Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. The origin, polarization and...
3. Macrophages in carcinogenesis...
4. TAM-targeted cancer therapy
5. Future Prospective
6. Concluding remarks
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(5):2235-2257. doi:10.7150/ijbs.106275 This issue Cite
Review
Harnessing Macrophages in Cancer Therapy: from Immune Modulators to Therapeutic Targets
Huabing Tan1,2,#, Meihe Cai3,#, Jincheng Wang4, Tao Yu5, Houjun Xia6, ![]() , Huanbin Zhao7,8,
, Huanbin Zhao7,8, ![]() , Xiaoyu Zhang9,
, Xiaoyu Zhang9, ![]()
1. Department of Infectious Diseases, Hepatology Institute, Renmin Hospital, Shiyan Key Laboratory of Virology, Hubei University of Medicine, Shiyan, Hubei Province, China.
2. General internal medicine, Wuhan Jinyintan Hospital, Tongji Medical College of Huazhong University of Science and Technology, Wuhan, China.
3. Department of Traditional Chinese Medicine, Zhushan Renmin Hospital, Zhushan, 442200, China.
4. Faculty of Medicine, Hokkaido University, Japan.
5. CAS Key Laboratory of Tissue Microenvironment and Tumor, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China.
6. Center for Cancer Immunology, Institute of Biomedicine and Biotechnology, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen, China.
7. Department of Pathophysiology, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
8. Present: Division of Pharmaceutical Sciences, Department of Pharmacy and Pharmaceutical Sciences, St. Jude Children's Research Hospital, Memphis, TN, USA.
9. Department of Gastrointestinal Surgery, Huai'an Second People's Hospital, The Affiliated Huai'an Hospital of Xuzhou Medical University, Huai'an, China.
#Co-first authors.
Received 2024-11-3; Accepted 2025-2-14; Published 2025-2-26
Abstract

Macrophages, as the predominant phagocytes, play an essential role in pathogens defense and tissue homeostasis maintenance. In the context of cancer, tumor-associated macrophages (TAMs) have evolved into cunning actors involved in angiogenesis, cancer cell proliferation and metastasis, as well as the construction of immunosuppressive microenvironment. Once properly activated, macrophages can kill tumor cells directly through phagocytosis or attack tumor cells indirectly by stimulating innate and adaptive immunity. Thus, the prospect of targeting TAMs has sparked significant interest and emerged as a promising strategy in immunotherapy. In this review, we summarize the diverse roles and underlying mechanisms of TAMs in cancer development and immunity and highlight the TAM-based therapeutic strategies such as inhibiting macrophage recruitment, inhibiting the differentiation reprogramming of TAMs, blocking phagocytotic checkpoints, inducing trained macrophages, as well as the potential of engineered CAR-armed macrophages in cancer therapy.
Keywords: macrophage, cancer immunity, immunotherapy, phagocytotic checkpoint, trained macrophage
1. Introduction
Tumorigenesis is a process of normal cells being transformed into cancer cells and characterized by uncontrolled tumor cell growth and impaired immune surveillance. The development and progression of tumors are influenced by a variety of factors. Primarily, oncogenic mutations and the activation of signaling pathways driven by these mutations play a key role [1-5]. Additionally, the interaction between tumor cells and the surrounding microenvironment significantly contributes to tumor growth. The tumor microenvironment (TME), a dynamic and complex milieu of various stromal cells around cancer cells, plays a critical role in tumor progression and treatment efficacy [6-10]. Tumor-associated macrophages (TAMs) are observed as the most abundant infiltrated immune cells in the TME [11]. As is known, macrophages are critical for inflammation, tissue repair, organ regeneration, and tissue homeostasis. By secreting growth factors, proteases, and cytokines, TAMs interact with other cell populations within tumors and are involved in pro-tumorigenic or anti-tumorigenic roles in various cancers [12, 13]. TAMs are extremely heterogeneous in TME which are determined by their ontogeny, intrinsic factors, and locations [14]. Throughout the different stages of malignant cancer, the sub-populations of TAMs are dynamically changed and are programed to increasingly adopt immune suppressive characteristics along with the tumor progression. The expansion of TAMs accelerates the formation of immunosuppressive TME driven by self-proliferation and monocyte differentiation [15]. In addition, tissue resident macrophages (TRMs) foster an anti-inflammatory conditions in organs which provide ideal niches for promoting metastasis, for example, peritoneal GATA6+ TRMs promote the ovarian cancer metastasis into the peritoneal cavity [16, 17] and liver [9]. Moreover, TAMs impede the CD8+ T cell mediated anti-tumor immune response, which is typically boosted by immune checkpoint blocking (ICB) [18, 19]. In summary, these data underscore the significant involvement of TAMs highly in shaping of the context of cancers during tumorigenesis.
With the application and innovation of multi-omics, more comprehensive insights into TAMs and their subpopulations within TME have been discovered. The phenotypes and functions of TAMs in tumor conditions are determined by transcriptional and epigenetic modulations [20, 21], which are greatly influenced by cytokines and metabolites released by cancer cells [22]. Understanding the diversity and contribution of TAMs to pathophysiological processes may provide new therapeutic targets for human cancers. Indeed, certain strategies designed to target TAMs have gained remarkable success in pre-clinic studies. However, the effectiveness of these strategies has been limited in clinical trials, highlighting that more precise mechanism and ingenious technologies should be further exploited in this field. In this review, we summarize the recent advancements in TAM research and aim to gain a comprehensive understanding of their roles in cancer immunity and therapy.
2. The origin, polarization and heterogeneity of TAMs
2.1 The origin of TAMs
First discovered by Ellie Metchnikoff, macrophages are a type of white blood cell that defends the host against pathogens through a process called phagocytosis and engages in innate and adaptive immunity by interacting with other immune cells [23]. It has long been held that macrophages originate from blood monocytes produced from myeloid progenitors in bone marrow (BM) [24]. Upon tissue injury, infection or carcinogenesis, these circulating monocytes are rapidly recruited to the corresponding site, where they differentiate into macrophages and accumulate in large amounts [25]. However, by lineage tracing and fate mapping technologies, cumulative evidence indicates that macrophages can also derive from embryonic progenitors originating from yolk sac or fetal liver, representing another major developmental path of macrophages in addition to monocyte differentiation [26, 27]. These embryonic-derived macrophages reside in organs (such as the brain, liver, and skin), proliferate, and maintain locally as TRMs throughout life, referring TRMs either in the liver as Kupffer cells or in the brain as microglia. TRMs can be classified into three subsets based on common life cycle properties and core gene signatures (Timd4, Lyve1, Folr2, and Ccr2) in most murine tissues: TLF+ macrophages (expressing TIM4 and/or LYVE1 and/or FOLR2), CCR2+ macrophages (TIM4-LYVE1-FOLR2-) and MHC-IIhi macrophages (TIM4-LYVE1-FOLR2-CCR2-). TLF+ macrophages are maintained through self-renewal with minimal monocyte input, while CCR2+ macrophages are almost entirely replaced by monocytes. MHC-IIhi macrophages, on the other hand, receive modest monocyte contribution, but are not continually replaced [27]. No matter what the origins are, colony stimulating factor 1 receptor (CSF1R) and its two ligands CSF1 and interleukin (IL)-34 are essential for the differentiation and expansion of macrophages [28]. Overall, macrophages are present in almost all tissues and exhibit complex phenotypic heterogeneity and functional diversity under various physiological and pathological conditions because of different developmental origins and tissues of residence.
In TME, infiltrated TAMs are also composed of both BM-derived macrophages and TRMs (Figure 1). Cancer cells can induce emergency myelopoiesis and expansion of bone marrow myeloid progenitors resulting in increased classical Ly6C+ monocytes [29]. BM-derived circulating peripheral monocytes are recruited into TME by cytokines and chemokines, such as CSF1, GM-CSF, IL-1β, SDF1α, VEGF and CCL2, and subsequently differentiate into TAMs [30-33]. In many cancers, these monocyte-derived macrophages are the main source of TAMs. For example, in a transgenic model of murine breast cancer, TAMs differentiated from monocytes are phenotypically distinct from the predominant mammary tissue macrophages in healthy mammary gland. Monocyte-derived TAMs gradually replace mammary tissue macrophages and promote tumor growth [15]. Additionally, retinoic acid, a metabolite of vitamin A1 produced by murine sarcoma tumor cells, selectively suppresses the DC-promoting transcription factor interferon regulatory factor-4 (IRF4) and drives intra-tumoral monocyte differentiation toward TAMs and away from DCs [34].
The origin of TAMs. TAMs derive from two main sources: tissue-resident macrophages and newly recruited monocyte-derived macrophages.
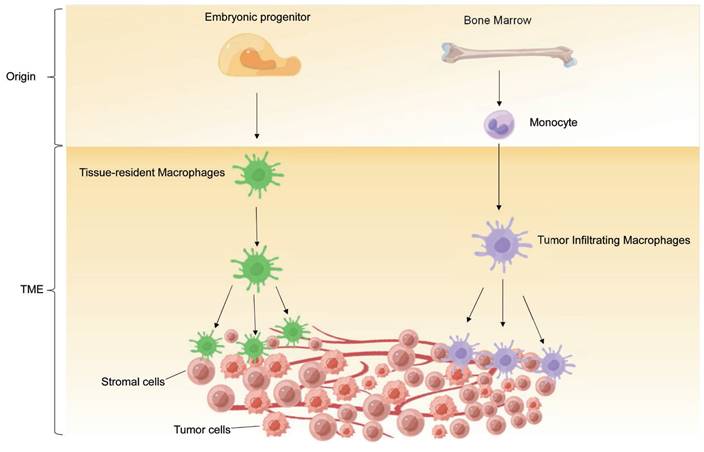
Meanwhile, the importance of TRMs in sustaining TAM levels and promoting tumor growth in certain types of cancers has been demonstrated by recent studies [17, 26, 35, 36]. TRMs are involved in defense, homeostasis, tissue integrity, and wound healing in healthy tissues. Although both embryonic-derived TRMs and monocyte-derived macrophages contribute to the accumulation of TAMs, it is not fully understood which TAMs population functions in regulating tumor progression. For instance, in a mouse model of breast cancer, depletion of TRMs did not reduce the tumor size, whereas depletion of circulating macrophages significantly decreased the tumor volume [15]. On the contrary, ablation of BM-derived macrophages did not disrupt tumor progression in a mouse model of pancreatic cancer, but depletion of TRMs dramatically reversed the trend [26]. Furthermore, in human breast cancer, FOLR2+ mammary resident macrophages in tumors, which are localized in perivascular areas in the tumor stroma, can efficiently prime effector CD8+ T cells and are correlated with patient survival [37].
It is noteworthy that TAM populations originating from different sources exhibit distinct temporal and spatial distribution in the TME. In the lung cancer model, macrophages from both origins were found to facilitate tumor growth and progression [38]. Moreover, at the early stage of non-small cell lung carcinoma (NSCLC), TRMs accumulated in close proximity to tumor cells and induced potent suppression of adaptive immunity mediated by regulatory T cell [36]. During tumor growth, TRMs undergo redistribution towards the periphery of the TME, which becomes dominated by monocyte-derived macrophages in both mouse and human NSCLC. This suggests that TRMs create a pro-tumorigenic niche for early NSCLC cells [36]. Nevertheless, these findings support the function complexity and diversity of TAMs, and further studies are needed to address the conundrum.
2.2 The polarization of TAMs
It's widely recognized that macrophages are highly plastic cells capable of undergoing specific polarization in different tissue environments. In response to different environmental signals, undifferentiated M0 macrophages which represent the unpolarized and resting state, can be polarized into two types: classically activated macrophages (M1) and alternatively activated macrophages (M2) [39]. M1 macrophages, triggered by interferon (IFN)-γ and bacterial lipopolysaccharide (LPS), exhibit increased levels of nitric oxide synthase (NOS) and reactive oxygen species (ROS). These M1 macrophages are considered as anti-tumor cells with secretion of inflammatory factors including IL-6, IL-1, and tumor necrosis factor-α (TNF-α), and promote adaptive immune response by highly expressing antigen presenting MHC complex [40]. By contrast, the M2 macrophages, polarized by IL-4, IL-13, and transforming growth factor β (TGF-β), are associated with the initiation, progression, metastasis, and immune evasion of tumors, by secreting anti-inflammatory cytokines such as IL-10, IL-4, and IL-13 [41]. Moreover, M2 macrophages are much more complex than M1, which can be further classified into M2a, M2b, M2c, and M2-like macrophages (Table 1) [42].
The different activators and biological functions of the M2 macrophage.
| Subgroups | Upstream activators | Functions |
|---|---|---|
| M2a | IL-4, IL-13 | Anti-inflammatory and tissue repair |
| M2b | IL-1β, TLR Ligands | Th2 activation and regulation of the immune response |
| M2c | IL-10, TGF-β, Glucocorticoids | Phagocytosis and immunosuppression |
| M2d | TLR Ligands, A2R agonists | Pro-tumor and angiogenesis |
Compared to the classic dual classification of macrophages, TAMs display greater phenotypic and functional diversity. In many cases, TAMs are considered as M2-like macrophages due to their similarities to M2 macrophage properties, such as high expression of ARG1, VEGF, CD206, CD204, and low expression of MHC-II [43]. The polarization of TAMs into M2-like phenotype can be induced by tumor-derived lactic acid, mediated by hypoxia-inducible factor 1α (HIF-1α) [44]. In addition, the high acidification of the TME caused by lactic acid accumulation, leads to the G protein-coupled receptor (GPCR)-dependent expression of the transcriptional repressor ICER in TAMs, promoting polarization of TAMs towards an M2-like phenotype and facilitating tumor growth [45]. However, studies also provide evidence suggesting that TAMs are a mixed population of cells expressing both M1 and M2 markers [46-48]. In the early stage of human lung cancer, a mixture of classical tissue monocytes and TAMs was observed with co-expression of M1/M2 markers, as well as T cell coinhibitory and costimulatory receptors [49]. These results indicate the complexity of TAMs and the limitation of classic M1/M2 classification.
