Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(4):1801-1817. doi:10.7150/ijbs.104067 This issue Cite
Research Paper
FBXW7 Directly Ubiquitinates and Degrades CTNNB1 Mediating the Suppression of ENKUR in Endometrial Cancer
YaHui Liu1,2#, Qian Wang2#, QiRun Guo2,#, Ying Zhu2, Li Lin2, ChunYan Yang3, Bin Gong1, Weiwei Yan1, RenTao Hou4, Yao Tang1, XiuQiong Wu1, Xinhui Liu5, BeiXian Zhou6, WeiYi Fang1,2, LuYun Shu3, SuiQun Guo2 ![]()
1. Oncology department, Southern Medical University Hospital of Integrated Traditional Chinese and Western Medicine, Southern Medical University, Guangzhou 510315, China.
2. Department of Obstetrics and Gynecology, The Third Affiliated Hospital/The Third Clinical Medicine School of Southern Medical University, Guangzhou 510315, China.
3. Department of Gynecology and Oncology, Huaihua Cancer Hospital, Huaihua 418100, China.
4. Department of radiotherapy, Shunde Hospital, Southern Medical University (The First People's Hospital of Shunde Foshan), 1# Jiazi Road, Foshan, 528300, Guangdong, China.
5. Department of Gastrointestinal Oncology, Shandong Cancer Hospital and Institute, Shandong First Medical University & Shandong Academy of Medical Science, Jinan 250117, China.
6. The People's Hospital of Gaozhou, Gaozhou 525200, China.
#These authors contributed equally to this work.
Received 2024-9-21; Accepted 2025-1-21; Published 2025-2-10
Abstract

Enkurin (ENKUR) is a tumor suppressor in some malignancies. However, its role in endometrial cancer (EC) remains unknown. Here, we firstly observed that reduced ENKUR expression promotes progression and poor prognosis in EC. Moreover,the overexpression of ENKUR suppressed the proliferation, migration, invasion, and intrahepatic dissemination of EC in vitro and in vivo. Repressing ENKUR expression by small-interfering RNA significantly reversed the inhibition of cell proliferation and invasion in vitro. We used co-immunoprecipitation combined with mass spectral analysis to identify the potential interactive proteins of ENKUR. Based on Gene Ontology analysis, we discovered that Wnt/β-catenin (Wnt/CTNNB1) signaling is a ENKUR-modulated key pathway. ENKUR binds to CTNNB1, significantly repressing its protein expression. Furthermore, ENKUR also binds to E3 ligase F-box and WD repeat domain containing 7 (FBXW7), a critical tumor suppressor. Interestingly, the latter binds to CTNNB1 and S502 of CTNNB1 is the key binding site, thereby increasing its protein ubiquitination and degradation. Finally, we confirmed that the predominant ubiquitination sites of CTNNB1 are located at K281 and K394. Transfection of ENKUR-overexpressing EC cells with CTNNB1 reversed the suppressive effects on tumor growth and invasion. ENKUR may be a tumor suppressor via recruiting FBXW7 to directly ubiquitinate and degrade CTNNB1 in EC.
Keywords: ENKUR, FBXW7, CTNNB1, ubiquitination, endometrial cancer
Introduction
Endometrial carcinoma (EC) is the most common gynecologic malignancy worldwide. It is classified into different subtypes, with endometrioid and serous carcinoma representing 80-85% and 5-10% of newly diagnosed cases, respectively[1, 2]. Despite great progress achieved in the treatment and diagnosis of EC[3-8], the incidence of advanced EC continues to increase[9]. However, the pathogenesis of EC remains to be clarified.
Enkurin, also known as the “enhanced nuclear factor kappa-B receptor” (ENKUR), is located at chromosomal band 10p12.1. It encodes a calmodulin that was originally identified using yeast two-hybrid screening in a study of transient receptor potential canonical (TRPC)[10]. ENKUR is a conserved protein of flagella and cilia[11]. Therefore, ENKUR protein is a necessary protein for sperm cilium movement and transport in the female fallopian tube, playing important roles in signal transduction and gamete fusion during the fertilization process of Cystoisospora suis[10, 12-14]. Additionally, an increase in ENKUR protein has been reported as an early diagnostic biomarker to predict the occurrence of noise-induced hearing loss at a very early stage[15]. At present, there are very few reports on the role of ENKUR in tumors. It has been identified as a novel marker for myeloproliferative neoplasms from platelet, megakaryocyte, and whole blood specimens[16]. Follow-up studies demonstrated that ENKUR is a tumor suppressor participating in the pathogenesis of colorectal cancer (CRC), lung adenocarcinoma (LUAD), hepatocellular carcinoma (HCC) and nasopharyngeal carcinoma (NPC)[17-21]. However, thus far, studies have not focused on the role of ENKUR in EC. In the present study, we detected the expression of ENKUR in EC, and evaluated its tumor suppressor role in human EC cells and nude mice.
Materials and methods
Tissue specimens
A total of 178 paraffin-embedded EC specimens and 23 endometrial epithelium samples were obtained from The Third Affiliated Hospital of Southern Medical University, Guangzhou City, China. Consent from the patients and approval from the Ethics Committee of the Third Affiliated Hospital of Southern Medical University were obtained before using the clinical samples for research purposes. Subcutaneous and intrahepatic dissemination tumors obtained from each group of nude mice (five mice per group) were immediately embedded in paraffin, sliced (thickness: 0.3µm), and subjected to hematoxylin-and-eosin (HE) and immunohistochemistry (IHC) staining.
IHC staining
ENKUR and proliferating cell nuclear antigen (PCNA) protein expression levels were detected by IHC staining. Immunochromogenic reagent (UltraSensitive sp mouse/rabbit) (MXB Biotechnologies, Fuzhou, China) and DAB kit (ZSGB BIO) were used for IHC staining. IHC was performed as previously described[22]. Two senior doctors in the dapartment of pathology evaluated the results of IHC staining. IHC scoring was based on a semiquantitative method according to the intensity and percentage of staining, as previously described[22]. The intensity of staining was scored using a scale ranging 0-3 (0=negative staining, 1=weakly positive staining, 2=moderately positive staining, and 3=strongly positive staining). The percentage of staining was estimated based on a scale ranging 0-4 (0=none, 1=positive staining in 1-25% of cancer cells, 2=positive staining in 26-50%; 3=positive staining in 51-75%; and 4=positive staining in 76-100%). The IHC score was determined by multiplying the intensity score by the percentage score; scores ≥6 and <6 denoted high and low expression, respectively. Details of the antibodies used are listed in Table S1.
Cell culture
RL95-2 and HEC-1-B cell lines obtained from the Cancer Research Institute of Southern Medical University (Guangzhou, China) were cultured in DMEM/MEM (Vivacell, Shanghai, China) supplemented with 1% penicillin-streptomycin (Biological Industries, Israel) and 10% fetal bovine serum (FBS) (Biowest, Nuaillé, France). HEK293T cells were acquired from the research group of Lukui Chen at the Integrated Hospital of Traditional Chinese Medicine of Southern Medical University, and were cultured in DMEM (Vivacell) containing 10% FBS (Biowest). All cells were incubated in 95% air and 5% CO2 at 37°C.
RNA extraction and reverse transcription-quantitative-polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the cell lines using the Total RNA Isolation Kit (Foregene, Chengdu, China). The concentration of total RNA was determined using Nanodrop 2000 (Thermo Fisher Scientific, Guangzhou, China). ENKUR and CTNNB1 mRNA was amplified using SYBR Premix Ex Taq, and reverse transcription was conducted using the PrimeScript RT reagent kit (Takara Biotechnology Co., Ltd., Dalian, China) to quantify ENKUR and CTNNB1 mRNA expression. Reverse transcription was performed at 37°C for 15 min and 85°C for 5 s. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin were used as internal controls for ENKUR and CTNNB1. The PCR primers used in the present study are listed in Table S2. Fold-changes for ENKUR and CTNNB1 mRNA expression levels were calculated using the 2-ΔΔCt method.
Lentivirus infection and transient transfection
Lentiviral particles carrying human ENKUR short hairpin RNA ENKUR and empty vector controls (PLV-Ctr) (Table S3) were constructed by GeneChem Company (Shanghai, China). RL95-2 and HEC-1-B cells were infected with lentiviral vectors with green fluorescent protein (Fig. S1C). Transient transfection using plasmids or small-interfering RNAs of FBXW7 and CTNNB1 were designed and synthesized by RiboBio Inc. (Guangzhou, China) (Table S3). Plasmids with His or Flag containing CTNNB1 mutant sequences were obtained from Miaoling Biology (Wuhan, China) (G42952, G42950, G42949, G42913, G42914). At 4-5 h prior to transfection, RL95-2, HEC-1-B or 293T cells were seeded into a plate (Nest Biotech, Shanghai, China) at 30-50% confluence. Next, the cells were transfected with small-interfering RNA (siRNA) or plasmids using Lipofectamine™ 3000 (Invitrogen Biotechnology, Guangzhou, China) for the following experiments, according to the instructions provided by the manufacturer.
Western blotting
Western blotting was performed using the Mini-PROTEAN Tetra- and Mini Trans-Blot (Bio-Rad, Hercules, CA, USA) systems, as previously described (https://www.westernblotprotocol.com).
Images were captured using a Chemiluminescence Imaging System (Minichemi, Beijing, China). We used antibodies against GAPDH, β-tubulin, ENKUR, CTNNB1, FBXW7, β-actin, hemagglutinin (HA), ubiquitin (UB), K48, Flag, and His, which are listed in Table S1.
MTT assays and colony formation assays
The MTT assay was conducted to detect cell viability according to the instructions provided by the manufacturer (Sigma-Aldrich, Shanghai, China). The optical density value was measured at 490 nm wavelength. EC cells were seeded at a density of 300-800 cells/well for colony formation. After 2 weeks of culture, the cells were washed with phosphate-buffered saline (PBS) and stained with hematoxylin solution. The number of colonies in each well was determined. All experiments were performed at least thrice.
