Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Background
The roles and regulation of...
Crosstalk between CSCs and...
Block PD-1/PD-L1 interruption to...
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(4):1819-1836. doi:10.7150/ijbs.101025 This issue Cite
Review
Breaking Immunosuppression to Enhance Cancer Stem Cell-Targeted Immunotherapy
Fang Zheng1#, Shan Zhang2#, Alfred E. Chang3, James J. Moon4, Max S. Wicha5, Shelley Xuelai Wang6, Junhui Chen7, Jixian Liu7 ![]() , Fanjun Cheng2
, Fanjun Cheng2 ![]() , Qiao Li3
, Qiao Li3 ![]()
1. Department of Pediatrics, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, Hubei Province, China.
2. Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, Hubei Province, China.
3. Department of Surgery, University of Michigan, Ann Arbor, Michigan 48109, USA.
4. Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, Michigan 48109, USA.
5. Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, USA.
6. Asian Academy of Aging Research & Translational Medicine, Shenzhen, China.
7. Peking University Shenzhen Hospital, Shenzhen, China.
# Fang Zheng and Shan Zhang contributed equally to this work.
Received 2024-7-16; Accepted 2024-12-3; Published 2025-2-10
Abstract

Cancer stem cell (CSC)-targeted immunotherapy has emerged as a novel strategy in cancer treatment in the past decade. However, its efficacy is significantly limited due to the existence of host immune suppressive activity. Specifically, programmed cell death ligand-1 (PD-L1) is overexpressed in CSCs, and PD-L1 overexpressed CSCs create immunosuppressive milieu via interacting with various immune cells in tumor microenvironments (TME). Hence, novel immunotherapeutic strategies targeting CSCs with concurrent immunosuppression interruption will be promising in enhancing anti-CSC effects. These include dendritic cell (DC) and nanodisc (ND)-based vaccines to present CSC antigens in the forms of CSC lysate, CSC-marker proteins, and CSC-derived peptides to induce anti-CSC immunity. In addition, CSC-directed bispecific antibodies (BiAbs) and antibody drug conjugates (ADCs) have been developed to target CSCs effectively. Furthermore, chimeric antigen receptor (CAR)-T cell therapy and natural killer (NK) cell-based therapy targeting CSCs have achieved progress in both solid and hematologic tumors, and inhibition of CSC associated signaling pathways has proven successful. In this review, we aimed to outline the roles and regulatory mechanisms of PD-L1 in the properties of CSCs; the crosstalk between CSCs and immunosuppressive cells in TME, and recent progress and future promises of immunosuppression blockage to enhance CSC-targeted immunotherapy.
Keywords: cancer stem cells, immunotherapy, immunosuppression, cancer vaccines, bispecific antibodies, antibody drug conjugates
Background
The concepts and clinical practices of blocking the interactions between tumor surface programmed cell death ligand-1 (PD-L1) and the programmed cell death protein 1 (PD-1) on the surface of effector T cells have revolutionized cancer immunotherapies. Anti-PD-1/PD-L1 therapies have become the standard therapy in many cancer types, including melanoma, non-small cell lung cancer, colorectal cancer, triple-negative breast cancer, head and neck squamous cell carcinoma, and hepatocellular carcinoma [1]. Despite advances in this field, only a minority of patients demonstrated benefits with anti-PD-1/PD-L1 therapy. Currently limitations of PD-L1 antibodies are mainly due to certain drawbacks such as tumor hyperprogression [2]; poor bioavailability; high cost and complicated processing, and immunogenic adverse reactions [3]. Increasing evidence indicates that PD-L1 is found not only on the tumor cell membrane but also within the cytoplasm, exosomes, and even nucleus. Based on these, it has been reported that the dynamic and spatial heterogeneous expression of PD-L1 in tumors is mainly responsible for the unsatisfactory efficacy of PD-L1 antibodies [4]. In addition, previous studies have demonstrated increased expression of PD-L1 in CSCs of multiple cancers, resulting in more aggressive resistance of CSCs to anti-PD-1/PD-L1 therapies, and CSC-intrinsic PD-L1 pathways take a nonnegligible role in cancer progression and metastasis in an immune independent way [5-11]. Intrinsic and acquired resistance to these agents represents a significant obstacle in their clinical application [12].
One putative theory for resistance to anti-PD-1/PD-L1 therapy was the existence of cancer stem cells (CSCs) in heterogeneous tumor microenvironment (TME). CSCs could be identified by surface protein markers; intracellular molecular signature including aldehyde dehydrogenase (ALDH) activity; stemness-related signaling pathways; drug carriers, and dynamic states of metabolome and cellular differentiation [13]. Tumor initiation, progression, metastasis, relapse, and therapeutic resistance endowed by CSCs make them vital targets for cancer immunotherapy. Moreover, the interactions of CSCs with multiple immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), regulatory dendritic cells (DCregs) and regulatory T cells (Tregs) in the TME further decrease the effectiveness of cancer immunotherapy [14].
Previous studies have demonstrated increased expression of PD-L1 in CSCs of multiple cancers such as non-small cell lung cancer, hepatocellular carcinoma, breast cancer, and colorectal cancer [5-10], which may partly explain more pronounced immune evasion features of the CSCs. Consequently, PD-L1-overexpressing CSCs create an immunosuppressive milieu by cooperating with other noncancerous cells in the TME [11]. Furthermore, CSC-intrinsic PD-L1 pathways take a nonnegligible role in cancer progression and metastasis in an immune independent way, adding more complexity to the mixed response of cancer patients to PD-1/PD-L1 blockade. PD-1/PD-L1 molecules also showed various expression on different types of immune cells in TME, exerting complicated impacts on the communications between the immune cells and CSCs. In this regard, concurrent CSC-targeted therapy with PD-1/PD-L1 blockage might lead to the development of more promising strategy in cancer immunotherapy. In this review, we aimed to outline the roles and regulatory mechanisms of PD-L1 in the properties of CSCs; the crosstalk between CSCs and immunosuppressive cells in TME; and the promise of PD-1/PD-L1 blockage to enhance CSC-targeted immunotherapies.
The roles and regulation of PD-L1 in CSCs
Characteristics of CSCs
To date, various markers have been used characterize CSCs in different cancer types, e.g., CD24, CD44, CD133, ALDH, ABCB5, EpCAM, LGR5, etc. CSCs are capable of proliferating extensively with a self-renewal ability, which enables them to initiate tumors in both immunocompromised and immunocompetent hosts [15, 16]. In contrast, xenotransplantation of non-CSCs into highly immunocompromised NSG mice resulted in tumor formation but not in immunocompetent models. These suggest that immune evasion represents a fundamental feature of CSCs. Antigen processing and presentation are impaired in CSCs as a result of low major histocompatibility complex (MHC) molecule expression as well as downregulation of other antigen processing molecules [17, 18]. In addition, downregulation of tumor-associated antigens (TAAs) was observed in CSCs, leading to inefficient recognition of CSCs by dendritic cells (DCs), which in turn resulted in reduced activation of T cells [19]. Compared with non-CSCs, CSCs retain a more intact DNA repair function that reduces immunogenicity. CSCs preferentially secrete various immune factors at higher levels than non-CSCs such as transforming growth factor β (TGF-β), IL-6, IL-1β and IL-10 in breast [20, 21], glioma [22], hepatic [23] and pancreatic [24] CSCs. These cytokines underlay CSC maintenance and immune suppression. Another clinically relevant property of CSCs is their resistance to traditional chemotherapies and radiotherapies. Unlike non-CSCs, CSCs tend to stay in dormant or quiescent state, protecting them from cytotoxic drugs targeting rapidly proliferating cancer cells [25]. High expression of efflux transporters and anti-apoptotic proteins and the presence of reactive oxygen species scavengers are associated with CSC drug resistance [26]. Development of strategies to target CSCs based on their distinct biological characters thus represents promising efforts in cancer immunotherapy.
PD-L1 expression in CSCs
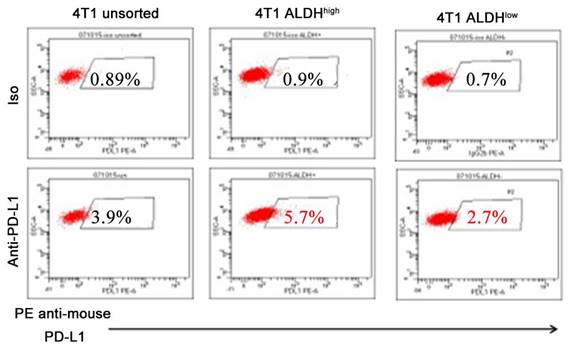
Accumulating literatures have illustrated that PD-L1 is highly expressed in CSCs in solid tumors, including hepatocellular carcinoma [5], gastric cancer [27], lung cancer [28], breast and colon cancer [29], pancreatic cancer [11], and melanoma [30] (Table 1). ALDH has been wildly used as a marker for CSCs. We observed that the expression levels of PD-L1 in ALDHhigh CSCs were significantly elevated, compared with those in ALDHlow non-CSCs by more than twofolds in murine breast cancer cell line (Figure 1). In endometrial carcinoma, high expression of PD-L1 in CD133+ CSCs was observed and found associated with stemness markers ALDH, OCT4, SOX2, NANOG [31]. In breast cancer and malignant mesothelioma, PD-L1 expression is also positively correlated with CSC markers such as CD44, ALDH, ALCAM [32, 33]. It was reported that high PD-L1 expression in pancreatic cancer tissues with high levels of CD44+/CD133+ CSCs predicts an unfavorable prognosis of pancreatic cancer [11]. It was also reported that high PD-L1 expression was strongly associated with high expression of the stemness markers CD44 and LGR5 in ovarian cancer [34]. These observations have suggested the prognostic value of PD-L1 expression in CSCs and emphasized the significance of targeting PD-L1high CSCs.
Higher levels of PD-L1 expression in CSCs than in non-CSCs.
| Cancer Type | Identification of CSCs by molecular markers | PD-L1 expression | Ref. | ||
|---|---|---|---|---|---|
| CSCs | Non-CSSs | Detection Method | |||
| Hepatoblastoma | CD34, CD90, OV-6, Vimentin | Positive | Negative | FC | 5 |
| Lung Adenocarcinoma | CD44 | High | Low | IHC | 9 |
| Pancreatic Cancer | CD44, CD133 | High | Low | IHC | 11 |
| Gastric Cancer | CD44, ALDH | High | Low | FC/WB | 24 |
| Breast and Colon Cancer | CD44, ALDH | High | Low | FC | 26 |
| Melanoma | ALDH | Positive | Negative | FC | 27 |
| Endometrial Cancer | CD133, ALDH, OCT4, SOX2, NANOG | High | Low | FC/WB/IF | 28 |
| Malignant Pleural Mesothelioma | ALCAM | High | Low | IHC | 30 |
| Ovarian Cancer | CD44, LGR5 | High | Low | IHC | 31 |
Expression of PD-L1 is higher on 4T1 ALDHhigh CSCs than on 4T1 ALDHlow non-CSCs. These figures represent flow cytometry assessments conducted by three members of our lab. Our lab, as well as many others, have successfully used the ALDEFLUOR Kit (StemCell Technologies) to isolate ALDHhigh vs ALDHlow cells [89, 93, 98]. Most importantly, we determined the stemness of ALDHhigh cells in our previous studies [89, 93, 110].
