Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. m6A modifiers
3. The pathogenesis role of m6A...
4. The interaction between m6A...
5. Conclusions and Perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(3):1187-1201. doi:10.7150/ijbs.104407 This issue Cite
Review
The Mechanism and Latest Progress of m6A Methylation in the Progression of Pancreatic Cancer
Ze-Hao Liu1,#, Peng Ma2,#, Ying He3,#, Yue-Feng Zhang4, Zuo Mou5, Ting Fang6, Wei Wang7, ![]() , Kai-Huan Yu8,
, Kai-Huan Yu8, ![]()
1. Department of Hepatobiliary Surgery, Renmin Hospital of Wuhan University, Wuhan, 430060, China.
2. Department of Hepatobiliary Surgery, East Hospital, Renmin Hospital of Wuhan University, Wuhan, 430060, China.
3. Department of Stomatology, Renmin Hospital of Wuhan University, Wuhan, 430060, China.
4. Department of Hepatobiliary Surgery, East Hospital, Renmin Hospital of Wuhan University, Wuhan, 430060, China.
5. Department of Hepatobiliary Surgery, Renmin Hospital of Wuhan University, Wuhan, 430060, China.
6. Department of Oncology, Renmin Hospital of Wuhan University, Wuhan, 430060, China.
7. Department of Hepatobiliary Surgery, East Hospital, Renmin Hospital of Wuhan University, Wuhan, 430060, China.
8. Department of Hepatobiliary Surgery, East Hospital, Renmin Hospital of Wuhan University, Wuhan, 430060, China.
#Contributed equally.
Received 2024-9-29; Accepted 2024-12-26; Published 2025-1-13
Abstract

Pancreatic cancer (PC), known as the "king of cancers," is characterized by an exceptionally low five-year survival rate, posing a formidable challenge to global public health. N6-methyladenosine (m6A) methylation is prevalent across various stages of eukaryotic RNA expression, including splicing, maturation, stability, translation, and localization, and represents a pivotal mechanism of epigenetic regulation. m6A methylation influences tumor initiation and progression by modulating post-transcriptional processes, playing a critical role in sustaining cancer cell stemness, promoting cell proliferation, and mediating drug resistance. Extensive research underscores the substantial contribution of m6A modifications to PC development. However, the multiplicity of m6A regulators and their intricate mechanisms of action complicate the landscape. This review aims to deepen the understanding of m6A's role in PC by delineating its involvement in four key areas of tumorigenesis: the hypoxic tumor microenvironment, metabolic reprogramming, immune microenvironment, and resistance mechanisms. Additionally, the review addresses the emerging frontier of m6A interactions with non-coding RNAs (ncRNAs), offering insights into the potential therapeutic and prognostic applications of m6A in the treatment and prognosis prediction of PC.
Keywords: Pancreatic cancer, RNA Methylation, m6A, Immune Evasion, RNA, Untranslated
1. Introduction
Pancreatic cancer (PC), known as the "king of cancers," remains a leading cause of cancer-related mortality. According to the latest data from the CA: A Cancer Journal for Clinicians, PC was the sixth leading cause of cancer deaths globally in 2022, accounting for 4.8% of all cancer fatalities worldwide. Mortality rates for PC have remained relatively stable across many countries in recent decades[1]. Projections indicate that by 2025, PC will become the third leading cause of cancer-related deaths in Europe, and by 2030, it is expected to rank second in the United States. As a highly aggressive malignancy with a poor prognosis, PC represents a significant public health burden[2-4]. The majority of PC cases present as pancreatic ductal adenocarcinoma (PDAC), which is marked by rapid growth, aggressive invasiveness, a high degree of malignancy, and typically lacks discernible early symptoms. This results in delayed diagnosis, often until the disease has reached an advanced stage or metastasis[5, 6]. Gemcitabine (GEM)-based combination chemotherapy remains the primary treatment for advanced PDAC; however, the emergence of drug resistance has severely hindered the efficacy of this regimen in recent years[7, 8]. Consequently, a deeper understanding of the molecular mechanisms driving PC initiation, progression, and drug resistance, along with insights into the role of the tumor microenvironment, is urgently needed.
N6-methyladenosine (m6A) modification, first identified in the 1970s, has garnered significant attention as the most prevalent modification of messenger RNA (mRNA) and long non-coding RNA (lncRNA) in eukaryotes[9]. m6A modification involves the methylation of adenosine at the N6 position, a reversible modification regulated by a dynamic interplay of methyltransferases ("writers"), demethylases ("erasers"), and RNA-binding proteins ("readers") that recognize and interpret the methylation marks[10]. m6A methylation is widespread in mammalian mRNA and has been identified in various RNA species, including transfer RNA (tRNA), ribosomal RNA (rRNA), circular RNA (circRNA), microRNA (miRNA), and lncRNA[11]. This modification plays a pivotal role in regulating gene expression in tumor cells by influencing RNA processes such as splicing, maturation, stability, translation, and localization, thereby controlling cancer progression[12]. Several studies have highlighted the essential role of m6A in cancer, particularly its influence on the proliferation, development, and metastasis of PC[13-15]. In a recent review, Wang et al. explored the epigenetic regulation of m6A regulators themselves, detailing post-translational modifications such as ubiquitination, SUMOylation, acetylation, methylation, phosphorylation, O-GlcNAcylation, ISGylation, and lactylation, all of which impact cancer progression[16]. However, comprehensive and up-to-date reviews on the mechanisms of m6A modification in PC remain scarce. This review aims to consolidate recent advances in m6A research in the context of PC, focusing on four key aspects: the hypoxic tumor microenvironment, metabolic reprogramming, immune microenvironment, and drug resistance mechanisms. Additionally, the review addresses emerging research on the interaction between non-coding RNAs (ncRNAs) and m6A methylation. By providing an in-depth understanding of m6A modification in PC, this review aims to contribute to the development of improved non-surgical treatment strategies for patients with PC, ultimately improving their clinical outcomes.
2. m6A modifiers
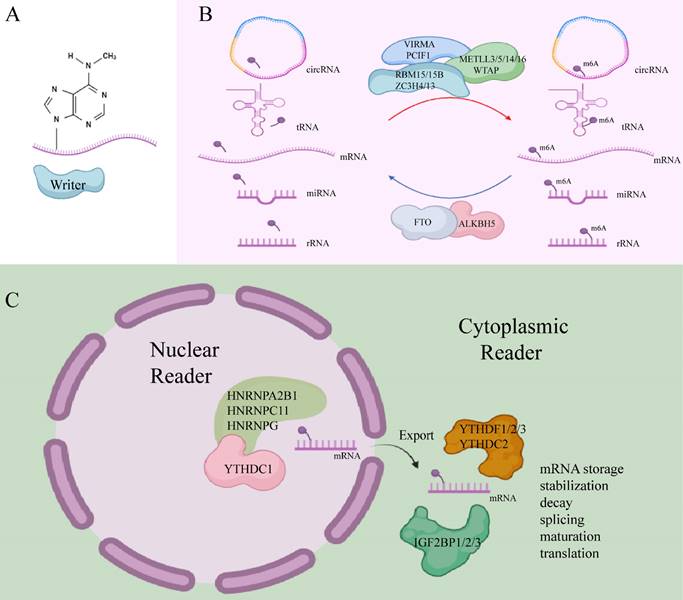
m6A writers, also referred to as methyltransferases, are enzymes that utilize S-adenosyl methionine (SAM) as a cofactor to catalyze N6-methylation by transferring a methyl group from SAM to the adenosine substrate[17]. These writers typically function as multifunctional complexes composed of several key components, including methyltransferase-like 3 (METTL3), methyltransferase-like 5 (METTL5), methyltransferase-like 14 (METTL14), methyltransferase-like 16 (METTL16), William's tumor 1-associated protein (WTAP), RNA-binding motif protein 15/15B (RBM15/15B), vir-like m6A methyltransferase-associated protein (VIRMA/KIAA1429), phosphorylated CTD-interacting factor 1 (PCIF1), and zinc finger CCCH-type containing proteins 4/13 (ZC3H4, ZC3H13)[18-20].
m6A erasers are demethylases responsible for removing m6A marks, thereby regulating the dynamic processes of m6A methylation. Notable m6A erasers include fat mass and obesity-related protein (FTO) and alpha-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5). These enzymes are frequently dysregulated in cancer and play pivotal roles in malignancy development[21, 22]. FTO primarily removes m6A methylation from mRNA in the nucleus, while ALKBH5 acts predominantly in the cytoplasm, where it catalyzes the demethylation of m6A marks by co-binding with nuclear speckles[23].
m6A readers are proteins that recognize m6A modifications, facilitating RNA-protein interactions and influencing regulatory pathways, such as RNA splicing, export, translation, and degradation[24]. These readers exhibit specific preferences for m6A methylation sites. Nuclear m6A readers include YTHDC1, HNRNPA2B1, HNRNPC11, and HNRNPG, while cytoplasmic m6A readers consist of YTHDF1, YTHDF2, YTHDF3, YTHDC2, as well as IGF2BP1, IGF2BP2, and IGF2BP3[11]. Recent research has identified additional m6A readers, such as the KH-type splicing regulatory protein (KHSRP), which plays an oncogenic role in PDAC progression[25]. New m6A readers have also been discovered, including fragile X mental retardation protein (FMRP), proline-rich coiled-coil 2A (PRRC2A), RNA-binding motif protein 33 (RBM33), and Eukaryotic Initiation Factor 3 (eIF3)[16]. Different m6A readers mediate diverse functions upon binding m6A-modified sites. For instance, YTHDF3 works with YTHDF1 to promote the translation of methylated RNA while also accelerating mRNA decay via interaction with YTHDF2. In contrast, IGF2BPs stabilize their target mRNAs in an m6A-dependent manner[26].
