Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. TGF-β signaling in...
2. Targeting TGF-β...
3. Targeting the modulators of...
Conclusions and prospects
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(4):1649-1665. doi:10.7150/ijbs.101548 This issue Cite
Review
Modulation of TGF-β signaling new approaches toward kidney disease and fibrosis therapy
Quan Hong1,2, Hyoungnae Kim1, Guang-Yan Cai2, Xiang-Mei Chen2, John Cijiang He1,3 ![]() , Kyung Lee1
, Kyung Lee1 ![]()
1. Department of Medicine, Division of Nephrology, Icahn School of Medicine at Mount Sinai, NY, USA.
2. Department of Nephrology, Chinese PLA General Hospital, Chinese PLA Institute of Nephrology, State Key Laboratory of Kidney Diseases, National Clinical Research Center of Kidney Diseases, Beijing, China.
3. James J. Peters Department of Veterans Affairs Medical Center, Bronx, NY, USA.
Received 2024-7-28; Accepted 2024-11-16; Published 2025-2-3
Abstract

The prevalence of chronic kidney disease (CKD) is increasing worldwide, posing a significant healthcare challenge. Despite the immense burden of CKD, optimal therapies remain limited in impact. Kidney fibrosis is a common mediator of all CKD progression, characterized by excessive extracellular matrix deposition and scarring of kidney parenchyma. Transforming growth factor-β (TGF-β) is a potent pro-fibrotic cytokine that signals through canonical and non-canonical pathways to promote kidney cell damage and fibrosis progression, thus garnering much interest as an optimal therapeutic target for CKD. However, the clinical translation of TGF-β inhibition in CKD and other disease settings has faced substantial challenges, particularly due to the highly pleiotropic effects of TGF-β in organ homeostasis and disease. Here, we review the kidney cell-specific biological effects of TGF-β signaling, discuss the current challenges in therapeutic targeting TGF-β in CKD, and provide the rationale for alternative targeting strategies of TGF-β signaling as potential approaches in CKD therapy. Selective inhibition of TGF-β signaling modulators to fine-tune TGF-β inhibition without a broad blockade may lead to new and safer treatments for CKD.
Keywords: Kidney fibrosis, CKD, DKD, TGF-β, ALK5, ALK1, TGFBR2, Smad3, HIPK2, LRG1.
Introduction
Chronic kidney disease (CKD) is estimated to affect more than 800 million individuals worldwide and is associated with high morbidity and mortality [1]. CKD tends to progress slowly to end-stage kidney disease (ESKD), requiring kidney replacement therapy (KRT) of dialysis or kidney transplantation as the main treatment to prolong survival. As the number of patients requiring KRT is expected to rise sharply over the next few decades [2], there is an urgent need to develop effective treatments for CKD and fibrosis to halt the progression to ESKD.
Transforming growth factor-β (TGF-β) is a well-established pathogenic driver of kidney disease development [3, 4]. Early human clinical studies have demonstrated the increased expression of TGF-β ligands in diseased kidneys, and numerous experimental studies have shown the causal link of TGF-β in inducing glomerulosclerosis and tubulointerstitial fibrosis. As such, its inhibition has garnered much interest as a therapeutic option for CKD, such as diabetic kidney disease (DKD) and focal segmental glomerulosclerosis (FSGS). However, the clinical translation of pharmacologic TGF-β inhibition has not been as successful in CKD as anticipated [5, 6]. While the lack of success of anti-TGF-β antibodies for kidney disease therapy may be multifactorial, a key factor may involve the complex and pleiotropic cellular actions of TGF-β that are carried out in a cell type- and context-dependent manner, posing a significant clinical challenge. For instance, while hyperactive TGF-β signaling exerts potent pro-fibrotic effects across multiple organ injuries, TGF-β is also required to regulate the inflammatory immune response, such that Tgfb1-null mice develop a multifocal inflammatory disease and early postnatal lethality [7, 8]. In the context of cancer progression, TGF-β has a biphasic function such that in the initial stages of tumorigenesis, it exerts antiproliferative and tumor-suppressive effects, but in later stages it contributes to malignant progression by promoting metastasis and chemoresistance [9, 10]. In the developing vasculature system, TGF-β signaling can elicit divergent outcomes. Depending on the engagement of specific type 1 TGF-β receptors and downstream cognate Smad signaling molecules [11], TGF-β can induce endothelial proliferation or inhibit proliferation to promote quiescence. Similarly, TGF-β signaling is an essential instructive component of nephrogenesis and kidney development [12-16], but its persistent signaling is a major driver of glomerulosclerosis and tubulointerstitial fibrosis in kidney disease settings. Moreover, its diverse kidney cell-specific effects, both homeostatic and pathogenic, continue to emerge in kidney injury response and disease progression. Therefore, the challenge to specifically restrain the TGF-β's deleterious role in kidney disease remains a critically unmet need. This review aims to provide an integrative conceptual framework on kidney cell- and context-specific roles of TGF-β, discuss the remaining challenges in therapeutic targeting of the signaling pathway, and provide a perspective on alternative approaches and opportunities in TGF-β therapy for kidney disease and fibrosis.
1. TGF-β signaling in kidney homeostasis and disease
1.1 Overview of TGF-β signaling
TGF-β proteins, belonging to a superfamily of cytokines that comprises a large group of structurally related growth factors [17], consist of three mammalian isoforms, TGF-β1, TGF-β2, and TGF-β3. Although TGF-β1 is the most studied isoform in kidney disease settings, all three isoforms have been detected in developing and adult human kidneys and shown to have similar potential to produce extracellular matrix in cultured kidney cells [18-20].
TGF-β molecules are synthesized and secreted into the extracellular matrix (ECM) in an inactive form as part of a latent complex containing TGF-β, the latency-associated peptide (LAP), and latent TGF-β binding protein (LTBP) [21] (Figure 1A). The latent TGF-β complex remains in the ECM until triggered by signals for its release. Once activated, TGF-βs elicit cellular responses through the heterotetrameric complex of type I and type II TGF-β receptors, TGFBR1 and TGFBR2. There are two mammalian type I receptors, ubiquitously expressed TGFBR1, also known as activin receptor-like kinase 5 (ALK5), and ALK1 (also known as ACVRL1), whose expression is largely restricted to endothelial cells. Constitutively active TGFBR2 dimers, upon TGF-β binding, oligomerize with type 1 receptor dimers, leading to their phosphorylation. The activated type I receptors, in turn, relay the signal through a family of receptor-regulated Smad (R-Smad) effector proteins (Smad1, 2, 3, 5, and 8). Phosphorylated R-Smads form complexes with co-Smad (Smad4) and are shuttled into the nucleus for transcriptional regulation on numerous target genes. Because of their relatively low DNA-binding affinity, Smad complexes cooperate with an extensive repertoire of high-affinity DNA-binding transcription factors and regulators to induce or repress gene expression [22]. Thus, the spatiotemporal expression and activity of transcriptional co-regulators allow the TGF-β signaling to elicit diverse biological outcomes in physiologic and pathologic contexts, albeit working through a common set of intracellular Smad proteins.
TGF-β signaling. A. Schematics of Smad-mediated canonical and Smad-independent noncanonical TGF-β signaling transduction are shown. Cell type and stage-specific TGF-β signaling is dictated by the expression of extracellular and intracellular signaling mediators and inhibitors and the combination of transcriptional repressors and activators present. B. TGF-β signaling in endothelial cells involving the interplay between two type 1 TGF-β receptors (ALK1 vs. ALK5) is shown.
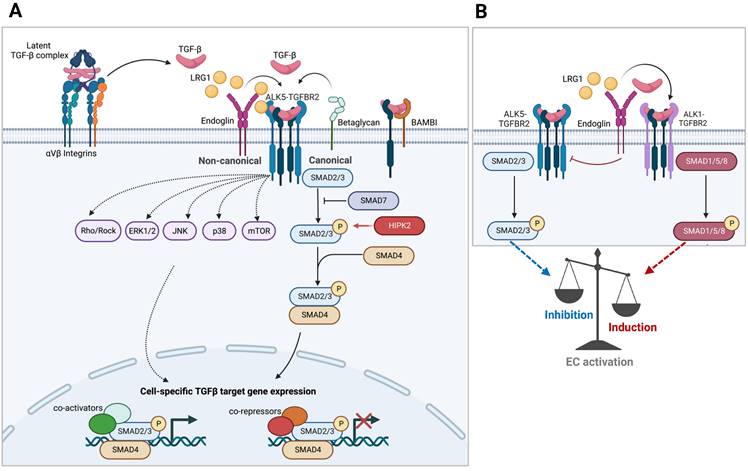
In addition to the canonical Smad-dependent signaling, activated TGF-β receptors can propagate Smad-independent, non-canonical signaling through intracellular effectors including mitogen-activated protein kinase (MAPK) family of proteins (ERK1/2, JNK, and p38 MAPK), phosphatidylinositol-3 kinase (PI3K), IκB kinase (IKK), and Src and Rho families of GTPases [23] (Figure 1A). The non-canonical mediators can signal independently or work in concert with Smads to generate highly context-dependent gene expression and physiologic responses. For instance, TGF-β-activated kinase 1 (TAK1), belonging to the MAP kinase kinase kinase (MAP3K) family, promotes Smad-dependent and -independent signaling in response to TGF-β. However, TAK1 signaling is not necessarily restricted to TGF-β stimuli, but it can also respond to pro-inflammatory cytokines, such as TNF-α , IL-1, and toll-like receptor (TLR) ligands, and acts as a crucial regulator of NF-κB activity in inflammatory and immune signaling pathways [24]. Thus, TAK1 can propagate both TGF-β signaling and mediate the interplay of crosstalk between TGF-β and inflammatory signaling pathways in a context-specific manner.
The fine-tuning of signaling amplitude, duration, and specificity of TGF-β is also achieved by the combinatorial actions of positive and negative regulatory feedback loops, mediated by post-translational modification of signaling protein or transcriptional induction [25]. For example, a set of genes rapidly induced in many cell types following TGF-β treatment encodes for inhibitory Smads (I-Smads), Smad6 and 7, as a part of a negative feedback mechanism. I-Smads inhibit the propagation of TGF-β signal transduction by interfering with R-Smad activation by competing for interaction with type 1 receptor and by recruitment of E3 ubiquitin ligases to promote degradation of type 1 receptor. TGF-β signaling also induces the expression of BMP and activin membrane-bound inhibitor (BAMBI), a pseudo-receptor with the extracellular domain related to type I receptor but lacking an intracellular kinase domain. It readily forms heterodimers with type I receptors, impeding their heteromeric complex formation with type II receptors to inhibit TGF-β signaling [26]. It also synergizes with I-Smad, Smad7, to inhibit the interaction between type I receptors and R-Smads [27]. Many miRNAs targeting the components of the TGF-β pathways and its downstream genes are also induced as a mechanism of feedback regulation. Conversely, in a positive feedback loop, TGF-β can further sustain and amplify its signaling by induction of genes in the signaling pathway, such as proteins involved in its activation from latency, such as integrins and matrix metalloproteinases as well as TGF-β itself, referred to as autoinduction. Thus, the feedback loops provide an additional cell- and context-dependent specificity of signaling output.
1.2 TGF-β signaling in kidney cell injury and disease
Increased TGF-β signaling in human CKD was observed more than 30 years ago [28-30], and numerous experimental models have well-established its pathogenic role in kidney disease and fibrosis. Examples of murine models include the transgenic mouse model with increased circulating TGF-β1 expression in the plasma (driven by albumin promoter in hepatocytes) that progressively developed glomerulosclerosis, interstitial fibrosis, and nephrotic syndrome [31] and transgenic mouse with a targeted increase of TGF-β1 in the juxtaglomerular apparatus (driven by Ren-1c promoter) that also developed progressive glomerular disease with reduced kidney function [32, 33]. Yet, the diverse kidney cell-specific effects of TGF-β continue to emerge and underscore the strong influence of cell type and context-dependent TGF-β signaling in kidney homeostasis and disease pathogenesis. In the following sections, select studies highlighting the kidney cell-specific effects of TGF-β are discussed, mainly gleaned from genetic manipulations of TGF-β signaling components in rodent models of CKD and fibrosis.
