Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. NO generation
3. NOS in health
4. NO scavenging
5. NO supplementation
6. Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(3):1097-1109. doi:10.7150/ijbs.105016 This issue Cite
Review
Regulation of nitric oxide generation and consumption
Husam M Abu-Soud1,2,3 ![]() , Olivia G Camp1, Jayanth Ramadoss1,2, Charalampos Chatzicharalampous4, George Kofinas4, Jason D Kofinas4
, Olivia G Camp1, Jayanth Ramadoss1,2, Charalampos Chatzicharalampous4, George Kofinas4, Jason D Kofinas4
1. Departments of Obstetrics and Gynecology, The C.S. Mott Center for Human Growth and Development, Wayne State University School of Medicine, Detroit, Michigan 48201, USA.
2. Department of Physiology, Wayne State University School of Medicine, Detroit, MI, 48201, USA.
3. Department of Microbiology, Immunology and Biochemistry, Wayne State University School of Medicine, Detroit, MI, 48201, USA.
4. Kofinas Fertility Group, 65 Broadway, 14th floor, New York, NY 10006, USA.
Received 2024-10-11; Accepted 2024-12-26; Published 2025-1-13
Abstract

Nitric oxide (NO), originally discovered for its role in cardiovascular function, is a key molecule in physiological processes including metabolism, neurotransmission (including memory, learning, neuroprotection and synaptic plasticity), immunity, reproduction, and much more. NO can be synthesized by the catalytic activity of the enzyme nitric oxide synthase (NOS), which is found biologically in three isoforms, or nonenzymatically based on simple reduction of nitrate and nitrite or by the NO-donor S-nitrosothiol (R-SNO). Importantly, the deficiency of NO has been noted in a wide range of pathologies including cardiovascular disease, cancer, erectile dysfunction, male and female infertility, and mitochondrial disease. While there are several pathways that can lead to a reduction in the bioavailability of NO (i.e., consumption, inhibition, and substrate competition) it is the conclusion of the authors that multiple pathways co-exist in pathological states. This article outlines for the first time the major pathways of NO generation, the importance of NO in health, NO scavenging and enzyme inhibition, and the potential benefits of supplementation.
Keywords: Nitric oxide deficiency, nitric oxide synthase, mammalian peroxidase, NOS inhibitors, ROS
1. Introduction
Nitric oxide (NO) is a signaling molecule and a powerful vasodilator. It plays a critical role in hormonal release stimulation and neurotransmission regulation [1, 2]. NO is not only generated enzymatically by nitric oxide synthases (NOSs) [neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS)] but also is generated non-enzymatically either by one and two electron reductions of nitrite (NO2-) and nitrate (NO3-), respectively or via the NO-donor S-nitrosothiol (R-SNO). The three NOS isoforms can be distinguished from each other by their primary sequence, post-translational modifications, cellular location, and tissue expression [3]. For example, eNOS and nNOS are constitutively expressed, whereas iNOS expression is induced. It has been suggested that crosstalk exists between the three isoforms to regulate NO production in both healthy conditions (physiological NO signaling) and in disease states (excessive NO generation). This is accomplished primarily through coordinated expression, subcellular localization, and activity levels [4, 5]. While there is ample evidence for crosstalk between the isoforms, a unifying concept of regulation and the factors that govern their concentrations in different biological compartments is lacking. Neuronal NOS is constitutively expressed in the brain and is made up of both particulate and soluble forms that qualifies these cells to handle various advanced responsibilities such as involvement in memory, modulation of learning, and neurogenesis [3]. The expression of nNOS can be found in the central nervous and peripheral nervous systems, as well as skeletal and smooth muscle cells, where it regulates synaptic transmission, central regulation of blood pressure, tone of smooth muscle in blood vessels, and vasodilation via peripheral nitrergic nerves [3]. Inducible NOS expression can be found in many cell types in response to lipopolysaccharide, cytokines, or other agents and contributes to the pathophysiology of inflammatory diseases and septic shock [3]. The primary role of iNOS is in the immune response where NO can break up or inactivate heme-containing enzymes or iron-sulfur clusters, modulate cysteine nitrosylation, or can react with superoxide (O2•-) to form peroxynitrite (ONOO-), a potent oxidant and nitrating agent [6]. Both NO and ONOO- can cause DNA breaks and damage by invading pathogens. Moreover, NO can activate the second messenger cyclic guanosine monophosphate (cGMP) through mediation of guanylate cyclase activity that then activates downstream signaling cascades [6]. Endothelial NOS is regularly expressed in endothelial cells where it functions mainly to keep blood vessels dilated, regulates blood pressure, displays several vasoprotective functions, and acts as an anti-atherosclerotic agent [3]. Endothelial cell eNOS activation is mediated by post-translational modification via its multi-site phosphorylation. For instance, ser1177eNOS is a widely investigated excitatory phosphorylation site in the enzyme's reductase domain, whereas thr495eNOS is an inhibitory site located in its calmodulin binding domain [7]. NO generated in endothelial cells diffuses rapidly to adjacent smooth muscle cells, which activates soluble guanylate cyclase (sGC) and allows the conversion of GTP to cyclic GMP, initiating a signaling cascade leading to vasodilation [8, 9].
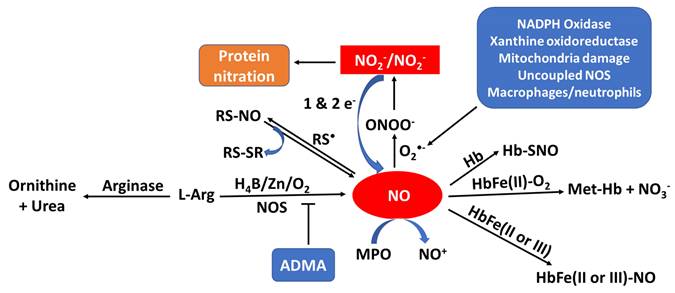
When sufficient NO is not produced, known as NO deficiency, there is an increased risk of several diseases like cancer, diabetes, cardiovascular, pulmonary diseases, mitochondrial diseases, infertility and heart disease [10-17]. NO deficiency can often be asymptomatic, while for others the deficiency comes with fatigue and decline in exercise capacity, poor vision, high blood pressure, slow wound healing, weakened immune system, and erectile dysfunction [18-23]. An increasing number of disorders are associated with reduced NO synthesis or elevated oxidative removal of NO into the arterial wall, frequently involving those linked with risk factors for atherosclerosis such as high blood pressure, high cholesterol, heart failure, smoking, and diabetes [24]. Regardless of whether the underlying etiology of endothelial imbalance is decreased levels of NO generation or enhancement of NO consumption, it has led to the investigation of numerous therapies to assess the reversibility of endothelial dysfunction by NO supplementation and the enhancement of the release of NO from the endothelium. NO supplementation and/or enhancing the release of NO from the endothelium significantly improves cardiac health, healing, erectile dysfunction, high blood pressure, and respiratory response [25]. Understanding the mechanisms that cause the deficiency of NO and the effects of low NO levels is essential for addressing this issue and improving overall health. There is considerable evidence, reviewed in this article, to suggest that multiple pathways lead to NO deficiency. As summarized in Figure 1, the bioavailability of NO can be regulated by one or more of the following: NOS uncoupling (deficiency of NOS substrate and/or cofactors), NO scavengers (e.g., oxyhemoglobin, mammalian peroxidases, and superoxide), NOS inhibitors (ADMA), NOS competitor (arginase II), and protein activators (soluble guanylate cyclase (sGC)). In the current review we will discuss the important role of NO in physiological endothelium, and we will highlight the consequences of this molecule in pathological states altering endothelial function [26].
Model outlining the pathways of NO production, consumption, and NOS inhibition and their byproducts. Sources of NO production include NOS, nitrite/nitrate (NO2-/NO3-) reduction, and R-SNO. Sources of consumption include superoxide (O2•-), hemoglobin (Hb), and myeloperoxidase (MPO). NOS inhibition occurs through arginase (enzymatic competition) or asymmetric dimethylarginine (ADMA).
2. NO generation
2a. Nitric oxide synthases
The enzymatic activity of the three NOSs requires the presence of flavins FMN and FAD, tetrahydrobiopterin (H4B), calmodulin (CaM), zinc ion (Zn), NADPH, L-arginine (L-Arg) and molecular oxygen (O2) [27] (Table 1a). In the NOS cycle, the electrons donated by NADPH are first delivered to the NOS flavins. In the presence of CaM, electrons transfer instantly to the NOS heme iron [28, 29] where the fully reduced form of NOS permits the heme iron to bind and activate molecular oxygen that catalyzes NO synthesis from L-Arg and H4B. L-Arg and the cofactors H4B and Zn play a central role in NOS coupling, with their deficiency resulting in the enzyme generating the free radical O2•- as a replacement of NO [3] (Table 1a).