Together, m6A writers and erasers facilitate the reversible methylation of m6A, while m6A readers participate in the post-transcriptional regulation of RNA (Figure 1). This coordinated system represents an emerging molecular mechanism for regulating gene expression at the post-transcriptional level. Increasing evidence links m6A modification to the initiation and progression of PC (Table 1).
m6A writer, eraser, and reader. (Created with BioRender.com).
The function of m6A regulators in pancreatic cancer
| Type | m6A regulators | Function | Target | Biological Function | Roles in PC | References |
|---|---|---|---|---|---|---|
| Writers | METTL3 | Catalyzes m6A modification | TLR4 (neutrophil) | Promote neutrophil activation | — | 19,74 |
| METTL3 | Catalyzes m6A modification | SOCS (T cell) | Promote naive T cells homeostasis, proliferation, and differentiation | antitumor | 19,69 | |
| METTL3 | Catalyzes m6A modification | STAT1 (macrophage) | Promote macrophage M1 polarization | antitumor | 19,66 | |
| METTL3 | Catalyzes m6A modification | ITGB1/HK2/BCAN-AS1/MALAT1/circMYO1C/FZR1/NUCB1/AREG/miR-380-3p/FOXD1-AS1 | Promote proliferation, metastasis, invasion, immune escape, glycolysis, drug resistance, and PNI | oncogenic | 19,27,30,56,78,79,88,90,99,100,103 | |
| METTL5 | Promotes the m6A methylation of 18 S rRNA | — | — | — | 16 | |
| METTL14 | Forms heterodimer with METTL3 to catalyze m6A modification | TGFB2/circSTX6/miR-380-3p | Promote proliferation, metastasis, and drug resistance | oncogenic | 19,50,53,100 | |
| METTL16 | Catalyzes m6A modification | — | — | — | 19 | |
| WTAP | Regulatory subunit of m6A methyltransferase and recruits METTL3 and METTL14 into the nuclear speckles | GBE1 | Promote proliferation | oncogenic | 19,49 | |
| RBM15 | Directs METTL3-METTL14 heterodimer to specific RNA sites | — | Inhibit macrophage infiltration and phagocytosis by macrophages | oncogenic | 19,63 | |
| RBM15B | Directs METTL3-METTL14 heterodimer to specific RNA sites | — | — | — | 19 | |
| VIRMA/KIAA1429 | Recruits METTL3 and METTL14, and direct m6A methylation at specific sites to induce mRNA splicing and RNA processing. | STRA6 | Promote metastasis | oncogenic | 19,38 | |
| PCIF1 | Catalyzes 5′ m6Am* methylation of capped mRNAs | — | — | — | 20 | |
| ZC3H4 | Mediates the m6A methylation of U2 snRNA to regulate pre-mRNA splicing | — | — | — | 16 | |
| ZC3H13 | Bridges WTAP to the mRNA-binding factor Nito; Anchors WTAP in the nucleus to enhance m6A modification | PHF10 | Promote drug resistance | oncogenic | 16,91 | |
| Erasers | FTO | Acts as m6A demethylase to promote mRNA splicing and translation | SNAI1/KDM5B/LINC01134/NEDD4 | Promote proliferation, drug resistance, and EMT | oncogenic | 19,32,51,86,87 |
| ALKBH5 | Removes m6A modification to promote mRNA nuclear processing and mRNA export | CXCR2/NLRP12/PTGER4 (neutrophil) | Promote neutrophil migration | — | 19,73 | |
| ALKBH5 | Removes m6A modification to promote mRNA nuclear processing and mRNA export | FZR1 | Inhibit drug resistance | antitumor | 19,88 | |
| ALKBH5 | Removes m6A modification to promote mRNA nuclear processing and mRNA export | HDAC4/FABP5/DDIT4-AS1 | Promote proliferation, metastasis, glycolysis, drug resistance and lipid metabolism | oncogenic | 19,45,52,89 | |
| Readers | YTHDC1 | Promotes mRNA splicing and transcriptional silencing | — | — | — | 19 |
| YTHDC2 | Improves the translation efficiency of target mRNA | — | — | — | 19 | |
| HNRNPA2B1 | Promotes primary miRNA processing and mRNA splicing | — | — | — | 19 | |
| HNRNPC11 | Mediates mRNA splicing and maturity | — | — | — | 16 | |
| HNRNPG | Mediates mRNA splicing and maturity | — | — | — | 16 | |
| YTHDF1 | Promotes mRNA translation initiation | transcripts encoding lysosomal proteases (DC) | Inhibit the cross-presentation of tumor antigens and the cross-priming of CD8+ T cells | oncogenic | 19,70 | |
| YTHDF1 | Promotes mRNA translation initiation | PHF10/FOXD1-AS1 | Promote drug resistance | oncogenic | 19,91,103 | |
| YTHDF2 | Promotes mRNA degradation | MAP2K4/MAP4K4/STAT1/PPAR- γ (macrophage) | Inhibit macrophage polarization and proinflammatory cytokine secretion | oncogenic | 19,65 | |
| YTHDF2 | Promotes mRNA degradation | PTEN/NEDD4/NUCB1 | Promote proliferation, migration, invasion, drug resistance, and the Warburg effect | oncogenic | 19,48,87,90 | |
| YTHDF2 | Promotes mRNA degradation | HDAC4/KDM5B/LINC01134 | Inhibit migration, glycolysis, and drug resistance | antitumor | 19,45,51,86 | |
| YTHDF3 | Interacts with YTHDF1 to promote mRNA translation or interacts with YTHDF2 to promote mRNA degradation | DICER1-AS1 | Promote proliferation and glycolysis | oncogenic | 19,47 | |
| IGF2BP1 | Promotes the stability and translation of mRNA | — | — | — | 19 | |
| IGF2BP2 | Promotes the stability and translation of mRNA | STRA6/TGFB2/PACERR/KLF12/C-Myc/PD-L1/CTSL | Promote proliferation, migration, invasion, glycolysis, and drug resistance | oncogenic | 19,38,50,64,79,93 | |
| IGF2BP3 | Promotes the stability and translation of mRNA | MIB1 (neutrophil) | Promote NET | — | 19,77 | |
| IGF2BP3 | Promotes the stability and translation of mRNA | GBE1 | Promote proliferation and stemness-like properties | oncogenic | 19,49 | |
| GEMIN5 | Recruiting eIF3 complex to promote mRNA translation | FZR1 | Promote drug resistance | oncogenic | 88 | |
| KHSRP | Promotes the stability and translation of mRNA | FAK | Promote proliferation, migration, and invasion | oncogenic | 25 | |
| eIF3 | Promotes mRNA translation | — | — | — | 19 | |
| FMRP | Promotes the nuclear export and stability of m6A-modified RNAs | — | — | — | 16 | |
| PRRC2A | Binds to a consensus GGACU motif in the Olig2 coding sequence to stabilize Olig2 mRNA | — | — | — | 16 | |
| RBM33 | Forms a complex with ALKBH5 and mediates m6 A demethylation of selected transcripts by regulating ALKBH5 substrate accessibility and activity | — | — | — | 16 |
m6Am: N6,2′-O-dimethyladenosine
3. The pathogenesis role of m6A modulation in PC
3.1 Tumor hypoxic microenvironment
Hypoxia is a common feature of solid tumors, and the extent of hypoxic regions in tumor tissues is a critical factor associated with poor survival outcomes in patients with PC[27]. The m6A modification, as part of the RNA regulatory landscape, undergoes methylation changes in response to hypoxic conditions, thereby influencing tumor initiation and progression[28]. For example, in breast cancer, lncRNA KB-1980E6.3 stabilizes c-Myc mRNA under hypoxic conditions by interacting with the m6A reader IGF2BP1, thus maintaining the stemness of cancer stem cells[29]. In the context of PC, hypoxia has been shown to stimulate the expression of the m6A writer METTL3, which activates the MAPK/ERK/PD-L1 signaling pathway by modulating the mRNA stability and expression of Integrin Beta 1 (ITGB1). This process promotes the proliferation, migration, and invasion of PC cells[30, 31]. Additionally, hypoxic conditions significantly reduce the expression of the lncRNA GATA6-AS1 in PDAC. GATA6-AS1 disrupts the stability of SNAI1 mRNA in an m6A-dependent manner by inhibiting FTO, thus suppressing hypoxia-induced PDAC progression and epithelial-mesenchymal transition (EMT)[32]. These findings suggest that targeting the inhibition of FTO expression under hypoxic conditions may be a potential strategy to alleviate or reverse hypoxia-induced EMT in PC.
