Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. An overview of the...
2. Mitochondrial quality control...
3. Mitochondrial metabolism in...
4. Clinical translation of...
5. Concluding remarks
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(5):2179-2200. doi:10.7150/ijbs.105446 This issue Cite
Review
Mitochondrial Regulation of Ferroptosis in Cancer Cells
Zilin Ding#, Zhiyu Li#, Kai Sun, Yi Liu, Zhou Fang, Shengrong Sun ![]() , Chenyuan Li
, Chenyuan Li ![]() , Zhong Wang
, Zhong Wang ![]()
Department of Breast & Thyroid Surgery, Renmin Hospital of Wuhan University, Wuhan, Hubei, People's Republic of China.
#Contributed equally.
Received 2024-10-18; Accepted 2025-2-11; Published 2025-2-24
Abstract

Ferroptosis is an iron-dependent nonapoptotic regulated cell death modality characterized by lethal levels of lipid peroxide accumulation and disrupted antioxidant systems. An increasing number of studies have revealed correlations between ferroptosis and the pathophysiology and treatment of cancer. Given the intricate involvement of mitochondria in ferroptosis, as suggested by previous studies, here, we review advances in understanding the roles of mitochondrial quality control and mitochondrial metabolism (including the roles of the TCA cycle, reactive oxygen species, iron metabolism, and lipid metabolism) in cancer-related ferroptosis and outline the molecular mechanism and clinical translation of mitochondria-related ferroptosis in cancer treatment. with the aim of promoting the precise utilization and prevention of ferroptosis in cancer therapeutics.
Keywords: ferroptosis, cancer, mitochondrial metabolism, mitochondrial quality control
Introduction
Ferroptosis is a nonapoptotic cell death modality characterized by lethal lipid peroxidation resulting from an imbalance in cellular metabolism and redox homeostasis, and it has attracted considerable attention in the scientific community because cells undergoing ferroptosis are morphologically different and the underlying mechanisms of ferroptosis differ from those of other known forms of regulated cell death, such as apoptosis and necrosis[1]. Recent studies have linked ferroptosis to various pathological conditions and diseases, and its important roles in cancer development and treatment are increasingly recognized[2], [3]. Notably, numerous types of therapy-resistant cancer cells, especially those that show high levels of mesenchymal and dedifferentiation characteristics are more susceptible to ferroptosis[4], [5], [6], [7]. Classic therapeutic agents, such as lapatinib and sorafenib, have been shown to induce ferroptosis and arrest tumor growth by increasing cellular iron levels or inhibiting cystine uptake[8], [9]. Therefore, the exploration of effective ferroptosis inducers or inhibitors may lead to a breakthrough in the search for treatments for cancer[10].
Mitochondria are cellular energy powerhouses involved in maintaining cell survival, defending against oxidative stress, and promoting cellular metabolic homeostasis. On one hand, mitochondria are sites of oxidative phosphorylation and large amounts of energy production, which is needed for metabolic activity. On the other hand, mitochondria are the major organelles responsible for reactive oxygen species (ROS) production and host fatty acid metabolism, iron metabolism, and many other important metabolic processes[11]. The external structure and cellular location of mitochondria are critical for their function and depend on a highly regulated quality control system, which involves, for example, continuous fission and fusion that govern the overall shape and timely removal of damaged mitochondria through mitophagy[11].
Given the central role of mitochondria in the regulation of cell death and the close association between ferroptosis and cellular metabolism, researchers have hypothesized that mitochondria may be involved in the regulation of ferroptosis. One of the characteristics of ferroptosis that initially distinguishes it from other types of regulated cell death is the difference in mitochondrial morphology, as observed via electron microscopy[1]. However, early studies revealed that cells with lost mitochondrial DNA or depleted mitochondria remained sensitive to ferroptosis[1], [12], while other studies concluded that mitochondrion-depleted HT-1080 cells appeared to be less sensitive to ferroptosis triggered by cysteine deprivation but showed no significant resistance to RSL3-induced ferroptosis[13]. In summary, increasing evidence suggests that the role of mitochondria in ferroptosis is context dependent.
This review discusses the known roles of mitochondria in regulating ferroptosis, especially amino acid metabolism, iron metabolism, and fatty acid metabolism, and presents the conflicting data and plausible explanations for the role of mitochondria in the regulation of ferroptosis in cancer.
1. An overview of the ferroptosis pathway
Iron overload is closely related to ferroptosis. The iron-mediated Fenton reaction and iron-dependent enzymes lipoxygenase (ALOX) and cytochrome P450 oxidoreductase (POR) play vital roles in lipid peroxidation. These factors partially explain the iron-dependent characteristic of ferroptosis. Iron homeostasis is achieved through the strict regulation of iron uptake, storage, utilization, and export. Transferrin is an iron-carrier protein in serum that transports extracellular iron into cells by interacting with transferrin receptor 1 (TFR1), thereby forming the labile redox-active iron pool required for ferroptosis[14]. Intracellular iron levels can be sensed by intracellular metal sensors, which respond to initiate the regulation of the levels of iron storage and transport, change the labile iron pool level, and ultimately increase or decrease the sensitivity of ferroptosis[15]. Fe3+ is stored by ferritin, which is composed of the ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1). Cytoplasmic ferritin levels can be downregulated by NCOA4-mediated ferritinophagy and the depletion of other autophagy-related genes, such as ATG5 and ATG7[16], [17], [18]. In addition, dihydroartemisinin has been shown to sensitize cancer cells to ferroptosis by inducing the lysosomal degradation of ferritin in an autophagy-independent manner[19]. The membrane protein ferroportin (FPN1) and Prominin2 (PROM2) establish the iron export pathways discovered thus far that maintain intracellular and plasma iron homeostasis in the presence of excess labile iron, endowing cancer cells with resistance to ferroptosis[20], [21]. Taken together, these data indicate that iron metabolism plays an essential role in ferroptosis governance through multiple mechanisms.
Cells are equipped with various defense mechanisms that prevent ferroptosis by stopping the lethal accumulation of lipid peroxides to counteract the increase in the degree of oxidative damage caused by metabolic activities associated with ferroptosis. Previous research has revealed three widely recognized defense mechanisms. First, the widely acknowledged canonical pathway has been shown to inhibit ferroptosis via the cystine/glutathione (GSH)/GPX4 signaling axis. System Xc- mediates the internalization of extracellular cystine in conjunction with the export of intracellular glutamate (Glu), a process that is required to maintain reduced GSH levels[7], [22]. Stable GSH levels ensure the activity of glutathione peroxidase 4 (GPX4), which is characterized mainly by its unique enzymatic function in the reduction of phospholipid hydroperoxides[7], [23]. The classical ferroptosis inhibitor erastin and RSL3 block system Xc- and GPX4 activities, respectively[22], [23]. Second, ferroptosis suppressor protein 1 (FSP1), previously known as apoptosis-inducing factor mitochondria-associated 2 (AIFM2), has been identified as a potent ferroptosis resistance factor. Mechanistically, FSP1 functions as an oxidoreductase to reduce coenzyme Q10 (CoQ or ubiquinone). In its fully reduced form, CoQH2 functions as a lipophilic free radical that traps the antioxidants that prevent the production of lipid peroxides[24], [25], [26], [27]. Third, ferroptosis in tumors with a specific genetic background is differentially regulated by sex hormones. Mechanistically, sex hormone signaling mediates the transcriptional upregulation of MBOAT1/2 to suppress ferroptosis in cancer cells, which selectively transfers MUFAs into lyso-PE, hence competitively reducing the PE-PUFA content[28]. Another study proposed that androgen receptor (AR) activates GPX4 expression in LAR tumors by directly binding to AR response elements located in the GPX4 promoter region[29]. Thus, AR lies at the nexus between PUFA metabolism and the GSH system. These novel findings underscore the concept that an integrated antioxidant system can effectively attenuate ferroptosis caused by excessive oxidative stress.
Ferroptosis ultimately depends on the catalytic oxidation of polyunsaturated fatty acids (PUFAs) in membrane phospholipids, resulting in the accumulation of lipid peroxides. PUFAs carry more than one unsaturated bond, which makes them vulnerable to the free radical attacks that induce lipid peroxidation. A recent study has revealed that PL-PUFA2, rather than PL-PUFA1, play a key role in the execution of ferroptosis. A selective reduction in PC-PUFA2 synthesis by MUFAs reverses the effect of free PUFAs, and thus MUFA supplementation prevents ferroptosis[30]. PUFA biosynthesis depends on certain lipid metabolic enzymes, such as acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). The oxidation of PUFAs is mediated mainly by enzymatic reactions and spontaneous nonenzymatic autooxidation. The mitochondrial electron transport chain (ETC), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), and myeloperoxidase (MPO) are the sources of some of the ROS needed for ferroptosis[31], [32], [33]. Nonenzymatic lipid peroxidation occurs through the Fenton reaction, which is a catalytic process in which Fe2+ reacts with H2O2 to produce Fe3+, HO·, and OH-, resulting in free radical ions that cause oxidative damage to various cellular components, including membrane lipids[34]. However, the enzymatic oxidation of PUFAs and subsequent ferroptosis are mediated in a cell type-dependent manner by different members of the ALOX family, such as ALOX12 and ALOX15[35], [36]. Alternatively, POR drives lipid peroxidation in an ALOX-independent manner, promoting ferroptosis across a wide spectrum of lineages and cell states[37]. Therefore, the suggestion that iron chelation or inactivation of ACSL4, LPCAT3, or POR blocks or considerably attenuates ferroptosis is reasonable.
2. Mitochondrial quality control and ferroptosis
One of the morphological features that distinguishes ferroptosis from other forms of cell death is the notable change in the mitochondrial ultrastructure during ferroptosis. Engineered HRAS-mutant BJeLR tumor cells treated with erastin presented morphological characteristics that included a smaller organelle size, condensed mitochondrial membranes, which increased their density, and a reduction in the number of mitochondrial cristae; however, these cells exhibited none of the characteristic morphologic features associated with necrosis (e.g., cytoplasmic and organelle swelling, and plasma membrane rupture), apoptosis (e.g., chromatin condensation and margination), or autophagy (e.g., the formation of double-membrane entrapment vesicles)[1].
Mitochondria have evolved multiple systems of quality control to ensure that the requisite number of functional mitochondria are available to meet the demands of cells[38]. Multilevel and well-orchestrated networks are involved in the maintenance of mitochondrial proteostasis, mitochondrial dynamics, mitophagy, and mitochondrial biogenesis (Figure 1).
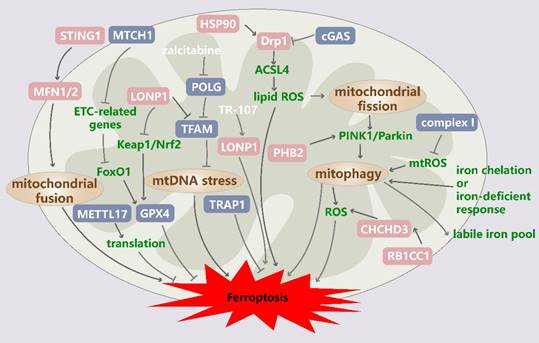
Mitochondrial quality control in ferroptosis. Zalcitabine targets POLG to induce the LONP1-dependent degradation of TFAM, thereby inducing mtDNA stress and subsequent ferroptosis. LONP1 is also involved in regulating ferroptosis mediated partly through the Keap1/Nrf2/GPX4 pathway. The RB1CC1-mediated upregulation of CHCHD3 stimulates MMP hyperpolarization and mtROS production to sensitize tumor cells to ferroptosis. TRAP1 plays a protective role in ferroptosis. STING1-MFN1/2 signaling triggers ferroptosis by inducing mitochondrial fusion. The upregulation of the HSP90/Drp1/ACSL4 pathway positively regulates ferroptosis via lipid ROS generation and altered mitochondrial morphology. MTCH1 deficiency downregulates mitochondrial ETC-related genes and promotes ferroptosis by triggering FoxO1-GPX4-axon-mediated retrograde signaling. METTL17 inhibition results in the impaired translation of mitochondrial protein-coding genes and increases ferroptosis sensitivity. Mitochondria-localized cGAS enhances DRP1 oligomerization to suppress ferroptosis. Mitochondrial fission is induced by increased ROS production, which drives PTEN-induced kinase 1 (PINK1)/Parkin-dependent mitophagy and ferroptosis. In addition, mitophagy is triggered by iron chelation or a cellular response to iron deficiency, and mitophagy tends to replenish the iron pool, ultimately leading to lipid peroxidation and ferroptosis. POLG: DNA polymerase gamma catalytic subunit; LONP1: Lon peptidase 1; TFAM: transcription factor A; Keap1: Kelch-like ECH-associated protein 1; Nrf2: NFE2-related factor 2; GPX4: glutathione peroxidase 4; RB1CC1: RB1-inducible coiled-coil 1; CHCHD3: coiled-coil-helix-coiled-coil-helix domain containing 3; TRAP1: tumor necrosis factor receptor-associated protein 1; STING1: stimulator of interferon response cGAMP interactor 1; MFN1/2: mitoferrin 1/2; HSP90: heat shock protein 90; Drp1: dynamin-related protein 1; ACSL4: acyl-CoA synthetase long-chain family member 4; MTCH1: mitochondrial carrier 1; PHB2: prohibitin 2.
2.1 Mitochondrial proteostasis
The mitochondrion-specific protein quality control system consists of chaperones and proteases that are critical for mitochondrial protein folding, which prevents the aggregation of misfolded proteins. Under overwhelming stress conditions, the mitochondrial unfolded protein response is activated to increase the expression of proteins involved in restoring mitochondrial proteostasis[39]. Previously, researchers had identified functional crosstalk between LONP1 and ClpP, which are two mitochondrial matrix proteases that cooperate to attenuate proteotoxic stress and protect mitochondrial functions to promote cancer cell survival[40]. The protein expression of both these factors is upregulated during ferroptosis and is accompanied by elevated mitochondrial ROS (mtROS) levels [41], [42]. Inhibition of LONP1 expression induces the activation of the Nrf2/Keap1 signaling pathway and GPX4 expression, and the ability of LONP1 to mediate the degradation of substrates involved in mitochondrial biogenesis induce mitochondrial DNA (mtDNA) stress and ferroptosis in human pancreatic ductal adenocarcinoma (PDAC) cells[43], [44]. These observed functions differ from the expected role of LONP1 in protecting cells from ferroptosis. Similarly, inhibiting ClpP in leukemic cells leads to abnormal protein accumulation and causes cell death[45], while the excessive activation of ClpP results in ETC degradation, which synergistically enhances the ferroptosis of leukemia cells in conjunction with GPX4 inhibition[46]. These results can be attributed to several reasons. LONP1 and ClpP are coexpressed at high levels in most cancers and share many target substrates, including proteins involved in oxidative phosphorylation, the TCA cycle, and amino acid and lipid metabolism[46]. These two proteases cooperate to maintain protein quality and target substrate activity to protect mitochondrial function, and their hyperactivation may lead to the excessive degradation of key mitochondrial components, ultimately activating the mitochondria-associated ferroptosis pathway and leading to irreversible cellular damage[40], [43], [44]. The therapeutic drug TR-107, a newly developed selective activator of ClpP, has shown encouraging preclinical efficacy against multiple malignancies. Mechanistically, TR-107 decreases the expression of proteins involved in the mitochondrial unfolded protein response and mitochondrial DNA transcription and translation, disrupts the mitochondrial respiratory chain, and upregulates mitophagy and ferroptosis[47]. In addition, tumor necrosis factor receptor-associated protein 1 (TRAP1), a member of the heat shock protein 90 (HSP90) chaperone family that resides mainly in mitochondria, has been shown to play a modest protective role in ferroptosis in melanoma cells[48]. Mechanistically, TRAP1 overexpression inhibits mPTP opening and increased ROS production, thereby preventing BAY-induced ferroptosis, while endogenous TRAP1 levels or TRAP1 activity are too low to prevent this outcome. However, in glioma, HSP90 binds to calcineurin and stimulates its activity, promotes the dephosphorylation of DRP1 at SER637, further binds to and stabilizes ACSL4, and ultimately sensitizes cells to ferroptosis[49]. Thus, the effects of HSP90 might depend on the context and molecular pathway.