Pathways of NO consumption and deficiency. Table outlining the ways in which NO bioavailability can be reduced through enzymatic uncoupling, enzymatic and non-enzymatic scavengers, enzyme inhibition, and enzyme competition with schematic representations of the mechanisms.
| NOS uncoupling | ||
|---|---|---|
| A. Substrate (L-Arg) and co-factors (H4B and/or Zn) deficiency | 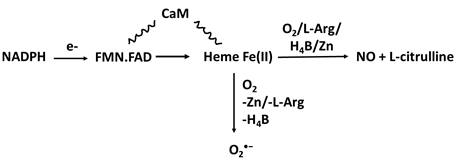 | NOS monomarization, Oxidative stress, Protein nitration; DNA damage and biomolecule modification including amino acids, proteins, enzymes, and cofactors; tyrosine nitration. |
| NO scaenger | Reaction | Effects |
| B. OxyHemoglobin | 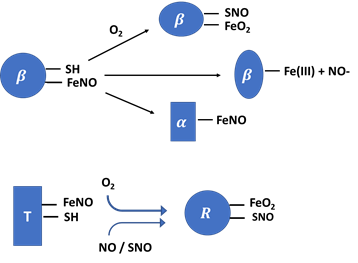 | Oxygen deficiency and Hb-SNO formation |
| C. Superoxide | 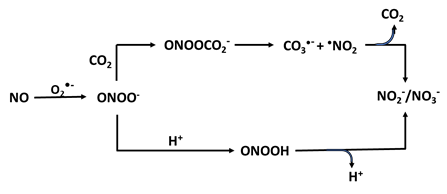 | Protein nitration; DNA damage and biomolecule modification including amino acids, proteins; enzyme and cofactor modification; tyrosine nitration |
| D. MPO | MPOFe(III or II) + NO → MPOFe ((III or II)-NO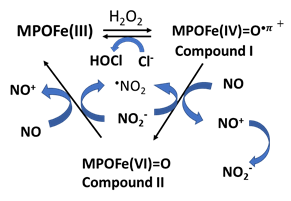 | Protein nitrosylation |
| NOS inhibitors | ||
| E. ADMA | 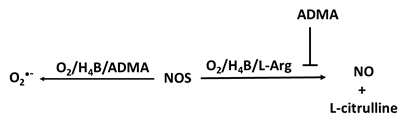 | Oxidative stress, DNA damage |
| NOS Competitor | ||
| F. Arginase II | 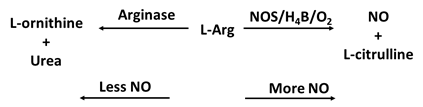 | O2•- generation |
2b. Nitrite /nitrate
NO can be generated non-enzymatically from the circulating pool of longer-lived NO metabolites including nitrite (NO2-), nitrate (NO3-), and S-nitroso and N-nitroso species providing an alternative pathway to supply bioactive NO in addition to typical NOS-derived NO. These metabolites can give rise to NO following reductive bioactivation and cooperates with tissue-bound storage forms of NO undergoing redox-activation to contribute to overall availability [30]. One essential contributor of NO2- is oral commensal bacteria that possess the ability to bioactivate NO3- [30]. Thus, the two major sources of NO3- supply are NO3- from oxidized endogenous NO or dietary intake [31, 32]. Nitrite acts as a key regulator of cellular signaling depending on the surrounding oxygen gradient [33]. Thus, during hypoxia and low pH situations when NO generation by the NOSs may be compromised, pathways for enzymatic and nonenzymatic reduction of NO2- to NO are enhanced [30] by elevation of NO3-—NO2-—NO activity. Due to the importance of nitrite in preserving cellular homeostasis, there must be a pathway in situ to maintain its total level in the face of fluctuating levels of dietary intake of nitrite/nitrate and alterations in eNOS activity secondary to fluctuations in blood flow [26]. Due to this essential role, dietary NO2- is necessary for various physiological activities and having a reservoir of NO2- and NO3- have positive biological NO-like functions. Thus, dietary NO2- plays an essential role in various physiological activities as an effective supplement of NO2- and NO in the human body [34].
2c. S-nitrosothiol, a NO donor
S-nitrosothiols (R-SNO) are NO donors that extend the lifetime of the active NO by essentially acting as a carrier for the relatively short-lived free radical. SNO is also essential in the localization and activity of enzymes and receptors by encouraging modulation of signaling pathways, axonal transport, synaptic plasticity, and protein assembly [35]. The two most commonly occurring and naturally abundant SNO forms are S-Nitroso-serum albumin and S-nitrosoglutathione (GSNO) [35]. They are both present in circulating blood and GSNO has also been found intracellularly [35].
S-nitrosolation can occur to any protein with a free thiol moiety but this process is often selective [36, 37]. The formation of R-SNO on proteins (namely S-nitrosation or S-nitrosylation) is a unique post-translational modification identified on proteins such as protein tyrosine phosphatases, NF-κB, IκB kinase, Ras, etc. This process is directed by peptide sequences around the cysteine residue, exogenous NO sources, or by compartmentalization of NO synthesis. SNO formation and release is influenced by the redox environment, oxygen and metal ion availability, and thiol reactivity [36]. For example, red blood cells release SNO under conditions of low-oxygen [38]. There are 3 known mechanisms in which the S-NO bond can be cleaved to release NO. First, copper(I), after Cu2+ reduction, can react with the nitrosothiol to form a sulfhydryl anion then regenerating Cu2+. Cu2+ can be reduced by ascorbate and free thiolate ions, or ascorbate can decompose SNO directly. In this second pathway of NO release, ascorbate concentrations greater than mM level, a free thiol and dehydroascorbate are generated upon NO release. Third, the S-NO bond may undergo homolytic cleavage by light (330-350 or 550-600 nm) to release NO and form disulfide bonds [35, 39]. Due to the different mechanisms of reduction and nature of RSNO that can alter the rate of NO liberation, concentration, and duration, the physiologic relevance of SNO is still under consideration.
3. NOS in health
NO plays a pivotal role in a wide variety of processes ranging from neurotransmission, vasodilation, and platelet function to immunity, penile erection, and modulation of metabolic function through regulation of vascular tone. This function is often attributed to the role of NO in activation of guanylate cyclase (GC). NO binds to GC-Fe(II) to form an unstable six coordinate ferrous-nitrosyl complex which converts to a five-coordinate GC-Fe(II)-NO complex by breaking the trans axial ligand bond, enabling the enzyme to catalyze the conversion of GTP to cGMP. This function is essential in vasculature homeostasis, where eNOS generated NO aids in tissue blood flow regulation, vascular remodeling, and endothelial protection against platelet aggregation and leukocyte adhesion [40, 41]. After synthesis, NO diffuses over 100 microns through tissues and enters red blood cells where it reacts with oxyhemoglobin [42]. Interestingly, NO removal by a reaction with Hb and O2•- does not result in the complete loss of NO-mediated signaling to vascular smooth muscle cells [43, 44], raising the possibility that alternative mechanisms exist for NO depletion.
A loss in NO production results in endothelial dysfunction and represents an early event in the development of hypertension. A disturbance in NOS activity has been attributed to the pathophysiology of heart failure in which it was reported under expression and/or uncoupled NOS activity which promotes the generation of O2•- instead of NO that results in dysfunctional calcium (Ca2+) handling, cardiac remodeling, hypertrophy, and thus, the development of heart failure [5, 45-48]. Conversely, overexpression of eNOS has shown to be protective in animal models [49]. Genetic predisposition to enhance NO signaling has been associated with reduced risk of coronary heart disease and stroke [50]. Optimal NO production is required, with a fine balance required for many heme and nonheme proteins via interaction with their metal centers [51, 52]. Hence, factors that affect rates of NO production and consumption are of significant interest.
Furthermore, nitrosative stress subsequent iNOS induction has been implicated in heart failure [53, 54]. NO is a potent scavenger of several radical intermediates such as alkoxyl radicals and lipid peroxyl [44, 55-58]. Similar processes likely occur in atherosclerotic lesions where lipid oxidation products are augmented [59]. Consistent with this notion, previous studies [60] demonstrated turnover-dependent consumption of NO by 15-lipoxygenase, an enzyme implicated in atherogenesis [61, 62]. NO overproduction has the ability to bind with hemoproteins heme iron at nearly diffusion-controlled rates to form the corresponding nitrosyl complex which inhibits their catalytic activity.