Advances in single cell omics and mass cytometry by time-of-flight (CyTOF) technologies have provided new approaches to analyze TAM states in more detail. scRNA-seq studies have been conducted in various cancers, including breast cancer, NSCLC, small-cell lung cancer, hepatocellular carcinoma (HCC), glioblastoma, colorectal cancer (CRC), renal cell carcinoma (RCC), and pan-cancer analysis [50-57]. These single cell studies have dissected TAMs into multiple distinct clusters based on transcriptomic profiles, which may have different functions in tumor progression. For example, MMP12-expressing TAMs in NSCLC, which do not resemble either M1 or M2 cells, are most strongly associated with a poor clinical outcome [58]; a high abundance of secreted phosphoprotein 1 (SPP1)-expressing TAMs is correlated with worse outcome in NSCLC, CRC, and pancreatic ductal adenocarcinoma (PDAC) [58]; inhibiting APOC1 promotes transformation of M2 macrophages into M1 phenotypic macrophage through the ferroptosis pathway, which reshapes the TME and improves anti-PD1 immunotherapy in HCC patients [59]; integrated analysis of bulk RNA and single-cell RNA sequencing databases reveals Complete Component 1q (C1Q) + TAMs as one major anti-tumor immune cell population in osteosarcoma patients [60]. In addition, macrophage subsets are found to show heterogeneous transcriptomic patterns among distinct tumor types with several tumor-enriched macrophage subsets were found: the ISG15+ TAMs upregulated multiple interferon-inducible genes, the SPP1+ TAMs and C1QC+ TAMs resembled dichotomous functional phenotypes of TAMs in CRC, LYVE1+ macrophages and NLRP3+ macrophages were preferentially enriched in non-cancer tissues and likely represented as pro-inflammatory TRMs clusters [21]. Similar to previous studies, a single-cell trajectory analysis of macrophages in gastric cancer reveals the existence of two distinct cell states: a proinflammatory "M1-like" state characterized by high CD163 and S100A12 expression, and an "M2-like" state of TAMs with elevated CD163 and FOLR2 expression [61]. Further research is needed to identify the phenotypic and functional similarities and the difference between TAM clusters in distinct cancers, in different stages of tumor progression, and in primary and metastatic cancers.
Besides, it is largely unknown how the spatial localization of TAMs within the tumor connects to phenotype and function of TAMs. The development of spatial transcriptomics tools also provides information on spatial distribution information of TAMs, adding a new dimension to our understanding of TAM function in different contexts of cancer. Spatial transcriptomics of TAMs infiltration in NSCLC reveals that TAMs enrichment in the TME is relevant to tumor cell resistance to ICB immunotherapy regardless of its PD-L1 status, which is mediated by CD27, ITGAM, and CCL5 gene expression upregulation within tumor compartment [62]. Spatial and single-cell analysis of human normal and cancer colorectal tissues elucidate co-localization of cancer cell with SPP1 + TAMs at the invasive front of tumor, where CRC cell secrets human leukocyte antigen G (HLA-G) to transform TAMs into macrophages with immunosuppressive feature and reduces cytotoxicity of ICB immunotherapy [63]. Likewise, the progress of these cutting-edge technologies will bring new insights and guide the research on the new cancer therapy methods by targeting the unique population of TAMs.
2.3 The heterogeneity of TAMs
Due to the multifaceted roles of macrophages in tissue homeostasis and tumor surveillance, the differentiation, activation, and regulation of macrophages within the microenvironment have become major research focuses. Currently, there are two main strategies that dominate the research on macrophages (Figure 2). The first involves using single-cell sequencing (scRNA-seq), a powerful tool to dissect the tumor heterogeneity [64], to categorize macrophages in normal or tumor tissues and functionally annotate the gene expression within each cluster. Building on this, in-depth functional studies are conducted using macrophage-specific genetically modified mice. This includes techniques like knocking out or knocking in specific genes in macrophages, followed by histological examination and functional analysis. Additionally, tumor transplantation models can be constructed on the basis of genetically modified mice to further investigate the impact of specific gene-regulated macrophage functions on tumor progression.
The multifaceted roles of macrophages and the approaches for functional study of macrophages. Created in BioRender. Zhao, H. (2025) https://BioRender.com/t36o292.
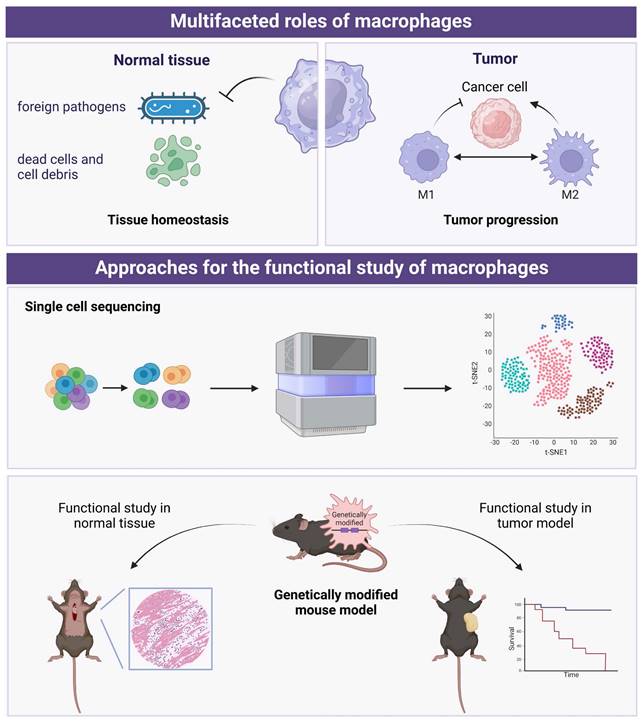
Recent scRNA-seq studies have shown that the traditional categorization of macrophages into M1 and M2 phenotypes is not as clear-cut as previously thought [65]. While M1 macrophages are generally associated with pro-inflammatory responses and M2 macrophages with anti-inflammatory responses, scRNA-seq analyses have revealed a more complex landscape of macrophage subpopulations. In a recent study, an extensive analysis of scRNA-seq data from myeloid cells in 380 samples spanning 15 different cancer types was conducted [21]. This analysis integrated newly collected data with eight previously published datasets, providing a comprehensive and expansive view of TAMs. By comparing monocytes and macrophages across multiple cancer types, the study consistently identified two distinct subsets of tumor-infiltrating monocytes (TIMs): CD14+ and CD16+ TIMs. Additionally, a subset of LYVE1+ interstitial macrophages were observed in non-cancerous tissues. Furthermore, the analysis revealed seven distinct clusters of TAMs, each characterized by specific marker gene expression patterns. These TAM clusters included INHBA+ TAMs, C1QC+ TAMs, ISG15+ TAMs, LNRP3+ TAMs, LYVE1+ TAMs, and SPP1+ TAMs. These findings shed light on the heterogeneity of TAM populations across various cancer types and non-cancerous tissues. This comprehensive scRNA-seq analysis provides valuable information for understanding the roles and potential therapeutic targets of TAMs in cancer progression and treatment response. In future studies, combined with single-cell sequencing data, new computational methods, such as unsupervised clustering approaches [66], can be considered to identify potential new subtypes of macrophages.
3. Macrophages in carcinogenesis and cancer immunity
Macrophages exert dual effects in carcinogenesis, with some promoting while others suppressing tumor growth [67, 68]. M1-like macrophages execute anti-tumor function by killing the tumor cells through cytotoxic activity directly, attacking cancer cells by cooperation with T cells through antigen present, or secreting cytokines to suppress tumor growth. However, most TAMs promote tumor growth and metastasis by secreting various factors and interacting with other cells in TME, leading to poor prognosis in multiple cancers including breast, cervix, bladder, brain, and prostate cancer [69-73]. Furthermore, TME converts M1-like macrophages to M2-like macrophages, which plays an important role in the development and progression of tumors. As discussed above, given the high plasticity and diversity of TAMs, it is crucial to fully understand the properties and functions of transcriptomic unique and spatial unique TAM clusters in regulating tumor initiation and development. Herein, we discuss the roles of TAMs in tumor cell proliferation, invasion, and metastasis, stimulating angiogenesis, tumor immunoevasion, and therapeutic resistance (Figure 3).
The role of macrophages in cancer development and therapy. (A) Proliferation; (B) Invasion and metastasis; (C) Angiogenesis; (D) Tumor immunity; (E) Therapeutic resistance.
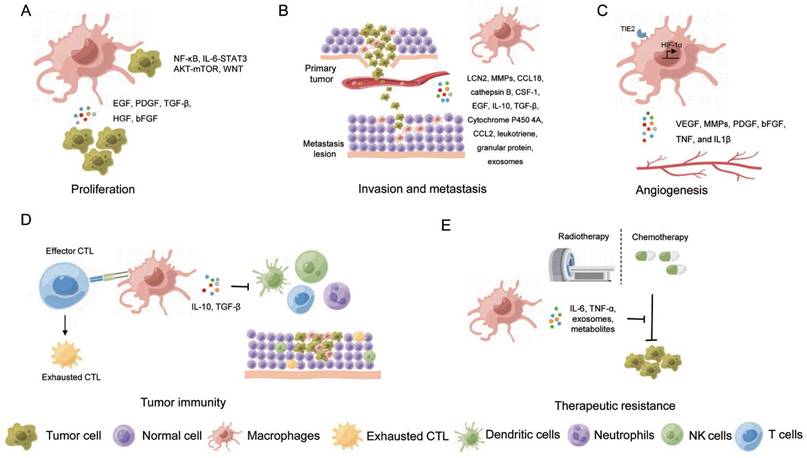
3.1 Anti-tumorigenic effects of TAMs
Macrophages are reported as the main phagocytic population within TME. By distinguishing cancer cells from normal cells, M1 type macrophages can directly engulf cancer cells by phagocytosis activity and indirectly eliminate tumor cells by inducing cancer cell death through secreting some molecules including ROS and NO or by activating other immune cells such as T cells and nature killer (NK) cells [74]. The potential tumor-suppressive role of TAMs has been studied in various tumor contexts. For instance, high infiltration of CD68+ TAMs has been associated with improved survival in colon, gastric, and endometrial cancer patients [75-77]. In a mouse model of CRC metastasis, depletion of Kupffer cells (TRMs in the liver) resulted in increased liver metastasis of CRC cells, suggesting an inhibitory function of macrophages in liver metastasis [78]. In melanoma, CD169+ macrophages have been shown to inhibit tumor growth by blocking the dissemination of tumor-derived extracellular vesicles [79]. In the single-cell analysis of TAMs, some M1-like TAM subsets and other newly identified TAM populations are correlated with better prognosis, providing further evidence for the existence of an anti-tumorigenic portion of TAMs within TME [80]. However, cancer cells have evolved mechanisms to escape uptake by TAMs with the expression of “don't eat me” signal genes such as CD47 and CD24, which disrupt the phagocytosis, and blocking CD47 or CD24 by antibodies can re-activate the macrophage mediated phagocytosis of tumor cells [81, 82].
Furthermore, M1-like TAMs can induce ferroptosis, an intracellular iron-dependent form of cell death, in cancer cells through various mechanisms. These include the release of proinflammatory cytokines, providing peroxides to trigger Fenton reactions, and activating CD8+ CTLs, with the latter being considered a major contributor to initiating ferroptosis in cancer cells [83, 84]. The activated CD8+ CTLs produce IFN-γ, which activates JAK/STAT1 pathway and downregulates the transcription of SLC3A2 and SLC7A11, two subunits of the glutamate-cysteine antiporter system xc- that involved in ferroptosis [85]. This action disables the GSH-dependent antioxidant system and consequently promotes tumor cell excessive lipid peroxidation and ferroptosis [85]. Additionally, during the respiratory burst, M1-like TAMs can release peroxides (H2O2) to trigger intracellular Fenton reaction and generate excessive ROS, therefore promoting tumor cell ferroptosis [86, 87]. Interestingly, ferroptosis products of dying cancer cell contrarily promotes TAMs switch into an M2-like pro-tumor phenotype via STAT3-dependent fatty acid oxidation and accelerates pancreatic adenocarcinomas [88], which suggests the crafty characteristics of tumors and the complicated crosstalk between TAMs and cancer cells.
Emerging evidence from scRNA-seq studies has shed light on the discovery of novel macrophage subtypes exhibiting remarkable potential in antitumor activities. One notable investigation found that the presence of CD74+ macrophages in hepatocellular carcinomas was strongly associated with improved prognosis and activation of immune response pathways [89]. Another study made a significant observation uncovering the role of LC3-associated phagocytosis, a distinct process from conventional autophagy, in driving TAMs to exert control over tumor growth [90]. This unique mechanism relies on the participation of tumor-infiltrating T cells and is dependent on the coordinated activation of stimulator of interferon response CGAMP Interactor 1 (STING) and type I interferon responses. In the context of breast cancer, single-cell studies have revealed the presence of a distinct population of folate receptor 2+ (FOLR2+) macrophages residing in the perivascular regions of the tumor stroma [91]. These macrophages engage in interactions with CD8+ T cells and demonstrate a remarkable ability to efficiently prime effector CD8+ T cells. Notably, a higher density of FOLR2+ macrophages within tumors is associated with improved patient survival, highlighting their potential as prognostic markers and their role in facilitating anti-tumor immune responses. In addition to these findings, recent research has highlighted the potential of targeting monoamine oxidase A (MAO-A) to modulate the polarization of TAMs [92]. MAO-A, an enzyme located in the mitochondrial membrane, has emerged as a promising therapeutic target due to its involvement in TAM function. Notably, compelling results have been observed in a preclinical study utilizing the B16 melanoma mouse model, in which the pharmacological inhibition of MAO-A enzymatic activity with commercially available inhibitors, commonly prescribed for neurological disorders, demonstrated significant efficacy. This inhibition of MAO-A activity resulted in a remarkable reduction in regulatory TAMs (Reg-TAMs) and a concomitant expansion of TAM subsets characterized by a proinflammatory signature.
3.2 TAMs promote carcinogenesis
Rather than exerting an anti-tumorigenic function, TAMs are broadly involved in tumor progression. TAMs collaborate with other immune cells and stromal cells, collectively constructing a special microenvironment for cancerous growth. Meanwhile, TAMs foster cancer progression by interacting with TME or by secreting growth factors such as epithelial growth factor (EGF), platelet-derived growth factor (PDGF), TGF-β, hepatocyte growth factor (HGF), basic fibroblast growth factor (bFGF) that stimulate tumor proliferation [93]. For example, in HCC, TAMs induced liver inflammation and subsequent carcinogenesis by releasing IL-6, IL-1β, TNF, HGF, CCL2, and other factors [94]. In human endometrial carcinoma, chemokine (C-X-C motif) ligand 8 (CXCL8) secreted by TAMs promoted tumor progression by suppressing the expression of estrogen receptors via homeobox B13 (HOXB13) [95]. In PDAC, IL-1β released by TAMs suppressed the expression of 15-hydroxyprostaglandin dehydrogenase (15-PGDH), an enzyme inversely associated with tumor advancement, presence of lymph node metastasis and nerve invasion, and poor prognosis of patients [96]. Increased colony-stimulating factors (CSFs) produced by TAMs has also been observed to be related to cancer development across a range of malignancies, including liver cancer, breast cancer, RCC, Hodgkin lymphoma, and ovarian cancer [97].