Intrinsic roles and regulation mechanisms of PD-L1 in CSCs
Although the interaction between tumor cell PD-L1 and T cell PD-1 has been widely characterized, the mechanisms regulating PD-L1 expression in cancers have been reviewed by Yamaguchi H, et al. [35]. However, the intrinsic role of PD-L1 in CSCs and its expression regulation are less defined. Notch3 was found to be overexpressed in PD-L1high breast cancer cells with the nature of stemness. The effect of Notch inhibitors on PD-L1 overexpression in CSCs was completely abrogated upon mTOR knockdown, demonstrating that overexpression of PD-L1 in CSCs was at least partly mediated by the Notch pathway through Notch3/mTOR axis [8]. CSC-intrinsic PD-L1 expression is also regulated via the PI3K/AKT/mTOR pathways as evidenced by the fact that the mTOR inhibition via Rapamycin decreased the cancer cell stemness in a similar fashion to PD-L1 knock down [36]. The transcription factors HIF-1α and HIF-2α activation triggered high level PD-L1 expression, which correlated with modulated self-renewal and tumorigenicity of CSCs [16]. It was also established that PD-L1 interacts with Frizzled 6 to activate β-catenin and form a positive feedback loop to promote CSC maintenance and expansion [37]. Epithelial-mesenchymal transition (EMT) is essential for enhanced tumor migratory and invasive capabilities. Previous studies reported that PD-L1 promotes EMT features and cancer cell stemness, and EMT enriches PD-L1 in CSCs by the EMT/β-catenin/STT3/PD-L1 signaling axis [6, 7]. From the perspective of epigenetically regulation, CSC-intrinsic PD-L1 is regulated by histone modifications [38]. Post-transcriptionally modulation of PD-L1 expression by microRNA is also found to regulate breast cancer cell stemness [39]. The direct binding of miR-873 to 3′-untranslated regions (UTR) of PD-L1 inhibited its expression, thus attenuated the stemness and chemoresistance of breast cancer cells, which depended on the downstream PI3K/Akt and ERK1/2 signaling pathways. Furthermore, CSCs surface PD-L1 could be regulated through protein modification such as ubiquitination and N-glycosylation [40, 41]. In many CSC models, it has been identified that PD-L1 is involved in the immune evasion mechanisms of CSCs. In breast cancer, Wnt signaling pathway regulates co-expressing PD-L1 with CD44v6 and ALDH1A1, and promotes the expression of CD200 and CD276, which facilitates the immune escape [42]. It was also reported that S100A14 inhibits STAT3-mediated PD-L1 expression, which negatively regulates colorectal cancer stemness and immune evasion [43]. In addition, EMT has been acknowledged to drive the enrichment of PD-L1 in CSCs and promote immune evasion [7]. These studies suggest complicated regulation mechanisms of CSC-intrinsic PD-L1 which warranties further investigation. Nevertheless, simultaneous targeting of CSC antigens as well as CSC signaling pathways while breaking down the immunosuppression may significantly enhance CSC-targeted immunotherapy.
Crosstalk between CSCs and immunosuppressive cells in TME
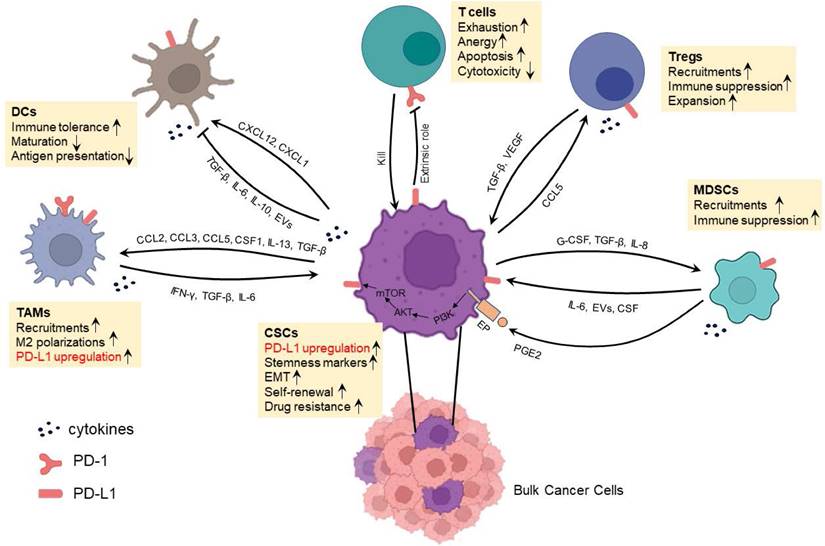
In addition to a direct binding between CSC surface PD-L1 and T cell surface PD-1 resulting in T cell exhaustion, anergy or apoptosis [44], CSCs also interact with immunosuppressive cells to achieve immune evasion and treatment resistance [45]. CSCs actively interact with various types of cells as shown in Figure 2 to induce their phenotype switching and function alternation, e.g., modulating the cytokine, chemokine secretion profiles. These immunosuppressive cells include DCregs, MDSCs, TAMs and Tregs. Recent studies have suggested that resistance to anti-PD-L1 therapy is in large part conferred by immunosuppressive cells. Of note, CSCs and these immunosuppressive cells express PD-1 and/or PD-L1 at various levels. In addition, MDSCs could induce the expression of PD-L1 in CSCs by secreting PGE2 followed by activating PI3K/AKT/mTOR pathway in CSCs. These phenomena usually result in the failure of antitumor effects of infiltrating T cells by extrinsic PD-1/PD-L1 axis thus adding rational to the simultaneous targeting of CSCs and the PD-1/PD-L1 axis in cancer immunotherapies. It is therefore important to understand the interaction between CSCs and immunosuppressive cells, particularly the impact of PD-L1 in these interactions.
Crosstalk between CSCs and immune or immunosuppressive cells which involves PD-1/PD-L1 interaction in the immunosuppressive TME.
Interaction between CSCs and DCregs
DCs have been implicated as key regulators that guide responses to immune checkpoint blockers (ICBs) in cancer immunotherapies. Recent studies have shown the plasticity of DCs in TME that a subset of DCs may alter their roles to immunosuppressive at advanced stages of tumor progression, thus termed as DCregs [46]. Moreover, there are growing appreciations of the protumor roles of reciprocal communications between CSCs and DCs. CSCs interfere with DC recruitment to the tumor site; impair their maturation and promote their differentiation into immunosuppressive DCs [23, 47, 48]. Grange et al. reported that CD105+ renal CSCs are able to impede the differentiation of DCs from monocytes to a greater extent than CD105- non-CSCs by a mechanism mainly related to the expression of human leukocyte antigen (HLA)-G released from extracellular vehicles (EVs) [48]. Zhong et al. indicated that TGF-β/AKT/Smad2 signaling in human liver cancer plays a critical role in cancer stemness and in impairing CD86 and MHC-II expression on DCs, which resulted in immune tolerance [23].
Studies also shown that PD-L1 on the surface of DCregs in immunosuppressive TME induces T-cell dysfunction [49]. Hence, anti-PD-L1 treatment blocking PD-L1 on both DCs and CSCs may explain the synergistic effects of DC vaccines combined with anti-PD-L1 treatment in mouse models [50, 51]. However, whether the interaction between CSCs and DCregs involves a direct PD-1/PD-L1 engagement is unclear.
Interaction between CSCs and MDSCs
MDSCs are a cluster of heterogeneous populations originated from myeloid cells with potent immunosuppressive effects and have emerged as important regulators of immune responses in cancers [52]. Increasing evidence have revealed sophisticated interactions between CSCs and MDSCs in TME [53]. In breast cancer, CSCs exhibit enhanced G-CSF production involving mTOR signaling and stimulate MDSC accumulation. In turn, MDSCs could increase CSC frequency through activating Notch in tumor cells, thus establishing a feed-forward loop [54]. Moreover, in clinical specimens of breast cancer, the presence of MDSCs correlates with the existence of CSCs. Further investigations manifested an increase in the ALDHhigh fraction with self-renewal capacity in human breast cancer cells after co-cultured with MDSCs, and the co-culture induced IL-6-dependent phosphorylation of STAT3 and activated Notch via nitric oxide (NO) in cancer cells [55]. Furthermore, MDSCs increased STAT3 phosphorylation; CD133 and CD44 expression and sphere formation of mouse and human colorectal cancer cell lines in vitro via secretion of exosomal S100A9 [56]. Indeed, STAT3 activity seems to be a key mediator in the crosstalk between CSCs and MDSCs. In epithelial ovarian cancer, MDSCs enhanced the stemness by inducing the CSF2/p-STAT3 signaling pathway [57]. Not only in solid cancers but in multiple myeloma, MDSCs endowed stemness qualities to malignant cells by inducing piRNA-823 expression and DNMT3B activation [58]. In addition, CD133+ melanoma CSCs activated TGF-β1 expression through modulating miRNA-92 and recruited immunosuppressive MDSCs in the tumor site [59]. Furthermore, glioblastoma patient-derived CSCs selectively drove MDSCs-mediated immune suppression by secreting high level of MIF which increased production of the immune-suppressive enzyme arginase-1 in MDSCs in a CXCR2-dependent manner [60].
Komura et al. [61] reported that co-culture MDSCs with ovarian cancer cells in vitro resulted in more ALDHhigh CSCs and enhanced expression of PD-L1 in ALDHhigh CSCs. This effect was achieved by activating the PI3K-AKT-mTOR pathway via the production of PGE2 by MDSCs. Treatment of ovarian cancer cells with rapamycin significantly inhibited this MDSC-mediated increase in PD-L1 expression. These studies demonstrate that MDSCs could induce PD-L1 expression in CSCs, suggesting that breaking the interaction between CSCs and MDSCs and/or blocking the PD-L1 expression in CSCs may help enhance CSC-targeted immunotherapy.
Interaction between CSCs and TAMs
TAMs can be classified as antitumor M1 and protumor M2 subtypes which represent the extremes of a wide spectrum of differentiation states. Moreover, the abundance of M2 in tumors is usually associated with poor prognosis mainly due to their immunosuppressive functions. TAMs secrete several cytokines including IFN-γ, VEGFA, TGF-β and IL-6 that manifest key mediators of their immunosuppressive functions and foster CSC phenotypes [62, 63]. CSCs recruit macrophages through the CC chemokines, the CXC chemokine subfamily, IL-33, and other soluble proteins [62]. Moreover, CSCs may impact the polarization of TAMs to protumor M2 state by secreting elevated levels of CCL2, CCL5, CSF1, IL-13, TGF-β, periostin and CCN4 than their non-CSC counterparts [62]. It was reviewed that various pro-inflammatory cytokines in TME, including IFN-γ, TGF-β, IL-6, IL-8, and IL-10 could upregulate the expression of PD-L1 and promote tumor progression [35]. In particular, CSC-secreted such cytokines can induce the polarization of macrophages to M2 TAMs, suggesting that CSCs could influence the expression of PD-L1 in TAM polarity. On the other hand, TAMs promote CSC stemness through chemokines; soluble protein molecules and extracellular vesicles [62]. For example, CCL5 derived from TAMs could promote prostate CSC self-renewal and cancer metastasis via activating β-catenin/STAT3 signaling [64]. These highlight the complexities of CSC-TAM crosstalk and underlay the necessity of simultaneously targeting TAM-induced immunosuppression and stemness phenotypes. Importantly, TAMs express both PD-1 and PD-L1 increasingly over time as developing from bone marrow monocytes to mature macrophages and the PD-1 and PD-L1 expression further increases with disease progression after being recruited to tumor tissues [65]. Zhu et al. observed that PD-1/PD-L1 could polarize TAMs to M2 phenotype [66]. Liu et al. reported that high PD-L1 expression in TAMs is associated with the level of PD-L1 in both tumor cells and infiltrating CD8+ T cells [67]. These studies suggest that breaking the interaction between CSCs and TAMs may benefit CSC-targeted immunotherapy.
Interaction between CSCs and Tregs
Napoletano et al. observed a positive correlation between the presence of CSCs and Tregs in cancers [68]. In general, Tregs are actively recruited into TME through various chemokines. Among different cancer types, diverse chemokines are preferentially secreted by CSCs vs. non-CSCs to attract Tregs. Sorted CD133+ ovarian CSCs showed elevated CCL5 production relative to non-CSCs. The interaction of CCL5 with its receptor CCR5 on Tregs in ovarian cancer patients resulted in Tregs recruitment. In return, recruited Tregs secret a higher level of IL-10 exerting pronounced immune-inhibitory function in CSC-enriched environments [69]. SOX2-overexpressing breast cancer cells activated NF-κB-CCL1 signaling to recruit Tregs which in turn upregulated the stemness of breast cancer cells evident by increased ALDHhigh population and enhanced stemness gene expression [70].