EdU incorporation
Cells were seeded into 96-well plates at 8,000 cells/well, and experiments were performed in triplicate for each condition in each group. At 6 h after seeding, the culture medium was changed to FBS-free or low-FBS medium for 24 h to synchronize the cell cycle. EdU staining was performed using the Cell-Light EdU Apollo567 in vitro Kit (Ribobio Inc., Guangzhou, China) and 4',6-Diamidino-2-phenylindole dihydrochloride (DAPI; 5 μg/mL) (Beyotime Biotechnology, Shanghai, China) according to the instructions provided by the manufacturer.
Flow cytometric analysis
To assess the cell cycle, a suspension of 1 million cells per milliliter was treated with 70% ethanol and stored at 4°C for a minimum of 12 h. Subsequently, the cells were exposed to RNase A at a concentration of 50 µg/mL at 37°C for 30 min. The DNA within the cells was stained using propidium iodide at a concentration of 50 µg/mL. The cell cycle distribution was determined using a LSRFortessa (BD Biosciences, Franklin Lakes, NJ, USA) flow cytometer equipped with fluorescence-activated cell sorting capabilities.
Wound-healing assay
EC cells transfected with plasmids, lentiviruses or siRNAs were seeded into six-well plates. We performed artificial wounds on cells using a 100 μL pipette tip, and the cells were subsequently washed. After incubation for 48 h in a humidified chamber, the degree of wound healing was determined to estimate the migratory ability of cells.
Transwell assay
Cells (1×105 cells/well) were seeded into a 24-well plate and washed twice with PBS. The upper chamber of the Transwell plate contained 100 μL of DMEM culture medium, while the lower chamber contained 500 μL of DMEM with 10% FBS. After culture at 37°C for 18-20 h, the cells that had not penetrated the membrane surface in the upper chamber were wiped off, and the remaining attached cells were washed thrice with PBS. Thereafter, the cells were fixed with paraformaldehyde for 10 min, stained with crystal violet for 15 min, and washed with PBS. Images of cell migration were captured under a microscope (Olympus Corporation, Tokyo, Japan) (100× magnification). Three fields of view were randomly selected to calculate the number of cells, with the average value denoting the number of cells penetrating the membrane.
Boyden assay
Initially, 24 µg/mL Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) was applied to precoat Transwell membranes (BD Biosciences) for 30 min. Next, a total of 1×105 cells were resuspended in 100 µL of serum-free DMEM culture medium and added to the upper chamber. Subsequently, DMEM culture medium containing 10% FBS was added to the lower chamber (500 µL). The filter was collected 24-30 h later, and staining with crystal violet solution for 3 min was performed. The cell capacity for invasion was assessed based on the average value obtained from three random fields.
Liquid chromatography-tandem mass spectrometry analysis and Gene Ontology (GO) enrichment analysis
The entire process of library construction and sequencing was performed at Shanghai Lifegenes Technology Co., Ltd. (Shanghai, China). Mass spectrometry analyses were performed at Shanghai Lifegenes Technology Co., Ltd, using 20 µg of proteins from the Co-immunoprecipitation (Co-IP) assays. GO enrichment analysis of differentially expressed genes was conducted using the clusterProfiler R package (v3.18.1)[23] (https://guangchuangyu.github.io/software/clusterProfiler). GO terms with p-values <0.05 denoted significant enrichment of differentially expressed genes.
In vivo tumorigenesis in nude mice
In vivo experiments were approved by the Animal Care and Use Committee of Southern Medical University and were performed in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. Nude mice (age: 4 weeks) were purchased from SPF (Beijing) Biotechnology Co., Ltd. (Beijing, China). The subcutaneous xenograft tumor model was constructed as follows[22]. A total of 5×106 EC cells in logarithmic growth were transfected with the ENKUR-overexpression plasmid or vector in 0.1 mL of Hanks' balanced salt solution (Chemical Book, Shanghai, China). Thereafter, they were subcutaneously injected into the mice (BALB/c; nu/nu; age: 4 weeks; sex: female; weight: 12-13 g; n=5 per group). The long and short diameters of the tumors were measured once every 3 days using a Vernier caliper. The tumor volume was calculated as follows: V=ab2/2 (a=tumor length, b=tumor width). At 21 days, the mice were sacrificed, and the tumors were removed, fixed using 4% paraformaldehyde, embedded in paraffin, and sectioned for subsequent detection (i.e., HE staining and PCNA staining)[22].
A mouse model of intrahepatic dissemination was constructed as follows. Mice (BALB/c; nu/nu; age: 4 weeks; sex: female; weight: 12-13 g; n=5 per group) were divided into four groups (i.e., RL95-2-Vector, RL95-2-ENKUR, HEC-1-B-Vector, and HEC-1-B-ENKUR cells) and anesthetized by intraperitoneal injection of 0.5% pentobarbital sodium (50 mg/kg; Sigma, Shanghai, China). The liver was exposed ,1×106 EC cells with stable ENKUR overexpression and control cells were resuspended in PBS and injected under the liver capsule using a needle, thereafter, the abdominal incision was sutured and disinfected. After 60 days, the mice were sacrificed, and the livers were isolated to observe intrahepatic dissemination tumors under fluorescence microscopy. Next, the intrahepatic dissemination tumors were fixed using 4% paraformaldehyde for observation.
Co-IP
Co-IP was carried out using a Pierce Co-IP kit (Thermo Scientific, Waltham, MA, USA). Briefly, total protein was extracted and quantified. A total of 1,000 mg of protein in 400 mL of supernatant was incubated with 10 mg of antibodies against ENKUR, CTNNB1, Flag, His, UB, FBXW7, and immunoglobulin G (IgG) for 12 h at 4°C. The beads (Bimake, Shanghai, China) were washed and eluted in sample buffer, and boiled for 5 min at 95°C. Immune complexes were subjected to western blotting analysis. Anti-IgG antibody was used as a negative control.
Immunofluorescence and confocal microscopy
Cells were separated and seeded on coverslips in a 48-well plate. After adherence, the cells were fixed using paraformaldehyde (4%) and permeabilized with Triton X-100 (0.2%). Subsequently, the cells were incubated with specific antibodies (i.e., against FBXW7, CTNNB1, and ENKUR) and counterstained with DAPI (0.2 mg/mL). Finally, images were captured under a Carl Zeiss LSM800 confocal laser scanning microscope (Zeiss, Germany).
Cycloheximide (CHX) chase and ubiquitination assays
CHX chase and ubiquitination assays were performed as previously described[19, 24]. CHX (50 µg/mL) (S7418; Selleck, Shanghai, China) was used to treat EC cells for various periods of time. Proteins were extracted using radioimmunoprecipitation assay lysis buffer, mixed with sodium dodecyl sulfate-loading buffer, boiled for 10 min, and subjected to immunoblotting. For the ubiquitination assays, cells were treated with MG132 (20 µM) (S2619; Selleck) for 12 h and proteins were harvested. Proteins were extracted using IP lysis buffer, and IP was performed with anti-CTNNB1 antibodies. Harvested proteins were resolved in sodium dodecyl sulfate-loading buffer, boiled for 10 min, and subjected to immunoblotting.
Protein-protein docking studies
Protein-protein docking was performed using the HDOCK server (http://hdock.phys.hust.edu.cn/). This docking program initially sampled the putative binding modes between the two proteins through a fast Fourier transform-based global search method 7. Thereafter, the sampled binding modes were evaluated with an improved iterative knowledge-based scoring function for protein-protein interactions[25-27]. The electron microscopy structures of CTNNB1 (Protein Data Bank [PDB] identifier: 8Y0G) as receptors and the X-ray crystallographic structures of FBXW7 (PDB identifier: 2OVP) were considered ligands for the protein-protein docking. These structures were recovered from the PDB (https://www.rcsb.org/). PyMol software (DeLano Scientific LLC, Shanghai, China) was used to visualize the docking results for the members with the highest scores.
Statistical analysis
Statistical analyses were performed with the SPSS 19.0 statistical software package (IBM Corp., Armonk, NY, USA). Data are expressed as the mean±standard deviation from at least three independent experiments. Comparisons were performed using a Student's t-test for two groups, one-way analysis of variance (ANOVA) for multiple groups, and a parametric generalized linear model with random effects for tumor growth. Survival analysis was performed using the Kaplan-Meier method. All statistical tests were two-sided. The p-values <0.05 indicated statistically significant differences.
