Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Mitochondria and its Function
Mitochondrial Quality Control
Association Between Common...
The Outcome of Impaired Quality...
Therapies on Regulating...
Conclusion
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(4):1767-1783. doi:10.7150/ijbs.107777 This issue Cite
Review
The Role of Mitochondrial Quality Control in Liver Diseases: Dawn of a Therapeutic Era
Jia Li, M.M.1#, Wenqin Liu, M.M.1#, Jie Zhang, M.D., Ph.D.1 ![]() , Chao Sun, M.D., Ph.D.1,2
, Chao Sun, M.D., Ph.D.1,2 ![]()
1. Department of Gastroenterology and Hepatology, Tianjin Medical University General Hospital, Anshan Road 154, Heping District, Tianjin 300052, China.
2. Department of Gastroenterology, Tianjin Medical University General Hospital Airport Hospital, East Street 6, Tianjin Airport Economic Area, Tianjin 300308, China.
# These authors contributed equally to this work.
Received 2024-11-28; Accepted 2025-1-28; Published 2025-2-10
Abstract

The liver is a vital metabolic organ that detoxifies substances, produces bile, stores nutrients, and regulates versatile metabolic processes. Maintaining normal liver cell function requires the prompt and delicate modulation of mitochondrial quality control (MQC), which encompasses a spectrum of processes such as mitochondrial fission, fusion, biogenesis, and mitophagy. Recent studies have shown that disruptions to this homeostatic status are closely linked to the advent and progression of a variety of acute and chronic liver diseases, including but not limited to alcohol-associated liver disease and metabolic dysfunction-associated fatty liver disease. However, the explicit mechanisms by which mitochondrial dysfunction impacts inflammatory pathways and cell death in the context of liver diseases remain unclear. In this narrative review, we provide a detailed description of MQC, analyze the mechanisms underpinning mitochondrial dysfunction induced by different detrimental insults, and further elucidate how imbalanced/disrupted MQC promotes the progression and aggravation of liver diseases, ultimately shedding light on the mitochondrion-centric therapeutic strategies for these pathophysiological entities.
Keywords: mitochondrial quality control, liver diseases, reactive oxygen species, mitophagy, mitochondrial dynamics, mtDNA, macrophage heterogeneity
Mitochondria and its Function
Mitochondria are double-membrane-bound organelles responsible for a myriad of essential cellular functions, including energy production, glucose and lipid metabolism, cell death, proliferation, and innate immune responses [1]. Mitochondria are crucial for cellular energy production, generating approximately 90% of cellular energy through oxidative phosphorylation (OXPHOS), producing adenosine triphosphate (ATP) [2]. In brief, energy substrates enter the mitochondrial matrix, where they are processed in the tricarboxylic acid cycle to generate electron carriers (nicotinamide adenine dinucleotide [NADH] and flavin adenine dinucleotide [FADH2]). These electron carriers then transfer electrons through the electron transport chain (ETC), driving the proton pumps to move protons from the matrix to the intermembrane space, thereby establishing an electrochemical gradient, also known as the mitochondrial transmembrane potential [3]. The proton gradient is employed by ATP synthase to phosphorylate adenosine diphosphate (ADP) into ATP. Finally, cytochrome c oxidase IV (COX IV), the terminal enzyme of the respiratory chain, accepts electrons from reduced cytochrome c molecules and transfers them to oxygen and protons, producing water as a byproduct [4]. Mitochondria are a prime example of endosymbiotic integration, coordinating cellular and organellar signal transduction pathways that are critical for adaptive responses and evolutionary processes. The cornerstone pertinent to this coordination refers to the Mitochondrial Information Processing System, a complex mechanism that interprets signals from ions, proteins, nutrients, and energy states, translating them into targeted genetic programs [5]. These programs, in turn, initiate metabolic reprogramming driving key cellular processes such as proliferation, differentiation, contractility, secretion, and apoptosis, all of which are fundamental for organismal survival and evolutionary fitness. However, the precise mechanisms by which these pathways interact within mitochondria to maintain regularly cellular homeostasis remain elusive. In addition to energy production, mitochondria are also involved in other critical cellular functions, including iron metabolism and the regulation of cell proliferation [6]. Furthermore, mitochondria play a pivotal role in controlling inflammation and the development of inflammation-related diseases by influencing innate immune responses [7].
Mitochondrial Quality Control
The MQC mechanism involves the intricate regulation of several processes, including dynamics (fission and fusion), biogenesis, and degradation, all of which are essential to maintain cellular homeostasis [8, 9].
1) Mitochondrial Dynamics (fission and fusion)
The fission (division) process is primarily mediated by dynamin-related protein 1 (Drp1), while mitochondrial fusion involves two dynamin-like GTPases, mitofusin 1 (MFN1) and mitofusin 2 (MFN2), as well as optic atrophy 1 (OPA1) and cardiolipin (CL), which mediate the fusion of the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM), respectively [10]. Mitochondrial fission separates a single mitochondrion into two smaller fragments, facilitating subsequent degradation, while fusion joins two adjacent mitochondria to orchestrate a larger organelle, preserving mitochondrial DNA (mtDNA) integrity [11-13]. Either abnormal mitochondrial fission or fusion has been reported in liver diseases such as non-alcoholic fatty liver disease (NAFLD). Previous studies using mouse models have shown that enhanced mitochondrial fission in hepatocytes is associated with the early stages of NAFLD, and that inhibiting this process can mitigate liver inflammation and fibrogenesis [14-18]. The relationship between mitochondrial fusion and the development of NAFLD has been demonstrated in both mice and human models [19-21]. Concordantly, the liver of mice and patients with NAFLD presented an increased expression of OPA1 and other proteins involved in mitochondrial function compared to controls. In the case of alcohol-associated liver diseases (ALD), a pronounced hyperactivation of the mitochondrial fission pathway was observed in patients, mice, and cells, characterized by a significant increase in the expression of Drp1 and its adapters/receptors, while the fusion pathway remained unaffected [22-24]. As for viral hepatitis, in vitro experiments have confirmed that hepatitis B virus (HBV)/hepatitis C virus (HCV)-induced mitochondrial fission may attenuate apoptosis and potentially contribute to persistent viral infection [25, 26].
2) Mitochondrial Biogenesis
Mitochondrial biogenesis is under management by certain nuclear transcription factors, including nuclear respiratory factor 1 (NRF1) and nuclear respiratory factor 2 (NRF2), as well as nuclear co-activators such as peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α). NRF1 and NRF2 regulate the expression of genes encoding mitochondrial respiratory subunits and mitochondrial transcription factor A (TFAM). TFAM binds to mtDNA at multiple sites, primarily accounting for both mtDNA maintenance and transcription initiation [27]. PGC-1α, which does not bind directly to DNA, is recruited to chromatin and acts as a pleiotropic regulator by interacting with nuclear receptors or activating transcription factors, thus promoting mitochondrial biogenesis [28, 29]. Mitochondrial biogenesis is crucial for maintaining cellular energy homeostasis and function, and its dysregulation is strongly linked to the pathogenesis of liver diseases, particularly NAFLD. In mice experiencing NAFLD and a well-established NAFLD cell model, the levels of sirtuin 1 (SIRT1) and PGC-1α were reduced, resulting in decreased cell viability, increased apoptosis, lipid accumulation, and reactive oxygen species (ROS) production [30].
3) Mitochondrial Degradation
There are multiple pathways involved in regulating MQC, functioning to remove or degrade specific mitochondrial components, portions of mitochondria, or entire mitochondria. The degradation of specific OMM proteins is mediated by the ubiquitin-proteasome system [31], while the degradation of IMM proteins depends on AAA ATPase family proteases, which are located on both sides of the IMM capable of degrading proteins within the matrix and intermembrane space [32]. More recently, a novel phenomenon called vesicle derived from the inner mitochondrial membrane (VDIM) has been identified to selectively remove damaged sections of IMM and likely maintains the structure of normal mitochondrial cristae, potentially protecting mitochondria from localized injury [33]. A portion of mitochondria can bud off as mitochondria-derived vesicles (MDVs), which serve to transport oxidized proteins to lysosomes or peroxisomes for further degradation [34]. Entirely damaged mitochondria can be encapsulated into microvesicles or migrasomes and secreted from cells as extracellular vesicles through processes such as mitocytosis and autophagic secretion of mitochondria [35, 36]. Furthermore, damaged mitochondria can be selectively eliminated via mitophagy, which involves autophagosomes, or through direct lysosome-mediated micromitophagy. Mitophagy is a well-known process in which excess or damaged mitochondria are selectively removed via autophagosomes. These autophagosomes, containing the mitochondria, eventually fuse with lysosomes, where the mitochondria are degraded through either PINK1-PRKN-dependent or -independent mitophagy pathways. Emerging evidence has shown that under certain conditions, mitochondria undergo morphological changes, forming specialized structures such as mitochondrial spheroids and mitochondria-lysosome-related organelles (MLROs), acquiring lysosomal markers for potential degradation. A novel mitochondrial structure observed in murine embryonic fibroblasts involves depolarized mitochondria undergoing morphological remodeling, adopting a ring- or C-shape, and forming ball-like spheroids with an internal lumen surrounded by membranes that contain cytosolic materials [37]. MLROs are derived from MDVs that fuse with lysosomes, which is negatively regulated by transcription factor EB (TFEB) and associated with mitochondrial protein degradation [38, 39]. Compared to VIDM, MLROs are capable of degrading all mitochondrial components, including the OMM, IMM, and matrix proteins [40]. Furthermore, the induction of MLROs is linked to the dedifferentiation of hepatocytes and adipocytes, which can be observed in mouse models of ALD and metabolic-associated fatty liver disease (MAFLD) [38, 41]. Hepatocyte dedifferentiation is commonly observed in the late stages of chronic liver diseases and contributes to liver failure, underscoring the potential relevance of MLROs in various liver diseases. Collectively, VDIM, mitochondrial spheroids and MLROs appear to form in response to oxidative mitochondrial damage independently of canonical mechanisms, representing novel mitochondrial dynamics. Since mitophagy is the main pathway of mitochondrial degradation, many studies have confirmed the relationship between mitophagy and liver disease, including NAFLD, ALD and viral hepatitis. Mitochondrial depolarization was observed in hepatocytes, indicating early mitochondrial dysfunction in NAFLD and subsequently triggering mitophagy, affirmative in cell, mouse, and human models [42-44]. Furthermore, it has been verified that acute/chronic alcohol exposure and HBV/HCV-induced mitochondrial mitophagy can mitigate hepatic injury and inflammation in multiple models [25, 26, 45-47].
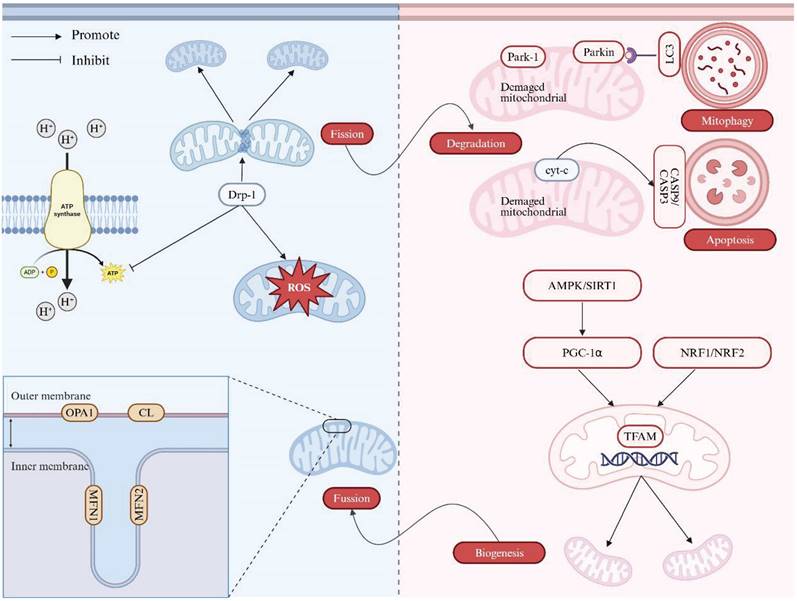
Together, these synergistic actions form a dynamic network that leverages mitochondrial “quality control” and restores homeostasis during energy deprivation or in response to mitochondrial damage. Figure 1 illustrates the regulation of mitochondria quality control. Impaired mitochondrial function and defective quality control mechanisms would contribute to the pathogenesis of various liver diseases, including NAFLD, ALD, and viral hepatitis. Table 1 summarizes the research progress on various types of MQC disorders and their association with hepatopathy.