TGF-β is critical for the communication between CSCs and Tregs through several mechanisms. Firstly, high level of TGF-β secreted by Tregs directly promotes cancer cell stemness [71]. Secondly, TGF-β indirectly participates in CSC formation and maintenance by inducing vascular endothelial growth factor (VEGF) which stimulates angiogenesis [72]. Moreover, Tregs could promote the M2-polarization of TAMs through TGF-β signaling, further contributing to the immunosuppressive niche and CSC properties as described above [73].
Fortunato et al. reported that PD-L1+ CSCs in non-small cell lung cancer are able to specifically increase the percentage of Tregs in culture, and this effect could be abrogated by CXCR4 inhibitors [74]. In accordance with these in vitro observations, inspections of clinical samples from metastatic lymph nodes of non-small cell lung cancer patients also proved the positive correlations of PD-L1+ CSCs with Tregs. One of the mechanisms underlying the negotiation between PD-L1+ CSCs and Tregs is that PD-L1 could augment Treg generation from naïve T cells by antagonizing the Akt-mTOR signaling cascade and thus enhance suppressive functions [75]. Taken together, these studies suggest that breaking the interaction between CSCs and Tregs by blocking the PD-L1 signaling pathway may enhance the overall efficacy of immunotherapy against CSCs.
Interactions between CSCs and NK cells and B cells
CSCs also interact with other immune cells in TME. Natural killer (NK) cells play a vital function in the immune system's defense against infections and tumor. Compared to normal cells, CSCs exhibit lower or no expression of MHC-I, which makes them more susceptible to recognition and elimination by NK cells [76]. It was reported that NK cells could directly eliminate CSCs through receptor/ligand connections [77, 78]. For example, CSCs express high levels of ULBP1, ULBP2, and MICA, which are NKG2D ligands [79, 80]. The NKG2D-NKG2D ligand interaction is the primary pathway through which NK cells exert their antitumor effects on CSCs [52]. In addition, NK cells can induce CSCs to differentiate into less-invasive and less-metastatic tumor cells [81]. Although CSCs are susceptible to recognition and elimination by NK cells, they can induce NK cell exhaustion or block NK cell recognition by modulating the expression of NKG2D ligands; immune checkpoints and immunosuppressive cytokines such as PD-L1, TGF-β and IL-10, which facilitate CSC immune evasion [82, 83]. The well-identified NKG2D ligands may serve as ideal markers in breaking immunosuppression to enhance CSC-targeted immunotherapy.
Tumor-infiltrating B cells have emerged as important players in cancer immunity and served as predictors of response to immunotherapy [84]. Notably, not all B cells possess antitumor capabilities, and a subset of B cells, e.g., Bregs, are immunosuppressive. The research on the interplay between B cells and CSCs is relatively limited. It was demonstrated that the specific binding of IgG produced by CSC-DC vaccine-primed B cells could directly target and kill CSCs [85]. However, other studies have indicated that a specific CSC-like B cell subpopulation exhibited self-renewal and multilineage differentiation capabilities in diffuse large B-cell lymphoma, which was regulated by HMGB3, SAP30, and E2F8 [86]. These findings suggest that the biological interaction between B cells and CSCs is a complex landscape, and warranties further investigation.
Block PD-1/PD-L1 interruption to enhance CSC-targeted immunotherapy
The concept of CSC represents a novel direction in CSC-targeted immunotherapy. PD-1 is predominantly expressed on the surface of antigen-stimulated T cells and transduces inhibitory signals capable of restraining the activity of these cells, while PD-L1 can be expressed by various cell types, including professional antigen-presenting cells, cancer cells and CSCs. In addition to the immunomodulatory actions of PD-L1 in CSCs, tumor intrinsic PD-L1 could directly promote the maintenance of CSC stemness [30]. Both CSCs and immunosuppressive cells express high levels of PD-L1 and they interact as reviewed in Part 2. This rationalizes the promises of combined anti-CSC strategy and PD-1/PD-L1 blockage in cancer immunotherapy.
CSC-targeted vaccines
The immune evasion property of CSCs leads to failures of immunosurveillance. CSC-targeted DC vaccines aim to reverse the ignorance of the immune system to CSCs by loading CSC-antigens onto the DCs as a vaccine to induce CSC-specific T cells and B cells/antibodies which in turn effectively eliminate the CSC antigen-bearing CSCs.
Several groups, including our own, have employed DC vaccines to induce immunological recognition and toxicity towards CSCs. Pellegatta and colleagues [87] attempted to generate CSC-DC vaccine by pulsing the lysate of CSC-enriched neurospheres (NS) from murine GL261 glioma cells onto DCs (NS-DC). Compared to adherent cell lysate pulsed DC vaccines (AC-DC), NS-DC cured more tumor significantly. Later, Xu et al. used both human samples and a syngeneic animal brain tumor model to investigate the immune response of glioblastoma-derived CSC-DC vaccine. Vaccination of DCs loaded with CSC lysate induced Th1 immune response and achieved a significant survival benefit in the brain tumor model [88].
Our group sorted murine ALDHhigh CSCs in two histologically different tumors (D5 melanoma and SCC7 squamous cell cancer) and the application of ALDHhigh CSC lysate-DC vaccine resulted in direct targeting of CSCs by both cellular and humoral immunity [89, 90]. Consistent findings have been obtained by designing CSC-DC vaccines by other CSC markers such as CD105, CD24/CD44 [91, 92].
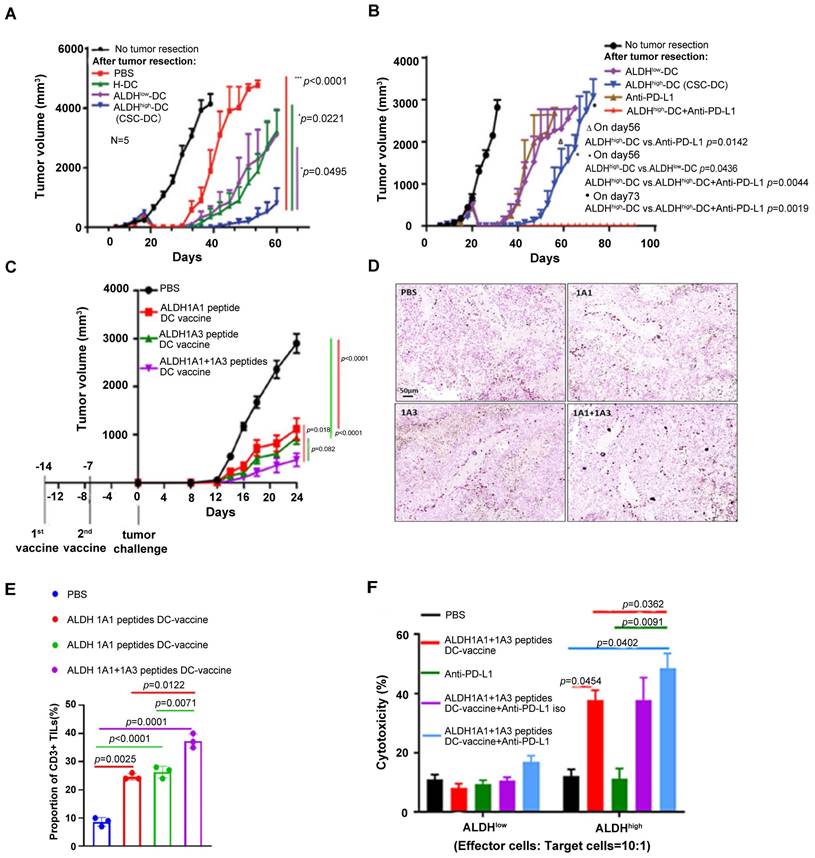
In a following up study in adjuvant setting, we found that the ALDHhigh CSC-DC vaccine significantly delayed tumor recurrence, resulting in significantly prolonged animal survival after surgical resection of the subcutaneous tumors (Figure 3A) [93]. Importantly, we found that ALDHhigh CSC-DC vaccination plus anti-PD-L1 administration significantly inhibited tumor relapse (Figure 3B) and further prolonged animal survival [93]. Further work in our group administrated ALDHhigh CSC-DC vaccine with both anti-PD-L1 and anti-CTLA-4. These triple therapies exerted more significant anti-melanoma immune effects than either CSC-DC vaccine alone or anti-PD-L1 plus anti-CTLA-4 [94]. These experiments clearly demonstrate that immunologically targeting CSCs with simultaneously blocking of the immunosuppressive components has the potential to significantly enhance the efficacy of CSC-targeted immunotherapy.
ALDHhigh CSC lysate-DC and ALDH peptides-DC vaccination with anti-PD-L1 therapy significantly inhibited cancer growth. (A) Vaccination of DCs pulsed with the lysate of ALDHhigh SCC7 CSCs (CSC-DC) significantly delayed tumor recurrence. Sourced from [93]. (B) Administration of anti-PD-L1 further significantly inhibited tumor recurrence in animals treated with ALDHhigh SCC7 CSC-DC vaccination (ALDHhigh-DC + Anti-PD-L1) after surgical resection of the subcutaneous SCC7 tumors as in (A). Animals remined tumor-free till day 90 when the experiment was terminated. Sourced from [93], reproduced with permission from American Association for Cancer Research publisher. (C) ALDH1A1+ALDH1A3 peptides-DC vaccine inhibited D5 tumor growth significantly more than single ALDH peptide-DC vaccine. Sourced from [98]. (D) ALDH peptide-DC vaccination treatment induced CD3+ TILs. Representative immunohistochemical images show CD3+ TILs in residual tumors harvested from the treated hosts. Sourced from [98] (E) Bar graph comparing the CD3+ TILs induced by different treatments as indicated. Sourced from [98]. (F) Cytotoxicity of splenic T cells isolated from D5-bearing mice treated in (C) (E/T = 10:1 ratio) against D5 ALDHhigh CSCs vs. ALDHlow non-CSCs. Sourced from [98], reproduced with permission from Springer Nature publisher.
Although preclinical models have endowed CSC lysate-DC vaccines with promising efficacy, the realistic plights remain in their clinical translation due to the difficulty to obtain adequate amounts of tumor tissues from each patient to make CSC lysate for vaccine preparation. To address this, CSC derived or associated proteins may represent an alternative source of CSC antigen. Integrin β4 (ITGB4) was identified as a receptor for the basement membrane protein laminin and involved in the regulation of CSCs in a variety of malignancies [95, 96]. We synthesized murine ITGB4 (mITGB4) protein, pulsed it to DCs to generate mITGB4-DC vaccine in murine breast cancer and head and neck squamous cancer models. mITGB4-DC vaccination inhibited both local tumor growth and lung metastases in both models, and addition of anti-PD-L1 administration significantly enhanced the therapeutic effectiveness [97].
More recently, we utilized ALDH1A1 and ALDH1A3 peptides derived from ALDH1 isoform to prime DCs and generated ALDH peptide(s)-DC vaccine [98]. ALDH1A1+1A3 dual peptides-DC vaccine mediated an additive anti-tumor effect compared to single peptide-DC vaccines in a D5 melanoma model (Figure 3C). PD-L1 blockade significantly enhanced ALDHhigh CSC-targeted vaccination [98]. This effect was associated with D5 ALDHhigh CSC specific cytotoxic T lymphocyte (CTL) activity (Figure 3D-F).