Results
Reduced ENKUR promotes EC progression
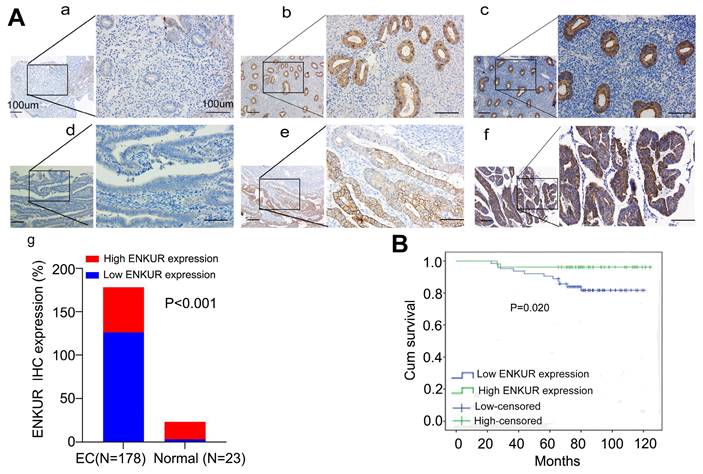
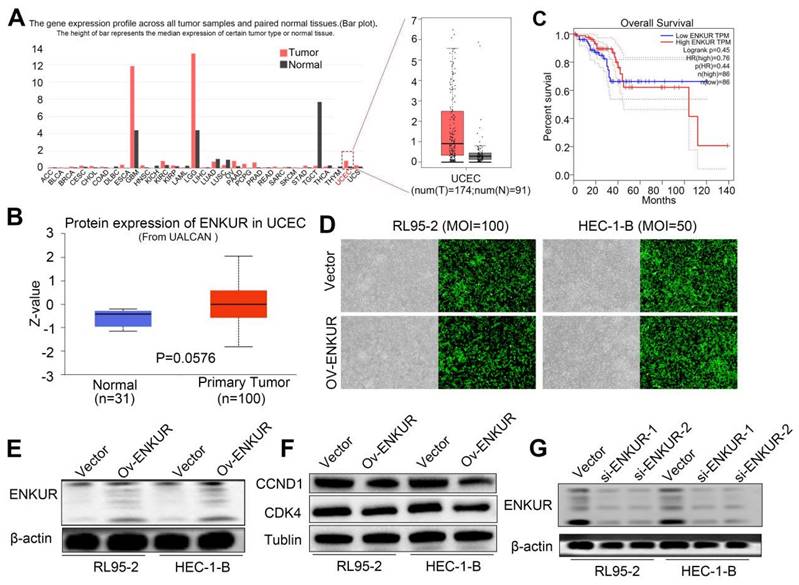
We initially sought to investigate whether ENKUR is involved in the pathogenesis of EC. Thus, we used the online Gene Expression Profiling Interactive Analysis (GEPIA) and UALCAN databases to predict changes in ENKUR expression. The data showed that both ENKUR mRNA and protein expression levels were upregulated in EC tissues compared with healthy endometrial and endometrial hyperplasia samples (Fig. S1A, B). Interestingly, we observed that high ENKUR expression predicts a favorable prognosis for patients with EC (Fig. S1C). We further confirmed that ENKUR expression is significantly downregulated in EC tissues compared with normal endometrial tissues by IHC (Fig. 1A and Table 1). The clinical characteristics of patients with EC are summarized in Table 2. There were no significant associations found between ENKUR expression and patient age, clinical stage, hormone levels, hypertension history, etc. Nevertheless, we observed that low ENKUR expression was associated with shorter patient survival than high ENKUR expression (log-rank test, p=0.020) (Fig. 1B). This evidence was consistent with the survival data obtained from the database analysis.
Lower expression of ENKUR promotes EC and is associated with a worse prognosis. (A) IHC staining of ENKUR in EC tissues and normal endometrial tissues. a: negative of ENKUR in normal endometrial tissues; b: positive of ENKUR in normal endometrial tissues; c: strongly positive of ENKUR in normal endometrial tissues; d: negative of ENKUR in EC samples; e: positive of ENKUR in EC tissues; f: strongly positive of ENKUR in EC tissues; g: a total 29.2% of endometrial cancer samples while 85% of normal endometrial tissues were strongly positive for ENKUR, ENKUR was significantly lower expressed in endometrial cancer compared with that in endometrial tissues (P<0.001). (B) A Kaplan-Meier survival analysis is shown for the overall survival of 178 EC patients on the basis of ENKUR expression. A log-rank test was used to calculate P-values, P=0.020. Scale bars, 100μm.
The expression of ENKUR in EC and Normal endometrium
| Group | Case (n) | ENKUR expression | P-value* | |
|---|---|---|---|---|
| Low | High | |||
| EC | 178 | 126 (70.8%) | 52(29.2%) | <0.001 |
| Normal endometrium | 23 | 3(15%) | 20 (85%) | |
EC: endometrial cancer
*χ2-test was applied to access the expression of ENKUR in EC and Normal
Correlation of ENKUR protein expression with clinical pathological parameters
| Parameters | Case | Low expression | High expression | P value |
|---|---|---|---|---|
| Age (y) | 0.077 | |||
| <54 | 78 | 60 | 18 | |
| ≥54 | 100 | 66 | 34 | |
| Hypertension history | 0.229 | |||
| With | 57 | 38 | 19 | |
| Without | 120 | 88 | 32 | |
| Hormonal levels | 0.230 | |||
| ER(+)PR(-) | 4 | 2 | 2 | |
| PR(+)ER(-) | 14 | 7 | 7 | |
| ER (-) PR(-) | 67 | 50 | 17 | |
| ER (+) PR(+) | 93 | 67 | 26 | |
| Clinical stages | 0.495 | |||
| I | 150 | 104 | 46 | |
| II | 20 | 15 | 5 | |
| III | 8 | 7 | 1 | |
| Histopathological grade | 0.947 | |||
| G1 | 32 | 23 | 9 | |
| G2 | 57 | 41 | 16 | |
| G3 | 89 | 62 | 27 |
ENKUR overexpression decreases EC cell growth in vitro and in vivo
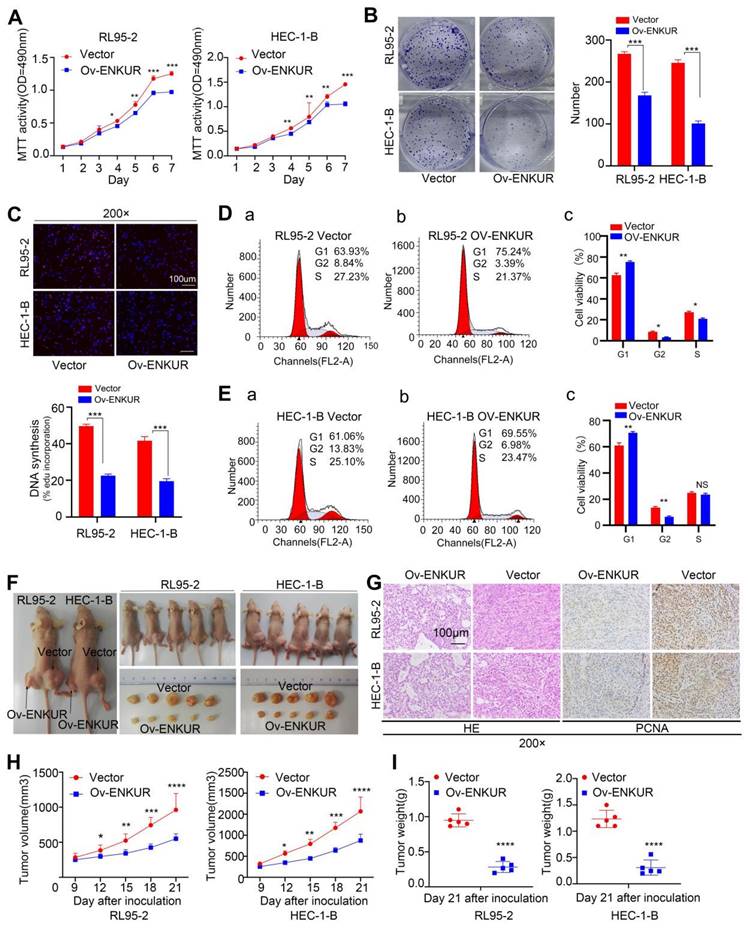
We also sought to verify the influence of ENKUR in EC cell proliferation. For this purpose, we used lentiviruses to establish stable ENKUR expression in EC cells (Fig. S1D). Significant ENKUR overexpression was confirmed in both cell lines (Fig. S1D, E). Subsequently, we carried out MTT, plate cloning, and EdU experiments. The results showed markedly reduced proliferation of EC cells overexpressing ENKUR compared with vector cells (Fig. 2A-C). Flow cytometry assay indicated that cell arrest was exhibited during the G1 phase in ENKUR overexpression EC cells (Fig. 2D, E). Western blotting indicated that in the ENKUR-overexpressing cell line, the protein levels of cyclin D1 (CCND1) and cyclin dependent kinase 4 (CDK4) were decreased (Fig. S1F). We also established a subcutaneous xenograft tumor model in nude mice to further verify the role of ENKUR in vivo (Fig. 2F). Xenograft tumors were verified by HE staining (Fig. 2G). IHC study of PCNA expression in xenograft tumors tissues also showed markedly reduced proliferation of ENKUR-overexpressing cells compared with vector cells (Fig. 2G). The tumor volume and weight were significantly lower in the ENKUR overexpression group versus the vector group (Fig. 2H-I).
ENKUR reduces EC proliferation in vitro and in vivo (Stable interference). The effects of ENKUR on the cell proliferation of RL95-2 and HEC-1-B cells were examined by (A) MTT assay, (B)Colony formation assay, (C) EdU experiments and (F) Xenograft tumour model in nude mice. (D-E) Flow cytometry analysis of EC cells showing G1 arrest of cell cycle. (F) Xenograft tumour model in nude mice. (G) Xenografts were stained with HE and subjected to immunohistochemistry for pcna expression (n=5 per group). (H)Tumor volume of xenografts in nude mice were measured (n=5 per group). (I)Tumor weights of xenografts in nude mice were measured (n=5 per group). Student's t test. Mean±SD. **p < 0.01, ***p < 0.001, ****p < 0.0001. Scale bars, 100µm.
Overexpression of ENKUR suppresses the migration, invasion and intrahepatic dissemination ability of EC cells
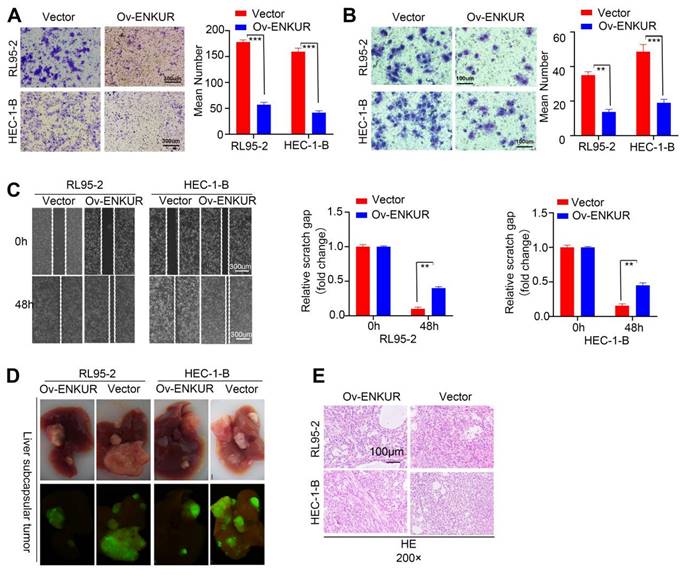
To assess the effect of ENKUR on the migratory and invasive capabilities of cells, Transwell invasion (Fig. 3A), Boyden chamber (Fig. 3B), and cell scratch (Fig. 3C) experiments were conducted. The results showed remarkable inhibition of the capacity of ENKUR-overexpressing cells for migration and invasion. In addition, western blotting showed downregulation of N-cadherin and Vimentin expression and upregulation of E-cadherin in ENKUR-overexpressing cells (Fig. S2A). To further verify the role of ENKUR in vivo, we established a intrahepatic dissemination model in nude mice. The numbers of intrahepatic dissemination tumors were calculated and compared. A significantly lower number of nude mice with intrahepatic dissemination was noted in the ENKUR-overexpressing group than the vector group (Fig. 3D). The intrahepatic dissemination tumors were dissected, embedded into wax blocks, and sliced. Subsequently, the sections were processed for HE staining (Fig. 3E). These data indicated that ENKUR inhibits tumor migration, invasion and intrahepatic dissemination in EC.