4. NO scavenging
There are several pathways that can decrease bioavailable NO. First, hemoglobin possesses the physiological ability to bind NO allowing hemoglobin to modulate NO activity, maintenance, and signaling function. NO can be depleted by myeloperoxidase (MPO), a pre-inflammatory enzyme, by binding to MPO ferric and ferrous heme iron, or it can be utilized as a one electron substrate for MPO Compounds I (Fe(IIV)=O + and Compound II (Fe(IV)=O complex) generating nitrosonium cation (NO+) [44, 52]. Asymmetric dimethylarginine (ADMA), a recognized NOS endogenous competitive inhibitor, also plays a role in the pathogenesis of lowering NO generation in several disorders [63]. ADMA is a competitive inhibitor of NOS binding to the L-Arg site, disturbing the function of the enzyme and resulting in the production of O2•- instead of NO. Below, we will discuss in detail the mechanisms in which NO can be depleted.
+ and Compound II (Fe(IV)=O complex) generating nitrosonium cation (NO+) [44, 52]. Asymmetric dimethylarginine (ADMA), a recognized NOS endogenous competitive inhibitor, also plays a role in the pathogenesis of lowering NO generation in several disorders [63]. ADMA is a competitive inhibitor of NOS binding to the L-Arg site, disturbing the function of the enzyme and resulting in the production of O2•- instead of NO. Below, we will discuss in detail the mechanisms in which NO can be depleted.
4a. Hemoglobin and NO scavenging
Nearly 92% of red blood cells are made up of hemoglobin (Hb), an oxygen-carrying protein that is responsible for their red color [64]. Hb is made up of a tetramer containing four protein globin subunits each with a heme prosthetic group allowing it to bind oxygen in two forms: the taut form (T-form) and the relaxed from (R-form) [65]. The T-form has a lower oxygen affinity while the R-form has a higher oxygen affinity, and with the ability to spontaneously change from one form into another, allows Hb to transfer molecular O2 from the lung to the outlying tissue, and carbon dioxide from tissue to the lung [66]. NO can also bind to the ferrous heme on the cysteine thiol at the beta-93 position of Hb forming the corresponding HbSNO complex (Table 1b). SNO-Hb can also be formed through a reaction of NO with a thiyl radical. Formation of SNO-Hb takes place when the Hb is in the R-state [67-71] and NO released from the complex is linked thermodynamically to the release of O2. Notably, the reactivity and function of this Cys is linked to the binding of oxygen (i.e.'thermodynamic linkage').
In its T-state, α-nitrosyl hemoglobin has NO binding to the heme iron which causes histidine and iron to break forming a five-coordinated Hb-Fe(II)-NO complex (one bound to the NO and four bound to the heme porphyrin ring) and this species can be detected directly by electron paramagnetic resonance (EPR) technique [72]. NO has a very high affinity for the ferrous heme, with a dissociation constant (Kd) of 10-10-10-11 M. NO can also bind to the methemoglobin to generate Hb-Fe(III)-NO complex with a considerably lower affinity (Kd=2.5 × 10-4 M) (Table 1b) [42]. The functional Hb that displays high capacity to deplete NO typically exist in two ferrous forms: oxygenated (oxy-Hb; Hb-Fe(II)-O2) and deoxygenated (Hb-Fe(II)). Hb ferrous (Fe (II)) displays high oxygen affinity with a carrying size ranging from 1.36 to 1.37 ml O2/gram of the protein. Methemoglobin (Fe-(III)), ferryl porphyrin radical cation, Hb-Fe(IV)=O+ π•), Compound I and (Hb-Fe(IV)=O), Compound II are all other forms of Hb with higher oxidative states that play a role in developing several undesired pathophysiologic disorders [73].
The encapsulation of hemoglobin in red blood cells has previously been shown to scavenge NO. The near diffusion reaction rate between NO and intraerythrocytic hemoglobin plays an essential role in NO bioavailability and alters homeostatic vascular function. One major pathway is through reaction with oxyhemoglobin (that occurs at a second order rate constant of 6-8 × 107 M-1 s-1) to produce met-hemoglobin and nitrate [74]. Through this reaction, the amount of nitrate made is biologically inactive, suggesting a major role of hemoglobin is to inhibit NO signaling. Despite the high affinity of oxyhemoglobin towards NO, several mechanisms have been documented by which hemoglobin may sustain, control, and even generate NO activity leading to NO maintenance of signaling function. These studies have shown that NO can be converted to several functional species including thiols nitrosation, lipid nitration, nitration, and nitrite generation that have been thought to be essential in signal transduction pathways within many different physiological processes [33, 42, 75-78]. Due to its essential role in many biological systems, loss of NO bioavailability leads to disease in several circumstances such as hemolytic anemias [79], older blood transfusions [80], and endothelial dysfunction that occurs in pathological conditions like metabolic syndrome, [81] as well as aging [82, 83]. In these circumstances, NO supplementation has additional effects that make NO an attractive therapeutic avenue for certain patients. Despite the significant amount of research done in this field, the argument regarding detailed mechanisms of NO activity protection and how Hb contributes to NO activity remains unanswered.
4b. Superoxide NO scavenger
NO is thought to act as a protective agent in endogenous anti-oxidative mechanisms by eliminating the toxic radical, O2•-, thereby helping in the maintenance of the required level of O2•- in normal circumstances. This provides a shielding function against the O2•-action in many organs including the kidney [84]. Mitochondrial damage is considered the main generator of intracellular pathologic levels of O2•- through NADPH oxidase, which originates in neutrophils, eosinophils, monocytes, and macrophages [85-88]. Xanthine oxidoreductase (XOR) another source of reactive oxygen species, converts hypoxanthine and xanthine to uric acid with immediate production of O2•- [88, 89]. Alternatively, uncoupled NOS mediated by the deficiency of the substrate or NOS cofactors allows NOS to generate O2•- instead of NO [28, 90]. In all cases, the majority of generated O2•- undergoes a nonenzymatic or superoxide dismutase (SOD)-catalyzed reaction generating hydrogen peroxide (H2O2). Imbalance between O2•- production and metabolism may mediate meiotic arrest and apoptotic cell death through activation of caspase-3 with DNA breaks and damage [91]. Similarly, compromised antioxidant machinery e.g., reduced glutathione, could affect optimal chromatin decondensation at fertilization and consequently alter gene expression [92, 93]. Similarly, DNA repair mechanisms could be altered as well, contributing further to DNA damage [92, 93].
The reaction of NO with O2•- occurs at near diffusion rate yielding ONOO- and accelerates NO depletion. ONOO- is capable to cross the erythrocyte membrane through anion channels [94, 95] and is classified as a strong oxidant, therefore, it reacts instantly with electron-rich groups including sulfhydryls [96], iron-sulfur centers [97], zinc-thiolates clusters [98], and the active site sulfhydryl in tyrosine phosphatases [99]. Due to the fact NO can readily diffuse through membranes, NO and superoxide do not have to be generated within the same cell to form ONOO-. Several studies have shown that accumulation of O2•- in biological systems can occur in NO deficient-environments and can cause variations in organ function [100]. It has been shown NOS inhibition elevates vascular O2•- generation both in rats and in humans, and such enhanced O2•- production was abolished by the use of a O2•- scavenger [101-103]. Earlier investigations have shown that during NO inhibition there is increased damage of kidney function in hypertensive animals [104-106]. Thus, the development of hypertension may in part be due to disproportion of NO and O2•- through their physiologic contributions to controlling normal kidney function [107].
The production of ONOO- from NO consumption is a key mechanism contributing to oxidative stress and is implicated in the pathogenesis of various chronic diseases, making further research in this area crucial for understanding its role in cellular damage. ONOO- is responsible for an increase in the NO2-: NO3- ratio [108, 109] (Table 1c). The pathway of the decay of ONOO- depends on two factors: the pH and bioavailability of carbon dioxide (CO2). At physiologic conditions, the CO2-catalyzed decomposition of ONOO- is significantly faster than the proton-catalyzed decomposition/isomerization to NO3- [110]. Many of the biological effects ascribed to NO are in reality mediated by ONOO- and subsequent formation of toxic free radical intermediates. In biological systems where CO2 levels are relatively high, the reaction rate with ONOO- is quite fast (rate constant of 3-6 × 104 M-1s-1). ONOO- and CO2 lead to the formation of a short-lived intermediate (lifetime, < 3 ms), nitrosoperoxycarbonate adduct (ONOOOCO2-), which decomposes in the absence of target molecules to NO3- and CO2 through rise of some highly oxidizing radical intermediates, •NO2- and •CO3- [111, 112] (Table 1c). These free radicals are much more toxic and attack many cellular components, enhancing nitrating capabilities compared with ONOO-/peroxynitrous acid (ONOOH), reacting with thiols and iron-sulfur centers, as well as initiating lipid peroxidation.
