987-992
16. Wang J, Song Y, Elsherif L. et al. Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation. Circulation. 2006;113(4):544-54
17. Gu J, Yan X, Dai X. et al. Metallothionein Preserves Akt2 Activity and Cardiac Function via Inhibiting TRB3 in Diabetic Hearts. Diabetes. 2020;69(2):267
18. Gu J, Cheng Y, Wu H. et al. Metallothionein Is Downstream of Nrf2 and Partially Mediates Sulforaphane Prevention of Diabetic Cardiomyopathy. Diabetes. 2017;66(2):529-42
19. Wang S, Gu J, Xu Z. et al. Zinc rescues obesity-induced cardiac hypertrophy via stimulating metallothionein to suppress oxidative stress-activated BCL10/CARD9/p38 MAPK pathway. J Cell Mol Med. 2017;21(6):1182-92
20. Zhou W, Young JL, Men H. et al. Sex differences in the effects of whole-life, low-dose cadmium exposure on postweaning high-fat diet-induced cardiac pathogeneses. Sci Total Environ. 2022;809:152176
21. Wiesolek HL, Bui TM, Lee JJ. et al. Intercellular Adhesion Molecule 1 Functions as an Efferocytosis Receptor in Inflammatory Macrophages. Am J Pathol. 2020;190(4):874-85
22. Carr RM. VCAM-1: closing the gap between lipotoxicity and endothelial dysfunction in nonalcoholic steatohepatitis. J Clin Invest. 2021;131(6):147556
23. Kanda H, Tateya S, Tamori Y. et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116(6):1494-505
24. Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283(2-3):65-87
25. Hu N, Zhang Y. TLR4 knockout attenuated high fat diet-induced cardiac dysfunction via NF-κB/JNK-dependent activation of autophagy. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):2001-11
26. Tong M, Saito T, Zhai P. et al. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ Res. 2019;124(9):1360-71
27. Beattie JH, Wood AM, Newman AM. et al. Obesity and hyperleptinemia in metallothionein (-I and -II) null mice. Proc Natl Acad Sci U S A. 1998;95(1):358-63
28. Lindeque JZ, Jansen van Rensburg PJ, Louw R. et al. Obesity and metabolomics: metallothioneins protect against high-fat diet-induced consequences in metallothionein knockout mice. Omics J Integr Biol. 2015;19(2):92-103
29. Wu H, Kong L, Cheng Y. et al. Metallothionein plays a prominent role in the prevention of diabetic nephropathy by sulforaphane via up-regulation of Nrf2. Free Radic Biol Med. 2015;89:431-42
30. Luijten IHN, Feldmann HM, von Essen G. et al. In the absence of UCP1-mediated diet-induced thermogenesis, obesity is augmented even in the obesity-resistant 129S mouse strain. Am J Physiol Endocrinol Metab. 2019;316(5):E729-40
31. Chu DT, Malinowska E, Jura M. et al. C57BL/6J mice as a polygenic developmental model of diet-induced obesity. Physiol Rep. 2017;5(7):e13093
32. Chou CH, Hung CS, Liao CW. et al. IL-6 trans-signalling contributes to aldosterone-induced cardiac fibrosis. Cardiovasc Res. 2018;114(5):690-702
33. Miyabe C, Miyabe Y, Bricio-Moreno L. et al. Dectin-2-induced CCL2 production in tissue-resident macrophages ignites cardiac arteritis. J Clin Invest. 2019;130:3610-24
34. Markó L, Henke N, Park JK. et al. Bcl10 mediates angiotensin II-induced cardiac damage and electrical remodeling. Hypertension. 2014;64(5):1032-9
35. Peterson MR, Getiye Y, Bosch L. et al. A potential role of caspase recruitment domain family member 9 (Card9) in transverse aortic constriction-induced cardiac dysfunction, fibrosis, and hypertrophy. Hypertens Res. 2020;43(12):1375-84
36. Shin SK, Cho HW, Song SE. et al. Oxidative stress resulting from the removal of endogenous catalase induces obesity by promoting hyperplasia and hypertrophy of white adipocytes. Redox Biol. 2020;37:101749
37. Zhou S, Wang J, Yin X. et al. Nrf2 expression and function, but not MT expression, is indispensable for sulforaphane-mediated protection against intermittent hypoxia-induced cardiomyopathy in mice. Redox Biol. 2018;19:11-21
38. Aimo A, Castiglione V, Borrelli C. et al. Oxidative stress and inflammation in the evolution of heart failure: From pathophysiology to therapeutic strategies. Eur J Prev Cardiol. 2020;27(5):494-510
39. Binder CJ. Lipid modification and lipid peroxidation products in innate immunity and inflammation. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(4):369-70
40. Liu RM, Desai LP. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015;6:565-77
41. Gu K, Xiang W, Zhang Y. et al. The association between serum zinc level and overweight/obesity: a meta-analysis. Eur J Nutr. 2019;58(8):2971-82
42. Zhao L, Zhang X, Shen Y. et al. Obesity and iron deficiency: a quantitative meta-analysis. Obes Rev. 2015;16(12):1081-93
43. Yang H, Liu CN, Wolf RM. et al. Obesity is associated with copper elevation in serum and tissues. Met Integr Biometal Sci. 2019;11(8):1363-71
44. Atrian S, Capdevila M. Metallothionein-protein interactions. Biomol Concepts. 2013;4(2):143-60
45. Wong DL, Yuan AT, Korkola NC. et al. Interplay between Carbonic Anhydrases and Metallothioneins: Structural Control of Metalation. Int J Mol Sci. 2020;21(16):E5697
46. Wang CT, Chang WT, Zeng WF. et al. Concentrations of calcium, copper, iron, magnesium, potassium, sodium and zinc in adult female hair with different body mass indexes in Taiwan. Clin Chem Lab Med. 2005;43(4):389-93
47. Antwi-Boasiako C, Dankwah GB, Aryee R. et al. Serum Iron Levels and Copper-to-Zinc Ratio in Sickle Cell Disease. Med Kaunas Lith. 2019;55(5):E180
48. Xu Y, Wei Y, Long T. et al. Association between urinary metals levels and metabolic phenotypes in overweight and obese individuals. Chemosphere. 2020;254:126763
49. Kahvaz MS, Soltani S, Soltani S. et al. Low Serum Levels of Selenium, Zinc, Iron, and Zinc/Copper Ratio in an Endemic Region of Cutaneous Leishmaniasis in Southwest Iran. Biol Trace Elem Res. 2021;199(4):1291-6
50. Mueller C, Schrag M, Crofton A. et al. Altered serum iron and copper homeostasis predicts cognitive decline in mild cognitive impairment. J Alzheimers Dis JAD. 2012;29(2):341-50
51. Viktorínová A, Toserová E, Krizko M. et al. Altered metabolism of copper, zinc, and magnesium is associated with increased levels of glycated hemoglobin in patients with diabetes mellitus. Metabolism. 2009;58(10):1477-82
52. Shokrzadeh M, Ghaemian A, Salehifar E. et al. Serum zinc and copper levels in ischemic cardiomyopathy. Biol Trace Elem Res. 2009;127(2):116-23
53. Valko M, Jomova K, Rhodes CJ. et al. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol. 2016;90(1):1-37
Author contact
![]() Corresponding author: Dr. Lu Cai, Pediatric Research Institute, Department of Pediatrics, University of Louisville, Louisville, KY, 40202, USA; E-mail: lu.caiedu.
Corresponding author: Dr. Lu Cai, Pediatric Research Institute, Department of Pediatrics, University of Louisville, Louisville, KY, 40202, USA; E-mail: lu.caiedu.