Cellular responses to hypoxia are primarily mediated by Hypoxia Inducible Factors (HIFs), a family of four subunits: HIF-1α, HIF-2α, HIF-3α, and HIF-1β. These HIF subunits form functional complexes composed of one HIF-α subunit and one HIF-1β subunit[33, 34]. HIFs play a significant role in PDAC progression. Specifically, HIF-2α expressed by cancer-associated fibroblasts (CAFs) within the tumor microenvironment (TME) influences tumor fibrosis, and the absence of CAF-HIF2 significantly inhibits the intratumoral recruitment of M2 macrophages and regulatory T cells (Tregs) and modestly reduced tumor fibrosis[35]. Numerous studies have demonstrated the relationship between HIFs and tumor progression, as well as their close association with m6A modifications. For example, in hepatocellular carcinoma (HCC), under hypoxic conditions, HIF-1α directly binds to the promoter region of the m6A reader YTHDF1, promoting the translation of autophagy-related genes such as ATG2A and ATG14, which drive autophagy[36]. HIF-1α also promotes metastasis in HCC through the METTL16/lnc-CSMD1-7/RBFOX2 axis[37]. In PC, methyltransferase VIRMA has been shown to target the m6A site of STRA6 mRNA, modifying it. IGF2BP2 then recognizes this m6A modification and enhances mRNA stability, leading to the activation of HIF-1α, which drives glycolysis in PDAC cells and contributes to poor prognosis[38]. Early glycolytic activation mediated by HIF-1α also promotes the migration of dendritic cells (DCs) to lymph nodes. However, stimulation by CC chemokine receptor 7 (CCR7) inhibits DC migration and affects PDAC progression by mediating m6A demethylation of lnc-Dpf3. This reduces the YTHDF2-m6A-dependent RNA degradation of lnc-Dpf3, which ultimately impacts the immune response within the TME[39, 40].
In summary, m6A methylation modifications play an essential role in regulating various signaling pathways and metabolic processes in PC cells, as well as in other cells within the TME under hypoxic conditions. Targeting the modulation of m6A progression, either by inhibiting or activating specific components of the m6A machinery, may offer promising non-surgical therapeutic strategies for patients with PC.
3.2 Tumor metabolic reprogramming
In 2011, Hanahan proposed six hallmarks of cancer and two enabling characteristics: Sustaining proliferative signaling, Resisting cell death, Inducing angiogenesis, Evading growth suppressors, Activating invasion and metastasis, Enabling replicative immortality, alongside Reprogramming Energy Metabolism and Evading Immune Destruction[41]. Tumor metabolic reprogramming arises from mutations in oncogenes and tumor suppressor genes, with alterations in metabolite levels impacting cellular signaling, epigenetic regulation, and gene expression[42]. The regulation of m6A methylation is crucial in tumor cell metabolism, influencing growth, proliferation, and chemoresistance[43].
PC, in particular, is characterized by a notably hypoxic microenvironment compared to other solid tumors[44]. In hypoxic PC cells, the overall mRNA m6A modification level, mediated by the eraser ALKBH5, is markedly reduced, which promotes glycolysis in an ALKBH5-dependent manner. This is mechanistically linked to a positive feedback loop involving ALKBH5, histone deacetylase type 4, and HIF1α[45]. Glutamate, in neuronal cells, enhances m6A modification of hexokinase 2 mRNA, increasing its expression via METTL3 upregulation, which in turn facilitates glycolysis in PDAC cells and supports perineural invasion, a hallmark of poor prognosis and decreased survival rates in PC[27, 46].
During glucose deprivation, miR-5586-5p induces the overexpression of the m6A reader YTHDF3, destabilizing DICER1 antisense RNA through m6A modification and promoting glycolysis and tumor progression in PC cells[47]. Similarly, studies have shown that circular RNA Hsa_circ_0007590 targets polypyrimidine tract binding protein 1 and upregulates the m6A reader protein YTHDF2, leading to PTEN mRNA degradation and activation of the PI3K/AKT/mTOR pathway. This reprograms glucose metabolism, driving the Warburg effect and enhancing the proliferation, migration, and invasion of PDAC cells[48]. Glycogen branching enzyme 1 (GBE1), which plays a critical role in glycogen metabolism, is significantly upregulated in PC and correlates with poor patient prognosis. This is attributed to m6A modifications by methyltransferase WTAP and the reader IGF2BP3, which enhance the stability and expression of GBE1 mRNA[49].
Advances in lipid metabolism research have also highlighted its significance in PDAC. During GEM-induced drug resistance, transforming growth factor-beta 2 (TGFB2) expression is gradually upregulated, potentially via METTL14-mediated m6A modification, which stabilizes TGFB2 post-transcriptionally and promotes lipid accumulation, contributing to GEM resistance[50]. The tumor suppressor SMAD4 (Mothers against decapentaplegic homolog 4) is inactivated in approximately 50%-60% of PDAC cases, disrupting the activation of the DLG1/YAP1 pathway. In this context, FTO enhances adipogenesis and lipid accumulation by stabilizing Lysine-specific demethylase 5B (KDM5B) and activating the DLG1/hippo-YAP pathway, which fosters stemness and proliferation of PDAC cells, reducing GEM sensitivity[51]. In pancreatic neuroendocrine neoplasms, upregulation of ALKBH5 plays a pivotal role in tumor growth and lipid metabolism, likely through the enhancement of FABP5 expression, which activates the mTOR signaling pathway, boosting lipid metabolism and proliferative capacity[52].
Abnormal amino acid (AA) metabolism plays a pivotal role in promoting tumor cell proliferation and invasion. For instance, METTL14 regulates the circular RNA circSTX6 and the 144-amino acid polypeptide encoded by circSTX6 through m6A-dependent mechanisms, thereby enhancing HCC proliferation and metastasis[53]. Glutamine (Gln), an essential and the most abundant AA in the blood, is integral to numerous biological processes that support cancer cell growth and proliferation, making it a critical component of cancer metabolism[54]. The rapid proliferation of PDAC relies on an atypical Gln utilization pathway; inhibition of this altered metabolism effectively suppresses PDAC growth, migration, and invasion[55]. In PC, SMAD nuclear-interacting protein 1 (SNIP1) recognizes and binds to m6A-modified lncRNA BCAN-AS1, preventing the ubiquitination and degradation of c-Myc, a process mediated by S-phase kinase-associated protein 2 (SKP2)[56]. Oncogenic levels of c-Myc, however, can drive transcriptional programs that promote glutaminolysis, creating cellular dependence on Gln as a bioenergetic substrate[57]. Reprogramming of Gln metabolism is a hallmark of cancer, with dysregulation of glutaminase (GLS) and glutamine synthetase (GS) identified as key events in tumor metabolic rewiring[58]. While there is limited research on the role of m6A in Gln metabolism in PC, emerging studies in other cancers have begun to elucidate this connection. For example, the m6A reader YTHDF1 promotes GLS expression in colon cancer, contributing to cisplatin resistance, while IGF2BP3 enhances glutamine and glutamic acid metabolism in cervical cancer cells by upregulating GLS expression, thereby facilitating immune evasion[59, 60]. Although the metabolic reprogramming of Gln is a characteristic of abnormal AA metabolism, research linking m6A modifications to this process in PC remains scarce. Drawing from studies in other malignancies could provide new avenues for targeted therapeutic strategies.
Metabolic reprogramming serves as a fundamental driver of tumor proliferation, migration, and invasion, with m6A methylation modifications playing a pivotal role in modulating metabolic pathways such as glucose, lipid, and AA metabolism. Targeting m6A regulation as a therapeutic strategy holds potential for benefiting patients with PC by prolonging survival and improving quality of life.
3.3 Tumor immune microenvironment
Studies proposed the association between the m6Ascore and poor overall survival as well as increased tumor recurrence in PDAC and other solid tumors, including colorectal and breast cancer. Higher m6Ascores were observed in basal-like (squamous) PDAC compared to classical PDAC. Tumors with elevated m6Ascores are characterized by diminished immune infiltration and T-cell exhaustion[61]. PC is generally regarded as an immune-impaired malignancy, where the development and activation of immune cells are heavily reliant on precise epigenetic regulation[62]. m6A mRNA methylation is an emerging post-transcriptional regulatory mechanism influencing gene expression, playing a significant role in the modulation of various immune cell functions[63].