Although it is necessary to remove unwanted ONOO- to neutralize its toxicity in a variety of pathologies such as cardiovascular and infertility disorders, stroke, diabetes, cancer, and neurodegenerative disorders, ONOO- could also play a role as a strong antimicrobial agent, and therefore, targeting the ONOO--detoxifying systems of microbes appears to be a decent strategy for infection control [113, 114]. Striking mechanisms are suggested by which enhanced ONOO-/NO, or related physiological changes may induce chronic fatigue syndrome, immune dysfunction, memory dysfunction, and multi-organ pain [115]. Several scavengers and neutralizers of ONOO- are currently available such as peroxiredoxins, metalloporphyrins, N-acetylcyteine, and dihydrolipoic acid [116]. Consequently, it is possible that in NO deficient diseases, there is an overproduction and accumulation of O2•- that lead to development of oxidative stress both in tissues and organs [117].
4c. Mammalian peroxidase and NO consumption
MPO, a highly expressed hemoprotein in neutrophils, has been shown to modulate the vascular signaling and vasodilatory functions of NO throughout acute inflammation [118]. NO functions to alter peroxidase catalytic activity by generating the labile nitrosoniom cation (NO+; lifetime 2 ns) which then decays to nitrite/nitrate [44, 119] (Table 1d). Acute endotoxemia and impaired endothelium in a rodent model, has been shown MPO accumulated in and around vascular endothelial cells after leukocyte degranulation in a dependent relaxant response, to which MPO-deficient mice were resistant [120]. Altered vascular responsiveness was attributed to catalytic scavenging of NO by MPO. Therefore, MPO can directly modulate vascular inflammatory responses through regulating NO bioavailability. This is because the mammalian peroxidase superfamily (MPO, EPO, and lactoperoxidase) have proven to be a potent scavenger of NO at sites of inflammation and cardiovascular disorders [44, 119]. Functional enzymes utilize H2O2 in the presence of halides (Cl-, Br-, I-) and pseudo (SCN-) to form a redox transient intermediate compound I, a ferryl π cation radical (Fe(IV)=O⋅+π) with a total formal heme charge of +5. Under these circumstances, the oxidation of halides and pseudo halide by compound I occurs through a single 2 e- transfer reaction, where the heme of the enzymes is reduced to ferric state and the corresponding hypohalous acid is formed [121, 122]. NO depletion is mediated through direct reaction of NO as 1e- physiological substrate and indirectly by radical-radical coupling reactions with peroxidase-produced free radical species. As a substrate, NO accelerates the formation and decay of compound II, the rate-limiting step in the peroxidase cycle, therefore, enhancing the overall rates of catalysis [44] (Table 1d). It also modulates the distribution of peroxidase intermediates, compounds I and II, available during catalysis, hence affecting the substrate selectivity of the enzymes. In contrast, high levels of NO binds ferric and ferrous forms of MPO to form the corresponding stable six-coordinated low spin, MPO-Fe(III)-NO and Fe(II)-NO complex, respectively, rendering them catalytically inactive [52]. Studies of the potential mechanism of interaction between the NO+ and DNA bases are crucial for the understanding of the progress of genetic modification. Importantly, it has been thought that the resistance of cancer cells and apoptosis is regulated by NO+-mediated S-nitrosylation of key enzymes [123].
4d. NOS inhibitor (ADMA)
All three isoforms of NOS can be inhibited by ADMA, released from myelin basic proteins and highly expressed in neuronal tissue. ADMA is thought to be involved in the pathogenesis of decreased NO production in several disorders as it is a competitive inhibitor that binds to the L-Arg site of NOSs, disturbing the function of the enzyme resulting in the generation of O2•- instead of NO [124] (Table 1e). It is estimated that around 300 μmol of ADMA/day is generated by humans, and plasma concentrations are estimated around 0.5-5 μmol/L [125]. The IC50 (half-maximal inhibitory concentration) for the three NOSs is dependent on the prevailing arginine concentration, and ADMA effects can be restored by adding excess L-Arg [126, 127]. Because of that, ADMA level has been identified as a marker and independent risk factor for development of atherosclerosis, cardiovascular death and all-cause mortality [128-131]. Notably, patients with hypertension who underwent Intensive Lifestyle Treatment (ILT) for six months showed improved in ADMA levels and showed that ADMA was dependent on the dietary inflammatory index (DII) content of the diet [132].
ADMA is typically degraded enzymatically through dimethylarginine dimethylaminohydrolase (DDAH), which occurs in two isoforms DDAH1 and DDAH2 [131]. DDAH1 is highly expressed in brain, suggesting specific function in this area. The presence of nNOS and DDAH1 in brain suggests that ADMA may have specific central nervous system (CNS) activity and be more than an unregulated metabolite [125]. DDAH2 is more abundant in the heart, lungs, and placenta [131, 133, 134]. An enhancement of plasma ADMA is associated with decreased NO in individuals with hypercholesterolemia, hypertension, polycystic ovary syndrome (PCOS) and atherosclerosis [124, 131, 135]. Serum ADMA levels have small variability during the menstrual cycle, with elevated amounts in the follicular phase and lowering levels in the luteal phase [135]. Utilizing dehydroepiandrosterone-induced PCOS rat (Sprague Dawley) model and the ovarian granulosa cell line, KGN, Li and colleagues [136] investigated the effect of the ADMA-dimethylarginine dimethylaminohydrolase 1 (DDAH1) pathway on redox status and ovarian apoptosis. These rats developed high levels of ADMA in serum and lower levels of DDAH1 expression in the ovaries. ADMA treatment of the KGN cells exposure to ADMA stimulate ROS accumulation which mediates apoptosis. Overexpression of DDAH1 enhanced cell viability, and reduce oxidative stress, and this effect was reversed in DDAH1 knockdown cells. Quantification of ADMA levels and redox status in serum specimens obtained from women (n=19) with PCOS and healthy individual (n=17) (controls) have shown that women with PCOS had increased serum ADMA levels and decreased glutathione peroxidase (GSH-PX) relative to controls. Collectively, these investigations highlight the involvement of enhanced ADMA levels and redox imbalance in PCOS, suggesting variations in the activity of DDAH that may restrict NO levels by modulating ADMA [137].
4e. NOS Competitor (arginase II)
Arginase is considered as a critical regulator of NOS that may contribute to the progression of multiple pathologies, including vascular disease, aging, cystic fibrosis, sickle cell disease [138]. Arginase, a crucial enzyme in the urea cycle, catalyzes the conversion of L-Arg to urea and L-ornithine [139]. The ability of arginase to compete with NOS for the same substrate, L-Arg, results in the inhibition of the generation of NO through NOS uncoupling (Table 1f). Under conditions of NOS uncoupling, such as in the absence of the substrate L-Arg and/or the cofactors zinc or H4B, the ferric resting NOS undergoes steady-state catalysis of NADPH oxidation. The NOS will still undergo heme iron-catalyzed O2 reduction but will generate superoxide and/or ONOO- instead of NO and no ferrous-nitrosyl complex was formed during study state catalysis of NOS [138]. The competition also causes restraint of the translation and stability of iNOS proteins and inhibition of iNOS activity throughout the urea production life cycle. Arginase also diverts the metabolism of L-Arg to L-ornithine and the generation of polyamines and L-proline which are important for smooth muscle cell growth and collagen synthesis [138]. Enhancement of arginase activity inhibits eNOS NO synthesis and may lead to endothelial dysfunction in several disorders including hypertension, diabetes, aging and ischemia-reperfusion, as initiation of arginase may encourage abnormal vessel wall remodeling and neointima formation [140]. Growing evidence suggests arginase may stimulate both endothelial and vascular smooth muscle cell dysfunction by altering the intracellular metabolism of L-Arg [138]. In circulation, the synthesis of NO by eNOS plays an important role in protecting vascular homeostasis by inhibiting vascular tone, platelet aggregation, as well as inflammation [41]. iNOS NO functions in an autocrine manner to limit collagen production and the medial expansion of smooth muscle cells by blocking cell growth and inspiring apoptosis [141]. Thus, arginase is characterized as a promising novel therapeutic target in the reversal of endothelial and smooth muscle cell dysfunction and for the prevention of vascular disease.