In summary, the maintenance of mitochondrial proteostasis protects cells from ferroptosis-related oxidative stress, but highly active proteases may induce ferroptosis. Further studies are needed to fully understand the exact mechanisms regulating mitochondrial quality control and their interconnections among proteostasis modules to develop effective cancer therapeutic strategies.
2.2 Mitochondrial dynamics
Mitochondrial fission relies on the interaction between the cytoplasmic proteins dynamin-related protein 1 (DRP1) and fission 1 (Fis1), which form a circular structure that surrounds and contracts a site on the outer membrane of mitochondria, ultimately producing smaller spherical mitochondria[50]. The regulation of this process is closely linked to ferroptosis. DRP1 knockdown triggers mitochondrial filamentation, attenuates the impaired MMP and reduces basal and maximal mitochondrial respiration, thus exerting protective effects on ferroptosis by preserving mitochondrial integrity and maintaining redox homeostasis[48]. The upregulation of the HSP90-DRP1-ACSL4 pathway positively regulates ferroptosis in glioma via lipid ROS generation and mitochondrial morphology alteration[49]. Mitochondrial fission is induced by increased ROS production and mitochondrial membrane potential abrogation, driving PTEN-induced kinase 1 (PINK1)/Parkin-dependent mitophagy and ferroptosis in non-small-cell lung cancer (NSCLC) cells, thus enhancing the anticancer effect of celastrol[51]. A consistent phenomenon was also observed in AML cells and malignant mesothelioma[52], [53]. In summary, increased ROS production tends to induce mitochondrial fission, which may induce mitophagy and ferroptosis. cGAS has been recognized as a cytosolic DNA sensor that activates innate immune responses through the production of the second messenger cGAMP and the adaptor protein STING. Recently, Qiu et al. reported the surprising finding that cGAS contains a mitochondrial targeting sequence (MTS) and is translocated to the OMM. Additionally, mitochondria-localized cGAS promotes cancer progression by suppressing ferroptosis in a manner dependent on an increase in DRP1 oligomerization instead of the canonical cGAS-STING pathway[54]. Notably, DRP1 is regulated by posttranslational modifications (such as ubiquitination and phosphorylation) and stress to function in mitochondria[49], [51], [55], [56]. Therefore, targeting a single gene to regulate ferroptosis through mitochondrial fission may provide new insights for cancer treatment.
Mitochondrial fusion is a cellular response that reduces mitochondrial stress induced by aging or damaged proteins and ROS; it allows damaged mitochondria to undergo functional repair and ameliorates the detrimental effects of heteroplasmic mtDNA mutations[57]. The coordinated fusion of inner and outer membranes is mainly driven by optic atrophy 1 (OPA1) and mitofusins (including MFN1 and MFN2). Because of these outcomes, mitochondrial fusion seems to relieve oxidative damage and protect cells from ferroptosis. Several lines of evidence suggest that defective mitochondrial quality control contributes to heightened mitochondrial fission, reduced mitochondrial fusion, and dysfunctional mitophagy, resulting in decreases in mitochondrial biogenesis and mitochondria pathway-mediated cell death[55], [56], [58], [59], [60], [61], [62], [63], [64], [65]. However, a study provided evidence showing that STING1/MFN1/2-mediated mitochondrial fusion promotes mitochondrial oxidative damage and subsequent ferroptosis in PANC1 cells independent of the mitochondrial DNA damage response and PINK1-dependent mitophagy[66]. This outcome may have been a result of multiple effects of mitochondrial dynamics, which regulate more processes than quality control; for example, mitochondrial dynamics affect systems involved in nutrient utilization and energy expenditure[67]. Excessive mitochondrial fission may destroy OXPHOS process, while increased mitochondrial fusion may generate mtROS during OXPHOS or increase the conversion of fatty acids to acetyl-CoA, leading to lipid peroxidation and ferroptosis[66]. Additionally, OPA1 has been reported to promote ferroptosis in osteosarcoma independent of mitochondrial fusion. Mechanistically, OPA1 deficiency promotes ferroptosis resistance by attenuating mtROS production and inducing the activating transcription factor 4 (ATF4)-dependent upregulation of the xCT-GSH-GPX4 pathway as part of an integrated stress response. This outcome highlights the role of OPA1 in modulating the mitochondrial structure and energetics (respiratory supercomplex assembly, cristae remodeling, and oxidative phosphorylation), in addition to mitochondrial fusion[68].
Therefore, mitochondrial fission and fusion are both reported to exert effects on ferroptosis.
2.3 Mitophagy
Mitophagy has been shown to facilitate tumorigenesis and the survival of various cancer cells by removing dysfunctional mitochondria[69]. It plays a pivotal role in reinstating cellular homeostasis under normal physiological and stress conditions by limiting mtROS production. Based on the role of mitophagy in response to increased mtROS production, a reasonable assumption is that mitophagy exerts a protective effect on ferroptosis. The therapeutic drug sappanone A has been reported to induce ferroptosis in NSCLC by regulating mitophagy and mitochondrial biogenesis. Mechanistically, sappanone A significantly inhibits PINK1/Parkin- and FUNDC1-mediated mitophagy, thereby disrupting downstream Nrf2-mediated mitochondrial energy metabolism (decreased ATP synthesis and mitochondrial respiratory function, as well as mPTP dysfunction) and mitochondrial biogenesis, ultimately activating ferroptosis mediated by the mitochondrial pathway[58]. However, increases in mitophagy and associated ROS production have been reported to lead to ferroptosis in melanoma cells and pancreatic cancer cells[48], [53]. These data suggest that enhanced mitophagy is a typical early response that promotes survival, while overwhelming or sustained mitochondrial damage can induce the pathological activation and acceleration of mitophagy, which leads to elevated mtROS production through a positive feedback loop, thus inducing ferroptosis[70]. As shown in a study by Basit F et al., mitochondrial complex I inhibition increases mtROS levels, stimulates mitophagy, and triggers an increase in mitophagy-dependent ROS production, ultimately leading to ferroptosis[48].
In terms of the mechanism, recent studies have focused mainly on the PINK1/Parkin pathway. PINK1 recruits Parkin, an E3 ubiquitin ligase, from the cytoplasm to mitochondria with a low membrane potential to initiate the autophagic degradation of damaged mitochondria[71]. In NSCLC cells, PINK1 knockdown reduced ubiquitinated protein levels in mitochondria, inhibited mitophagy and prevented ferroptosis[51]. In addition, prohibitin 2 (PHB2), a conserved inner mitochondria membrane (IMM) protein with functions in development, lifespan regulation, and diverse mitochondrial processes, and has been identified as a crucial mitophagy-related receptor protein during Parkin-mediated mitophagy in mammalian cells[51]. In mtDNA-depleted 143B human osteosarcoma (ρ0) cells, PHB2 expression was significantly downregulated after H2O2 treatment, contributing to an elevation in aquaporin expression and an associated increase in mitochondrial membrane permeability, thereby increasing cellular sensitivity to ferroptosis[72]. In addition to ubiquitin-dependent mitophagy, receptor-mediated mitophagy also plays a role in ferroptosis-related oxidative stress. For example, BNIP3- and NIX-mediated mitophagy defects increase mtROS levels, which synergistically increase the sensitivity of HeLa cells to ferroptosis with impaired Nrf2-driven antioxidant enzyme systems[73]. FUNDC1-mediated mitophagy plays a similar role in protecting against ferroptosis[58]. In addition, PINK1/Parkin-independent mitophagy has been reported to cause ferroptosis[53], [74]. Following CA9 inhibition in a hypoxic environment, malignant mesothelioma cells exhibit an iron-deficient response via accelerated iron intake processes and low stored iron levels, ultimately leading to lipid peroxidation and ferroptosis[53]. Mitophagy may be involved in the increase in the labile iron pool to some extent by releasing iron from numerous iron-sulfur clusters (ISCs) involved in oxidative phosphorylation[53]. Another study showed that iron chelation led to mitophagy independent of PINK/Parkin pathway activity. This induction of mitophagy may not be related to the damage response, but may recycle iron to maintain other essential iron-dependent functions[75].
In summary, mitophagy may function during ferroptosis by regulating energy metabolism or increasing the levels of metabolic substrates.
2.4 Mitochondrial biogenesis
Human mitochondrial DNA encodes a portion of the ETC components involved in the oxidative phosphorylation needed for ATP synthesis. mtDNA integrity is critical for maintaining the cellular energy supply and cell cycle and for programmed cell death.
Multiple transcription factors and coactivators have been shown to orchestrate nuclear and mitochondrial gene expression during mitochondrial biogenesis. Nrf2 was the first nuclear transcription factor shown to be involved in the transcription of several mitochondrial genes encoding subunits of mitochondrial respiratory chain complexes and heme biosynthetic enzymes that localize to the mitochondrial matrix[76]. A study has shown that the upregulated Nrf2-dependent transcription of heme oxygenase-1 (HO-1) promotes ferroptosis in colorectal cancer (CRC) cells[77]. However, another study showed that the inhibition of the Nrf2/HO-1 axis promotes ferroptosis in KRAS-mutant CRC cells[78]. These inconsistent results suggest that the involvement of Nrf2 in mitochondrial biogenesis plays a context-dependent role in ferroptosis. In fact, Nrf2 is a key hub gene involved in the regulation of ferroptosis, orchestrating multiple effects on mitochondrial biogenesis, mitochondrial fission and fusion, and mitophagy[79]. Mitochondrial transcription factor A (TFAM) stabilizes mtDNA and regulates the amount of mtDNA by forming nucleoids[80]. The degradation of TFAM has been shown to induce mtDNA stress and macroautophagy/autophagy-dependent ferroptosis in human pancreatic cancer cells[44]. In addition, Huang C, et al. found that TFAM complementation completely reversed mitochondrial network disorganization, oxidative DNA damage and ferroptosis induced by the impaired ability of PDAC- and HCC-derived cells to respond to stress[81]. In conclusion, TFAM, a key regulator of mitochondrial biogenesis, can be considered an antagonist of ferroptosis.
Researchers found that cytochrome c oxidase subunit 7a (COX7A1), an important complex in the mitochondrial ETC, increased the sensitivity of NSCLC cells to CDI ferroptosis, accompanied by the downregulation of PINK, Parkin, PGC-1α (mitochondrial biogenesis activator), TFAM, DRP1, and MFN1, indicating its inhibition of mitochondrial biogenesis, mitophagy, and mitochondrial fusion and fission[82]. This result may be explained by the fact that these mitochondrial processes occur dynamically, with one mechanism dominating after counteracting another. As part of the mitochondrial inner membrane structure regulatory complex, coiled-coil-helix-coiled-coil-helix domain containing 3 (CHCHD3) plays an important role in increasing the stability and cristae morphology of mitochondria. Xue, Xiangfei et al. reported that RB1-inducible coiled-coil 1 (RB1CC1)-mediated upregulation of CHCHD3 stimulates MMP hyperpolarization and mtROS production to sensitize tumor cells to ferroptosis[83]. Mitochondrial carrier 1 (MTCH1) is a mitochondrial outer membrane protein insertase. Its deficiency downregulates mitochondrial ETC-related genes, thereby disrupting mitochondrial OXPHOS and governing ferroptosis by triggering FoxO1-GPX4-axon-mediated retrograde signaling[84]. These results highlight the important roles of the abnormal expression and regulation of genes involved in maintaining mitochondrial integrity and respiratory function in the process of ferroptosis.
Epigenetic modulation also plays an important role in controlling mitochondrial function. Li H, et al. found that the inhibition of the mitochondrial protein METTL17 or its interacting proteins significantly reduced mitochondrial RNA methylation, leading to the impaired translation of mitochondrial protein-coding genes and increased ferroptosis sensitivity[85]. However, ρ0 cells displayed the same sensitivity to ferroptosis inducers as parental cells[1]. This outcome may be attributable to a compensatory response to the prolonged lack of mitochondria in which the metabolism in these cells is fundamentally rewired, and thus, mitochondrion-independent ferroptosis is regulated by a mechanism that differs from that of the parental cells[13].
Despite some inconsistent results, the modulation of mitochondrial biogenesis can be concluded to be closely related to ferroptosis. Further in-depth investigations of the mechanisms of the interactions among these processes of mitochondrial dynamics and ferroptosis may lead to new ideas for the prevention and treatment of cancer.