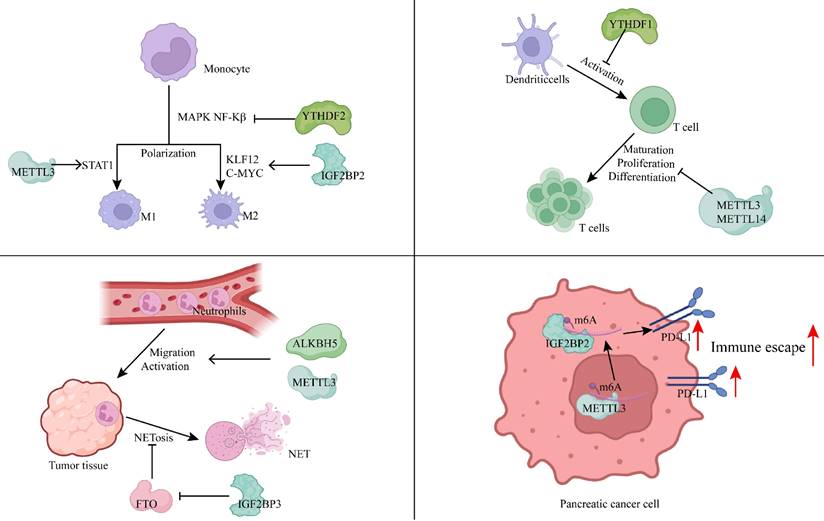
Macrophage polarization serves as a key indicator of the immune status within tumors and is linked to poor prognosis in patients with PDAC. In research conducted by Wang et al. from our team, inhibition of the m6A writer RBM15 was shown to increase macrophage infiltration, promoting phagocytosis of PC cells by macrophages, thereby establishing RBM15 as an independent prognostic factor for PC[63]. The lncRNA-PACERR, which binds to IGF2BP2, enhances the stability of KLF12 and c-Myc in the cytoplasm of tumor-associated macrophages (TAMs) in an m6A-dependent manner. PACERR, by interacting with miR-671-3p, activates the KLF12/p-AKT/c-Myc signaling pathway, increases the number of M2-polarized macrophages, and promotes the proliferation, invasion, and migration of tumor cells[64]. The m6A reader YTHDF2 inhibits MAPK and NF-κB signaling pathways by downregulating MAP2K4, MAP4K4, STAT1, and PPAR-γ expression, preventing macrophage polarization and secretion of pro-inflammatory cytokines[65]. m6A also participates in the M1 polarization of TAMs, with the m6A methyltransferase METTL3 being selectively upregulated during M1 polarization. METTL3 mediates the methylation of STAT1 mRNA, significantly enhancing its stability and expression, thereby driving M1 polarization of macrophages[66]. Depletion of METTL3 in TAMs enhances both M1 and M2-like TAMs, as well as Treg infiltration into tumors, remodeling the tumor microenvironment and promoting tumor growth and metastasis[67].
Dong et al. developed an m6A scoring system, discovering a significant correlation between the m6Ascore and various immune cell types, including CD4+ T cells, Tregs, type 1 T helper cells (Th1), γδ T cells, natural killer T cells (NKT), activated DCs, CD56+ NK cells, and NK cells. Notably, high expression of RBM15 showed a positive correlation with lymphocyte counts, and silencing the RBM15 gene inhibited PC cell proliferation, migration, and metastasis[68]. In METTL3-deficient naive T cells, the m6A modification of suppressor of cytokine signaling (SOCS) family gene mRNAs (such as SOCS1, SOCS3, and CISH) was diminished, leading to slower mRNA decay compared to wild-type naive T cells. This resulted in elevated mRNA and protein levels, inhibition of IL-7-mediated STAT5 activation, and disruption of naive T cell homeostasis, proliferation, and differentiation[69]. The m6A reader YTHDF1 in DCs enhances antigen degradation, limiting the presentation of tumor neoantigens to T cells and inhibiting T cell-mediated immune responses[70].
Neutrophils, the most abundant immune cells in human blood circulation, are essential in defending against microbial infections and play a significant role in regulating both innate and adaptive immunity. They interact with a wide range of immune cells, including macrophages, mesenchymal stem cells, DCs, NK cells, and B and T cells, forming complex bidirectional relationships within tissues[71]. In recent years, neutrophils have attracted increased attention due to their cancer-promoting roles. They contribute to cancer progression through mechanisms such as DNA damage induction, angiogenesis promotion, and immune suppression[72]. m6A methylation plays a critical role in neutrophil functions, including migration, activation, and the formation of neutrophil extracellular traps (NETs). For example, the m6A eraser ALKBH5 enhances neutrophil migration by increasing the expression of CXCR2 and NLRP12 while decreasing the expression of PTGER4, TNC, and WNK1[73]. Upon reaching the tissues, inflammatory stimuli activate neutrophils, where METTL3-mediated m6A methylation boosts TLR4 expression, regulating neutrophil activation[74]. Activated neutrophils can release NETs during the process of NETosis[75]. These NETs mediate proteolytic remodeling of laminin, which subsequently enhances cancer cell proliferation and metastasis[76]. Additionally, upregulation of IGF2BP3 in neutrophils enhances the expression of MIB1, an E3 ubiquitin-protein ligase that promotes FTO degradation via the ubiquitin-proteasome pathway. This results in increased m6A-mediated NET formation[77]. Thus, m6A modification plays a pivotal role in neutrophil migration, activation, and NET formation, which subsequently influences tumor initiation and progression.
Moreover, m6A modification influences immune evasion mechanisms in tumor cells by regulating immune-related signaling pathways. METTL3, for instance, positively regulates the expression of lncRNA MALAT1 in PC cells, which enhances PD-L1 expression and modulates tumor immune surveillance[78]. Additionally, METTL3 facilitates the circularization of circMYO1C, which targets the m6A site of PD-L1 mRNA. By interacting with IGF2BP2, circMYO1C stabilizes PD-L1 mRNA, promoting immune evasion in PDAC[79]. In bladder and breast cancers, METTL3-mediated m6A modification of PD-L1 mRNA stabilizes the mRNA in an IGF2BP1- or IGF2BP3-dependent manner[80, 81]. However, in non-small cell lung cancer, METTL3 has been shown to increase the expression of pro-tumorigenic chemokines, including CXCL1, CXCL5, and CCL20, while destabilizing PD-L1 mRNA in an m6A-dependent manner[82]. This suggests that the regulatory role of m6A on immune signaling can vary, and even be opposite, across different cancer types, emphasizing the need to tailor approaches for studying m6A mechanisms based on specific cancer contexts.
Immune evasion is a hallmark of tumorigenesis, facilitating tumor proliferation, migration, and invasion. m6A methylation is involved in nearly every aspect of immune evasion, including macrophage development and polarization, T cell proliferation and differentiation, neutrophil migration and activation, and the transduction of immune signaling pathways (Figure 2). Targeting m6A modifications in these processes with therapeutic agents may offer promising strategies for patients who are resistant or have low response rates to conventional chemotherapy.
m6A methylation modification modulates the immune microenvironment by regulating immune cells and immune pathways. (Created with BioRender.com).
3.4 Tumor resistance mechanisms
Chemotherapy, with GEM as a cornerstone due to its cytotoxic effects on DNA synthesis, is the primary treatment modality for PDAC. GEM-based therapy has remained the first-line treatment for PDAC since its approval in 1997[83]. However, both primary and acquired resistance to chemotherapy have emerged as significant challenges, extending beyond traditional cytotoxic chemotherapy to targeted therapies and immunomodulatory approaches[84]. Understanding the role of m6A methylation modifications in resistance mechanisms holds substantial promise for alleviating this issue and potentially guiding the development of novel therapeutic strategies.
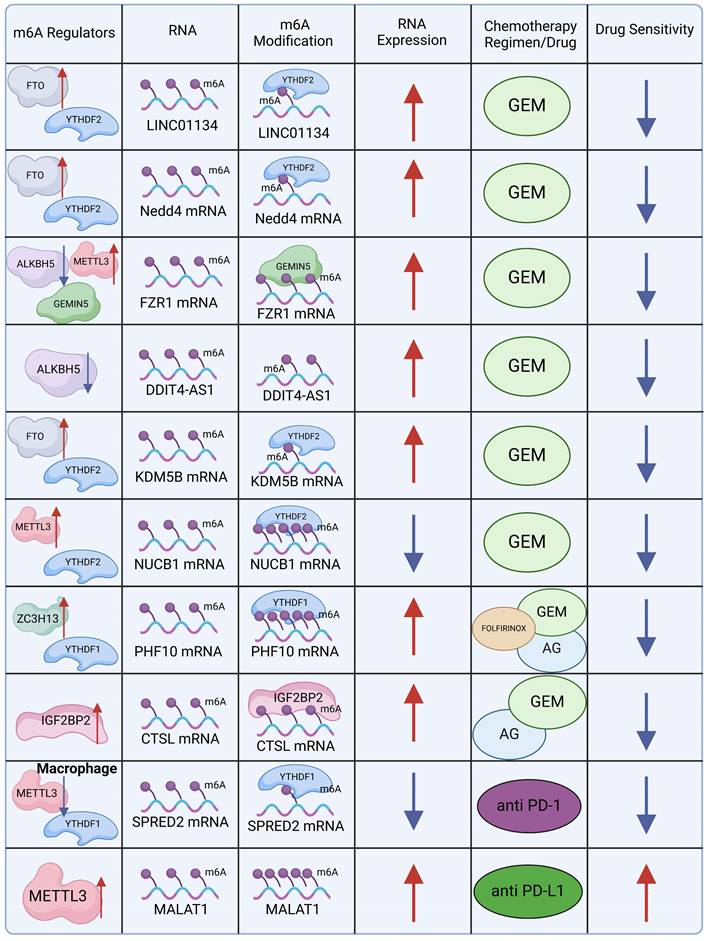
GEM-based combination chemotherapy regimens, often paired with irinotecan, cisplatin, oxaliplatin, and other agents, have provided systemic treatment options with meaningful benefits for patients with advanced or metastatic PDAC[85]. Nevertheless, patients who exhibit resistance or insensitivity to GEM—the cornerstone of these combination therapies—experience significantly reduced clinical benefits. Investigating the mechanistic role of m6A in GEM resistance could provide new insights into overcoming this challenge. The m6A eraser FTO plays a key role in this context by maintaining the stability of long intergenic non-protein coding RNA 1134 (LINC01134) mRNA. FTO reduces the abundance of m6A modifications on LINC01134, preventing RNA degradation after m6A reader YTHDF2 binds to the m6A sites. LINC01134 regulates the WNT signaling pathway through competitive binding to miR-140-3p, promoting PDAC resistance to GEM[86]. In a similar vein, FTO and YTHDF2 cooperatively stabilize the mRNA of Neuronal Precursor Cell-Expressed Developmentally Downregulated 4 (NEDD4), which regulates the PTEN/PI3K/AKT pathway, contributing to GEM resistance in PDAC[87].