Furthermore, interactions between cancer stem cells and TAMs are reported to promote tumorigenesis as well [98]. For example, TAMs promoted stem cell-like properties of cancer cells by activating the nuclear transcription factor-κB (NFκB) pathway in colon cancer and breast cancer, the IL-6-STAT3 (Signal transducer and activator of transcription 3) pathway in HCC and the AKT-mTOR pathway in RCC [99]. Even in 3D engineered microenvironments, TAMs intensified the stem-like properties and malignant phenotypes of ovarian cancer cells through the WNT pathway [100].
In addition to the effects of TAMs on cancer cells, the latter reciprocally polarize TAMs towards states in favor of tumor progression by producing cytokines, chemokines, and metabolites. For instance, substances such as succinate, histamine, CSF1, E3 ubiquitin protein ligase COP1, and β-glucosylceramide released by cancer cells can modulate metabolic state and induce ER stress of TAMs, thereby escalating the generation of pro-tumor TAMs [58, 101-105]. In glioblastoma, periostin secreted by tumor stem cells recruited monocyte-derived macrophages from peripheral blood and polarized them into M2-like TAMs to promote tumorigenesis [106]. Overall, the mechanisms underlying TAMs promoting tumorigenesis are extremely diverse and display context dependency.
In the context of HCC, a specific subset of M2 macrophages characterized by high expression of C-C motif chemokine ligand 18 (CCL18) and the transcription factor CAMP responsive element modulator (CREM) has been identified through scRNA-seq [107]. These M2 macrophages are believed to play pivotal roles in tumor progression. Understanding the association between M2 macrophages, CCL18 expression, and CREM provides valuable insights into the underlying mechanisms driving HCC development and paves the way for targeted interventions to combat this aggressive cancer. In another notable study, a tumor-specific macrophage subpopulation marked by the upregulation of triggering receptor expressed on myeloid cells 2 (TREM2)/apolipoprotein E (APOE)/complement C1q (C1Q) markers has been discovered and validated using advanced imaging techniques [108]. This subset of macrophages demonstrated a significant enrichment in tumors from patients who experienced recurrence following surgery, specifically in clear cell renal cell carcinoma (ccRCC). The identification of these TREM2/APOE/C1Q-positive macrophages not only offers a potential prognostic biomarker for ccRCC recurrence but also presents a promising target for therapeutic strategies aimed at preventing tumor relapse. Collectively, these studies shed light on the diverse, context-dependent roles of macrophages in the tumor microenvironment and their considerable impact on cancer progression.
3.3 TAMs enhance tumor metastasis
In the theatre of oncology, studies have revealed the critical roles of TAMs in stimulating tumor invasion and metastasis. TAMs can orchestrate a hostile breakout, releasing an arsenal of weapons including matrix metalloproteinases (MMPs), cathepsin, urokinase, protease, and matrix remodeling enzymes. These molecular saboteurs disrupt cell-cell and cell-matrix junction, enabling the escape and invasion of cancer cells [109-113]. The plot thickens when sphingosine-1-phosphate (S1P), released by apoptotic tumor cells, stimulated TAMs to secrete lipocalin-2 (LCN2), further propelling tumor metastasis [114]. Similarly, in a RCC model undergoing IL-2/anti-CD40 immunotherapy, macrophage-dependent NO in the tumor microenvironment was essential to regulate the activity of MMPs and the expression of adhesion molecules, which was the basis for metastasis [115]. TAMs are also capable of igniting cancer cell invasions in other ways. TAMs-derived CCL18 activated the interaction between integrin and receptor membrane-associated phosphatidylinositol transfer protein 3 (PITPNM3) to promote the metastasis of breast cancer [116]. TAM-produced cathepsin B has also been shown to enhance breast cancer cell invasion in a lung-metastasis model. The consumption of glucose fuels enhances hexosamine biosynthetic pathway (HBP) and O-GlcNAcylation of cathepsin B in TAMs, which supported cancer metastasis [117]. The positive feedback between tumor cells and TAMs triggered tumor cells to secrete CSF-1, stimulating TAMs to secrete epidermal growth factor (EGF), which also accelerated tumor invasion and metastasis by destroying the matrix [118-120]. Additionally, TAMs have a hand in regulating epithelial-mesenchymal transformation (EMT), a well-known bioprocess intrinsically linked with tumor metastasis. In pancreatic cancer, for example, TAMs are revealed to bolster EMT and foster cancer metastasis by reducing the expression of E-cadherin via activating the TLR4/IL-10 signaling pathway [121, 122]. In another study, TAMs were demonstrated to promote EMT and metastasis of liver cancer and CRC through secreting TGF-β [123].
In addition to the assistance in metastasis at primary sites, studies have shown that TAMs play a crucial role in preparing a favorable landing strip for migratory cancer cells, assisting these rogue cells to seed in distal tissues or organs. For instance, cytochrome P450 4A released by TAMs, fostered the formation of pre-metastasis niche and the trend of M1 polarization [124]. In lung metastasis models of breast cancer and melanoma, monocytes were recruited by CCL2 produced by cancer cells to differentiate into macrophages, creating a pre-metastatic niche for tumor cells [125, 126]. In a liver metastasis model of PDAC, TAMs derived inflammatory monocytes, are able to secrete granular protein to transform the resident hepatic stellate cells into myofibroblasts to support cancer cell implantation [127]. Interestingly, TRMs can also facilitate cancer metastasis into the tissue, due to their anti-inflammatory property. For example, Alveolar macrophages, TRMs in the lung, promoted lung metastasis of HCC and breast cancer by secreting leukotriene and suppressing the Th1 response [128]. Peritoneal TRMs promoted ovarian cancer metastasis into the peritoneal cavity by driving the spheroid formation [129]. Besides, TAMs are affected by exosomes produced by tumor cells, for example, exosome-educated macrophages boosted liver metastasis of pancreatic cancer through TGF-β secretion [130].
Recent advances in single-cell research have provided invaluable insights into tumor microenvironment modulation and the intricate relationship between macrophages and cancer cell invasion. These studies not only highlight the impact of macrophages on cancer cell behavior but also spotlight their potential as therapeutic targets. For instance, a lung cancer study revealed that the depletion of tissue-resident macrophages led to significant changes in the tumor microenvironment, curbing tumor invasiveness and growth [36]. These alterations included a decrease in regulatory T cell numbers and a shift in their phenotype, accompanied by an accumulation of CD8+ T cells. Furthermore, the relocation of tissue-resident macrophages from the tumor core to the periphery during tumor progression indicated their dynamic role in lung cancer development. Another study focused on glioblastoma demonstrated the ability of macrophages to induce a transition of glioblastoma cells into mesenchymal-like states [131]. This transition was mediated by the secretion of oncostatin M by macrophages, which interacted with its receptors on glioblastoma cells, activating the signal transducer and activator of transcription 3 (STAT3) signaling pathway. Importantly, the acquisition of mesenchymal-like states in glioblastoma cells was associated with increased expression of a mesenchymal program in macrophages and enhanced cytotoxicity of T cells. These findings highlight the extensive alterations in the immune microenvironment orchestrated by macrophages and underscore their potential therapeutic implications.
3.4 TAMs enhance angiogenesis
In solid tumors, TME is typically characterized by a state of hypoxia, an essential element for tumor angiogenesis, which is recognized as one of the hallmarks of cancer [132]. A wealth of research has evidenced that hypoxic TME steers the recruited macrophages towards an M2-like state, inciting TAMs to unleash pro-angiogenic factors such as vascular endothelial growth factor (VEGF), HIF, MMP, PDGF, bFGF, TNF, and IL-1β [133, 134]. For example, in breast and colon cancer, TAMs were positively correlated with VEGF level and microvascular density [135, 136]. TAM-induced MMP9 has been found to be a strong ally in promoting tumor angiogenesis in ovarian cancer, while TAM-derived thymine phosphorylase (TP) has been implicated in fostering tumor angiogenesis in gastric cancer [137, 138]. Another piece of this intricate puzzle is the role of WNT7b, produced by TAMs, which ratchets up the expression of VEGF-A in vascular endothelial cells, thereby enhancing angiogenesis in breast cancer [139]. The significance of HIF expression in TAMs can also not be overstated for tumor angiogenesis, as supported by the observation that knockout of HIF-1α in TAMs resulted in curtailed angiogenesis and a reduction in tumor burden in breast cancer. Additionally, TIE2-expressing monocytes, a particular breed of TAM existed both in human peripheral blood and tumors, has been noted to fuel tumor angiogenesis and tumor growth in endometriosis lesions, pancreatic cancer, and ovarian cancer [140, 141].
3.5 TAMs in tumor immunity
In addition to their direct effects on cancer cells, TAMs function as pro-tumorigenic cells by attenuating cancer immunity to construct an immunosuppressive microenvironment for cancer cell growth in several ways [142, 143]. As phagocytes, TAMs compete with dendritic cells and degrade tumor-associated antigens (TAA) in TME [144]. Meanwhile, antigen presentation activity is abnormal in TAMs, resulting in inhibition of adaptive immune response and thereby facilitating tumor immune evasion. This is evident where the transcription factor IRF8 is required for cancer cell antigen presentation by monocyte-derived TAMs, which was essential for promoting cytotoxic T lymphocyte (CTL) exhaustion within the tumor. Notably, TAM-specific IRF8 deletion prevented exhaustion of cancer cell-reactive CTLs and suppressed tumor growth [145].
TAMs also impede the anti-tumor activity of tumor-infiltrating natural killer cells and T cells and synergize with myeloid-derived suppressor cells (MDSCs), tumor-associated dendritic cells, and neutrophils to foster an immunosuppressive tumor microenvironment [142, 144]. T cell immunity is suppressed by TAMs as evidenced by the unleashed T cell response upon TAM blockage in several cancers [146, 147]. Recruitment of CD8+ T cells was blocked by TAMs in TME of HCC through the inhibition of CXCL9 and CXCL10, meanwhile CD8+ T cell activation and proliferation were attenuated through regulating L-arginine by ARG1 and inducible NO synthase (iNOS) in lung cancer and lymphoma [44, 148]. TAM-secreted cytokines including IL-10 and TGF-β, inhibited T cell proliferation and differentiation and promoted T cell exhaustion [149, 150].
Furthermore, TAMs contribute to the blockade of cytotoxic activities in T cells, natural killer T cells, and natural killer cells on account of the high expression of immune checkpoint ligands on TAMs such as programmed cell death protein ligand 1 (PD-L1), programmed cell death protein 1 (PD-1), B7-H4, and cytotoxic T-lymphocyte-associated protein 4 (CTLA4), which leads to reinforced tumor growth [147, 151-153]. For example, in mouse models of colon cancer and breast cancer, M2-like TAMs expressed high levels of PD-1, which not only reduced the anti-tumor function of T cells but also inhibited the phagocytosis of macrophages and promoted the growth of tumors [154]. Additionally, a wealth of data reveals that TAMs can also mediate T cell depletion in TME. The activated antigen-specific Fas+CD8+ T cells undergo apoptosis following their interaction with FasL+CD11b+F4/80+ monocyte-derived macrophages within the liver, which systemically depleted the peripheral T cell numbers and diminished tumoral T cell diversity and function by siphoning activated CD8+ T cells from circulation [18]. Importantly, TAMs and M-MDSCs, but not cancer cells, consumed the most glucose per cell in TME and maintain robust glucose metabolism [155], implying that TAMs could trigger T cell death by glucose deprivation and lactate production [156]. Taken together, all these results highlight macrophage as a central player of the immunosuppressive TME through regulating the recruitment and the function of multiple immune subtypes.
3.6 TAMs in therapeutic resistance
Growing studies have indicated the significant roles of TAMs in chemo- or radio-resistance. Generally, TAMs contribute to therapeutic resistance either by promoting cell survival and cancer cell stemness or by shielding cancer cells from death. For example, TAMs have been reported to activate the STAT3 signaling pathway in cancer cells by producing cytokines such as IL-6 and TNF-α, which enhanced the resistance to chemotherapy in various cancer cells [157, 158]. It was also shown that TAMs can secret exosomes containing microRNAs and metabolites that are implicated in chemotherapy resistance in different type of cancers including ovarian cancer, gastric cancer, and PDAC [130, 159, 160]. In addition, blockage of TAMs or certain factors secreted by TAMs has shown to improve the radiotherapy sensitivity in head and neck cancer as well as breast cancer [161, 162].
4. TAM-targeted cancer therapy
Conventional cancer treatments, including surgical resection and kinase inhibitors, frequently encounter challenges such as tumor relapses and drug resistance [163, 164]. This underscores the urgent need to develop novel therapeutic strategies for more effective cancer therapy. Given the importance of TAMs in tumor progression and immune response, targeting TAMs as a potential cancer therapeutic strategy has aroused great interests. Numerous approaches are either being developed or are under active research for different types of cancer, with many clinical trials currently underway. These strategies are designed either through inhibiting the pro-tumorigenic function or boosting the anti-tumorigenic capabilities of TAMs by manipulating the mass, the states, and the activity of TAMs. This discussion will center on the current macrophage-targeting therapies, that can be broadly divided as follows: inhibition of monocyte/macrophage recruitment, depletion of macrophages, reprogramming and engineering of TAMs, and other therapies (Figure 4).
TAM-targeted cancer therapy. (A) Inhibition of TAMs differentiation; (B) Elimination of TAMs; (C) Reprogramming and engineering of TAMs; (D) Blocking phagocytotic checkpoints; (E) Application of trained macrophage.