3. Mitochondrial metabolism in ferroptosis
3.1 Tricarboxylic acid (TCA) cycle regulation in ferroptosis
The TCA cycle is a central metabolic pathway in the mitochondrial matrix, and it is a ubiquitous cycle in aerobic organisms and the nexus of the glucose, lipid, and amino acid metabolic pathways in the body. In the TCA cycle, acetyl-CoA derived from carbohydrates, fatty acids, amino acids, and ketone bodies is oxidized, and NADH and FADH2 are produced for use in the ETC; eight enzymes are involved in the TCA cycle[86]. The mitochondrial protein pyruvate dehydrogenase kinase 4 (PDK4) functions as a ferroptosis inhibitor in an acetyl CoA carboxylase (ACC)-dependent manner, blocking pyruvate dehydrogenase-dependent pyruvate oxidation and subsequent citric acid and fatty acid production in PDAC cells to ultimately inhibit glucose-dependent ferroptosis[87]. In contrast, although pyruvate also enters the TCA cycle through pyruvate carboxylase (PC)-mediated oxaloacetate production, PC knockdown failed to downregulate ferroptosis[87]. Thus, the maintenance of the TCA cycle may contribute to ferroptosis by switching the glucose metabolism machinery to increase fatty acid synthesis. Mitochondrial NADP+-dependent isocitrate dehydrogenase (IDH2) catalyzes the conversion of isocitrate to α-ketoglutarate (α-KG), the first oxidative decarboxylation reaction in the TCA cycle. The downregulation of IDH2 sensitizes cancer cells to ferroptosis by reducing the mitochondrial NADPH pool, which is a major site of mitochondrial GSH turnover[88]. α-Ketoglutarate dehydrogenase (α-KGDH) converts α-KG to succinyl-CoA, and when its E3 component DLD is silenced, lipid peroxidation and cystine deprivation-induced ferroptosis in head and neck cancer cells is inhibited via a mechanism involving glutaminolysis[89]. Glutamine (Gln) is a nonessential amino acid and major fuel source for energy production and lipid biosynthesis for respiration mediated by glutaminolysis in proliferating cells. Tumor cells exhibit a Gln dependence that is rarely seen in normal cells because glutaminolysis replenishes the number of TCA cycle intermediates and supports the biosynthetic processes required for tumor growth[90]. Thus, glutaminolysis has been implicated in ferroptosis regulation in tumor cells[13], [91]. Cystine deprivation failed to induce lipid ROS accumulation or ferroptosis in the absence of Gln or after glutaminolysis blockade, and this outcome was offset by supplementation with α-KG and its downstream metabolites, such as succinate, fumarate, and malate[13], [91]. Previous studies revealed that ferroptosis was abolished by blocking the production of components involved in a series of reactions involved glutaminolysis; these components included SLC1A5 and SLC38A1 (involved in Gln intake), GLS2 (converts Gln to Glu), and transaminase and GLUD1 (converts Glu to α-KG)[91], [92]. Additionally, ASS1, a key enzyme participating in the urea cycle, promotes the reductive carboxylation of Gln, blocks Gln from entering the TCA cycle, reduces mitochondrial lipid ROS production, and promotes de novo MUFA synthesis through a mechanism dependent on reductive pathway-derived acetyl-CoA, thereby conferring ferroptosis resistance to cancer cells[93]. These data highlight that the anaplerotic reactions mediated by glutaminolysis are indispensable for cysteine deprivation-induced (CDI) ferroptosis. In addition, mitochondrial pyruvate carrier 1 (MPC1) in the IMM internalizes pyruvate into mitochondria. MPC1 suppression increases the vulnerability of cancer cells to ferroptosis, which retained their mesenchymal cell traits and maintained glutaminolysis[94]. Taken together, two distinct upstream pathways (pyruvate oxidation and Gln anaplerosis) may increase ferroptosis sensitivity via their TCA cycle-related activities, through which they not only mediate ROS production but also promote fatty acid synthesis[87].
In another biological role, the TCA cycle provides energy for the body. Through acetyl-CoA, which the initiates the cycle, electrons are transferred to the ETC, promoting the production of ATP through oxidative phosphorylation. AMP-activated protein kinase (AMPK), an energy sensor whose activity is modulated based on the cellular ADP:ATP ratio, plays a dual role in regulating ferroptosis via its phosphorylation of substrates. For example, glucose starvation prevents ferroptosis in mouse embryonic fibroblasts (MEFs) by activating AMPK-mediated phosphorylation of acetyl-CoA[95]. In contrast, AMPK-mediated phosphorylation of beclin 1 (BECN1) plays an autophagy-independent role in promoting ferroptosis by directly blocking system Xc- activity in CRC cells[96]. Additionally, AMPKα1/α2 knockdown in PANC1 cells or MEFs leads to distinct cellular sensitivity to erastin- or RSL3-induced death under high-glucose or glucose starvation conditions, indicating that the role of AMPK in ferroptosis is cell type-dependent and that AMPK is involved in the complex relationship between ferroptosis and the cellular energy state[87]. Recently, research revealed that activated AMPK may inhibit ferroptosis by regulating pyrimidinosome formation. Mechanistically, stress-activated AMPK dissociates from the pyrimidinosome to increase pyrimidine flux up to the biosynthesis of DHOA/OA. Then, DHOA can be safely secreted from cells to relieve reductive stress or converted to OA by DHODH[97], [98]. However, AMPK activators and DHODH inhibitors can synergistically induce ferroptosis in cancer cells; thus, the active AMPK-promoted pyrimidinosome-mediated ferroptosis defense appears to serve as a protective pathway to prevent self-jury, and the DHODH-mediated metabolic conversion potentially plays a key role in antagonizing AMPK-related stresses[99]. The TCA cycle supports the electron transport activity of protein complexes located in the IMM. As shown by Gao et al., the inhibition of ETC mitochondrial complexes I, II, III, and IV inhibited ROS accumulation and ferroptosis induced by cystine starvation and erastin activity in HT-1080 cells, but the GPX4 inhibitor RSL3 had no effect[13]. The different outcomes may be related to the subcellular localization of erastin and RSL3; notably, erastin has been shown to target voltage-dependent anion-selective channels (VDACs) in mitochondria, while GPX4 is the most downstream component in the defensive antiferroptotic pathway and is critical for lipid ROS clearance.
In summary, the TCA cycle is involved in the regulation of ferroptosis through its role as a major metabolic hub and its function in altering the energy status (Figure 2).
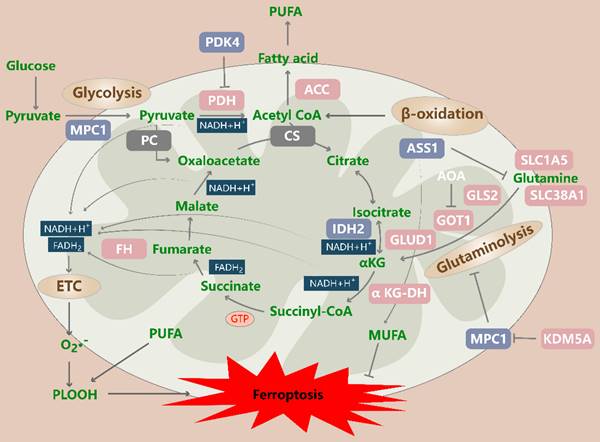
Regulation of the tricarboxylic acid (TCA) cycle in ferroptotic cells. The TCA cycle drives ferroptosis by transferring electrons to the electron transport chain (ETC), which generates ROS, as well as by promoting ACC-mediated fatty acid synthesis. Glycolysis, β-oxidation, and glutaminolysis produce TCA cycle precursors, and this production is suppressed by PDK4-mediated glucose metabolism and increased by enhanced glutaminolysis and exogenous supplementation with TCA cycle substrates. In addition, MPC1 inhibits ferroptosis in part by inhibiting glycolysis and glutaminolysis. ACC: acetyl-CoA carboxylase; PDK4: pyruvate dehydrogenase kinase 4; MPC1: mitochondrial pyruvate carrier 1; PDH: pyruvate dehydrogenase; PLOOH: phospholipid hydroperoxide; PUFA: polyunsaturated fatty acid; PC: pyruvate carboxylase; CS: citrate synthase; GLS2: glutaminase 2; GOT1: glutamic-oxaloacetic transaminase 1; GLUD1: glutamate dehydrogenase 1; IDH2: isocitrate dehydrogenase (NADP[+]) 2; FH: fumarate hydratase; α-KGDH: alpha-ketoglutarate dehydrogenase subunit E2; KDM5A: lysine demethylase 5A; ETC: electron transport chain; SLC1A5: solute carrier family 1 member 5; SLC38A1: solute carrier family 38 member 1.
3.2 Association of mitochondrial ROS production with ferroptosis
ROS are produced during oxidative stress; these ROS include superoxide anions (O2·-), hydrogen peroxides (H2O2), hydroxyl radicals (HO·), lipid hydroperoxides, and peroxy radicals, which mediate intracellular signal transduction. They are mainly produced by NOX and the ETC in endogenous biological systems[32], [100].
An increase in the levels of mitochondrial ROS facilitates ferroptosis, which can be inhibited by mitochondrion-targeted antioxidants (Figure 3).
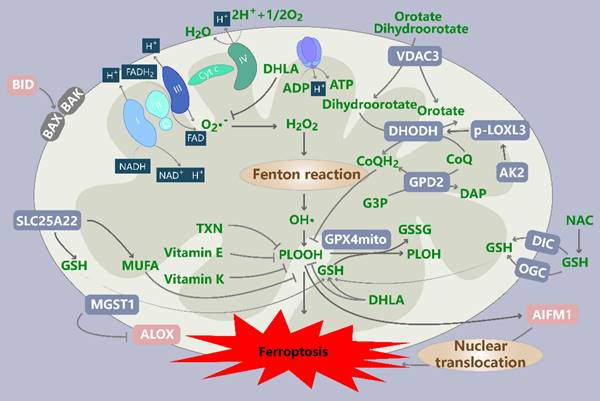
Association of mitochondrial ROS production with ferroptosis. GPX4, DHODH, GPD2, AK2 and MGST1 are mitochondrial antioxidant enzymes that play important roles in protecting cells from ferroptosis. Nonenzymatic substances also play pivotal roles. OMM-localized VDAC3 serves as a ferroptosis suppressor and provides internal access for DHOA and OA. SLC25A22, a driver of ferroptosis resistance, mediates the production of GSH and MUFAs. DIC and OGC transport glutathione (GSH) into mitochondria, and their inhibition can lead to GSH depletion and an increased ferroptosis rate. TXN, vitamin E, vitamin K, dihydrolipoic acid (DHLA) and N-acetylcysteine (NAC) also possess antioxidant activities. In addition, the release of AIFM1 induced by mitochondrial oxidative damage promotes ferroptosis after its translocation to the nucleus. BID mediates mitochondrial dysfunction, mtROS production and ferroptosis. DHODH: dihydroorotate dehydrogenase; G3P: glycerol-3-phosphate; GPD2: G3P dehydrogenase 2; AK2: mitochondrial adenylate kinase 2; MGST1: microsomal glutathione S-transferase 1; ALOX: lipoxygenase; DIC/SLC25A10: solute carrier family 25 member 10; OGC/SLC25A11: solute carrier family 25 member 11; TXN: thioredoxin; DHLA: dihydrolipoic acid; NAC: N-acetylcysteine; AIFM1/AIF: apoptosis-inducing factor mitochondria-associated 1; BID: BH3-interacting domain death agonist.
3.2.1 Mitochondrion-targeted ROS scavengers
First, mitochondrial lipid ROS levels are increased significantly and induce ferroptosis in erastin-treated HT-1080 cells and MEFs, as measured by the C11 BODIPY 581/591 staining intensity and the quantitative analysis of malondialdehyde (MDA), mitoPeDPP and MitoSOX levels[13]. An imbalance between oxidative stress levels and antioxidant system activity drives ferroptosis. Therefore, mitochondrion-targeted ROS scavengers such as mitoTEMPO, mitoquinone (MitoQ), and XJB-5-131 function as exogenous antioxidant mechanisms in oxidative stress resistance and reverse ferroptosis in a variety of cancer cell types and animal models[98], [101].
3.2.2 Mitochondrial antioxidant enzymes
Second, several mitochondrial antioxidant enzymes play vital roles in inhibiting ferroptosis. GPX4cyto is a pivotal component of the cellular antioxidant defense system, and studies have shown that the mitochondrial form GPX4mito plays a key role in mitigating mitochondrial oxidative damage during ferroptosis[102]. DHODH mediates the oxidation of dihydroorotate to yield orotate, which is coupled with the reduction of CoQ to CoQH2. The combination of DHODH knockdown and GPX4 inactivation, but not either alone, substantially induced mitochondrial lipid peroxidation and ferroptosis in cancer cells, which was reversed by the restoration of GPX4mito but not GPX4cyto[98]. These data suggest that DHODH is an antioxidant component that functions in parallel with GPX4mito in mitochondria. Similar to DHODH deficiency, cell death induced by VDAC3 depletion is partially prevented by ferrostatin-1 and is more potent when combined with uridine supplementation, as the OMM-localized VDAC3 protein serves as a platform to integrate the pyrimidinosome and provide internal access for DHOA and OA[99]. In addition, AMPK agonists and DHODH inhibitors synergistically increase lipid ROS levels and induce ferroptosis in cancer cells, and low expression of the AMPK protein renders cancer cells more dependent on DHODH-mediated ferroptosis defenses[99]. Thus, DHODH serves as a hub connecting substance metabolism, energy metabolism, and defense systems. IMM-localized glycerol-3-phosphate (G3P) dehydrogenase 2 (GPD2) exerts an effect similar to that of DHODH in that it catalyzes G3P oxidation, which is coupled with the reduction of CoQ to CoQH2, which functions as a radical-trapping antioxidant in mitochondria that suppresses ferroptosis. The supplementation of cancer cells with G3P attenuates ferroptosis triggered by GPX4 inhibitors in a GPD2-dependent manner[103]. As a member of the lysyl-oxidase (LOX) family, LOXL3 can be transported to mitochondria in a manner dependent on OMM-localized TOM20. Researchers found that mitochondrial adenylate kinase 2 (AK2) phosphorylates LOXL3-S704 to enhance lysyl oxidase activity and block DHODH-K344 ubiquitination, in turn conferring resistance to chemotherapy-induced ferroptosis[104]. In addition, microsomal glutathione S-transferase 1 (MGST1), a membrane-bound transferase located mainly in mitochondria, the endoplasmic reticulum (ER), the plasma membrane, and peroxisomes that plays a clear role in the conjugation of electrophiles and oxidative stress protection, has also been reported to bind to ALOX5, limiting ALOX5-mediated lipid peroxidation and subsequent ferroptosis in an Nrf2-dependent manner in pancreatic cancer cells[105]. In conclusion, mitochondrial antioxidant enzymes play roles in defensive antiferroptotic responses by eliminating lipid peroxidation damage (directly or indirectly by producing free radical scavengers) or blocking the enzymatic oxidation of PUFA-phospholipids (PLs).
3.2.3 Nonenzymatic mitochondrial antioxidants
Third, a variety of nonenzymatically generated substances are involved in the antioxidant system. As a core contributor to ferroptosis, GSH is not produced in mitochondria and cannot penetrate the IMM but is transported into mitochondria by potential carrier proteins, such as the dicarboxylate carrier (DIC, SLC25A10) and the oxoglutarate carrier (OGC, SLC25A11). SLC25A22 (a mitochondrial glutamate transporter)-dependent NAPDH synthesis mediates the production of GSH[106]. Low GSH levels in mitochondria can also reduce the S-glutathionylation rate of mitochondrial proteins, especially the ETC complexes and TCA cycle enzymes, and diminish their activity. This feedback loop further increases ROS generation and GSH consumption in mitochondria[107]. N-acetylcysteine (NAC) is the acetylated precursor of GSH and shows strong antioxidant activity. NAC has been reported to block the increases in ROS production and ferroptosis in hepatocellular cancer (HCC) and PDAC cells[81], [108]. In addition, biological antioxidants such as CoQ, lipoic acid and its reduced form dihydrolipoic acid (DHLA), thioredoxin (TXN), and vitamins E and K have all been reported to play protective roles in ferroptosis[109], [110], [111], [112], [113]. Notably, most of the major regulatory factors of the aforementioned endogenous antioxidant defense system are, at least in part, associated with Nrf2; these enzymes include GPX4[114] and MGST1[115], as well as vitamin E[116]. In summary, antioxidants such as vitamin E antagonize the Nrf2-Keap1 interaction and lead to the stabilization and activation of Nrf2[116]. Moreover, Nrf2 maintains redox homeostasis by modulating antioxidant-response element (ARE)-dependent transcription and the expression of antioxidant defense enzymes, playing vital roles in lipid peroxidation and ferroptosis prevention[114], [117].