GEMIN5, an m6A mediator identified as a m6A reader, interacts with m6A-modified Fizzy-Related Protein 1 (FZR1) and recruits the eIF3 translation initiation complex, enhancing FZR1 translation. The upregulation of FZR1 maintains the quiescent G0-G1 state in PDAC cells, thereby reducing GEM sensitivity[88]. Another m6A-driven resistance mechanism involves ALKBH5-mediated m6A modification, which promotes the overexpression of the antisense transcript of DNA damage-inducible transcript 4 (DDIT4), encoding the lncRNA DDIT4-AS1. This overexpression enhances PDAC stemness and diminishes GEM chemosensitivity by destabilizing DDIT4 and activating the mTOR pathway[89]. Furthermore, FTO-induced upregulation of KDM5B expression, achieved by stabilizing KDM5B mRNA, contributes to PDAC resistance. KDM5B targets DLG1, which in turn promotes YAP1 nuclear translocation, inducing de novo lipogenesis and contributing to chemotherapy resistance[51]. In PDAC, METTL3 also regulates m6A methylation of the 5′ untranslated region (UTR) of nucleobindin 1 (NUCB1). The m6A reader YTHDF2 binds to these m6A modifications, leading to the downregulation of NUCB1. Notably, NUCB1 plays an essential role in inhibiting PDAC cell proliferation and enhancing GEM efficacy[90]. DNA damage repair represents a major obstacle to the efficacy of chemotherapy in PDAC. Knocking out ZC3H13 downregulates m6A methylation of PHF10, a subunit of the PBAF chromatin remodeling complex. This reduces PHF10 translation in a YTHDF1-dependent manner, inhibiting homologous recombination repair of DNA double-strand breaks[91].
In a Phase III clinical trial, GEM combined with erlotinib, a human epidermal growth factor receptor type 1 (HER1/EGFR)-targeted agent, demonstrated superior efficacy compared to GEM with placebo, significantly extending overall survival[92]. This highlights the potential of exploring how m6A modifications can augment the sensitivity of targeted therapies or mitigate drug resistance. A study investigating the Nab-paclitaxel and GEM (AG) regimen revealed that Sulindac (K-80003), a drug inhibiting the PI3K/Akt pathway, sensitizes AG through circRNA cFAM124A[93]. Notably, cFAM124A was shown to enhance CTSL expression in an m6A-dependent manner via IGF2BP2, resulting in aberrant activation of the PI3K/Akt pathway and increased resistance to GEM. This underscores the importance of investigating whether combining IGF2BP2 inhibitors with current first-line chemotherapy regimens can provide clinical benefits for patients.
m6A also plays a critical role in immunotherapy. Studies have demonstrated that PD-1 checkpoint blockade therapy exhibits diminished efficacy in METTL3-deficient mice, positioning METTL3 as a potential target for tumor immunotherapy[67]. Similarly, improved therapeutic outcomes have been observed with PD-L1 checkpoint blockade in YTHDF1-deficient DCs[70]. In contrast, the loss of YTHDF2 in CD8 T cells accelerates tumor progression and impairs response to PD-1 blockade, both in mice and humans[94]. In PC, PD-1 and PD-L1 expression is closely associated with several m6A regulators, with the strongest correlations found between PD-1 and RBM15, and between PD-L1 and WTAP[78]. Furthermore, METTL3 upregulation in PC elevates lncRNA MALAT1 levels, which in turn enhances PD-L1 expression, potentially enhancing the efficacy of PD-L1-directed immunotherapy.
The exploration of PDAC resistance mechanisms has progressively revealed the essential role of m6A modifications in mediating resistance to therapies (Figure 3). m6A modifications are implicated across cytotoxic, targeted, and immunomodulatory therapies, suggesting that targeted modulation of m6A could improve the efficacy of existing chemotherapy and extend survival in patients with PDAC.
Molecular mechanism of m6A in chemotherapy resistance. (Created with BioRender.com).
4. The interaction between m6A and ncRNA in PC
Approximately 75% of the human genome is transcribed into RNA, with only 3% transcribed into protein-coding mRNA[95]. ncRNAs, including miRNA, lncRNA, circRNA, and PIWI-interacting RNA (piRNA), perform essential physiological functions in processes such as apoptosis, metastasis, invasion, migration, and cellular proliferation across various cancers[96]. The m6A methylation of ncRNAs plays a critical role in regulating both pro-tumorigenic and tumor-suppressive mechanisms during different stages of cancer progression.
MiRNAs, a subclass of ncRNAs, consist of short RNA molecules, typically 19 to 25 nucleotides in length, that regulate post-transcriptional silencing of target genes. A single miRNA can target hundreds of mRNAs, influencing the expression of numerous functionally interacting genes[97]. MiRNAs are central to carcinogenesis, classified into two categories: tumor-suppressive miRNAs and oncogenic miRNAs. Dysregulation of either category contributes to tumorigenesis[98]. Amphoteric regulatory protein (AREG) serves as an independent prognostic marker in PC, with METTL3 enhancing AREG mRNA stability via m6A methylation. In contrast, miR-33a-3p inhibits METTL3, thereby suppressing PC proliferation, migration, and invasion[99]. Additionally, miR-380-3p in PC is enriched with m6A modifications. Silencing METTL3 and METTL14 to remove these modifications synergistically reduces miR-380-3p expression in PC cells, thus inhibiting cell proliferation, migration, and epithelial-mesenchymal transition[100]. While miRNAs regulate m6A modifications, m6A can also directly modify miRNAs to influence their biological functions.
LncRNAs, which are typically longer than 200 nucleotides and can extend beyond 100,000 nucleotides, do not encode proteins[101]. Increasing evidence highlights the critical roles of lncRNAs in various biological processes associated with human diseases. Meta-analyses indicate that lncRNA dysregulation is linked to the overall survival of patients with PC, suggesting their potential as biomarkers for diagnosis and prognosis in PC[102]. As noted earlier, the interaction between m6A modifications and lncRNAs plays a pivotal role in the development and progression of PC. In PC, METTL3 and YTHDF1 promote the upregulation of lncRNA FOXD1-AS1 levels via m6A-dependent modifications, enhancing tumorigenesis and the self-renewal of cancer stem cells[103]. Cao et al. constructed an m6A-related lncRNA model using TCGA data, analyzing the impact of m6A-related lncRNAs on immune function, including effects on cytolytic activity, inflammation, T cell co-inhibition, checkpoint regulation, and T cell co-stimulation in tumors[104]. The m6A modification of lncRNA ANRIL regulates GEM resistance in PC by modulating the splicing of ANRIL by Serine/arginine-rich splicing factor 3 (SRSF3)[105]. The interplay between m6A methylation and lncRNAs is involved in nearly every process governing PC development, including cell proliferation, migration, immune evasion, and drug resistance.
circRNA, a class of RNA molecules with a circular structure, performs a wide range of functions by acting as molecular sponges to regulate cellular activities. It can bind to proteins, modulate gene expression as a trans-acting factor, and influence RNA methylation levels, including m6A modifications[106]. For instance, circMYO1C targets the m6A site on PD-L1 RNA, enhancing its stability in collaboration with IGF2BP2, thereby facilitating immune evasion in PDAC[79]. Recent studies have further highlighted the role of m6A modifications in circRNA-mediated regulation of innate immunity. Exogenous circRNAs serve as potent adjuvants, promoting antigen-specific T cell activation, antibody production, and anti-tumor immunity in vivo. However, m6A modifications suppress immune gene activation and diminish the adjuvant effects of circRNAs[107]. The Ye team developed a bioinformatics tool, Circm6A, which identified 8,807 m6A-circRNAs. In PDAC tumor tissues, m6A-circRNAs exhibit hypermethylation compared to normal tissues. These hypermethylated m6A-circRNAs are implicated in the dysregulation of several critical cancer-related pathways through the formation of circRNA-mRNA co-expression networks[108].
Moreover, exosomes, which carry a variety of ncRNAs, have emerged as key biomolecules in intercellular communication[109]. The potential of exosomes to deliver ncRNAs to PDAC tissues, thereby influencing cancer progression via m6A methylation modifications, presents an intriguing avenue for therapeutic intervention, particularly for patients with advanced PDAC.