ENKUR decreased the invasion, migration, and intrahepatic dissemination of EC cells. (A) Transwell invasion, (B) Boyden chamber, and (C) cell scratch experiment showed that the capacities of ENKUR-overexpressing cells for migration and invasion were remarkably inhibited. (D) Intrahepatic dissemination model in nude mice, representative liver images for each group (E) HE staining of excised tumor tissues. Student's t-test. Data are presented as the mean±standard deviation. **p<0.01, ***p<0.001, ****p<0.0001. Original magnification: 40×. Scale bars: 100 µm, 300 µm. Each bar represents the mean±standard deviation of three independent experiments.
Targeting ENKUR restores cell growth, migration, and invasion
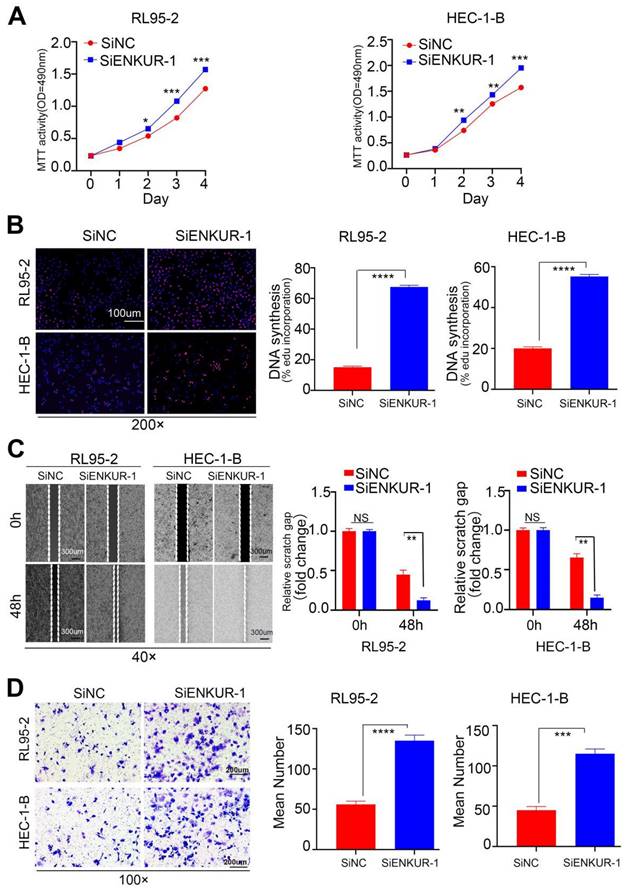
To further investigate the actions of ENKUR on EC, RL95-2 and HEC-1B cell lines were transfected with ENKUR siRNAs (Fig. S1G). The siRNA fragment #1 was selected for subsequent experiments. Next, MTT (Fig. 4A) and EdU experiments (Fig. 4B) were performed to verify changes in the proliferative ability of cells. The findings showed that targeting ENKUR cells significantly increased the number of proliferating EC cells. Cell scratch (Fig. 4C) and Transwell invasion (Fig. 4D) experiments showed that the migratory and invasive capabilities of targeted ENKUR cells were remarkably inhibited.
Silencing ENKUR restores cell growth, migration, and invasion. The effects of targeting ENKUR on the cell proliferation of RL95-2 and HEC-1-B cells were examined using (A). MTT assay and (B) EdU experiments. The effects of targeting ENKUR on the migratory and invasive abilities of RL95-2 and HEC-1-B cells were examined using (C) Cell scratch and (D) Transwell invasion experiments. Student's t-test. Data are presented as the mean±standard deviation. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Original magnification: 100×, 200×. Scale bars: 100 µm, 300 µm. Each bar represents the mean±standard deviation of three independent experiments.
ENKUR combines with CTNNB1
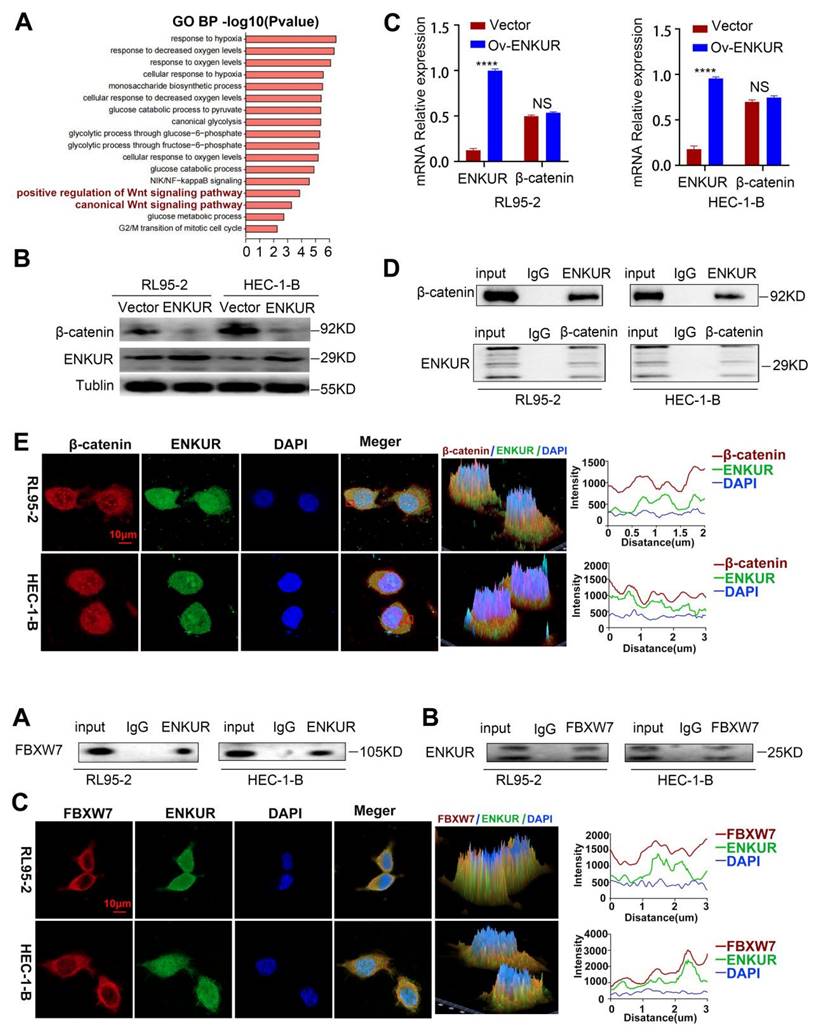
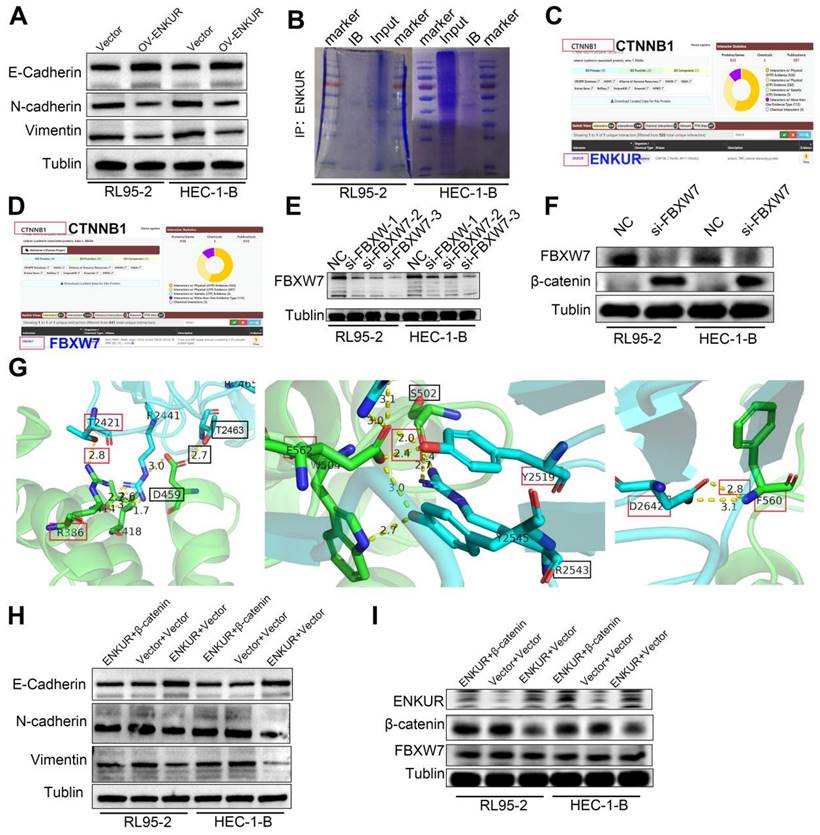
We attempted to elucidate the inhibition mechanisms of ENKUR in EC. Co-IP of cells with stable ENKUR overexpression and mass spectrometric analysis were performed to identify the partners that interact with ENKUR in EC (Fig. S2B). Furthermore, GO pathway enrichment analysis was performed on differential proteins. The analysis showed that ENKUR was involved in cell response through the canonical Wnt signaling pathway, etc. (Fig. 5A). Western blotting experiments revealed a marked decrease in the expression of canonical Wnt signaling pathway-related protein CTNNB1 in ENKUR-overexpressing EC cells (Fig. 5B). However, there was no significant alteration in CTNNB1 mRNA expression (Fig. 5C). BioGRID database analysis predicted that CTNNB1 interacts with ENKUR (Fig. S2C). In addition, Co-IP experiments were conducted in RL95-2 and HEC-1-B cells to confirm their interaction (Fig. 5D). Immunofluorescence staining also revealed colocalization of ENKUR with CTNNB1 in the cytoplasm and nucleus of EC cells; of note, cytoplasm localization was more pronounced (Fig. 5E). Overall, these results suggest that ENKUR binds with CTNNB1 in EC cells.