5. NO supplementation
Several studies have shown NO supplementation provides a remarkably enhanced respiratory response, which significantly elevates the speed throughout phase II of pulmonary O2 utilization at the start of moderately intense endurance exercise. Other functions include better recovery after major injury or trauma, prevention of the common cold, reduction in the side-effects of memory loss, and the effective healing of diabetic foot ulcers [25]. Increasing the intake of vegetables high in NO3- such as celery, arugula, beetroot, and spinach, can help boost NO in the body, as well as participating in normal exercise that in turn improves endothelial performance and, therefore, natural NO production [142]. NO supplementation through direct NO inhalation (approved by the US Food and Drug Administration (FDA)), or through a L-Arg/citrulline or dietary supplementation for improving NOS or enhancing NO bioavailability may help improve cardiac health, reduce erectile dysfunction, improve ovulation, enhance performance during exercise, reduce high blood pressure during pregnancy, and improve healing processes and the respiratory response [25]. For example NO inhalation and NO generated by electrochemical reduction of NO2- using copper catalyst (copper (II)-tri(2-pyridylmethyl) amine (Cu(II)TPMA) complex) have been used as a mediator, for insistent pulmonary hypertension of newborn babies (PPHN) [143]. This treatment has been shown to improve oxygenation and reduce the requirement for higher-risk extracorporeal membrane oxygenation (ECMO) therapy. Direct inhalation NO not only promotes privileged pulmonary vasodilation and lowers pulmonary vascular resistance, but also has a beneficial effect on treatment of other illnesses including pneumonia, stroke, and acute respiratory distress syndrome (ARDS) [144]. Inhalation has recently been used as antiseptic agent in the treatment of cystic fibrosis and tuberculosis, and as an anti-inflammatory agent to modulate immune response and promote survival in patients with malaria [145]. Inhalation NO has also been shown to provide neuroprotection and ease brain damage [146]. NO supplementation improves lung function in cystic fibrosis patients undergoing treatment for altitude sickness [25].
It is important to note that rare side effects can occur with NO supplementation such as headache, nausea, diarrhea, stomach pain, and heat-palpitations and may cause low blood pressure (hypotension), methemoglobinemia (a rare blood disorder where the blood can't carry oxygen effectively), decreased platelet aggregation (increased bleeding risk), pulmonary inflammation due to the formation of nitrogen dioxide, surfactant dysfunction, and potential for toxicity in sensitive individuals, particularly newborns with certain heart conditions; abrupt discontinuation of NO therapy can also lead to rebound pulmonary hypertension in some cases [25, 147]. Supplementation may not be beneficial for those with kidney disease, herpes, and after a person has had a heart attack and it can interfere with certain medications including those for blood pressure. It is recommended to speak with a physician first before starting nitric oxide supplements.
6. Conclusion
NO is essential in health as an ideal signaling molecule allowing different cells to communicate and coordinate tissue functions providing the body with the ability to facilitate adaptive changes. Thus, dysregulation in NO production, function, and signaling has been implicated in a variety of pathologies with recent research investigating targeted treatments. While physiological sources of NO include both enzymatic and non-enzymatic pathways, disturbance of either can give rise to an altered REDOX state. Enhanced oxidative stress can negatively affect NOS-NO and cGMP-sGC system through multiple pathways including NOS uncoupling, NO scavenging, and sGC oxidation [40]. Understanding the different pathways that cause NO deficiency either by direct NO consumption, or disturbance in NO production by the three NOS isoforms is essential to understanding NO signaling. Importantly, based on the information presented, it is the authors understanding that multiple pathways of NO consumption likely exist simultaneously thus contributing to poorer prognosis in diseases such as cardiovascular disease, in which bioavailable NO is reduced both by the production of MPO and accumulation of free radicals. Therefore, progressing therapeutic options such as through dietary supplements that can overcome NO deficiency such as substrate supplements (i.e., arginine or citrulline), nitrite/nitrate enriched food that may be a good alternative as an arginine precursor, or nutritional adaptations that can support the stability of NO justifies further investigation. Consequently, it is of importance to understand how the imbalances of this crucial molecule can affect a patient's health and clarify conflicting signs and symptoms, with therapeutic treatments supporting medical nutrition.
Abbreviations
NO: Nitric oxide; NOSs: nitric oxide synthases; nNOS: neuronal NOS; iNOS: inducible NOS; eNOS: endothelial NOS; NO2-: nitrite; NO3-: nitrate; R-SNO: S-nitrosothiol; ONOO-: peroxynitrite; cGMP: cyclic guanosine monophosphate; MPO: myeloperoxidase; ADMA: asymmetric dimethylarginine; O2•-: superoxide; H4B: tetrahydrobiopterin; CaM: calmodulin; L-Arg: l-arginine; Zn: zinc; GC: guanylate cyclase; Hb: hemoglobin; Hb-oxy: oxy-hemoglobin; CO2: carbon dioxide; PCOS: polycystic ovary syndrome.
Acknowledgements
Funding support by NIH AA23520 and HL151497.
Competing Interests
The authors have declared that no competing interest exists.
References
1. van Faassen EE, Bahrami S, Feelisch M, Hogg N, Kelm M, Kim-Shapiro DB. et al. Nitrite as regulator of hypoxic signaling in mammalian physiology. Medicinal research reviews. 2009;29:683-741
2. Green SJ, Mellouk S, Hoffman SL, Meltzer MS, Nacy CA. Cellular mechanisms of nonspecific immunity to intracellular infection: cytokine-induced synthesis of toxic nitrogen oxides from L-arginine by macrophages and hepatocytes. Immunology letters. 1990;25:15-9
3. Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. European heart journal. 2012;33:829-37 37a-37d
4. Persichini T, Mariotto S, Suzuki H, Butturini E, Mastrantonio R, Cantoni O. et al. Cross-Talk Between NO Synthase Isoforms in Neuro-Inflammation: Possible Implications in HIV-Associated Neurocognitive Disorders. Current medicinal chemistry. 2016;23:2706-14
5. Lundberg JO, Weitzberg E. Nitric oxide signaling in health and disease. Cell. 2022;185:2853-78
6. Cinelli MA, Do HT, Miley GP, Silverman RB. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Medicinal research reviews. 2020;40:158-89
7. Ramadoss J, Pastore MB, Magness RR. Endothelial caveolar subcellular domain regulation of endothelial nitric oxide synthase. Clinical and experimental pharmacology & physiology. 2013;40:753-64
8. Murad F, Rapoport RM, Fiscus R. Role of cyclic-GMP in relaxations of vascular smooth muscle. Journal of cardiovascular pharmacology. 1985;7(Suppl 3):S111-8
9. Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. The open cardiovascular medicine journal. 2010;4:302-12
10. Almannai M, El-Hattab AW. Nitric Oxide Deficiency in Mitochondrial Disorders: The Utility of Arginine and Citrulline. Frontiers in molecular neuroscience. 2021;14:682780
11. Luo Y, Zhu Y, Basang W, Wang X, Li C, Zhou X. Roles of Nitric Oxide in the Regulation of Reproduction: A Review. Frontiers in endocrinology. 2021;12:752410
12. Choudhari SK, Chaudhary M, Bagde S, Gadbail AR, Joshi V. Nitric oxide and cancer: a review. World journal of surgical oncology. 2013;11:118
13. Suresh V, Reddy A. Dysregulation of nitric oxide synthases during early and late pathophysiological conditions of diabetes mellitus leads to amassing of microvascular impedement. Journal of diabetes and metabolic disorders. 2021;20:989-1002
14. Raddino R, Caretta G, Teli M, Bonadei I, Robba D, Zanini G. et al. Nitric oxide and cardiovascular risk factors. Heart international. 2007;3:18