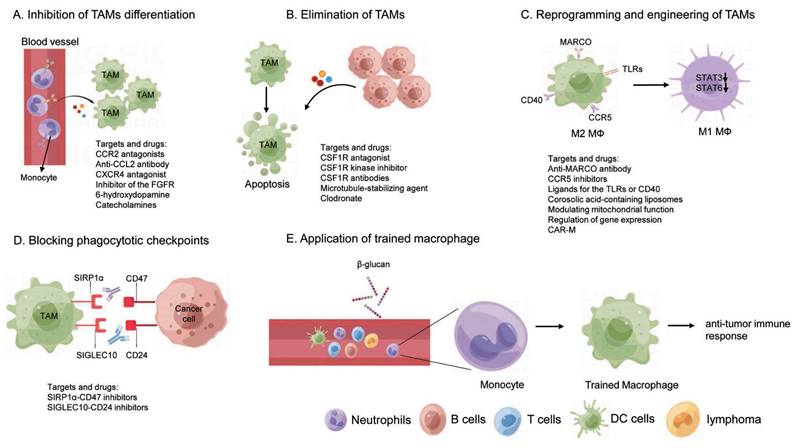
4.1 Inhibition of monocyte/macrophage recruitment
The strategy of inhibiting the recruitment of monocyte into tumor tissue holds promise, as TAMs are predominantly derived from circulating monocyte precursors. Chemokine ligand 2 (CCL2) is essential for the recruitment and localization of monocytes into tumors [165], making the targeting CCL2 and its receptors CCR2 a viable method for curtailing monocyte infiltration and TAM production. In preclinical models, blocking CCL2-CCR2 signaling by genetic approach or small molecular inhibitors resulted in reduced tumor growth and metastasis and improved the efficacies of chemotherapy, radiation therapy, and immunotherapy in HCC [166-168]. In a mouse model of pancreatic cancer, CCR2 antagonists blocked the mobilization of CCR2-positive monocytes from bone marrow into tumors, thereby limiting TAM production and curbing tumor growth and metastasis [169]. Carlumab, an anti-CCL2 monoclonal antibody, has demonstrated promising results in preventing the development of certain cancers in mouse models [170]. Several small molecular inhibitors and antibodies aimed to disrupt the CCL2-CCR2 axis are under clinical trial. The CXCL12-CXCR4 pathway is another potential target to decrease TAM recruitment, the blockade of which mobilized CD8+ T cells to the tumor and reduced TAM accumulation in multiple cancers [171-173]. Meanwhile, a peptide antagonist of CXCR4, named as motixafortide, is currently under teste in ongoing clinical trials [174].
Other molecules such MAC-1 (CD11b/CD18) and fibroblast growth factor receptor (FGFR) have also been reported as potential targets. For example, inhibition of MAC-1 has been shown to enhance tumor response to radiation therapy by reducing myeloid cell recruitment, consequently attenuating squamous cell carcinoma growth [175]. Likewise, AZD4547, an inhibitor of the FGFR tyrosine kinase family, has been observed to block the FGFR in a lung adenocarcinoma mouse model, resulting in robust TAM elimination and tumor regression, rendering this receptor a potential therapeutic target [176]. The potential of targeting 6-hydroxydopamine catecholamines, CSF-1R, and CD88 for cancer therapy in lung cancer and colon cancer as well [28, 177, 178].
4.2 Reduction and clearance of TAM
4.2.1 Inhibition of TAMs differentiation
As discussed above, CSF1R is the key factor for TAM survival and proliferation and is highly expressed across all TAM states. This makes the interruption of the CSF1-CSF1R axis a promising method to reduce TAMs.
Firstly, the inhibition of CSF1-CSF1R signaling has resulted in substantial cell apoptosis of TAMs and improvement in T cell response in many tumor models [179-181]. The small molecule CSF1R antagonist named PLX3397 (Pexidartinib), has been found to penetrate the blood-brain barrier and significantly reduce the amount of tumor-associated microglia, thereby preventing tumor invasion in a glioblastoma mouse model [182]. In murine breast-to-brain metastasis models, the combination of BLZ945, an inhibitor of CSF1R, and AC4-130, an inhibitor of CSF2Rb-STAT5 signaling, has proven effective in controlling tumor growth, normalizing of microglia activation states, and mitigating neuronal damage [183]. For advanced ovarian cancer patients, GW2580, a CSF1R kinase inhibitor, has been reported to inhibit macrophage function, reduce M2 macrophage infiltration, and significantly decrease the number of ascites [184]. In addition to compounds, CSF-1R antibodies (such as Emactuzumab) are also developed to block the CSF1-CSF1R pathway and have proved its efficacy in diffuse-type giant cancer cells [185, 186]. Secondly, the efficacy of chemotherapy or ICB has been found to be improved when applied to block the CSF1-CSF1R axis. For example, docetaxel (microtubule-stabilizing agent) coupled with anti-CSF1R led to TAM depletion in a murine epithelial ovarian cancer model [187]. Finally, many ongoing CSF1-CSF1R targeting trials are evaluating their anti-tumor efficacy either alone or in combination with other drugs such as chemotherapy agents or immune checkpoint inhibitors.
4.2.2 Elimination of TAMs
Macrophages always undergo transcriptionally and epigenetically remodeling to adapt to the local microenvironment. Targeting the intrinsic regulators of TAMs provides a specific way to deplete the tissue specific TAMs without defects induced by general depletion of monocytes/macrophages. In peritoneal cavity, for example, transcription factor GATA6 is critical for the peritoneal macrophage differentiation and maintenance [188, 189]. The depletion of GATA6 in peritoneal TRMs induces cell apoptosis and number loss, indicating that targeting GATA6 can be used to eliminate peritoneal TAMs. Retinoid X receptors (RXRs) determine the identity of peritoneal TRMs by regulating the chromatin accessibility of GATA6. RXRs deficiency impairs neonatal expansion of the large peritoneal macrophages (LPMs) pool and reduces the survival of adult LPMs through excessive lipid accumulation. Depletion of RXR diminished LPMs accumulation in ovarian cancer and strongly inhibits tumor progression in mice [190].
Novel artificial materials have been developed to eliminate the TAMs as well. For example, trabectedin and lurbinectedin could reverse the immunosuppression effect of TAMs through depleting macrophages in the TME. However, these two chemicals inevitably caused side effects due to unselectively macrophage consumption, potentially disturbing immune homeostasis [191]. The clodronate liposome, a non-nitrogen bisphosphonates which elicits toxic effects on macrophages via phagocytosis, has been used to deplete TAMs in vivo, resulting in reduced tumor growth in PDAC [192] and ovarian cancer metastasis [16]. Depletion of TAMs with clodronate has also been shown to prevent aerobic glycolysis and tumor hypoxia, improving tumor response to chemotherapy [193]. Moreover, as a result of TAM depletion, PD-L1 expression, as well as T-cell infiltration, is significantly increased in aerobic cancer cells, which dramatically promoted the antitumor efficacy of PD-L1 antibodies [193]. Zoledronate, a third-generation nitrogen-containing bisphosphonate, has been shown to exhibit selective cytotoxicity towards TAMs, impairing differentiation of monocytes into TAMs and to reducing the infiltration of TAMs, which finally resulted in decreased tumor angiogenesis and inhibited tumor progression [194].
Furthermore, therapies using Fc domain enhanced anti-TREM2 monoclonal antibody have been developed to promote anti-tumor immunity by eliminating and modulating TAM populations, which leads to enhanced CD8+ TIL infiltration and effector function [195]. In addition, chimeric antigen receptor (CAR) T cells, genetically modified to express receptors that recognize TAMs-specific antigens, are designed to eliminate TAMs. In an ovarian cancer study, both mouse and human FRβ-specific CAR T cells recognized and depleted the FRβ+ TAMs, interrupting ovarian cancer metastasis [196].
4.3 Reprogramming of TAMs
Macrophages demonstrate a high degree of plasticity, enabling them to adapt to variable microenvironments. This adaptability paves the way for the reprogramming of TAMs into a tumoricidal phenotype, thereby restoring their anti-tumor effects [197]. The reprogramming of M2-like TAMs into M1-like TAMs within the TME has shown promising results. Several surface markers of TAM can be targeted to switch their phenotypes, such as the scavenger receptor MARCO, toll-like receptors (TLRs), CD40, or CCR5 [198-201].
In models of breast and colon carcinoma as well as melanoma, an anti-MARCO monoclonal antibody has been developed and has exhibited anti-tumor effects in some cases through reprogramming TAMs to pro-inflammatory phenotypes and enhancing tumor immune responses [198]. Similarly, CCR5 inhibitors such as maraviroc, vicriviroc, TAK-779, and anibamine have shown anti-tumor effects in mouse model of multiple cancers and are tested clinically in breast cancer, colon cancer and PDAC [202]. In addition, specific ligands for the TLRs or CD40 have also been identified to activate M1 macrophages. The TLR7 agonist imiquimod has been approved by the FDA for topical treatment of superficial basal cell carcinoma [203]. TLR3 agonist poly-ICLC, which activates the NFκB pathway and anti-tumor immunity, is under clinical test for glioma [204]. Paclitaxel decreases tumor growth by reprogramming TAMs to an M1 subtype in a TLR4-dependent manner [205]. Anti-CD40 antibodies have shown significant anti-tumor activity as single agents in several preclinical models including PDAC and breast cancer [206-208]. Combined administration of monophosphoryl lipid A (MPLA) and IFN-γ stimulates type I IFN signaling in breast cancer, which reprogramed CD206+ TAMs to iNOS+ TAMs, resulting in cytotoxic T cell activation through macrophage-secreted IL-12 and TNF-α, finally reduction of primary tumor growth and metastasis [209].
Specific pathways involving anti-inflammatory responses can also be modified to reshape TAMs. For example, by specifically targeting STAT3 through CD163-targeted corosolic acid-containing liposomes, M1-like TAMs were reprogrammed, resulting in a decrease in IL-10 expression and increase in pro-inflammatory TNF-α [210]. Similarly, it has been shown that several synthetic molecules (AS1517499, TMC-264, A771726) inhibited STAT6, one of the major signal transducers activated by IL-13 and involved in M2 polarization, leading to inhibited TAMs transformation and tumor progression in a mouse model of breast cancer [211]. Furthermore, inhibiting STAT6 transcriptional activity by enhancing STAT6 acetylation suppresses TAMs M2-like polarization, reshapes TME into a tumor-suppressive state, and represses tumor progression in melanoma [212]. PI3Kγ, a key macrophage lipid kinase, selectively drives immunosuppressive transcriptional programming in macrophages which promotes tumor immune invasion [213, 214]. PI3Kγ signaling in TAMs inhibits NFκB activation and stimulates CCAAT/enhancer binding protein (C/EBP)-β activation through AKT and mammalian target of rapamycin (mTOR), thereby induces a transcriptional program of immunosuppression [213]. Genetic depletion of Pik3cg or selective pharmacologic targeting of PI3Kγ by IPI-549 reprogramed TAMs, reshaped the TME, and promoted CTL-mediated tumor regression [213-215].
A few other strategies are studied likewise to manipulate TAMs toward to M1-like states. Modulating macrophage mitochondrial function could be considered as an approach to activating TAMs reprogramming. Under hypoxia condition, nuclear-encoded mitochondrial pyruvate dehydrogenase beta gene expression is attenuated by promoting Nuclear Respiratory Factor 1 (NRF1) degradation, dampening hypoxia-mediated NRF1 degradation decreases the Warburg effect and promotes M1 polarization of TAM, promoting tumor cells to become more sensitive to apoptosis through a FADD-dependent manner [216]. Depletion of NF-κB effector molecule Gadd45b in myeloid cells recovered the activation of pro-inflammatory TAMs and increased intratumor immune infiltration, thereby diminishing HCC and ovarian cancer oncogenesis in mouse [217]. For NSCLC patients, disrupting Angptl2, a secreted inflammatory glycoprotein, may be an effective strategy to re-educate TAM polarization and reprogramming of M2-like TAMs to M1-like TAMs [218].
4.4 Blocking phagocytotic checkpoints
The therapeutic exploitation of innate immune clearance of dying cancer cells has emerged as an exciting new area of cancer immunotherapy. Similar to the immune checkpoints on T cells, several phagocytotic checkpoints on macrophages have been identified to modulate the tumor-associated antigens uptake, presentation, and degradation. Targeting these phagocytotic checkpoints is critical for tumor clearance and type I IFN immune response. Some cancer cells express “don't eat me” signal ligands such as CD47 and CD24, which can be recognized by TAM receptors such as SIPR1a (for CD47) and SIGLEC10 (for CD24), effectively blocking the attack from TAMs. Interrupting SIPR1α-CD47 or SIGLEC10-CD24 axis by CD47 or CD24 antibodies stimulated TAMs to phagocytose cancer cells and enhanced antitumor T cell responses in mouse models [81, 219, 220]. Furthermore, a phase I trial involving an anti-CD47 antibody Hu5F9-G4 demonstrated partial remissions in two patients with ovarian/fallopian tube cancers for 5.2 and 9.2 months [221]. As a general marker of embryonic-derived TRMs [222-224], T cell immunoglobulin and mucin domain-containing molecule-4 (TIM4) mediates the uptake of apoptotic cell by recognizing phosphatidylserine (PS). Interestingly, TIM4+ cavity TAMs sequester and impair CD8+ T cells proliferation through the recognition between TIM4 and PS, which is elevated on activated T cells. Hence, the TIM4 blockade abrogated this sequestration, restored T cell proliferation, and thus enhanced anti-tumor efficacy in models of anti-PD-1 therapy and adoptive T cell therapy in mice [19]. Additionally, TIM4-mediated uptake and degradation of dying tumor cells are important for the immune evasion via the canonical autophagy due to reduced antigen presentation [225]. Besides, TIM4 functions with LC3-associated phagocytosis (LAP) to promote immune tolerance and blockage of TIM4 with antibody releases the STING-mediated type I interferon responses in TAMs [90]. Consistently, blockade of phagocytic receptor MerTK with antibody also resulted in accumulation of apoptotic cells within tumors and triggered a type I interferon response which stimulated T cell activation and synergized with anti-PD-1 or anti-PD-L1 therapy [226].