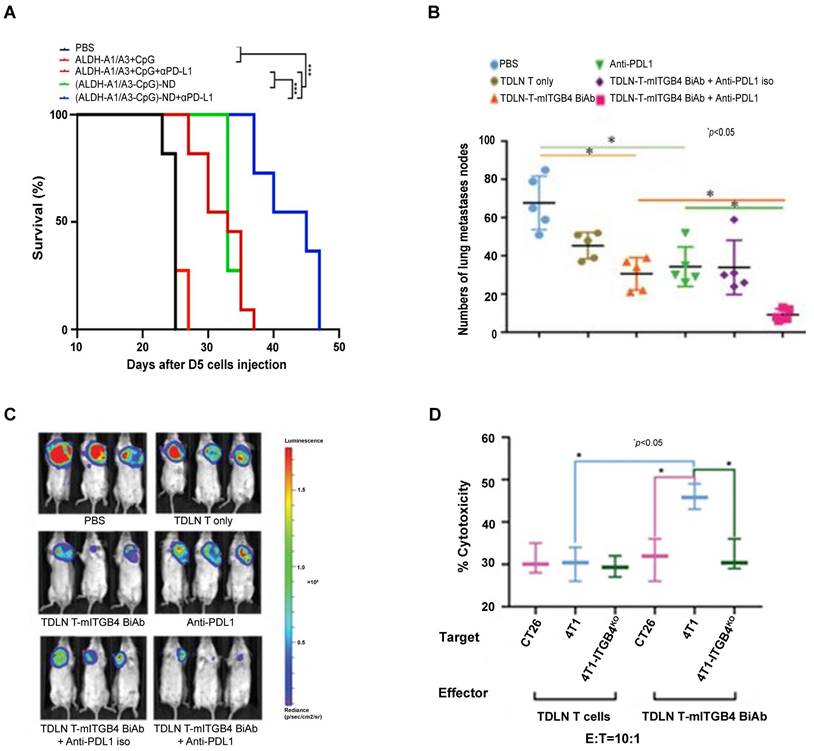
To develop “off-the-shelf” CSC vaccines for cancer patients, we evaluated nano-vaccine systems in animal models [99]. In initial studies, we co-loaded ALDH1A1 and/or ALDH1A3 peptides along with CpG (a TLR-9 agonist) to the synthetic high-density lipoprotein (sHDL) nanodiscs developed by our collaborator James Moon at the University of Michigan [100, 101]. These nanodiscs efficiently delivered ALDH peptides to tumor-draining lymph nodes (TDLNs) and induced significant T cell responses against ALDHhigh CSCs. Compared with soluble peptide vaccination, nanodiscs vaccination significantly slowed down tumor growth and prolonged the animal survival in D5 murine melanoma model. Of note, anti-PD-L1 therapy concurrently administrated with the ALDH peptide(s)-ND vaccine significantly enhanced the suppression on tumor growth and further prolonged the animal survival (Figure 4A) [102]. This work represents the first attempt to developing an off-the-shelf nanoparticle-based vaccine strategy against CSCs to avoid the invasive DC collection from patients.
Anti-PD-L1 enhanced anti-CSC immunity induced by ALDH peptide-ND vaccine and mITGB4 BiAb-armed TDLN T cells. (A) Overall survival of C57BL/6 mice inoculated s.c. in the flank with D5 tumor cells and immunized with (ALDH-A1/A3-CpG)-ND nanoparticles or a soluble mixture of ALDH-A1/A3 and CpG (control). A subset of mice also received i.p. administration of anti-PD-L1 to enhance antitumor immunity. Sourced from [102]. Copyright 2020 American Chemical Society. Reprinted with permission. (B) mITGB4 BiAb-TDLN T cells significantly suppressed the metastases in therapeutic 4T1 model. Source from [97]. (C) mITGB4 BiAb-armed TDLN T cells significantly inhibited tumor growth in the therapeutic 4T1 model, which was enhanced by anti-PD-L1. Sourced from [97]. (D) mITGB4 BiAb-armed TDLN T cells (TDLN T-mITGB4 BiAb) mediated greater cytotoxicity to ITGB4-expressing 4T1 cells than to 4T1-ITGB4 knockout (4T1-ITGB4KO) cells or ITGB4-negative CT26 cells in vitro. Sourced from [97], reproduced with permission from American Association for Cancer Research publisher.
In our recent studies, we explored additional novel immunogenic peptide epitopes identified from CSC-associated transcription factors including SOX2 and NANOG. We synthesized two Sox2 and two Nanog-derived immunogenic peptides and co-loaded them along with the ALDH 1A1 and 1A3 peptides to the sHDL and formulated multi-CSC peptides-nanodisc cocktail vaccine [103]. As a result, the multi-CSC peptides-nanodisc vaccine reduced tumor growth and extended animal survival significantly more than ALDH1A1/1A3 peptides-ND. We anticipate that immune suppression disruption, e.g., administration of anti-PD-L1 and/or anti-CTLA-4 may further augment the efficacy of this multi-CSC peptides-ND vaccine in CSC-targeted immunotherapy.
Taken together, CSC-targeted vaccines, based either on DCs or on NDs, have made breaking through progress in the past decade. The presentable CSC antigens have been optimized from CSC lysate to CSC associated proteins, and now to CSC marker protein-derived antigenic peptides. Multiple investigators, including us highlighted the benefits to block immunosuppressive signals to enhance CSC-targeted cellular and humoral immunity. This may lead to the development of novel immunotherapeutic regiment to treat cancer patient in clinic.
Bispecific Ab targeting CSCs
Bispecific antibodies (BiAbs) against T cell markers and tumor cell markers bind to CD3 on T cells and antigens on tumor cells, resulting in the recruitment of T cells to the tumor. This is followed by T cell activation and degranulation and tumor cell elimination. T cell-engaging BiAbs have shown substantial effects in several hematological malignancies [104]. However, T cell-engaging BiAbs in solid tumors have been less developed, partially due to the paucity of target molecules expressed on the solid tumor cell surfaces, which may lead to off-tumor toxicity. Catumaxomab (CD3-EpCAM BiAb) was the first approved T cell redirecting antibody for the treatment of malignant ascites [105], and it has been involved in clinic trials in several solid tumors including gastric cancer (NCT00464893), ovarian cancer (NCT01815528), and bladder cancer (NCT04819399). In attempt to specifically target CSCs, our group generated an anti-CD3/anti-CD133 BiAb that bound to effector cytokine-induced killer (CIK) cells (BiAb-CIK) to target CD133high CSCs [106]. In both mouse models of pancreatic and hepatic cancer, adoptive transfer of BiAb-CIK cells significantly inhibited CD133high tumor growth than that by CIK cells or BiAb alone. On the other hand, resistance to BiAbs was observed in clinic, which was found at least partially due to the inhibitory effect on the T cell-tumor cell interaction through the PD-1/PD-L1 axis, leading to T cell dysfunction and exhaustion. Preclinical and clinical studies have shown that blockade of PD-1/PD-L1 axis benefits the antitumor activity of T cell-redirecting BiAbs [107, 108]. In our effort to target ITGB4high CSCs, we synthesized BiAb using anti-mITGB4 and anti-CD3 monoclonal antibodies and utilized it to arm TDLN T cells and generated mITGB4 BiAb-TDLN T cells. When transferred into 4T1 or SCC7 tumor-bearing mouse hosts [97], these T cells specifically targeted ITGB4high CSCs and conferred host anti-CSC immunity, resulting in significant inhibition of local tumor growth and lung metastases, and this effect was significantly boosted by co-administration of anti-PD-L1 (Figure 4B-D).
Antibody drug conjugate (ADC) targeting CSCs
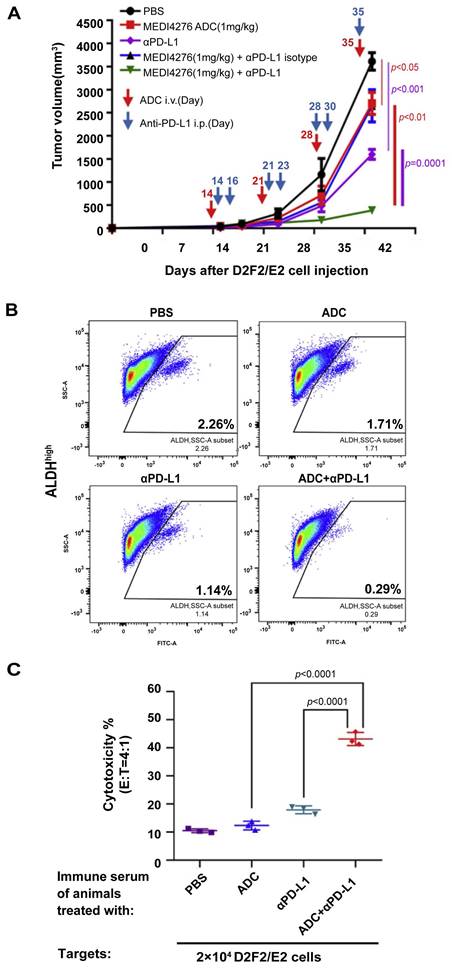
Antibody drug conjugate (ADC) represents an effective combination of target specificity and toxicity via the unique structure consisting of antibody (or antibody fragment), chemical linker, and cytotoxic payload. Human epidermal growth factor receptor 2 (HER2) has been one of the pivotal hotspots in ADC targets for its overexpression contributing to tumorigenic growth and CSC population increase in breast cancer [109]. We tested MEDI4276 ADC against engineered human HER2-expressing murine breast cancers D2F2/E2 and HER2-4T1 in immunocompetent mouse models [110]. MEDI4276 ADC demonstrated effective and specific anti-tumor activity in HER2-expressing cancer models in vivo, and anti-PD-L1 therapy significantly augmented the therapeutic efficacy of MEDI4276 ADC (Figure 5A). Importantly, these effects are associated with significant targeting of HER2+ALDHhigh CSCs, resulting in reduction of this cell subset by ~90% post ADC + anti-PD-L1 therapy vs. PBS control (Figure 5B). Co-administration of anti-PD-L1 and MEDI4276 ADC significantly increased anti-tumor immunity of tumor-infiltrating lymphocytes (TILs) and host splenocytes. Furthermore, we identified HER2-targeted ADC treatment induced humoral immunity (Figure 5C) as well as T cell anti-tumor activity. These studies have demonstrated the benefits of anti-PD-L1 in ADC immunotherapy targeting CSCs.
PD-l/PD-L1 blockade benefited HER2-targeted ADC immunotherapy to induce host immunity against CSCs. (A) Anti-PD-L1 significantly enhanced the efficacy of HER2-targeted ADC inhibiting D2F2/E2 tumor growth. (B) Evaluation of ALDHhigh D2F2/E2 CSC frequencies in residual tumors after indicated treatment. (C) HER2-targeted ADC and anti-PD-L1 therapy induced significant humoral immunity against HER2-expressing tumor cells. Antibody-dependent cellular cytotoxicity (ADCC) assays were performed using the immune serum collected from the animals subjected to treatment as indicated to test IgG-mediated cytotoxicity against D2F2/E2 tumor cells. Source from [110], reproduced with permission from Elsevier publisher.
CSC-targeted chimeric antigen receptor (CAR)-T cell therapy
CAR-T cells are engineered T cells which express an artificial receptor specific for TAAs, leading to TAA-specific targeting and killing of cancer cells. CSC surface markers including CD44, EpCAM, CD47, CD123, CD133, TRKB have been reported as targets in CAR-T therapies in preclinical studies, and some of them are recently tested in clinical trials [111-115]. A preclinical study indicated that anti-CD133 CAR-T cells exhibited pronounced killing efficiency of cisplatin-exposed CD133+ gastric cancer cells. Moreover, cisplatin and anti-CD133 CAR-T combination treatment inhibited gastric cancer progression with diminished CD133+ stem cell-like cell infiltration in mouse models [114]. Phase I/II clinical trial (NCT02541370) has reported feasibility, controllable toxicities, and effective activity of CD133-directed CAR-T in patients with CD133+ hepatocellular carcinoma, pancreatic carcinomas, and colorectal carcinomas [116, 117].
Oncolytic adenovirus constructed to express a PD-L1 blocking mini-antibody successfully blocked the interactions between PD-1 and PD-L1, and increased the killing effect of HER2 CAR-T cells in vitro, and co-administration of the oncolytic adenovirus with HER2 CAR-T cells into xenograft prostate cancer models prolonged survival [118]. More recently, Yamaguchi et al. demonstrated that PD-L1 blockade during the CD19 CAR-T cell transfer altered the M2 macrophages to more M1 like subsets, thus indirectly improved the antitumor activities of the CAR-T cells [119].