ENKUR interacts with CTNNB1. (A) GO pathway based on the differential interaction protein expression of cell lines (RL95-2, HEC-1-B) with ENKUR or vector group. (B)CTNNB1 were detected in the ENKUR overexpression EC cells by WB experiments, tublin served as a loading control. (C) The transcript levels of CTNNB1 mRNA were measured by RT-PCR. (D) Interactions of ENKUR and CTNNB1 in RL95-2 and HEC-1-B cells were detected by Co-IP analysis. (E)The colocalization of ENKUR and CTNNB1 were visualized by immunofluorescence staining in EC cells. Scale bar, 10 µm.
ENKUR interacts with FBXW7
We further investigated the regulatory mechanisms of ENKUR in EC. Co-IP experiments were conducted in RL95-2 and HEC-1-B cells to verify the interaction between ENKUR and FBXW7 (Fig. 6A, B). Immunofluorescence staining also revealed colocalization of ENKUR and FBXW7 in the cytoplasm of EC cells (Fig. 6C). Overall, these results suggest that ENKUR interacts with FBXW7 in EC cells.
ENKUR interacts with FBXW7. (A and B) Co-IP analysis was performed to assess the interaction of ENKUR and FBXW7 in RL95-2 and HEC-1-B cells. (C) Colocalization of ENKUR and FBXW7 was evaluated by immunofluorescence staining. Scale bar: 10 µm.
FBXW7 interacts with CTNNB1
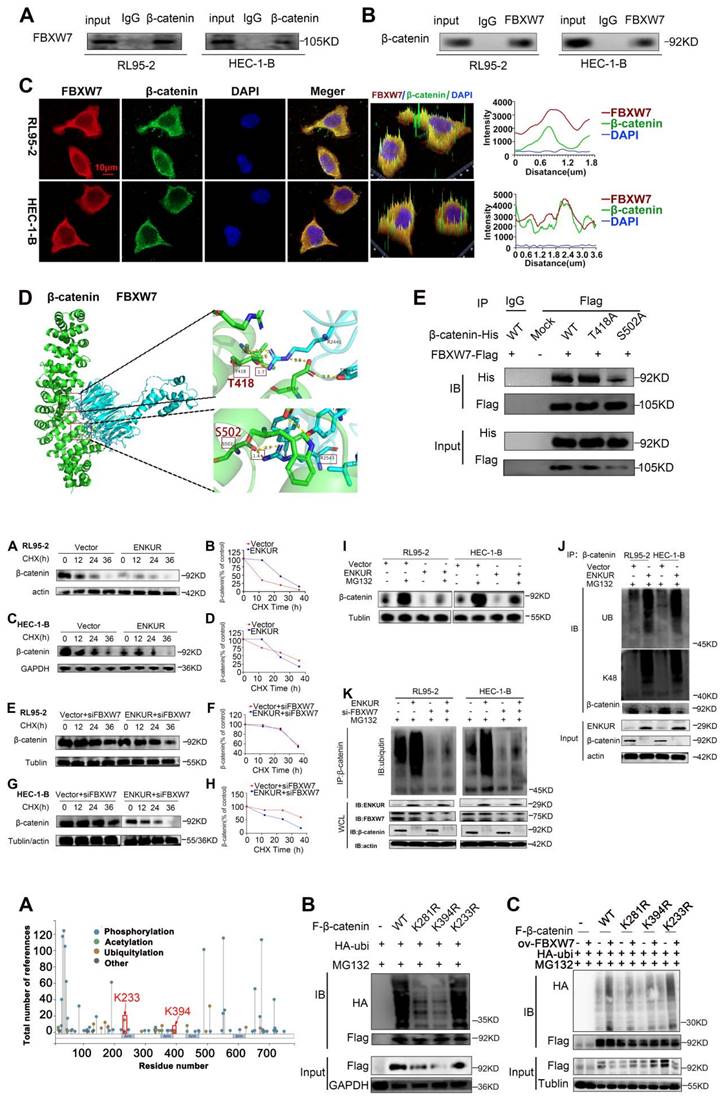
BioGRID database analysis was conducted to evaluate CTNNB1 as a potential FBXW7-binding protein (Fig. S2D). Co-IP assay and immunofluorescence staining showed cytoplasmic colocalization of CTNNB1 and FBXW7 proteins in EC cells (Fig. 7A-C). Western blotting experiments revealed a marked increase in the expression of CTNNB1 in FBXW7 -deducing EC cells (Fig. S2E-F). To further investigate the potential interaction of CTNNB1 and FBXW7, protein-protein docking was carried out using the HDOCK Server. Hydrogen bonding interactions were formed between the T2421, Y2519, T2463, and D2642 of FBXW7 with R386, E562, D459, and F560 of CTNNB1, respectively (Fig. S2G). In addition, the distances between the acceptor and donor heavy atoms of the two hydrogen bonds were both less than 3 Å, indicating bond stability. We selected the two binding sites with the shortest distance on CTNNB1 (i.e., T418 and S502), which correspond to R2441 and R2543 on FBXW7, respectively (Fig. 7D). We mutated these two sites to alanine residues (T418A, S502A) to test their stability and function. The results of Co-IP assays revealed a significant reduction in the binding affinity between FBXW7 and the CTNNB1 S502 mutant compared with the wild-type (WT) (Fig. 7E). Cells transfected with pCMV-CTNNB1(human)-WT-8×His-Neo served as the CTNNB1-WT group. These results suggested that CTNNB1 mutation at the S502 site has a significant impact on the CTNNB1-FBXW7 protein interaction.
FBXW7 binds to CTNNB1. (A and B) Co-IP analysis was performed to assess the interaction of FBXW7 and CTNNB1 in RL95-2 and HEC-1-B cells. (C) Colocalization of CTNNB1 and FBXW7 was evaluated by immunofluorescence staining. (D) Detailed binding sites between FBXW7 and CTNNB1. Key residues of FBXW7 (cyan) and CTNNB1 (green) are displayed as sticks. Hydrogen bonds are displayed in yellow dash lines and the distances (acceptor to donor heavy atom) of hydrogen bonds are labeled. (E) HEK293T cells were transfected with the mutants of CTNNB1 and analyzed by immunoprecipitation using an anti-Flag antibody. Scale bar: 10 µm.
ENKUR ubiquitinates and degrades CTNNB1 via recruiting FBXW7
Ubiquitination analysis was performed to further investigate the influence of ENKUR on CTNNB1. CHX chase assay revealed that the CTNNB1 half-life was significantly shorter in ENKUR-overexpressing cells versus vector cells (Fig. 8A, C). Moreover, CHX chase assay results showed that decreased expression of FBXW7 reduced the effect of ENKUR overexpression on CTNNB1 protein levels (Fig. 8E, G). In addition, CTNNB1 downregulation induced by ENKUR overexpression was reversed following treatment with MG132 for 12 h (Fig. 8I). Thereafter, the results of immunoprecipitation and western blotting analyses determined that, depending on FBXW7, the overexpression of ENKUR promoted K48-linked CTNNB1 ubiquitination (Fig. 8J, K). These results suggested that ENKUR mediated the ubiquitination and degradation of K48-linked CTNNB1 through the recruitment of FBXW7.
ENKUR accelerates CTNNB1 ubiquitination through FBXW7 recruitment. (A,C) CHX chase analysis of CTNNB1 protein half-life in ENKUR over-expressing and Vector group in RL95-2 and HEC-1-B cells. CHX (50 µg/ml). (E,G) CHX chase analysis detected the effects of FBXW7 knockdown on protein stability of CTNNB1. (B,D,F,H) The half-life curves of CTNNB1 protein in RL95-2 and HEC-1-B cells. (I) The effects of MG132 (20 µM) treatment on the stability of the CTNNB1 protein in the Vector and ENKUR overexpression groups. (J) Co-IP and western blotting assays was detected the effect of ENKUR overexpression on CTNNB1 ubiquitination at K48. (K) Co-IP and western blotting assays were used to examine the effect of ENKUR overexpression and treated with siFBXW7 on the ubiquitination level of β-catenin.
FBXW7 ubiquitinates and degrades CTNNB1 at the K281 and K394 sites
We further investigated the mechanism by which FBXW7 catalyzed the ubiquitination and degradation of CTNNB1. PhosphoSitePlus and GPS-UBER databases were used to search for potential ubiquitin-modified amino acid sites in CTNNB1. The results suggested that K233, K394, and K281 are potential CTNNB1 ubiquitination sites (Fig. 9A and Table 3). Plasmids with mutations of lysine to arginine at the potential ubiquitination sites were constructed to prevent ubiquitination. Next, 293T cells were transfected with plasmids expressing HA-UB, K233R, K394R, and K281R of CTNNB1, and wild-type β-catenin (CTNNB1-WT); cells transfected with pCMV-CTNNB1(human)-WT-3×FLAG-Neo served as the CTNNB1-WT group. The results showed that cells transfected with CTNNB1-WT and CTNNB1 with K233R generated a normal ubiquitination signal, whereas those with mutated K394 and K281 ubiquitination sites underwent remarkably reduced ubiquitination modification (Fig. 9B). Subsequently, we observed that FBXW7 significantly reduced CTNNB1 ubiquitination levels in the K394- and K281-mutant groups compared with the CTNNB1-WT group. These findings suggested that K394 and K281 of CTNNB1 are important for FBXW7-mediated CTNNB1 ubiquitination, and that mutations of these sites can significantly reduce UB modification catalyzed by FBXW7 (Fig. 9C). These results demonstrated that K394 and K281 are key regulatory sites through which FBXW7 mediates CTNNB1 ubiquitination modification.