4.5 Application of trained macrophage
The application of trained immunity in macrophages provides a potential strategy for cancer treatment. Traditionally, innate immunity has been understood to react rapidly and nonspecifically upon encountering a pathogen, without building up immunological memory akin to adaptive immunity. However, studies have shown that prototypical innate immune cells (such as monocytes, macrophages, or natural killer cells) have the potential for increased responsiveness upon secondary stimulation, a phenomenon termed “trained immunity” [227, 228]. Contrary to the stringent antigen/pathogen specificity of adaptive immunity, trained innate immune cells can trigger systemically enhanced immune responses to a variety of heterologous stimulants after primary stimulation [228, 229]. Capitalizing on this characteristic, trained immunity has been leveraged to disrupt the immunosuppressive TME and boost the systemic anti-tumor response via pre-stimulating the myeloid cells. For example, trained immunity induced by pre-treatment of mice with β-glucan, a fungal-derived prototypical agonist of trained immunity, has been associated with transcriptomic and epigenetic rewiring of granulopoiesis and neutrophil reprogramming toward an anti-tumor phenotype [230]. Meanwhile, β-glucan also attracts circulating monocyte/macrophages influx into the pancreas with features of trained immunity to exert anti-tumor functions [231]. Furthermore, the metabolite S1P mediated whole β-glucan particle (WGP) induced trained immunity in lung interstitial macrophages, leading to inhibition of tumor metastasis and prolonged survival in multiple mouse models of metastasis. Application of WGP-trained BM-derived macrophages through adoptive transfer reduced tumor lung metastasis [232]. Interestingly, a recent study also observed that acute respiratory viral infections induced trained immunity in lung tissue-resident alveolar macrophages. These macrophages are poised to exert long-lasting tissue-specific anti-tumor immune response [233], suggesting that trained immunity in macrophage can provide a reprogrammed and persistent activation of immune response. Consequently, a designed nano-therapy has been developed to specifically induce trained immunity with nanoparticle MTP10-HDL in a B16F10 mouse melanoma model to overcome the immunosuppressive tumor microenvironment and synergize with immune checkpoint inhibitors [234]. Therefore, creating and modulating the trained immunity in monocyte/macrophage should enhance the anti-tumor immune responses, which might be a novel and promising immunotherapy against advanced cancer and metastasis.
4.6 The potential of engineered CAR-macrophages in cancer therapy
Earlier research focused on macrophage functions and their anti-tumor properties, but recent studies have shifted toward utilizing macrophages directly as therapeutic tools (Figure 5). The laboratory methods to obtain macrophages involve isolating mononuclear cells or monocytes from bone marrow or peripheral blood, and then stimulating, amplifying, and differentiating them in vitro (e.g., with GM-CSF and IFN-γ). A recent study used induced pluripotent stem cells (iPSCs) to obtain macrophages after in-vitro differentiation [235]. Based on this, macrophages can be further armed with chimeric antigen receptors (CARs), adding a second signal within the macrophages. Similar to CAR-T cells, macrophages armed with CARs offers several benefits: firstly, CAR can precisely target and kill tumors by recognizing tumor-specific antigens on their surface; secondly, it can act as an antigen-presenting cell to prime and activate T cells; and thirdly, further genetic modification of macrophages may enhance their cytokine secretion capabilities, thereby improving their tumor-killing effectiveness.
The application of CAR-armed macrophages in cancer therapy. Created in BioRender. Zhao, H. (2025) https://BioRender.com/x76e687.
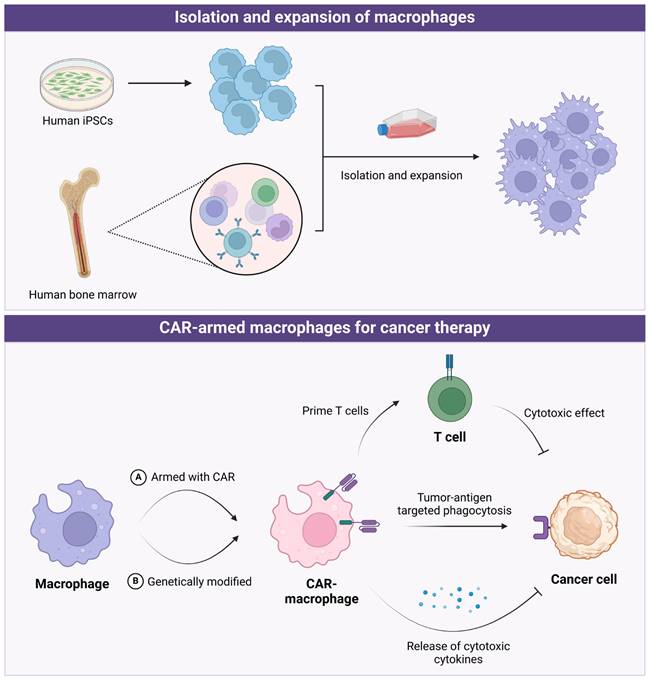
Based on the ability of macrophages to clear pathogens and antigens, engineered macrophages by modifying antigen receptors on macrophages have also been developed, known as CAR-M (chimeric antigen receptor macrophage) cells. Macrophages engineered with targeted CARs can enhance its antigen presentation and phagocytic capacity, through which CAR-M cells could recognize antigens expressed specifically on cancer cells, therefore attacking and eliminating malignant cells. Zhang Jin's team developed CAR-expressing macrophages using iPSCs as the cell source, referred to as first-generation CD3ζ-based CAR-macrophages (iMACs)[236]. Building on this, they further developed iMACs with toll-like receptor 4 intracellular TIR (Toll/IL-1R) domain-containing CARs and M1 polarization characteristics, which demonstrated enhanced orthogonal phagocytosis, polarization, and superior antitumor functions in treating solid tumors [235]. Yizhao Chen and colleagues developed CAR-M targeting HER2 and CD47, demonstrating their inhibitory effects on HER2 or CD47-positive ovarian cancer in vitro and in vivo [237]. The study preliminarily confirmed that these effects are primarily due to phagocytosis, the promotion of adaptive immunity, and modulation of the tumor microenvironment [237]. Another recent preclinical study by Zahir Shah and colleagues demonstrated that iPSC-derived CAR-M targeting the tumor antigen PSCA exhibit strong antitumor activity against human pancreatic solid tumors both in vitro and in vivo [238]. Genetically engineered CAR-M targeting HER2 decreased tumor burden in a mouse model [239, 240]. Delivery of Adenovirus-delivered CAR to macrophages transforms M2 macrophages into M1 polarization, reshaping TME and amplifying anti-tumor cytotoxicity of T cells, which inhibits lung cancer metastasis during ovarian cancer treatment [240].
The majority of CAR-M strategies are currently in pre-clinical trials, with some already progressing to clinical trials. As an example, the first-in-human multi-center trial utilizing CAR-M carrying an adenoviral vector Ad5f35 targeting HER2 in various HER2-overexpressing solid tumors is currently in Phase I of interventional clinical trials (NCT04660929, estimated completion time: 2024-12), this has demonstrated promising results in effectively targeting solid tumors (https://classic.clinicaltrials.gov/ct2/show/NCT04660929). The phase I clinical trial results of the CAR-M product (CT-0508) demonstrate its preliminary safety, tolerability, and manufacturing feasibility for HER2+ tumors [241]. All the above studies elucidate CAR-M is anticipated to emerge as the forefront of tumor immunotherapy.
5. Future Prospective
Macrophages are important innate immune cells that play critical roles in clearing pathogens and maintaining tissue homeostasis. As the dominant myeloid cells infiltrate TME, TAMs influence cancer progression and immune response through multiple routes. Co-existence of two distinguished polarizations of TAMs displays spatial and temporal distribution in different types of cancer. M1-like TAMs activate the immune system and suppress tumor progression, whereas M2-like TAMs suppress the immune system to promote tumor development. Cancer cells and other infiltrated cells in TME tend to repress the anti-tumorigenic function and activate the pro-tumorigenic effects of TAMs, which provides a potential approach to take advantage of the M2-like TAMs by switching their polarization to M1-like.
High plasticity is the core characteristic of macrophages, giving rise to phenotypic diversity and functional complexity of TAMs. Although macrophage infiltration is a shared property in different tumors, substantial differences in TAM phenotypes and roles are observed in tumors arising in or disseminating to different tissues. As proof, while TAM infiltration is correlated with poor prognosis in majority tumors, there are noteworthy exceptions such as primary CRC. Advanced technologies have identified increasing subgroups of TAMs and progressively expanded our understanding of TAMs beyond the simple dual classification. This certainly leads to many open questions for future studies. First, functional specificity of unique TAM subsets needs to be elucidated at single cell level, especially in different genetic and tumor contexts. Second, mechanisms underlying TAM regulation on tumor development at primary site and metastatic lesions need more comprehensive analyses since the tissue intrinsic properties vary a lot. Third, spatial distribution of TAMs and their corresponding function within tumors should be explored. Last but not the least, more studies are needed to decipher the master transcriptional and epigenetic regulators accounting for pro- or anti- tumorigenic function of TAMs. These explorations will shed new insights into the fundamental biology of TAM and cancer immunotherapies targeting TAMs.
Given the high infiltration of TAMs in TME, approaches are developed for cancer treatment by depleting macrophages. Despite scientific advancements and promising preclinical studies, the translation of TAM-targeting therapies into effective clinical applications is still challenging. One of the reasons could be the heterogeneous nature of macrophages, which exhibit diverse phenotypes even within the same tumor. Another challenge is related to drug delivery. Many TAM-targeting agents fail to reach the tumor site due to the physiological barriers within the TME. Advanced drug delivery systems, such as nanoparticle-based delivery, are currently being explored to improve drug bioavailability. The side effects of these methods should be evaluated properly since macrophages are widespread and essential for normal tissue homeostasis.
TAMs are mainly replenished by the circulating myeloid precursor pool, which gives rise to the exploitation of cancer therapy by TAM recruitment disruption. One feasible idea is that we could make use of the strong attraction of macrophages to tumor tissues, to engineer T cells to overcome the poor recruitment of T cells at tumor site. This thought requires a profound understanding of the molecular basis of TAM recruitment and may broaden the application of CAR T cells in cancer immunotherapy. While high plasticity makes reprogramming TAMs operable, TAM heterogeneity is also the obstacle for TAM targeting drugs. Rather than bulk TAMs, targeting a key small portion of TAMs could be more effective with reduced side effects, which might be a future direction. Reprogramming macrophages towards antitumor phenotypes, rather than tumor suppressive ones, represents a promising direction, even though the potential for macrophage subset reprogramming has just been uncovered. Although TAM-targeting methods are still at the early stage, investigation into mechanisms of resistance to TAM-based immunotherapies is urgently needed as very limited data is available currently. The plasticity of macrophages allows them to switch phenotypes under different conditions, potentially contributing to drug resistance. Additionally, TAM-targeted cancer prevention and vaccine strategies should be considered, given the crucial roles of TAMs in cancer initiation, progression, and the formation of an immunosuppressive tumor microenvironment.
With the recent advancement of CAR-armed macrophage technology, its clinical potential still requires thorough evaluation through both preclinical and clinical trials. We would like to emphasize that the successful integration of CAR-macrophages with other therapies, such as CAR-T cells, in future clinical applications will depend on several key factors: (1) the ability of CAR-macrophages to sustain potent and long-lasting anti-tumor activity. As we know, one major issue with CAR-T cells in clinical applications is their tendency to become exhausted, leading to a loss of sustained functionality in some patients [10]. Could CAR-M cells face similar challenges? (2) whether the toxicity and side effects associated with CAR-macrophages are manageable and potentially lower than those of CAR-T cells; and (3) the identification of additional tumor-specific surface antigens suitable for effective CAR-macrophage targeting.
The current TAM-targeting approaches face several limitations, and several challenges need to be addressed to better understand the roles of TAMs in cancer, including: (1) clarifying the tumor heterogeneity which may complicate the development of universal therapies targeting TAMs; (2) further understanding the complexity of TAM polarization, because TAMs can exist in a range of activation states (M1, M2, etc.), and this plasticity makes it challenging to target TAMs effectively without disrupting their beneficial roles in tissue homeostasis and immune regulation; (3) further understanding the molecular mechanisms that influence the function of TAMs, such as TAM-associated metabolites that promote tumor progression and TAM-specific transcriptional and epigenetic factors, as well as surface markers, to distinguish between pro- and anti-tumoral TAM subsets; (4) elucidating the detailed mechanisms underlying TAM-mediated immunosuppression in the tumor microenvironment, for example, how TAMs interact with other immune cells and tumor cells, and whether we use certain molecular signatures to predict the efficacy of therapies targeting TAMs?[242] (5) developing novel delivery systems to enhance drug penetration for efficient targeting of TAMs; and (6) further understanding the resistance mechanisms of TAM-targeting therapies, for examples, the upregulation of alternative pathways or through the recruitment of other immune cells that compensate for TAM depletion or modulation.
6. Concluding remarks
In this review, we summarize the origins and polarization of tumor-associated macrophages (TAMs), discuss their role in regulating tumor development and immunity, and highlight the latest strategies in TAM-targeting cancer immunotherapy. The inherent heterogeneity of TAMs allows them to interact with various cells and participate in tumorigenesis and cancer immunity through diverse mechanisms, providing numerous opportunities for developing TAM-targeting therapies. However, for these strategies to be successfully translated into clinical practice, a more comprehensive and precise understanding of TAM heterogeneity and plasticity is essential. While several compounds, antibodies, and TAM engineering approaches have been developed, further supportive testing is needed to evaluate their clinical potential, both alone and in combination with other therapies, across different cancer contexts. Ongoing basic, translational, and clinical research will open new avenues for innovative therapeutic interventions, with promising outcomes expected in the future.
Abbreviations
APOE: apolipoprotein E; bFGF: basic fibroblast growth factor; BM: bone marrow; CAR: chimeric antigen receptor; CCL: C-C motif chemokine ligand; ccRCC: clear cell renal cell carcinoma; CRC: colorectal cancer; CREM: CAMP responsive element modulator; CSF1R: colony stimulating factor 1 receptor; CTLA4: cytotoxic T-lymphocyte-associated protein 4; CTL: cytotoxic T lymphocyte; CXCL8: chemokine ligand 8; EGF: epithelial growth factor; EMT: epithelial-mesenchymal transformation; FOLR2+: folate receptor 2+; GPCR: G protein-coupled receptor; HBP: hexosamine biosynthetic pathway; HCC: hepatocellular carcinoma; HGF: hepatocyte growth factor; HIF-1α: hypoxia-inducible factor 1α; HLA-G: human leukocyte antigen G; HOXB13: homeobox B13; IFN: interferon; LAP: LC3-associated phagocytosis; LCN2: lipocalin-2; IL: interleukin; iNOS: inducible NO synthase; IRF4: interferon regulatory factor-4; LPMs: large peritoneal macrophages; LPS: lipopolysaccharide; MAO-A: monoamine oxidase A; MDSCs: myeloid-derived suppressor cells; MMPs: matrix metalloproteinases; MPLA: monophosphoryl lipid A; NFκB: nuclear transcription factor-κB; NK: nature killer; NOS: nitric oxide synthase; NRF1: Nuclear Respiratory Factor 1; NSCLC: non-small cell lung carcinoma; PDAC: pancreatic ductal adenocarcinoma; PDGF: platelet-derived growth factor; PD-L1: programmed cell death protein ligand 1; PD-1: programmed cell death protein 1; PITPNM3: phosphatidylinositol transfer protein 3; PS: phosphatidylserine; RCC: renal cell carcinoma; ROS: reactive oxygen species; STAT3: Signal transducer and activator of transcription 3; STING: stimulator of interferon response CGAMP Interactor 1; SPP1: secreted phosphoprotein 1; S1P: sphingosine-1-phosphate; TAA: tumor-associated antigens; TAMs: Tumor-associated macrophages; TIM4: T cell immunoglobulin and mucin domain-containing molecule-4; TLRs: toll-like receptors; TP: thymine phosphorylase; TGF-β: transforming growth factor β; TIMs: tumor-infiltrating monocytes; TME: tumor microenvironment; TNF-α: tumor necrosis factor-α; TREM2: triggering receptor expressed on myeloid cells 2; TRMs: tissue resident macrophages; VEGF: vascular endothelial growth factor; WGP: whole β-glucan particle; 15-PGDH: 15-hydroxyprostaglandin dehydrogenase.