1.2.1. Epithelial cells
Tubular epithelial cells
Epithelial-to-mesenchymal transition (EMT) is an integral process during development and is tightly regulated in a spatiotemporal manner. Partial EMT also occurs during epithelial injury as a mechanism of wound healing. Nevertheless, its deregulation underlies organ fibrosis and cancer progression in pathologic contexts. TGF-β is among the potent inducers of EMT via Smad-dependent and independent pathways and by cooperative crosstalk with other EMT-inducing pathways, such as Notch, Wnt, Hedgehog, and TAZ/YAP [34]. The actions of TGF-β on kidney tubular epithelial cells (TECs) in vivo, evidenced by TEC-specific overexpression of TGF-β1 or constitutively active TGFBR1, are associated with partial EMT, deregulated proliferation and cell cycle arrest, and apoptosis and tubular atrophy, all of which promote kidney fibrosis in concert with neighboring interstitial cells [35-37]. Similarly, TEC-specific overexpression of leucine-rich-α-2-glycoprotein-1 (LRG1), a secreted glycoprotein that potentiates TGF-β signaling, exacerbated unilateral ureteral obstruction (UUO)- and aristolochic acid-induced kidney fibrosis in mice [38]. Interestingly, in diabetic Akita mice, proximal tubule-specific Tgfb1 overexpression not only induced tubulointerstitial fibrosis but significantly augmented albuminuria without apparent change in the GFR, which was associated with decreased megalin expression in the proximal tubules [39]. This is consistent with the earlier observation that TGF-β1 can reduce megalin expression in cultured tubular cells and that TGF-β inhibition with soluble TGFBR2 (sTβRII.Fc) treatment reduces albumin excretion in streptozotocin (STZ)-induced diabetic rats [40].
Nevertheless, notwithstanding the abundance of evidence of hyperactive TGF-β signaling in driving tubulointerstitial fibrosis, emerging evidence also indicates its involvement in tubular homeostasis and appropriate response to acute kidney injury and repair. For instance, the conditional ablation of Tgfbr2 in proximal tubules, driven by γ-glutamyl transferase (γGT)-Cre system, conferred protection against tubular apoptosis induced by mercuric chloride [41], but the same mouse model challenged to aristolochic acid-induced injury showed worsened renal damage and fibrosis development, characterized by increased proximal tubular mitochondrial injury, oxidative stress and metabolic shifting toward aerobic glycolysis, and Th17 inflammatory response [42, 43]. Similarly, the loss of Tgfbr2 or Smad2 in collecting duct cells, driven by Ksp-Cre, worsened UUO-induced tubular damage and fibrosis, which paradoxically was associated with increased TGF-β availability [44, 45]. However, the global loss of Smad3 protected against UUO-induced fibrosis [46]. Thus, these seemingly contrasting results highlight the potential difference in the response to TGF-β inhibition between tubular segments (proximal versus distal), between acute and chronic injury, and potentially even between TGF-β signaling components (e.g., Smad2 vs. Smad3). Indeed, while often acting in concert, differential or even antagonistic roles between Smad2 and Smad3 have been observed in early development gene transcription by forkhead DNA-binding protein [47], cytostatic growth inhibition in epithelial cells [48], breast cancer cell metastasis [49], growth or migration properties in pancreatic adenocarcinoma cells [50], and vertebrate neurogenesis [51]. These context-specific opposing effects between Smad2 and Smad3 are thought to be driven by their differential recruitment of transcriptional co-factors and targeting of regulatory elements [52].
Podocytes
As a highly specialized epithelial cell type in the kidney, podocytes are an integral component of the glomerular filtration barrier, and their injury and loss are critical determinants in the development of glomerular diseases. Mechanisms of TGF-β-mediated podocyte injury, as delineated by in vitro studies, include epithelial-to-mesenchymal transition (EMT) and de-differentiation [53-55], cytoskeletal alterations that allow their detachment from the glomerular basement membrane [56], and apoptosis [57-59]. The glomerular phenotypes of transgenic mice with increased circulating TGF-β1 included progressive glomerulosclerosis marked by reduced Wilms' tumor 1 (WT1) expression and podocyte apoptosis [31, 55, 57] . In line with these observations, the induction of podocyte-specific overexpression of constitutively active TGF-β type I receptor (Tgfbr1) in mice induces podocytopathy and glomerular disease development, eventually leading to kidney failure [60]. The podocyte damage caused in the transgenic mice also resulted in significant mitochondrial dysfunction and oxidative stress damage in the neighboring glomerular endothelial cells (GECs), mediated by podocyte-derived endothelin 1 (EDN1). Notably, mitigating the mitochondrial injury in GECs also attenuated TGFBR1-induced podocyte loss in transgenic mice, underscoring the intricate involvement of crosstalk between signaling pathways and between glomerular cells and in kidney disease development.
In the context of diabetic kidney disease (DKD), the study by Hathaway et al. demonstrated the disease-promoting effects of increased TGF-β1 and the disease-preventing effects of TGF-β1 suppression [39]. Using a murine model of graded expression of Tgfb1 (10% hypomorph to 300% hypermorph) in type 1 diabetic Akita mice, the authors demonstrated that the global suppression of TGF-β1 levels prevents DKD development, while increased expression markedly exacerbates it akin to advanced human DKD [39]. They also showed that the podocyte-specific TGF-β1 increase markedly worsened diabetes-induced mesangial matrix expansion, GBM thickening, podocyte foot process effacement, and kidney function impairment in Akita mice. Podocyte-specific deletion of the negative modulator of TGF-β signaling, BAMBI, similarly accentuated podocyte loss and worsened diabetic glomerulopathy in type 1 diabetic eNOS-deficient mice [61]. Conversely, conditional deletion of Smad4 in podocytes attenuated DKD in diabetic eNOS-deficient mice [62], and global Smad3 loss also protected from podocyte injury caused by high fat diet-induced obesity in mice [63], further corroborating the importance of podocyte TGF-β signaling in promoting diabetic glomerulopathy.
1.2.2. Endothelial cells
Glomerular angiogenesis, hypertrophy, and hyperfiltration are manifestations of early DKD and are associated with disease pathogenesis [64]. Recent experimental evidence implicates TGF-β to be among the vascular factors promoting glomerular angiogenesis and endothelial dysfunction in early diabetic kidneys [61, 65, 66]. Indeed, the global loss of type 1 TGF-β receptors (TGFBR1 or ALK1) or type 2 receptor (TGFBR2) in mice leads to early embryonic lethality due to severe vasculature development defects [67-69]. In vitro studies have delineated that TGF-β signaling can induce either pro- or anti-angiogenic effects based on the intricate balance between the opposing actions of ALK1 and ALK5, such that ALK1-mediated activation Smad1/5 promotes endothelial proliferation and migration to promote angiogenesis, while ALK5-mediated Smad2/3 activation promotes endothelial quiescence or apoptosis in specific contexts [70-73] (Figure 1B). The discriminate activation of ALK1 versus ALK5 in endothelial cells by TGF-β is contextually determined in part by cell-specific expression of co-receptors and extracellular signal modulators, such as co-receptor endoglin and secreted molecule LRG1 that preferentially engage with TGF-β and ALK1 complex [74-76]. Moreover, although ALK1-Smad1/5 can antagonize ALK5-Smad2/3 signaling, ALK5 activity is also required for optimal ALK1 activity in endothelial cells in vitro [77], suggesting a highly dynamic and integrated regulation of endothelial TGF-β signaling in vivo. Therefore, it is not surprising that the activation of both Smad1/5 and Smad2/3 are increased in GECs of diabetic mice [65]. Endothelial-specific loss of BAMBI, a negative modulator of TGF-β signaling, resulted in aggravated diabetic glomerulopathy in STZ-induced diabetic mice, marked by GEC proliferation and increased oxidative stress [61]. Similarly, a global loss of LRG1, a secreted glycoprotein whose expression is highest in GECs among endothelial cells in murine kidneys, reduced diabetes-induced GEC proliferation and oxidative stress injury to attenuate DKD [65, 66]. Notably, despite the increased number of GECs in diabetic mice, gene expression analysis indicated that proliferation and apoptosis were occurring concurrently and that both processes were attenuated by reducing TGF-β signaling [66, 78, 79]. Thus, it is likely that the balance of Smad1/5 and Smad2/3 pathways promotes diabetic GEC injury and contributes to DKD progression. As advanced kidney diseases are marked by capillary rarefaction, whether and how TGF-β signaling affects GEC loss in advanced DKD remains to be better elucidated.
TGF-β signaling is also implicated in endothelial-to-mesenchymal transition (EndMT) and peritubular capillary rarefaction, a common feature of progressive kidney disease and fibrosis progression. The induction of constitutively active TGFBR1 expression in endothelial cells in transgenic mice triggered cutaneous, visceral, and microvascular fibrosis, including in the kidney [80]. Conversely, partial ablation of pan-endothelial Tgfbr2 attenuated interstitial fibrosis following folic acid- and UUO-induced kidney injury in mice, which was associated with reduced EndMT and improved microvascular density and patency, and with reduced Smad2 activation but enhanced Smad1/5 activation in peritubular capillary cells [81]. Similarly, the inhibition of Smad3 in streptozotocin-induced diabetic mice reduced EndMT of peritubular capillaries and interstitial matrix deposition [82]. Thus, these findings support the notion that the overactive TGF-β signaling in injured kidneys promotes EndMT and peritubular capillary rarefaction mediated by Smad2/3 activation to promote inflammation and fibrosis development in CKD. Interestingly, ALK1 haploinsufficiency worsened UUO-induced tubulointerstitial fibrosis in mice [83] and was characterized by reduced Smad1/5 signaling in cells expressing myofibroblast markers (α-SMA and S100A4). However, as global ALK1 heterozygous null mice were used in the study and multiple cell types can express these markers in injured kidneys, further validations are required to elucidate the role of TGF-β/ALK1 specifically in the regulation of peritubular capillary stability in response to kidney injury in vivo. Similar to the context of vasculature development, temporally regulated ALK1 and ALK5 signaling are both likely required in peritubular capillaries for the appropriate response to kidney injury and fibrosis development.
1.2.3. Mesangial cells and myofibroblasts
Mesangial cells
Derived from stromal mesenchyme, the mesangial cells exhibit overlapping features of various stromal cells, such as pericytes, fibroblasts, and vascular smooth muscle cells [84, 85]. As such, in disease states, mesangial cell activation and matrix expansion orchestrate the development of glomerulosclerosis [86]. TGF-β is a potent regulator of mesangial matrix synthesis and degradation, and mice with increased TGF-β expression or glomerular signaling invariably develop glomerulosclerosis with evident mesangial expansion [31, 39, 60]. In the context of diabetic kidneys, in vitro studies showed that high glucose conditions can induce mesangial proliferation and further induce TGF-β expression [87-89], and in vivo administration of anti-TGF-β antibody or the interference with canonical TGF-β signaling (by overexpression of inhibitory Smad7, Smad3 loss, or reduction in TGFBR2) attenuates mesangial matrix expansion and diabetic glomerulopathy in mice [90-93]. However, TGF-β can also confer cytoprotection of mesangial cells against cellular insults through TAK1 and p38 activation and autophagy induction [94]. TGF-β-mediated autophagy also participated in collagen I degradation, highlighting the importance of specificity in TGF-β signaling mediators in modulating biological outcomes.
Myofibroblasts
Myofibroblasts are the central matrix-producing cells in the kidney for wound healing and fibrosis development. Various cellular sources are indicated in the origin of activated myofibroblasts in kidney disease, including kidney resident fibroblasts, pericytes, bone marrow-derived mesenchymal cells, macrophages, tubular epithelial cells, and endothelial cells [95, 96]. Among these, several studies indicate that fibroblasts and pericytes are the major sources from which matrix-producing myofibroblasts originate [97-99]. TGF-β is the principal initiator of matrix accumulation by the myofibroblasts, and its interference is strongly associated with a reduction in interstitial fibrosis in experimental models. For instance, the genetic ablation of Tgfbr2 in fibroblasts, driven by α-SMA-Cre or PDGFR-β-Cre, attenuated UUO-induced kidney fibrosis in mice [100, 101]. Similarly, Smad2 ablation in fibroblasts by FSP1-Cre reduced interstitial fibrosis in diabetic mice, which was associated with reduced Smad3 and TGF-β levels [102]. However, some experimental evidence indicates that TGF-β blockade in interstitial cells may not sufficiently reduce kidney fibrosis development [103] and that collagen accumulation is a necessary reparative response against kidney injury [104]. While further clarity is required to elucidate the discrepant outcomes between experimental models, they also underscore the necessary role of myofibroblast activation as an adaptive process in wound healing response to acute kidney injury, while persistent and uncontrolled myofibroblast activation promotes deleterious fibrosis development in the setting of ongoing tubular insult and vascular rarefaction [105-107].