5. Conclusions and Perspectives
In recent years, epigenetic regulation has garnered increasing attention in cancer research, with RNA m6A modification emerging as a pivotal area of investigation. Numerous studies have highlighted its role in various biological processes of PC, often referred to as the "king of cancers." To underscore the significance of m6A modification in PC, this review explores its involvement in four key aspects of tumor progression: the hypoxic microenvironment, metabolic reprogramming, immune microenvironment, and resistance mechanisms (Fig. 4). m6A modifications are ubiquitous, promoting tumor adaptation to hypoxic conditions while inhibiting immune cell recruitment in such environments. Additionally, m6A modifications participate in metabolic reprogramming in PC, including the well-characterized Warburg effect. Crucially, m6A also regulates the immune microenvironment of PC, influencing immune evasion through interactions with various immune cells, including T cells, B cells, NK cells, DCs, and neutrophils, as well as immune-related pathways such as PD-L1 expression. Furthermore, m6A plays a key role in drug resistance mechanisms, whether in cytotoxic, targeted, or immunomodulatory therapies. This review also introduces the interaction between m6A modifications and ncRNAs. Despite the fact that most ncRNAs do not encode proteins, recent studies have shown that they are essential in regulating cancer cell proliferation, migration, and invasion, interacting with m6A methylation to jointly govern PC progression.
Extensive role of m6A methylation modification in pancreatic cancer. (Created with BioRender.com).
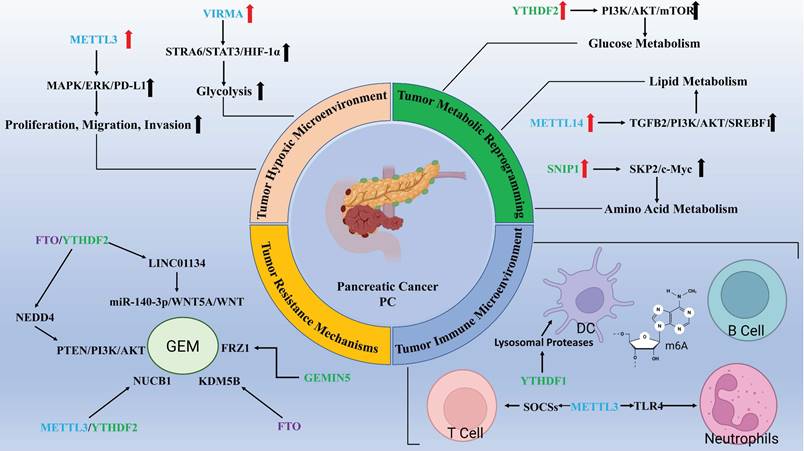
However, it is evident that research on m6A in PC remains in its early stages. Our understanding of the detailed mechanisms behind m6A modification in PC is still limited. This review primarily addresses the regulation of m6A methylation by m6A regulators. In reality, mutations in m6A sites may alter RNA m6A modifications, disrupting post-transcriptional regulation and contributing to tumorigenesis[15]. Moreover, mutations or alterations in m6A regulators can lead to abnormal cellular m6A levels or disrupt biological functions mediated by m6A methylation[16]. The interaction between m6A and other epigenetic factors also plays a significant role in PC progression. Therefore, future research should not be constrained by existing knowledge but should aim for more comprehensive and integrative studies. This will involve incorporating the complexity of m6A modifications into a unified model, identifying novel biomarkers and therapeutic targets, and exploring their potential clinical applications.
In conclusion, m6A-mediated epigenetic regulation stands as a key frontier in PC research. However, the intricate mechanisms of m6A demand systematic, in-depth investigations to elucidate its interactions with the malignant progression of PC, thereby paving the way for more personalized treatment approaches in clinical settings.
Abbreviations
SAM: S-adenosyl methionine; HIF: Hypoxia Inducible Factor; MAPK: Mitogen-activated protein kinase; CAFs: Cancer-associated fibroblasts; ERK: Extracellular signal-regulated kinase; PTEN: Phosphatase and tensin homolog deleted on chromosome ten; PD-L1: Programmed cell death-Ligand 1; PI3K: Phosphatidylinositol 3-kinase; ITGB1: Integrin Beta 1; mTOR: Mammalian target of rapamycin; GBE1: Glycogen branching enzyme 1; SMAD: Mothers against decapentaplegic homolog; DLG1: Discs Large Homolog 1; YAP1: Yes-Associated Protein 1; KDM5B: Lysine-specific demethylase 5B; AA: Amino acid; SNIP1: SMAD nuclear-interacting protein 1; SKP2: S phase kinase-associated protein 2; KLF12: Kruppel-like factor 12; NF-κB: Nuclear factor-kappa B; PPAR-γ: Peroxisome proliferator-activated receptor γ; STAT1: Signal transducer and activator of transcription 1; SOCS: Suppressor of cytokine signaling; CXCR2: CXC-Chemokine Receptor 2; NLRP12: NOD-like receptors family pyrin domain containing 12; PTGER4: Prostaglandin E Receptor 4; TNC: Tenascin C; WNK1: WNK lysine deficient protein kinase 1; NETs: Neutrophil extracellular traps; CXCL1: CXC-chemokine ligand 1; CCL20: C-C chemokine ligand 20; NUCB1: Nucleobindin-1; NEDD4: Neuronal precursor cell-expressed developmentally downregulated 4; FABP5: Fatty Acid Binding Protein 5.
Acknowledgements
Funding
This study was supported by grants from the National Natural Science Foundation of China (Grant No. 82303077 and Grant No.82401150) and the Open Project of Hubei Key Laboratory (Grant No. 2023KFZZ002).
Author contributions
ZeHao Liu: Writing - original draft and Conceptualization; Peng Ma, YueFeng Zhang: Investigation and Methodology; Ying He, Wei Wang: Funding acquisition and Resources; Zuo Mou, Ting Fang: Software and Visualization; KaiHuan Yu: Writing - review and editing, Project administration, and Supervision.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913-21
3. Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet. 2020;395:2008-20
4. Ferlay J, Partensky C, Bray F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016;55:1158-60
5. Cai J, Chen H, Lu M, Zhang Y, Lu B, You L. et al. Advances in the epidemiology of pancreatic cancer: Trends, risk factors, screening, and prognosis. Cancer Lett. 2021;520:1-11
6. Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV. et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022
7. Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15:333-48
8. Kim MP, Gallick GE. Gemcitabine resistance in pancreatic cancer: picking the key players. Clin Cancer Res. 2008;14:1284-5
9. Liu N, Pan T. N6-methyladenosine-encoded epitranscriptomics. Nat Struct Mol Biol. 2016;23:98-102
10. Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19:88
11. Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z. et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74
12. Yue SW, Liu HL, Su HF, Luo C, Liang HF, Zhang BX. et al. m6A-regulated tumor glycolysis: new advances in epigenetics and metabolism. Mol Cancer. 2023;22:137
13. Hu X, Lei X, Guo J, Fu W, Sun W, Lu Q. et al. The Emerging Role of RNA N6-Methyladenosine Modification in Pancreatic Cancer. Front Oncol. 2022;12:927640
14. Chen S, Ren H, Zhang X, Chang L, Wang Z, Wu H. et al. Research advances of N6-methyladenosine in diagnosis and therapy of pancreatic cancer. J Clin Lab Anal. 2022;36:e24611
15. Li J, Wang F, Liu Y, Wang H, Ni B. N(6)-methyladenosine (m(6)A) in pancreatic cancer: Regulatory mechanisms and future direction. Int J Biol Sci. 2021;17:2323-35
16. Wang Y, Wang Y, Patel H, Chen J, Wang J, Chen ZS. et al. Epigenetic modification of m(6)A regulator proteins in cancer. Mol Cancer. 2023;22:102
17. Oerum S, Meynier V, Catala M, Tisné C. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res. 2021;49:7239-55
18. Han X, Zhu Y, Ke J, Zhai Y, Huang M, Zhang X. et al. Progression of m(6)A in the tumor microenvironment: hypoxia, immune and metabolic reprogramming. Cell Death Discov. 2024;10:331
19. An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21:14
20. Sendinc E, Valle-Garcia D, Dhall A, Chen H, Henriques T, Navarrete-Perea J. et al. PCIF1 Catalyzes m6Am mRNA Methylation to Regulate Gene Expression. Mol Cell. 2019;75:620-30.e9
21. Qu J, Yan H, Hou Y, Cao W, Liu Y, Zhang E. et al. RNA demethylase ALKBH5 in cancer: from mechanisms to therapeutic potential. J Hematol Oncol. 2022;15:8
22. Li Y, Su R, Deng X, Chen Y, Chen J. FTO in cancer: functions, molecular mechanisms, and therapeutic implications. Trends Cancer. 2022;8:598-614
23. Lan Q, Liu PY, Bell JL, Wang JY, Hüttelmaier S, Zhang XD. et al. The Emerging Roles of RNA m(6)A Methylation and Demethylation as Critical Regulators of Tumorigenesis, Drug Sensitivity, and Resistance. Cancer Res. 2021;81:3431-40
24. Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616-24