Collectively, these studies have suggested that CAR-T cells can be generated to mediate CSC-specific targeting, and this effort can be modulated by concurrent immunosuppression blockage. This may help develop more potent CAR-T cell therapy both for hematologic malignancies and for solid tumors.
CSC-targeted NK cell therapy
NK cells simultaneously express inhibitory and activating receptors maintaining the subtle balance of transmitted signals after encountering target cells. These inhibitory receptors recognize various forms of MHC-I molecules on target cells [120]. Reduction of MHC-I molecules on CSCs leads to more sensitivity to NK cells [77, 121, 122]. Additionally, CSCs express higher levels of the ligands for NK cell activating receptors (such as NKG2D, NKp44 and NKp30) compared to non-CSCs [123-127]. In this context, NK cell-mediated killing represents a promising approach for targeting CSCs [76].
Correspondingly, experiments on human breast, colon, melanoma, and glioblastoma revealed that NK cells could identify and eliminate CSCs in solid tumors [79, 128-130]. Preclinical studies demonstrated that several CSC-specific antigens (e.g., GD2, HER2, CD133, PSCA, CLDN6)-targeted CAR-NK cells displayed superior anti-tumor activity [131-136]. For example, CLDN6 was deemed as related to cancer stemness [136], and Li et al. recently reported that CLDN6 targeted CAR-NK cells could specifically kill CLDN6+ ovarian cancer cells in vitro [137]. Furthermore, CLDN6 targeted CAR-NK cells successfully eliminated ovarian cancer cells in subcutaneous and intraperitoneal tumor models. However, CSCs can evade NK cell-mediated killing through interactions with prominent inhibitory receptors on NK cells such as PD-1, NKG2A, and KIRs [83, 138, 139]. Rationally, combining ICBs with NK cell-based therapy may enhance anti-CSC responses. In this case, Li et al. found that CLDN6-targeted CAR-NK cells induced PD-L1 expression on the surface of tumor cells, and these PD-L1+ tumor cells were resistant to CAR-NK cells. More importantly, they revealed that combined with anti-PD-L1 synergistically enhanced the antitumor efficacy of CLDN6-targeted CAR-NK cells [137].
Targeting CSC associated signaling pathways
Well-characterized signaling pathways that regulate the maintenance and survival of CSCs have proved as effective targets for CSCs, such as Hedgehog (Hh), Wnt/β-catenin, TGF-β and Notch pathways [140]. For instance, Hh pathway is activated in basal cell carcinoma, gastrointestinal tract cancer, prostate cancer, breast cancer, glioblastoma, leukemia, and myeloma [141]. Several Hh inhibitors were tested in different cancer types in clinic trials. Two FDA approved orally agents, vismodegib (GDC-0449) and sonidegib (LDE225), have demonstrated remarkable efficacies in locally advanced and metastatic basal cell carcinoma [142, 143]. An ongoing clinical trial combining vismodegib with anti-PD-L1 administration (NCT05538091) is promising by concurrent immunosuppression blockage. The canonical Wnt/β-catenin pathway regulates stem cell pluripotency and determines the fate of cell differentiation. Correspondingly, PRI-724, an inhibitor of β-catenin, reduced drug resistance and CSC phenotypes in triple-negative breast cancer and downregulated SOX2 and CD44 expression in preclinical settings [144, 145]. In addition, Osawa et al. described that combining PRI-724 with anti-PD-L1 treatment resulted in regression of tumor growth in a mouse model of colon cancer and provoked more profound antitumor CD8+ T cell response compared with each monotherapy [146]. TGF-β pathway inhibitors have also been actively developed [147, 148]. It is reasonable to anticipate that dual blockade of TGF-β and PD-L1 with bifunctional fusion proteins targeting TGF-β and PD-L1 [149, 150], or co-administration of TGF-β receptor inhibitor galunisertib with the PD-L1 antibody durvalumab [151] will lead to more effective breaking down of the host immunosuppression, thus enhancing cancer immunotherapy targeting CSC-associated signaling pathways.
Challenges and future prospects
Recently, the term “CSC plasticity” was proposed, which refers to the ability of these cells to switch between stem-like and differentiated states [152]. For example, remarkable plasticity in the intestine has been found that various cell types can dedifferentiate into Lgr5+ cells to replenish the stem cell pool during perturbations to stem cells, thus acting as a hindrance to CSC-targeting therapy [153]. In the current CSC-targeted immunotherapy, the majority of studies have identified CSCs based on established stem cell markers including ALDH, CD105, CD24, CD44, CD133, CD47, SOX2 and NANOG. However, this approach may increase the risk with off-target effects in CSC-targeting therapy, given the similarities in cell surface markers and stemness programs between adult stem cells and CSCs. In addition, CSCs exhibited significant resistance to chemotherapy; molecularly targeted therapy, and immunotherapy. CSCs require multiple mechanisms to escape immune surveillance. It was shown that CSCs with elevated EMT programs exhibit resistance to T cell cytotoxicity [154]. Wnt/β-catenin stemness signaling in melanoma hinders the attraction of CD103+ dendritic cells, resulting in reduced T cell infiltration [155]. Alternatively, CSCs can elevate the level of inhibitory receptors, e.g., PD-L1, CD47, and CD206, to evade immune response [152]. Together, these challenges have suggested the limitations of translating CSC-targeted therapy to clinical applications for cancer patient treatment.
Given the immense heterogeneity and the plasticity of CSCs, accurate targeting of intrinsical CSC subset in tumor progress needs to be addressed. As single-cell profiling technologies advance in genomic, proteomic, and metabolic realms, coupled with spatial information [156, 157], they may provide hopeful approaches to discern the complex evolutionary changes of CSCs during the development of cancer. To address the immune evasion mechanisms of CSCs, research efforts may focus on pinpointing and targeting of molecular mechanisms that orchestrate the immune evasion of CSCs. As reviewed above, CSCs interact with the immunosuppressive cells in TME, such as DCregs, MDSCs, TAMs, Tregs, and Bregs to achieve immune evasion and diminish the efficacy of CSC-targeted immunotherapy. Destroying these interactions may help augment the effectiveness of such immunotherapy. For example, combination of oncolytic adenoviruses with anti-PD-L1 and anti-CTLA-4 synergistically enhanced the antitumor effect by recruiting CD8+ T cells and memory T cells, reducing the number of regulatory T cells, and promoting the polarization of TAMs from the M2 to the M1 phenotype [158]. This strongly suggests that CSC-targeted immunotherapy with concurrent blockage of the immunosuppressive TME may represent an avenue to enhance the effectiveness to eliminate CSCs in clinic.
Several clinical trials and case studies targeting CSCs are summarized in Table 2. A phase I/II Trial of CSC-derived mRNA-transfected DC vaccine in glioblastoma patients showed that vaccination against CSCs was safe, well-tolerated, and prolonged progression-free survival (NCT00846456) [159]. In another phase I study using lysate derived from an allogeneic GBM stem-like cell line to pulse autologous DCs was safe and well tolerated [160]. However, additional challenges remain in the translation of this strategy. For instance, which tumor antigen source triggers the most potent immune response? We have reported that ALDHhigh CSC lysis-DC vaccine [89], integrin β4 (a protein marker of CSCs)-DC vaccine [97], and ALDH peptide(s)-DC vaccine [98] respectively induced significant antitumor immunity. In recent years, several novel PD-L1 inhibitors including antibodies, small molecule inhibitors, and bifunctional small molecules are patented [35]. The selection of the most appropriate PD-L1 inhibitor provides the opportunity and remains a challenge as well. Addressing these issues is crucial in future endeavors for the clinic practice to break immunosuppression to enhance CSC-targeted immunotherapy.
Clinical Trials Targeting CSCs with Immunotherapies.
| NCT number/ Case | Agent | Target | Cancer Type | Phase (N) | Primary endpoint | Results | Ref. |
|---|---|---|---|---|---|---|---|
| NCT00846456 | DC vaccine with mRNA from GSCs | GSCs | Glioblastoma | Phase I/II (n=20) | AEs | Safe and well tolerated; prolonged PFS vs. controls | [159] |
| NCT02010606 | DC vaccine pulsed with GSC lysate | GSCs | Newly diagnosed/ recurrent Glioblastoma | Phase I (n=36) | AEs | Safe and well tolerated; improved PFS/OS vs. controls | [169] |
| NCT01189968 | Demcizumab + standard chemotherapy | CSCs | Metastatic non- squamous NSCLC | Phase I (n=46) | MTD | safe and well tolerated; 50% response rate | [170] |
| NCT03113643 | SL-401 + Azacitidine or Azacitidine/ Venetoclax | CSCs | AML, BPDCN, high-risk MDS | Phase I (n=72) | MTD | Safe; 69% response rate | [171] |
| NCT02074046 | Pancreatic CSC vaccine | CSCs | Pancreatic cancer | Phase I/II (n=90) | AEs | Safe and well tolerated; effectively activated CSC immune responses | [172] |
| NCT03030612 | Cusatuzumab | LSCs | Newly diagnosed AML, high risk MDS | Phase I/II (n=12) | Toxicity; ORR | ORR=100%, reduced LSC number | [173] |
| NCT03222674 | Anti-CLL1 CAR-T cells | LSCs | Pediatric R/R-AML | Phase I/II (n=8) | Treatment response; safety and tolerability | Well-tolerated; high targeting efficacy | [174] |
| Case | Anti -CLL1 CAR-T cells | LSCs | Secondary AML | n=1 | Therapeutic response; MRD; CRS | CR> 10 months | [175] |
| Case | CART123 + Haplo-HSCT | LSCs | FUS-ERG+ AML, post-allo-HSCT relapse | n=1 | Treatment response; CAR-T expansion; toxicity | Reduced AML blasts; full donor chimerism; myeloid implantation. | [176] |
GSCs: Glioma stem cells; AE: adverse event; PFS: Progression-free survival; OS: Overall survival; NSCLC: Non-Small Cell Lung Cancer; MTD: Maximum tolerated dose; AML: Acute myeloid leukemia; BPDCN: Blastic plasmacytoid dendritic cell neoplasm; MDS: Myelodysplastic syndrome; LSCs: Leukemic stem cells; ORR: Overall response rate; R/R-AML: Relapsed/Refractory Acute Myeloid Leukemia; CLL1:C-type lectin-like molecule 1; MRD: Minimal residual disease; CRS: Cytokine release syndrome; CR: Complete remission; Haplo-HSCT: Haploidentical hematopoietic stem cell transplantation; FUS-ERG+ AML: Fused in Sarcoma and Erythroblast Transformation-Specific Acute Myeloid Leukemia; allo-HSCT: Allogeneic Hematopoietic Stem Cell Transplantation.
Indeed, new therapeutic approaches are being explored day by day. These include oncolytic viruses and cancer nanomedicine. Increasing ongoing pre-clinical trials demonstrated that oncolytic viruses could successfully abrogate CSCs [161-163]. It was also reported that PD-L1 promotes oncolytic virus infection via a metabolic shift that inhibits the type I IFN pathway [164]. This suggested the facilitate to explore the PD-1/PD-L1 interaction to modulate the oncolytic virotherapy. In addition, with the rapid development of nanotechnology, novel drug delivery systems specifically designed to target CSCs are on the rise. It was reported that CSC-targeted nanomedicine revealed the potential to overcome stemness-associated chemoresistance by multiple strategies, e.g. nanomedicine with ligand modification by polysaccharide, peptide, antibody or aptamer; co-loading the nano particles with chemotherapeutic and chemopotentiators, such as CSC-eliminating agent, chemosensitizer, self-renewal inhibitor, and differentiation-inducing agent [165-168]. However, efficacious and unique stimuli for clinical practice with these encouraging novel strategies remain to be further explored.
Conclusion
CSCs are heterogenous and plastic in different cancer types for disease initiation, progression, and relapse by escaping from immune surveillance and creating immunosuppressive microenvironment. Nevertheless, multiple anti-CSC immunological strategies have been developed including CSC specific vaccines, BiAbs, ADCs, and CAR-T cells as well as inhibitors for CSC associated signaling pathways. Accumulating literatures, including our own work, demonstrated that immune checkpoint blockade during these CSC-targeted immunotherapies could achieve more potent effectiveness. Despite this, more efforts are needed to improve our grasp over the intact roles of PD-L1 in CSCs, their involvement in the interactions with TME, and additional and more potent strategies to block the immunosuppression, which may pave the road for more precise development and utilizations of immune checkpoint blockades along with CSC-directed treatment in clinic.