1.2.4. Immune cells
Early studies have demonstrated a strong immunomodulatory role of TGF-β in mice lacking TGF-β1, which manifested multiorgan inflammation phenotype reminiscent of autoimmune disorder [7, 8]. Many studies have since demonstrated the wide-ranging effects of TGF-β on various facets of the innate and adaptive immune system [108, 109]. The effects of TGF-β's actions on immune dynamics in kidney disease are only briefly discussed here, but the readers are referred to an excellent recent review that covers this area [110].
Akin to the myofibroblast activation discussed above, the initiation of the inflammatory response is also triggered as a reparative process during the early phases of kidney injury [111, 112]. However, its persistent and maladaptive inflammatory response becomes a pathogenic component driving kidney disease and fibrosis development. Thus, it is not surprising that TGF-β regulation of immune cells is associated with both pro- and anti-inflammatory responses in kidney disease and fibrosis. For instance, an aspect of TGF-β's actions on monocytes is facilitating their recruitment to the site of injury and the production of pro-inflammatory mediators [108, 109]. The genetic deletion of Tgfbr2 in macrophages (by CD11b-Cre) reduced macrophage infiltration and ensuing fibrosis development after acute kidney injury in mice [113]. However, TGF-β also acts as an anti-inflammatory mediator by inhibiting the secretion of inflammatory molecules and promoting the alternatively activated M2 macrophage phenotype. In this manner, the adoptive transfer of macrophages modified ex vivo by IL-10/TGF-β ameliorated adriamycin-induced nephropathy in mice attenuated renal inflammation, which was associated with decreased CD4+ T cell proliferation and infiltration and increased regulatory T cells [114, 115]. This is consistent with several studies demonstrating the renoprotective function of M2 macrophages in various kidney disease models [116-118]. In the context of kidney fibrosis, some evidence also indicates that a specific subset of macrophages may contribute to the matrix-producing myofibroblast pool through the macrophage-to-mesenchymal transition [119-122]. In addition to macrophage/monocytes, the pleiotropic effects of TGF-β are evidenced by its regulation of lymphocytes, dendritic cells, natural killer cells, mast cells, and granulocytes to influence the initiation and resolution of inflammatory responses and immune surveillance [108, 109]. An active area of research in anti-TGF-β therapy is being pursued in oncology to restore the antitumor immune response in cancer [9, 10].
In the experimental models of anti-glomerular basement membrane glomerulonephritis (anti-GBM GN), conflicting results have been observed with the blockade of TGF-β signaling in vivo. In the study by Zhou et al., the administration of the soluble extracellular domain of TGFBR2 improved renal function and ameliorated crescent formation and interstitial fibrosis [123], whereas anti-TGF-β antibody administration exacerbated the disease in the study by Mesnard et al. [124]. Moreover, overexpression of the latent form of TGF-β in mice also attenuated anti-GBM GN [125]. These seemingly contradictory findings are likely due to the complex role of TGF-β in multiple cell types, including immune cells in CKD.
2. Targeting TGF-β signaling in kidney disease and fibrosis: past and current approaches
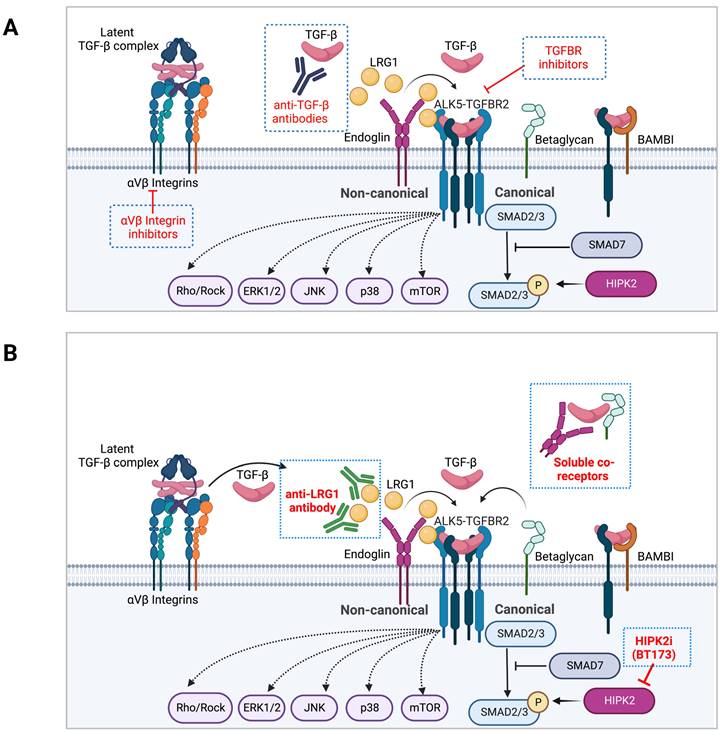
As TGF-β exerts a wide range of cellular effects in various organ systems, the therapeutic modalities to dampen its effects in CKD would need to consider the balanced duality of its homeostatic and pathogenic functions of TGF-β. Drugs targeting the TGF-β pathway comprise those that inhibit its biosynthesis, activation from latency, ligand and receptor binding, receptor kinase activity, and downstream canonical and non-canonical signaling mediators. The recent advancements in anti-TGF-β therapies include function-blocking monoclonal antibodies, ligand traps, small molecule inhibitors, and antisense oligonucleotides (AONs), all currently in clinical evaluation for cancer therapy [9, 10]. In this section, various approaches and modalities of TGF-β interventions in kidney disease and fibrosis development are discussed (Figure 2A).
Regulation of TGF-β signaling modulators as an alternative therapeutic approach in CKD. A. Examples of current approaches in TGF-β signaling inhibitors are shown. B. Inhibitors of TGF-β signaling modulators are shown on the left as an approach to dampen the hyperactive TGF-β signaling in disease settings without a complete blockade.
2.1 Targeting TGF- β1 activation from latency
As TGF-β is secreted in its latent form and bound to latent TGF-β binding proteins (LTBPs), its release from its latent complex via proteolytic cleavage, mechanical forces applied by the ECM, and integrin-mediated release provides additional layers of spatiotemporal control of TGF-β signaling. In this process, αv-containing integrins (αvβ1, αvβ3, αvβ5, αvβ6, and αvβ8) that selectively bind to the Arg-Gly-Asp (RGD) motif have emerged as a crucial regulator of TGF-β activation in vivo [126]. Pan-αv integrin small molecule inhibitors (CWHM-12/CWHM-680) were shown to attenuate renal fibrosis in mice (CWHM-12 in obstructed kidneys; CWHM-680 in aristolochic acid nephropathy) [127, 128]. However, as αv integrins are expressed in many cell types with wide-ranging functions, molecules targeting pan-αv integrin heterodimers may be susceptible to off-target effects [129]. Inhibitors of specific αv heterodimers showed efficacy of TGF-β inhibition and disease progression in several experimental models. Small molecule inhibitor of αvβ1 integrin, compound 8, reduced UUO- and adenine-mediated renal fibrosis development in mice [130], and αvβ3 inhibitor MK-0429, originally developed for osteoporosis, attenuated albuminuria and renal fibrosis in ZSF1 diabetic rat model [131]. Inhibition of αvβ6 with a function-blocking monoclonal antibody attenuated renal fibrosis and inflammation in Col4a3-deficient Alport syndrome mice [132]. Consistent with these findings, the genetic ablation of Itgb6 protected against renal fibrosis in Col4a3-deficient mice and mice with obstructed kidneys [132, 133]. However, Itgb6 loss in mice also leads to exaggerated lung and skin inflammation and the development of emphysema [134, 135], highlighting the physiological importance of integrins in immune modulation. Importantly, the partial inhibition of αvβ6 at a lower dose of monoclonal antibody attenuated bleomycin-induced pulmonary fibrosis in mice without exacerbating inflammation [130]. No clinical outcome data yet exists for αv integrin inhibitor in kidney disease, as the phase II study of humanized monoclonal anti-αvβ6 antibody STX-100 (Stromedix) in chronic allograft was withdrawn (NCT0087876), and the results for phase II study of monoclonal anti- αvβ3 antibody VPI-2690B (Vascular Pharmaceuticals) for in diabetic nephropathy (NCT02251067) have not been reported.
A recent study demonstrated that a monoclonal antibody, LTBP-49247, which binds selectively to TGF-β1 in latent complex with LTBPs, reduces renal fibrosis in Col4a3-null mouse model of Alport syndrome and the rat adenine model of kidney disease [136]. The antibody was shown to not bind to other TGF-β isoforms, free LTBPs, or GARP- or LRCC33- bound TGF-β1 complex. As Tgfb2 and Tgfb3 knockout mice have cardiovascular development defects, whereas Tgfb1-null mice are born normally, and latent TGF-β1 are bound to GARP/LRCC33 as a latent form in immune cells, specific blockade of LTBP-bound TGF-β1 may be better tolerated.
2.2. Targeting TGF-β ligand
Active TGF-β signaling can further augment its signaling by autoinduction and increasing its transcript expression [137, 138], leading to a vicious cycle of signal amplification in pathogenic settings. To block the TGF-β signaling at the level of its biosynthesis, antisense oligonucleotides (AONs) targeting the translation initiation of Tgfb1 mRNA have been used to repress ECM accumulation in rodent models of anti-Thy1 GN, diabetic nephropathy (DN), and UUO-mediated interstitial fibrosis [139-141]. Pirfenidone, an anti-inflammatory and anti-fibrotic small molecule approved for the treatment of idiopathic pulmonary fibrosis (IPF), is thought to exert its effects in part by suppressing TGF-β production, although its mechanism of action is not entirely understood. Phase 2 trial is ongoing to assess the efficacy of pirfenidone in preventing CKD progression (NCT04258397).
To block the engagement of TGF-β from binding and activating its cognate receptors, ligand traps have been utilized by the introduction of soluble forms of TGF-β-interacting proteins and monoclonal antibodies against TGF-β. The fusion protein consisting of TGFBR2 ectodomain and human immunoglobulin Fc domain was utilized and shown to attenuate the development of kidney disease in several experimental models that include anti-Thy1 GN, anti-GBM GN, diabetic nephropathy, and Alport syndrome [40, 123, 132, 142]. In addition, abundant pre-clinical evidence had also emerged to demonstrate the efficacy of neutralizing antibodies against TGF-β1 or all isoforms - their use attenuated kidney injury or fibrosis development in models of glomerulonephritis [143, 144], DKD in type 1 and type 2 diabetes [90, 145, 146], glomerular injury in salt-sensitive hypertension [147], puromycin aminonucleoside nephropathy (PAN)[148], UUO- or cyclosporin A-induced tubular injury and fibrosis [149-152], and chronic allograft rejection [153]. However, the efficacy of anti-TGF-β antibody Fresolimumab (1D11), a monoclonal antibody that recognizes all three TGF-β isoforms, was significantly diminished in its renoprotection if initiated during the advanced stages of DN in streptozotocin-injected rats [146]. In addition, in PAN rodent model, while a lower dose of 1D11 (0.5mg/kg) was effective in attenuating glomerulosclerosis and tubulointerstitial fibrosis, this effect was largely abrogated with a higher dose (5.0mg/kg) [148], suggesting a narrow therapeutic window. In clinical settings, although Fresolimumab(1D11) was well tolerated for the treatment of focal and segmental glomerulosclerosis (FSGS) [154], it failed to achieve the pre-specified efficacy endpoints for proteinuria reduction in a randomized, double-blind phase II clinical trial (NCT01665391) [5]. Similarly, the monoclonal antibody against TGF-β1, LY2382770, failed to show significant improvement in renal function and proteinuria in DN patients in the randomized double-blind phase II clinical trial (NCT11133801)[6]. The lack of success of TGF-β1 antibodies in these studies is likely multi-factorial, as even the experimental models mentioned above have demonstrated varying results based on the timing of the initiation and dosage required for optimal therapeutic intervention [146, 148].