15. Tonelli AR, Haserodt S, Aytekin M, Dweik RA. Nitric oxide deficiency in pulmonary hypertension: Pathobiology and implications for therapy. Pulmonary circulation. 2013;3:20-30
16. Dutta S, Sengupta P. The Role of Nitric Oxide on Male and Female Reproduction. The Malaysian journal of medical sciences: MJMS. 2022;29:18-30
17. Ivy JL. Inorganic Nitrate Supplementation for Cardiovascular Health. Methodist DeBakey cardiovascular journal. 2019;15:200-6
18. Erdinest N, London N, Ovadia H, Levinger N. Nitric Oxide Interaction with the Eye. Vision (Basel, Switzerland). 2021;5:29
19. Malone-Povolny MJ, Maloney SE, Schoenfisch MH. Nitric Oxide Therapy for Diabetic Wound Healing. Advanced healthcare materials. 2019;8:e1801210
20. da Silva GM, da Silva MC, Nascimento DVG, Lima Silva EM, Gouvêa FFF, de França Lopes LG. et al. Nitric Oxide as a Central Molecule in Hypertension: Focus on the Vasorelaxant Activity of New Nitric Oxide Donors. Biology. 2021;10:1041
21. Bogdan C. Nitric oxide and the immune response. Nature immunology. 2001;2:907-16
22. Husmann F, Bruhn S, Mittlmeier T, Zschorlich V, Behrens M. Dietary Nitrate Supplementation Improves Exercise Tolerance by Reducing Muscle Fatigue and Perceptual Responses. Frontiers in physiology. 2019;10:404
23. Burnett AL. The role of nitric oxide in erectile dysfunction: implications for medical therapy. Journal of clinical hypertension (Greenwich, Conn). 2006;8:53-62
24. Poznyak AV, Sadykhov NK, Kartuesov AG, Borisov EE, Melnichenko AA, Grechko AV. et al. Hypertension as a risk factor for atherosclerosis: Cardiovascular risk assessment. Frontiers in cardiovascular medicine. 2022;9:959285
25. Kiani AK, Bonetti G, Medori MC, Caruso P, Manganotti P, Fioretti F. et al. Dietary supplements for improving nitric-oxide synthesis. Journal of preventive medicine and hygiene. 2022;63:E239-e45
26. Bryan NS. Nitrite in nitric oxide biology: cause or consequence? A systems-based review. Free radical biology & medicine. 2006;41:691-701
27. Stuehr DJ. Enzymes of the L-arginine to nitric oxide pathway. The Journal of nutrition. 2004;134:2748S-51S discussion 65S-67S
28. Abu-Soud HM, Stuehr DJ. Nitric oxide synthases reveal a role for calmodulin in controlling electron transfer. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:10769-72
29. Stuehr DJ, Abu-Soud HM, Rousseau DL, Feldman PL, Wang J. Control of electron transfer in neuronal nitric oxide synthase by calmodulin, substrate, substrate analogs, and nitric oxide. Advances in pharmacology (San Diego, Calif). 1995;34:207-13
30. Weitzberg E, Hezel M, Lundberg JO. Nitrate-nitrite-nitric oxide pathway: implications for anesthesiology and intensive care. Anesthesiology. 2010;113:1460-75
31. Hidaka M, Gotoh A, Shimizu T, Minamisawa K, Imamura H, Uchida T. Visualization of NO3⁻/NO2⁻ Dynamics in Living Cells by Fluorescence Resonance Energy Transfer (FRET) Imaging Employing a Rhizobial Two-component Regulatory System. The Journal of biological chemistry. 2016;291:2260-9
32. Lundberg JO, Carlström M, Weitzberg E. Metabolic Effects of Dietary Nitrate in Health and Disease. Cell metabolism. 2018;28:9-22
33. Bryan NS, Fernandez BO, Bauer SM, Garcia-Saura MF, Milsom AB, Rassaf T. et al. Nitrite is a signaling molecule and regulator of gene expression in mammalian tissues. Nature chemical biology. 2005;1:290-7
34. Ma L, Hu L, Feng X, Wang S. Nitrate and Nitrite in Health and Disease. Aging and disease. 2018;9:938-45
35. Frost MC, Reynolds MM, Meyerhoff ME. Polymers incorporating nitric oxide releasing/generating substances for improved biocompatibility of blood-contacting medical devices. Biomaterials. 2005;26:1685-93
36. Miersch S, Mutus B. Protein S-nitrosation: biochemistry and characterization of protein thiol-NO interactions as cellular signals. Clinical biochemistry. 2005;38:777-91
37. van Faassen E, Vanin AF. Low-molecular-weight S-nitrosothiols. Radicals for Life. 2007:173-99
38. Premont RT, Reynolds JD, Zhang R, Stamler JS. Red Blood Cell-Mediated S-Nitrosohemoglobin-Dependent Vasodilation: Lessons Learned from a beta-Globin Cys93 Knock-In Mouse. Antioxid Redox Signal. 2021;34:936-61
39. Singh RJ, Hogg N, Joseph J, Kalyanaraman B. Mechanism of nitric oxide release from S-nitrosothiols. The Journal of biological chemistry. 1996;271:18596-603
40. Daiber A, Xia N, Steven S, Oelze M, Hanf A, Kröller-Schön S. et al. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease. International journal of molecular sciences. 2019;20:187
41. Tran N, Garcia T, Aniqa M, Ali S, Ally A, Nauli SM. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: in Physiology and in Disease States. American journal of biomedical science & research. 2022;15:153-77
42. Helms C, Kim-Shapiro DB. Hemoglobin-mediated nitric oxide signaling. Free radical biology & medicine. 2013;61:464-72
43. White CR, Darley-Usmar V, Berrington WR, McAdams M, Gore JZ, Thompson JA. et al. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:8745-9
44. Abu-Soud HM, Hazen SL. Nitric oxide is a physiological substrate for mammalian peroxidases. The Journal of biological chemistry. 2000;275:37524-32
45. Silberman GA, Fan TH, Liu H, Jiao Z, Xiao HD, Lovelock JD. et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010;121:519-28
46. Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G. et al. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet (London, England). 2004;363:1365-7
47. Haywood GA, Tsao PS, von der Leyen HE, Mann MJ, Keeling PJ, Trindade PT. et al. Expression of inducible nitric oxide synthase in human heart failure. Circulation. 1996;93:1087-94
48. Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B. et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. The Journal of clinical investigation. 2005;115:1221-31
49. Loyer X, Gómez AM, Milliez P, Fernandez-Velasco M, Vangheluwe P, Vinet L. et al. Cardiomyocyte overexpression of neuronal nitric oxide synthase delays transition toward heart failure in response to pressure overload by preserving calcium cycling. Circulation. 2008;117:3187-98
50. Emdin CA, Khera AV, Klarin D, Natarajan P, Zekavat SM, Nomura A. et al. Phenotypic Consequences of a Genetic Predisposition to Enhanced Nitric Oxide Signaling. Circulation. 2018;137:222-32
51. Martell JD, Li H, Doukov T, Martásek P, Roman LJ, Soltis M. et al. Heme-coordinating inhibitors of neuronal nitric oxide synthase. Iron-thioether coordination is stabilized by hydrophobic contacts without increased inhibitor potency. Journal of the American Chemical Society. 2010;132:798-806
52. Abu-Soud HM, Hazen SL. Nitric oxide modulates the catalytic activity of myeloperoxidase. The Journal of biological chemistry. 2000;275:5425-30
53. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY. et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351-6
54. Guo Y, Wen J, He A, Qu C, Peng Y, Luo S. et al. iNOS contributes to heart failure with preserved ejection fraction through mitochondrial dysfunction and Akt S-nitrosylation. Journal of advanced research. 2023;43:175-86
55. O'Donnell VB, Chumley PH, Hogg N, Bloodsworth A, Darley-Usmar VM, Freeman BA. Nitric oxide inhibition of lipid peroxidation: kinetics of reaction with lipid peroxyl radicals and comparison with alpha-tocopherol. Biochemistry. 1997;36:15216-23
56. Goss SP, Hogg N, Kalyanaraman B. The effect of nitric oxide release rates on the oxidation of human low density lipoprotein. The Journal of biological chemistry. 1997;272:21647-53
57. Jessup W, Mohr D, Gieseg SP, Dean RT, Stocker R. The participation of nitric oxide in cell free- and its restriction of macrophage-mediated oxidation of low-density lipoprotein. Biochimica et biophysica acta. 1992;1180:73-82
58. Hayashi K, Noguchi N, Niki E. Action of nitric oxide as an antioxidant against oxidation of soybean phosphatidylcholine liposomal membranes. FEBS letters. 1995;370:37-40
59. Suarna C, Dean RT, May J, Stocker R. Human atherosclerotic plaque contains both oxidized lipids and relatively large amounts of alpha-tocopherol and ascorbate. Arteriosclerosis, thrombosis, and vascular biology. 1995;15:1616-24