3.3 Mitochondrial iron metabolism and ferroptosis
Mitochondria are not only hubs of cellular energy metabolism but also the main organelles involved in intracellular iron regulation (Figure 4). Iron must cross the outer mitochondrial membrane (OMM) and IMM to enter the mitochondrial matrix, where it is involved in various metabolic activities. Mitochondrial iron is involved mainly in the biosynthesis of heme and ISCs, and it is stored by mitochondrial ferritin (FtMt). The dependence of cancer cells on iron to maintain their rapid growth rate makes them more sensitive to the dysregulation of iron metabolism. During rapid cell proliferation, more iron may be imported into mitochondria to meet the increasing demand for cofactors[118]. However, excessive mitochondrial iron levels can induce ROS generation or abnormal iron-containing enzyme activity, promoting ferroptosis.
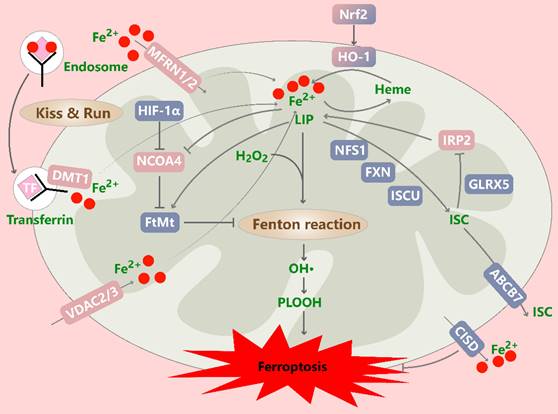
Mitochondrial iron metabolism in ferroptotic cells. Iron can be transported into mitochondria through MFRN1/2, VDAC2/3 and “kiss and run” pathways, is mainly used for the synthesis of heme and iron-sulfur clusters (ISCs), and is also stored in mitochondrial ferritin. Excess iron promotes ferroptosis through the Fenton reaction. The HO-1-mediated degradation of heme promotes Fe2+ release and regulates ferroptosis in a cell type-dependent manner. Interference with the biosynthesis (NFS1, FXN, and ISCU), transfer (GLRX5), and export (ABCB7) of ISCs enhances ferroptosis. In addition, FtMt levels can be regulated by HIF-1α and NCOA4. CISD mediates Fe2+ transport out of mitochondria, thereby inhibiting ferroptosis. MFRN1/2: mitoferrin 1/2; VDAC2/3: voltage-dependent anion-selective channel 2/3; HO-1: heme oxygenase-1; NFS1: cysteine desulfurase; FXN: frataxin; ISCU: iron-sulfur cluster assembly enzyme; GLRX5: glutaredoxin 5; FtMt: mitochondrial ferritin; NCOA4: nuclear receptor coactivator 4; CISD: CDGSH iron sulfur domain.
3.3.1 Iron transport
Several molecules involved in mitochondrial iron transport have been found to contribute to ferroptosis. First, VDACs function as gatekeepers for the entry and exit of metabolites and ions between the cytosol and the mitochondria. Erastin treatment induces VDAC2/3 opening and is associated with mitochondrial iron accumulation and iron-dependent ferroptosis. Recent research has shown that VDAC3 ubiquitination increases the ferroptosis sensitivity of gastric cancer cells due to the loss of control over iron ion entry into mitochondria[119]. Second, iron transport across the IMM can be mediated by the membrane transporter mitoferrin 1 (Mfrn1) and its homolog mitoferrin 2 (Mfrn2), also known as the mitochondrial solute carrier family members SLC25A37 and SLC25A28, respectively[120]. In vitro and in vivo evidence suggest that Mfrn1 acts as a tumor suppressor to regulate mitochondrial iron-induced ferroptosis in HCC[121]. A previously study revealed the Mfrn2-dependent mitochondrial iron uptake induced iron-dependent mitochondrial dysfunction and the subsequent killing of human head and neck squamous carcinoma cells[122]. However, Mfrn1/2-mediated mitochondrial iron accumulation promotes osteosarcoma carcinogenesis, and high expression of Mfrn1 in GBM tumor tissues increases the level of mitochondrial iron and is associated with a poor prognosis[123], [124]. Overall, the expression and function of Mfrn1/2 may vary depending on the cell types and conditions[123]. Alternatively, mitochondria directly ingest iron through transient interactions with endosomes containing iron-bound transferrin (in a “kiss and run” process)[125]. Endosome docking is mediated by divalent metal transporter 1 (DMT1) located in the OMM, and DMT1 knockdown blocks erastin-induced ferroptosis by boosting GSH antioxidant defenses and mitophagy[126]. Another pathway apparently induces DMT1-independent iron uptake, but its characteristics remain to be elucidated[127].
Previous localization studies showed that CDGSH iron sulfur domain 1 (CISD1, also called mitoNEET) is an integral protein in the OMM that has been reported to regulate VDAC1 activity in a redox-dependent manner, leading to pore closure and likely disrupting the flow of metabolites through VDACs[128]. The depletion of CISD1 drove iron-mediated intramitochondrial lipid peroxidation and ferroptosis in HCC cells[129]. In addition, given the role of VDAC1 in the regulation of mitophagy and the ability of CISD1 to mediate iron and ROS homeostasis in mitochondria, CISD1 has been hypothesized to regulate mitophagy-related ferroptosis by targeting VDAC1[130], [131]. CISD2 and CISD3 are both members of the NEET protein family and have been proposed to prevent ferroptosis in cancer cells[132], [133], [134]. Mechanistically, CISD2 inhibition leads to cellular autophagy, which has been implicated in the activation of ferritinophagy, resulting in an increased cellular labile iron pool[133]. Moreover, studies have shown that CISD3 depletion leads to metabolic reprogramming toward glutaminolysis, which is required for fueling mitochondrial oxidative phosphorylation, and that ferroptosis induced by CISD3 knockdown can be rescued by mitophagy, which eliminates damaged mitochondria[132]. In summary, NEET protein family members regulate ferroptosis through various mechanisms and are worthy of further exploration.
In summary, abnormal iron transport disrupts mitochondrial iron levels and ROS homeostasis, ultimately promoting ferroptosis.
3.3.2 Iron utilization
HO-1 is an ER-anchored enzyme that metabolizes heme into oxidant-promoting ferrous iron, carbon monoxide and the antioxidant biliverdin[135]. HO-1 plays an essential role in clear cell renal cell carcinoma cell ferroptosis, and the mechanism involves the release of free iron mediated by HO-1-induced heme degradation, contributing to the generation of oxidized lipids in the mitochondrial membrane[136], and it can be increased by the nuclear translocation of Nrf2[114]. This process is independent of the iron-regulating action of the canonical hepcidin/ferroportin axis. Similar results have been reported in studies of acute myeloid leukemia cells, human lung cancer cells, CRC cells and human breast cancer cells[77], [137], [138], [139], [140]. These data showed that high expression of HO-1 positively regulated ferroptosis through the modulation of iron and ROS levels, making it an important target for mediating the adverse effects of ferroptosis induction[141]. However, a previous study revealed that reduced HO-1 levels mediated synergistic lysosomotropic agent- and tyrosine kinase inhibitor-induced ferroptosis in glioblastoma cells[142]. Moreover, a decrease in HO-1 levels was not a consequence of decreased Nrf2 expression but due to proteasome degradation[142]. Similarly, HO-1 protected HCC and melanoma cells from ferroptosis[143], [144], [145]. This protective effect may be attributable to the HO-1 byproducts biliverdin and bilirubin; this effect was similar to that reported by another study in which GSH and bilirubin showed complementary antioxidant activities, and GSH protected water-soluble proteins and bilirubin protected lipids from oxidation[146]. In summary, these inconsistent results regarding the level of HO-1 expression observed during ferroptosis may be interpreted as context and cell type dependent.
Mitochondrial ISCs are modular cofactors that perform essential cellular functions. These clusters are synthesized via a multistep process involving the multisubunit mitochondrial machinery. The first step of ISC biosynthesis is the synthesis of [2Fe-2S] clusters on highly conserved scaffold proteins, with the disruption of this process increasing the iron load and promoting ferroptosis. For example, cysteine desulfurase 1 (NFS1) is an essential enzyme in eukaryotes that extracts sulfur from cysteine and makes it available for ISC biosynthesis, the suppression of which activates a robust iron starvation response mediated by iron-responsive element-binding protein 2 (IREB2, also known as IRP2); in conjunction with GSH synthesis inhibition, IREB2 activation drives ferroptosis in vitro and slows lung tumor growth[147], [148]. Moreover, frataxin and iron-sulfur cluster assembly enzyme (ISCU) are important proteins participating in this ferroptosis-inducing process, and their inhibition both significantly impedes ISC assembly and increases the ferroptosis rate by accelerating free iron accumulation in cancer cells[149], [150], [151]. Second, [2Fe-2S] is released from scaffold proteins and binds to transporters. Interference with this process affects ferroptosis sensitivity. For instance, glutaredoxin 5 (GLRX5) is an essential protein engaged in mitochondrial ISC transfer to IRP, m-aconitase, and ferrochelatase. The inhibition of GLRX5 expression predisposes cisplatin-resistant head and neck cancer cells to ferroptosis[152]. Third, the abnormal function of the ISC export machinery may cause ferroptosis. The IMM protein ABCB7 is a core component of the ISC export machinery and is directly involved in the export of ISCs synthesized in mitochondria. A recent study has reported that nonapoptotic cell death induced by ABCB7 depletion is partially mediated by ROS production triggered in response to changes in iron metabolism; however, additional characterization of this process is required to determine whether this death modality is, in fact, ferroptosis[153]. These data suggest that proteins involved in ISC assembly and export may play roles in ferroptosis by regulating iron metabolism.
3.3.3 Iron storage
FtMt is an iron storage protein located in mitochondria that plays a significant role in modulating cellular iron metabolism. FtMt exerts a protective effect on ferroptosis triggered by erastin in SH-SY5Y neuroblastoma cells[154], [155]. Nuclear receptor coactivator 4 (NCOA4) is a selective cargo receptor that binds to ferritin and mediates its delivery to autophagosomes, which subsequently fuse with lysosomes, leading to ferritin degradation and concomitant iron release[156]. HERC2-mediated ubiquitylation of NCOA4 depends on iron, which ensures that the NCOA4 liberated from HERC2 under iron-deficient conditions contributes to ferritinophagy to replenish the iron pool[156]. Inhibition of NCOA4 suppresses ferritin degradation and ferroptosis in cancer cells[18]. In addition, the mRNA level of NCOA4 in macrophages is reduced under hypoxic conditions, thereby preventing ferroptosis, while NCOA4 and FtMt levels in fibrosarcoma cells do not respond to a decrease in the oxygen level[155]. In conclusion, blocking the binding of NCOA4 to FtMt, inactivating NCOA4, and reducing the stability of NCOA4 may protect cancer cells from ferroptosis by preventing ferritinophagy and maintaining the stored iron level.
3.4 Mitochondrial lipid metabolism in ferroptotic cells
The accumulation of lipid peroxides is a key step in ferroptosis (Figure 5). Initially, ACSF2 and CS were identified as high-confidence genes required for erastin-induced ferroptosis, and their silencing conferred broad protective effects on ferroptosis in a variety of cell lines[1]. Since these discoveries were made, research on mitochondrial fatty acid metabolism has also been reported. For example, we found that the role of mitochondrial fatty acid metabolism in ferroptosis is reflected mainly by the yield of specific lipid precursors needed for lipid peroxidation and substrates needed for energy metabolism (as described above).
Mitochondrial lipid metabolism in ferroptotic cells. ACSF2 is encoded by a high-confidence gene and induces erastin-induced ferroptosis. CPT1/2 mediate the entry of long-chain acyl-CoA into mitochondria. CPT1 activity is inhibited by ACC-mediated malonyl-CoA production and restored by ADPN, affecting the ferroptosis sensitivity of cells. Increased ACC phosphorylation limits its activity. The DECR1-mediated promotion of polyunsaturated fatty acid (PUFA) β-oxidation confers ferroptosis resistance to cancer cells, but acetyl-CoA produced via fatty acid oxidation (FAO) enters the TCA cycle and affects ferroptosis sensitivity. The LD-specific marker PLIN2 and tumor protein D52, which are critical for lipid storage, exert protective effects on ferroptosis. In contrast, the expression of proteins involved in the fusion of LDs and mitochondria and subsequent lipophagy (RAB7A, PGRMC1) induces stored PUFA release and lipid peroxidation, leading to ferroptosis. ACSF2: acyl-CoA synthetase family member 2; CPT1/2: diacylglycerol cholinephosphotransferase 1/2; ADPN: adiponectin; DECR1: 2,4-dienoyl-CoA reductase 1; CACT/SLC25A20: solute vector family 25 member 20; TPD52: tumor protein D52; PLIN2: Perilipin2; PGRMC1: progesterone receptor membrane component 1.
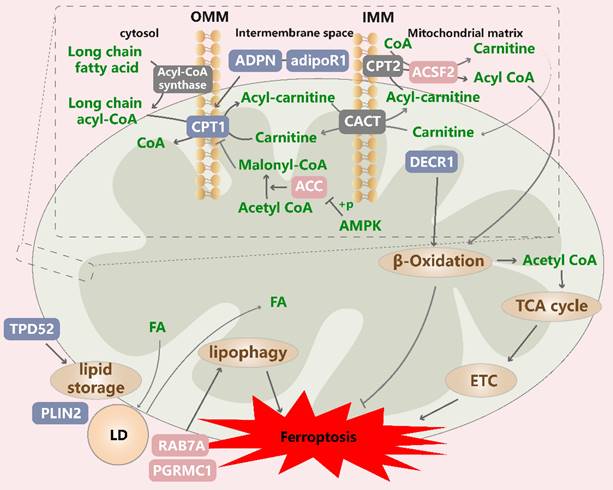
First, a recently identified a specific lipid class, PC-PUFA2, has been shown to specifically interact with the mitochondrial ETC, enabling the initiation of ROS production in mitochondria and lipid peroxidation in the ER, thereby driving ferroptosis[30]. These fatty acids are catalyzed to produce acyl-CoA outside mitochondria. Long-chain acyl-CoA cannot directly permeate the IMM and thus is unavailable for oxidative decomposition; therefore, it is transported into mitochondria by L-carnitine. Carnitine palmitoyltransferase 1 (CPT1) is critical for this step, and its suppression enhances ferroptosis[111], [157]. When fatty acid oxidation (FAO) cannot proceed normally, other oxidative stress-related factors play relatively dominant roles, leading to fatty acid peroxidation. As an insulin-sensitizing hormone, adiponectin is an important factor in the regulatory networks involved in lipid metabolism and blood glucose homeostasis. Notably, adiponectin needs to bind to its cognate receptors to exert its biological effects. Adiponectin was shown to rescue ferroptosis induced by a fatty acid oxidation/peroxide imbalance by restoring CPT1 activity in individuals with gestational diabetes; however, whether this finding can be generalized to tumor cells remains to be investigated[158]. AdipoR1 is a verified adiponectin receptor, and its knockdown remarkedly inhibits the activation of the Nrf2/SLC7A11 pathway and increases the ferroptosis rate of HCC cells[159]. Malonyl-CoA, a substrate in fatty acid synthesis, is an inhibitor of CPT1 and is synthesized from ACC1/2[160]. Notably, increased ACC phosphorylation inhibits ACC activity, thereby protecting cells from ferroptosis, while its suppression inhibits ferroptosis driven by various inducers[95], [161].