2.3 Targeting TGF-β receptors
Experimental approaches to TGF-β receptor inhibition in kidney disease include small-molecule kinase inhibitors of ALK5 (TGFBR1) and function-blocking monoclonal antibodies against TGFBR2. Various ATP-competitive kinase inhibitors of ALK5 have shown efficacy in reducing renal fibrosis in rodent models. IN-1130 (IC50 of 5.3nM in inhibiting ALK5-mediated Smad3 phosphorylation) impeded the UUO-induced renal fibrosis development in rats [154], and R-268712 (IC50 of 2.5nM) attenuated glomerulosclerosis in anti-Thy1 nephritis and UUO-induced fibrosis in rats [155]. SB-431542, AZ12601011, and GW788388 are select kinase inhibitors of ALK4, ALK5, and ALK7 (IC50 is 94nM for SB-431542 and 18nM for AZ12601011 and GW788388 in ALK5 inhibition) [156-159]. Use of SB-431542 was shown to mitigate tubulointerstitial fibrosis in mouse UUO kidneys [150]; AZ12601011 in fibrosis development following UUO and ischemic reperfusion injury in rats [158]; and GW788388 in diabetes-induced renal fibrosis in db/db mice [160]. Vactosertib (TEW-7197, MedPacto) is a select kinase inhibitor of ALK2, ALK4, and ALK5 (IC50 of 11nM for ALK5), which demonstrated a favorable safety profile and antitumor efficacy in phase I trial for advanced refractory solid tumors [161, 162] (NCT02160106). Vactosertib reduced the UUO-induced tubulointerstitial fibrosis development in mice and attenuated diabetic glomerulopathy in db/db mice when treated for 10 weeks [163, 164]. Similarly, monoclonal antibodies against TGFBR2 as a broad TGFβ inhibition are actively pursued in various areas of cancer therapy. In the context of kidney fibrosis, a monoclonal antibody developed against TGFBR2 reduced mesangial and interstitial matrix accumulation in the model of anti-Thy1 nephritis [165]. There are currently no active clinical studies of anti-TGFBR2 monoclonal antibodies for kidney disease.
However, targeting the TGF-β ligands or receptors that result in pan-TGF-β inhibition requires considerable caution in the long-term treatment of chronic diseases, such as CKD, as circumventing the potential side effects remains a significant hurdle to overcome. For instance, the anti-TGFBR2 monoclonal antibody (LY3022859, Eli Lilly) therapy for advanced solid tumors in the phase I trial was hampered in assessing the maximum tolerated dose for efficacy beyond 25mg dose level due to the uncontrolled cytokine release, despite the prophylactic therapy with corticosteroid and antihistamine (NCT01646203) [166]. Treatment with the pan-TGF-β antibody, fresolimumab, was associated with the development of cutaneous carcinomas in phase I study of patients with melanoma or renal cell carcinoma [167, 168]. In preclinical studies, long-term exposure to pan-TGF-β antibodies and small molecule inhibitors of ALK5, such as AZ12601011, demonstrated cardiovascular toxicity with heart valve thickening, inflammation, hemorrhage, and stromal hyperplasia [157, 169, 170]. These findings are consistent with the phenotype of mice with postnatal Tgfbr2 deletion in smooth muscle cells that displayed pronounced aortopathy [171, 172], indicating that the chronic and broad pharmacologic blockade of TGF-β can inadvertently lead to cardiovascular disease. Thus, balancing the therapeutic window and systemic off-target effects remains a significant challenge in pan-TGF-β inhibition for CKD.
2.4. Targeting the downstream TGF-β signaling mediators
Compared to inhibitors of TGF-β ligand and receptors, there are far fewer inhibitors of TGF-β signaling mediators in clinical testing or development. However, several experimental models have demonstrated the efficacy of Smad3 inhibition in attenuating kidney disease and ensuing fibrosis in various mouse models, consistent with findings of genetic ablation of Smad3 in different kidney cells, as discussed above. SIS3 is a purported Smad3-specific inhibitor, which reduced TGF-β-induced phosphorylation of Smad3 but not Smad2 in cultured fibroblasts [173]. However, a recent study using Smad2- and Smad3-null cell lines indicates that SIS3 may be a broader inhibitor of Smad2/3 complex [174]. In kidney injury in vivo, SIS3 delayed the development of diabetic nephropathy in streptozotocin-induced type 1 diabetic mice and type 2 diabetic db/db mice, and the inhibition of Smad3 in SIS3-treated db/db mouse kidneys was associated with increased Smad7 expression and suppression of NF-κB-mediated inflammation [82, 175]. SIS3 also attenuated high fat-induced proteinuria and podocyte injury [63] and suppressed UUO-induced tubulointerstitial fibrosis development [176], demonstrating the therapeutic efficacy of SIS3 in reducing kidney injury and fibrosis in experimental models. As systemic Smad3 loss is associated with impaired mucosal immunity, chronic infection, and metastatic colorectal cancer in mice [177-179], whether the long-term use of SIS3 would be associated with any undesired effects remains to be determined.
In addition to the canonical Smad-mediated pathways, TGF-β receptors also activate other non-canonical signaling pathways such as PI3-AKT and ERK/MAPK, albeit at lower levels than by receptor tyrosine kinases (RTKs), as well as stress-activated MAPKs, p38 MAPK and JNK [23]. Selective inhibitors of p38 or JNK (NPC31145, NPC31169, FR167653, and SB203580 for p38; SP600125, CC401 and CC930 for JNK) have been shown to reduce podocytes injury, inflammation and fibrosis in animal models of anti-glomerular basement membrane (GBM) glomerulonephritis, ureteral obstruction, and diabetic nephropathy [180-187]. However, p38 MAPK inhibitors are adversely associated with hepatotoxicity, rash, dizziness and inhibition of erythropoietin production to affect erythropoiesis [188, 189]. Studies have shown that p38 MAPK deletion mice have apparent renal structural abnormalities, including proximal tubule dilation, vacuolar degeneration, focal interstitial fibrosis, and inflammation [190], whereas JNK1 and JNK2 double knockout leads to early embryonic lethality due to neural tube defects in mice [191].
Apoptosis signal-regulating kinase 1 (ASK1) is an upstream signaling kinase of p38 MAPK and JNK in kidney diseases [192]. Previous studies in animal models of kidney diseases have shown that ASK1 promotes p38 MAPK activation induced by oxidative stress, whereas ASK1 deficiency or ASK1-selective inhibition reduces p38 MAPK and JNK activation and improves kidney injury in mouse models of CKD and fibrosis [193-195]. The phase 2 clinical trial to evaluate the safety and efficacy of selonsertib, an ASK1 selective inhibitor, in patients with moderate to advanced diabetic nephropathy showed no dose-dependent adverse effects by 48 weeks, but the results did not meet the primary endpoint of change in eGFR [196]. A post hoc analysis nevertheless indicated that selonsertib slowed the kidney function decline after adjusting the confounding eGFR differences. A subsequent clinical trial evaluating the safety and efficacy of selonsertib in moderate to advanced DKD (MOSAIC, NCT04026165) demonstrated a slower eGFR decline compared to placebo but also raised potential safety concerns for acute kidney injury [197].
In addition to these cellular effectors of canonical and non-canonical signaling, various intracellular signaling mediators intersect with the TGF-β signaling pathway, thus serving as drug targets to intercept the TGF-β signaling. One such example is glycogen synthase kinase-3β (GSK-3β) [23], and recent evidence highlights a permissive effect of GSK-3β on the profibrogenic plasticity of renal tubular epithelial cells and its contribution to renal fibrogenesis in progressive CKD. Although initially identified as a regulator of glucose metabolism, GSK-3β is now understood to be a multi-functional kinase that impacts numerous biological processes. In cultured rodent tubular epithelial cells and fibroblasts, TGF-β1 can increase GSK-3β expression, and its increased expression and activity have been observed in fibrotic kidneys [198, 199]. Pharmacological inhibition of GSK3 abolished TGF-β1-induced Smad3 activation in vitro and reduced kidney fibrosis in animal models of ischemia-reperfusion and folic acid-induced injuries [198, 199]. However, currently existing inhibitors of GSK-3 have poor GSK-3β selectivity and suboptimal potency and are associated with chronic toxicities [200]. Notably, several in vivo studies, as described above, indicated that micro-dosed lithium (one-third to one-half of neurobiological dose) bypasses the potential neurological or nephrotoxicity associated with higher doses of lithium but is still effective at conferring renoprotection against AKI and glomerular diseases in mice [199, 201-203]. These results strongly indicate that lithium may be safely repurposed as a pragmatic and affordable treatment for diverse types of CKD [204].
3. Targeting the modulators of TGF-β signaling as alternative approaches in CKD
Although working through a common set of receptors and a limited number of intracellular effector proteins, TGF-β signaling can achieve wide-ranging cellular and biological effects in organismal development, tissue homeostasis, and disease pathogenesis. Such multifaceted repertoire of signaling capacity and its potency relies on the cell type and context-specific expression of modifiers and regulators at various steps of the signaling cascade, such as 1) regulators involved in the activation of latent TGF-β complex, 2) cell surface regulators of TGF-β and receptor complex formation, 3) regulators of Smad activation and nuclear translocation, 4) transcription factors/co-regulators that interact with Smad protein to dictate context-specific gene expression, and 5) mediators that integrate crosstalk with other cellular signaling pathways. Furthermore, TGF-β-induced genes can provide a feedback loop to either amplify or limit the magnitude and duration of the signaling output. Importantly, select modifiers can significantly amplify or limit the magnitude and duration of TGF-β signaling output in disease settings, but their loss is not detrimental at tissue or organismal levels, in contrast to the loss of TGF-β ligands or receptors that lead to severe developmental or postnatal defects. Therefore, targeting such regulators may dampen the hyperactive TGF-β signaling in disease settings to thwart the disease progression (Figure 2B).
3.1. Modulators of ligand-receptor interaction
An early point of TGF-β signaling regulation occurs by the cell surface molecules that exert context-dependent augmentation or inhibition of the active TGF-β ligand binding with its cognate receptors, such as auxiliary TGF-β co-receptors (e.g., betaglycan, endoglin, and BAMBI), small leucine-rich proteoglycans (SLRPs) that can bind and sequester TGF-β ligands (e.g., biglycan and decorin), and secreted molecules that participate in TGF-β and receptor engagement (e.g., connective tissue growth factor and LRG1). A considerable influence of the above cell surface signaling modifiers on the TGF-β signaling in disease pathogenesis has been demonstrated in various models of kidney injury and disease. As SLRPs and pseudo-receptor BAMBI restrain TGF-β signaling by binding to and saturating out active TGF-β ligands, their loss markedly worsens renal fibrosis and diabetic nephropathy progression in mice [61, 205-208]. Conversely, the increased soluble form of recombinant decorin attenuated the glomerular matrix accumulation in the rat model of anti-Thy1 nephritis [209, 210], and soluble recombinant betaglycan also limited the progression of diabetic nephropathy in db/db mice [211]. However, the genetic ablation of co-receptors, endoglin and betaglycan, are also associated with detrimental defects in mice [212-215], and targeting the soluble co-receptors or SLRPs may face challenges in fine-tuning the hyperactive TGF-β signaling without a complete blockade, similarly as with soluble TGFR2 or neutralizing TGF-β antibodies that are associated with adverse toxicities.
The expression of secreted factors that regulate TGF-β signaling, such as connective tissue growth factor (CTGF) and LRG1, are often elevated in pathological conditions such as atherosclerosis, inflammatory conditions, fibrosis, and various forms of cancer [216, 217]. CTGF itself is a gene induced by TGF-β signaling, and it exerts a broad range of biological influences beyond its involvement in TGF-β ligand recruitment [220]. LRG1, as mentioned above, interacts with both type 1 and 2 receptors and co-receptor endoglin to facilitate their complex formation to enhance downstream signaling [76]. Notably, elevated levels of circulating CTGF and LRG1 levels are both associated with worse renal outcomes in DKD patients [65, 218, 219], and are causal in kidney disease pathogenesis. Thus, podocyte-specific CTGF overexpression worsened diabetic nephropathy [222], whereas reducing CTGF expression by specific antisense oligonucleotides (ASO) improved kidney function and reduced the mesangial matrix expansion in mouse models of type 1 and 2 diabetes [221]. However, CTGF inhibition may not be limited to TGF-β signaling attenuation, as it also exerts a broad range of biological influences, and its global knockout results in early perinatal lethality in mice with defective organ development and respiratory failure [212, 223]. In contrast, the global LRG1 expression does not affect embryonic development or survival in adult mice, but is effective in curtailing the progression of DKD [65, 66] and tubulointerstitial fibrosis in vivo [38]. Recently, LRG1-neutralizing antibody, Magacizumab, was shown to curtail TGF-β-mediated retinal vascular leakage and for anti-tumor activity in mice [224-226]. Thus, it remains to be determined whether LRG1 blockade would be of greater utility than pan-TGF-β inhibition, particularly in kidney disease settings.