25. Xu Z, Zhou Y, Liu S, Zhao H, Chen Z, Li R. et al. KHSRP Stabilizes m6A-Modified Transcripts to Activate FAK Signaling and Promote Pancreatic Ductal Adenocarcinoma Progression. Cancer Res. 2024;84:3602-3616
26. Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother. 2019;112:108613
27. Li F, He C, Yao H, Zhao Y, Ye X, Zhou S. et al. Glutamate from nerve cells promotes perineural invasion in pancreatic cancer by regulating tumor glycolysis through HK2 mRNA-m6A modification. Pharmacol Res. 2023;187:106555
28. Zhuang Y, Liu K, He Q, Gu X, Jiang C, Wu J. Hypoxia signaling in cancer: Implications for therapeutic interventions. MedComm (2020). 2023;4:e203
29. Zhu P, He F, Hou Y, Tu G, Li Q, Jin T. et al. A novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene. 2021;40:1609-27
30. Lu Y, Chen Q, Zhu S, Gong X. Hypoxia promotes immune escape of pancreatic cancer cells by lncRNA NNT-AS1/METTL3-HuR-mediated ITGB1 m(6)A modification. Exp Cell Res. 2023;432:113764
31. Lu Y, Hu J, Sun W, Li S, Deng S, Li M. MiR-29c inhibits cell growth, invasion, and migration of pancreatic cancer by targeting ITGB1. Onco Targets Ther. 2016;9:99-109
32. Zhou Y, Zhou X, Ben Q, Liu N, Wang J, Zhai Y. et al. GATA6-AS1 suppresses epithelial-mesenchymal transition of pancreatic cancer under hypoxia through regulating SNAI1 mRNA stability. J Transl Med. 2023;21:882
33. Hu J, Li X, Yang L, Li H. Hypoxia, a key factor in the immune microenvironment. Biomed Pharmacother. 2022;151:113068
34. Wigerup C, Påhlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. 2016;164:152-69
35. Garcia Garcia CJ, Huang Y, Fuentes NR, Turner MC, Monberg ME, Lin D. et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology. 2022;162:2018-31
36. Li Q, Ni Y, Zhang L, Jiang R, Xu J, Yang H. et al. HIF-1α-induced expression of m6A reader YTHDF1 drives hypoxia-induced autophagy and malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct Target Ther. 2021;6:76
37. Wang Y, Yang Y, Yang Y, Dang Y, Guo Z, Zhuang Q. et al. Hypoxia induces hepatocellular carcinoma metastasis via the HIF-1α/METTL16/lnc-CSMD1-7/RBFOX2 axis. iScience. 2023;26:108495
38. Yang K, Zhong Z, Zou J, Liao JY, Chen S, Zhou S. et al. Glycolysis and tumor progression promoted by the m(6)A writer VIRMA via m(6)A-dependent upregulation of STRA6 in pancreatic ductal adenocarcinoma. Cancer Lett. 2024;590:216840
39. Guak H, Al Habyan S, Ma EH, Aldossary H, Al-Masri M, Won SY. et al. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat Commun. 2018;9:2463
40. Liu J, Zhang X, Chen K, Cheng Y, Liu S, Xia M. et al. CCR7 Chemokine Receptor-Inducible lnc-Dpf3 Restrains Dendritic Cell Migration by Inhibiting HIF-1α-Mediated Glycolysis. Immunity. 2019;50:600-15.e15
41. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
42. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200
43. Gu Y, Wu X, Zhang J, Fang Y, Pan Y, Shu Y. et al. The evolving landscape of N(6)-methyladenosine modification in the tumor microenvironment. Mol Ther. 2021;29:1703-15
44. Tan Z, Xu J, Zhang B, Shi S, Yu X, Liang C. Hypoxia: a barricade to conquer the pancreatic cancer. Cell Mol Life Sci. 2020;77:3077-83
45. Liu X, Feng M, Hao X, Gao Z, Wu Z, Wang Y. et al. m6A methylation regulates hypoxia-induced pancreatic cancer glycolytic metabolism through ALKBH5-HDAC4-HIF1α positive feedback loop. Oncogene. 2023;42:2047-60
46. Jurcak NR, Rucki AA, Muth S, Thompson E, Sharma R, Ding D. et al. Axon Guidance Molecules Promote Perineural Invasion and Metastasis of Orthotopic Pancreatic Tumors in Mice. Gastroenterology. 2019;157:838-50.e6
47. Hu Y, Tang J, Xu F, Chen J, Zeng Z, Han S. et al. A reciprocal feedback between N6-methyladenosine reader YTHDF3 and lncRNA DICER1-AS1 promotes glycolysis of pancreatic cancer through inhibiting maturation of miR-5586-5p. J Exp Clin Cancer Res. 2022;41:69
48. Zheng D, Chen W, Peng J, Huang X, Zhang S, Zhuang Y. Hsa_circ_0007590/PTBP1 complex reprograms glucose metabolism by reducing the stability of m(6)A-modified PTEN mRNA in pancreatic ductal adenocarcinoma. Cancer Gene Ther. 2024;31:1090-102
49. Jin W, Yao Y, Fu Y, Lei X, Fu W, Lu Q. et al. WTAP/IGF2BP3-mediated GBE1 expression accelerates the proliferation and enhances stemness in pancreatic cancer cells via upregulating c-Myc. Cell Mol Biol Lett. 2024;29:97
50. Ma MJ, Shi YH, Liu ZD, Zhu YQ, Zhao GY, Ye JY. et al. N6-methyladenosine modified TGFB2 triggers lipid metabolism reprogramming to confer pancreatic ductal adenocarcinoma gemcitabine resistance. Oncogene. 2024;43:2405-20
51. Wang Y, Liu S, Wang Y, Li B, Liang J, Chen Y. et al. KDM5B promotes SMAD4 loss-driven drug resistance through activating DLG1/YAP to induce lipid accumulation in pancreatic ductal adenocarcinoma. Cell Death Discov. 2024;10:252
52. Chen J, Ye M, Bai J, Gong Z, Yan L, Gu D. et al. ALKBH5 enhances lipid metabolism reprogramming by increasing stability of FABP5 to promote pancreatic neuroendocrine neoplasms progression in an m6A-IGF2BP2-dependent manner. J Transl Med. 2023;21:741
53. Lu J, Ru J, Chen Y, Ling Z, Liu H, Ding B. et al. N(6) -methyladenosine-modified circSTX6 promotes hepatocellular carcinoma progression by regulating the HNRNPD/ATF3 axis and encoding a 144 amino acid polypeptide. Clin Transl Med. 2023;13:e1451
54. Xu R, Yang J, Ren B, Wang H, Yang G, Chen Y. et al. Reprogramming of Amino Acid Metabolism in Pancreatic Cancer: Recent Advances and Therapeutic Strategies. Front Oncol. 2020;10:572722
55. Ma Y, Li Y, Ling S, Li X, Kong B, Hu M. et al. Loss of heterozygosity for Kras(G12D) promotes REDD1-dependent, non-canonical glutamine metabolism in pancreatic ductal adenocarcinoma. Biochem Biophys Res Commun. 2020;526:880-8
56. Wu G, Su J, Zeng L, Deng S, Huang X, Ye Y. et al. LncRNA BCAN-AS1 stabilizes c-Myc via N(6)-methyladenosine-mediated binding with SNIP1 to promote pancreatic cancer. Cell Death Differ. 2023;30:2213-30
57. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK. et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782-7
58. Matés JM, Campos-Sandoval JA, Márquez J. Glutaminase isoenzymes in the metabolic therapy of cancer. Biochim Biophys Acta Rev Cancer. 2018;1870:158-64
59. Chen P, Liu XQ, Lin X, Gao LY, Zhang S, Huang X. Targeting YTHDF1 effectively re-sensitizes cisplatin-resistant colon cancer cells by modulating GLS-mediated glutamine metabolism. Mol Ther Oncolytics. 2021;20:228-39
60. Zhou T, Xiao Z, Lu J, Zhang L, Bo L, Wang J. IGF2BP3-mediated regulation of GLS and GLUD1 gene expression promotes treg-induced immune escape in human cervical cancer. Am J Cancer Res. 2023;13:5289-305
61. Zhou Z, Zhang J, Xu C, Yang J, Zhang Y, Liu M. et al. An integrated model of N6-methyladenosine regulators to predict tumor aggressiveness and immune evasion in pancreatic cancer. EBioMedicine. 2021;65:103271
62. Torphy RJ, Schulick RD, Zhu Y. Understanding the immune landscape and tumor microenvironment of pancreatic cancer to improve immunotherapy. Mol Carcinog. 2020;59:775-82
63. Wang W, He Y, Yao LC, Yuan Y, Lu C, Xiong LK. et al. Identification of m6A modification patterns and RBM15 mediated macrophage phagocytosis in pancreatic cancer: An integrative analysis. Biochim Biophys Acta Mol Basis Dis. 2024;1870:167304
64. Liu Y, Shi M, He X, Cao Y, Liu P, Li F. et al. LncRNA-PACERR induces pro-tumour macrophages via interacting with miR-671-3p and m6A-reader IGF2BP2 in pancreatic ductal adenocarcinoma. J Hematol Oncol. 2022;15:52
65. Chen L, Gao Y, Xu S, Yuan J, Wang M, Li T. et al. N6-methyladenosine reader YTHDF family in biological processes: Structures, roles, and mechanisms. Front Immunol. 2023;14:1162607
66. Liu Y, Liu Z, Tang H, Shen Y, Gong Z, Xie N. et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation of STAT1 mRNA. Am J Physiol Cell Physiol. 2019;317:C762-c75
67. Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D. et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. 2021;12:1394