Regulation of mitochondria quality control. The mitochondrial quality control mechanism involves mitochondrial fission, fusion, biogenesis, and degradation. (1) Mitochondrial fission and fusion. The fission process is mediated by Drp1, while mitochondrial fusion is regulated by MFN1, MFN2 and OPA1. (2) Mitochondrial biogenesis. Mitochondrial biogenesis is under management by certain nuclear transcription factors, including NRF1 and NRF2, as well as nuclear co-activators such as PGC-1α. (3) Mitochondrial degradation. Mitochondrial can be removed via canonical macro-autophagy and PINK1- PARKIN-mediated mitophagy or other forms of mitochondrial removal. Note: Drp1: dynamin-related protein 1; ROS: reactive oxygen species; OPA1: optic atrophy 1; CL: cardiolipin; MFN1: mitofusin 1; MFN2: mitofusin 2; LC3: light chain 3; Cyt c: cytochrome c; CASP9: caspase 9; CASP3: caspase 3; AMPK: AMP-activated protein kinase; SIRT1: sirtuin1; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1α; NRF1: nuclear respiratory factor 1; NRF2: nuclear respiratory factor 2; TFAM: transcription factor A. Created with BioRender.com.
The association between various types of mitochondrial quality control disorders and liver diseases
| Type | Liver Diseases | Species | Study Protocol | Outcomes | Reference |
|---|---|---|---|---|---|
| Fission | NAFLD | Mice | Mice were fed a standard diet or choline-deficient, L-amino acid-defined diet with vehicle or mitochondrial division inhibitor-1. | Mitochondrial fission increase in hepatocytes was linked to early-stage NASH, including steatosis, liver inflammation, and hepatocyte ballooning. | [14] |
| NASH | Mice | NASH mice were fed an MCD diet for 12 weeks to induce severe NASH, while control mice received a standard diet. | Compared to the control group, NASH liver organoids exhibited abnormal mitochondrial and organoid morphology, along with elevated levels of DRP1, mitochondrial mitogen protein, and ROS production. | [15] | |
| ALD | Mice/ VL-17A cell | Wild-type (WT) mice and liver-specific Drp1 knockout (KO) mice were divided into two groups and fed either a control diet or an ethanol diet. The human hepatoma VL-17A cell was treated with or without 100 mmol/L ethanol for 3 or 14 days. | Ethanol-fed mice exhibited exacerbated hepatic megamitochondria compared to wild-type mice on the same diet, while also showing reduced ethanol-induced toxicity. Prolonged ethanol exposure in VL-17A cells reduced Drp1 translocation, whereas Drp1 inactivation in the absence of ethanol resulted in hyperfused megamitochondria. | [23] | |
| AH | Human | Hepatic expression of mitochondria-shaping proteins (MSPs) was assessed in alcoholic hepatitis (AH) patients using DNA microarray (n = 15) and RT-PCR (n = 32). Drp1 activation was studied in mitochondria from ALD patient liver biopsies (n = 8). Alcohol effects on mitochondrial dynamics and MSP expression were examined in precision-cut liver slices (PCLS) exposed to ethanol (50-250 mM) for 24 hours. | AH patients exhibited hyperactivation of the fragmentation pathway, with increased Drp1 and its adapters/receptors. Drp1 translocation to mitochondria was induced in severe ALD patients and mildly affected in PCLS after short-term EtOH exposure. | [22] | |
| Fusion | NAFLD | Human | By employing a multimodal imaging approach, the structure and morphometric parameters of normal and giant mitochondria were analyzed in four patients. | Giant mitochondria, resulting from mitochondrial fusion, exhibit impaired function due to a reduced surface area-to-volume ratio and disorganization of the inner and cristae membranes. | [21] |
| NAFLD | Mice/ Human | Male mice selectively lacking OPA1 were fed a high-fat diet for 20 weeks. The human transcriptomic data were retrieved from the gene expression omnibus (GEO) repository. | Mice lacking OPA1 were protected from hepatic steatosis and obesity due to reduced lipid absorption. The liver of NAFLD subjects showed increased OPA1 expression and elevated levels of proteins involved in mitochondrial function compared to controls. | [19] | |
| Biogenesis | NAFLD | Mice/ HepG2 cell | Six-week-old mice were randomly assigned to the control (ordinary diet) or HFD group (high-fat diet) for 8 weeks, with eight animals per group. HepG2 cells were treated with 1.5 mM oleic acid (OA) for 48 hours to induce NAFLD in the cell model, and SIRT-1 induction was monitored. | In mice with NAFLD, the levels of SIRT-1 and PGC-1α were reduced. In a well-established NAFLD cell model, exposure of the HepG2 hepatocyte cell line to OA led to decreased cell viability, increased apoptosis, lipid accumulation, and reactive oxygen species production. | [30] |
| ALD | Mice | Mice were divided into two groups: intragastric alcohol-fed and control. They were fed a high-fat diet, with or without alcohol, starting at 22.7 g/kg/day and gradually increasing to 38.4% of total caloric intake after 4 weeks. | Intragastric alcohol feeding in mice led to 1) altered mitochondrial morphology with increased elongated mitochondria and 2) enhanced mitochondrial biogenesis in the liver, associated with upregulation of PGC-1α. | [141] | |
| Degradation | NAFLD | Mice/AML 12 cell | Eight-week-old male Nrf2+/+ (WT) and Nrf2-/- mice were randomly assigned to the Ctrl, sulforaphane (SF), HFD, or HFD + SF groups. AML12 cells were treated with the indicated concentration of mixed fatty acid (FA), palmitic acid (PA), stearic acid (SA), palmitoleic acid (PO), or oleic acid for 24h. | HFD increased KEAP1 and decreased NRF2 in the liver of Nrf2+/+ mice, while altering autophagy-related proteins in an NRF2-independent manner. Autophagosome formation was reduced in both HFD mouse liver tissue and FA-treated AML12 cells. HFD and SFAs (FA, PA, SA), but not MUFAs (PO, OA), suppressed autophagosome biogenesis. | [42] |
| NASH | Mice | C57BL/6 mice were fed a Western diet (high fat, fructose and cholesterol) for 2 weeks, 2 months and 6 months. | After 2 weeks on a Western diet, mitochondrial depolarization (mtDepo) was observed in 50-70% of hepatocytes, indicating early mitochondrial dysfunction in NASH, which triggered mitophagy and increased mitophagic markers at 2 and 6 months, while concurrently impairing autophagic processing. | [43] | |
| NASH | Human | Liver samples were collected from 130 adults with clinical obesity undergoing bariatric surgery at the University of Missouri Hospital, grouped into No disease/control (CTRL, n = 13), Nonalcoholic fatty liver (NAFL, n = 34), Borderline-NASH (B-NASH, n = 27), and Definite-NASH (D-NASH, n = 56). | Hepatic markers of macro-autophagy, including ULK1, were significantly lower in D-NASH compared to CTRL. | [44] | |
| ALD | Mice | The WT and Parkin KO mice were treated with alcohol by the acute-binge and Gao-binge models. | Parkin KO mice exhibited reduced mitophagy, β-oxidation, mitochondrial respiration, and cytochrome c oxidase activity following acute alcohol treatment, compared to WT mice. | [142] | |
| CHB/ CHC/ NASH | Human | Patients with chronic hepatitis B (CHB; n = 146) were compared to patients with chronic hepatitis C (CHC; n = 33), NASH (n = 12), and healthy controls (n = 24). | PRKN mRNA was significantly increased in patients with CHB, CHC, or NASH compared to controls. | [47] | |
| Fission and Mitophagy | HCV infection | Huh7 cell | Huh7 cells were cultured, and cell culture-derived HCV of JC1 and JFH1 strains were used. | HCV-induced mitochondrial fission and mitophagy serve to attenuate apoptosis. | [25] |
| HBV infection | Huh7 cell/ HepG2 cell/ HepAD38 cell | Cells were cultured under appropriate conditions and transfected with siRNA for further study. | HBV and its HBx protein promote mitochondrial fragmentation and mitophagy by inducing Drp1 expression and Parkin translocation. | [26] |
Note: NAFLD: nonalcoholic fatty liver disease; NASH: nonalcoholic steatohepatitis; MCD: methionine- and choline deficient diet; Drp1: dynamin-related protein 1; ROS: reactive oxygen species; ALD: alcohol-associated liver diseases; WT: wild-type; KO: knockout; AH: alcoholic hepatitis; MSPs: mitochondria-shaping proteins; PCLS: precision-cut liver slices; HFD: high-fat-diet; OA: oleic acid; SIRT-1: sirtuin 1; PGC-1α: proliferator-activated receptor gamma coactivator 1α; OPA1: optic atrophy 1; GEO: gene expression omnibus; SF: sulforaphane; FA: fatty acid; PA: palmitic acid; SA: stearic acid; PO: palmitoleic acid; KEAP1: kelch-like ECH-associated protein 1; NRF2: nuclear factor erythroid 2-related factor 2; SFAs: saturated fatty acids; MUFAs: monounsaturated fatty acids; mtDepo: mitochondrial depolarization; CTRL: no disease/control; NAFL: nonalcoholic fatty liver; B-NASH: borderline-NASH; D-NASH: definite-NASH; ULK1: UNC-52-like kinase 1; CHB: chronic hepatitis B; CHC: chronic hepatitis C; PRKN: parkinson juvenile disease protein; HCV: hepatitis C virus; HBV: hepatitis B virus; Hbx: hepatitis B virus X protein.
Association Between Common Causes of Liver and Diseases Mitochondrial Dysfunction
Virus Infection
The primary pathogenic mechanism underlying long-term infection on account of HBV involves the induction of mitochondrial dysfunction, including alterations in mitochondrial morphology and dynamics. Ultrastructural analysis of hepatocytes using transmission electron microscopy revealed that HBV infection leads to cytoskeletal disruption and mitochondrial morphological abnormalities, such as the loss of typical tubular or circular shape, disappearance of cristae, and mitochondrial swelling. These changes pertaining to mitochondrial dynamics upregulate Drp-1 expression and promote Parkin translocation, thereby shifting the balance of mitochondrial dynamics toward augmented fission and mitophagy. The enhanced mitochondrial division can reduce ATP production and disrupt calcium flux [48]. AMP-activated protein kinase (AMPK), a highly conserved sensor of intracellular adenosine nucleotide levels, is activated in alignment with increased AMP or ADP, which result from decreased ATP production. AMP/ADP binds directly to the γ regulatory subunits of AMPK, causing a conformational change that promotes its activating phosphorylation [49, 50]. Additionally, phosphorylation of Thr172 is required for AMPK activation, which is mediated by the serine/threonine kinases or Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CAMKK2) in response to AMP/ADP and calcium flux, respectively [51, 52].