3.2 HIPK2, a modulator of TGF-β/Smad3 signaling potency
Downstream of the receptor activation, a substantial influence of TGF-β/Smad3 signaling is exerted by homeodomain interacting protein 2 (HIPK2) [227, 228]. In addition to potentiation of TGF-β signaling, HIPK2 acts as a multifunctional regulator of diverse signaling pathways that include Wnt/β-catenin, Notch, p53, and Hippo signaling [227, 229-233]. While HIPK2 protein expression is low at basal levels in the adult kidney, its expression is significantly increased in CKD of various etiologies [228]. Genetic deletion of HIPK2 attenuated renal fibrosis development in various mouse models of renal fibrosis, which was associated with attenuation of TGF-β/Smad3-, Wnt/β-catenin-, and Notch-targeted genes [228, 234], and conversely, its overexpression results in exacerbated kidney injuries in experimental models of CKD and tubulointerstitial fibrosis [234-237]. Notably, Liu et al. reported the development of a small molecule inhibitor of HIPK2, BT173, that allosterically blocks its interaction with Smad3 without altering its kinase activity [238]. BT173 effectively reduced UUO- and HIV-induced renal fibrosis in vivo, which was associated with reduced Smad3 phosphorylation and repression of TGF-β-responsive matrix gene expression [238]. More recently, BT173 analogs have been further developed for optimal physicochemical properties for clinical testing, which reduced renal fibrosis in mouse CKD models of HIVAN and Alport syndrome [239, 240]. Importantly, similar to LRG1 knockout, global loss of HIPK2 does not result in developmental or immune deregulation phenotypes observed in knockout mice lacking major TGF-β signaling components. Thus, targeting HIPK2-Smad3 interaction with allosteric HIPK2 inhibitors, such as BT173, may effectively mitigate kidney fibrosis without a systemic TGF-β blockade as an alternative therapeutic strategy.
Conclusions and prospects
Although much progress has been made in understanding TGF-β signaling in various disease contexts in the past two decades, the cell type- and disease context-specific determinants of TGF-β signaling that help define its diversity, versatility, and complexity in biological output remain to be better understood. Thus, despite the obvious potential of therapeutic targeting of TGF-β in organ fibrosis, the clinical approach to optimize the therapeutic window while minimizing the adverse effects remains a significant challenge. Nevertheless, these challenges should not preclude the remaining potential of its targeting as a fibrosis therapy but underscore a need for alternative approaches for improved specificity. In CKD settings, specific antagonisms of TGF-β signaling modulators have demonstrated effectiveness as therapeutic targets, such as LRG1 and HIPK2. As these modulators function to tip the balance toward excessive pathological signaling, their blockade is an attractive option to attenuate CKD progression and fibrosis without a complete blockade of TGF-β signaling. Therefore, future therapies aimed toward an effective combination of strategies to block the modulators of TGF-β signaling potency and duration may lead to the more successful generation of new CKD and renal fibrosis treatments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kovesdy CP. Epidemiology of chronic kidney disease: an update 2022. Kidney Int Suppl (2011). 2022;12:7-11
2. Liyanage T, Ninomiya T, Jha V, Neal B, Patrice HM, Okpechi I. et al. Worldwide access to treatment for end-stage kidney disease: a systematic review. Lancet. 2015;385:1975-82
3. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325-38
4. Li L, Fu H, Liu Y. The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat Rev Nephrol. 2022;18:545-57
5. Vincenti F, Fervenza FC, Campbell KN, Diaz M, Gesualdo L, Nelson P. et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int Rep. 2017;2:800-10
6. Voelker J, Berg PH, Sheetz M, Duffin K, Shen T, Moser B. et al. Anti-TGF-beta1 Antibody Therapy in Patients with Diabetic Nephropathy. J Am Soc Nephrol. 2017;28:953-62
7. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M. et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693-9
8. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC. et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770-4
9. Colak S, Ten Dijke P. Targeting TGF-beta Signaling in Cancer. Trends Cancer. 2017;3:56-71
10. Tauriello DVF, Sancho E, Batlle E. Overcoming TGFbeta-mediated immune evasion in cancer. Nat Rev Cancer. 2022;22:25-44
11. Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20:556-67
12. Plisov SY, Yoshino K, Dove LF, Higinbotham KG, Rubin JS, Perantoni AO. TGF beta 2, LIF and FGF2 cooperate to induce nephrogenesis. Development. 2001;128:1045-57
13. Bush KT, Sakurai H, Steer DL, Leonard MO, Sampogna RV, Meyer TN. et al. TGF-beta superfamily members modulate growth, branching, shaping, and patterning of the ureteric bud. Dev Biol. 2004;266:285-98
14. Sims-Lucas S, Caruana G, Dowling J, Kett MM, Bertram JF. Augmented and accelerated nephrogenesis in TGF-beta2 heterozygous mutant mice. Pediatr Res. 2008;63:607-12
15. Kim SI, Lee SY, Wang Z, Ding Y, Haque N, Zhang J. et al. TGF-beta-activated kinase 1 is crucial in podocyte differentiation and glomerular capillary formation. J Am Soc Nephrol. 2014;25:1966-78
16. Rowan CJ, Li W, Martirosyan H, Erwood S, Hu D, Kim YK. et al. Hedgehog-GLI signaling in Foxd1-positive stromal cells promotes murine nephrogenesis via TGFbeta signaling. Development. 2018;145:de159947
17. Hinck AP, Mueller TD, Springer TA. Structural Biology and Evolution of the TGF-beta Family. Cold Spring Harb Perspect Biol. 2016;8:a022103
18. Ito Y, Goldschmeding R, Kasuga H, Claessen N, Nakayama M, Yuzawa Y. et al. Expression patterns of connective tissue growth factor and of TGF-beta isoforms during glomerular injury recapitulate glomerulogenesis. Am J Physiol Renal Physiol. 2010;299:F545-58
19. Yamamoto T, Noble NA, Cohen AH, Nast CC, Hishida A, Gold LI. et al. Expression of transforming growth factor-beta isoforms in human glomerular diseases. Kidney Int. 1996;49:461-9
20. Yu L, Border WA, Huang Y, Noble NA. TGF-beta isoforms in renal fibrogenesis. Kidney Int. 2003;64:844-56
21. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T. et al. Latent TGF-beta structure and activation. Nature. 2011;474:343-9
22. Hill CS. Transcriptional Control by the SMADs. Cold Spring Harb Perspect Biol. 2016;8:a022079
23. Zhang YE. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb Perspect Biol. 2017;9:a022129
24. Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci. 2012;33:522-30
25. Yan X, Xiong X, Chen YG. Feedback regulation of TGF-beta signaling. Acta Biochim Biophys Sin (Shanghai). 2018;50:37-50
26. Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massague J. et al. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature. 1999;401:480-5
27. Yan X, Lin Z, Chen F, Zhao X, Chen H, Ning Y. et al. Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-beta signaling. J Biol Chem. 2009;284:30097-104
28. Yoshioka K, Takemura T, Murakami K, Okada M, Hino S, Miyamoto H. et al. Transforming growth factor-beta protein and mRNA in glomeruli in normal and diseased human kidneys. Lab Invest. 1993;68:154-63
29. Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci U S A. 1993;90:1814-8
30. Waldherr R, Noronha IL, Niemir Z, Kruger C, Stein H, Stumm G. Expression of cytokines and growth factors in human glomerulonephritides. Pediatr Nephrol. 1993;7:471-8
31. Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Bottinger EP. et al. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest. 1996;74:991-1003
32. Krag S, Osterby R, Chai Q, Nielsen CB, Hermans C, Wogensen L. TGF-beta1-induced glomerular disorder is associated with impaired concentrating ability mimicking primary glomerular disease with renal failure in man. Lab Invest. 2000;80:1855-68
33. Wogensen L, Nielsen CB, Hjorth P, Rasmussen LM, Nielsen AH, Gross K. et al. Under control of the Ren-1c promoter, locally produced transforming growth factor-beta1 induces accumulation of glomerular extracellular matrix in transgenic mice. Diabetes. 1999;48:182-92
34. Derynck R, Muthusamy BP, Saeteurn KY. Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Curr Opin Cell Biol. 2014;31:56-66
35. Koesters R, Kaissling B, Lehir M, Picard N, Theilig F, Gebhardt R. et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 2010;177:632-43
36. Gentle ME, Shi S, Daehn I, Zhang T, Qi H, Yu L. et al. Epithelial cell TGFbeta signaling induces acute tubular injury and interstitial inflammation. J Am Soc Nephrol. 2013;24:787-99
37. Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL. et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21:998-1009
38. Hong Q, Cai H, Zhang L, Li Z, Zhong F, Ni Z. et al. Modulation of transforming growth factor-beta-induced kidney fibrosis by leucine-rich ⍺-2 glycoprotein-1. Kidney Int. 2022;101:299-314
39. Hathaway CK, Gasim AM, Grant R, Chang AS, Kim HS, Madden VJ. et al. Low TGFbeta1 expression prevents and high expression exacerbates diabetic nephropathy in mice. Proc Natl Acad Sci U S A. 2015;112:5815-20
40. Russo LM, del Re E, Brown D, Lin HY. Evidence for a role of transforming growth factor (TGF)-beta1 in the induction of postglomerular albuminuria in diabetic nephropathy: amelioration by soluble TGF-beta type II receptor. Diabetes. 2007;56:380-8
41. Gewin L, Vadivelu S, Neelisetty S, Srichai MB, Paueksakon P, Pozzi A. et al. Deleting the TGF-beta receptor attenuates acute proximal tubule injury. J Am Soc Nephrol. 2012;23:2001-11
42. Nlandu-Khodo S, Neelisetty S, Phillips M, Manolopoulou M, Bhave G, May L. et al. Blocking TGF-beta and beta-Catenin Epithelial Crosstalk Exacerbates CKD. J Am Soc Nephrol. 2017;28:3490-503
43. Kayhan M, Vouillamoz J, Rodriguez DG, Bugarski M, Mitamura Y, Gschwend J. et al. Intrinsic TGF-beta signaling attenuates proximal tubule mitochondrial injury and inflammation in chronic kidney disease. Nat Commun. 2023;14:3236
44. Gewin L, Bulus N, Mernaugh G, Moeckel G, Harris RC, Moses HL. et al. TGF-beta receptor deletion in the renal collecting system exacerbates fibrosis. J Am Soc Nephrol. 2010;21:1334-43
45. Meng XM, Huang XR, Chung AC, Qin W, Shao X, Igarashi P. et al. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol. 2010;21:1477-87
46. Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486-94
47. Labbe E, Silvestri C, Hoodless PA, Wrana JL, Attisano L. Smad2 and Smad3 positively and negatively regulate TGF beta-dependent transcription through the forkhead DNA-binding protein FAST2. Mol Cell. 1998;2:109-20
48. Kim SG, Kim HA, Jong HS, Park JH, Kim NK, Hong SH. et al. The endogenous ratio of Smad2 and Smad3 influences the cytostatic function of Smad3. Mol Biol Cell. 2005;16:4672-83
49. Petersen M, Pardali E, van der Horst G, Cheung H, van den Hoogen C, van der Pluijm G. et al. Smad2 and Smad3 have opposing roles in breast cancer bone metastasis by differentially affecting tumor angiogenesis. Oncogene. 2010;29:1351-61
50. Ungefroren H, Groth S, Sebens S, Lehnert H, Gieseler F, Fandrich F. Differential roles of Smad2 and Smad3 in the regulation of TGF-beta1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: control by Rac1. Mol Cancer. 2011;10:67
51. Miguez DG, Gil-Guinon E, Pons S, Marti E. Smad2 and Smad3 cooperate and antagonize simultaneously in vertebrate neurogenesis. J Cell Sci. 2013;126:5335-43