Next, acyl-CoA entering the mitochondrial matrix undergoes β oxidation, resulting in the formation of acetyl-CoA and acyl-CoA. 2,4-Dienoyl-CoA reductase 1 (DECR1) is the rate-limiting enzyme in a PUFA β oxidation auxiliary pathway, and its knockdown induces the significant accumulation of phospholipid hydroperoxides, increases the levels of mitochondrial oxidative stress, and induces ferroptosis in prostate cancer cells[162]. Another study revealed that DECR1 knockdown induces ER stress and sensitizes castration-resistant prostate cancer (CRPC) cells to ferroptosis, highlighting the importance of DECR1 in tumor progression and the development of therapeutic resistance[163]. In contrast, DECR1 knockdown does not affect saturated fatty acid and palmitate metabolism in mitochondria[162]. These results suggest that the induction of FAO can confer ferroptosis resistance to cancer cells, but the acetyl-CoA produced via FAO can enter the TCA cycle, where it interacts with other pathways to influence ferroptosis sensitivity.
Lipid droplets (LDs) protect cells from PUFA oxidation because free fatty acids are transferred to their hydrophobic core, thereby defending against oxidative stress[164]. This action may also explain the mechanism through which LDs inhibit ferroptosis. One study indicated that the overexpression of the LD-specific marker Perilipin2 (PLIN2) in gastric carcinoma cells increased number of LDs, reduced the intracellular ROS content and inhibited ferroptosis, which was interpreted as the relative hypoxia of cells due to a high lipid content, thereby inhibiting the occurrence of oxidative stress[165]. In addition, the inhibition of tumor protein D52, which is critical for lipid storage, decreased the number of LDs and increased the ferroptosis rate[166]. In contrast, lipophagy, which degrades intracellular LDs through autophagy, drives cytosolic fatty acids into mitochondria via the fusion of LDs and mitochondria, resulting in simultaneous FAO and LD formation in cells[167], [168]. Lipophagy has been reported to release stored PUFAs and induce lipid peroxidation and ferroptosis in vivo, which was inhibited by the knockdown of the LD cargo receptor RAB7A[166], and induced by ionizing radiation[169]. Progesterone receptor membrane component 1 (PGRMC1) can alter lipid metabolism, promote autophagy and mediate chemotherapy resistance. One study showed that PGRMC1 affected lipophagy and FAO rates by anchoring LDs to mitochondria, and that its expression in paclitaxel-tolerant persister cancer cells increased the sensitivity to ferroptosis inducers[170]. These results suggest that the dynamic balance of LD formation and degradation exerts effects on ferroptosis. Notably, considering lipid metabolic reprogramming characterized by increased levels of free fatty acids, numbers of LDs and rate of FAO in resistant cancer cells, the modulation of LD formation and degradation may be a potential strategy to eradicate persister cells that survive conventional or targeted chemotherapy by increasing their susceptibility to ferroptosis[170].
4. Clinical translation of antitumor effects
Traditional therapies have advantages and limitations, such as lower antitumor activity at low doses, toxic side effects due to off-target effects when applied at high doses, and the chemoresistance of tumor cells caused by adaptive tolerance. Ferroptosis, an adaptive mechanism to eliminate malignant cells, offers new possibilities for tumor suppression. Here, we outline several tumor therapeutic strategies based on mitochondria-associated ferroptosis-related mechanisms, which can be categorized as follows: first, targeting one or more ferroptosis targets within the mitochondria; second, a synergistic enhancement of efficacy by combining agents; and third, inducing ferroptosis can improve the efficacy of immunotherapy and enhance antitumor immune responses.
4.1 Targeting one or more ferroptosis biomarkers within the mitochondria
As mentioned above, agents target certain mitochondrial processes, such as GPX4, DHODH, iron metabolism pathway, etc., to increase intracellular oxidative stress or attenuate the ferroptosis defense system, thereby inducing the ferroptosis of tumor cells and blocking tumor cell growth. For example, commonly used ferroptosis inducers such as ML-162 and RSL3 reduce GPX4 activity to induce the ferroptosis of cancer cells and have been applied in the treatment of advanced treatment-resistant prostate cancer, ccRCC, HCC, TNBC and other cancers[171], [172], [173], [174]. Natural product derivatives, synthetic drugs and nanoparticle-encapsulated molecules have also been extensively studied and shown to exert antitumor effects by affecting GPX4 translation, proteolysis and protein modification[173], [174], [175], [176], [177], [178]. Notably, Gan H, et al. developed a theranostic ferroptosis inducer (IR780-SPhF) that enables the simultaneous imaging and therapy of TNBC by directly targeting mitochondria to monitor and deplete mitochondrial GSH levels. IR780-SPhF exhibited stronger anticancer potency than cyclophosphamide, indicating that mitochondria-targeted ferroptosis inducers may represent promising candidates for precision cancer therapy[179], [180].
Moreover, a few Chinese herbs, engineered compounds and autophagy modulators have been characterized as ferroptosis inducers that are able to enhance mitochondrial dysfunction, thereby exerting anticancer effects on NSCLC, HCC, breast cancer stem cells[58], [180], [181], [182], [183], [184]. For example, andrographolide (ADE), a major effective ingredient of Andrographis paniculata, has been shown to increase mtROS release, MMP depolarization, mitochondrial ATP reduction and ferroptosis, repressing NSCLC cell growth, which could be rescued by a mitochondria-targeted antioxidant[181]. These results emphasize the involvement of mitochondrial oxidative stress and disrupted energy metabolism in the induction of ferroptosis-mediated anticancer effects by these agents.
Additionally, novel mitochondria-targeted nanosystems have played an eye-opening role in precision cancer therapy. These nanosystems efficiently deliver significant amounts of CO, iron, ferroptosis inducers, and photosensitizers to mitochondria to induce mtROS production and finally drive ferroptosis, and can be utilized in combination with nonclassical physiotherapy[185], [186], [187], [188], [189].
4.2 Synergistic enhancement of efficacy by combining agents
Combination therapy shows promise in overcoming compensatory mechanisms and unwanted off-target effects. For example, brequinar has been reported to disrupt the ferroptosis defense system of GPX4low tumors, inhibiting their growth by inhibiting mitochondrial DHODH. For GPX4high tumors, DHODH inhibition increased cellular GSH levels, thus making GPX4 more effective in suppressing ferroptosis[98]. Brequinar has been tested in large clinical trials for treating advanced melanoma, gastrointestinal cancer, and squamous cell carcinoma of the head or neck[190], [191], [192]. The combination of DHODH inhibitors with sulfasalazine, oxaliplatin and cisplatin has been shown to sensitize cells to ferroptosis and reverse chemoresistance[98], [104], [193]. Moreover, the therapeutic agent NL-1, an inhibitor of the mitochondrial iron transporter CISD1, can accelerate sorafenib-induced lipid peroxidation and ferroptosis by promoting mitochondrial iron accumulation, which increases the efficacy of sorafenib in HCC[194]. Notably, several iron-containing nanoparticle formulations have been developed to load ferroptosis inducers into tumors, accelerating the destruction of antioxidant systems and the accumulation of lipid peroxides, resulting in the synergistic inhibition of tumor growth[195], [196].
Some unconventional pathway regulators have also shown promising efficacy in combination with ferroptosis inducers, and they usually act synergistically upstream and downstream. Xue, Xiangfei et al. reported that FDA-approved c-Jun N-terminal kinase (JNK) agonists reinforced RB1CC1 nuclear translocation and sensitized cells to ferroptosis. Coadministering imidazole ketone erastin with four different JNK activators in mouse models resulted in an increase in anti-tumorigenic efficacy[83]. The combination with autophagy activators has also been described to play a role in sensitizing cells to the anticancer potency of ferroptosis inducers, given that the autophagy-ferroptosis-ROS-autophagy loop has been described as an amplifier of ferroptosis under certain conditions[82], [197]. For example, rapamycin alleviated the blockade of autophagy flux induced by COX7A1, further increasing the lethality of ferroptosis inducers in NSCLC cells[82].
In contrast to normal cells, most cancer cells exhibit increased aerobic glycolysis and impaired oxidative phosphorylation, also known as the Warburg effect[198]. In combination with 2DG, which mimics glucose starvation, the antiglioma effect of chloramphenicol was enhanced, which disrupted mitochondrial ribosomes and triggered mitochondrial oxidative stress and ferroptosis[199]. This result may be because the glucose starvation state makes mitochondria dominant. This mechanism by which metabolic reprogramming enhances the antitumor effects of combination therapy could provide new insights into the development of new combination regimens.
In addition, the mitochondria-targeted nanodelivery system mentioned above combined with photodynamic and sonodynamic physiotherapy is expected to achieve simultaneous monitoring and treatment.
4.2 Inducing ferroptosis can improve the efficacy of immunotherapy and enhance antitumor immune responses
In recent years, immune checkpoint blockade (ICB) has been a breakthrough strategy for the treatment of cancers[200]. On one hand, ferroptosis shows potential for increasing the responsivity of ICB by inducing immunogenic cell death (ICD)[201]. On the other hand, triggering ferroptosis in the tumor microenvironment to deplete immunosuppressive cells can also reverse immunotherapy resistance. Specifically, during ferroptosis, intracellular damage-associated molecular patterns (DAMPs), such as calreticulin (CRT), adenosine triphosphate (ATP), high mobility group box 1 (HMGB1), and mtDNA, accumulate or are relocalized[201]. The increased levels of DAMPs in the tumor microenvironment could further enhance ICB. Therefore, amplifying the effect of ferroptosis to drive DAMP production could be a viable strategy to induce ICD and enhance the antitumor immunity of ICB[202]. For example, manganese has been reported to induce ferroptosis in cancer cells through type-I IFN-dependent inhibition of mitochondrial DHODH. A recent study showed uncontrolled tumor growth and metastasis in Mn-deficient mice, while the cellular cGAS-STING pathway was activated after Mn supplementation, which increased antigen delivery and increased the activation of CD8+ T cells and NK cells to exert antitumor effects[203]. Similarly, mitochondrial-targeting liposomes and nanoparticles have also been elaborately developed for drug delivery, activating the cGAS-STING pathway and triggering ICD. Their antitumor activity has been observed in bladder cancer and lung cancer[202], [204]. These data suggest that ferroptosis promotes the specific activation of STING pathway represents a potent strategy to increase the efficacy of immunotherapy in tumors. Additionally, previous studies have fabricated cinnamaldehyde dimers (CDC) into lipid-like materials to form dimersomes, which deplete GSH and deliver therapeutic agents to enhance ferroptosis and the efficacy of immunotherapy in breast cancer[205]. Yang F, et al. suggested that the combination of GPX4 inhibitors and anti-PD1 possessed greater therapeutic efficacy than the monotherapy for tumors resembling LAR in biological characteristics, which was attributed to LAR tumors eminently utilizing the GSH system for detoxification[29].
In addition to activating tumor cell immunogenicity, the inhibition of tumor cell immune escape also helps to enhance the immune response. Targeting PD-1/PD-L1 is a common strategy used in ICB. Researchers found that the mitochondrial translocator protein (TSPO) inhibits ferroptosis by enhancing the Nrf2-dependent antioxidant defense system and promotes HCC immune evasion by upregulating PD-L1 expression through Nrf2-mediated transcription. Notably, the TSPO inhibitor PK11195 administrated in combination with anti-PD-1 antibody exerted a synergistic antitumor effect on a mouse model[206]. Moreover, the active molecule B2 derived from a novel mitochondrion-targeting DHODH inhibitor can downregulate PD-L1 expression and alleviate immunosuppression by decreasing the expression of the mitochondria-related protein MTHFD2, thereby promoting ferroptosis in melanoma cells[207].
In addition to tumor cell-intrinsic approaches, triggering the ferroptosis of immune suppressor cells in the tumor microenvironment is a viable approach to increase immunotherapy efficacy. A study of melanoma showed that tumor growth was suppressed in mice with a Treg-specific deletion of GPX4 but antitumor immune responses were enhanced without inducing autoimmunity[208]. In a study of lung cancer, researchers developed zero-valent-iron nanoparticles (ZVI-NPs), which can simultaneously target the tumor microenvironment and cancer cells. Specifically, they accumulate preferentially in tumor and lung tissues, causing mitochondrial dysfunction and ferroptosis in lung cancer cells, and target the iron homeostasis of immune cells. ZVI-NPs significantly regulated macrophage polarization from the immunosuppressive M2 phenotype to the antitumor M1 phenotype and decreased the proportion of Tregs, augmenting antitumor immunity[189]. To date, several nanoparticles have been shown to inhibit tumor growth by reprogramming the TME and promoting tumor ferroptosis[209], [210]. Therefore, the induction of ferroptosis in immunosuppressive cells might be an innovative approach to potentiate the efficacy of immunotherapy.
The insufficient immunogenicity of tumor cells, sensitization of immune cells to ferroptosis by oxidative stress, and immunosuppressive TME are the causes of the poor response to tumor immunotherapy. Therefore, based on the biological characteristics of the immune TME, researchers can determine whether the application of combinations of immunotherapy and ferroptosis-targeting therapies is appropriate to improve efficacy.
5. Concluding remarks
Research into the mechanism underlying the promotion lipid peroxidation and ferroptosis and the sophisticated molecular mechanism regulating the antioxidant defense system has led to a greatly improved understanding. Interference with ferroptosis via key regulators exerts potential therapeutic effects on cancer cells. The discovery of a complex metabolic regulatory network and the pluripotency of ferroptosis inhibitors with different targets has established a blueprint for us to use in developing therapies.