Acknowledgements
Funding
This work was supported by the National Natural Science Foundation of China (No. 82273208 to H. Xia), National Natural Science Foundation of China for Young Scientists (No.32200632), Guangdong University Characteristic Innovation Project (Natural Science 2022KTSCX222 to H. Xia), and Shenzhen Science and Technology Program (JCYJ20220818101601004 to H. Xia).
Author contributions
Huabing Tan, Meihe Cai, Jincheng Wang and Tao Yu: Original draft, Visualization. Houjun Xia, Huanbin Zhao and Xiaoyu Zhang: Review & editing, Supervision.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liu P, Jiao B, Zhang R, Zhao H, Zhang C, Wu M. et al. Palmitoylacyltransferase Zdhhc9 inactivation mitigates leukemogenic potential of oncogenic Nras. Leukemia. 2016;30:1225-8
2. Liu P, Jiao B, Zhang RH, Zhao HB, Zhang C, Wu M. et al. Palmitoyl Acyltransferase DHHC9 Is Required for Efficient Induction of Leukemia By Oncogenic NRAS. Blood. 2014 124
3. Zhao H, Liu P, Zhang R, Wu M, Li D, Zhao X. et al. Roles of palmitoylation and the KIKK membrane-targeting motif in leukemogenesis by oncogenic KRAS4A. J Hematol Oncol. 2015;8:132
4. Zhao HB, Ren RB. Differential roles of palmitoylation on oncogenic potential of NRAS and KRAS4A. Cancer Research. 2015 75
5. Fu RJ, He W, Wang XB, Li L, Zhao HB, Liu XY. et al. DNMT1-maintained hypermethylation of Kruppel-like factor 5 involves in the progression of clear cell renal cell carcinoma. Cell Death Dis. 2017;8:e2952
6. Blanco-Fernandez B, Gaspar VM, Engel E, Mano JF. Proteinaceous Hydrogels for Bioengineering Advanced 3D Tumor Models. Adv Sci (Weinh). 2021;8:2003129
7. Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T. et al. Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP ProteinsAdipocyte-Derived Lipids Drive Melanoma Progression via FATP. Cancer Discov. 2018;8:1006-25
8. Braun DA, Street K, Burke KP, Cookmeyer DL, Denize T, Pedersen CB. et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell. 2021;39:632-48 e8
9. Hossain M, Shim R, Lee WY, Sharpe AH, Kubes P. Gata6(+) resident peritoneal macrophages promote the growth of liver metastasis. Nat Commun. 2022;13:4406
10. Zhu YH, Tan HB, Wang JC, Zhuang HW, Zhao HB, Lu XJ. Molecular insight into T cell exhaustion in hepatocellular carcinoma. Pharmacological Research. 2024;203:107161
11. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286-91
12. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nature Reviews Drug Discovery. 2022;21:799-820
13. Wang S, Liu R, Yu Q, Dong L, Bi Y, Liu G. Metabolic reprogramming of macrophages during infections and cancer. Cancer Lett. 2019;452:14-22
14. Bleriot C, Chakarov S, Ginhoux F. Determinants of Resident Tissue Macrophage Identity and Function. Immunity. 2020;52:957-70
15. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K. et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921-5
16. Xia H, Li S, Li X, Wang W, Bian Y, Wei S. et al. Autophagic adaptation to oxidative stress alters peritoneal residential macrophage survival and ovarian cancer metastasis. JCI insight. 2020;5:e141115
17. Etzerodt A, Moulin M, Doktor TK, Delfini M, Mossadegh-Keller N, Bajenoff M. et al. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J Exp Med. 2020;217:e20191869
18. Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE. et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med. 2021;27:152-64
19. Chow A, Schad S, Green MD, Hellmann MD, Allaj V, Ceglia N. et al. Tim-4(+) cavity-resident macrophages impair anti-tumor CD8(+) T cell immunity. Cancer Cell. 2021;39:973-88 e9
20. Niu Y, Chen J, Qiao Y. Epigenetic Modifications in Tumor-Associated Macrophages: A New Perspective for an Old Foe. Front Immunol. 2022;13:836223
21. Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184:792-809 e23
22. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019;30:36-50
23. Metchnikoff E. Lectures on the comparative pathology of inflammation delivered at the Pasteur Institute in 1891. Рипол Классик; 1893.
24. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37:208-20
25. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762-74
26. Zhu Y, Herndon JM, Sojka DK, Kim K-W, Knolhoff BL, Zuo C. et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. 2017;47:323-38 e6
27. Dick SA, Wong A, Hamidzada H, Nejat S, Nechanitzky R, Vohra S. et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol. 2022;7:eabf7777
28. Hume DA, MacDonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119:1810-20
29. Garner H, de Visser KE. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat Rev Immunol. 2020;20:483-97
30. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G. et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212:1043-59
31. Ma RY, Zhang H, Li XF, Zhang CB, Selli C, Tagliavini G. et al. Monocyte-derived macrophages promote breast cancer bone metastasis outgrowth. J Exp Med. 2020;217:e20191820
32. Fu WB, Xu L, Liu J, Han XF, Wang JY, Wang XP. et al. G-CSF Upregulates the Expression of Aquaporin-9 through C/EBP-Beta to Enhance the Cytotoxic Activity of Arsenic Trioxide to Acute Myeloid Leukemia Cells. Blood. 2021;138:195
33. Fu W, Zhu G, Xu L, Liu J, Han X, Wang J. et al. G-CSF upregulates the expression of aquaporin-9 through CEBPB to enhance the cytotoxic activity of arsenic trioxide to acute myeloid leukemia cells. Cancer Cell Int. 2022;22:195
34. Devalaraja S, To TKJ, Folkert IW, Natesan R, Alam MZ, Li M. et al. Tumor-Derived Retinoic Acid Regulates Intratumoral Monocyte Differentiation to Promote Immune Suppression. Cell. 2020;180:1098-114 e16
35. Zhang N, Kim SH, Gainullina A, Erlich EC, Onufer EJ, Kim J. et al. LYVE1+ macrophages of murine peritoneal mesothelium promote omentum-independent ovarian tumor growth. J Exp Med. 2021;218:e20210924
36. Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM. et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021;595:578-84
37. Nalio Ramos R, Missolo-Koussou Y, Gerber-Ferder Y, Bromley CP, Bugatti M, Nunez NG. et al. Tissue-resident FOLR2(+) macrophages associate with CD8(+) T cell infiltration in human breast cancer. Cell. 2022;185:1189-207 e25
38. Loyher PL, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z. et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med. 2018;215:2536-53
39. Italiani P, Boraschi D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front Immunol. 2014;5:514
40. Liu JY, Geng XF, Hou JX, Wu GS. New insights into M1/M2 macrophages: key modulators in cancer progression. Cancer Cell International. 2021;21:1-7
41. Wang J, Mi S, Ding M, Li X, Yuan S. Metabolism and polarization regulation of macrophages in the tumor microenvironment. Cancer Lett. 2022;543:215766
42. Zhang B, Su YC, Zhou JC, Zheng YF, Zhu DH. Toward a Better Regeneration through Implant-Mediated Immunomodulation: Harnessing the Immune Responses. Advanced Science. 2021;8:2100446
43. Malla R, Padmaraju V, Kundrapu DB. Tumor-associated macrophages: Potential target of natural compounds for management of breast cancer. Life Sci. 2022;301:120572
44. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559-63
45. Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N. et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. 2018;19:1319-29
46. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A. et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome biology. 2017;18:1-14
47. Kim W, Khan SK, Liu Y, Xu R, Park O, He Y. et al. Hepatic Hippo signaling inhibits protumoural microenvironment to suppress hepatocellular carcinoma. Gut. 2018;67:1692-703
48. Sorensen MD, Dahlrot RH, Boldt HB, Hansen S, Kristensen BW. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol Appl Neurobiol. 2018;44:185-206
49. Singhal S, Stadanlick J, Annunziata MJ, Rao AS, Bhojnagarwala PS, O'Brien S. et al. Human tumor-associated monocytes/macrophages and their regulation of T cell responses in early-stage lung cancer. Sci Transl Med. 2019;11:eaat1500
50. Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT. et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8:15081
51. Wu F, Fan J, He Y, Xiong A, Yu J, Li Y. et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat Commun. 2021;12:2540
52. Stewart CA, Gay CM, Xi Y, Sivajothi S, Sivakamasundari V, Fujimoto J. et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat Cancer. 2020;1:423-36
53. Zheng H, Pomyen Y, Hernandez MO, Li C, Livak F, Tang W. et al. Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology. 2018;68:127-40
54. Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P. et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017;21:1399-410
55. Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL. et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet. 2017;49:708-18
56. Kim KT, Lee HW, Lee HO, Song HJ, Jeong da E, Shin S. et al. Application of single-cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol. 2016;17:80
57. Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, McFarland JM. et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet. 2020;52:1208-18
58. Pittet MJ, Michielin O, Migliorini D. Clinical relevance of tumour-associated macrophages. Nature Reviews Clinical Oncology. 2022;19:402-21
59. Hao X, Zheng Z, Liu H, Zhang Y, Kang J, Kong X. et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 2022;56:102463
60. Tu J, Wang D, Zheng X, Liu B. Single-cell RNA datasets and bulk RNA datasets analysis demonstrated C1Q+ tumor-associated macrophage as a major and antitumor immune cell population in osteosarcoma. Front Immunol. 2023;14:911368
61. Kumar V, Ramnarayanan K, Sundar R, Padmanabhan N, Srivastava S, Koiwa M. et al. Single-Cell Atlas of Lineage States, Tumor Microenvironment, and Subtype-Specific Expression Programs in Gastric Cancer. Cancer Discov. 2022;12:670-91
62. Larroquette M, Guegan JP, Besse B, Cousin S, Brunet M, Le Moulec S. et al. Spatial transcriptomics of macrophage infiltration in non-small cell lung cancer reveals determinants of sensitivity and resistance to anti-PD1/PD-L1 antibodies. J Immunother Cancer. 2022;10:e003890
63. Ozato Y, Kojima Y, Kobayashi Y, Hisamatsu Y, Toshima T, Yonemura Y. et al. Spatial and single-cell transcriptomics decipher the cellular environment containing HLA-G+ cancer cells and SPP1+ macrophages in colorectal cancer. Cell Rep. 2023;42:111929
64. Huang X, Li YZ, Zhang JL, Yan L, Zhao HB, Ding L. et al. Single-cell systems pharmacology identifies development-driven drug response and combination therapy in B cell acute lymphoblastic leukemia. Cancer Cell. 2024;42:552-567.e6
65. Mariottoni P, Jiang SW, Prestwood CA, Jain V, Suwanpradid J, Whitley MJ. et al. Single-Cell RNA Sequencing Reveals Cellular and Transcriptional Changes Associated With M1 Macrophage Polarization in Hidradenitis Suppurativa. Front Med (Lausanne). 2021;8:665873
66. Li Z, Zhao H, Yang W, Maillard M, Yoshimura S, Hsiao YC. et al. Molecular and pharmacological heterogeneity of ETV6::RUNX1 acute lymphoblastic leukemia. Nat Commun. 2025;16:1153
67. Chanmee T, Ontong P, Konno K, Itano N. Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers (Basel). 2014;6:1670-90
68. Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol. 2017;10:58
69. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ. et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One. 2012;7:e50946
70. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N. et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology. 2013;2:e26968
71. Hanada T, Nakagawa M, Emoto A, Nomura T, Nasu N, Nomura Y. Prognostic value of tumor-associated macrophage count in human bladder cancer. Int J Urol. 2000;7:263-9
72. Lissbrant IF, Stattin P, Wikstrom P, Damber JE, Egevad L, Bergh A. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int J Oncol. 2000;17:445-51
73. Andersen RS, Anand A, Harwood DSL, Kristensen BW. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers (Basel). 2021;13:4255
74. Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol. 2020;11:583084
75. Yang CG, Wei C, Wang SY, Shi DD, Zhang CX, Lin XB. et al. Elevated CD163/CD68 Ratio at Tumor Invasive Front is Closely Associated with Aggressive Phenotype and Poor Prognosis in Colorectal Cancer. Int J Biol Sci. 2019;15:984-98
76. Su CY, Fu XL, Duan W, Yu PW, Zhao YL. High density of CD68+ tumor-associated macrophages predicts a poor prognosis in gastric cancer mediated by IL-6 expression. Oncol Lett. 2018;15:6217-24
77. Ohno S, Ohno Y, Suzuki N, Kamei T, Koike K, Inagawa H. et al. Correlation of histological localization of tumor-associated macrophages with clinicopathological features in endometrial cancer. Anticancer Res. 2004;24:3335-42
78. Matsumura H, Kondo T, Ogawa K, Tamura T, Fukunaga K, Murata S. et al. Kupffer cells decrease metastasis of colon cancer cells to the liver in the early stage. Int J Oncol. 2014;45:2303-10
79. Pucci F, Garris C, Lai CP, Newton A, Pfirschke C, Engblom C. et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science. 2016;352:242-6
80. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. 2021;6:75
81. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW. et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572:392 -+
82. Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R. et al. CD47 Is Upregulated on Circulating Hematopoietic Stem Cells and Leukemia Cells to Avoid Phagocytosis. Cell. 2009;138:271-85
83. Emam M, Tabatabaei S, Sargolzaei M, Mallard B. Response to Oxidative Burst-Induced Hypoxia Is Associated With Macrophage Inflammatory Profiles as Revealed by Cellular Genome-Wide Association. Front Immunol. 2021;12:688503
84. Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166:S4-8
85. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270-4
86. Haschka D, Hoffmann A, Weiss G. Iron in immune cell function and host defense. Semin Cell Dev Biol. 2021;115:27-36