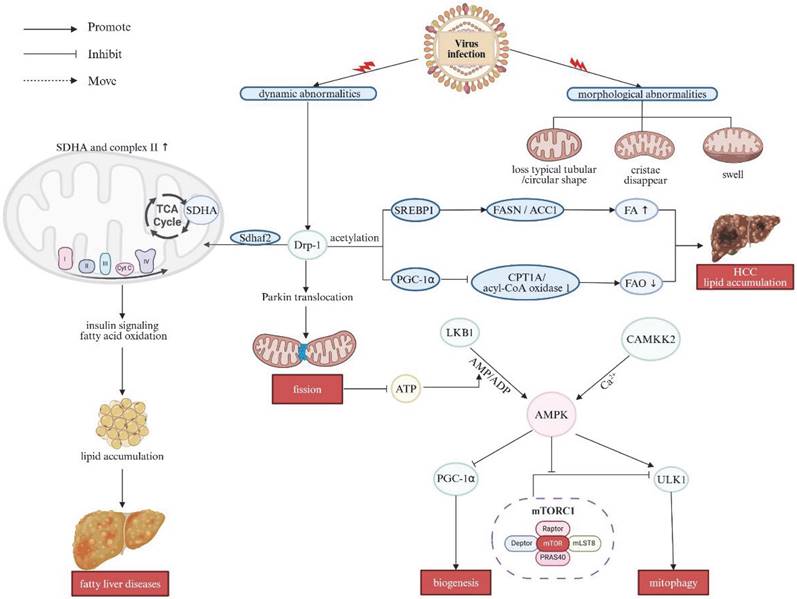
Once activated, AMPK can stimulate autophagy through a dual mechanism: by activating UNC-52-like kinase 1 (ULK1) alongside directly suppressing the inhibitory effect with regard to the mechanistic target of rapamycin complex 1 (mTORC1) complex on ULK1 [53, 54]. Furthermore, Ser616-phosphorylated Drp1 forms a complex with autophagosomes marked by Ras-related protein 9 (Rab9) in a ULK1-dependent manner, thereby further triggering autophagic activities [55]. This shift contributes to suppress innate immune responses in host cells and facilitates viral replication, giving rising to the persistence of infection. Furthermore, overexpression of Drp1 enhanced de novo fatty acid (FA) synthesis and repressed fatty acid oxidation (FAO) by promoting the acetylation of sterol regulatory element binding protein 1 (SREBP1) and PGC-1α. The increase in SREBP1 subsequently upregulated the expression of lipogenic genes, that is, fatty acid synthase (FASN) and acetyl-CoA carboxylase 1 (ACC1), thereby fostering deleterious lipid accumulation in the hepatocellular carcinoma (HCC) cells. Meanwhile, PGC-1α inhibited FAO by downregulating the expression of carnitine palmitoyl-transferase 1A (CPT1A) and acyl-CoA oxidase 1. Therefore, activation of mitochondrial fission significantly accelerated the proliferative alongside metastatic behaviors of HCC cells both in vitro and in vivo [56]. Sdhaf2, a protein essential for the flavination of succinate dehydrogenase subunit-A (SDHA) gene and the assembly of complex II, facilitates mitochondrial localization of this complexes through interaction with Drp1 [57]. Overexpressed Drp1 also enhances the function of SDHA and complex II in the ETC and the trichloroacetic acid (TCA) cycle, thereby impairing insulin signaling and FAO [58, 59]. This perturbation may aggravate the fat accumulation, which is a key pathogenic reason/precipitating factor in various liver diseases [15]. These multiple effects on MQC following viral infection are summarized in Figure 2.
The effects on mitochondrial quality control after viral infection. Long-term HBV infection primarily induces mitochondrial dysfunction, including alterations in mitochondrial morphology and dynamics. HBV infection causes cytoskeletal disruption and mitochondrial abnormalities such as the loss of tubular or circular shapes, disappearance of cristae, and mitochondrial swelling. These changes in mitochondrial dynamics upregulate Drp1 expression and promote Parkin translocation, shifting the balance toward increased mitochondrial fission and mitophagy. Drp1 overexpression enhances de novo FA synthesis and suppresses FAO by promoting acetylation of SREBP1 and PGC-1α, leading to elevated SREBP1 levels that upregulate lipogenic gene expression, contributing to lipid accumulation in HCC cells. Additionally, overexpressed Drp1 enhances the function of SDHA and complex II in the ETC and TCA cycle, impairing insulin signaling and FAO, further exacerbating fat accumulation. LKB1 and CAMKK2 activate AMPK, stimulating autophagy through a dual mechanism: by activating ULK1 and directly inhibiting the mTORC1 complex's suppressive effect on ULK1. Note: Drp1: dynamin-related protein 1; SDHA: succinate dehydrogenase subunit-A; TCA: trichloroacetic acid; SREBP1: sterol regulatory element binding protein 1; FASN: fatty acid synthase; ACC1: acetyl-CoA carboxylase 1; FA: fatty acid; FAO: fatty acid oxidation; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1α; CPT1A: carnitine palmitoyl-transferase 1A; HCC: hepatocellular carcinoma; ATP: adenosine triphosphate; ULK1: UNC-52-like kinase 1; mTORC1: mechanistic target of rapamycin complex 1. Created with BioRender.com.
Alcohol Consumption
Excessive alcohol consumption is a primary and major cause of ALD, a significant global health concern with limited therapeutic options. It is highlighted that alcohol can interrupt mitochondrial function, disrupt ATP production and trigger a burst of oxidative stress, which in consequence culminates in evidently cellular damage and inflammation. Both morphological and functional alterations of mitochondria have been implicated in the pathogenesis of ALD [60, 61]. Alcohol impacts the balance between mitochondrial fission/fusion, leading to structural changes such as the formation of megamitochondria and fragmented mitochondria. Increased megamitochondria have been pathologically related to reduced Drp1 expression, arising from decreased TFEB activation [23, 62]. Otherwise, alcohol also stimulates p53-mediated induction of Drp1 expression, exacerbating mitochondrial fission and instigating troublesome mitochondrial dysfunction [61]. Chronic alcohol exposure initially stimulates mitochondrial biogenesis, resulting in increased expression of components regarding the mitochondrial ETC and enhanced mitochondrial respiration. This response is connected with the upregulation of key factors such as PGC-1α and mitochondrial TFAM, indispensible for mtDNA replication. This process is further dictated by regulators like SIRT1 and AMPK located as upstream cascade, leading to enhanced mitochondrial function. However, prolonged binge disrupts mitochondrial biogenesis by increasing the NADH/NAD+ ratio, which reduces SIRT1 and AMPK activity, impairing the PGC-1α pathway and ultimately giving rise to mitochondrial dysfunction (Figure 3A) [61].
Relationship between mitochondrial dysfunction and binge drinking. (A)The effects of alcohol consumption on mitochondrial morphology and dynamics. Alcohol disrupts mitochondrial fission/fusion balance, leading to enlarged mitochondria and fragmentation. It decreases TFEB activation, reducing Drp1 expression and promoting larger mitochondria. Alcohol also activates p53, increasing Drp1 expression and enhancing mitochondrial fission. Chronic alcohol exposure upregulates PGC-1α and TFAM, essential for mitochondrial DNA replication, boosting mitochondrial ETC components and respiration. This process is regulated by upstream factors like SIRT1 and AMPK. However, prolonged binge drinking increases the NADH/NAD+ ratio, reducing SIRT1 and AMPK activity and impairing the PGC-1α pathway, disrupting mitochondrial biogenesis. (B) Alcohol metabolism and its effects on mitochondrial function. Alcohol is metabolized in the liver via oxidative and non-oxidative pathways. In the oxidative pathway, ethanol is first converted to acetaldehyde, which is then metabolized by ALDH2 to acetate. Acetate is further converted to acetyl-CoA, entering the TCA cycle to produce citrate. Citrate is exported to the cytosol via SLC25A1 and converted by ACLY and ACC to malonyl-CoA, which inhibits CPT1 and impairs fatty acid transport into mitochondria, promoting lipid accumulation in hepatocytes. Chronic ethanol consumption inhibits mitochondrial respiratory complex protein synthesis and damages mtDNA, increasing ROS production and causing cell damage. In the non-oxidative pathway, alcohol is converted to FAEEs, destabilizing mitochondrial membranes, disrupting electron flow, and inhibiting OXPHOS. Note: Drp1: dynamin-related protein 1; TFEB: transcription factor EB; MFN1: mitofusin 1; MFN2: mitofusin 2; NADH: nicotinamide adenine dinucleotide; SIRT1: sirtuin 1; AMPK: AMP-activated protein kinase; PGC-1: peroxisome proliferator-activated receptor gamma coactivator 1α; TFAM: transcription factor A; ETC: electron transport chain; FAEEs: fatty acid ethyl esters; ALDH2: acetaldehyde dehydrogenase 2; TCA: trichloroacetic acid; ACC: acetyl-CoA carboxylase; ACLY: ATP citrate lyase; CPT1: CPT1A, carnitine palmitoyl-transferase 1; SLC25A1: mitochondrial citrate carrier. Created with BioRender.com.
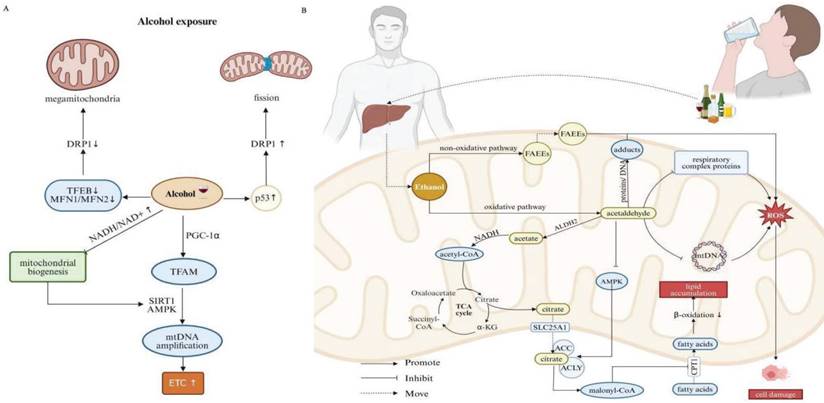
Alcohol is primarily metabolized in the liver through two main pathways: oxidative and non-oxidative manner. While only a small fraction of alcohol undergoes non-oxidative metabolism, it is still threatening to fitness. In particular, alcohol is converted into fatty acid ethyl esters (FAEEs), which can destabilize the mitochondrial membranes and disrupt electron flow in the respiratory chain, ultimately inhibiting OXPHOS [63, 64]. The oxidative pathway is the preliminary step by which ethanol is converted into acetaldehyde. Acetaldehyde is a highly reactive metabolite that can form adducts with proteins and DNA, especially within mitochondria, whose extensive accumulation leads to overwhelming oxidative stress and liver injury [63, 65]. Damaged mitochondria may exhibit altered redox signaling pathways, further aggravating acetaldehyde-induced oxidative stress and cellular injury. Acetaldehyde is subsequently metabolized by acetaldehyde dehydrogenase 2 (ALDH2), an enzyme predominantly expressed in mitochondria, toward less harmful substance, that is, acetate and NADH. Acetate is then converted into acetyl-CoA, entering the TCA cycle, to produce citrate. Citrate is transported out of the mitochondria into the cytosol through the mitochondrial citrate carrier (SLC25A1), where it undergoes serially enzymatic reactions involving ATP citrate lyase (ACLY) and ACC, responsible for the formation of malonyl-CoA. Malonyl-CoA inhibits carnitine palmitoyl transferase 1 (CPT1), a key enzyme involved in the transport of FAs into the mitochondria. Ethanol exposure also suppresses AMPK activity, which further fosters ACC activity and increases malonyl-CoA levels. Intriguingly, ethanol enhances the sensitivity of CPT1 to malonyl-CoA [66]. As a result, the reduced activity of CPT1 diminishes mitochondrial β-oxidation, promoting de novo lipogenesis and contributing to lipid accumulation in hepatocytes [61]. Moreover, chronic ethanol consumption inhibits the synthesis of mitochondrial respiratory complex proteins. Accordingly, resultant reduced capacity concerning OXPHOS, combined with elevated reducing pressure, orchestrate conditions conducive to ROS production [67]. MtDNA is particularly vulnerable to oxidative stress due to its proximity to the IMM, known as the primary intracellular source of ROS. Ethanol-induced damage to mtDNA, if not adequately, effectively or promptly repaired, negatively impacts cellular energy metabolism, further augmenting cellular damage (Figure 3B) [68].
Taken together, alcohol impairs mitochondrial FA oxidation and OXPHOS, disproportionating cellular energy and contributing to the development of hepatic steatosis along with relevant cellular damage.