Glucose, lipids, amino acid and iron metabolism, which provide energy for tumor cells to support growth and maintain iron homeostasis, are mitochondrial processes. When the energy status is abnormal in tumor cells or these cells are exposed to exogenous stimuli, a variety of metabolic activities in mitochondria may cause oxidative stress and promote ferroptosis[13], [95]. However, mitochondria are equipped with potent antiferroptotic defense factors, such as GPX4mito and DHODH-derived CoQ[25]. An impaired mitochondrial antiferroptotic defensive system cannot eliminate lipid peroxides, which can accumulate to lethal levels in mitochondria, and this imbalance between oxidative stress and the antioxidant system can lead to ferroptosis[102]. In summary, because of metabolic abnormalities and damage to mitochondria during ferroptosis in different models, an increasing number of studies on mitochondrial quality control pathways have proposed that mitochondria are intricately involved in the regulation of ferroptosis.
Mitochondria link ferroptosis with other classical death modalities, such as autophagy and apoptosis, suggesting a new perspective for exploring the complex mechanisms underlying various death modalities. For example, the aforementioned VDACs can be antagonized by erastin, resulting in channel opening, an elevated MMP, increased mtROS generation and ferroptosis[211]. Previous studies have shown that VDACs are key proteins involved in the mPTP for mitochondria-mediated apoptosis, facilitating the influx of proapoptotic proteins into cytosol via mitochondrial channels[211]. These effluents, such as mtDNA, are sensed by the cGAS/STING pathway and initiate inflammatory signaling and immune responses[212]. In situ Raman spectroscopy revealed that erastin may induce apoptosis and accelerate the release of Cyt c from mitochondria. However, Cyt c can trigger ferroptosis by initiating Fenton-like reactions and lipid peroxidation[213]. In addition, recent studies have found that iron in melanoma cells acts as a sensitizing agent for ROS, and the induced oxidation of the OMM protein TOM20 recruits Bax to mitochondria, thereby triggering subsequent Caspase3-GSDME-mediated pyroptosis[214]. Indeed, iron can trigger different types of cell death, depending on the activation of different signaling pathways. Moreover, Hsp90 has been identified as a coregulatory node of necroptosis and ferroptosis. The necroptosis inhibitor CDDO inhibits necroptosis and ferroptosis by targeting HSP90[215]. Another study showed that RIPK3, a key molecule involved in necroptosis, has a dual role when responding to drug-induced organ damage. It not only phosphorylates MLKL to induce necroptosis but also phosphorylates FSP1 to inhibit its enzymatic activity, thereby promoting ferroptosis[216]. Moreover, RIPK3 can promote mitochondrial energy metabolism and upregulate the expression of mitochondrial NADPH oxidase 4 to induce mitochondrial damage[217]. The simultaneous occurrence of necroptosis and ferroptosis may be related to acute tissue injury. Studies have shown that autophagy is involved in various pathways of ferroptosis (ferritinophagy, lipophagy, chaperone-mediated autophagy, etc.). Autophagy activates ferroptosis by selectively degrading antiferroptotic factors or increasing substrates to promote the accumulation of lipid peroxides[218], [219]. On the other hand, oxidative stress and lipid peroxidation products are powerful inducers of autophagy, while excessive autophagy further amplifies the ferroptosis signal[218], [219]. As a key protein involved in various signal transduction pathways, AMPK activates autophagy by negatively regulating mTOR. Several inhibitors and drugs can induce ferroptosis and autophagy by inhibiting mTOR or activating the AMPK pathway, as mentioned above; rapamycin further enhances the anticancer efficacy of ferroptosis inducers[82]. Taken together, since proteins and signal transduction pathways are shared between different cell death pathways, they provide subtle and plastic control of the cell fate in response to specific stresses, thus providing opportunities for therapeutic interventions to direct the cell death response.
Researchers have attempted to translate the current research on the mechanism by which mitochondria regulate cancer-related ferroptosis into clinical applications to address the issues of drug resistance, off-target effects, and the side effects of traditional chemotherapy or immunotherapy. Mitochondrial function is closely related to processes such as stress, metabolic reprogramming, and autophagy. Therefore, the corresponding regulators could work together with ferroptosis inducers in cancer therapy. The combination of ferroptosis modulators and immunotherapy is a highly discussed strategy for cancer treatment. However, its clinical translation is worrisome due to its double-edged sword effect. Ferroptosis may target both tumor cells and antitumor immune cells. Therefore, tailoring ferroptosis-related therapies to the heterogeneity of ferroptosis in different cancer cells, elucidating TME properties suitable for this therapy, and developing more specific targeted therapies may provide important insights into the design of effective combinatorial strategies against cancer.
The limitations are mentioned above. First, the intricate and dynamic network of mitochondrial regulation of ferroptosis involves multiple pathways, and thus identifying the dominant pathway according to the properties of the cell can help in the design of targeted therapeutic strategies. Second, although a variety of therapeutic approaches are under development, most of them have only been tested in animal experiments rather than large-scale clinical trials, and thus much more work is needed before clinical translation.
Abbreviations
ROS: reactive oxygen; ALOX: lipoxygenase; POR: cytochrome P450 oxidoreductase; TFR1: transferrin receptor 1; FTL: ferritin light chain; FTH1: ferritin heavy chain 1; FPN1: ferroportin; PROM2: Prominin2; GSH: glutathione; Glu: glutamate; GPX4: glutathione peroxidase 4; FSP1: ferroptosis suppressor protein 1; AIFM2: apoptosis-inducing factor mitochondria-associated 2; CoQ: coenzyme Q10; AR: androgen receptor; PUFAs: polyunsaturated fatty acids; ACSL4: acyl-CoA synthetase long-chain family member 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; ETC: electron transport chain; NADPH: nicotinamide adenine dinucleotide phosphate; MPO: myeloperoxidase; PDAC: pancreatic ductal adenocarcinoma; TRAP1: tumor necrosis factor receptor-associated protein 1; HSP90: heat shock protein 90; DRP1: dynamin-related protein 1; Fis1: fission 1; PINK1: PTEN-induced kinase 1; NSCLC: non-small-cell lung cancer; MTS: mitochondrial targeting sequence; OPA1: optic atrophy 1; MFN: mitofusin; ATF4: activating transcription factor 4; PHB2: prohibitin 2; IMM: inner mitochondria membrane; ISCs: iron-sulfur clusters; HO-1: heme oxygenase-1; CRC: colorectal cancer; TFAM: transcription factor A; COX7A1: cytochrome c oxidase subunit 7a; PGC-1α: mitochondrial biogenesis activator; CHCHD3: coiled-coil-helix-coiled-coil-helix domain containing 3; MTCH1: mitochondrial carrier 1; ACC: acetyl CoA carboxylase; PC: pyruvate carboxylase; IDH2: isocitrate dehydrogenase; α-KG: α-ketoglutarate; Gln: glutamine; MPC1: mitochondrial pyruvate carrier 1; AMPK: AMP-activated protein kinase; MEFs: embryonic fibroblasts; BECN1: beclin 1; VDACs: voltage-dependent anion-selective channels; H2O2: hydrogen peroxide; MDA: malondialdehyde; G3P: glycerol-3-phosphate; LOX: lysyl-oxidase; AK2: adenylate kinase 2; ER: endoplasmic reticulum; PLs: PUFA-phospholipids; DIC: dicarboxylate carrier; OGC: oxoglutarate carrier; NAC: N-acetylcysteine; HCC: hepatocellular cancer; DHLA: reduced form dihydrolipoic acid; TXN: thioredoxin; ARE: antioxidant-response element; OMM: outer mitochondrial membrane; FtMt: mitochondrial ferritin; Mfrn: mitoferrin; DMT1: divalent metal transporter 1; NFS1: cysteine desulfurase 1; IRP2: iron-responsive element-binding protein 2; ISCU: iron-sulfur cluster assembly enzyme; GLRX5: glutaredoxin 5; NCOA4: nuclear receptor coactivator 4; CPT1: carnitine palmitoyltransferase 1; FAO: fatty acid oxidation; DECR1: 2,4-Dienoyl-CoA reductase 1; CRPC: castration-resistant prostate cancer; LD: lipid droplet; PLIN2: Perilipin2; PGRMC1: progesterone receptor membrane component 1; ICB: immune checkpoint blockade; ICD: immunogenic cell death; DAMPs: intracellular damage-associated molecular patterns; ATP: adenosine triphosphate; HMGB1: high mobility group box 1.
Acknowledgements
This research was support by the National Natural Science Foundation of China (Grant No. 82203374), the Fundamental Research Funds for the Central Universities (Grant No. 2042022kf1081 and 2042023kf0020) and the Open Project of Hubei Key Laboratory (No. 2023KFZZ008).
Funding
This work was partially supported by a National Natural Science Foundation of China (NSFC) grant (Grant No. 82203374) and a grant from the Fundamental Research Funds for the Central Universities (No. 2042022kf1081) to Zhong Wang, a grant from the Fundamental Research Funds for the Central Universities (No. 2042023kf0020) and a grant from the Open Project of Hubei Key Laboratory (No. 2023KFZZ008) to Zhiyu Li.
Consent for publication
All authors approved the final manuscript and the submission to the journal.
Author contributions
Zilin Ding and Zhiyu Li collected, read relevant literature and wrote the article. Kai Sun, Yi Liu and Zhou Fang collected relative literatures. Chenyuan Li and Zhong Wang guided this article. Shengrong Sun guided the mentality and this article. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dixon SJ, Lemberg KM, Lamprecht MR. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072
2. Mou Y, Wang J, Wu J. et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12(1):34
3. Su Y, Zhao B, Zhou L. et al. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020;483:127-136
4. Hangauer MJ, Viswanathan VS, Ryan MJ. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551(7679):247-250
5. Tsoi J, Robert L, Paraiso K. et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell. 2018;33(5):890-904.e5
6. Viswanathan VS, Ryan MJ, Dhruv HD. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453-457
7. Friedmann Angeli JP, Krysko D V, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19(7):405-414
8. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7(7):e2307
9. Louandre C, Ezzoukhry Z, Godin C. et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133(7):1732-1742
10. Liang C, Zhang X, Yang M, Dong X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv Mater. 2019;31(51):e1904197
11. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145-1159
12. Gaschler MM, Hu F, Feng H, Linkermann A, Min W, Stockwell BR. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem Biol. 2018;13(4):1013-1020
13. Gao M, Yi J, Zhu J. et al. Role of Mitochondria in Ferroptosis. Mol Cell. 2019;73(2):354-363.e3
14. Feng H, Schorpp K, Jin J. et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020;30(10):3411-3423.e7
15. Chen PH, Wu J, Ding CKC. et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020;27(3):1008-1022
16. Gryzik M, Asperti M, Denardo A, Arosio P, Poli M. NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim Biophys Acta Mol Cell Res. 2021;1868(2):118913
17. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021-1032
18. Hou W, Xie Y, Song X. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425-1428
19. Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27(1):242-254
20. Geng N, Shi BJ, Li SL. et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci. 2018;22(12):3826-3836
21. Brown CW, Amante JJ, Chhoy P. et al. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev Cell. 2019;51(5):575-586.e4
22. Dixon SJ, Patel D, Welsch M. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3(3):e02523
23. Yang WS, Sriramaratnam R, Welsch ME. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1-2):317-331
24. Doll S, Freitas FP, Shah R. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693-698
25. Bersuker K, Hendricks JM, Li Z. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688-692
26. Kraft VAN, Bezjian CT, Pfeiffer S. et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci. 2020;6(1):41-53
27. Soula M, Weber RA, Zilka O. et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16(12):1351-1360
28. Liang D, Feng Y, Zandkarimi F. et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell. 2023;186(13):2748-2764.e22
29. Yang F, Xiao Y, Ding JH. et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023;35(1):84-100.e8
30. Qiu B, Zandkarimi F, Bezjian CT. et al. Phospholipids with two polyunsaturated fatty acyl tails promote ferroptosis. Cell. 2024;187(5):1177-1190.e18
31. Yang WH, Ding CKC, Sun T. et al. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019;28(10):2501-2508.e4
32. Yang WH, Huang Z, Wu J, Ding CKC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol Cancer Res. 2020;18(1):79-90
33. Yee PP, Wei Y, Kim SY. et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun. 2020;11(1):5424
34. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054-2081
35. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966-E4975
36. Doll S, Proneth B, Tyurina YY. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91-98
37. Wenzel SE, Tyurina YY, Zhao J. et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell. 2017;171(3):628-641.e26
38. Pickles S, Vigié P, Youle RJ. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol. 2018;28(4):R170-R185
39. Quiles JM, Gustafsson ÅB. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front Physiol. 2020;11:515
40. Lee YG, Kim HW, Nam Y. et al. LONP1 and ClpP cooperatively regulate mitochondrial proteostasis for cancer cell survival. Oncogenesis. 2021;10(2):18
41. Li MD, Fu L, Lv BB. et al. Arsenic induces ferroptosis and acute lung injury through mtROS-mediated mitochondria-associated endoplasmic reticulum membrane dysfunction. Ecotoxicol Environ Saf. 2022;238:113595
42. Ishizawa J, Zarabi SF, Davis RE. et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell. 2019;35(5):721-737.e9