87. Zhou Z, Xu B, Hu N, Guo Z, Bao W, Shao B. et al. Targeting the Macrophage-Ferroptosis Crosstalk: A Novel Insight into Tumor Immunotherapy. Front Biosci (Landmark Ed). 2022;27:203
88. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ. et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16:2069-83
89. Xiao N, Li K, Zhu X, Xu B, Liu X, Lei M. et al. CD74(+) macrophages are associated with favorable prognosis and immune contexture in hepatocellular carcinoma. Cancer Immunol Immunother. 2022;71:57-69
90. Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC. et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell. 2018;175:429-41 e16
91. Ramos RN, Missolo-Koussou Y, Gerber-Ferder Y, Bromley CP, Bugatti M, Núñez NG. et al. Tissue-resident FOLR2 macrophages associate with CD8 T cell infiltration in human breast cancer. Cell. 2022;185:1189 -+
92. Wang YC, Wang X, Yu J, Ma F, Li Z, Zhou Y. et al. Targeting monoamine oxidase A-regulated tumor-associated macrophage polarization for cancer immunotherapy. Nat Commun. 2021;12:3530
93. Kogure A, Kosaka N, Ochiya T. Cross-talk between cancer cells and their neighbors via miRNA in extracellular vesicles: an emerging player in cancer metastasis. J Biomed Sci. 2019;26:7
94. Arvanitakis K, Koletsa T, Mitroulis I, Germanidis G. Tumor-Associated Macrophages in Hepatocellular Carcinoma Pathogenesis, Prognosis and Therapy. Cancers (Basel). 2022;14:226
95. Tong H, Ke JQ, Jiang FZ, Wang XJ, Wang FY, Li YR. et al. Tumor-associated macrophage-derived CXCL8 could induce ERalpha suppression via HOXB13 in endometrial cancer. Cancer Lett. 2016;376:127-36
96. Arima K, Komohara Y, Bu L, Tsukamoto M, Itoyama R, Miyake K. et al. Downregulation of 15-hydroxyprostaglandin dehydrogenase by interleukin-1beta from activated macrophages leads to poor prognosis in pancreatic cancer. Cancer Sci. 2018;109:462-70
97. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39-51
98. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. Journal of Biomedical Science. 2018;25:1-18
99. Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L. et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014;352:160-8
100. Raghavan S, Mehta P, Xie YY, Lei YL, Mehta G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. Journal for Immunotherapy of Cancer. 2019;7:1-15
101. Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL. et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol Cell. 2020;77:213-27 e5
102. Chryplewicz A, Scotton J, Tichet M, Zomer A, Shchors K, Joyce JA. et al. Cancer cell autophagy, reprogrammed macrophages, and remodeled vasculature in glioblastoma triggers tumor immunity. Cancer Cell. 2022;40:1111-27 e9
103. Chen YC, Lai YS, Hsuuw YD, Chang KT. Withholding of M-CSF Supplement Reprograms Macrophages to M2-Like via Endogenous CSF-1 Activation. Int J Mol Sci. 2021;22:3532
104. Wang XQ, Tokheim C, Gu SS, Wang BB, Tang Q, Li YH. et al. CRISPR screens identify the E3 ligase as a modulator of macrophage infiltration and cancer immunotherapy target. Cell. 2021;184:5357 -+
105. Di Conza G, Tsai CH, Gallart-Ayala H, Yu YR, Franco F, Zaffalon L. et al. Tumor-induced reshuffling of lipid composition on the endoplasmic reticulum membrane sustains macrophage survival and pro-tumorigenic activity. Nat Immunol. 2021;22:1403-15
106. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J. et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170-82
107. Song G, Shi Y, Zhang M, Goswami S, Afridi S, Meng L. et al. Global immune characterization of HBV/HCV-related hepatocellular carcinoma identifies macrophage and T-cell subsets associated with disease progression. Cell Discov. 2020;6:90
108. Obradovic A, Chowdhury N, Haake SM, Ager C, Wang V, Vlahos L. et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell. 2021;184:2988-3005 e16
109. Hagemann T, Robinson SC, Schulz M, Trümper L, Balkwill FR, Binder C. Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-α dependent up-regulation of matrix metalloproteases. Carcinogenesis. 2004;25:1543-9
110. Li R, Zhou R, Wang H, Li W, Pan M, Yao X. et al. Gut microbiota-stimulated cathepsin K secretion mediates TLR4-dependent M2 macrophage polarization and promotes tumor metastasis in colorectal cancer. Cell Death Differ. 2019;26:2447-63
111. Hildenbrand R, Jansen C, Wolf G, Bohme B, Berger S, von Minckwitz G. et al. Transforming growth factor-beta stimulates urokinase expression in tumor-associated macrophages of the breast. Lab Invest. 1998;78:59-71
112. Arnold SA, Rivera LB, Miller AF, Carbon JG, Dineen SP, Xie Y. et al. Lack of host SPARC enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis Model Mech. 2010;3:57-72
113. Akkari L, Gocheva V, Kester JC, Hunter KE, Quick ML, Sevenich L. et al. Distinct functions of macrophage-derived and cancer cell-derived cathepsin Z combine to promote tumor malignancy via interactions with the extracellular matrix. Genes Dev. 2014;28:2134-50
114. Jung M, Oren B, Mora J, Mertens C, Dziumbla S, Popp R. et al. Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci Signal. 2016;9:ra64
115. Weiss JM, Ridnour LA, Back T, Hussain SP, He P, Maciag AE. et al. Macrophage-dependent nitric oxide expression regulates tumor cell detachment and metastasis after IL-2/anti-CD40 immunotherapy. J Exp Med. 2010;207:2455-67
116. Chen J, Yao Y, Gong C, Yu F, Su S, Chen J. et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell. 2011;19:541-55
117. Shi Q, Shen Q, Liu Y, Shi Y, Huang W, Wang X. et al. Increased glucose metabolism in TAMs fuels O-GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer Cell. 2022;40:1207-22 e10
118. Leek RD, Hunt NC, Landers RJ, Lewis CE, Royds JA, Harris AL. Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. Journal of Pathology. 2000;190:430-6
119. Sousa S, Maatta J. The role of tumour-associated macrophages in bone metastasis. J Bone Oncol. 2016;5:135-8
120. Wyckoff J, Wang WG, Lin EY, Wang YR, Pixley F, Stanley ER. et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Research. 2004;64:7022-9
121. Yao RR, Li JH, Zhang R, Chen RX, Wang YH. M2-polarized tumor-associated macrophages facilitated migration and epithelial-mesenchymal transition of HCC cells via the TLR4/STAT3 signaling pathway. World J Surg Oncol. 2018;16:9
122. Liu CY, Xu JY, Shi XY, Huang W, Ruan TY, Xie P. et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Laboratory Investigation. 2013;93:844-54
123. Cai JH, Xia LM, Li JL, Ni SC, Song HY, Wu XB. Tumor-Associated Macrophages Derived TGF-β-Induced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells through Smad2,3-4/Snail Signaling Pathway. Cancer Research and Treatment. 2019;51:252-66
124. Chen XW, Yu TJ, Zhang J, Li Y, Chen HL, Yang GF. et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene. 2017;36:5045-57
125. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T. et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515:130-3
126. Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A. et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest. 2013;123:1371-81
127. Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A. et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nature cell biology. 2016;18:549-60
128. Nosaka T, Baba T, Tanabe Y, Sasaki S, Nishimura T, Imamura Y. et al. Alveolar Macrophages Drive Hepatocellular Carcinoma Lung Metastasis by Generating Leukotriene B(4). J Immunol. 2018;200:1839-52
129. Yin M, Li X, Tan S, Zhou HJ, Ji W, Bellone S. et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J Clin Invest. 2016;126:4157-73
130. Yin Z, Ma TT, Huang BW, Lin LH, Zhou Y, Yan JH. et al. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-β signaling pathway. Journal of Experimental & Clinical Cancer Research. 2019;38:1-20
131. Hara T, Chanoch-Myers R, Mathewson ND, Myskiw C, Atta L, Bussema L. et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell. 2021;39:779 -+
132. Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12:31-46
133. Medrek C, Pontén F, Jirström K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. Bmc Cancer. 2012;12:1-9
134. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. 2014;5:75
135. Valković T, Dobrila F, Melato M, Sasso F, Rizzardi C, Jonjić N. Correlation between vascular endothelial growth factor, angiogenesis, and tumor-associated macrophages in invasive ductal breast carcinoma. Virchows Archiv. 2002;440:583-8
136. Marech I, Ammendola M, Sacco R, Sammarco G, Zuccala V, Zizzo N. et al. Tumour-associated macrophages correlate with microvascular bed extension in colorectal cancer patients. J Cell Mol Med. 2016;20:1373-80
137. Huang S, Van Arsdall M, Tedjarati S, McCarty M, Wu W, Langley R. et al. Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J Natl Cancer Inst. 2002;94:1134-42
138. Takebayashi Y, Miyadera K, Akiyama S, Hokita S, Yamada K, Akiba S. et al. Expression of thymidine phosphorylase in human gastric carcinoma. Jpn J Cancer Res. 1996;87:288-95
139. Yeo EJ, Cassetta L, Qian BZ, Lewkowich I, Li JF, Stefater JA 3rd. et al. Myeloid WNT7b mediates the angiogenic switch and metastasis in breast cancer. Cancer Res. 2014;74:2962-73
140. Wang XJ, Zhu QY, Lin YY, Wu L, Wu XL, Wang K. et al. Crosstalk between TEMs and endothelial cells modulates angiogenesis and metastasis via IGF1-IGF1R signalling in epithelial ovarian cancer. Br J Cancer. 2017;117:1371-82
141. Lewis CE, De Palma M, Naldini L. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res. 2007;67:8429-32
142. Guo Q, Jin Z, Yuan Y, Liu R, Xu T, Wei H. et al. New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy. J Immunol Res. 2016;2016:9720912
143. Valilou SF, Keshavarz-Fathi M, Silvestris N, Argentiero A, Rezaei N. The role of inflammatory cytokines and tumor associated macrophages (TAMs) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev. 2018;39:46-61
144. Lohela M, Casbon AJ, Olow A, Bonham L, Branstetter D, Weng N. et al. Intravital imaging reveals distinct responses of depleting dynamic tumor-associated macrophage and dendritic cell subpopulations. Proc Natl Acad Sci U S A. 2014;111:E5086-95
145. Nixon BG, Kuo F, Ji L, Liu M, Capistrano K, Do M. et al. Tumor-associated macrophages expressing the transcription factor IRF8 promote T cell exhaustion in cancer. Immunity. 2022;55:2044-58 e5
146. Chen W, Ma T, Shen X-n, Xia X-f, Xu G-d, Bai X-l. et al. Macrophage-Induced Tumor Angiogenesis Is Regulated by the TSC2-mTOR PathwayTSC2-mTOR Regulates Macrophage-Induced Tumor Angiogenesis. Cancer research. 2012;72:1363-72
147. Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P. et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203:871-81
148. Petty AJ, Li A, Wang X, Dai R, Heyman B, Hsu D. et al. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J Clin Invest. 2019;129:5151-62
149. Gu Y, Yang J, Ouyang X, Liu W, Li H, Yang J. et al. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur J Immunol. 2008;38:1807-13
150. Cao Q, Wang Y, Zheng D, Sun Y, Wang Y, Lee VW. et al. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol. 2010;21:933-42
151. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-9
152. Horlad H, Ma C, Yano H, Pan C, Ohnishi K, Fujiwara Y. et al. An IL-27/Stat3 axis induces expression of programmed cell death 1 ligands (PD-L1/2) on infiltrating macrophages in lymphoma. Cancer Sci. 2016;107:1696-704
153. Yu GT, Bu LL, Zhao YY, Mao L, Deng WW, Wu TF. et al. CTLA4 blockade reduces immature myeloid cells in head and neck squamous cell carcinoma. Oncoimmunology. 2016;5:e1151594
154. Kondo A, Yamashita T, Tamura H, Zhao W, Tsuji T, Shimizu M. et al. Interferon-gamma and tumor necrosis factor-alpha induce an immunoinhibitory molecule, B7-H1, via nuclear factor-kappaB activation in blasts in myelodysplastic syndromes. Blood. 2010;116:1124-31
155. Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR. et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. 2021;593:282-8
156. Xia H, Wang W, Crespo J, Kryczek I, Li W, Wei S. et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naive T cell apoptosis and affects tumor immunity. Sci Immunol. 2017;2:eaan4631
157. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G. et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393-404
158. Lo SZY, Steer JH, Joyce DA. TNF-α renders macrophages resistant to a range of cancer chemotherapeutic agents through NF-κB-mediated antagonism of apoptosis signalling. Cancer Letters. 2011;307:80-92
159. Zhu X, Shen H, Yin X, Yang M, Wei H, Chen Q. et al. Macrophages derived exosomes deliver miR-223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J Exp Clin Cancer Res. 2019;38:81
160. Zheng P, Chen L, Yuan X, Luo Q, Liu Y, Xie G. et al. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J Exp Clin Cancer Res. 2017;36:53
161. Chen X, Fu E, Lou H, Mao X, Yan B, Tong F. et al. IL-6 induced M1 type macrophage polarization increases radiosensitivity in HPV positive head and neck cancer. Cancer Lett. 2019;456:69-79
162. Rahal OM, Wolfe AR, Mandal PK, Larson R, Tin S, Jimenez C. et al. Blocking Interleukin (IL)4- and IL13-Mediated Phosphorylation of STAT6 (Tyr641) Decreases M2 Polarization of Macrophages and Protects Against Macrophage-Mediated Radioresistance of Inflammatory Breast Cancer. Int J Radiat Oncol Biol Phys. 2018;100:1034-43
163. Hakoda H, Ichida A, Hasegawa K. Advances in systemic therapy leading to conversion surgery for advanced hepatocellular carcinoma. Biosci Trends. 2025;18:525-34
164. Momose H, Kudo S, Yoshida T, Hasui N, Matsuki R, Kogure M. et al. Trends in the treatment of advanced pancreatic cancer. Biosci Trends. 2024;18:224-32
165. Mizutani K, Sud S, McGregor NA, Martinovski G, Rice BT, Craig MJ. et al. The Chemokine CCL2 Increases Prostate Tumor Growth and Bone Metastasis through Macrophage and Osteoclast Recruitment. Neoplasia. 2009;11:1235-42