Abbreviations
CSCs: cancer stem cells; PD-L1: programmed cell death ligand-1; ALDH: aldehyde dehydrogenase; Hh: hedgehog; NK: natural killer; MDSCs: myeloid-derived suppressor cells; TAMs: tumor-associated macrophages; DCregs: regulatory dendritic cells; Tregs: regulatory T cells; MHC: major histocompatibility complex; TAAs: tumor associated antigens; TGF-β: transforming growth factor β; EMT: epithelial-mesenchymal transition; UTR: untranslated regions; DCs: dendritic cells; ICBs: immune checkpoint blockers; HLA: human leukocyte antigen; EVs: extracellular vehicles; PMN: polymorphonuclear; NO: nitric oxide; VEGF: vascular endothelial growth factor; ITGB4: integrin β4; mITGB4: murine ITGB4; CTL: cytotoxic T lymphocyte; sHDL: synthetic high-density lipoprotein; CIK: cytokine-induced killer; TDLNs: tumor-draining lymph nodes; BiAbs: bispecific antibodies; HER2: human epidermal growth factor receptor 2; TILs: tumor-infiltrating lymphocytes; ADCs: antibody drug conjugates; CAR: chimeric antigen receptor; ADCC: antibody-dependent cellular cytotoxicity.
Acknowledgements
The authors would like to acknowledge Ning Ning, Yangyang Hu, Shasha Ruan, Alireza Hassani Najafabadi, Leiming Xia, Fei Liao and Marisa E. Aikins for their contributions to the research projects in the past years.
Funding
This work was supported by the National Key Research and Development Program of China (Fanjun Cheng No.2020YFC2006000) and the National Natural Science Foundation of China (Fang Zheng No. 81974426).
Consent for publication
All authors have read and approved the submitted version.
Availability of data and material
The data that support the findings of this study are available from the corresponding author QL upon reasonable request.
Author contributions
QL and FC contributed to conception and design of the study. FZ and SZ wrote the draft of the manuscript. AC, JM, MW, SW, JC, JL and QL reviewed and edited the manuscript. FC and FZ acquired the fundings.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sun Q, Hong Z, Zhang C. et al. Immune checkpoint therapy for solid tumours: clinical dilemmas and future trends. Signal Transduct Target Ther. 2023;8:320
2. Kim CG, Kim KH, Pyo KH. et al. Hyperprogressive disease during PD-1/PD-L1 blockade in patients with non-small-cell lung cancer. Ann Oncol. 2019;30:1104-1113
3. Javed SA, Najmi A, Ahsan W. et al. Targeting PD-1/PD-L-1 immune checkpoint inhibition for cancer immunotherapy: success and challenges. Front Immunol. 2024;15:1383456
4. Wang Y, Zhou Y, Yang L. et al. Challenges Coexist with Opportunities: Spatial Heterogeneity Expression of PD-L1 in Cancer Therapy. Adv Sci (Weinh). 2024;11:e2303175
5. Lee-Theilen M, Fadini DD, Hadhoud JR. et al. Hepatoblastoma Cancer Stem Cells Express PD-L1, Reveal Plasticity and Can Emerge upon Chemotherapy. Cancers (Basel). 2022;14:5825
6. Koh YW, Han J-H, Haam S. Expression of PD-L1, cancer stem cell and epithelial-mesenchymal transition phenotype in non-small cell lung cancer. Pathology. 2021;53:239-246
7. Hsu J-M, Xia W, Hsu Y-H. et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. 2018;9:1908
8. Mansour FA, Al-Mazrou A, Al-Mohanna F. et al. PD-L1 is overexpressed on breast cancer stem cells through notch3/mTOR axis. Oncoimmunology. 2020;9:1729299
9. Zhang C, Wang H, Wang X. et al. CD44, a marker of cancer stem cells, is positively correlated with PD-L1 expression and immune cells infiltration in lung adenocarcinoma. Cancer Cell Int. 2020;20:583
10. Calderaro J, Rousseau B, Amaddeo G. et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship With clinical and pathological features. Hepatology. 2016;64:2038-2046
11. Hou Y-C, Chao Y-J, Hsieh M-H. et al. Low CD8⁺ T Cell Infiltration and High PD-L1 Expression Are Associated with Level of CD44⁺/CD133⁺ Cancer Stem Cells and Predict an Unfavorable Prognosis in Pancreatic Cancer. Cancers. 2019;11:541
12. Ouyang P, Wang L, Wu J. et al. Overcoming cold tumors: a combination strategy of immune checkpoint inhibitors. Front Immunol. 2024;15:1344272
13. Zeng Z, Fu M, Hu Y. et al. Regulation and signaling pathways in cancer stem cells: implications for targeted therapy for cancer. Mol Cancer. 2023;22:172
14. Wu B, Shi X, Jiang M. et al. Cross-talk between cancer stem cells and immune cells: potential therapeutic targets in the tumor immune microenvironment. Mol Cancer. 2023;22:38
15. Quintana E, Shackleton M, Sabel MS. et al. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593-598
16. Golan H, Shukrun R, Caspi R. et al. In Vivo Expansion of Cancer Stemness Affords Novel Cancer Stem Cell Targets: Malignant Rhabdoid Tumor as an Example. Stem Cell Rep. 2018;11:795-810
17. Di Tomaso T, Mazzoleni S, Wang E. et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res. 2010;16:800-813
18. Wei J, Barr J, Kong L-Y. et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010;9:67-78
19. Tsuchiya H, Shiota G. Clinical and Biological Implications of Cancer Stem Cells in Hepatocellular Carcinoma. Yonago Acta Med. 2021;64:1-11
20. Shipitsin M, Campbell LL, Argani P. et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259-273
21. Kim S-Y, Kang JW, Song X. et al. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal. 2013;25:961-969
22. Lottaz C, Beier D, Meyer K. et al. Transcriptional profiles of CD133+ and CD133- glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 2010;70:2030-2040
23. Zhong M, Zhong C, Cui W. et al. Induction of tolerogenic dendritic cells by activated TGF-β/Akt/Smad2 signaling in RIG-I-deficient stemness-high human liver cancer cells. BMC cancer. 2019;19:439
24. Nomura A, Gupta VK, Dauer P. et al. NFκB-Mediated Invasiveness in CD133+ Pancreatic TICs Is Regulated by Autocrine and Paracrine Activation of IL1 Signaling. Mol Cancer Res. 2018;16:162-172
25. Kirkland JL. Tumor dormancy and disease recurrence. Cancer Metastasis Rev. 2023;42:9-12
26. Fatma H, Siddique HR. Cancer cell plasticity, stem cell factors, and therapy resistance: how are they linked? Cancer Metastasis Rev. 2024;43:423-440
27. Sun L, Huang C, Zhu M. et al. Gastric cancer mesenchymal stem cells regulate PD-L1-CTCF enhancing cancer stem cell-like properties and tumorigenesis. Theranostics. 2020;10:11950-11962
28. Raniszewska A, Vroman H, Dumoulin D. et al. PD-L1+ lung cancer stem cells modify the metastatic lymph-node immunomicroenvironment in nsclc patients. Cancer Immunol Immunother. 2021;70:453-461
29. Wu Y, Chen M, Wu P. et al. Increased PD-L1 expression in breast and colon cancer stem cells. Clin Exp Pharmacol Physiol. 2017;44:602-604
30. Zheng F, Dang J, Zha H. et al. PD-L1 Promotes Self-Renewal and Tumorigenicity of Malignant Melanoma Initiating Cells. Biomed Res Int. 2017;2017:1293201
31. Yin S, Guo Ye, Wen X. et al. Increased expression of PD-L1 in endometrial cancer stem-like cells is regulated by hypoxia. Front Biosci (Landmark Ed). 2022;27:23
32. Polónia A, Pinto R, Cameselle-Teijeiro JF. et al. Prognostic value of stromal tumour infiltrating lymphocytes and programmed cell death-ligand 1 expression in breast cancer. J Clin Pathol. 2017;70:860-867
33. Inaguma S, Lasota J, Wang Z. et al. Expression of ALCAM (CD166) and PD-L1 (CD274) independently predicts shorter survival in malignant pleural mesothelioma. Hum Pathol. 2018;71:1-7
34. Alwosaibai K, Aalmri S, Mashhour M. et al. PD-L1 is highly expressed in ovarian cancer and associated with cancer stem cells populations expressing CD44 and other stem cell markers. BMC cancer. 2023;23:13
35. Yamaguchi H, Hsu JM, Yang WH. et al. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat Rev Clin Oncol. 2022;19:287-305
36. Almozyan S, Colak D, Mansour F. et al. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int J Cancer. 2017;141:1402-1412
37. Fu L, Fan J, Maity S. et al. PD-L1 interacts with Frizzled 6 to activate β-catenin and form a positive feedback loop to promote cancer stem cell expansion. Oncogene. 2022;41:1100-1113
38. Darvin P, Sasidharan Nair V, Elkord E. PD-L1 Expression in Human Breast Cancer Stem Cells Is Epigenetically Regulated through Posttranslational Histone Modifications. J Oncol. 2019;2019:3958908
39. Gao L, Guo Q, Li X. et al. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. EBioMedicine. 2019;41:395-407
40. Li C-W, Lim S-O, Xia W. et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632
41. Chen M, Sharma A, Lin Y. et al. Insluin and epithelial growth factor (EGF) promote programmed death ligand 1(PD-L1) production and transport in colon cancer stem cells. BMC cancer. 2019;19:153
42. Castagnoli L, Cancila V, Cordoba-Romero SL. et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene. 2019;38:4047-4060
43. Min HY, Cho J, Sim JY. et al. S100A14: A novel negative regulator of cancer stemness and immune evasion by inhibiting STAT3-mediated programmed death-ligand 1 expression in colorectal cancer. Clin Transl Med. 2022;12:e986
44. Mortezaee K, Majidpoor J. Mechanisms of CD8+ T cell exclusion and dysfunction in cancer resistance to anti-PD-(L)1. Biomed Pharmacother. 2023;163:114824
45. Borlongan MC, Saha D, Wang H. Tumor Microenvironment: A Niche for Cancer Stem Cell Immunotherapy. Stem Cell Rev Rep. 2024;20:3-24
46. Katopodi T, Petanidis S, Charalampidis C. et al. Tumor-Infiltrating Dendritic Cells: Decisive Roles in Cancer Immunosurveillance, Immunoediting, and Tumor T Cell Tolerance. Cells. 2022;11:3183
47. Liang S, Ristich V, Arase H. et al. Modulation of dendritic cell differentiation by HLA-G and ILT4 requires the IL-6-STAT3 signaling pathway. Proc Natl Acad Sci U S A. 2008;105:8357-8362
48. Grange C, Tapparo M, Tritta S. et al. Role of HLA-G and extracellular vesicles in renal cancer stem cell-induced inhibition of dendritic cell differentiation. BMC cancer. 2015;15:1009
49. Del Prete A, Salvi V, Soriani A. et al. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell Mol Immunol. 2023;20:432-447
50. Xu L, Zhang Y, Tian K. et al. Apigenin suppresses PD-L1 expression in melanoma and host dendritic cells to elicit synergistic therapeutic effects. J Exp Clin Cancer Res. 2018;37:261
51. Hassannia H, Ghasemi Chaleshtari M, Atyabi F. et al. Blockage of immune checkpoint molecules increases T-cell priming potential of dendritic cell vaccine. Immunology. 2020;159:75-87
52. Lasser SA, Ozbay Kurt FG, Arkhypov I. et al. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat Rev Clin Oncol. 2024;21:147-164
53. Ding G, Yu H, Jin J. et al. Reciprocal relationship between cancer stem cells and myeloid-derived suppressor cells: implications for tumor progression and therapeutic strategies. Future Oncol. 2024;20:215-228