Obesity and Metabolic Derangement to Liver
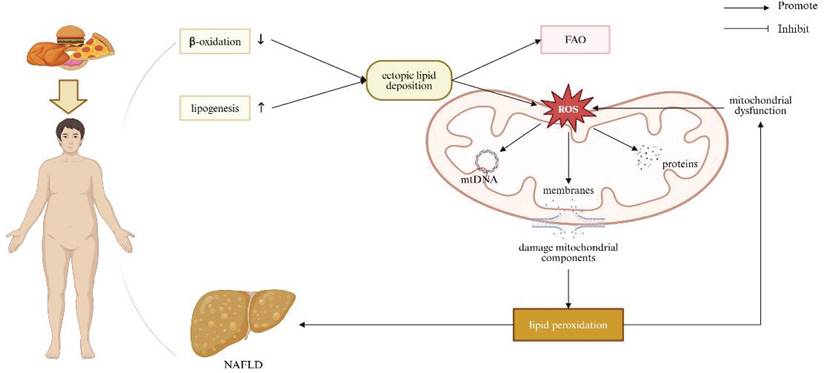
Obesity induced by high-fat or high-cholesterol dietary intakes is associated with dysregulated lipid homeostasis, leading to ectopic lipid deposition in non-adipocyte cells, such as hepatocytes [69]. Reduced β-oxidation and increased lipogenesis result in lipid accumulation within haptic parenchyma. Excessive hepatocellular lipids simultaneously stimulate mitochondrial FAO and ROS production. This creates a vicious cycle, wherein damaged mitochondria become dysfunctional, primarily on account of hepatic mitochondrial structural defects and an imbalance in mitochondrial dynamics, indicative of cristae loss, rarefied matrix, reduced levels of OPA-1 and MFN1/2, and increased expression of Drp-1. These alterations impair OXPHOS and further increase ROS production [70]. Excessive ROS generation affects mitochondrial components, including membranes, proteins, and mtDNA, and in consequence enhances lipid peroxidation. Cell membranes are significantly susceptible to radical-induced damage due to their abundance pertaining to polyunsaturated FA. ROS-mediated lipid peroxidation occurs in the circumstance of membrane phospholipids interacting with ROS, leading to the oxidation of unsaturated lipid chains and the formation of hydro peroxided lipids and alkyl radicals. This peroxidation negatively impacts membrane structure, compromising their fluidity and integrity [71]. A previous study demonstrated that rats challenged by a high-fat diet (HFD) for 6 weeks exhibited changes in mitochondrial oxidative stress markers, including increased hydrogen peroxide production and reduced activity of aconitase and superoxide dismutase [72, 73]. Another study consistently indicated that rats exposed to a 14-week HFD exhibited significantly increased serum levels of 8-hidroxy-2-deoxyguanosine in relation to overwhelming oxidative stress systemically [74]. Moreover, emerging evidence has implicated that obesity-dictated ROS production can induce DNA damage by repressing the expression of genes involved in DNA repair [75-77]. Defective mitophagy has been observed in both in vivo and in vitro models experiencing NAFLD [78-81]. It has been proved that hepatocyte-specific deletion of Parkin exacerbates fatty liver disease and insulin resistance in the context of HFD-fed mice [82]. Hepatic mitophagy was reduced in obese mice with fatty liver, uncovering connection between loss of hepatic mitophagy and the advent of NAFLD [83]. Collectively, mitochondrial dysfunction exacerbates lipid homeostasis disruption, contributing to the progression of NAFLD (Figure 4).
Relationship between mitochondrial dysfunction and obesity/NAFLD. Obesity caused by high-fat or high-cholesterol diets disrupts lipid homeostasis, leading to reduced β-oxidation and increased lipogenesis, resulting in lipid accumulation in the liver. Excess hepatocellular lipids stimulate mitochondrial FAO and ROS production. Elevated ROS damages mitochondrial components, including membranes, proteins, and mtDNA, promoting lipid peroxidation and contributing to the development of obesity/NAFLD. Note: FAO: fatty acid oxidation; NAFLD: non-alcoholic fatty liver disease. Created with BioRender.com.
The Outcome of Impaired Quality Control of Mitochondrial and Liver Diseases
Oxidative Stress and Liver Diseases
Mitochondria are essential double-membrane organelles responsible for aerobic respiration in addition to cellular metabolism. When mitochondrial dysfunction occurs, it leads to excessive production of ROS, which is regarded as one of the primary factors contributing to the pathogenesis of various liver injuries [84, 85].
Nuclear factor-kappa B (NF-κB) is a family of transcription factors that plays a central role in inflammatory activities and immunoregulatory actions [86]. Under normal conditions, NF-κB is sequestered in the cytoplasm by inhibitor of kappa B (IκB). However, overproduction of ROS triggers the phosphorylation, ubiquitination, and proteasome-mediated degradation of IκB, thereby allowing NF-κB to translocate into the nucleus [87]. Nuclear NF-κB is capable of instigating the expression of genes involved in inflammation and survival, such as manganese superoxide dismutase and anti-caspase proteins to exert anti-apoptotic effects. In contrast, sustained elevation of cellular ROS levels can lead to the upregulation of pro-apoptotic cytokines, including interleukin-1β (IL-1β), interleukin-18 (IL-18), and tumor necrosis factor-α (TNF-α), which promote hepatocyte apoptosis and necrosis [7]. Moreover, ROS can activate the tumor suppressor protein p53, subsequently, stimulate the expression of Bax, a potent stimulator of mitochondrial pore formation. This process can trigger the mitochondrial permeability transition and facilitate the cell's apoptotic machinery [88]. Given the delicate balance between anti- and pro-apoptotic signals, a mild increase in cellular ROS does not necessarily lead to liver cell death, but, excessive, overwhelming and frustrating ROS accumulation can cause liver damage and lesions [89].
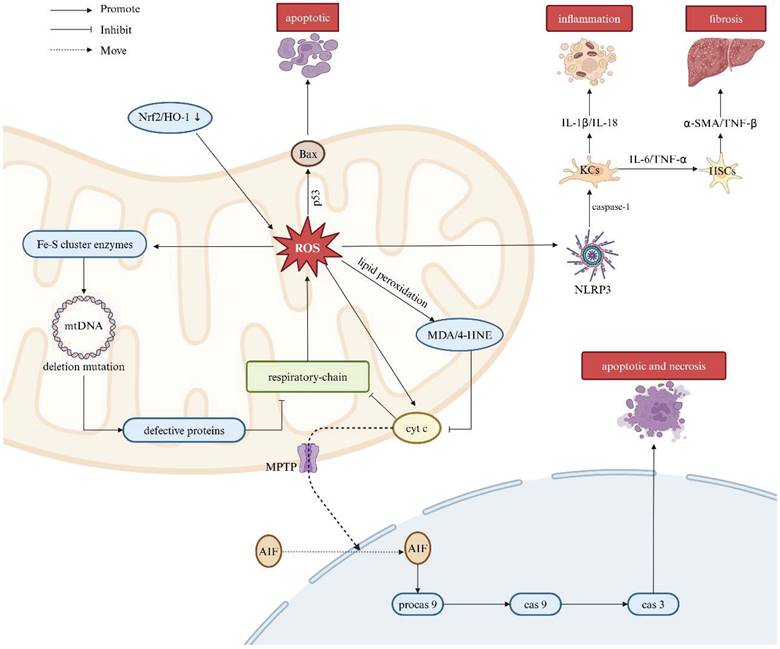
Accumulating evidence suggests that defects in the respiratory chain are a key determinant of mitochondrial dysfunction, leading to increased production of ROS [90]. Massive ROS generation, in turn, triggers lipid peroxidation, resulting in the formation of malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE). These lipid peroxidation products further inhibit the respiratory chain by disrupting cytochrome c oxidase activity in mitochondrial complex IV, thereby establishing a self-sustaining loop of oxidative stress. Additionally, ROS can damage mtDNA through deletion mutations and influence iron-sulfur (Fe-S) cluster enzymes in the respiratory chain [91]. Relative to nuclear DNA, mtDNA is dramatically vulnerable to ROS-induced injury on account of its lack of protective histones in addition to limited and insufficient DNA repairment [92]. Extensive mtDNA damage results in the production of defective mitochondrial proteins, compromising the integral respiratory chain and exacerbating oxidative stress [93]. Mitochondria-derived ROS further contribute to lipid peroxidation by oxidizing unsaturated lipids. MDA, a byproduct of lipid oxidation, impairs components of the ETC, thereby promoting subsequent ROS generation from the mitochondria [94]. The NRF2 is a basic leucine zipper transcription factor that regulates the expression of cytoprotective and antioxidant genes, thereby protecting cells from injurious oxidation induced by inflammation [95]. However, excessive ROS production can lead to the downregulation of the NRF2/heme oxygenase-1 (HO-1) signaling pathway, further impairing the cell's antioxidant defense system. On the other hand, mitochondrial ROS production can induce the mitochondrial permeability transition pore (PTP), which increases mitochondrial outer membrane permeabilization. Collectively, aforesaid process facilitates the release of intermembrane space proteins, such as cytochrome c, into the cytosol. Cytochrome c then enhances the translocation of apoptosis-inducing factor (AIF) to the nucleus and activates procaspase-9, forming the apoptosome complex. In this regard, the apoptosome cleaves procaspase-9 into its active form, which subsequently activates effector caspase-3, initiating both apoptotic and necrotic pathways [96].
Moreover, ROS serve as activators of the NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome, responsible for the self-cleavage of pro-caspase-1 and activation of Kupffer cells (KCs). Activated KCs release proinflammatory cytokines, including IL-1β and IL-18, thereby aggravating the inflammatory response [97]. Furthermore, KCs also secret other cytokines such as IL-6 and TNF-α, which activate hepatic stellate cells (HSCs), consequently promoting collagen production and contributing to the development of liver fibrosis [98, 99]. The overproduction of ROS and subsequent damage to mitochondrial proteins and mtDNA can lead to irreversible mitochondrial membrane depolarization and a decrease in mitochondrial membrane potential (Δψm) known as hallmarks pertinent to mitochondrial dysfunction. Under these conditions, protective mitophagy is capable of degrading damaged mitochondria, thereby limiting the release of mtDNA and the production of free radicals. This process also suppresses certain inflammatory response driven by damage-associated molecular patterns (DAMPs) and assists in mitigating secondary liver injury (Figure 5).
Association between ROS and liver diseases. Mitochondrial dysfunction leads to excessive ROS production, a key factor in liver injury pathogenesis. The respiratory chain is central to this dysfunction, driving increased ROS generation. ROS triggers lipid peroxidation, forming MDA and 4-HNE, which impair the respiratory chain by disrupting cytochrome c oxidase in complex IV. This enhances cytochrome c-mediated AIF translocation to the nucleus, activating procaspase-9 and initiating apoptosis. ROS also damage mtDNA, impairing respiratory chain function and exacerbating oxidative stress. Additionally, ROS activate p53, promoting Bax expression, mitochondrial pore formation, and cell apoptosis. ROS activate the NLRP3 inflammasome, leading to proinflammatory cytokine release from KCs, which promotes liver inflammation and fibrosis through HSC activation. Note: ROS: reactive oxygen species; Nrf2: nuclear factor erythroid 2-related factor 2; HO-1: heme oxygenase-1; BAX: BCL-2-associated X protein; Fe-S: iron-sulfur; MDA: malondialdehyde; 4-HNE: 4-hydroxynonenal; MOMP: mitochondrial outer membrane permeabilization; AIF: apoptosis-inducing factor; Procas 9: procaspase-9; Cas 9: caspase-9; Cas 3: caspase-3; NLRP3: NOD-like receptor thermal protein domain associated protein 3; KCs: Kupffer cells; HSCs: hepatic stellate cells; IL-6: interleukin-6; IL-1β: interleukin-1β; IL-18: interleukin-18; TNF-α: tumor necrosis factor-α; TNF-β: tumor necrosis factor-β; α-SMA: alpha-smooth muscle actin. Created with BioRender.com.