The predicted binding sites between CTNNB1 and FBXW7. (A) PhosphoSitePlus analysis showing potential ubiquitin-modified amino acid sites in CTNNB1. (B) Comparison of ubiquitination modification between wild-type CTNNB1 and CTNNB1 with different mutations. (C) Effect of FBXW7 on CTNNB1 ubiquitination at the K233R, K394R, and K281R sites.
ID:sp|P35222|CTNB1_HUMAN Catenin beta-1 OS=Homo sapiens OX=9606 GN=CTNNB1 PE=1 SV=1 (from https://gpsuber.biocuckoo.cn/wsresult.php)
| Position | Code | Kinase | Peptide | Score | Cutoff |
|---|---|---|---|---|---|
| 158 | K | General | LATRAIPELTKLLNDEDQVVV | 0.4163 | 0.2655 |
| 233 | K | General | HHREGLLAIFKSGGIPALVKM | 0.4839 | 0.2655 |
| 281 | K | General | MAVRLAGGLQKMVALLNKTNV | 0.5891 | 0.2655 |
| 335 | K | General | VNIMRTYTYEKLLWTTSRVLK | 0.3478 | 0.2655 |
| 345 | K | General | KLLWTTSRVLKVLSVCSSNKP | 0.278 | 0.2655 |
| 354 | K | General | LKVLSVCSSNKPAIVEAGGMQ | 0.4002 | 0.2655 |
| 394 | K | General | TLRNLSDAATKQEGMEGLLGT | 0.6038 | 0.2655 |
| 666 | K | General | AAVLFRMSEDKPQDYKKRLSV | 0.3441 | 0.2655 |
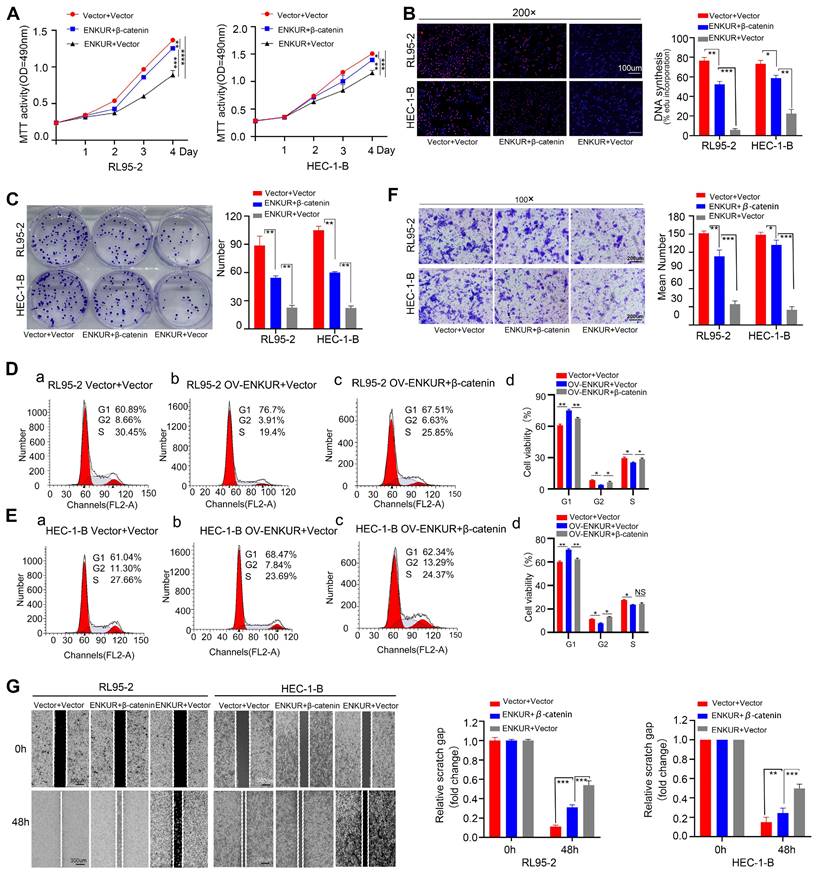
Overexpression of CTNNB1 restores ENKUR-induced repression of cell growth, migration, and invasion
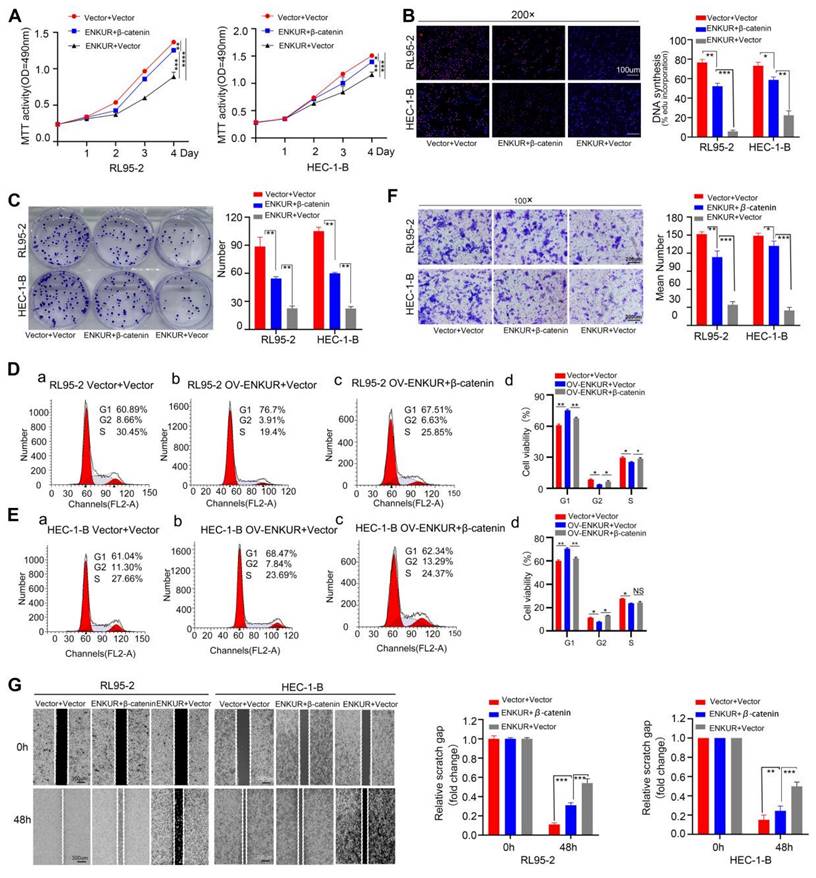
To confirm the relationship between ENKUR and CTNNB1, CTNNB1 was overexpressed in ENKUR-overexpressing EC lines (ENKUR+CTNNB1 group) by plasmid transfection. MTT analysis, plate clone formation assay, and EdU assay showed that the number of proliferating EC cells was partly restored in ENKUR+CTNNB1 EC (Fig. 10A-C). Flow cytometry findings indicated a restoration in the transition from G1 phase to S phase/G2 phase in ENKUR+CTNNB1 EC (Fig. 10D, E). Similarly, cell scratch and Transwell invasion experiments demonstrated that the cell capacities for migration and invasion were partly restored in ENKUR+CTNNB1 EC (Fig. 10F-J). In addition, western blotting showed upregulation of CTNNB1, N-cadherin and Vimentin expression and downregulation of E-cadherin; however, the ENKUR and FBXW7 levels were not altered (Fig. S2H-I). These data indicated that ENKUR inhibits EC proliferation, growth, and invasion through CTNNB1.
Overexpression of CTNNB1 restores ENKUR-induced repression in cell growth, migration, and invasion. The effects of transfection of RL95-2 and HEC-1-B OV-ENKUR cells with CTNNB1 on cell proliferation were examined by (A) MTT assay, (B) EdU experiments and (C) Colony formation assays. The effects of transfection of RL95-2 and HEC-1-B OV-ENKUR cells with CTNNB1 on the cell cycle were examined by (D, E) Flow cytometry. The effects of transfection of RL95-2 and HEC-1-B OV-ENKUR cells with CTNNB1 on cell migration and invasion were examined by (F) Transwell invasion experiments and (G)Cell scratch. Student's t-test. Data are presented as the mean±standard deviation. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Original magnification: 100×, 200×. Scale bars: 100 µm, 300 µm. Each bar represents the mean±standard deviation of three independent experiments.
Discussion
Rapid cell proliferation and metastasis are responsible for the uncontrolled progression of EC. Previous studies have identified numerous factors that regulate EC initiation and development, such as p53, phosphatase and tensin homolog (PTEN), telomere and telomerase-associated proteins, CTNNB1, adipokines, hormone receptor biomarkers (such as estrogen receptor, progesterone receptor and androgen receptor mismatch repair deficient), high microsatellite instability, polymerase epsilon (POLE) exonuclease domain mutated, and other potential biomarkers[7, 28-39]. Nonetheless, these abnormal changes in genes partly explain the occurrence, development, and prognosis of EC.
Our previous studies demonstrated that the tumor suppressor ENKUR participates in the occurrence, development, and chemosensitity of certain tumors by modulating different genes. These effects include stabilization of p53 expression to suppress NPC metastasis[20], suppression of myosin heavy chain 9-mediated (MYH9-mediated) c-Myc deubiquitination in LUAD[18], antagonism of CTNNB1/c-Jun/MYH9/USP7 pathway through an increase in c-Myc ubiquitylation degradation to suppress cell cycle and epithelial-to-mesenchymal transition signals in HCC[19].