43. Wang H, Liu C, Zhao Y. et al. Inhibition of LONP1 protects against erastin-induced ferroptosis in Pancreatic ductal adenocarcinoma PANC1 cells. Biochem Biophys Res Commun. 2020;522(4):1063-1068
44. Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2021;17(4):948-960
45. Cole A, Wang Z, Coyaud E. et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell. 2015;27(6):864-876
46. Akiyama H, Zhao R, Ostermann LB. et al. Correction: Mitochondrial regulation of GPX4 inhibition-mediated ferroptosis in acute myeloid leukemia. Leukemia. 2024;38(4):926
47. Giarrizzo M, LaComb JF, Patel HR. et al. TR-107, an Agonist of Caseinolytic Peptidase Proteolytic Subunit, Disrupts Mitochondrial Metabolism and Inhibits the Growth of Human Colorectal Cancer Cells. Mol Cancer Ther. 2024;23(12):1761-1778
48. Basit F, Van Oppen LMPE, Schöckel L. et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8(3):e2716
49. Miao Z, Tian W, Ye Y. et al. Hsp90 induces Acsl4-dependent glioma ferroptosis via dephosphorylating Ser637 at Drp1. Cell Death Dis. 2022;13(6):548
50. Schmitt K, Grimm A, Dallmann R. et al. Circadian Control of DRP1 Activity Regulates Mitochondrial Dynamics and Bioenergetics. Cell Metab. 2018;27(3):657-666.e5
51. Liu M, Fan Y, Li D. et al. Ferroptosis inducer erastin sensitizes NSCLC cells to celastrol through activation of the ROS-mitochondrial fission-mitophagy axis. Mol Oncol. 2021;15(8):2084-2105
52. Wei Y, Liu W, Wang R. et al. Propionate promotes ferroptosis and apoptosis through mitophagy and ACSL4-mediated ferroptosis elicits anti-leukemia immunity. Free Radic Biol Med. 2024;213:36-51
53. Li Z, Jiang L, Chew SH, Hirayama T, Sekido Y, Toyokuni S. Carbonic anhydrase 9 confers resistance to ferroptosis/apoptosis in malignant mesothelioma under hypoxia. Redox Biol. 2019;26:101297
54. Qiu S, Zhong X, Meng X. et al. Mitochondria-localized cGAS suppresses ferroptosis to promote cancer progression. Cell Res. 2023;33(4):299-311
55. Chang X, Zhou S, Liu J. et al. Zishen Tongyang Huoxue decoction (TYHX) alleviates sinoatrial node cell ischemia/reperfusion injury by directing mitochondrial quality control via the VDAC1-β-tubulin signaling axis. J Ethnopharmacol. 2024;320:117371
56. Chang X, Li Y, Liu J. et al. ß-tubulin contributes to Tongyang Huoxue decoction-induced protection against hypoxia/reoxygenation-induced injury of sinoatrial node cells through SIRT1-mediated regulation of mitochondrial quality surveillance. Phytomedicine. 2023;108:154502
57. Chan DC. Mitochondrial Dynamics and Its Involvement in Disease. Annu Rev Pathol. 2020;15:235-259
58. Wang J, Zhuang H, Yang X. et al. Exploring the Mechanism of Ferroptosis Induction by Sappanone A in Cancer: Insights into the Mitochondrial Dysfunction Mediated by NRF2/xCT/GPX4 Axis. Int J Biol Sci. 2024;20(13):5145-5161
59. Zhang X, Zhou H, Chang X. Involvement of mitochondrial dynamics and mitophagy in diabetic endothelial dysfunction and cardiac microvascular injury. Arch Toxicol. 2023;97(12):3023-3035
60. Chang X, Liu R, Li R, Peng Y, Zhu P, Zhou H. Molecular Mechanisms of Mitochondrial Quality Control in Ischemic Cardiomyopathy. Int J Biol Sci. 2023;19(2):426-448
61. Chang X, Zhou S, Liu J. et al. Zishenhuoxue decoction-induced myocardial protection against ischemic injury through TMBIM6-VDAC1-mediated regulation of calcium homeostasis and mitochondrial quality surveillance. Phytomedicine. 2024;132:155331
62. Chang X, Zhang Q, Huang Y. et al. Quercetin inhibits necroptosis in cardiomyocytes after ischemia-reperfusion via DNA-PKcs-SIRT5-orchestrated mitochondrial quality control. Phytother Res. 2024;38(5):2496-2517
63. Wang J, Zhuang H, Jia L. et al. Nuclear receptor subfamily 4 group A member 1 promotes myocardial ischemia/reperfusion injury through inducing mitochondrial fission factor-mediated mitochondrial fragmentation and inhibiting FUN14 domain containing 1-depedent mitophagy. Int J Biol Sci. 2024;20(11):4458-4475
64. Pu X, Zhang Q, Liu J. et al. Ginsenoside Rb1 ameliorates heart failure through DUSP-1-TMBIM-6-mediated mitochondrial quality control and gut flora interactions. Phytomedicine. 2024;132:155880
65. Pang B, Dong G, Pang T. et al. Emerging insights into the pathogenesis and therapeutic strategies for vascular endothelial injury-associated diseases: focus on mitochondrial dysfunction. Angiogenesis. 2024;27(4):623-639
66. Li C, Liu J, Hou W, Kang R, Tang D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front Cell Dev Biol. 2021;9:698679
67. Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17(4):491-506
68. Liang FG, Zandkarimi F, Lee J. et al. OPA1 promotes ferroptosis by augmenting mitochondrial ROS and suppressing an integrated stress response. Mol Cell. 2024;84(16):3098-3114.e6
69. Schofield JH, Schafer ZT. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid Redox Signal. 2021;34(7):517-530
70. Su L, Zhang J, Gomez H, Kellum JA, Peng Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy. 2023;19:401-414
71. Matsuda N, Sato S, Shiba K. et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211-221
72. Takashi Y, Tomita K, Kuwahara Y. et al. Mitochondrial dysfunction promotes aquaporin expression that controls hydrogen peroxide permeability and ferroptosis. Free Radic Biol Med. 2020;161:60-70
73. Yamashita SI, Sugiura Y, Matsuoka Y. et al. Mitophagy mediated by BNIP3 and NIX protects against ferroptosis by downregulating mitochondrial reactive oxygen species. Cell Death Differ. 2024;31(5):651-661
74. Rademaker G, Boumahd Y, Peiffer R. et al. Myoferlin targeting triggers mitophagy and primes ferroptosis in pancreatic cancer cells. Redox Biol. 2022;53:102324
75. Allen GFG, Toth R, James J, Ganley IG. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013;14(12):1127-1135
76. Scarpulla RC. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann N Y Acad Sci. 2008;1147:321-334
77. Wei R, Zhao Y, Wang J. et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int J Biol Sci. 2021;17(11):2703-2717
78. Yang J, Mo J, Dai J. et al. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021;12(11):1079
79. Chen QM. Nrf2 for protection against oxidant generation and mitochondrial damage in cardiac injury. Free Radic Biol Med. 2022;179:133-143
80. Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7(1-2):39-44
81. Huang C, Santofimia-Castaño P, Liu X. et al. NUPR1 inhibitor ZZW-115 induces ferroptosis in a mitochondria-dependent manner. Cell Death Discov. 2021;7(1):269
82. Feng Y, Xu J, Shi M. et al. COX7A1 enhances the sensitivity of human NSCLC cells to cystine deprivation-induced ferroptosis via regulating mitochondrial metabolism. Cell Death Dis. 2022;13(11):988
83. Xue X, Ma L, Zhang X. et al. Tumour cells are sensitised to ferroptosis via RB1CC1-mediated transcriptional reprogramming. Clin Transl Med. 2022;12(2):e747
84. Wang X, Ji Y, Qi J. et al. Mitochondrial carrier 1 (MTCH1) governs ferroptosis by triggering the FoxO1-GPX4 axis-mediated retrograde signaling in cervical cancer cells. Cell Death Dis. 2023;14(8):508
85. Li H, Yu K, Hu H. et al. METTL17 coordinates ferroptosis and tumorigenesis by regulating mitochondrial translation in colorectal cancer. Redox Biol. 2024;71:103087
86. Kang W, Suzuki M, Saito T, Miyado K. Emerging Role of TCA Cycle-Related Enzymes in Human Diseases. Int J Mol Sci. 2021;22(23):13057
87. Song X, Liu J, Kuang F. et al. PDK4 dictates metabolic resistance to ferroptosis by suppressing pyruvate oxidation and fatty acid synthesis. Cell Rep. 2021;34(8):108767
88. Kim H, Lee JH, Park JW. Down-regulation of IDH2 sensitizes cancer cells to erastin-induced ferroptosis. Biochem Biophys Res Commun. 2020;525(2):366-371
89. Shin D, Lee J, You JH, Kim D, Roh JL. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. 2020;30:101418
90. Jin L, Alesi GN, Kang S. Glutaminolysis as a target for cancer therapy. Oncogene. 2016;35(28):3619-3625
91. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59(2):298-308
92. Luo M, Wu L, Zhang K. et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018;25(8):1457-1472
93. Hu Q, Dai J, Zhang Z. et al. ASS1-Mediated Reductive Carboxylation of Cytosolic Glutamine Confers Ferroptosis Resistance in Cancer Cells. Cancer Res. 2023;83(10):1646-1665
94. You JH, Lee J, Roh JL. Mitochondrial pyruvate carrier 1 regulates ferroptosis in drug-tolerant persister head and neck cancer cells via epithelial-mesenchymal transition. Cancer Lett. 2021;507:40-54
95. Lee H, Zandkarimi F, Zhang Y. et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225-234
96. Song X, Zhu S, Chen P. et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X c- Activity. Curr Biol. 2018;28(15):2388-2399.e5
97. Wang Y, Bai C, Ruan Y. et al. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat Commun. 2019;10(1):201
98. Mao C, Liu X, Zhang Y. et al. Author Correction: DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;596(7873):E13
99. Yang C, Zhao Y, Wang L. et al. De novo pyrimidine biosynthetic complexes support cancer cell proliferation and ferroptosis defence. Nat Cell Biol. 2023;25(6):836-847
100. Dächert J, Ehrenfeld V, Habermann K, Dolgikh N, Fulda S. Targeting ferroptosis in rhabdomyosarcoma cells. Int J Cancer. 2020;146(2):510-520
101. Jelinek A, Heyder L, Daude M. et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic Biol Med. 2018;117:45-57
102. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. 2021;220(9):e202105043
103. Wu S, Mao C, Kondiparthi L, Poyurovsky M V, Olszewski K, Gan B. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc Natl Acad Sci U S A. 2022;119(26):e2121987119
104. Zhan M, Ding Y, Huang S. et al. Lysyl oxidase-like 3 restrains mitochondrial ferroptosis to promote liver cancer chemoresistance by stabilizing dihydroorotate dehydrogenase. Nat Commun. 2023;14(1):3123
105. Kuang F, Liu J, Xie Y, Tang D, Kang R. MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem Biol. 2021;28(6):765-775.e5
106. Liu Y, Wang Y, Lin Z, Kang R, Tang D, Liu J. SLC25A22 as a Key Mitochondrial Transporter Against Ferroptosis by Producing Glutathione and Monounsaturated Fatty Acids. Antioxid Redox Signal. 2023;39(1-3):166-185
107. Jang S, Chapa-Dubocq XR, Tyurina YY. et al. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. 2021;45:102021
108. DeHart DN, Fang D, Heslop K, Li L, Lemasters JJ, Maldonado EN. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem Pharmacol. 2018;148:155-162
109. Camiolo G, Tibullo D, Giallongo C. et al. α-Lipoic Acid Reduces Iron-induced Toxicity and Oxidative Stress in a Model of Iron Overload. Int J Mol Sci. 2019;20(3):609
110. Zhou Q, Meng Y, Li D. et al. Ferroptosis in cancer: From molecular mechanisms to therapeutic strate-gies. Signal Transduct Target Ther. 2024;9(1):55
111. Kagan VE, Mao G, Qu F. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81-90
112. Chen Y, Yang Z, Wang S. et al. Boosting ROS-Mediated Lysosomal Membrane Permeabilization for Cancer Ferroptosis Therapy. Adv Healthc Mater. 2023;12(6):e2022150
113. Mishima E, Ito J, Wu Z. et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608(7924):778-783
114. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107
115. Dodson M, Anandhan A, Zhang DD. MGST1, a new soldier of NRF2 in the battle against ferroptotic death. Cell Chem Biol. 2021;28(6):741-742
116. Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2-an update. Free Radic Biol Med. 2014;66:36-44
117. Chen GH, Song CC, Pantopoulos K, Wei XL, Zheng H, Luo Z. Mitochondrial oxidative stress mediated Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic Biol Med. 2022;180:95-107
118. Sandoval-Acuña C, Torrealba N, Tomkova V. et al. Targeting Mitochondrial Iron Metabolism Suppresses Tumor Growth and Metastasis by Inducing Mitochondrial Dysfunction and Mitophagy. Cancer Res. 2021;81(9):2289-2303
119. Huang G, Xiang Z, Wu H. et al. The lncRNA BDNF-AS/WDR5/FBXW7 axis mediates ferroptosis in gastric cancer peritoneal metastasis by regulating VDAC3 ubiquitination. Int J Biol Sci. 2022;18(4):1415-1433
120. Shaw GC, Cope JJ, Li L. et al. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440(7080):96-100
121. Zhang T, Sun L, Hao Y. et al. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat Cancer. 2022;3(1):75-89
122. Hung HI, Schwartz JM, Maldonado EN, Lemasters JJ, Nieminens AL. Mitoferrin-2-dependent mitochondrial iron uptake sensitizes human head and neck squamous carcinoma cells to photodynamic therapy. J Biol Chem. 2013;288(1):677-686
123. Ali MY, Griguer CE, Flor S, Oliva CR. Mitoferrin-1 Promotes Proliferation and Abrogates Protein Oxidation via the Glutathione Pathway in Glioblastoma. Antioxidants (Basel). 2023;12(2):349
124. Ni S, Kuang Y, Yuan Y, Yu B. Mitochondrion-mediated iron accumulation promotes carcinogenesis and Warburg effect through reactive oxygen species in osteosarcoma. Cancer Cell Int. 2020;20(1):399
125. Das A, Nag S, Mason AB, Barroso MM. Endosome-mitochondria interactions are modulated by iron release from transferrin. J Cell Biol. 2016;214(7):831-45
126. Song Q, Peng S, Sun Z, Heng X, Zhu X. Temozolomide Drives Ferroptosis via a DMT1-Dependent Pathway in Glioblastoma Cells. Yonsei Med J. 2021;62(9):843-849
127. Wolff NA, Garrick MD, Zhao L, Garrick LM, Ghio AJ, Thévenod F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci Rep. 2018;8(1):211
128. Lipper CH, Stofleth JT, Bai F. et al. Redox-dependent gating of VDAC by mitoNEET. Proc Natl Acad Sci U S A. 2019;116(40):19924-19929
129. Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. 2016;478(2):838-844
130. Sohn YS, Tamir S, Song L. et al. NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc Natl Acad Sci U S A. 2013;110(36):14676-14681
131. Geisler S, Holmström KM, Skujat D. et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119-131
132. Li Y, Wang X, Huang Z. et al. CISD3 inhibition drives cystine-deprivation induced ferroptosis. Cell Death Dis. 2021;12(9):839
133. Kim EH, Shin D, Lee J, Jung AR, Roh JL. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018;432:180-190
134. Homma T, Kobayashi S, Fujii J. Cysteine preservation confers resistance to glutathione-depleted cells against ferroptosis via CDGSH iron sulphur domain-containing proteins (CISDs). Free Radic Res. 2020;54(6):397-407