166. Li XG, Yao WB, Yuan Y, Chen PZ, Li B, Li JQ. et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157-67
167. Teng K-Y, Han J, Zhang X, Hsu S-H, He S, Wani NA. et al. Blocking the CCL2-CCR2 Axis Using CCL2-Neutralizing Antibody Is an Effective Therapy for Hepatocellular Cancer in a Mouse Model. Molecular cancer therapeutics. 2017;16:312-22
168. Tu MM, Abdel-Hafiz HA, Jones RT, Jean A, Hoff KJ, Duex JE. et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun Biol. 2020;3:720
169. Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D. et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin Cancer Res. 2013;19:3404-15
170. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nature Reviews Clinical Oncology. 2017;14:399-416
171. Seo YD, Jiang X, Sullivan KM, Jalikis FG, Smythe KS, Abbasi A. et al. Mobilization of CD8+ T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic CancerCXCR4 and PD-1 Blockade in Human Pancreatic Cancer. Clin Cancer Res. 2019;25:3934-45
172. Li X, Bu W, Meng L, Liu X, Wang S, Jiang L. et al. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp Cell Res. 2019;378:131-8
173. Beider K, Bitner H, Leiba M, Gutwein O, Koren-Michowitz M, Ostrovsky O. et al. Multiple myeloma cells recruit tumor-supportive macrophages through the CXCR4/CXCL12 axis and promote their polarization toward the M2 phenotype. Oncotarget. 2014;5:11283-96
174. Hidalgo M, Macarulla T, Semenisty V, Borazanci E, Feliu J, Ponz-Sarvise M. et al. Abstract CT177: A multi-center phase 2a trial of the CXCR4 inhibitor motixafortide (BL-8040)(M) in combination with pembrolizumab (P) and chemotherapy (C), in patients with metastatic pancreatic adenocarcinoma (mPDAC). Cancer Research. 2021;81:CT177-CT
175. Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A. 2010;107:8363-8
176. Hegab AE, Ozaki M, Kameyama N, Gao J, Kagawa S, Yasuda H. et al. Effect of FGF/FGFR pathway blocking on lung adenocarcinoma and its cancer-associated fibroblasts. J Pathol. 2019;249:193-205
177. Xia Y, Wei Y, Li ZY, Cai XY, Zhang LL, Dong XR. et al. Catecholamines contribute to the neovascularization of lung cancer via tumor-associated macrophages. Brain Behav Immun. 2019;81:111-21
178. Piao C, Zhang WM, Li TT, Zhang CC, Qiu S, Liu Y. et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp Cell Res. 2018;366:127-38
179. Zhu Y, Yang J, Xu D, Gao XM, Zhang Z, Hsu JL. et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68:1653-66
180. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J. et al. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer research. 2014;74:5057-69
181. Escamilla J, Schokrpur S, Liu C, Priceman SJ, Moughon D, Jiang Z. et al. CSF1 Receptor Targeting in Prostate Cancer Reverses Macrophage-Mediated Resistance to Androgen Blockade Therapy. Cancer research. 2015;75:950-62
182. Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL. et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene. 2017;36:6049-58
183. Klemm F, Mockl A, Salamero-Boix A, Alekseeva T, Schaffer A, Schulz M. et al. Compensatory CSF2-driven macrophage activation promotes adaptive resistance to CSF1R inhibition in breast-to-brain metastasis. Nat Cancer. 2021;2:1086-101
184. Moughon DL, He H, Schokrpur S, Jiang ZK, Yaqoob M, David J. et al. Macrophage Blockade Using CSF1R Inhibitors Reverses the Vascular Leakage Underlying Malignant Ascites in Late-Stage Epithelial Ovarian Cancer. Cancer research. 2015;75:4742-52
185. Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA. et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 2015;16:949-56
186. Machiels JP, Gomez-Roca C, Michot JM, Zamarin D, Mitchell T, Catala G. et al. Phase Ib study of anti-CSF-1R antibody emactuzumab in combination with CD40 agonist selicrelumab in advanced solid tumor patients. J Immunother Cancer. 2020;8:e001153
187. Lu X, Meng T. Depletion of tumor-associated macrophages enhances the anti-tumor effect of docetaxel in a murine epithelial ovarian cancer. Immunobiology. 2019;224:355-61
188. Gautier EL, Ivanov S, Williams JW, Huang SC, Marcelin G, Fairfax K. et al. Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J Exp Med. 2014;211:1525-31
189. Rosas M, Davies LC, Giles PJ, Liao CT, Kharfan B, Stone TC. et al. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science. 2014;344:645-8
190. Casanova-Acebes M, Menendez-Gutierrez MP, Porcuna J, Alvarez-Errico D, Lavin Y, Garcia A. et al. RXRs control serous macrophage neonatal expansion and identity and contribute to ovarian cancer progression. Nat Commun. 2020;11:1655
191. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M. et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249-62
192. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C. et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity. 2017;47:323-38 e6
193. Jeong H, Kim S, Hong BJ, Lee CJ, Kim YE, Bok S. et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res. 2019;79:795-806
194. Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114:623-33
195. Binnewies M, Pollack JL, Rudolph J, Dash S, Abushawish M, Lee T. et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 2021;37:109844
196. Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O'Connor RS. et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun. 2021;12:877
197. Laplagne C, Domagala M, Le Naour A, Quemerais C, Hamel D, Fournie JJ. et al. Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression. Int J Mol Sci. 2019;20:4719
198. Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Ostling J. et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016;15:2000-11
199. Sommariva M, Le Noci V, Storti C, Bianchi F, Tagliabue E, Balsari A. et al. Activation of NK cell cytotoxicity by aerosolized CpG-ODN/poly(I:C) against lung melanoma metastases is mediated by alveolar macrophages. Cellular Immunology. 2017;313:52-8
200. Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M. et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell. 2016;29:587-601
201. Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNγ and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic CarcinomaMonocytes Regulate Fibrosis in Pancreatic Carcinoma. Cancer Discov. 2016;6:400-13
202. Qi B, Fang Q, Liu S, Hou W, Li J, Huang Y. et al. Advances of CCR5 antagonists: From small molecules to macromolecules. Eur J Med Chem. 2020;208:112819
203. Stockfleth E, Trefzer U, Garcia-Bartels C, Wegner T, Schmook T, Sterry W. The use of Toll-like receptor-7 agonist in the treatment of basal cell carcinoma: an overview. Br J Dermatol. 2003;149(Suppl 66):53-6
204. De Waele J, Verhezen T, van der Heijden S, Berneman ZN, Peeters M, Lardon F. et al. A systematic review on poly(I:C) and poly-ICLC in glioblastoma: adjuvants coordinating the unlocking of immunotherapy. J Exp Clin Cancer Res. 2021;40:213
205. Wanderley CW, Colon DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA. et al. Paclitaxel Reduces Tumor Growth by Reprogramming Tumor-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer research. 2018;78:5891-900
206. Yasmin-Karim S, Bruck PT, Moreau M, Kunjachan S, Chen GZ, Kumar R. et al. Radiation and Local Anti-CD40 Generate an Effective in situ Vaccine in Preclinical Models of Pancreatic Cancer. Front Immunol. 2018;9:2030
207. Wingett DG, Vestal RE, Forcier K, Hadjokas N, Nielson CP. CD40 is functionally expressed on human breast carcinomas: Variable inducibility by cytokines and enhancement of Fas-mediated apoptosis. Breast Cancer Research and Treatment. 1998;50:27-36
208. Ghamande S, Hylander BL, Oflazoglu E, Lele S, Fanslow W, Repasky EA. Recombinant CD40 ligand therapy has significant antitumor effects on CD40-positive ovarian tumor xenografts grown in SCID mice and demonstrates an augmented effect with cisplatin. Cancer Research. 2001;61:7556-62
209. Sun L, Kees T, Almeida AS, Liu B, He XY, Ng D. et al. Activating a collaborative innate-adaptive immune response to control metastasis. Cancer Cell. 2021;39:1361-74 e9
210. Andersen MN, Etzerodt A, Graversen JH, Holthof LC, Moestrup SK, Hokland M. et al. STAT3 inhibition specifically in human monocytes and macrophages by CD163-targeted corosolic acid-containing liposomes. Cancer Immunol Immunother. 2019;68:489-502
211. Binnemars-Postma K, Bansal R, Storm G, Prakash J. Targeting the Stat6 pathway in tumor-associated macrophages reduces tumor growth and metastatic niche formation in breast cancer. FASEB J. 2018;32:969-78
212. Yu T, Gan S, Zhu Q, Dai D, Li N, Wang H. et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat Commun. 2019;10:4353
213. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S. et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature. 2016;539:437-42
214. Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P. et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016;6:870-85
215. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH. et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. 2016;539:443-7
216. Ma B, Cheng H, Mu C, Geng G, Zhao T, Luo Q. et al. The SIAH2-NRF1 axis spatially regulates tumor microenvironment remodeling for tumor progression. Nat Commun. 2019;10:1034
217. Verzella D, Bennett J, Fischietti M, Thotakura AK, Recordati C, Pasqualini F. et al. GADD45β Loss Ablates Innate Immunosuppression in Cancer. Cancer research. 2018;78:1275-92
218. Wei X, Nie S, Liu H, Sun J, Liu J, Li J. et al. Angiopoietin-like protein 2 facilitates non-small cell lung cancer progression by promoting the polarization of M2 tumor-associated macrophages. Am J Cancer Res. 2017;7:2220-33
219. Freile JA, Avtenyuk NU, Corrales MG, Lourens HJ, Huls G, van Meerten T. et al. CD24 Is a Potential Immunotherapeutic Target for Mantle Cell Lymphoma. Biomedicines. 2022;10:1175
220. Haddad F, Daver N. Targeting CD47/SIRPalpha in Acute Myeloid Leukemia and Myelodysplastic Syndrome: Preclinical and Clinical Developments of Magrolimab. J Immunother Precis Oncol. 2021;4:67-71
221. Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D. et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J Clin Oncol. 2019;37:946-53
222. Shaw TN, Houston SA, Wemyss K, Bridgeman HM, Barbera TA, Zangerle-Murray T. et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J Exp Med. 2018;215:1507-18
223. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J. et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. 2019;363:eaau0964
224. Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG. et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20:29-39
225. Baghdadi M, Yoneda A, Yamashina T, Nagao H, Komohara Y, Nagai S. et al. TIM-4 glycoprotein-mediated degradation of dying tumor cells by autophagy leads to reduced antigen presentation and increased immune tolerance. Immunity. 2013;39:1070-81
226. Zhou Y, Fei M, Zhang G, Liang WC, Lin W, Wu Y. et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity. 2020;52:357-73 e9
227. Divangahi M, Aaby P, Khader SA, Barreiro LB, Bekkering S, Chavakis T. et al. Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat Immunol. 2021;22:2-6
228. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG. et al. Trained immunity: A program of innate immune memory in health and disease. Science. 2016;352:aaf1098
229. Netea MG, Joosten LAB. Trained Immunity and Local Innate Immune Memory in the Lung. Cell. 2018;175:1463-5
230. Kalafati L, Kourtzelis I, Schulte-Schrepping J, Li X, Hatzioannou A, Grinenko T. et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell. 2020;183:771-85 e12
231. Geller AE, Shrestha R, Woeste MR, Guo H, Hu X, Ding C. et al. The induction of peripheral trained immunity in the pancreas incites anti-tumor activity to control pancreatic cancer progression. Nat Commun. 2022;13:759
232. Ding C, Shrestha R, Zhu X, Geller AE, Wu S, Woeste MR. et al. Inducing trained immunity in pro-metastatic macrophages to control tumor metastasis. Nat Immunol. 2023;24:239-54
233. Wang T, Zhang J, Wang Y, Li Y, Wang L, Yu Y. et al. Influenza-trained mucosal-resident alveolar macrophages confer long-term antitumor immunity in the lungs. Nat Immunol. 2023;24:423-38
234. Priem B, van Leent MMT, Teunissen AJP, Sofias AM, Mourits VP, Willemsen L. et al. Trained Immunity-Promoting Nanobiologic Therapy Suppresses Tumor Growth and Potentiates Checkpoint Inhibition. Cell. 2020;183:786-801 e19
235. Lei A, Yu H, Lu S, Lu H, Ding X, Tan T. et al. A second-generation M1-polarized CAR macrophage with antitumor efficacy. Nat Immunol. 2024;25:102-16
236. Zhang L, Tian L, Dai X, Yu H, Wang J, Lei A. et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol. 2020;13:153
237. Chen Y, Zhu X, Liu H, Wang C, Chen Y, Wang H. et al. The application of HER2 and CD47 CAR-macrophage in ovarian cancer. J Transl Med. 2023;21:654
238. Shah Z, Tian L, Li Z, Jin L, Zhang J, Li Z. et al. Human anti-PSCA CAR macrophages possess potent antitumor activity against pancreatic cancer. Cell Stem Cell. 2024;31:803-17 e6
239. Castellarin M, Sands C, Da T, Scholler J, Graham K, Buza E. et al. A rational mouse model to detect on-target, off-tumor CAR T cell toxicity. JCI insight. 2020;5:e136012
240. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38:947-53
241. Reiss KA, Angelos MG, Dees EC, Yuan Y, Ueno NT, Pohlmann PR. et al. CAR-macrophage therapy for HER2-overexpressing advanced solid tumors: a phase 1 trial. Nat Med. 2025
242. Chen L, Yin G, Wang Z, Liu Z, Sui C, Chen K. et al. A predictive radiotranscriptomics model based on DCE-MRI for tumor immune landscape and immunotherapy in cholangiocarcinoma. Biosci Trends. 2024;18:263-76
Author contact
![]() Corresponding authors: Xiaoyu Zhang: zhangxiaoyu_eycom. Huanbin Zhao: huanbinzhaocom or huanbin.zhaoorg. Houjun Xia: Hj.xiaac.cn.
Corresponding authors: Xiaoyu Zhang: zhangxiaoyu_eycom. Huanbin Zhao: huanbinzhaocom or huanbin.zhaoorg. Houjun Xia: Hj.xiaac.cn.