52. Brown KA, Pietenpol JA, Moses HL. A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF-beta signaling. J Cell Biochem. 2007;101:9-33
53. Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol. 2008;172:299-308
54. Herman-Edelstein M, Thomas MC, Thallas-Bonke V, Saleem M, Cooper ME, Kantharidis P. Dedifferentiation of immortalized human podocytes in response to transforming growth factor-beta: a model for diabetic podocytopathy. Diabetes. 2011;60:1779-88
55. Sakairi T, Abe Y, Kopp JB. TGF-beta1 reduces Wilms' tumor suppressor gene expression in podocytes. Nephrol Dial Transplant. 2011;26:2746-52
56. Dessapt C, Baradez MO, Hayward A, Dei Cas A, Thomas SM, Viberti G. et al. Mechanical forces and TGFbeta1 reduce podocyte adhesion through alpha3beta1 integrin downregulation. Nephrol Dial Transplant. 2009;24:2645-55
57. Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P. et al. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807-16
58. Wada T, Pippin JW, Terada Y, Shankland SJ. The cyclin-dependent kinase inhibitor p21 is required for TGF-beta1-induced podocyte apoptosis. Kidney Int. 2005;68:1618-29
59. Wu DT, Bitzer M, Ju W, Mundel P, Bottinger EP. TGF-beta concentration specifies differential signaling profiles of growth arrest/differentiation and apoptosis in podocytes. J Am Soc Nephrol. 2005;16:3211-21
60. Daehn I, Casalena G, Zhang T, Shi S, Fenninger F, Barasch N. et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest. 2014;124:1608-21
61. Lai H, Chen A, Cai H, Fu J, Salem F, Li Y. et al. Podocyte and endothelial-specific elimination of BAMBI identifies differential transforming growth factor-beta pathways contributing to diabetic glomerulopathy. Kidney Int. 2020;98:601-14
62. Li J, Sun YBY, Chen W, Fan J, Li S, Qu X. et al. Smad4 promotes diabetic nephropathy by modulating glycolysis and OXPHOS. EMBO Rep. 2020;21:e48781
63. Sun YB, Qu X, Howard V, Dai L, Jiang X, Ren Y. et al. Smad3 deficiency protects mice from obesity-induced podocyte injury that precedes insulin resistance. Kidney Int. 2015;88:286-98
64. Mohandes S, Doke T, Hu H, Mukhi D, Dhillon P, Susztak K. Molecular pathways that drive diabetic kidney disease. J Clin Invest. 2023;133:e165654
65. Hong Q, Zhang L, Fu J, Verghese DA, Chauhan K, Nadkarni GN. et al. LRG1 Promotes Diabetic Kidney Disease Progression by Enhancing TGF-beta-Induced Angiogenesis. J Am Soc Nephrol. 2019;30:546-62
66. Wang X, Sun Z, Fu J, Fang Z, Zhang W, He JC. et al. LRG1 loss effectively restrains glomerular TGF-beta signaling to attenuate diabetic kidney disease. Mol Ther. 2024;32:3177-3193
67. Oshima M, Oshima H, Taketo MM. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol. 1996;179:297-302
68. Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet. 2000;26:328-31
69. Larsson J, Goumans MJ, Sjostrand LJ, van Rooijen MA, Ward D, Leveen P. et al. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001;20:1663-73
70. Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK. et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:2626-31
71. Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002;21:1743-53
72. Schulick AH, Taylor AJ, Zuo W, Qiu CB, Dong G, Woodward RN. et al. Overexpression of transforming growth factor beta1 in arterial endothelium causes hyperplasia, apoptosis, and cartilaginous metaplasia. Proc Natl Acad Sci U S A. 1998;95:6983-8
73. Leksa V, Godar S, Schiller HB, Fuertbauer E, Muhammad A, Slezakova K. et al. TGF-beta-induced apoptosis in endothelial cells mediated by M6P/IGFII-R and mini-plasminogen. J Cell Sci. 2005;118:4577-86
74. Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M. et al. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004;23:4018-28
75. ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nature reviews Molecular cell biology. 2007;8:857-69
76. Wang X, Abraham S, McKenzie JAG, Jeffs N, Swire M, Tripathi VB. et al. LRG1 promotes angiogenesis by modulating endothelial TGF-beta signalling. Nature. 2013;499:306-11
77. Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C. et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817-28
78. Fu J, Akat KM, Sun Z, Zhang W, Schlondorff D, Liu Z. et al. Single-Cell RNA Profiling of Glomerular Cells Shows Dynamic Changes in Experimental Diabetic Kidney Disease. J Am Soc Nephrol. 2019;30:533-45
79. Fu J, Wei C, Zhang W, Schlondorff D, Wu J, Cai M. et al. Gene expression profiles of glomerular endothelial cells support their role in the glomerulopathy of diabetic mice. Kidney Int. 2018;94:326-45
80. Wermuth PJ, Carney KR, Mendoza FA, Piera-Velazquez S, Jimenez SA. Endothelial cell-specific activation of transforming growth factor-beta signaling in mice induces cutaneous, visceral, and microvascular fibrosis. Lab Invest. 2017;97:806-18
81. Xavier S, Vasko R, Matsumoto K, Zullo JA, Chen R, Maizel J. et al. Curtailing endothelial TGF-beta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J Am Soc Nephrol. 2015;26:817-29
82. Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y. et al. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59:2612-24
83. Munoz-Felix JM, Lopez-Novoa JM, Martinez-Salgado C. Heterozygous disruption of activin receptor-like kinase 1 is associated with increased renal fibrosis in a mouse model of obstructive nephropathy. Kidney Int. 2014;85:319-32
84. Chung JJ, Goldstein L, Chen YJ, Lee J, Webster JD, Roose-Girma M. et al. Single-Cell Transcriptome Profiling of the Kidney Glomerulus Identifies Key Cell Types and Reactions to Injury. J Am Soc Nephrol. 2020;31:2341-54
85. Avraham S, Korin B, Chung JJ, Oxburgh L, Shaw AS. The Mesangial cell - the glomerular stromal cell. Nat Rev Nephrol. 2021;17:855-64
86. Schlondorff D, Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol. 2009;20:1179-87
87. Nahman NS Jr, Leonhart KL, Cosio FG, Hebert CL. Effects of high glucose on cellular proliferation and fibronectin production by cultured human mesangial cells. Kidney Int. 1992;41:396-402
88. Oh JH, Ha H, Yu MR, Lee HB. Sequential effects of high glucose on mesangial cell transforming growth factor-beta 1 and fibronectin synthesis. Kidney Int. 1998;54:1872-8
89. Kolm-Litty V, Tippmer S, Haring HU, Schleicher E. Glucosamine induces translocation of protein kinase C isoenzymes in mesangial cells. Exp Clin Endocrinol Diabetes. 1998;106:377-83
90. Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522-30
91. Kim HW, Kim BC, Song CY, Kim JH, Hong HK, Lee HS. Heterozygous mice for TGF-betaIIR gene are resistant to the progression of streptozotocin-induced diabetic nephropathy. Kidney Int. 2004;66:1859-65
92. Wang A, Ziyadeh FN, Lee EY, Pyagay PE, Sung SH, Sheardown SA. et al. Interference with TGF-beta signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol. 2007;293:F1657-65
93. Chen HY, Huang XR, Wang W, Li JH, Heuchel RL, Chung AC. et al. The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes. 2011;60:590-601
94. Ding Y, Kim JK, Kim SI, Na HJ, Jun SY, Lee SJ. et al. TGF-beta1 protects against mesangial cell apoptosis via induction of autophagy. J Biol Chem. 2010;285:37909-19
95. Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ. Diverse origins of the myofibroblast-implications for kidney fibrosis. Nat Rev Nephrol. 2015;11:233-44
96. Mack M, Yanagita M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015;87:297-307
97. Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617-27
98. Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV. et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85-97
99. Kuppe C, Ibrahim MM, Kranz J, Zhang X, Ziegler S, Perales-Paton J. et al. Decoding myofibroblast origins in human kidney fibrosis. Nature. 2021;589:281-6
100. LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C. et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047-53
101. Fuchs MAA, Broeker KAE, Schrankl J, Burzlaff N, Willam C, Wagner C. et al. Inhibition of transforming growth factor beta1 signaling in resident interstitial cells attenuates profibrotic gene expression and preserves erythropoietin production during experimental kidney fibrosis in mice. Kidney Int. 2021;100:122-37
102. Loeffler I, Liebisch M, Allert S, Kunisch E, Kinne RW, Wolf G. FSP1-specific SMAD2 knockout in renal tubular, endothelial, and interstitial cells reduces fibrosis and epithelial-to-mesenchymal transition in murine STZ-induced diabetic nephropathy. Cell Tissue Res. 2018;372:115-33
103. Neelisetty S, Alford C, Reynolds K, Woodbury L, Nlandu-Khodo S, Yang H. et al. Renal fibrosis is not reduced by blocking transforming growth factor-beta signaling in matrix-producing interstitial cells. Kidney Int. 2015;88:503-14
104. Buchtler S, Grill A, Hofmarksrichter S, Stockert P, Schiechl-Brachner G, Rodriguez Gomez M. et al. Cellular Origin and Functional Relevance of Collagen I Production in the Kidney. J Am Soc Nephrol. 2018;29:1859-73
105. Kaissling B, Lehir M, Kriz W. Renal epithelial injury and fibrosis. Biochim Biophys Acta. 2013;1832:931-9
106. Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J Am Soc Nephrol. 2015;26:1765-76
107. Arai H, Yanagita M. Janus-Faced: Molecular Mechanisms and Versatile Nature of Renal Fibrosis. Kidney360. 2020;1:697-704
108. Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annual review of immunology. 2014;32:51-82
109. Sanjabi S, Oh SA, Li MO. Regulation of the Immune Response by TGF-beta: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol. 2017;9:a022236
110. Tang PC, Chan AS, Zhang CB, Garcia Cordoba CA, Zhang YY, To KF. et al. TGF-beta1 Signaling: Immune Dynamics of Chronic Kidney Diseases. Front Med (Lausanne). 2021;8:628519
111. Meng XM, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol. 2014;10:493-503
112. Anders HJ. Immune system modulation of kidney regeneration-mechanisms and implications. Nat Rev Nephrol. 2014;10:347-58
113. Chung S, Overstreet JM, Li Y, Wang Y, Niu A, Wang S. et al. TGF-beta promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI insight. 2018;3:e123563
114. Cao Q, Wang Y, Zheng D, Sun Y, Wang Y, Lee VW. et al. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol. 2010;21:933-42
115. Lu J, Cao Q, Zheng D, Sun Y, Wang C, Yu X. et al. Discrete functions of M2a and M2c macrophage subsets determine their relative efficacy in treating chronic kidney disease. Kidney Int. 2013;84:745-55
116. Wang Y, Wang YP, Zheng G, Lee VW, Ouyang L, Chang DH. et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007;72:290-9
117. Zheng D, Wang Y, Cao Q, Lee VW, Zheng G, Sun Y. et al. Transfused macrophages ameliorate pancreatic and renal injury in murine diabetes mellitus. Nephron Exp Nephrol. 2011;118:e87-99
118. Cao Q, Wang C, Zheng D, Wang Y, Lee VW, Wang YM. et al. IL-25 induces M2 macrophages and reduces renal injury in proteinuric kidney disease. J Am Soc Nephrol. 2011;22:1229-39
119. Nikolic-Paterson DJ, Wang S, Lan HY. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int Suppl (2011). 2014;4:34-8
120. Wang S, Meng XM, Ng YY, Ma FY, Zhou S, Zhang Y. et al. TGF-beta/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget. 2016;7:8809-22
121. Meng XM, Wang S, Huang XR, Yang C, Xiao J, Zhang Y. et al. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016;7:e2495
122. Tang PM, Zhang YY, Xiao J, Tang PC, Chung JY, Li J. et al. Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage-myofibroblast transition. Proc Natl Acad Sci U S A. 2020;117:20741-52