In the present study, we continued to investigate the role of ENKUR in EC. Although ENKUR mRNA and protein expression levels did not show significant differences between EC and normal endometrium tissues, higher ENKUR expression is predictive of a favorable outcome in patients with EC based on the GEPIA and UALCAN database analyses. However, these differences did not reach statistical significance with possible reasons including the detection of EC at an early stage and the high 5-year relative survival rate (>85%) in those undergoing surgical removal of EC[2, 40]. Unexpectedly, we observed that ENKUR protein is significantly down-regulated in EC tissues compared with control endometrial epithelial tissues. In addition, we noted that reduced ENKUR expression results in a shorter overall survival time of patients with EC. These results are similar to our previous data in NPC and HCC, as well as those obtained from the GEPIA database[19, 20]. Collectively, these findings suggest that ENKUR (as a tumor suppressor) is involved in the pathogenesis of EC.
Subsequently, we investigated the mechanism underlying the effects of ENKUR in EC. Data obtained from a series of experiments showed that ENKUR overexpression in EC cells decreased cell proliferation, migration, invasion, and intrahepatic dissemination, arrested the cell cycle in the G1 phase, suppressed the CCND1, CDK4 and epithelial-to-mesenchymal transition signals. These results further supported the notion that ENKUR may act as tumor suppressor in EC. This is consistent with previous reports on the role of ENKUR in CRC, LUAD, HCC etc.[19-21].
In previous studies, ENKUR-mediated suppression of phosphatidylinositol 3 kinase/protein kinase B (PI3K/AKT) signaling reduced CRC cell proliferation and invasion. In addition, it was reported that ENKUR suppresses LUAD cell proliferation and invasion by directly interacting with PI3K downstream, and may be involved in the PI3K/AKT and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling pathways[17]. Our previous studies also showed that ENKUR or its enkurin domain binds to FBXW7, CTNNB1 and MYH9, thereby inhibiting carcinogenic factors c-JUN and p53. In turn, this effect leads to the suppression of tumor growth, invasion, migration and metastasis[18-20]. In addition, ENKUR could be induced by the small molecule chemical compound cinobufotalin and overcome resistance to cisplatin and sorafenib in NPC and HCC[18-20]. However, the molecular mechanism underlying the suppressive effects of ENKUR on the occurrence and progression of EC has not been elucidated.
In the present investigation, we used Co-IP and mass spectrometry analysis to identify proteins that interact with ENKUR; this analysis yielded 285 potential interactive proteins. Subsequently, we conducted GO pathway enrichment analysis for these proteins. We found that ENKUR is significantly involved in the classical Wnt signaling pathway. It is well established that CTNNB1 is an important component in the classical Wnt signaling pathway[41-45]. The aberrant activation of the Wnt/CTNNB1 signaling pathway promoted the proliferation, invasion, migration and epithelial-to-mesenchymal transition of EC cells[46-48]. Previous studies demonstrated that ENKUR binds to CTNNB1, and suppresses its nuclear transfer through interaction of CTNNB1 with the IQ motif domain in HCC and NPC[19, 20]; this process does not induce changes in the total CTNNB1 protein levels. It has been shown that ENKUR binds to CTNNB1 to indirectly decrease its protein levels in LUAD[18].
Next, we identified that ENKUR interacts with CTNNB1 and colocalized in the cytoplasm and nucleus. Furthermore, we found that the CTNNB1 protein levels were markedly decreased in ENKUR-overexpressing cells, whereas they were increased in ENKUR-knockdown cells. However, there was no significant change in CTNNB1 mRNA levels observed after ENKUR overexpression, indicating that ENKUR might modulate CTNNB1 protein through post-transcriptional modifications. However, the mechanism by which ENKUR regulates the levels of CTNNB1 protein in EC remains unknown.
FBXW7, a member of the SCF E3 ligase family, functions as a tumor suppressor in some tumors via targeting multiple transcriptional activators and proto-oncogenes for UB-mediated degradation[49, 50]. Interestingly, the BioGRID analysis predicted that FBXW7 protein interacts with CTNNB1 protein. Our results confirmed that FBXW7 binds to CTNNB1 and colocalizes in the cytoplasm. In addition, CTNNB1 protein levels were up-regulated following the knockdown of FBXW7. These results are in accordance with previously reported observations in studies[51, 52]. However, whether ENKUR recruits FBXW7 to directly ubiquitinate and degrade CTNNB1 is currently unknown. Subsequently, the results of molecular docking assay predicted that FBXW7 binds to CTNNB1. Furthermore, we observed a reduction in the binding affinity between CTNNB1 and FBXW7 after mutating the S502 residue. Finally, we confirmed that the ENKUR-mediated ubiquitination and degradation of CTNNB1 is dependent on the recruitment of FBXW7. In addition, the mutation of K394 and K281 sites markedly reduced the levels of CTNNB1 ubiquitination induced by FBXW7.
In summary, our findings demonstrated that ENKUR reduces EC proliferation, migration, invasion, and intrahepatic dissemination by the recruitment of FBXW7 to directly facilitate the ubiquitination and degradation of CTNNB1. The present data are the first to demonstrate that ENKUR is a tumor suppressor in EC, and may be an important target for EC therapy.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Funding
This study was supported by the Open Fund of Guangdong Provincial Key Laboratory of Research and Development in Traditional Chinese Medicine (No. KFKT02-005), the Special Research Platform Project of Traditional Chinese Medicine Bureau of Guangdong Province (No. 20233005), Hunan Provincial Natural Science Foundation (No. 2021JJ30532, 2021JJ30533), the Foundation of Medical Science and Technology Research of Guangdong Province (A2021286), National Natural Science Foundation of China (No. 82203074) and the 74th batch of grant from China Postdoctoral Science Foundation (Certificate Number: 2023M741570).
Data availability
All relevant data are available from the authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bell DW, Ellenson LH. Molecular Genetics of Endometrial Carcinoma. Annu Rev Pathol. 2019;14:339-67
2. Kuhn TM, Dhanani S, Ahmad S. An Overview of Endometrial Cancer with Novel Therapeutic Strategies. Curr Oncol. 2023;30:7904-19
3. Bogani G, Monk BJ, Powell MA, Westin SN, Slomovitz B, Moore KN. et al. Adding immunotherapy to first-line treatment of advanced and metastatic endometrial cancer. Ann Oncol. 2024;35:414-28
4. Kobayashi-Kato M, Fujii E, Asami Y, Ahiko Y, Hiranuma K, Terao Y. et al. Utility of the revised FIGO2023 staging with molecular classification in endometrial cancer. Gynecol Oncol. 2023;178:36-43
5. McCluggage WG, Bosse T, Gilks CB, Howitt BE, McAlpine JN, Nucci MR. et al. FIGO 2023 endometrial cancer staging: too much, too soon? Int J Gynecol Cancer. 2024;34:138-43
6. Zannoni GF, Santoro A, Arciuolo D, Travaglino A, Angelico G, Bragantini E. et al. Endometrial cancer and 2023 FIGO staging system: Not too soon, but maybe too much? Gynecol Oncol. 2024;189:98-100
7. Yang Y, Wu SF, Bao W. Molecular subtypes of endometrial cancer: Implications for adjuvant treatment strategies. Int J Gynaecol Obstet. 2024;164:436-59
8. Berek JS, Matias-Guiu X, Creutzberg C, Fotopoulou C, Gaffney D, Kehoe S. et al. FIGO staging of endometrial cancer: 2023. J Gynecol Oncol. 2023;34:e85
9. Concin N, Creutzberg CL, Vergote I, Cibula D, Mirza MR, Marnitz S. et al. ESGO/ESTRO/ESP Guidelines for the management of patients with endometrial carcinoma. Virchows Arch. 2021;478:153-90
10. Sutton KA, Jungnickel MK, Wang Y, Cullen K, Lambert S, Florman HM. Enkurin is a novel calmodulin and TRPC channel binding protein in sperm. Dev Biol. 2004;274:426-35
11. Sigg MA, Menchen T, Lee C, Johnson J, Jungnickel MK, Choksi SP. et al. Evolutionary Proteomics Uncovers Ancient Associations of Cilia with Signaling Pathways. Dev Cell. 2017;43:744-62.e11
12. Feix AS, Cruz-Bustos T, Ruttkowski B, Joachim A. Inhibition of sexual stage-specific proteins results in reduced numbers of sexual stages and oocysts of Cystoisospora suis (Apicomplexa: Coccidia) in vitro. Int J Parasitol. 2022;52:829-41
13. Jungnickel MK, Sutton KA, Baker MA, Cohen MG, Sanderson MJ, Florman HM. The flagellar protein Enkurin is required for mouse sperm motility and for transport through the female reproductive tract. Biol Reprod. 2018;99:789-97
14. Beech DJ. Canonical transient receptor potential 5. Handb Exp Pharmacol. 2007(179):109-23
15. Tumane RG, Thakkar L, Pingle SK, Jain RK, Jawade AA, Raje DV. Expression of serum proteins in noise induced hearing loss workers of mining based industry. J Proteomics. 2021;240:104185