60. O'Donnell VB, Taylor KB, Parthasarathy S, Kühn H, Koesling D, Friebe A. et al. 15-Lipoxygenase catalytically consumes nitric oxide and impairs activation of guanylate cyclase. The Journal of biological chemistry. 1999;274:20083-91
61. Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF. et al. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. The Journal of clinical investigation. 1999;103:1597-604
62. Steinberg D. At last, direct evidence that lipoxygenases play a role in atherogenesis. The Journal of clinical investigation. 1999;103:1487-8
63. Guo X, Xing Y, Jin W. Role of ADMA in the pathogenesis of microvascular complications in type 2 diabetes mellitus. Frontiers in endocrinology. 2023;14:1183586
64. Pittman RN. Integrated Systems Physiology: From Molecule to Function to Disease. Regulation of Tissue Oxygenation. San Rafael (CA): Morgan & Claypool Life Sciences Copyright © 2011 by Morgan & Claypool Life Sciences. 2011
65. Marengo-Rowe AJ. Structure-function relations of human hemoglobins. Proceedings (Baylor University Medical Center). 2006;19:239-45
66. Patel S, Jose A, Mohiuddin SS. Physiology, Oxygen Transport And Carbon Dioxide Dissociation Curve. StatPearls. Treasure Island (FL) ineligible companies. StatPearls Publishing Copyright © 2024, StatPearls Publishing LLC. 2024
67. Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221-6
68. Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J. et al. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science (New York, NY). 1997;276:2034-7
69. Gow AJ, Stamler JS. Reactions between nitric oxide and haemoglobin under physiological conditions. Nature. 1998;391:169-73
70. Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622-6
71. Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annual review of physiology. 2005;67:99-145
72. Premont RT, Singel DJ, Stamler JS. The enzymatic function of the honorary enzyme: S-nitrosylation of hemoglobin in physiology and medicine. Molecular aspects of medicine. 2022;84:101056
73. Chintagari NR, Jana S, Alayash AI. Oxidized Ferric and Ferryl Forms of Hemoglobin Trigger Mitochondrial Dysfunction and Injury in Alveolar Type I Cells. American journal of respiratory cell and molecular biology. 2016;55:288-98
74. Joshi MS, Ferguson TB Jr, Han TH, Hyduke DR, Liao JC, Rassaf T. et al. Nitric oxide is consumed, rather than conserved, by reaction with oxyhemoglobin under physiological conditions. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10341-6
75. Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106:675-83
76. Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S. et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nature medicine. 2003;9:1498-505
77. Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nature reviews Drug discovery. 2008;7:156-67
78. Batthyany C, Schopfer FJ, Baker PR, Durán R, Baker LM, Huang Y. et al. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. The Journal of biological chemistry. 2006;281:20450-63
79. Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO 3rd, Schechter AN. et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature medicine. 2002;8:1383-9
80. Donadee C, Raat NJ, Kanias T, Tejero J, Lee JS, Kelley EE. et al. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation. 2011;124:465-76
81. Frisbee JC. Reduced nitric oxide bioavailability contributes to skeletal muscle microvessel rarefaction in the metabolic syndrome. American journal of physiology Regulatory, integrative and comparative physiology. 2005;289:R307-r16
82. Labinskyy N, Csiszar A, Veress G, Stef G, Pacher P, Oroszi G. et al. Vascular dysfunction in aging: potential effects of resveratrol, an anti-inflammatory phytoestrogen. Current medicinal chemistry. 2006;13:989-96
83. Kunz J. Initial lesions of vascular aging disease (arteriosclerosis). Gerontology. 2000;46:295-9
84. Wink DA, Cook JA, Pacelli R, Liebmann J, Krishna MC, Mitchell JB. Nitric oxide (NO) protects against cellular damage by reactive oxygen species. Toxicology letters. 1995;82-83:221-6
85. Canton M, Sánchez-Rodríguez R, Spera I, Venegas FC, Favia M, Viola A. et al. Reactive Oxygen Species in Macrophages: Sources and Targets. Frontiers in immunology. 2021;12:734229
86. Kowaltowski AJ, Vercesi AE. Mitochondrial damage induced by conditions of oxidative stress. Free radical biology & medicine. 1999;26:463-71
87. Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. The Biochemical journal. 1973;134:707-16
88. Goud AP, Goud PT, Diamond MP, Gonik B, Abu-Soud HM. Reactive oxygen species and oocyte aging: role of superoxide, hydrogen peroxide, and hypochlorous acid. Free radical biology & medicine. 2008;44:1295-304
89. Guérin P, El Mouatassim S, Ménézo Y. Oxidative stress and protection against reactive oxygen species in the pre-implantation embryo and its surroundings. Human reproduction update. 2001;7:175-89
90. Rosen GM, Tsai P, Weaver J, Porasuphatana S, Roman LJ, Starkov AA. et al. The role of tetrahydrobiopterin in the regulation of neuronal nitric-oxide synthase-generated superoxide. The Journal of biological chemistry. 2002;277:40275-80
91. Tatemoto H, Sakurai N, Muto N. Protection of porcine oocytes against apoptotic cell death caused by oxidative stress during In vitro maturation: role of cumulus cells. Biology of reproduction. 2000;63:805-10
92. Fraga CG, Motchnik PA, Shigenaga MK, Helbock HJ, Jacob RA, Ames BN. Ascorbic acid protects against endogenous oxidative DNA damage in human sperm. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:11003-6
93. Aitken J, Fisher H. Reactive oxygen species generation and human spermatozoa: the balance of benefit and risk. BioEssays: news and reviews in molecular, cellular and developmental biology. 1994;16:259-67
94. Macfadyen AJ, Reiter C, Zhuang Y, Beckman JS. A novel superoxide dismutase-based trap for peroxynitrite used to detect entry of peroxynitrite into erythrocyte ghosts. Chemical research in toxicology. 1999;12:223-9
95. Denicola A, Souza JM, Radi R. Diffusion of peroxynitrite across erythrocyte membranes. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3566-71
96. Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. The Journal of biological chemistry. 1991;266:4244-50
97. Castro L, Rodriguez M, Radi R. Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. The Journal of biological chemistry. 1994;269:29409-15
98. Crow JP, Beckman JS, McCord JM. Sensitivity of the essential zinc-thiolate moiety of yeast alcohol dehydrogenase to hypochlorite and peroxynitrite. Biochemistry. 1995;34:3544-52
99. Takakura K, Beckman JS, MacMillan-Crow LA, Crow JP. Rapid and irreversible inactivation of protein tyrosine phosphatases PTP1B, CD45, and LAR by peroxynitrite. Archives of biochemistry and biophysics. 1999;369:197-207
100. Burton GJ, Jauniaux E. Oxidative stress. Best practice & research Clinical obstetrics & gynaecology. 2011;25:287-99
101. Chen Q, Wang Q, Zhu J, Xiao Q, Zhang L. Reactive oxygen species: key regulators in vascular health and diseases. British journal of pharmacology. 2018;175:1279-92
102. Münzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. Journal of the American College of Cardiology. 2017;70:212-29
103. Papaharalambus CA, Griendling KK. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends in cardiovascular medicine. 2007;17:48-54
104. Dedeoglu IO, Springate JE. Effect of nitric oxide inhibition on kidney function in experimental renovascular hypertension. Clinical and experimental hypertension (New York, NY: 1993). 2001;23:267-75
105. Hammad FT, Lubbad L, Al-Salam S, Yasin J, Meeran MFN, Ojha S. et al. The Effect of Hypertension on the Recovery of Renal Dysfunction following Reversal of Unilateral Ureteral Obstruction in the Rat. International journal of molecular sciences. 2023;24:7365
106. Bidani AK, Polichnowski AJ, Loutzenhiser R, Griffin KA. Renal microvascular dysfunction, hypertension and CKD progression. Current opinion in nephrology and hypertension. 2013;22:1-9
107. Kopkan L, Majid DS. Enhanced superoxide activity modulates renal function in NO-deficient hypertensive rats. Hypertension (Dallas, Tex: 1979). 2006;47:568-72