123. Zhou A, Ueno H, Shimomura M, Tanaka R, Shirakawa T, Nakamura H. et al. Blockade of TGF-beta action ameliorates renal dysfunction and histologic progression in anti-GBM nephritis. Kidney Int. 2003;64:92-101
124. Mesnard L, Keller AC, Michel ML, Vandermeersch S, Rafat C, Letavernier E. et al. Invariant natural killer T cells and TGF-beta attenuate anti-GBM glomerulonephritis. J Am Soc Nephrol. 2009;20:1282-92
125. Huang XR, Chung AC, Zhou L, Wang XJ, Lan HY. Latent TGF-beta1 protects against crescentic glomerulonephritis. J Am Soc Nephrol. 2008;19:233-42
126. Worthington JJ, Klementowicz JE, Travis MA. TGFbeta: a sleeping giant awoken by integrins. Trends Biochem Sci. 2011;36:47-54
127. Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH. et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617-24
128. Basta J, Robbins L, Stout L, Prinsen MJ, Griggs DW, Rauchman M. Pharmacologic inhibition of RGD-binding integrins ameliorates fibrosis and improves function following kidney injury. Physiol Rep. 2020;8:e14329
129. Schnittert J, Bansal R, Storm G, Prakash J. Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Adv Drug Deliv Rev. 2018;129:37-53
130. Chang Y, Lau WL, Jo H, Tsujino K, Gewin L, Reed NI. et al. Pharmacologic Blockade of alphavbeta1 Integrin Ameliorates Renal Failure and Fibrosis In Vivo. J Am Soc Nephrol. 2017;28:1998-2005
131. Zhou X, Zhang J, Haimbach R, Zhu W, Mayer-Ezell R, Garcia-Calvo M. et al. An integrin antagonist (MK-0429) decreases proteinuria and renal fibrosis in the ZSF1 rat diabetic nephropathy model. Pharmacol Res Perspect. 2017;5:e00354
132. Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH. et al. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol. 2007;170:110-25
133. Ma LJ, Yang H, Gaspert A, Carlesso G, Barty MM, Davidson JM. et al. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6(-/-) mice. Am J Pathol. 2003;163:1261-73
134. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J. et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319-28
135. Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G. et al. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature. 2003;422:169-73
136. Jackson JW, Frederick CS Jr, Pal A, Coricor G, Boston C, Brueckner CT. et al. An antibody that inhibits TGF-beta1 release from latent extracellular matrix complexes attenuates the progression of renal fibrosis. Sci Signal. 2024;17:eadn6052
137. Van Obberghen-Schilling E, Roche NS, Flanders KC, Sporn MB, Roberts AB. Transforming growth factor beta 1 positively regulates its own expression in normal and transformed cells. J Biol Chem. 1988;263:7741-6
138. Kim SJ, Angel P, Lafyatis R, Hattori K, Kim KY, Sporn MB. et al. Autoinduction of transforming growth factor beta 1 is mediated by the AP-1 complex. Mol Cell Biol. 1990;10:1492-7
139. Akagi Y, Isaka Y, Arai M, Kaneko T, Takenaka M, Moriyama T. et al. Inhibition of TGF-beta 1 expression by antisense oligonucleotides suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 1996;50:148-55
140. Isaka Y, Tsujie M, Ando Y, Nakamura H, Kaneda Y, Imai E. et al. Transforming growth factor-beta 1 antisense oligodeoxynucleotides block interstitial fibrosis in unilateral ureteral obstruction. Kidney Int. 2000;58:1885-92
141. Han DC, Hoffman BB, Hong SW, Guo J, Ziyadeh FN. Therapy with antisense TGF-beta1 oligodeoxynucleotides reduces kidney weight and matrix mRNAs in diabetic mice. Am J Physiol Renal Physiol. 2000;278:F628-34
142. Isaka Y, Akagi Y, Ando Y, Tsujie M, Sudo T, Ohno N. et al. Gene therapy by transforming growth factor-beta receptor-IgG Fc chimera suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 1999;55:465-75
143. Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor beta 1. Nature. 1990;346:371-4
144. Fukasawa H, Yamamoto T, Suzuki H, Togawa A, Ohashi N, Fujigaki Y. et al. Treatment with anti-TGF-beta antibody ameliorates chronic progressive nephritis by inhibiting Smad/TGF-beta signaling. Kidney Int. 2004;65:63-74
145. Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M. et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97:8015-20
146. Benigni A, Zoja C, Campana M, Corna D, Sangalli F, Rottoli D. et al. Beneficial effect of TGFbeta antagonism in treating diabetic nephropathy depends on when treatment is started. Nephron Exp Nephrol. 2006;104:e158-68
147. Dahly-Vernon AJ, Sharma M, McCarthy ET, Savin VJ, Ledbetter SR, Roman RJ. Transforming growth factor-beta, 20-HETE interaction, and glomerular injury in Dahl salt-sensitive rats. Hypertension. 2005;45:643-8
148. Ma LJ, Jha S, Ling H, Pozzi A, Ledbetter S, Fogo AB. Divergent effects of low versus high dose anti-TGF-beta antibody in puromycin aminonucleoside nephropathy in rats. Kidney Int. 2004;65:106-15
149. Miyajima A, Chen J, Lawrence C, Ledbetter S, Soslow RA, Stern J. et al. Antibody to transforming growth factor-beta ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int. 2000;58:2301-13
150. Wu CF, Chiang WC, Lai CF, Chang FC, Chen YT, Chou YH. et al. Transforming growth factor beta-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am J Pathol. 2013;182:118-31
151. Islam M, Burke JF Jr, McGowan TA, Zhu Y, Dunn SR, McCue P. et al. Effect of anti-transforming growth factor-beta antibodies in cyclosporine-induced renal dysfunction. Kidney Int. 2001;59:498-506
152. Ling H, Li X, Jha S, Wang W, Karetskaya L, Pratt B. et al. Therapeutic role of TGF-beta-neutralizing antibody in mouse cyclosporin A nephropathy: morphologic improvement associated with functional preservation. J Am Soc Nephrol. 2003;14:377-88
153. Guan Q, Li S, Gao S, Chen H, Nguan CY, Du C. Reduction of chronic rejection of renal allografts by anti-transforming growth factor-beta antibody therapy in a rat model. Am J Physiol Renal Physiol. 2013;305:F199-207
154. Moon JA, Kim HT, Cho IS, Sheen YY, Kim DK. IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 2006;70:1234-43
155. Terashima H, Kato M, Ebisawa M, Kobayashi H, Suzuki K, Nezu Y. et al. R-268712, an orally active transforming growth factor-beta type I receptor inhibitor, prevents glomerular sclerosis in a Thy1 nephritis model. Eur J Pharmacol. 2014;734:60-6
156. Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD. et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65-74
157. Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, Powell S. et al. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol. 2011;39:916-24
158. Spender LC, Ferguson GJ, Hughes GD, Davies BR, Goldberg FW, Herrera B. et al. Preclinical Evaluation of AZ12601011 and AZ12799734, Inhibitors of Transforming Growth Factor beta Superfamily Type 1 Receptors. Mol Pharmacol. 2019;95:222-34
159. Gellibert F, de Gouville AC, Woolven J, Mathews N, Nguyen VL, Bertho-Ruault C. et al. Discovery of 4-4-[3-(pyridin-2-yl)-1H-pyrazol-4-yl]pyridin-2-yl-N-(tetrahydro-2H- pyran-4-yl)benzamide (GW788388): a potent, selective, and orally active transforming growth factor-beta type I receptor inhibitor. J Med Chem. 2006;49:2210-21
160. Petersen M, Thorikay M, Deckers M, van Dinther M, Grygielko ET, Gellibert F. et al. Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008;73:705-15
161. Jin CH, Krishnaiah M, Sreenu D, Subrahmanyam VB, Rao KS, Lee HJ. et al. Discovery of N-((4-([1,2,4]triazolo[1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)-2-fluoroaniline (EW-7197): a highly potent, selective, and orally bioavailable inhibitor of TGF-beta type I receptor kinase as cancer immunotherapeutic/antifibrotic agent. J Med Chem. 2014;57:4213-38
162. Jung SY, Hwang S, Clarke JM, Bauer TM, Keedy VL, Lee H. et al. Pharmacokinetic characteristics of vactosertib, a new activin receptor-like kinase 5 inhibitor, in patients with advanced solid tumors in a first-in-human phase 1 study. Invest New Drugs. 2020;38:812-20
163. Park SA, Kim MJ, Park SY, Kim JS, Lee SJ, Woo HA. et al. EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-beta/Smad and ROS signaling. Cellular and molecular life sciences: CMLS. 2015;72:2023-39
164. Ha KB, Sangartit W, Jeong AR, Lee ES, Kim HM, Shim S. et al. EW-7197 Attenuates the Progression of Diabetic Nephropathy in db/db Mice through Suppression of Fibrogenesis and Inflammation. Endocrinol Metab (Seoul). 2022;37:96-111
165. Kasuga H, Ito Y, Sakamoto S, Kawachi H, Shimizu F, Yuzawa Y. et al. Effects of anti-TGF-beta type II receptor antibody on experimental glomerulonephritis. Kidney Int. 2001;60:1745-55
166. Tolcher AW, Berlin JD, Cosaert J, Kauh J, Chan E, Piha-Paul SA. et al. A phase 1 study of anti-TGFbeta receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2017;79:673-80
167. Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M. et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PloS one. 2014;9:e90353
168. Lacouture ME, Morris JC, Lawrence DP, Tan AR, Olencki TE, Shapiro GI. et al. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor beta by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol Immunother. 2015;64:437-46
169. Mitra MS, Lancaster K, Adedeji AO, Palanisamy GS, Dave RA, Zhong F. et al. A Potent Pan-TGFbeta Neutralizing Monoclonal Antibody Elicits Cardiovascular Toxicity in Mice and Cynomolgus Monkeys. Toxicol Sci. 2020;175:24-34
170. Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST. et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015;9:4479-99
171. Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J. et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124:755-67
172. Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN. et al. Postnatal Deletion of the Type II Transforming Growth Factor-beta Receptor in Smooth Muscle Cells Causes Severe Aortopathy in Mice. Arterioscler Thromb Vasc Biol. 2015;35:2647-56
173. Jinnin M, Ihn H, Tamaki K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol Pharmacol. 2006;69:597-607
174. Itoh Y, Sawaguchi T, Fu H, Omata C, Saitoh M, Miyazawa K. Indole-derived compound SIS3 targets a subset of activated Smad complexes. J Biochem. 2023;173:283-91
175. He H, Wang H, Chen X, Zhong Y, Huang XR, Ma RC. et al. Treatment for type 2 diabetes and diabetic nephropathy by targeting Smad3 signaling. International journal of biological sciences. 2024;20:200-17
176. Zhang Y, Meng XM, Huang XR, Lan HY. The preventive and therapeutic implication for renal fibrosis by targetting TGF-beta/Smad3 signaling. Clin Sci (Lond). 2018;132:1403-15
177. Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703-14
178. Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H. et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18:1280-91
179. Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol Cell Biol. 1999;19:2495-504
180. Zhou J, Zhong J, Huang Z, Liao M, Lin S, Chen J. et al. TAK1 mediates apoptosis via p38 involve in ischemia-induced renal fibrosis. Artif Cells Nanomed Biotechnol. 2018;46:1016-25
181. Jin N, Wang Q, Zhang X, Jiang D, Cheng H, Zhu K. The selective p38 mitogen-activated protein kinase inhibitor, SB203580, improves renal disease in MRL/lpr mouse model of systemic lupus. Int Immunopharmacol. 2011;11:1319-26
182. Sugiyama N, Kohno M, Yokoyama T. Inhibition of the p38 MAPK pathway ameliorates renal fibrosis in an NPHP2 mouse model. Nephrol Dial Transplant. 2012;27:1351-8
183. Peerapen P, Thongboonkerd V. p38 MAPK mediates calcium oxalate crystal-induced tight junction disruption in distal renal tubular epithelial cells. Sci Rep. 2013;3:1041