54. Welte T, Kim IS, Tian L. et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18:632-644
55. Peng D, Tanikawa T, Li W. et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016;76:3156-3165
56. Wang Y, Yin K, Tian J. et al. Granulocytic Myeloid-Derived Suppressor Cells Promote the Stemness of Colorectal Cancer Cells through Exosomal S100A9. Adv Sci (Weinh). 2019;6:1901278
57. Li X, Wang J, Wu W. et al. Myeloid-derived suppressor cells promote epithelial ovarian cancer cell stemness by inducing the CSF2/p-STAT3 signalling pathway. FEBS J. 2020;287:5218-5235
58. Ai L, Mu S, Sun C. et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol Cancer. 2019;18:88
59. Shidal C, Singh NP, Nagarkatti P. et al. MicroRNA-92 Expression in CD133+ Melanoma Stem Cells Regulates Immunosuppression in the Tumor Microenvironment via Integrin-Dependent Activation of TGFβ. Cancer Res. 2019;79:3622-3635
60. Wang G, Lu X, Dey P. et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016;6:80-95
61. Komura N, Mabuchi S, Shimura K. et al. The role of myeloid-derived suppressor cells in increasing cancer stem-like cells and promoting PD-L1 expression in epithelial ovarian cancer. Cancer Immunol Immunother. 2020;69:2477-2499
62. Luo S, Yang G, Ye P. et al. Macrophages Are a Double-Edged Sword: Molecular Crosstalk between Tumor-Associated Macrophages and Cancer Stem Cells. Biomolecules. 2022;12:850
63. Wang L, Zhang L, Zhao L. et al. VEGFA/NRP-1/GAPVD1 axis promotes progression and cancer stemness of triple-negative breast cancer by enhancing tumor cell-macrophage crosstalk. Int J Biol Sci. 2024;20:446-463
64. Huang R, Wang S, Wang N. et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating β-catenin/STAT3 signaling. Cell Death Dis. 2020;11:234
65. Noguchi T, Ward JP, Gubin MM. et al. Temporally Distinct PD-L1 Expression by Tumor and Host Cells Contributes to Immune Escape. Cancer Immunol Res. 2017;5:106-117
66. Zhu Z, Zhang H, Chen B. et al. PD-L1-Mediated Immunosuppression in Glioblastoma Is Associated With the Infiltration and M2-Polarization of Tumor-Associated Macrophages. Front Immunol. 2020;11:588552
67. Liu Y, Zugazagoitia J, Ahmed FS. et al. Immune Cell PD-L1 Colocalizes with Macrophages and Is Associated with Outcome in PD-1 Pathway Blockade Therapy. Clin Cancer Res. 2020;26:970-977
68. Napoletano C, Bellati F, Ruscito I. et al. Immunological and Clinical Impact of Cancer Stem Cells in Vulvar Cancer: Role of CD133/CD24/ABCG2-Expressing Cells. Anticancer Res. 2016;36:5109-5116
69. You Y, Li Y, Li M. et al. Ovarian cancer stem cells promote tumour immune privilege and invasion via CCL5 and regulatory T cells. Clin Exp Immunol. 2018;191:60-73
70. Xu Y, Dong X, Qi P. et al. Sox2 Communicates with Tregs Through CCL1 to Promote the Stemness Property of Breast Cancer Cells. Stem Cells. 2017;35:2351-2365
71. Schober M, Fuchs E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-β and integrin/focal adhesion kinase (FAK) signaling. Proc Natl Acad Sci U S A. 2011;108:10544-10549
72. Gupta S, Joshi K, Wig JD. et al. Intratumoral FOXP3 expression in infiltrating breast carcinoma: Its association with clinicopathologic parameters and angiogenesis. Acta Oncol. 2007;46:792-797
73. Vilbois S, Xu Y, Ho P-C. Metabolic interplay: tumor macrophages and regulatory T cells. Trends Cancer. 2024;10:242-255
74. Fortunato O, Belisario DC, Compagno M. et al. CXCR4 Inhibition Counteracts Immunosuppressive Properties of Metastatic NSCLC Stem Cells. Front Immunol. 2020;11:02168
75. Francisco LM, Salinas VH, Brown KE. et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015-3029
76. Guo F, Zhang Y, Bai L. et al. Natural killer cell therapy targeting cancer stem cells: Old wine in a new bottle. Cancer Lett. 2023;570:216328
77. Chovatiya N, Kaur K, Huerta-Yepez S. et al. Inability of ovarian cancers to upregulate their MHC-class I surface expression marks their aggressiveness and increased susceptibility to NK cell-mediated cytotoxicity. Cancer Immunol Immunother. 2022;71:2929-2941
78. Bushnell GG, Sharma D, Wilmot HC. et al. Natural Killer Cell Regulation of Breast Cancer Stem Cells Mediates Metastatic Dormancy. Cancer Res. 2024;84:3337-3353
79. Yin T, Wang G, He S. et al. Human cancer cells with stem cell-like phenotype exhibit enhanced sensitivity to the cytotoxicity of IL-2 and IL-15 activated natural killer cells. Cell Immunol. 2016;300:41-45
80. Du Y, Pollok KE, Shen J. Unlocking Glioblastoma Secrets: Natural Killer Cell Therapy against Cancer Stem Cells. Cancers (Basel). 2023;15:5836
81. Frazao A, Rethacker L, Messaoudene M. et al. NKG2D/NKG2-Ligand Pathway Offers New Opportunities in Cancer Treatment. Front Immunol. 2019;10:661
82. Duan S, Guo W, Xu Z. et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol Cancer. 2019;18:29
83. Pesce S, Greppi M, Grossi F. et al. PD/1-PD-Ls Checkpoint: Insight on the Potential Role of NK Cells. Front Immunol. 2019;10:1242
84. Ma J, Wu Y, Ma L. et al. A blueprint for tumor-infiltrating B cells across human cancers. Science. 2024;384:eadj4857
85. Lu L, Tao H, Chang AE. et al. Cancer stem cell vaccine inhibits metastases of primary tumors and induces humoral immune responses against cancer stem cells. Oncoimmunology. 2015;4:e990767
86. Liu F, Zheng J, Yang G. et al. Unraveling the enigma of B cells in diffuse large B-cell lymphoma: unveiling cancer stem cell-like B cell subpopulation at single-cell resolution. Front Immunol. 2023;14:1310292
87. Pellegatta S, Poliani PL, Corno D. et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006;66:10247-10252
88. Xu Q, Liu G, Yuan X. et al. Antigen-specific T-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells. 2009;27:1734-1740
89. Ning N, Pan Q, Zheng F. et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. 2012;72:1853-1864
90. Teitz-Tennenbaum S, Wicha MS, Chang AE. et al. Targeting cancer stem cells via dendritic-cell vaccination. Oncoimmunology. 2012;1:1401-1403
91. Zhang X-F, Weng D-S, Pan K. et al. Dendritic-cell-based immunotherapy evokes potent anti-tumor immune responses in CD105+ human renal cancer stem cel ls. Mol Carcinog. 2017;56:2499-2511
92. Dashti A, Ebrahimi M, Hadjati J. et al. Dendritic cell based immunotherapy using tumor stem cells mediates potent antitumor immune responses. Cancer Lett. 2016;374:175-185
93. Hu Y, Lu L, Xia Y. et al. Therapeutic Efficacy of Cancer Stem Cell Vaccines in the Adjuvant Setting. Cancer Res. 2016;76:4661-4672
94. Zheng F, Dang J, Zhang H. et al. Cancer Stem Cell Vaccination With PD-L1 and CTLA-4 Blockades Enhances the Eradication of Melanoma Stem Cells in a Mouse Tumor Model. J Immunother. 2018;41:361-368
95. Guo L, Mohanty A, Singhal S. et al. Targeting ITGB4/SOX2-driven lung cancer stem cells using proteasome inhibitors. iScience. 2023;26:107302
96. Bierie B, Pierce SE, Kroeger C. et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc Natl Acad Sci U S A. 2017;114:E2337-E2346
97. Ruan S, Lin M, Zhu Y. et al. Integrin β4-Targeted Cancer Immunotherapies Inhibit Tumor Growth and Decrease Metastasis. Cancer Res. 2020;80:771-783
98. Liao F, Zhang J, Hu Y. et al. Efficacy of an ALDH peptide-based dendritic cell vaccine targeting cancer stem cells. Cancer Immunol Immunother. 2022;71:1959-1973
99. Scheetz L, Park KS, Li Q. et al. Engineering patient-specific cancer immunotherapies. Nat Biomed Eng. 2019;3:768-782
100. Kuai R, Li D, Chen YE. et al. High-Density Lipoproteins: Nature's Multifunctional Nanoparticles. ACS nano. 2016;10:3015-3041
101. Kuai R, Ochyl LJ, Bahjat KS. et al. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16:489-496
102. Hassani Najafabadi A, Zhang J, Aikins ME. et al. Cancer Immunotherapy via Targeting Cancer Stem Cells Using Vaccine Nanodiscs. Nano Lett. 2020;20:7783-7792
103. Aikins ME, Qin Y, Dobson HE. et al. Cancer stem cell antigen nanodisc cocktail elicits anti-tumor immune responses in melanoma. J Control Release. 2022;351:872-882
104. van de Donk NWCJ, Zweegman S. T-cell-engaging bispecific antibodies in cancer. Lancet. 2023;402:142-158
105. Wei J, Yang Y, Wang G. et al. Current landscape and future directions of bispecific antibodies in cancer immunotherapy. Front Immunol. 2022;13:1035276
106. Huang J, Li C, Wang Y. et al. Cytokine-induced killer (CIK) cells bound with anti-CD3/anti-CD133 bispecific antibodies target CD133(high) cancer stem cells in vitro and in vivo. Clin Immunol. 2013;149:156-168
107. Geng Q, Jiao P. Anti-PD-L1-Based Bispecific Antibodies Targeting Co-Inhibitory and Co-Stimulatory Molecules for Cancer Immunotherapy. Molecules. 2024;29:454
108. Chang C-H, Wang Y, Li R. et al. Combination Therapy with Bispecific Antibodies and PD-1 Blockade Enhances the Antitumor Potency of T Cells. Cancer Res. 2017;77:5384-5394
109. von Arx C, De Placido P, Caltavituro A. et al. The evolving therapeutic landscape of trastuzumab-drug conjugates: Future perspectives beyond HER2-positive breast cancer. Cancer Treat Rev. 2023;113:102500
110. Xia L, Wen L, Qin Y. et al. HER2-targeted antibody-drug conjugate induces host immunity against cancer stem cells. Cell Chem Biol. 2021;28:610-624.e5
111. Golubovskaya V, Berahovich R, Zhou H. et al. CD47-CAR-T Cells Effectively Kill Target Cancer Cells and Block Pancreatic Tumor Growth. Cancers. 2017;9:139
112. Liang D, Tang J, Sun B. et al. Novel CAR-T cells targeting TRKB for the treatment of solid cancer. Apoptosis. 2024;29:2183-2196
113. Kondo T, Ando M, Nagai N. et al. The NOTCH-FOXM1 Axis Plays a Key Role in Mitochondrial Biogenesis in the Induction of Human Stem Cell Memory-like CAR-T Cells. Cancer Res. 2020;80:471-483
114. Han Y, Sun B, Cai H. et al. Simultaneously target of normal and stem cells-like gastric cancer cells via cisplatin and anti-CD133 CAR-T combination therapy. Cancer Immunol Immunother. 2021;70:2795-2803
115. Lontos K, Wang Y, Joshi SK. et al. Metabolic reprogramming via an engineered PGC-1α improves human chimeric antigen receptor T-cell therapy against solid tumors. J Immunother Cancer. 2023;11:e006522
116. Wang Y, Chen M, Wu Z. et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology. 2018;7:e1440169
117. Feng KC, Guo YL, Liu Y. et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol. 2017;10:4
118. Tanoue K, Rosewell Shaw A, Watanabe N. et al. Armed Oncolytic Adenovirus-Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2017;77:2040-2051
119. Yamaguchi Y, Gibson J, Ou K. et al. PD-L1 blockade restores CAR T cell activity through IFN-γ-regulation of CD163+ M2 macrophages. J Immunother Cancer. 2022;10:e004400