16. Seetharam SM, Liu Y, Wu J, Fechter L, Murugesan K, Maecker H. et al. Enkurin: a novel marker for myeloproliferative neoplasms from platelet, megakaryocyte, and whole blood specimens. Blood Adv. 2023;7:5433-45
17. Ma Q, Lu Y, Lin J, Gu Y. ENKUR acts as a tumor suppressor in lung adenocarcinoma cells through PI3K/Akt and MAPK/ERK signaling pathways. J Cancer. 2019;10:3975-84
18. Liu JH, Yang HL, Deng ST, Hu Z, Chen WF, Yan WW. et al. The small molecule chemical compound cinobufotalin attenuates resistance to DDP by inducing ENKUR expression to suppress MYH9-mediated c-Myc deubiquitination in lung adenocarcinoma. Acta Pharmacol Sin. 2022;43:2687-95
19. Hou R, Li Y, Luo X, Zhang W, Yang H, Zhang Y. et al. ENKUR expression induced by chemically synthesized cinobufotalin suppresses malignant activities of hepatocellular carcinoma by modulating beta-catenin/c-Jun/MYH9/USP7/c-Myc axis. Int J Biol Sci. 2022;18:2553-67
20. Hou R, Liu X, Yang H, Deng S, Cheng C, Liu J. et al. Chemically synthesized cinobufagin suppresses nasopharyngeal carcinoma metastasis by inducing ENKUR to stabilize p53 expression. Cancer Lett. 2022;531:57-70
21. Ma Q, Lu Y, Gu Y. ENKUR Is Involved in the Regulation of Cellular Biology in Colorectal Cancer Cells via PI3K/Akt Signaling Pathway. Technol Cancer Res Treat. 2019;18:1533033819841433
22. Liu Y, Li X, Zhang Y, Tang Y, Fang W, Liu X. et al. NAP1L1 targeting suppresses the proliferation of nasopharyngeal carcinoma. Biomed Pharmacother. 2021;143:112096
23. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284-7
24. Zhang Z, Xu P, Hu Z, Fu Z, Deng T, Deng X. et al. CCDC65, a Gene Knockout that leads to Early Death of Mice, acts as a potentially Novel Tumor Suppressor in Lung Adenocarcinoma. Int J Biol Sci. 2022;18:4171-86
25. Yan Y, Tao H, He J, Huang SY. The HDOCK server for integrated protein-protein docking. Nat Protoc. 2020;15:1829-52
26. Mustapha T, B S, Zubair T, Patil RB, Bhongade BA, Sangshetti JN. et al. In vitro and in silico investigation of effects of antimicrobial peptides from Solanaceae plants against rice sheath blight pathogen Rhizoctinia solani. PLoS One. 2024;19:e0302440
27. Hessel SS, Dwivany FM, Zainuddin IM, Wikantika K, Celik I, Emran TB. et al. A computational simulation appraisal of banana lectin as a potential anti-SARS-CoV-2 candidate by targeting the receptor-binding domain. J Genet Eng Biotechnol. 2023;21:148
28. Moar K, Pant A, Saini V, Maurya PK. Potential biomarkers in endometrial cancer: a narrative review. Biomarkers. 2023;28:358-71
29. Hutt S, Tailor A, Ellis P, Michael A, Butler-Manuel S, Chatterjee J. The role of biomarkers in endometrial cancer and hyperplasia: a literature review. Acta Oncol. 2019;58:342-52
30. Travaglino A, Raffone A, Raimondo D, Reppuccia S, Ruggiero A, Arena A. et al. Prognostic significance of CTNNB1 mutation in early stage endometrial carcinoma: a systematic review and meta-analysis. Arch Gynecol Obstet. 2022;306:423-31
31. Kobayashi KM, Asami Y, Takayanagi D, Matsuda M, Shimada Y, Hiranuma K. et al. Clinical impact of genetic alterations of CTNNB1 in patients with grade 3 endometrial endometrioid carcinoma. Cancer Sci. 2022;113:1712-21
32. Deng L, Liang H, Han Y. Cyclooxygenase-2 and beta-Catenin as Potential Diagnostic and Prognostic Markers in Endometrial Cancer. Front Oncol. 2020;10:56
33. Jamieson A, Vermij L, Kramer C, Jobsen JJ, Jürgemlienk-Schulz I, Lutgens L. et al. Clinical Behavior and Molecular Landscape of Stage I p53-Abnormal Low-Grade Endometrioid Endometrial Carcinomas. Clin Cancer Res. 2023;29:4949-57
34. Button L, Rogers B, Thomas E, Bradfield A, Alnafakh R, Drury J. et al. Telomere and Telomerase-Associated Proteins in Endometrial Carcinogenesis and Cancer-Associated Survival. Int J Mol Sci. 2022;23:626
35. Navaridas R, Vidal-Sabanés M, Ruiz-Mitjana A, Altés G, Perramon-Güell A, Yeramian A. et al. In Vivo Intra-Uterine Delivery of TAT-Fused Cre Recombinase and CRISPR/Cas9 Editing System in Mice Unveil Histopathology of Pten/p53-Deficient Endometrial Cancers. Adv Sci (Weinh). 2023;10:e2303134
36. Ruiz-Mitjana A, Vidal-Sabanés M, Navaridas R, Perramon-Güell A, Yeramian A, Nicholson-Sabaté N. et al. Metformin exhibits antineoplastic effects on Pten-deficient endometrial cancer by interfering with TGF-β and p38/ERK MAPK signalling. Biomed Pharmacother. 2023;168:115817
37. Kaysudu I, Gungul TB, Atici S, Yilmaz S, Bayram E, Guven G. et al. Cholesterol biogenesis is a PTEN-dependent actionable node for the treatment of endocrine therapy-refractory cancers. Cancer Sci. 2023;114:4365-75
38. Yadav A, Sistla A, Swain M, Gowrishankar S, Padua M, Modi T. et al. To study the expression of estrogen, progesterone receptor and p53 immunohistochemistry markers in subtyping endometrial carcinoma. Indian J Pathol Microbiol. 2024;67:62-7
39. Moatamed NA, Vahdatshariatpanahi S, Gjertson DW, Sachs CR, Kang Y, Ostrzega N. et al. Androgen receptor and its correlation with estrogen and progesterone receptors, aimed for identification of cases for future anti-androgen therapy in endometrial cancers. PLoS One. 2023;18:e0291361
40. Saris DH, Smith A, Brensinger C, Kim SH, Haggerty AF, Latif N. et al. Disparities in cancer-specific and overall survival in black women with endometrial cancer: A Medicare-SEER study. Gynecol Oncol Rep. 2022;40:100922
41. Zhang X, Yu X. Crosstalk between Wnt/beta-catenin signaling pathway and DNA damage response in cancer: a new direction for overcoming therapy resistance. Front Pharmacol. 2023;14:1230822
42. Chuan J, Li W, Pan S, Jiang Z, Shi J, Yang Z. Progress in the development of modulators targeting Frizzleds. Pharmacol Res. 2024;206:107286
43. Li C, Furth EE, Rustgi AK, Klein PS. When You Come to a Fork in the Road, Take It: Wnt Signaling Activates Multiple Pathways through the APC/Axin/GSK-3 Complex. Cells. 2023;12:2256
44. Chen X, Tu J, Yang M, Wang Y, Liu B, Qiu H. et al. RUNX1-MUC13 Interaction Activates Wnt/β-Catenin Signaling Implications for Colorectal Cancer Metastasis. Int J Biol Sci. 2024;20:4999-5026
45. Tian Y, Ai M, Liu C, Wu Y, Khan M, Wang B. et al. Upregulated Long Non-coding RNA Lnc-MRPL39-2:1 Induces the Growth and Invasion of Nasopharyngeal Carcinoma by Binding to HuR and Stabilizing β-Catenin mRNA. Int J Biol Sci. 2023;19:2349-65
46. Tian Y, Lai T, Li Z, Mao M, Jin Y, Liu Y. et al. Role of non-coding RNA intertwined with the Wnt/beta-catenin signaling pathway in endometrial cancer (Review). Mol Med Rep. 2023;28:150
47. Goyal A, Murkute SL, Bhowmik S, Prasad CP, Mohapatra P. Belling the "cat": Wnt/beta-catenin signaling and its significance in future cancer therapies. Biochim Biophys Acta Rev Cancer. 2024;1879:189195
48. Sun X, Xing G, Zhang C, Lu K, Wang Y, He X. Knockdown of Trop2 inhibits proliferation and migration and induces apoptosis of endometrial cancer cells via AKT/β-catenin pathway. Cell Biochem Funct. 2020;38:141-8
49. Baxter JS, Brough R, Krastev DB, Song F, Sridhar S, Gulati A. et al. Cancer-associated FBXW7 loss is synthetic lethal with pharmacological targeting of CDC7. Mol Oncol. 2024;18:369-85
50. Sailo BL, Banik K, Girisa S, Bordoloi D, Fan L, Halim CE. et al. FBXW7 in Cancer: What Has Been Unraveled Thus Far? Cancers (Basel). 2019;11:246
51. Jiang JX, Sun CY, Tian S, Yu C, Chen MY, Zhang H. Tumor suppressor Fbxw7 antagonizes WNT signaling by targeting beta-catenin for degradation in pancreatic cancer. Tumour Biol. 2016;37:13893-902
52. Zhong L, Pan Y, Shen J. FBXW7 inhibits invasion, migration and angiogenesis in ovarian cancer cells by suppressing VEGF expression through inactivation of β-catenin signaling. Exp Ther Med. 2021;21:514
Author contact
![]() Corresponding authors: ChunYan Yang, Department of Gynecology and Oncology, Huaihua Cancer Hospital,Huaihua 418100, China. Electronic address: 2354429374com. BeiXian Zhou, PhD, The People's Hospital of Gaozhou, Gaozhou 525200, China. Electronic address: zbeixiancom. WeiYi Fang, PhD, Southern Medical University Hospital of Integrated Traditional Chinese and Western Medicine, Southern Medical University, 510315 Guangzhou, China. Electronic address: fangweiyi1975com. LuYun Shu, The Second People's Hospital of Huaihua. Electronic address: 596927786com. SuiQun Guo, Department of Obstetrics and Gynecology, The Third Affiliated Hospital/The Third Clinical Medicine School of Southern Medical University, Guangzhou 510315, China. Electronic address: guosq2005com.
Corresponding authors: ChunYan Yang, Department of Gynecology and Oncology, Huaihua Cancer Hospital,Huaihua 418100, China. Electronic address: 2354429374com. BeiXian Zhou, PhD, The People's Hospital of Gaozhou, Gaozhou 525200, China. Electronic address: zbeixiancom. WeiYi Fang, PhD, Southern Medical University Hospital of Integrated Traditional Chinese and Western Medicine, Southern Medical University, 510315 Guangzhou, China. Electronic address: fangweiyi1975com. LuYun Shu, The Second People's Hospital of Huaihua. Electronic address: 596927786com. SuiQun Guo, Department of Obstetrics and Gynecology, The Third Affiliated Hospital/The Third Clinical Medicine School of Southern Medical University, Guangzhou 510315, China. Electronic address: guosq2005com.