Abnormal Mitophagy and Liver Diseases
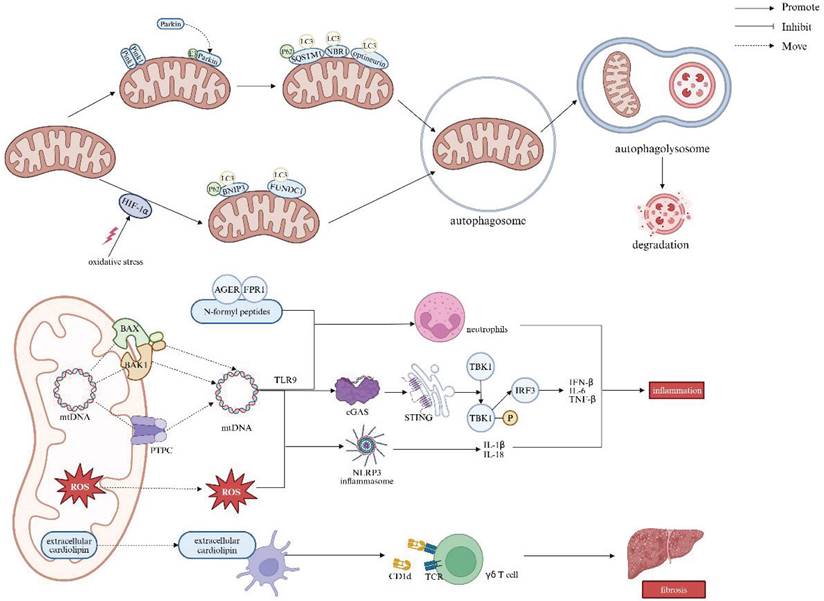
Cellular mitophagy is mediated through the Pink1/Parkin, BCL2 interacting protein 3/NIP3-like protein X (BNIP3L/Nix), and FUN14 domain-containing 1 (FUNDC1) pathways, with the Pink1/Parkin pathway being the most extensively studied in mammals [100]. In healthy mitochondria, Pink1 is typically undetectable, but it accumulates on the mitochondrial outer membrane in response to membrane depolarization. This accumulation recruits Parkin from the cytosol to specific damaged mitochondria, wherein activating Parkin's E3 ubiquitin ligase. Parkin then ubiquitinates mitochondrial outer membrane proteins, which facilitates the recruitment of autophagy adaptors such as sequestosome 1 (SQSTM1/P62), neighbor of BRCA1 (NBR1) gene, and optineurin. These adaptors bind to the autophagy-associated protein microtubule-associated protein 1 light chain 3 (LC3) at autophagosomes, leading to mitochondrial degradation [101]. In addition to Pink1/Parkin, newly discovered mitochondrial outer-membrane proteins, BNIP3 and FUNDC1, also serve as important receptors for hypoxia-induced mitophagy [102, 103]. Upon binding to LC3, these proteins enhance the autophagosome formation, in which a bilayer membrane envelops the mitochondrion. The autophagosome then fuses with a lysosome to orchestrate an autophagolysosome, the site for mitochondrion degradation. Prior studies have shown that the regulation of BNIP3 and FUNDC1 is dependent on the stabilization of hypoxia-inducible factor 1-alpha (HIF-1α) [104, 105]. However, oxidative stress can disrupt HIF-1α stabilization, impairing HIF-1α-mediated BNIP3/FUNDC1 mitophagy, and thus exacerbating liver injury [106].
Inhibition of mitophagy promotes the cytoplasmic release of mtDNA in macrophages, leading to the activation of the cGAS/stimulator of interferon genes (STING)/NLRP3 signaling pathway. mtDNA can escape from mitochondria across the pores formed by BCL-2-associated X protein (BAX) and BCL-2 antagonist/killer 1 (BAK1), or via the mitochondrial permeability transition pore complex (PTPC). This mtDNA leakage activates cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS), which in turn triggers stimulator of interferon response cGAMP interactor 1 (STING1) signaling and the subsequent production of pro-inflammatory cytokines, including interferon-β (IFN-β), IL-6, and TNF-α. Undoubtfully, these processes may exacerbate hepatic injury and fibrosis [107-109]. Moreover, upon release from dysfunctional mitochondria, both mtDNA and ROS can induce secretory IL-1β and IL-18 in the way of inflammasome activation. Additionally, extracellular mtDNA along with other mitochondrial components, such as N-formyl peptides, which accumulate during regulated cell death, can activate and boost neutrophils. This occurs through the recognition of mtDNA by Toll-like receptor 9 (TLR9), and N-formyl peptides by the advanced glycosylation end product-specific receptor (AGER) as well as formyl peptide receptor 1 (FPR1), respectively. Finally, extracellular CL, which is typically confined to the IMM, can be presented by dendritic cells on the major histocompatibility complex class I-like molecule CD1d, giving rise to the activation of γδ T cells [109].
The accumulation of dysfunctional mitochondria crushing the clearance capacity of mitophagy leads to impaired mitophagy, which in turn triggers inflammatory and fibrotic responses by promoting the release of mt-DAMPs and mtDNA. In immune cells, defective autophagy also results in overloaded autophagosomal and autolysosomal contents serving as inflammatory mediators, thereby activating pro-inflammatory signaling pathways. Moreover, mt-DAMPs, including mtDNA, bind to and activate TLR9 on collagen-producing HSCs. In this regard, direct activation of TLR9 subsequently induces HSCs activation and fibrosis (Figure 6) [62].
Relationship between mitophagy and liver diseases. Mitophagy is mediated by the Pink1/Parkin, BNIP3L/Nix, and FUNDC1 pathways, which promote autophagosome formation and fusion with lysosomes for mitochondrial degradation. Inhibition of mitophagy causes mtDNA release in macrophages, activating the cGAS/STING/NLRP3 pathway. MtDNA leaks through BAX/BAK1 pores or the mitochondrial PTPC, triggering cGAS and STING1 signaling, leading to pro-inflammatory cytokine production (e.g., IFN-β, IL-6, TNF-α). Both mtDNA and ROS activate inflammasomes, inducing IL-1β and IL-18 secretion. N-formyl peptides released during cell death enhance neutrophil activation. Extracellular cardiolipin activates γδ T cells via dendritic cell presentation on CD1d, promoting liver fibrosis. Note: LC3: light chain 3; HIF-1α: hypoxia-inducible factor 1-alpha; BNIP3: BCL2 interacting protein 3; FUNDC1: FUN14 domain-containing 1; BAX: BCL-2-associated X protein; BAK1: BCL-2 antagonist/killer 1; PTPC: permeability transition pore complex; ROS: reactive oxygen species; AGER: advanced glycosylation end product-specific receptor; FPR1: formyl peptide receptor 1; TLR9: Toll-like receptor 9; cGAS: cyclic GMP-AMP synthase; STING: stimulator of interferon response cGAMP interactor; TBK1: TANK-binding kinase 1; IRF3: interferon regulatory factor 3; NLRP3: NOD-like receptor thermal protein domain associated protein 3; TCR: T-cell receptor; IL-6: interleukin-6; IL-1β: interleukin-1β; IL-18: interleukin-18; TNF-β: tumor necrosis factor-β; IFN-β: interferon-β. Created with BioRender.com.
Spatial/Temporal Dimension of Macrophage Heterogeneity in Liver Diseases
Liver macrophages, particularly KCs, exhibit significant heterogeneity in response to different stimuli, which can adopt either pro-inflammatory M1-like or anti-inflammatory M2-like phenotypes, depending on the microenvironment and pathological conditions [110]. Macrophage polarization, influenced by mitochondrial dynamics, contributes significantly to liver inflammation and fibrosis [111-113]. Mitochondrial dysfunction is a key factor in the pathogenesis of liver diseases, especially in macrophages. In the context of liver inflammation, damaged mitochondria in macrophages release mtDNA and ROS, which activate inflammasomes and contribute to the production of pro-inflammatory cytokines. This inflammatory cascade not only exacerbates liver injury but also accelerates the progression of fibrosis [114-116]. The relationship between MQC and macrophage heterogeneity further complicates liver disease progression. In NAFLD and liver fibrosis, mitochondrial dysfunction in macrophages can skew the macrophage polarization towards a pro-inflammatory M1 phenotype [111, 117]. This polarization amplifies the release of cytokines like TNF-α and IL-1β, which perpetuate liver injury and fibrosis. Conversely, efficient MQC can promote the polarization of macrophages toward an anti-inflammatory M2 phenotype, which aids in tissue repair and resolution of inflammation. Recent studies through single-cell RNA sequencing have revealed that macrophages exhibit a considerably phenotypic diversity than previously established M1/M2 paradigm, underscoring the spatial and time dimension of macrophage heterogeneity in the liver [118]. In terms of spatial dimension, the predominant macrophage type in different liver regions may be linked to specific liver diseases. For example, TREM2+ CD9+ scar-associated macrophages can be observed in human liver cirrhosis exhibiting a profibrogenic gene signature [119], while TREM2-expressing macrophages have been identified in the circumstance of liver cancer [120]. Moreover, the localization of macrophages in distinct hepatic zones may influence their mitochondrial dynamics, thereby affecting their roles in inflammation and repair. Notably, positioned within liver sinusoids and extending through the space of Disse to hepatocyte layers, KCs are strategically placed to detect changes in the bloodstream and hepatocyte stress, which supports their involvement in immune tolerance and acute responses [121]. Additionally, previous studies have addressed that in conditions like NAFLD, there is a shift from a KC-dominant to a monocyte-derived macrophages (MoMFs)-dominant liver macrophage pool, particularly in the portal areas, strongly correlating with disease progression markers [122, 123]. Regarding the temporal aspect, liver macrophages exhibit dynamic responses to disruptions in homeostasis, with MoMFs migrating to injury sites to clear debris and initiate tissue repair. While KCs are relatively immobile, chemokine (C-C motif) receptor 2 positive MoMFs can rapidly transit through healthy liver circulation but slow down and accumulate at sites of active inflammation upon injury [124, 125]. Thus, the heterogeneity of macrophages in the liver and the critical role of MQC in maintaining macrophage function are central to the development and progression of liver diseases.
Therapies on Regulating Mitochondrial Quality Control to Protect Liver Diseases
Given mitochondrial function as a cornerstone in the pathogenesis of different liver diseases, therapeutic strategies aimed at regulating MQC held great promise. Here, we discuss potential strategies for therapeutically targeting MQC in liver diseases using small-molecule approaches (Table 2). Mdivi-1, an effective inhibitor of mitochondrial fission, prevented the self-assembly of DRP1 into oligomers and reduced its GTPase activity. In studies conducted by Zhang et al. in 2022 and 2024, it was found that Mdivi-1 can inhibit the proinflammatory responses by decreasing the level of phosphorylation of DRP1 at serine 616 site and attenuating the activation of STING signaling in KCs. [126, 127] It is highlighted that AICAR and PXL770 are the agonist of the AMPK pathway. In 2019, Hu et al. showed that the gene expression levels of transforming growth factor-β1 (TGF-β1), alpha-smooth muscle actin, and collagen 1 in HSCs are significantly decreased after AICAR treatment in the bile duct ligation mouse model prone to liver fibrosis and portal hypertension. Moreover, AICAR can alleviate portal pressure without changing systemic hemodynamics and alleviate liver cirrhosis in this model. [128] Another investigation conducted by Fouqueray et al. revealed that PXL770 can inhibit de novo lipogenesis and improve glycemic parameters and indices regarding insulin sensitivity by administering 12 overweight/obese patients with NAFLD and insulin resistance with PXL770. [128] Peroxisome proliferator-activated receptors (PPARs) are ligand-inducible transcription factors that regulate various genes, including PGC-1α [129]. Yatsuga et al. proved that PPAR agonists, such as Bezafibrate and Thiazolidinediones, enhance mitochondrial biogenesis in the manner of binding to and activating PPARs [130]. Idebenone, a structural analogue of coenzyme Q10, exhibits spatially restricted partial agonistic activity towards both PPARα and PPARγ. Tiefenbach et al. implementing a mouse model of type 2 diabetes found that idebenone alleviates fatty liver disease by binding to PPAR-α and regulating lipid metabolism [131]. Cilostazol, a 2-oxyquinolone derivative, exerts its effects by inhibiting phosphodiesterase III, leading to increased cyclic adenosine monophosphate (cAMP) levels [132]. In 2015, Joe et al. demonstrated that cilostazol upregulates the expression of PGC-1α, NRF1, and TFAM in hepatocytes, increases COX III/IV expression in addition to mtDNA content, thereby alleviating ischemia/reperfusion-induced hepatic mitochondrial damage [133].