184. de Borst MH, Prakash J, Sandovici M, Klok PA, Hamming I, Kok RJ. et al. c-Jun NH2-terminal kinase is crucially involved in renal tubulo-interstitial inflammation. J Pharmacol Exp Ther. 2009;331:896-905
185. Lim AK, Ma FY, Nikolic-Paterson DJ, Ozols E, Young MJ, Bennett BL. et al. Evaluation of JNK blockade as an early intervention treatment for type 1 diabetic nephropathy in hypertensive rats. Am J Nephrol. 2011;34:337-46
186. Ma FY, Flanc RS, Tesch GH, Han Y, Atkins RC, Bennett BL. et al. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J Am Soc Nephrol. 2007;18:472-84
187. Xue M, Sun H, Xu R, Wang Y, Guo J, Li X. et al. GADD45B Promotes Glucose-Induced Renal Tubular Epithelial-Mesenchymal Transition and Apoptosis via the p38 MAPK and JNK Signaling Pathways. Front Physiol. 2020;11:1074
188. Iwano S, Asaoka Y, Akiyama H, Takizawa S, Nobumasa H, Hashimoto H. et al. A possible mechanism for hepatotoxicity induced by BIRB-796, an orally active p38 mitogen-activated protein kinase inhibitor. J Appl Toxicol. 2011;31:671-7
189. Ma FY, Liu J, Nikolic-Paterson DJ. The role of stress-activated protein kinase signaling in renal pathophysiology. Braz J Med Biol Res. 2009;42:29-37
190. Maruyama M, Yagasaki Y, Sudo T, Osada H. Renal abnormalities in mice caused by insufficiency of p38alpha. J Recept Signal Transduct Res. 2003;23:173-83
191. Gerits N, Kostenko S, Moens U. In vivo functions of mitogen-activated protein kinases: conclusions from knock-in and knock-out mice. Transgenic Res. 2007;16:281-314
192. Tesch GH, Ma FY, Nikolic-Paterson DJ. ASK1: a new therapeutic target for kidney disease. Am J Physiol Renal Physiol. 2016;311:F373-81
193. Amos LA, Ma FY, Tesch GH, Liles JT, Breckenridge DG, Nikolic-Paterson DJ. et al. ASK1 inhibitor treatment suppresses p38/JNK signalling with reduced kidney inflammation and fibrosis in rat crescentic glomerulonephritis. J Cell Mol Med. 2018;22:4522-33
194. Ma FY, Tesch GH, Nikolic-Paterson DJ. ASK1/p38 signaling in renal tubular epithelial cells promotes renal fibrosis in the mouse obstructed kidney. Am J Physiol Renal Physiol. 2014;307:F1263-73
195. Chen A, Xu J, Lai H, D'Agati VD, Guan TJ, Badal S. et al. Inhibition of apoptosis signal-regulating kinase 1 mitigates the pathogenesis of human immunodeficiency virus-associated nephropathy. Nephrol Dial Transplant. 2021;36:430-41
196. Chertow GM, Pergola PE, Chen F, Kirby BJ, Sundy JS, Patel UD. et al. Effects of Selonsertib in Patients with Diabetic Kidney Disease. J Am Soc Nephrol. 2019;30:1980-90
197. Heerspink HJL, Perkovic V, Tuttle KR, Pergola PE, Mahaffey KW, Patel UD. et al. Selonsertib in Patients with Diabetic Kidney Disease: A Phase 2b Randomized Active Run-In Clinical Trial. J Am Soc Nephrol. 2024;35:1726-1736
198. Singh SP, Tao S, Fields TA, Webb S, Harris RC, Rao R. Glycogen synthase kinase-3 inhibition attenuates fibroblast activation and development of fibrosis following renal ischemia-reperfusion in mice. Dis Model Mech. 2015;8:931-40
199. Chen B, Wang P, Liang X, Jiang C, Ge Y, Dworkin LD. et al. Permissive effect of GSK3beta on profibrogenic plasticity of renal tubular cells in progressive chronic kidney disease. Cell Death Dis. 2021;12:432
200. Arciniegas Ruiz SM, Eldar-Finkelman H. Glycogen Synthase Kinase-3 Inhibitors: Preclinical and Clinical Focus on CNS-A Decade Onward. Front Mol Neurosci. 2021;14:792364
201. Fang Y, Chen B, Liu Z, Gong AY, Gunning WT, Ge Y. et al. Age-related GSK3beta overexpression drives podocyte senescence and glomerular aging. J Clin Invest. 2022;132:e141848
202. Lu M, Wang P, Qiao Y, Jiang C, Ge Y, Flickinger B. et al. GSK3beta-mediated Keap1-independent regulation of Nrf2 antioxidant response: A molecular rheostat of acute kidney injury to chronic kidney disease transition. Redox Biol. 2019;26:101275
203. Zhang J, Anshul F, Malhotra DK, Jaume J, Dworkin LD, Gong R. Microdose Lithium Protects against Pancreatic Islet Destruction and Renal Impairment in Streptozotocin-Elicited Diabetes. Antioxidants (Basel). 2021;10:138
204. Gong R, Wang P, Dworkin L. What we need to know about the effect of lithium on the kidney. Am J Physiol Renal Physiol. 2016;311:F1168-F71
205. Schaefer L, Macakova K, Raslik I, Micegova M, Grone HJ, Schonherr E. et al. Absence of decorin adversely influences tubulointerstitial fibrosis of the obstructed kidney by enhanced apoptosis and increased inflammatory reaction. Am J Pathol. 2002;160:1181-91
206. Schaefer L, Mihalik D, Babelova A, Krzyzankova M, Grone HJ, Iozzo RV. et al. Regulation of fibrillin-1 by biglycan and decorin is important for tissue preservation in the kidney during pressure-induced injury. Am J Pathol. 2004;165:383-96
207. Merline R, Lazaroski S, Babelova A, Tsalastra-Greul W, Pfeilschifter J, Schluter KD. et al. Decorin deficiency in diabetic mice: aggravation of nephropathy due to overexpression of profibrotic factors, enhanced apoptosis and mononuclear cell infiltration. J Physiol Pharmacol. 2009;60(Suppl 4):5-13
208. Fan Y, Li X, Xiao W, Fu J, Harris RC, Lindenmeyer M. et al. BAMBI elimination enhances alternative TGF-beta signaling and glomerular dysfunction in diabetic mice. Diabetes. 2015;64:2220-33
209. Border WA, Noble NA, Yamamoto T, Harper JR, Yamaguchi Y, Pierschbacher MD. et al. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature. 1992;360:361-4
210. Isaka Y, Brees DK, Ikegaya K, Kaneda Y, Imai E, Noble NA. et al. Gene therapy by skeletal muscle expression of decorin prevents fibrotic disease in rat kidney. Nat Med. 1996;2:418-23
211. Juarez P, Vilchis-Landeros MM, Ponce-Coria J, Mendoza V, Hernandez-Pando R, Bobadilla NA. et al. Soluble betaglycan reduces renal damage progression in db/db mice. Am J Physiol Renal Physiol. 2007;292:F321-9
212. Stenvers KL, Tursky ML, Harder KW, Kountouri N, Amatayakul-Chantler S, Grail D. et al. Heart and liver defects and reduced transforming growth factor beta2 sensitivity in transforming growth factor beta type III receptor-deficient embryos. Mol Cell Biol. 2003;23:4371-85
213. Walker KA, Sims-Lucas S, Caruana G, Cullen-McEwen L, Li J, Sarraj MA. et al. Betaglycan is required for the establishment of nephron endowment in the mouse. PloS one. 2011;6:e18723
214. Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343-51
215. Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG. et al. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534-7
216. Ramazani Y, Knops N, Elmonem MA, Nguyen TQ, Arcolino FO, van den Heuvel L. et al. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018;68-69:44-66
217. Camilli C, Hoeh AE, De Rossi G, Moss SE, Greenwood J. LRG1: an emerging player in disease pathogenesis. J Biomed Sci. 2022;29:6
218. Nguyen TQ, Tarnow L, Jorsal A, Oliver N, Roestenberg P, Ito Y. et al. Plasma connective tissue growth factor is an independent predictor of end-stage renal disease and mortality in type 1 diabetic nephropathy. Diabetes care. 2008;31:1177-82
219. Liu JJ, Pek SLT, Ang K, Tavintharan S, Lim SC, study SD. Plasma Leucine-Rich alpha-2-Glycoprotein 1 Predicts Rapid eGFR Decline and Albuminuria Progression in Type 2 Diabetes Mellitus. J Clin Endocrinol Metab. 2017;102:3683-91
220. Abreu JG, Ketpura NI, Reversade B, De Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol. 2002;4:599-604
221. Guha M, Xu ZG, Tung D, Lanting L, Natarajan R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007;21:3355-68
222. Yokoi H, Mukoyama M, Mori K, Kasahara M, Suganami T, Sawai K. et al. Overexpression of connective tissue growth factor in podocytes worsens diabetic nephropathy in mice. Kidney Int. 2008;73:446-55
223. Baguma-Nibasheka M, Kablar B. Pulmonary hypoplasia in the connective tissue growth factor (Ctgf) null mouse. Dev Dyn. 2008;237:485-93
224. Javaid F, Pilotti C, Camilli C, Kallenberg D, Bahou C, Blackburn J. et al. Leucine-rich alpha-2-glycoprotein 1 (LRG1) as a novel ADC target. RSC Chem Biol. 2021;2:1206-20
225. Kallenberg D, Tripathi V, Javaid F, Pilotti C, George J, Davis S. et al. A Humanized Antibody against LRG1 that Inhibits Angiogenesis and Reduces Retinal Vascular Leakage. bioRxiv. 2021
226. Singhal M, Gengenbacher N, Abdul Pari AA, Kamiyama M, Hai L, Kuhn BJ. et al. Temporal multi-omics identifies LRG1 as a vascular niche instructor of metastasis. Sci Transl Med. 2021;13:eabe6805
227. Zhang J, Pho V, Bonasera SJ, Holtzman J, Tang AT, Hellmuth J. et al. Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nat Neurosci. 2007;10:77-86
228. Jin Y, Ratnam K, Chuang PY, Fan Y, Zhong Y, Dai Y. et al. A systems approach identifies HIPK2 as a key regulator of kidney fibrosis. Nat Med. 2012;18:580-8
229. Wei G, Ku S, Ma GK, Saito S, Tang AA, Zhang J. et al. HIPK2 represses beta-catenin-mediated transcription, epidermal stem cell expansion, and skin tumorigenesis. Proc Natl Acad Sci U S A. 2007;104:13040-5
230. Lee W, Andrews BC, Faust M, Walldorf U, Verheyen EM. Hipk is an essential protein that promotes Notch signal transduction in the Drosophila eye by inhibition of the global co-repressor Groucho. Dev Biol. 2009;325:263-72
231. Zhang Q, Yoshimatsu Y, Hildebrand J, Frisch SM, Goodman RH. Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell. 2003;115:177-86
232. Nardinocchi L, Puca R, Givol D, D'Orazi G. Counteracting MDM2-induced HIPK2 downregulation restores HIPK2/p53 apoptotic signaling in cancer cells. FEBS Lett. 2010;584:4253-8
233. Chen J, Verheyen EM. Homeodomain-interacting protein kinase regulates Yorkie activity to promote tissue growth. Curr Biol. 2012;22:1582-6
234. Xiao W, E J, Bao L, Fan Y, Jin Y, Wang A. et al. Tubular HIPK2 is a key contributor to renal fibrosis. JCI insight. 2020;5:e136004
235. Oh HJ, Kato M, Deshpande S, Zhang E, Das S, Lanting L. et al. Inhibition of the processing of miR-25 by HIPK2-Phosphorylated-MeCP2 induces NOX4 in early diabetic nephropathy. Sci Rep. 2016;6:38789
236. Xu L, Li X, Zhang F, Wu L, Dong Z, Zhang D. EGFR drives the progression of AKI to CKD through HIPK2 overexpression. Theranostics. 2019;9:2712-26
237. Chang X, Zhen X, Liu J, Ren X, Hu Z, Zhou Z. et al. The antihelmenthic phosphate niclosamide impedes renal fibrosis by inhibiting homeodomain-interacting protein kinase 2 expression. Kidney Int. 2017;92:612-24
238. Liu R, Das B, Xiao W, Li Z, Li H, Lee K. et al. A Novel Inhibitor of Homeodomain Interacting Protein Kinase 2 Mitigates Kidney Fibrosis through Inhibition of the TGF-beta1/Smad3 Pathway. J Am Soc Nephrol. 2017;28:2133-43
239. Lee K, Feng Y, Chen Y, Beresis RT, Drakas R, He JC. A Novel Allosteric HIPK2 Inhibitor Attenuates Renal Fibrosis with Superior Pharmacokinetic, Selectivity, and Safety Profiles. American Society of Nephrology, Kidney Week. 2021 Abstract. 2021: PO2474
240. Lee K, Feng Y, Chen Y, Beresis RT, Drakas R, He JC. Mechanistic Evaluation and In Vivo Validation of a Novel HIPK2 Inhibitor as Anti-Fibrosis Therapy for Kidney Disease. American Society of Nephrology, Kidney Week 2022 Abstract, FR-PO946. 2022;33:585
Author contact
![]() Corresponding author: Kyung Lee, PhD or John Cijiang He, MD, PhD, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1243, New York, NY 10029, Tel: 212-241-9522, Email: kim.leeedu or cijiang.heedu.
Corresponding author: Kyung Lee, PhD or John Cijiang He, MD, PhD, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1243, New York, NY 10029, Tel: 212-241-9522, Email: kim.leeedu or cijiang.heedu.