108. Bartesaghi S, Radi R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox biology. 2018;14:618-25
109. Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD. et al. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Archives of biochemistry and biophysics. 1992;298:431-7
110. Carballal S, Bartesaghi S, Radi R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochimica et biophysica acta. 2014;1840:768-80
111. Squadrito GL, Pryor WA. Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite, and carbon dioxide. Free radical biology & medicine. 1998;25:392-403
112. Banerjee J, Shaeib F, Maitra D, Saed GM, Dai J, Diamond MP. et al. Peroxynitrite affects the cumulus cell defense of metaphase II mouse oocytes leading to disruption of the spindle structure in vitro. Fertility and sterility. 2013;100:578-84.e1
113. Sharifi-Rad M, Anil Kumar NV, Zucca P, Varoni EM, Dini L, Panzarini E. et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Frontiers in physiology. 2020;11:694
114. Uribe-Querol E, Rosales C. Control of Phagocytosis by Microbial Pathogens. Frontiers in immunology. 2017;8:1368
115. Missailidis D, Annesley SJ, Fisher PR. Pathological Mechanisms Underlying Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Diagnostics (Basel, Switzerland). 2019;9:80
116. Wang Q, Zennadi R. The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants (Basel, Switzerland). 2021;10:1608
117. Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V. et al. Oxidative Stress: Harms and Benefits for Human Health. Oxidative medicine and cellular longevity. 2017;2017:8416763
118. Lau D, Baldus S. Myeloperoxidase and its contributory role in inflammatory vascular disease. Pharmacology & therapeutics. 2006;111:16-26
119. Abu-Soud HM, Khassawneh MY, Sohn JT, Murray P, Haxhiu MA, Hazen SL. Peroxidases inhibit nitric oxide (NO) dependent bronchodilation: development of a model describing NO-peroxidase interactions. Biochemistry. 2001;40:11866-75
120. Golubinskaya V, Brandt-Eliasson U, Gan LM, Kjerrulf M, Nilsson H. Endothelial function in a mouse model of myeloperoxidase deficiency. BioMed research international. 2014;2014:128046
121. Podrez EA, Abu-Soud HM, Hazen SL. Myeloperoxidase-generated oxidants and atherosclerosis. Free radical biology & medicine. 2000;28:1717-25
122. Davies MJ. Myeloperoxidase-derived oxidation: mechanisms of biological damage and its prevention. Journal of clinical biochemistry and nutrition. 2011;48:8-19
123. Salvatori L, Spallotta F, Gaetano C, Illi B. Pillars and Gaps of S-Nitrosylation-Dependent Epigenetic Regulation in Physiology and Cancer. Life (Basel, Switzerland). 2021;11:1424
124. Böger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the "L-arginine paradox" and acts as a novel cardiovascular risk factor. The Journal of nutrition. 2004;134:2842S-7S discussion 53S
125. Kielstein A, Tsikas D, Galloway GP, Mendelson JE. Asymmetric dimethylarginine (ADMA)-a modulator of nociception in opiate tolerance and addiction? Nitric oxide: biology and chemistry. 2007;17:55-9
126. Aykul S, Martinez-Hackert E. Determination of half-maximal inhibitory concentration using biosensor-based protein interaction analysis. Analytical biochemistry. 2016;508:97-103
127. Vallance P, Leiper J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1023-30
128. Lu TM, Ding YA, Lin SJ, Lee WS, Tai HC. Plasma levels of asymmetrical dimethylarginine and adverse cardiovascular events after percutaneous coronary intervention. European heart journal. 2003;24:1912-9
129. Valkonen VP, Päivä H, Salonen JT, Lakka TA, Lehtimäki T, Laakso J. et al. Risk of acute coronary events and serum concentration of asymmetrical dimethylarginine. Lancet (London, England). 2001;358:2127-8
130. Meinitzer A, Seelhorst U, Wellnitz B, Halwachs-Baumann G, Boehm BO, Winkelmann BR. et al. Asymmetrical dimethylarginine independently predicts total and cardiovascular mortality in individuals with angiographic coronary artery disease (the Ludwigshafen Risk and Cardiovascular Health study). Clinical chemistry. 2007;53:273-83
131. Sibal L, Agarwal SC, Home PD, Boger RH. The Role of Asymmetric Dimethylarginine (ADMA) in Endothelial Dysfunction and Cardiovascular Disease. Current cardiology reviews. 2010;6:82-90
132. Vamvakis A, Gkaliagkousi E, Lazaridis A, Grammatikopoulou MG, Triantafyllou A, Nikolaidou B. et al. Impact of Intensive Lifestyle Treatment (Diet Plus Exercise) on Endothelial and Vascular Function, Arterial Stiffness and Blood Pressure in Stage 1 Hypertension: Results of the HINTreat Randomized Controlled Trial. Nutrients. 2020;12:1326
133. Liu YR, Wang JQ, Huang ZG, Chen RN, Cao X, Zhu DC. et al. Histone deacetylase-2: A potential regulator and therapeutic target in liver disease (Review). International journal of molecular medicine. 2021;48:131
134. Nair PC, Mangoni AA, Rodionov RN. Redefining the biological and pathophysiological role of dimethylarginine dimethylaminohydrolase 2. Trends in molecular medicine. 2024;30:552-561
135. Awonuga AO, Camp OG, Abu-Soud HM. A review of nitric oxide and oxidative stress in typical ovulatory women and in the pathogenesis of ovulatory dysfunction in PCOS. Reproductive biology and endocrinology: RB&E. 2023;21:111
136. Li T, Zhang T, Wang H, Zhang Q, Gao H, Liu R. et al. The ADMA-DDAH1 axis in ovarian apoptosis of polycystic ovary syndrome. The Journal of steroid biochemistry and molecular biology. 2023;225:106180
137. Bódis J, Várnagy A, Sulyok E, Kovács GL, Martens-Lobenhoffer J, Bode-Böger SM. Negative association of L-arginine methylation products with oocyte numbers. Human reproduction (Oxford, England). 2010;25:3095-100
138. Durante W, Johnson FK, Johnson RA. Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clinical and experimental pharmacology & physiology. 2007;34:906-11
139. Caldwell RW, Rodriguez PC, Toque HA, Narayanan SP, Caldwell RB. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiological reviews. 2018;98:641-65
140. Li Z, Wang L, Ren Y, Huang Y, Liu W, Lv Z. et al. Arginase: shedding light on the mechanisms and opportunities in cardiovascular diseases. Cell death discovery. 2022;8:413
141. Kolpakov V, Gordon D, Kulik TJ. Nitric oxide-generating compounds inhibit total protein and collagen synthesis in cultured vascular smooth muscle cells. Circulation research. 1995;76:305-9
142. Olas B. The Cardioprotective Role of Nitrate-Rich Vegetables. Foods (Basel, Switzerland). 2024;13:691
143. Ren H, Wu J, Xi C, Lehnert N, Major T, Bartlett RH. et al. Electrochemically modulated nitric oxide (NO) releasing biomedical devices via copper(II)-Tri(2-pyridylmethyl)amine mediated reduction of nitrite. ACS applied materials & interfaces. 2014;6:3779-83
144. Nasrullah A, Virk S, Shah A, Jacobs M, Hamza A, Sheikh AB. et al. Acute Respiratory Distress Syndrome and the Use of Inhaled Pulmonary Vasodilators in the COVID-19 Era: A Narrative Review. Life (Basel, Switzerland). 2022;12:1766
145. Bergmark B, Bergmark R, Beaudrap PD, Boum Y, Mwanga-Amumpaire J, Carroll R. et al. Inhaled nitric oxide and cerebral malaria: basis of a strategy for buying time for pharmacotherapy. The Pediatric infectious disease journal. 2012;31:e250-4
146. Jung P, Ha E, Zhang M, Fall C, Hwang M, Taylor E. et al. Neuroprotective role of nitric oxide inhalation and nitrite in a Neonatal Rat Model of Hypoxic-Ischemic Injury. PloS one. 2022;17:e0268282
147. Carter SJ, Gruber AH, Raglin JS, Baranauskas MN, Coggan AR. Potential health effects of dietary nitrate supplementation in aging and chronic degenerative disease. Medical hypotheses. 2020;141:109732
Author contact
![]() Corresponding author: Husam Abu-Soud, Ph.D, Department of Obstetrics and Gynecology, Wayne State University School of Medicine, The C.S. Mott Center for Human Growth and Development, 275 E. Hancock Detroit, MI 48201, Tel.: 313 577-6178, Fax: 313 577-8554, E-Mail: habusoudwayne.edu.
Corresponding author: Husam Abu-Soud, Ph.D, Department of Obstetrics and Gynecology, Wayne State University School of Medicine, The C.S. Mott Center for Human Growth and Development, 275 E. Hancock Detroit, MI 48201, Tel.: 313 577-6178, Fax: 313 577-8554, E-Mail: habusoudwayne.edu.