Pharmacological targeting of mitochondrial quality control
| Target Type | Molecule Name | Functional Mechanism | Model | Clinical Trial | ||||
|---|---|---|---|---|---|---|---|---|
| Species | Period | Outcome | Status | Period | Identifier | |||
| Fission | Mdivi-1 | Inhibit the activation of DRP1 signaling | Mouse [127] | 8 weeks | Reduced hepatic steatosis | |||
| Biogenesis | AICAR | Stimulate AMPK | Rat [128] | 2 weeks | Decreased hepatic fibrogenesis | |||
| PXL770 | Stimulate AMPK | Phase Ⅰ /Completed | 5 weeks | NCT05441904 | ||||
| Bezafibrate /Thiazolidinediones | Bind to and activate PPARs | Mouse [130] | 6 weeks | Improved all liver functions and reduced oxidative stress | PhaseⅢ /Completed | 24 months | NCT01654731 | |
| Idebenone | Bind with PPAR-α | Mouse [131] | 3 weeks | Reversed fatty liver development | PhaseⅡ /Active | 48 weeks | NCT04669158 | |
| Cilostazol | Increase the expression of PGC-1α and the content of mtDNA | Mouse [133] | 12 weeks | Reduced liver lipid accumulation | PhaseⅡ /Active | 12 weeks | NCT04761848 | |
| Quercetin | Scavenge mitochondrial ROS and promote PGC-1α-mediated biogenesis | L02 cell [134] | 24 hours | Downregulated redox status, lipid droplets, and LPO release. Restored damaged mitochondrial membrane potential, repaired mtDNA damage, and improved mitochondrial dynamics | Phase I /Completed | 4 weeks | NCT01438320 | |
| Urolithin A | Promote mitophagy and activate Nrf2/ARE signaling | Mouse [135] | 48 hours | Reduced oxidative stress | Phase II /Completed | 4 Months | NCT03283462 | |
| Mitophagy | Metformin | Activate AMPK and restore the expression of UQCRC2 | Mouse [136] | 10 days | Reduced ethanol-induced liver injury | Phase Ⅳ /Completed | 48 weeks | NCT00546442 |
| TMP | Restore UQCRC2 expression | Mouse [137] | 4 weeks | Decreased oxidative stress and hepatic inflammation | ||||
| Diisopropylfluorophosphate (DFP) | Induce mitophagy by iron chelation | Mouse [139] | 2 weeks | Decreased hepatic inflammation | Phase Ⅳ /Completed | NCT01767103 | ||
| Icaritin | Recover Δψm and inhibit mTOR complex I | Mouse [140] | 1 week | Prolonged survival time of mice at the advanced stage of HCC | Phase Ⅱ /Completed | NCT01972672 | ||
Note: Drp1: dynamin-related protein 1; AICAR: 5-Aminoimidazole-4-carboxamide 1-β-D-ribofuranoside; AMPK: AMP-activated protein kinase; PPARs: peroxisome proliferator-activated receptors; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1α; mtDNA: mitochondrial DNA; ROS: reactive oxygen species; LPO: lipid peroxidation; Nrf2: nuclear factor erythroid 2-related factor 2; ARE: antioxidant response element; TMP: tetramethylpyrazine; UQCRC2: ubiquinol-cytochrome c reductase core protein 2; mTOR: mechanistic target of rapamycin; HCC: hepatocellular carcinoma.
Quercetin, a dietary phytochemical abundant in lots of fruits and vegetables, encompasses anti-inflammatory and antioxidant properties. Zhao et al. employed L02 cells to establish an in vitro model with ethanol-induced hepatocyte pyroptosis, indicated that quercetin protects hepatocytes by scavenging mitochondrial ROS and promoting PGC-1α-governed mitochondrial homeostasis [134]. Urolithin A represents a metabolite of ellagitannin-derived compounds derived from the gastrointestinal microbiota processing. One study reported that UA promotes mitophagy and activates the Nrf2/ARE signaling pathway, consequently, reducing oxidative stress in the context of acetaminophen-induced liver damage [135]. Ubiquinol-cytochrome c reductase core protein 2 (UQCRC2) constitutes a component of the mitochondrial ETC. Lu et al. implicated that activation of AMPK by metformin restores the expression of UQCRC2 through the activation of the NRF2 pathway, further enhancing Parkin recruitment to the mitochondria and promoting mitophagy, ultimately mitigating liver injury [136]. Tetramethylpyrazine (TMP), an extract from Ligusticum wallichii Franch, can promotes mitophagy by upregulating UQCRC2 levels, thereby bringing protective effects against alcohol-induced liver injury, inflammation, and ROS overproduction [137]. Collectively, these findings suggest that restoring UQCRC2 expression may enhance Parkin-mediated mitophagy, offering potential therapeutic benefits for liver diseases. Tong et al. demonstrated that Diisopropylfluorophosphate treatment, an FDA-approved orally active iron chelator [138], inhibits the ferroptosis markers ACSL4 and ALOX15 in MAFLD mouse models, while ameliorates hepatic inflammation via the induction of mitophagy [139]. In 2022, Yu et al. found that icaritin and doxorubicin remodel the immunosuppressive tumor microenvironment and trigger robust immune memory response, efficiently improving anti-HCC effect at an early stage in mouse HCC model. Their findings unveil an anti-HCC mechanism concerning icaritin on mitophagy and provide an effective immune-based therapeutic strategy in the context of HCC [140]. Overall, pharmacological targeting MQC shows promise as a potential treatment for liver diseases, although many studies are still at the animal or cell model stage.
Conclusion
MQC plays a crucial role in maintaining liver health and preventing the onset and progression of liver diseases. Understanding the intricate mechanisms with respect to MQC across distinct pathophysiological conditions undisputedly opens up new avenues for therapeutic interventions. Future researches focusing on mitochondrial-targeted treatments could offer novel strategies to improve the prevention and management of liver diseases, in hopes of achieving better clinical outcome.
Abbreviations
ACC: acetyl-CoA Carboxylase; ACC1: acetyl-CoA carboxylase 1; ADP: adenosine diphosphate; AGER: advanced glycosylation end product-specific receptor; AIF: apoptosis-inducing factor; ALD: alcohol-associated liver diseases; ALDH2: acetaldehyde dehydrogenase 2; AMPK: AMP-activated protein kinase; ATP: adenosine triphosphate; BAK1: BCL-2 antagonist/killer 1; BAX: BCL-2-associated X protein; BNIP3L: BCL2 interacting protein 3; cAMP: cyclic adenosine monophosphate; CAMKK2: calcium/calmodulin-dependent protein kinase kinase 2; cGAS: cyclic guanosine monophosphate-adenosine monophosphate synthase; CL: cardiolipin; COX IV: cytochrome c oxidase IV; CPT1: carnitine palmitoyl transferase 1; CPT1A: carnitine palmitoyl-transferase 1A; DAMPs: damage-associated molecular patterns; Drp1: dynamin-related protein 1; ETC: electron transport chain; FA: fatty acid; FADH2: flavin adenine dinucleotide; FAEEs: fatty acid ethyl esters; FAO: fatty acid oxidation; FASN: fatty acid synthase; Fe-S: Iron-sulfur; FPR1: formyl peptide receptor 1; FUNDC1: FUN14 domain-containing 1; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HFD: high-fat diet; HIF-1α: hypoxia-inducible factor 1-alpha; 4-HNE: 4-hydroxynonenal; HO-1: heme oxygenase-1; HSCs: hepatic stellate cells; IFN-β: interferon-β; IL-1β: interleukin-1β; IL-18: interleukin-18; IMM: inner mitochondrial membrane; IκB: inhibitor of kappa B; KCs: Kupffer cells; LC3: light chain 3; MAFLD: metabolic-associated fatty liver disease; MDA: malondialdehyde; MDVs: mitochondria-derived vesicles; MFN1: mitofusin 1; MFN2: mitofusin 2; MLROs: mitochondria-lysosome-related organelles; MoMFs: monocyte-derived macrophages; MQC: mitochondrial quality control; mtDNA: mitochondrial DNA; mTORC1: mechanistic target of rapamycin complex 1; NADH: nicotinamide adenine dinucleotide; NAFLD: non-alcoholic fatty liver disease; NBR1: neighbor of BRCA1; NF-κB: nuclear factor-kappa B; NLRP3: NOD-like receptor thermal protein domain associated protein 3; Nix: NIP3-like protein X; NRF1: nuclear respiratory factor 1; NRF2: nuclear respiratory factor 2; OMM: outer mitochondrial membrane; OPA1: optic atrophy 1; OXPHOS: oxidative phosphorylation; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1α; PPARs: peroxisome proliferators-activated receptors; PTP: permeability transition pore; PTPC: permeability transition pore complex; Rab9: ras-related protein 9; ROS: reactive oxygen species; SDHA: ruccinate dehydrogenase subunit-A; SIRT1: sirtuin 1; SLC25A1: mitochondrial citrate carrier; SQSTM1: sequestosome 1; SREBP1: sterol regulatory element binding protein 1; STING: stimulator of interferon genes; TCA: trichloroacetic acid; TFAM: transcription factor A; TFEB: transcription factor EB; TGF-β1: transforming growth factor-β1; TLR9: toll-like receptor 9; TNF-α: tumor necrosis factor-α; TMP: tetramethylpyrazine; ULK1: UNC-52-like kinase 1; UQCRC2: ubiquinol-cytochrome c reductase core protein 2; VDIM: vesicle derived from the inner mitochondrial membrane.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ma X, Niu M, Ni HM, Ding WX. Mitochondrial dynamics, quality control, and mtDNA in alcohol-associated liver disease and liver cancer. Hepatology. 2024
2. Li R, Toan S, Zhou H. Role of mitochondrial quality control in the pathogenesis of nonalcoholic fatty liver disease. Aging (Albany NY). 2020;12:6467-85
3. Vercellino I, Sazanov LA. The assembly, regulation and function of the mitochondrial respiratory chain. Nat Rev Mol Cell Biol. 2022;23:141-61
4. Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun. 2017;482:426-31
5. Picard M, Shirihai OS. Mitochondrial signal transduction. Cell Metab. 2022;34:1620-53
6. Horbay R, Bilyy R. Mitochondrial dynamics during cell cycling. Apoptosis. 2016;21:1327-35
7. Mansouri A, Gattolliat CH, Asselah T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology. 2018;155:629-47
8. Larson-Casey JL, He C, Carter AB. Mitochondrial quality control in pulmonary fibrosis. Redox Biol. 2020;33:101426
9. Vazquez-Calvo C, Suhm T, Büttner S, Ott M. The basic machineries for mitochondrial protein quality control. Mitochondrion. 2020;50:121-31
10. Li YJ, Cao YL, Feng JX, Qi Y, Meng S, Yang JF. et al. Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat Commun. 2019;10:4914
11. Chan DC. Mitochondrial Dynamics and Its Involvement in Disease. Annu Rev Pathol. 2020;15:235-59
12. Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265-87
13. Sidarala V, Zhu J, Levi-D'Ancona E, Pearson GL, Reck EC, Walker EM. et al. Mitofusin 1 and 2 regulation of mitochondrial DNA content is a critical determinant of glucose homeostasis. Nat Commun. 2022;13:2340
14. Miyao M, Kawai C, Kotani H, Minami H, Abiru H, Hamayasu H. et al. Mitochondrial fission in hepatocytes as a potential therapeutic target for nonalcoholic steatohepatitis. Hepatol Res. 2022;52:1020-33
15. Elbadawy M, Tanabe K, Yamamoto H, Ishihara Y, Mochizuki M, Abugomaa A. et al. Evaluation of the efficacy of mitochondrial fission inhibitor (Mdivi-1) using non-alcoholic steatohepatitis (NASH) liver organoids. Front Pharmacol. 2023;14:1243258
16. Quan Y, Shou D, Yang S, Cheng J, Li Y, Huang C. et al. Mdivi1 ameliorates mitochondrial dysfunction in non-alcoholic steatohepatitis by inhibiting JNK/MFF signaling. J Gastroenterol Hepatol. 2023;38:2215-27