120. Wu S-Y, Fu T, Jiang Y-Z. et al. Natural killer cells in cancer biology and therapy. Mol Cancer. 2020;19:120
121. Bui VT, Tseng HC, Kozlowska A. et al. Augmented IFN-gamma and TNF-alpha Induced by Probiotic Bacteria in NK Cells Mediate Differentiation of Stem-Like Tumors Leading to Inhibition of Tumor Growth and Reduction in Inflammatory Cytokine Release; Regulation by IL-10. Front Immunol. 2015;6:576
122. Tallerico R, Garofalo C, Carbone E. A New Biological Feature of Natural Killer Cells: The Recognition of Solid Tumor-Derived Cancer Stem Cells. Front Immunol. 2016;7:179
123. Kim GR, Ha G-H, Bae J-H. et al. Metastatic colon cancer cell populations contain more cancer stem-like cells with a higher susceptibility to natural killer cell-mediated lysis compared with primary colon cancer cells. Oncol Lett. 2015;9:1641-1646
124. Voutsadakis IA. Expression and function of immune ligand-receptor pairs in NK cells and cancer stem cells: therapeutic implications. Cell Oncol (Dordr). 2018;41:107-121
125. Grossenbacher SK, Canter RJ, Murphy WJ. Natural killer cell immunotherapy to target stem-like tumor cells. J Immunother Cancer. 2016;4:19
126. Tallerico R, Todaro M, Di Franco S. et al. Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol. 2013;190:2381-2390
127. Kaur K, Nanut MP, Ko M-W. et al. Natural killer cells target and differentiate cancer stem-like cells/undifferentiated tumors: strategies to optimize their growth and expansion for effective cancer immunotherapy. Curr Opin Immunol. 2018;51:170-180
128. Ames E, Canter RJ, Grossenbacher SK. et al. NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. J Immunol. 2015;195:4010-4019
129. Castriconi R, Daga A, Dondero A. et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol. 2009;182:3530-3539
130. Schmohl JU, Felices M, Oh F. et al. Engineering of Anti-CD133 Trispecific Molecule Capable of Inducing NK Expansion and Driving Antibody-Dependent Cell-Mediated Cytotoxicity. Cancer Res Treat. 2017;49:1140-1152
131. Yilmaz A, Cui H, Caligiuri MA. et al. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. 2020;13:168
132. Kailayangiri S, Altvater B, Spurny C. et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology. 2017;6:e1250050
133. Boissel L, Betancur-Boissel M, Lu W. et al. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology. 2013;2:e26527
134. Zhang C, Burger MC, Jennewein L. et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J Natl Cancer Inst. 2016;108:djv375
135. Klapdor R, Wang S, Hacker U. et al. Improved Killing of Ovarian Cancer Stem Cells by Combining a Novel Chimeric Antigen Receptor-Based Immunotherapy and Chemotherapy. Hum Gene Ther. 2017;28:886-896
136. Medrano-Gonzálezl PA, Cruz-Villegas F, Alarcón Del Carmen A. et al. Claudin-6 increases SNAI1, NANOG and SOX2 gene expression in human gastric adenocarcinoma AGS cells. Mol Biol Rep. 2022;49:11663-11674
137. Li J, Hu H, Lian H. et al. NK-92MI Cells Engineered with Anti-claudin-6 Chimeric Antigen Receptors in Immunotherapy for Ovarian Cancer. Int J Biol Sci. 2024;20:1578-1601
138. Wang B, Wang Q, Wang Z. et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014;74:5746-5757
139. Paczulla AM, Rothfelder K, Raffel S. et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. 2019;572:254-259
140. Liu Y, Wang H. Biomarkers and targeted therapy for cancer stem cells. Trends Pharmacol Sci. 2024;45:56-66
141. Zhang Y, Beachy PA. Cellular and molecular mechanisms of Hedgehog signalling. Nat Rev Mol Cell Biol. 2023;24:668-687
142. Dummer R, Guminksi A, Gutzmer R. et al. Long-term efficacy and safety of sonidegib in patients with advanced basal cell carcinoma: 42-month analysis of the phase II randomized, double-blind BOLT study. Br J Dermatol. 2020;182:1369-1378
143. Sekulic A, Migden MR, Basset-Seguin N. et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: final update of the pivotal ERIVANCE BCC study. BMC cancer. 2017;17:332
144. Ring A, Nguyen C, Smbatyan G. et al. CBP/β-Catenin/FOXM1 Is a Novel Therapeutic Target in Triple Negative Breast Cancer. Cancers. 2018;10:525
145. Chan KC, Chan LS, Ip JCY. et al. Therapeutic targeting of CBP/β-catenin signaling reduces cancer stem-like population and synergistically suppresses growth of EBV-positive nasopharyngeal carcinoma cells with cisplatin. Sci Rep. 2015;5:9979
146. Osawa Y, Kojika E, Nishikawa K. et al. Programmed cell death ligand 1 (PD-L1) blockade attenuates metastatic colon cancer growth in cAMP-response element-binding protein (CREB)-binding protein (CBP)/β-catenin inhibitor-treated livers. Oncotarget. 2019;10:3013-3026
147. Yamazaki T, Gunderson AJ, Gilchrist M. et al. Galunisertib plus neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer: a single-arm, phase 2 trial. Lancet Oncol. 2022;23:1189-1200
148. Platzbecker U, Germing U, Götze KS. et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18:1338-1347
149. Redman JM, Friedman J, Robbins Y. et al. Enhanced neoepitope-specific immunity following neoadjuvant PD-L1 and TGF-β blockade in HPV-unrelated head and neck cancer. J Clin Invest. 2022;132:e161400
150. Feng J, Tang D, Wang J. et al. SHR-1701, a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, for Recurrent or Metastatic Cervical Cancer: A Clinical Expansion Cohort of a Phase I Study. Clin Cancer Res. 2022;28:5297-5305
151. Melisi D, Oh D-Y, Hollebecque A. et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J Immunother Cancer. 2021;9:e002068
152. Loh JJ, Ma S. Hallmarks of cancer stemness. Cell Stem Cell. 2024;31:617-639
153. Jadhav U, Saxena M, O'Neill NK. et al. Dynamic Reorganization of Chromatin Accessibility Signatures during Dedifferentiation of Secretory Precursors into Lgr5+ Intestinal Stem Cells. Cell Stem Cell. 2017;21:65-77.e5
154. Corgnac S, Damei I, Gros G. et al. Cancer stem-like cells evade CD8+CD103+ tumor-resident memory T (TRM) lymphocytes by initiating an epithelial-to-mesenchymal transition program in a human lung tumor model. J Immunother Cancer. 2022;10:e004527
155. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231-235
156. Li R, Liu X, Huang X. et al. Single-cell transcriptomic analysis deciphers heterogenous cancer stem-like cells in colorectal cancer and their organ-specific metastasis. Gut. 2024;73:470-484
157. Holton E, Muskovic W, Powell JE. Deciphering cancer cell state plasticity with single-cell genomics and artificial intelligence. Genome Med. 2024;16:36
158. Zhang H, Xie W, Zhang Y. et al. Oncolytic adenoviruses synergistically enhance anti-PD-L1 and anti-CTLA-4 immunotherapy by modulating the tumour microenvironment in a 4T1 orthotopic mouse model. Cancer Gene Ther. 2022;29:456-465
159. Vik-Mo EO, Nyakas M, Mikkelsen BV. et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. 2013;62:1499-1509
160. Maeng HM, Moore BN, Bagheri H. et al. Phase I Clinical Trial of an Autologous Dendritic Cell Vaccine Against HER2 Shows Safety and Preliminary Clinical Efficacy. Front Oncol. 2021;11:789078
161. Tong Y, You L, Liu H. et al. Potent antitumor activity of oncolytic adenovirus expressing Beclin-1 via induction of autophagic cell death in leukemia. Oncotarget. 2013;4:860-874
162. Hu JC, Coffin RS, Davis CJ. et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12:6737-6747
163. Yang H, Peng T, Li J. et al. Treatment of colon cancer with oncolytic herpes simplex virus in preclinical models. Gene Ther. 2016;23:450-459
164. Hodgins JJ, Abou-Hamad J, O'Dwyer CE. et al. PD-L1 promotes oncolytic virus infection via a metabolic shift that inhibits the type I IFN pathway. J Exp Med. 2024;221:e20221721
165. Liu H, Liu M, Zhao Y. et al. Nanomedicine strategies to counteract cancer stemness and chemoresistance. Explor Target Antitumor Ther. 2023;4:630-656
166. Dzobo K, Sinkala M. Cancer Stem Cell Marker CD44 Plays Multiple Key Roles in Human Cancers: Immune Suppression/Evasion, Drug Resistance, Epithelial-Mesenchymal Transition, and Metastasis. Omics. 2021;25:313-332
167. Singh AK, Verma A, Singh A. et al. Salinomycin inhibits epigenetic modulator EZH2 to enhance death receptors in colon cancer stem cells. Epigenetics. 2021;16:144-161
168. Pan Y, Zhou S, Li Y. et al. Novel dendritic polyglycerol-conjugated, mesoporous silica-based targeting nanocarriers for co-delivery of doxorubicin and tariquidar to overcome multidrug resistance in breast cancer stem cells. J Control Release. 2021;330:1106-1117
169. Hu JL, Omofoye OA, Rudnick JD. et al. A Phase I Study of Autologous Dendritic Cell Vaccine Pulsed with Allogeneic Stem-like Cell Line Lysate in Patients with Newly Diagnosed or Recurrent Glioblastoma. Clin Cancer Res. 2022;28:689-696
170. McKeage MJ, Kotasek D, Markman B. et al. Phase IB Trial of the Anti-Cancer Stem Cell DLL4-Binding Agent Demcizumab with Pemetrexed and Carboplatin as First-Line Treatment of Metastatic Non-Squamous NSCLC. Target Oncol. 2018;13:89-98
171. Lane AA, Garcia JS, Raulston EG. et al. Phase 1b trial of tagraxofusp in combination with azacitidine with or without venetoclax in acute myeloid leukemia. Blood Adv. 2024;8:591-602
172. Lin M, Yuan YY, Liu SP. et al. Prospective study of the safety and efficacy of a pancreatic cancer stem cell vaccine. J Cancer Res Clin Oncol. 2015;141:1827-1833
173. Riether C, Pabst T, Höpner S. et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med. 2020;26:1459-1467
174. Zhang H, Bu C, Peng Z. et al. Characteristics of anti-CLL1 based CAR-T therapy for children with relapsed or refractory acute myeloid leukemia: the multi-center efficacy and safety interim analysis. Leukemia. 2022;36:2596-2604
175. Zhang H, Gan W-T, Hao W-G. et al. Successful Anti-CLL1 CAR T-Cell Therapy in Secondary Acute Myeloid Leukemia. Front Oncol. 2020;10:685
176. Yao S, Jianlin C, Yarong L. et al. Donor-Derived CD123-Targeted CAR T Cell Serves as a RIC Regimen for Haploidentical Transplantation in a Patient With FUS-ERG+ AML. Front Oncol. 2019;9:1358
Author contact
![]() Corresponding authors: Qiao Li, E-mail: qiaoliumich.edu, Department of Surgery, University of Michigan, Ann Arbor, Michigan 48109, USA. Fanjun Cheng, E-mail: chengfanjun001com, Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China. Jixian Liu, E-mail: liujixiancom, Peking University Shenzhen Hospital, Shenzhen, China.
Corresponding authors: Qiao Li, E-mail: qiaoliumich.edu, Department of Surgery, University of Michigan, Ann Arbor, Michigan 48109, USA. Fanjun Cheng, E-mail: chengfanjun001com, Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China. Jixian Liu, E-mail: liujixiancom, Peking University Shenzhen Hospital, Shenzhen, China.