135. Gottlieb Y, Truman M, Cohen LA, Leichtmann-Bardoogo Y, Meyron-Holtz EG. Endoplasmic reticulum anchored heme-oxygenase 1 faces the cytosol. Haematologica. 2012;97(10):1489-1493
136. Han S, Lin F, Qi Y. et al. HO-1 Contributes to Luteolin-Triggered Ferroptosis in Clear Cell Renal Cell Carcinoma via Increasing the Labile Iron Pool and Promoting Lipid Peroxidation. Oxid Med Cell Longev. 2022;2022:3846217
137. Lai X, Sun Y, Zhang X. et al. Honokiol Induces Ferroptosis by Upregulating HMOX1 in Acute Myeloid Leukemia Cells. Front Pharmacol. 2022;13:897791
138. Lou JS, Zhao LP, Huang ZH. et al. Ginkgetin derived from Ginkgo biloba leaves enhances the therapeutic effect of cisplatin via ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR wild-type non-small-cell lung cancer. Phytomedicine. 2021;80:153370
139. Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018;416:124-137
140. Consoli V, Sorrenti V, Pittalà V. et al. Heme Oxygenase Modulation Drives Ferroptosis in TNBC Cells. Int J Mol Sci. 2022;23(10):5709
141. Chiang SK, Chen SE, Chang LC. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int J Mol Sci. 2018;20(1):39
142. Villalpando-Rodriguez GE, Blankstein AR, Konzelman C, Gibson SB. Lysosomal Destabilizing Drug Siramesine and the Dual Tyrosine Kinase Inhibitor Lapatinib Induce a Synergistic Ferroptosis through Reduced Heme Oxygenase-1 (HO-1) Levels. Oxid Med Cell Longev. 2019;2019:9561281
143. Sun X, Ou Z, Chen R. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173-184
144. Liao Y, Jia X, Ren Y. et al. Suppressive role of microRNA-130b-3p in ferroptosis in melanoma cells correlates with DKK1 inhibition and Nrf2-HO-1 pathway activation. Hum Cell. 2021;34(5):1532-1544
145. Zheng X jin, Chen W lin, Yi J. et al. Apolipoprotein C1 promotes glioblastoma tumorigenesis by reducing KEAP1/NRF2 and CBS-regulated ferroptosis. Acta Pharmacol Sin. 2022;43:2977-2992
146. Sedlak TW, Saleh M, Higginson DS, Paul BD, Juluri KR, Snyder SH. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc Natl Acad Sci U S A. 2009;106(13):5171-5176
147. Terzi EM, Sviderskiy VO, Alvarez SW, Whiten GC, Possemato R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci Adv. 2021;7(22):eabg4302
148. Alvarez SW, Sviderskiy VO, Terzi EM. et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551(7682):639-643
149. Stemmler TL, Lesuisse E, Pain D, Dancis A. Frataxin and mitochondrial FeS cluster biogenesis. J Biol Chem. 2010;285(35):26737-26743
150. Du J, Zhou Y, Li Y. et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020;32:101483
151. Du J, Wang T, Li Y. et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356-369
152. Lee J, You JH, Shin D, Roh JL. Inhibition of Glutaredoxin 5 predisposes Cisplatin-resistant Head and Neck Cancer Cells to Ferroptosis. Theranostics. 2020;10(17):7775-7786
153. Kim JY, Kim JK, Kim H. ABCB7 simultaneously regulates apoptotic and non-apoptotic cell death by modulating mitochondrial ROS and HIF1α-driven NFκB signaling. Oncogene. 2020;39(9):1969-1982
154. Wang YQ, Chang SY, Wu Q. et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front Aging Neurosci. 2016;8:308
155. Fuhrmann DC, Mondorf A, Beifuß J, Jung M, Brüne B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020;36:101670
156. Masaldan S, Clatworthy SAS, Gamell C. et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018;14:100-115
157. Ma L, Chen C, Zhao C. et al. Targeting carnitine palmitoyl transferase 1A (CPT1A) induces ferroptosis and synergizes with immunotherapy in lung cancer. Signal Transduct Target Ther. 2024;9(1):64
158. Zheng Y, Hu Q, wu J. Adiponectin ameliorates placental injury in gestational diabetes mice by correcting fatty acid oxidation/peroxide imbalance-induced ferroptosis via restoration of CPT-1 activity. Endocrine. 2022;75(3):781-793
159. Feng H, Liu Y, Gan Y. et al. AdipoR1 Regulates Ionizing Radiation-Induced Ferroptosis in HCC cells through Nrf2/xCT Pathway. Oxid Med Cell Longev. 2022;2022:1-14
160. Schlaepfer IR, Joshi M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology. 2020;161(2):bqz046
161. Shimada K, Skouta R, Kaplan A. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497-503
162. Nassar ZD, Mah CY, Dehairs J. et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. Elife. 2020;9:1-34
163. Blomme A, Ford CA, Mui E. et al. 2,4-dienoyl-CoA reductase regulates lipid homeostasis in treatment-resistant prostate cancer. Nat Commun. 2020;11(1):2508
164. Chen X, Yu C, Kang R, Kroemer G, Tang D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021;28(4):1135-1148
165. Sun X, Yang S, Feng X. et al. The modification of ferroptosis and abnormal lipometabolism through overexpression and knockdown of potential prognostic biomarker perilipin2 in gastric carcinoma. Gastric Cancer. 2020;23(2):241-259
166. Bai Y, Meng L, Han L. et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508(4):997-1003
167. Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32(6):678-692
168. Benador IY, Veliova M, Liesa M, Shirihai OS. Mitochondria Bound to Lipid Droplets: Where Mitochondrial Dynamics Regulate Lipid Storage and Utilization. Cell Metab. 2019;29(4):827-835
169. Yang P, Li J, Zhang T. et al. Ionizing radiation-induced mitophagy promotes ferroptosis by increasing intracellular free fatty acids. Cell Death Differ. 2023;30(11):2432-2445
170. You JH, Lee J, Roh JL. PGRMC1-dependent lipophagy promotes ferroptosis in paclitaxel-tolerant persister cancer cells. J Exp Clin Cancer Res. 2021;40(1):350
171. Ghoochani A, Hsu EC, Aslan M. et al. Ferroptosis Inducers Are a Novel Therapeutic Approach for Advanced Prostate Cancer. Cancer Res. 2021;81(6):1583-1594
172. Zou Y, Palte MJ, Deik AA. et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617
173. Ni W, Li Y, Liang L. et al. Tumor Microenvironment-Responsive Nanodrug for Clear-Cell Renal Cell Carcinoma Therapy via Triggering Waterfall-Like Cascade Ferroptosis. J Biomed Nanotechnol. 2022;18(2):327-342
174. Chen Y, Shang H, Wang C. et al. RNA-Seq Explores the Mechanism of Oxygen-Boosted Sonodynamic Therapy Based on All-in-One Nanobubbles to Enhance Ferroptosis for the Treatment of HCC. Int J Nanomedicine. 2022;17:105-123
175. Ding Y, Chen X, Liu C. et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. 2021;14(1):19
176. Zhou L, Chen J, Li R. et al. Metal-Polyphenol-Network Coated Prussian Blue Nanoparticles for Synergistic Ferroptosis and Apoptosis via Triggered GPX4 Inhibition and Concurrent In Situ Bleomycin Toxification. Small. 2021;17(47):e2103919
177. Qian B, Che L, Du ZB. et al. Protein phosphatase 2A-B55β mediated mitochondrial p-GPX4 dephosphorylation promoted sorafenib-induced ferroptosis in hepatocellular carcinoma via regulating p53 retrograde signaling. Theranostics. 2023;13(12):4288-4302
178. Song H, Liang J, Guo Y. et al. A potent GPX4 degrader to induce ferroptosis in HT1080 cells. Eur J Med Chem. 2024;265:116110
179. Gan H, Huang X, Luo X. et al. A Mitochondria-Targeted Ferroptosis Inducer Activated by Glutathione-Responsive Imaging and Depletion for Triple Negative Breast Cancer Theranostics. Adv Healthc Mater. 2023;12(22):e2300220
180. Tian L, Ji H, Wang W. et al. Mitochondria-targeted pentacyclic triterpenoid carbon dots for selective cancer cell destruction via inducing autophagy, apoptosis, as well as ferroptosis. Bioorg Chem. 2023;130:106259
181. Jiaqi L, Siqing H. et al. Andrographolide promoted ferroptosis to repress the development of non-small cell lung cancer through activation of the mitochondrial dysfunction. Phytomedicine. 2023;109:154601
182. Yang R, Gao W, Wang Z. et al. Polyphyllin I induced ferroptosis to suppress the progression of hepatocellular carcinoma through activation of the mitochondrial dysfunction via Nrf2/HO-1/GPX4 axis. Phytomedicine. 2024;122:155135
183. Cosialls E, Pacreau E, Duruel C. et al. mTOR inhibition suppresses salinomycin-induced ferroptosis in breast cancer stem cells by ironing out mitochondrial dysfunctions. Cell Death Dis. 2023;14(11):744
184. Noè R, Inglese N, Romani P. et al. Organic Selenium induces ferroptosis in pancreatic cancer cells. Redox Biol. 2023;68:102962
185. Zhu L, Wang J, Tang X. et al. Efficient Magnetic Nanocatalyst-Induced Chemo- and Ferroptosis Synergistic Cancer Therapy in Combination with T1-T2 Dual-Mode Magnetic Resonance Imaging Through Doxorubicin Delivery. ACS Appl Mater Interfaces. 2022;14(3):3621-3632
186. Wang J, Zhao Z, Liu Y. et al. “Mito-Bomb”: a novel mitochondria-targeting nanosystem for ferroptosis-boosted sonodynamic antitumor therapy. Drug Deliv. 2022;29(1):3111-3122
187. Liu N, Lin Q, Huang Z. et al. Mitochondria-Targeted Prodrug Nanoassemblies for Efficient Ferroptosis-Based Therapy via Devastating Ferroptosis Defense Systems. ACS Nano. 2024;18:7945-7958
188. Chen S, Yang J, Liang Z. et al. Synergistic Functional Nanomedicine Enhances Ferroptosis Therapy for Breast Tumors by a Blocking Defensive Redox System. ACS Appl Mater Interfaces. 2023;15(2):2705-2713
189. Hsieh CH, Hsieh HC, Shih FS. et al. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics. 2021;11(14):7072-7091
190. Natale R, Wheeler R, Moore M. et al. Multicenter phase II trial of brequinar sodium in patients with advanced melanoma. Ann Oncol. 1992;3(8):659-660
191. Urba S, Doroshow J, Cripps C. et al. Multicenter phase II trial of brequinar sodium in patients with advanced squamous-cell carcinoma of the head and neck. Cancer Chemother Pharmacol. 1992;31(2):167-169
192. Moore M, Maroun J, Robert F. et al. Multicenter phase II study of brequinar sodium in patients with advanced gastrointestinal cancer. Invest New Drugs. 1993;11(1):61-65
193. Jiang M, Song Y, Liu H, Jin Y, Li R, Zhu X. DHODH Inhibition Exerts Synergistic Therapeutic Effect with Cisplatin to Induce Ferroptosis in Cervical Cancer through Regulating mTOR Pathway. Cancers (Basel). 2023;15(2):546
194. Hu X, Zhang P, Li S. et al. Mitochondrial GCN5L1 acts as a novel regulator for iron homeostasis to promote sorafenib sensitivity in hepatocellular carcinoma. J Transl Med. 2024;22(1):593
195. Huang A, Li Q, Shi X. et al. An iron-containing nanomedicine for inducing deep tumor penetration and synergistic ferroptosis in enhanced pancreatic cancer therapy. Mater Today Bio. 2024;27:101132
196. Zhang Y, Song Q, Zhang Y. et al. Iron-Based Nanovehicle Delivering Fin56 for Hyperthermia-Boosted Ferroptosis Therapy Against Osteosarcoma. Int J Nanomedicine. 2024;19:91-107
197. Lee S, Hwang N, Seok BG, Lee S, Lee SJ, Chung SW. Autophagy mediates an amplification loop during ferroptosis. Cell Death Dis. 2023;14(7):464
198. Wang Y, Patti GJ. The Warburg effect: a signature of mitochondrial overload. Trends Cell Biol. 2023;33(12):1014-1020
199. Miki K, Yagi M, Noguchi N. et al. Induction of glioblastoma cell ferroptosis using combined treatment with chloramphenicol and 2-deoxy-D-glucose. Sci Rep. 2023;13(1):10497
200. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350-1355
201. Demuynck R, Efimova I, Naessens F, Krysko D V. Immunogenic ferroptosis and where to find it? J Immunother Cancer. 2021;9(12):e003430
202. Ding Q, Tang W, Li X. et al. Mitochondrial-targeted brequinar liposome boosted mitochondrial-related ferroptosis for promoting checkpoint blockade immunotherapy in bladder cancer. J Control Release. 2023;363:221-234
203. Zhang S, Kang L, Dai X. et al. Manganese induces tumor cell ferroptosis through type-I IFN dependent inhibition of mitochondrial dihydroorotate dehydrogenase. Free Radic Biol Med. 2022;193(Pt 1):202-212
204. Liang JL, Jin XK, Zhang SM. et al. Specific activation of cGAS-STING pathway by nanotherapeutics-mediated ferroptosis evoked endogenous signaling for boosting systemic tumor immunotherapy. Sci Bull (Beijing). 2023;68(6):622-636
205. Zhou Z, Liang H, Yang R. et al. Glutathione Depletion-Induced Activation of Dimersomes for Potentiating the Ferroptosis and Immunotherapy of “Cold” Tumor. Angew Chem Int Ed Engl. 2022;61(22):1455741
206. Zhang D, Man D, Lu J. et al. Mitochondrial TSPO Promotes Hepatocellular Carcinoma Progression through Ferroptosis Inhibition and Immune Evasion. Adv Sci (Weinh). 2023;10(15):2206669
207. Hai Y, Fan R, Zhao T. et al. A novel mitochondria-targeting DHODH inhibitor induces robust ferroptosis and alleviates immune suppression. Pharmacol Res. 2024;202:107115
208. Xu C, Sun S, Johnson T. et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021;35(11):109235
209. Hu S, Ma J, Su C. et al. Engineered exosome-like nanovesicles suppress tumor growth by reprogramming tumor microenvironment and promoting tumor ferroptosis. Acta Biomater. 2021;135:567-581
210. Jiang Q, Wang K, Zhang X. et al. Platelet Membrane-Camouflaged Magnetic Nanoparticles for Ferroptosis-Enhanced Cancer Immunotherapy. Small. 2020 16(22)
211. Scharstuhl A, Mutsaers HAM, Pennings SWC, Russel FGM, Wagener FADTG. Involvement of VDAC, Bax and ceramides in the efflux of AIF from mitochondria during curcumin-induced apoptosis. PLoS One. 2009;4(8):e6688
212. Flores-Romero H, Dadsena S, García-Sáez AJ. Mitochondrial pores at the crossroad between cell death and inflammatory signaling. Mol Cell. 2023;83(6):843-856
213. Tang J, Zhu J, Xie H. et al. Mitochondria-Specific Molecular Crosstalk between Ferroptosis and Apoptosis Revealed by In Situ Raman Spectroscopy. Nano Lett. 2024;24(7):2384-2391
214. Zhou B, Zhang J yuan, Liu X shuo. et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28(12):1171-1185
215. Wu Z, Geng Y, Lu X. et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116(8):2996-3005
216. Lai K, Wang J, Lin S. et al. Sensing of mitochondrial DNA by ZBP1 promotes RIPK3-mediated necroptosis and ferroptosis in response to diquat poisoning. Cell Death Differ. 2024;31(5):635-650
217. Morgan MJ, Kim YS. Roles of RIPK3 in necroptosis, cell signaling, and disease. Exp Mol Med. 2022;54(10):1695-1704
218. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89-100
219. Li J, Liu J, Xu Y. et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy. 2021;17(11):3361-3374
Author contact
![]() Corresponding authors: Shengrong Sun, Email: sun137com; Chenyuan Li, Email: lcyfeifeiedu.cn; Zhong Wang, Email: zhongwangchnedu.cn.
Corresponding authors: Shengrong Sun, Email: sun137com; Chenyuan Li, Email: lcyfeifeiedu.cn; Zhong Wang, Email: zhongwangchnedu.cn.