17. Steffen J, Ngo J, Wang SP, Williams K, Kramer HF, Ho G. et al. The mitochondrial fission protein Drp1 in liver is required to mitigate NASH and prevents the activation of the mitochondrial ISR. Mol Metab. 2022;64:101566
18. Takeichi Y, Miyazawa T, Sakamoto S, Hanada Y, Wang L, Gotoh K. et al. Non-alcoholic fatty liver disease in mice with hepatocyte-specific deletion of mitochondrial fission factor. Diabetologia. 2021;64:2092-107
19. Da Dalt L, Moregola A, Svecla M, Pedretti S, Fantini F, Ronzio M. et al. The inhibition of inner mitochondrial fusion in hepatocytes reduces non-alcoholic fatty liver and improves metabolic profile during obesity by modulating bile acid conjugation. Cardiovasc Res. 2024;119:2917-29
20. Kunlayawutipong T, Apaijai N, Tepmalai K, Kongkarnka S, Leerapun A, Pinyopornpanish K. et al. Imbalance of mitochondrial fusion in peripheral blood mononuclear cells is associated with liver fibrosis in patients with metabolic dysfunction-associated steatohepatitis. Heliyon. 2024;10:e27557
21. Shami GJ, Cheng D, Verhaegh P, Koek G, Wisse E, Braet F. Three-dimensional ultrastructure of giant mitochondria in human non-alcoholic fatty liver disease. Sci Rep. 2021;11:3319
22. Palma E, Riva A, Moreno C, Odena G, Mudan S, Manyakin N. et al. Perturbations in Mitochondrial Dynamics Are Closely Involved in the Progression of Alcoholic Liver Disease. Alcohol Clin Exp Res. 2020;44:856-65
23. Palma E, Ma X, Riva A, Iansante V, Dhawan A, Wang S. et al. Dynamin-1-Like Protein Inhibition Drives Megamitochondria Formation as an Adaptive Response in Alcohol-Induced Hepatotoxicity. Am J Pathol. 2019;189:580-9
24. Das S, Hajnóczky N, Antony AN, Csordás G, Gaspers LD, Clemens DL. et al. Mitochondrial morphology and dynamics in hepatocytes from normal and ethanol-fed rats. Pflugers Arch. 2012;464:101-9
25. Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG. et al. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci U S A. 2014;111:6413-8
26. Kim SJ, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013;9:e1003722
27. Picca A, Lezza AM. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion. 2015;25:67-75
28. Liu L, Li Y, Wang J, Zhang D, Wu H, Li W. et al. Mitophagy receptor FUNDC1 is regulated by PGC-1α/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 2021;22:e50629
29. Dominy JE, Puigserver P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol. 2013;5:a015008
30. Jiang Y, Chen D, Gong Q, Xu Q, Pan D, Lu F. et al. Elucidation of SIRT-1/PGC-1α-associated mitochondrial dysfunction and autophagy in nonalcoholic fatty liver disease. Lipids Health Dis. 2021;20:40
31. Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol. 2011;23:476-82
32. Leonhard K, Guiard B, Pellecchia G, Tzagoloff A, Neupert W, Langer T. Membrane protein degradation by AAA proteases in mitochondria: extraction of substrates from either membrane surface. Mol Cell. 2000;5:629-38
33. Prashar A, Bussi C, Fearns A, Capurro MI, Gao X, Sesaki H. et al. Lysosomes drive the piecemeal removal of mitochondrial inner membrane. Nature. 2024;632:1110-7
34. Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA. et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22:135-41
35. Jiao H, Jiang D, Hu X, Du W, Ji L, Yang Y. et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. 2021;184:2896-910.e13
36. Tan HWS, Lu G, Dong H, Cho YL, Natalia A, Wang L. et al. A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nat Commun. 2022;13:3720
37. Ding WX, Li M, Biazik JM, Morgan DG, Guo F, Ni HM. et al. Electron microscopic analysis of a spherical mitochondrial structure. J Biol Chem. 2012;287:42373-8
38. Ma X, Ni HM, Ding WX. Perspectives of mitochondria-lysosome-related organelle in hepatocyte dedifferentiation and implications in chronic liver disease. eGastroenterology. 2024 2
39. Ma X, Manley S, Qian H, Li Y, Zhang C, Li K. et al. Mitochondria-lysosome-related organelles mediate mitochondrial clearance during cellular dedifferentiation. Cell Rep. 2023;42:113291
40. Ma X, Ding WX. Quality control of mitochondria involves lysosomes in multiple definitive ways. Autophagy. 2024;20:2599-601
41. Chao X, Wang S, Ma X, Zhang C, Qian H, Williams SN. et al. Persistent mTORC1 activation due to loss of liver tuberous sclerosis complex 1 promotes liver injury in alcoholic hepatitis. Hepatology. 2023;78:503-17
42. Liu P, Anandhan A, Chen J, Shakya A, Dodson M, Ooi A. et al. Decreased autophagosome biogenesis, reduced NRF2, and enhanced ferroptotic cell death are underlying molecular mechanisms of non-alcoholic fatty liver disease. Redox Biol. 2023;59:102570
43. Krishnasamy Y, Gooz M, Li L, Lemasters JJ, Zhong Z. Role of mitochondrial depolarization and disrupted mitochondrial homeostasis in non-alcoholic steatohepatitis and fibrosis in mice. Int J Physiol Pathophysiol Pharmacol. 2019;11:190-204
44. Moore MP, Cunningham RP, Meers GM, Johnson SA, Wheeler AA, Ganga RR. et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology. 2022;76:1452-65
45. Samuvel DJ, Li L, Krishnasamy Y, Gooz M, Takemoto K, Woster PM. et al. Mitochondrial depolarization after acute ethanol treatment drives mitophagy in living mice. Autophagy. 2022;18:2671-85
46. Ma Y, Chai H, Ding Q, Qian Q, Yan Z, Ding B. et al. Hepatic SIRT3 Upregulation in Response to Chronic Alcohol Consumption Contributes to Alcoholic Liver Disease in Mice. Front Physiol. 2019;10:1042
47. Loureiro D, Tout I, Narguet S, Bed CM, Roinard M, Sleiman A. et al. Mitochondrial stress in advanced fibrosis and cirrhosis associated with chronic hepatitis B, chronic hepatitis C, or nonalcoholic steatohepatitis. Hepatology. 2023;77:1348-65
48. Cho HM, Ryu JR, Jo Y, Seo TW, Choi YN, Kim JH. et al. Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol Cell. 2019;73:364-76.e8
49. Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D. et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230-3
50. Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S. et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433-5
51. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA. et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329-35
52. Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM. et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9-19
53. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W. et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456-61
54. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132-41
55. Saito T, Nah J, Oka SI, Mukai R, Monden Y, Maejima Y. et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest. 2019;129:802-19
56. Wu D, Yang Y, Hou Y, Zhao Z, Liang N, Yuan P. et al. Increased mitochondrial fission drives the reprogramming of fatty acid metabolism in hepatocellular carcinoma cells through suppression of Sirtuin 1. Cancer Commun (Lond). 2022;42:37-55
57. Rasheed M, Tarjan G. Succinate Dehydrogenase Complex: An Updated Review. Arch Pathol Lab Med. 2018;142:1564-70
58. Hederstedt L. Structural biology. Complex II is complex too. Science. 2003;299:671-2
59. Zhou Z, Ma A, Moore TM, Wolf DM, Yang N, Tran P. et al. Drp1 controls complex II assembly and skeletal muscle metabolism by Sdhaf2 action on mitochondria. Sci Adv. 2024;10:eadl0389
60. Ma X, McKeen T, Zhang J, Ding WX. Role and Mechanisms of Mitophagy in Liver Diseases. Cells. 2020;9:837
61. Thoudam T, Gao H, Jiang Y, Huda N, Yang Z, Ma J. et al. Mitochondrial quality control in alcohol-associated liver disease. Hepatol Commun. 2024;8:e0534
62. Lemasters JJ, Zhong Z. Mitophagy in hepatocytes: Types, initiators and role in adaptive ethanol metabolism☆. Liver Res. 2018;2:125-32
63. Williams JA, Ding WX. A Mechanistic Review of Mitophagy and Its Role in Protection against Alcoholic Liver Disease. Biomolecules. 2015;5:2619-42
64. Park SH, Seo W, Xu MJ, Mackowiak B, Lin Y, He Y. et al. Ethanol and its Nonoxidative Metabolites Promote Acute Liver Injury by Inducing ER Stress, Adipocyte Death, and Lipolysis. Cell Mol Gastroenterol Hepatol. 2023;15:281-306
65. Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longev. 2010;3:178-85
66. Rumgay H, Murphy N, Ferrari P, Soerjomataram I. Alcohol and Cancer: Epidemiology and Biological Mechanisms. Nutrients. 2021;13:3173
67. Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct. 2012;2012:646354
68. Luther J, Khan S, Gala MK, Kedrin D, Sridharan G, Goodman RP. et al. Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation. Proc Natl Acad Sci U S A. 2020;117:11667-73
69. Snel M, Jonker JT, Schoones J, Lamb H, de Roos A, Pijl H. et al. Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. Int J Endocrinol. 2012;2012:983814
70. Domínguez-Pérez M, Simoni-Nieves A, Rosales P, Nuño-Lámbarri N, Rosas-Lemus M, Souza V. et al. Cholesterol burden in the liver induces mitochondrial dynamic changes and resistance to apoptosis. J Cell Physiol. 2019;234:7213-23
71. Yadav DK, Kumar S, Choi EH, Chaudhary S, Kim MH. Molecular dynamic simulations of oxidized skin lipid bilayer and permeability of reactive oxygen species. Sci Rep. 2019;9:4496
72. Mollica MP, Lionetti L, Moreno M, Lombardi A, De Lange P, Antonelli A. et al. 3,5-diiodo-l-thyronine, by modulating mitochondrial functions, reverses hepatic fat accumulation in rats fed a high-fat diet. J Hepatol. 2009;51:363-70
73. Vial G, Dubouchaud H, Couturier K, Cottet-Rousselle C, Taleux N, Athias A. et al. Effects of a high-fat diet on energy metabolism and ROS production in rat liver. J Hepatol. 2011;54:348-56
74. Petito G, Cioffi F, Silvestri E, De Matteis R, Lattanzi D, de Lange P. et al. 3,5-Diiodo-L-Thyronine (T2) Administration Affects Visceral Adipose Tissue Inflammatory State in Rats Receiving Long-Lasting High-Fat Diet. Front Endocrinol (Lausanne). 2021;12:703170
75. Kidane D, Chae WJ, Czochor J, Eckert KA, Glazer PM, Bothwell AL. et al. Interplay between DNA repair and inflammation, and the link to cancer. Crit Rev Biochem Mol Biol. 2014;49:116-39
76. Xu J, Cao K, Li Y, Zou X, Chen C, Szeto IM. et al. Bitter gourd inhibits the development of obesity-associated fatty liver in C57BL/6 mice fed a high-fat diet. J Nutr. 2014;144:475-83
77. Cioffi F, Giacco A, Petito G, de Matteis R, Senese R, Lombardi A. et al. Altered Mitochondrial Quality Control in Rats with Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD) Induced by High-Fat Feeding. Genes (Basel). 2022;13:315
78. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121-35
79. Liu P, Lin H, Xu Y, Zhou F, Wang J, Liu J. et al. Frataxin-Mediated PINK1-Parkin-Dependent Mitophagy in Hepatic Steatosis: The Protective Effects of Quercetin. Mol Nutr Food Res. 2018;62:e1800164
80. Zhou T, Chang L, Luo Y, Zhou Y, Zhang J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019;21:101120
81. Wang L, Liu X, Nie J, Zhang J, Kimball SR, Zhang H. et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology. 2015;61:486-96