68. Dong H, Zhang H, Mao X, Liu S, Xu W, Zhang Y. RBM15 Promates the Proliferation, Migration and Invasion of Pancreatic Cancer Cell Lines. Cancers (Basel). 2023;15:1084
69. Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J. et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548:338-42
70. Han D, Liu J, Chen C, Dong L, Liu Y, Chang R. et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566:270-4
71. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519-31
72. Xiong S, Dong L, Cheng L. Neutrophils in cancer carcinogenesis and metastasis. J Hematol Oncol. 2021;14:173
73. Liu Y, Song R, Zhao L, Lu Z, Li Y, Zhan X. et al. m(6)A demethylase ALKBH5 is required for antibacterial innate defense by intrinsic motivation of neutrophil migration. Signal Transduct Target Ther. 2022;7:194
74. Luo S, Liao C, Zhang L, Ling C, Zhang X, Xie P. et al. METTL3-mediated m6A mRNA methylation regulates neutrophil activation through targeting TLR4 signaling. Cell Rep. 2023;42:112259
75. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191-218
76. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME. et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361:eaao4227
77. Dai W, Tian R, Yu L, Bian S, Chen Y, Yin B. et al. Overcoming therapeutic resistance in oncolytic herpes virotherapy by targeting IGF2BP3-induced NETosis in malignant glioma. Nat Commun. 2024;15:131
78. Song Z, Wang X, Chen F, Chen Q, Liu W, Yang X. et al. LncRNA MALAT1 regulates METTL3-mediated PD-L1 expression and immune infiltrates in pancreatic cancer. Front Oncol. 2022;12:1004212
79. Guan H, Tian K, Luo W, Li M. m(6)A-modified circRNA MYO1C participates in the tumor immune surveillance of pancreatic ductal adenocarcinoma through m(6)A/PD-L1 manner. Cell Death Dis. 2023;14:120
80. Ni Z, Sun P, Zheng J, Wu M, Yang C, Cheng M. et al. JNK Signaling Promotes Bladder Cancer Immune Escape by Regulating METTL3-Mediated m6A Modification of PD-L1 mRNA. Cancer Res. 2022;82:1789-802
81. Wan W, Ao X, Chen Q, Yu Y, Ao L, Xing W. et al. METTL3/IGF2BP3 axis inhibits tumor immune surveillance by upregulating N(6)-methyladenosine modification of PD-L1 mRNA in breast cancer. Mol Cancer. 2022;21:60
82. Yu H, Liu J, Bu X, Ma Z, Yao Y, Li J. et al. Targeting METTL3 reprograms the tumor microenvironment to improve cancer immunotherapy. Cell Chem Biol. 2024;31:776-91.e7
83. Heinemann V. Gemcitabine: progress in the treatment of pancreatic cancer. Oncology. 2001;60:8-18
84. Beatty GL, Werba G, Lyssiotis CA, Simeone DM. The biological underpinnings of therapeutic resistance in pancreatic cancer. Genes Dev. 2021;35:940-62
85. Wang J, Yang J, Narang A, He J, Wolfgang C, Li K. et al. Consensus, debate, and prospective on pancreatic cancer treatments. J Hematol Oncol. 2024;17:92
86. Lu J, Yang Y, Liu X, Chen X, Song W, Liu Z. FTO-mediated LINC01134 stabilization to promote chemoresistance through miR-140-3p/WNT5A/WNT pathway in PDAC. Cell Death Dis. 2023;14:713
87. Lin K, Zhou E, Shi T, Zhang S, Zhang J, Zheng Z. et al. m6A eraser FTO impairs gemcitabine resistance in pancreatic cancer through influencing NEDD4 mRNA stability by regulating the PTEN/PI3K/AKT pathway. J Exp Clin Cancer Res. 2023;42:217
88. Su J, Li R, Chen Z, Liu S, Zhao H, Deng S. et al. N 6-methyladenosine Modification of FZR1 mRNA Promotes Gemcitabine Resistance in Pancreatic Cancer. Cancer Res. 2023;83:3059-76
89. Zhang Y, Liu X, Wang Y, Lai S, Wang Z, Yang Y. et al. The m(6)A demethylase ALKBH5-mediated upregulation of DDIT4-AS1 maintains pancreatic cancer stemness and suppresses chemosensitivity by activating the mTOR pathway. Mol Cancer. 2022;21:174
90. Hua YQ, Zhang K, Sheng J, Ning ZY, Li Y, Shi WD. et al. NUCB1 Suppresses Growth and Shows Additive Effects With Gemcitabine in Pancreatic Ductal Adenocarcinoma via the Unfolded Protein Response. Front Cell Dev Biol. 2021;9:641836
91. Huang C, Zhou S, Zhang C, Jin Y, Xu G, Zhou L. et al. ZC3H13-mediated N6-methyladenosine modification of PHF10 is impaired by fisetin which inhibits the DNA damage response in pancreatic cancer. Cancer Lett. 2022;530:16-28
92. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S. et al. Erlotinib Plus Gemcitabine Compared With Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2023;41:4714-20
93. Xie CK, Liao CY, Lin HY, Wu YD, Lu FC, Huang XX. et al. Sulindac (K-80003) with nab-paclitaxel and gemcitabine overcomes drug-resistant pancreatic cancer. Mol Cancer. 2024;23:215
94. Zhang H, Luo X, Yang W, Wu Z, Zhao Z, Pei X. et al. YTHDF2 upregulation and subcellular localization dictate CD8 T cell polyfunctionality in anti-tumor immunity. Nat Commun. 2024;15:9559
95. Kimura T. [Non-coding Natural Antisense RNA: Mechanisms of Action in the Regulation of Target Gene Expression and Its Clinical Implications]. Yakugaku Zasshi. 2020;140:687-700
96. Yan H, Bu P. Non-coding RNA in cancer. Essays Biochem. 2021;65:625-39
97. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141:1202-7
98. Daoud AZ, Mulholland EJ, Cole G, McCarthy HO. MicroRNAs in Pancreatic Cancer: biomarkers, prognostic, and therapeutic modulators. BMC Cancer. 2019;19:1130
99. Su X, Lai T, Tao Y, Zhang Y, Zhao C, Zhou J. et al. miR-33a-3p regulates METTL3-mediated AREG stability and alters EMT to inhibit pancreatic cancer invasion and metastasis. Sci Rep. 2023;13:13587
100. Jiang Z, Song X, Wei Y, Li Y, Kong D, Sun J. N(6)-methyladenosine-mediated miR-380-3p maturation and upregulation promotes cancer aggressiveness in pancreatic cancer. Bioengineered. 2022;13:14460-71
101. Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760-74
102. Seyed Hosseini E, Nikkhah A, Sotudeh A, Alizadeh Zarei M, Izadpanah F, Nikzad H. et al. The impact of LncRNA dysregulation on clinicopathology and survival of pancreatic cancer: a systematic review and meta-analysis (PRISMA compliant). Cancer Cell Int. 2021;21:447
103. Ouyang L, Sun MM, Zhou PS, Ren YW, Liu XY, Wei WY. et al. LncRNA FOXD1-AS1 regulates pancreatic cancer stem cell properties and 5-FU resistance by regulating the miR-570-3p/SPP1 axis as a ceRNA. Cancer Cell Int. 2024;24:4
104. Cao PW, Liu L, Li ZH, Cao F, Liu FB. Prognostic Value of Drug Targets Predicted Using Deep Bioinformatic Analysis of m6A-Associated lncRNA-Based Pancreatic Cancer Model Characteristics and Its Tumour Microenvironment. Front Genet. 2022;13:853471
105. Wang ZW, Pan JJ, Hu JF, Zhang JQ, Huang L, Huang Y. et al. SRSF3-mediated regulation of N6-methyladenosine modification-related lncRNA ANRIL splicing promotes resistance of pancreatic cancer to gemcitabine. Cell Rep. 2022;39:110813
106. Li H. circRNA: a promising all-around star in the future. Epigenomics. 2023;15:677-85
107. Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L. et al. N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol Cell. 2019;76:96-109.e9
108. Ye Y, Feng W, Zhang J, Zhu K, Huang X, Pan L. et al. Genome-wide identification and characterization of circular RNA m(6)A modification in pancreatic cancer. Genome Med. 2021;13:183
109. Sha G, Zhang W, Jiang Z, Zhao Q, Wang D, Tang D. Exosomal non-coding RNA: A new frontier in diagnosing and treating pancreatic cancer: A review. Int J Biol Macromol. 2024;263:130149
Author contact
![]() Corresponding authors: Kai-Huan Yu, Department of Hepatobiliary Surgery, East Hospital, Renmin Hospital of Wuhan University, Wuhan, 430060, Hubei Province, China; E-mail: rm001823edu.cn. Wei Wang, Department of Hepatobiliary Surgery, East Hospital, Renmin Hospital of Wuhan University, Wuhan, 430060, Hubei Province, China; E-mail: rm003855edu.cn.
Corresponding authors: Kai-Huan Yu, Department of Hepatobiliary Surgery, East Hospital, Renmin Hospital of Wuhan University, Wuhan, 430060, Hubei Province, China; E-mail: rm001823edu.cn. Wei Wang, Department of Hepatobiliary Surgery, East Hospital, Renmin Hospital of Wuhan University, Wuhan, 430060, Hubei Province, China; E-mail: rm003855edu.cn.