82. Edmunds LR, Xie B, Mills AM, Huckestein BR, Undamatla R, Murali A. et al. Liver-specific Prkn knockout mice are more susceptible to diet-induced hepatic steatosis and insulin resistance. Mol Metab. 2020;41:101051
83. Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II. et al. Measuring In Vivo Mitophagy. Mol Cell. 2015;60:685-96
84. Seen S. Chronic liver disease and oxidative stress - a narrative review. Expert Rev Gastroenterol Hepatol. 2021;15:1021-35
85. Liu Y, Huang Y, Xu C, An P, Luo Y, Jiao L. et al. Mitochondrial Dysfunction and Therapeutic Perspectives in Cardiovascular Diseases. Int J Mol Sci. 2022;23:16053
86. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103-15
87. Arra M, Swarnkar G, Ke K, Otero JE, Ying J, Duan X. et al. LDHA-mediated ROS generation in chondrocytes is a potential therapeutic target for osteoarthritis. Nat Commun. 2020;11:3427
88. Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol. 2019;25:101084
89. Torres S, Segalés P, García-Ruiz C, Fernández-Checa JC. Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Cells. 2022;11:1475
90. Zong Y, Li H, Liao P, Chen L, Pan Y, Zheng Y. et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024;9:124
91. Tell G, Vascotto C, Tiribelli C. Alterations in the redox state and liver damage: hints from the EASL Basic School of Hepatology. J Hepatol. 2013;58:365-74
92. Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018;592:728-42
93. Chen P, Yao L, Yuan M, Wang Z, Zhang Q, Jiang Y. et al. Mitochondrial dysfunction: A promising therapeutic target for liver diseases. Genes Dis. 2024;11:101115
94. Zheng J, Conrad M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020;32:920-37
95. Mahmoud AM, Mohammed HM, Khadrawy SM, Galaly SR. Hesperidin protects against chemically induced hepatocarcinogenesis via modulation of Nrf2/ARE/HO-1, PPARγ and TGF-β1/Smad3 signaling, and amelioration of oxidative stress and inflammation. Chem Biol Interact. 2017;277:146-58
96. Wang Y, Qi H, Liu Y, Duan C, Liu X, Xia T. et al. The double-edged roles of ROS in cancer prevention and therapy. Theranostics. 2021;11:4839-57
97. Kurachi K, Suzuki S, Sakaguchi T, Yokoi Y, Konno H, Baba S. et al. Kupffer cells modulate splenic interleukin-10 production in endotoxin-induced liver injury after partial hepatectomy. J Hepatol. 2003;38:193-9
98. Zhai L, Pei H, Yang Y, Zhu Y, Ruan S. NOX4 promotes Kupffer cell inflammatory response via ROS-NLRP3 to aggravate liver inflammatory injury in acute liver injury. Aging (Albany NY). 2022;14:6905-16
99. Zhao Y, Wang Z, Feng D, Zhao H, Lin M, Hu Y. et al. p66Shc Contributes to Liver Fibrosis through the Regulation of Mitochondrial Reactive Oxygen Species. Theranostics. 2019;9:1510-22
100. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F. et al. Molecular definitions of autophagy and related processes. Embo j. 2017;36:1811-36
101. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85:257-73
102. Delgado JM, Shepard LW, Lamson SW, Liu SL, Shoemaker CJ. The ER membrane protein complex restricts mitophagy by controlling BNIP3 turnover. Embo j. 2024;43:32-60
103. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P. et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177-85
104. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J. et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570-81
105. Zhang W, Guo C, Li Y, Wang H, Wang H, Wang Y. et al. Mitophagy mediated by HIF-1α/FUNDC1 signaling in tubular cells protects against renal ischemia/reperfusion injury. Ren Fail. 2024;46:2332492
106. Sun Y, Wen F, Yan C, Su L, Luo J, Chi W. et al. Mitophagy Protects the Retina Against Anti-Vascular Endothelial Growth Factor Therapy-Driven Hypoxia via Hypoxia-Inducible Factor-1α Signaling. Front Cell Dev Biol. 2021;9:727822
107. Wang Q, Bu Q, Liu M, Zhang R, Gu J, Li L. et al. XBP1-mediated activation of the STING signalling pathway in macrophages contributes to liver fibrosis progression. JHEP Rep. 2022;4:100555
108. Wu NN, Wang L, Wang L, Xu X, Lopaschuk GD, Zhang Y. et al. Site-specific ubiquitination of VDAC1 restricts its oligomerization and mitochondrial DNA release in liver fibrosis. Exp Mol Med. 2023;55:269-80
109. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol. 2023;23:159-73
110. Verdeguer F, Aouadi M. Macrophage heterogeneity and energy metabolism. Exp Cell Res. 2017;360:35-40
111. Tao R, Han M, Yuan W, Xiao F, Huang J, Wang X. et al. Fibrinogen-like protein 2 promotes proinflammatory macrophage polarization and mitochondrial dysfunction in liver fibrosis. Int Immunopharmacol. 2023;117:109631
112. Wang Y, Guo Y, Xu Y, Wang W, Zhuang S, Wang R. et al. HIIT Ameliorates Inflammation and Lipid Metabolism by Regulating Macrophage Polarization and Mitochondrial Dynamics in the Liver of Type 2 Diabetes Mellitus Mice. Metabolites. 2022;13:14
113. Guan D, Huang P, Liu X, Li Q, Zhang X, Liu N. et al. Deficiency of myeloid NPC1 exacerbates liver injury and fibrosis by impairing macrophage efferocytosis. J Adv Res. 2024:S2090-1232(24)00539-3.
114. Wu D, Xu J, Zhang Y, Wang Y, Bai Y, Zhan X. et al. tBHQ mitigates fatty liver ischemia-reperfusion injury by activating Nrf2 to attenuate hepatocyte mitochondrial damage and macrophage STING activation. Int Immunopharmacol. 2024;138:112515
115. Gao L, Zuo XL, Dong LL, Zhou SF, Wang ZJ, Duan YS. et al. Hepatocyte mitochondrial DNA mediates macrophage immune response in liver injury induced by trichloroethylene. Ecotoxicol Environ Saf. 2024;276:116317
116. Lee JH, Seo KH, Yang JH, Cho SS, Kim NY, Kim JH. et al. CCCP induces hepatic stellate cell activation and liver fibrogenesis via mitochondrial and lysosomal dysfunction. Free Radic Biol Med. 2024;225:181-92
117. Ma Z, Xie K, Xue X, Li J, Yang Y, Wu J. et al. Si-Wu-Tang attenuates hepatocyte PANoptosis and M1 polarization of macrophages in non-alcoholic fatty liver disease by influencing the intercellular transfer of mtDNA. J Ethnopharmacol. 2024;328:118057
118. Guillot A, Tacke F. Spatial dimension of macrophage heterogeneity in liver diseases. eGastroenterology. 2023 1
119. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT. et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512-8
120. Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G. et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54:1883-900.e5
121. Bonnardel J, T'Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L. et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity. 2019;51:638-54.e9
122. Guillot A, Winkler M, Silva Afonso M, Aggarwal A, Lopez D, Berger H. et al. Mapping the hepatic immune landscape identifies monocytic macrophages as key drivers of steatohepatitis and cholangiopathy progression. Hepatology. 2023;78:150-66
123. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A. et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185:379-96.e38
124. Filliol A, Saito Y, Nair A, Dapito DH, Yu LX, Ravichandra A. et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature. 2022;610:356-65
125. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T. et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology. 2016;64:1667-82
126. Zhang Q, Wei J, Liu Z, Huang X, Sun M, Lai W. et al. STING signaling sensing of DRP1-dependent mtDNA release in kupffer cells contributes to lipopolysaccharide-induced liver injury in mice. Redox Biol. 2022;54:102367
127. Zhang Q, Liu Z, Huang X, Heng X, Wu J, Chen Z. et al. MDIVI-1 ALLEVIATES SEPSIS-INDUCED LIVER INJURY BY INHIBITING STING SIGNALING ACTIVATION. Shock. 2024;62:95-102
128. Hu L, Su L, Dong Z, Wu Y, Lv Y, George J. et al. AMPK agonist AICAR ameliorates portal hypertension and liver cirrhosis via NO pathway in the BDL rat model. J Mol Med (Berl). 2019;97:423-34
129. Jamwal S, Blackburn JK, Elsworth JD. PPARγ/PGC1α signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharmacol Ther. 2021;219:107705
130. Yatsuga S, Suomalainen A. Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum Mol Genet. 2012;21:526-35
131. Tiefenbach J, Magomedova L, Liu J, Reunov AA, Tsai R, Eappen NS. et al. Idebenone and coenzyme Q(10) are novel PPARα/γ ligands, with potential for treatment of fatty liver diseases. Dis Model Mech. 2018;11:dmm034801
132. Kherallah RY, Khawaja M, Olson M, Angiolillo D, Birnbaum Y. Cilostazol: a Review of Basic Mechanisms and Clinical Uses. Cardiovasc Drugs Ther. 2022;36:777-92
133. Joe Y, Zheng M, Kim HJ, Uddin MJ, Kim SK, Chen Y. et al. Cilostazol attenuates murine hepatic ischemia and reperfusion injury via heme oxygenase-dependent activation of mitochondrial biogenesis. Am J Physiol Gastrointest Liver Physiol. 2015;309:G21-9
134. Zhao X, Wang C, Dai S, Liu Y, Zhang F, Peng C. et al. Quercetin Protects Ethanol-Induced Hepatocyte Pyroptosis via Scavenging Mitochondrial ROS and Promoting PGC-1α-Regulated Mitochondrial Homeostasis in L02 Cells. Oxid Med Cell Longev. 2022;2022:4591134
135. Gao Z, Yi W, Tang J, Sun Y, Huang J, Lan T. et al. Urolithin A protects against acetaminophen-induced liver injury in mice via sustained activation of Nrf2. Int J Biol Sci. 2022;18:2146-62
136. Lu X, Xuan W, Li J, Yao H, Huang C, Li J. AMPK protects against alcohol-induced liver injury through UQCRC2 to up-regulate mitophagy. Autophagy. 2021;17:3622-43
137. Zhou Y, Wu R, Wang X, Jiang Y, Xu W, Shao Y. et al. Activation of UQCRC2-dependent mitophagy by tetramethylpyrazine inhibits MLKL-mediated hepatocyte necroptosis in alcoholic liver disease. Free Radic Biol Med. 2022;179:301-16
138. Fiorillo M, Tóth F, Brindisi M, Sotgia F, Lisanti MP. Deferiprone (DFP) Targets Cancer Stem Cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production. Cells. 2020;9:1529
139. Tong J, Lan XT, Zhang Z, Liu Y, Sun DY, Wang XJ. et al. Ferroptosis inhibitor liproxstatin-1 alleviates metabolic dysfunction-associated fatty liver disease in mice: potential involvement of PANoptosis. Acta Pharmacol Sin. 2023;44:1014-28
140. Yu Z, Guo J, Hu M, Gao Y, Huang L. Icaritin Exacerbates Mitophagy and Synergizes with Doxorubicin to Induce Immunogenic Cell Death in Hepatocellular Carcinoma. ACS Nano. 2020;14:4816-28
141. Han D, Ybanez MD, Johnson HS, McDonald JN, Mesropyan L, Sancheti H. et al. Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: biogenesis, remodeling, and functional alterations. J Biol Chem. 2012;287:42165-79
142. Williams JA, Ni HM, Ding Y, Ding WX. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am J Physiol Gastrointest Liver Physiol. 2015;309:G324-40
Author contact
![]() Corresponding authors: Jie Zhang, M.D., Ph.D., E-mail: Zhangjie_xhkedu.cn; Fax: +86-022-27813550; Chao Sun, M.D., Ph.D., E-mail: chaosunedu.cn; Telephone: +8613820846027; Fax: +86-022-27813550.
Corresponding authors: Jie Zhang, M.D., Ph.D., E-mail: Zhangjie_xhkedu.cn; Fax: +86-022-27813550; Chao Sun, M.D., Ph.D., E-mail: chaosunedu.cn; Telephone: +8613820846027; Fax: +86-022-27813550.