Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and methods
3. Results
4. Discussion
5. Conclusion
Supplementary Material
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(7):3030-3044. doi:10.7150/ijbs.103971 This issue Cite
Research Paper
Theaflavins attenuate iron overload-induced liver oxidative injury by inhibiting hepatocyte ferroptosis
Junzhou Chen1,2,#, Mingdao Mu3,#, Xin Lai1,2, Chen Liu1,2, Yuheng Luo1,2, Jun He1,2, Bing Yu1,2, Quyuan Wang1,2, Huifen Wang1,2, Daiwen Chen1,2, ![]() , Aimin Wu1,2,
, Aimin Wu1,2, ![]()
1. Institute of Animal Nutrition, Sichuan Agricultural University, Chengdu 611130, China.
2. Key Laboratory for Animal Disease-resistance Nutrition of China Ministry of Education, Sichuan Agricultural University, Chengdu 611130, China.
3. School of Medicine, Southeast University, Nanjing 210096, China.
#These authors contributed equally to this work.
Received 2024-9-21; Accepted 2025-1-2; Published 2025-4-21
Abstract

Liver, as a major iron storage organ, is particularly sensitive to oxidative stress-induced damage stemming from iron overload. Thus, antioxidant therapies are often used to reverse oxidative stress-induced tissues damage, however, the cellular mechanisms remain enigmatic. This study investigated the protective effects and mechanisms of theaflavins, a nature production from tea, against oxidative damage in iron overload hepatocytes and mouse liver. Iron overload disrupted iron metabolism in hepatocytes by activating inflammation and enhancing HO-1 expression, leading to hepatic ferroptosis and serious liver damage. Additionally, iron overload inhibited Xc- system (SLC7A11 and SLC3A2), decreasing GSH synthesis, ultimately further induced ferroptosis. Intriguingly, theaflavins supplementation robustly counteracted iron overload-induced ferroptosis and subsequent liver damage. Notably, inhibition of HO-1 and activation of Xc- system provided the mechanistic insights into theaflavins inhibition of hepatocytes ferroptosis. Take together, these results highlight ferroptosis as an inducer of iron overload-induced liver damage, which is inhibited by theaflavins. This nature product form tea represents a potential therapeutic approach to attenuating organ damage in iron overload individuals.
Keywords: iron overload, ferroptosis, liver injury, theaflavins, antioxidant
1. Introduction
Iron is a vital nutrient that plays a pivotal role in various physiological processes, including oxygen transport, energy metabolism, and cellular proliferation [1, 2]. However, an abundance of iron in the body, referred to as iron overload, can result in significant organ damage, especially in the liver [3]. The liver is the primary organ responsible for iron storage and regulation, making it highly susceptible to iron overload-induced injury [4]. Mounting evidence suggests a close association between iron overload and a wide spectrum of liver diseases, such as hereditary hemochromatosis [3], nonalcoholic fatty liver disease, and hepatitis C virus infection [4]. Moreover, patients with chronic liver disease often exhibit disturbed iron homeostasis, further exacerbating the progression of liver injury [5].
Recent years have witnessed substantial advancements in unraveling the molecular processes responsible for liver damage caused by iron overload. One of the key pathways implicated in this process is ferroptosis, a novel form of cell death closely linked with free iron (Fe2+)-mediated lipid peroxidation [6]. Ferroptosis is triggered by the accumulation of reactive oxygen species (ROS) and lipid peroxidation products, leading to catastrophic cell membrane damage and ultimately cell death [7, 8]. Emerging evidence indicates that ferroptosis is intricately linked to various biological processes, including iron metabolism, amino acid and polyunsaturated fatty acid metabolism, and the biosynthesis of glutathione (GSH), NADPH, and coenzyme Q10 [9]. Disruption of these pathways has been shown to contribute to the onset and progression of ferroptosis [10]. Notably, ferroptosis has been implicated in the pathogenesis of several liver disorders associated with iron overload [8]. Therefore, targeting ferroptosis represents a promising therapeutic strategy for mitigating iron overload-induced liver injury [11].
Theaflavins is a group of natural polyphenolic compounds derived from black and oolong teas [12]. Theaflavins is formed during the fermentation process of tea leaves, where catechins, such as epicatechin and epigallocatechin-3-gallate, undergo oxidation by polyphenol oxidases and peroxidases [13]. The resulting theaflavins possesses potent antioxidant properties attributed to their unique structural features [13]. Meanwhile, theaflavins commonly found in black tea, are frequently highlighted in scientific literature for their ability to chelate iron ions effectively. This chelation property allows theaflavins to bind to free iron, thereby reducing iron's availability to participate in harmful reactions such as the Fenton reaction, which can lead to oxidative stress and cellular damage [14]. Accumulating evidence suggests that theaflavins exerts a wide range of biological activities, including neuroprotective, cardioprotective, nephroprotective and anti-inflammatory, antimicrobial effects [15]. These pleiotropic actions of theaflavins are mediated through the modulation of various cellular signaling pathways, such as the activation of nuclear factor erythroid 2-related factor 2 (Nrf2)/Kelch-like ECH-associated protein 1 (Keap1) signaling cascade and the inhibition of mitogen-activated protein kinase (MAPK) signaling [16]. While the liver-protective properties of theaflavins have been documented, their capacity to mitigate iron overload-induced liver damage by suppressing ferroptosis remain largely uninvestigated.
This study aimed to explore the protective effects of theaflavins against liver damage caused by iron overload and to elucidate the underlying molecular mechanisms, with a particular emphasis on ferroptosis. Our findings revealed that iron overload triggered liver injury by activating ferroptosis in hepatocytes. Activation of HO-1 and inhibition of system Xc- (SLC3A2 and SLC7A11) provided a mechanistic explanation for this phenomenon. Remarkably, theaflavins demonstrated a pronounced inhibitory effect on ferroptosis by downregulating HO-1 expression and activating system Xc-, thereby protecting iron overload-induced liver injury. These findings highlight the therapeutic potential of theaflavins as a natural agent for the prevention and treatment of iron overload-related liver disease.
2. Materials and methods
2.1. Chemicals and antibodies
Theaflavins (purity ≥ 98%) were sourced from Teaturn Bio-pharmaceutical Co., Ltd (Fig. 1A). The addition dosage of theaflavins is based on previous research [17]. Primary antibodies targeting TfR1, Fpn, FTH/L, GPX4, SLC7A11 and HO-1 were acquired from Abcam, while β-actin was procured from Santa Cruz Biotechnology.
Effects of theaflavins supplementation on growth performance and blood parameters in mice fed high-iron diets. (A) Structural formula of theaflavins. (B) Overview of the experimental design and treatment concentrations. (C) Body weight of mice in four groups measured throughout the experiment. Mouse body weight was recorded weekly for the first three weeks and daily after initiating gavage administration. (D) Survival curves of mice at 8 weeks of the experiment. (E) Red blood cell count (RBC). (F) Hemoglobin (HGB). (G) Hematocrit (HCT). (H) Mean corpuscular volume (MCV). (I) Mean corpuscular hemoglobin (MCH). (J) Serum iron level. (K) Unsaturated iron binding capacity (UIBC). (L) Total iron binding capacity (TIBC). (M) Transferrin saturation (TF). Statistical significance is indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
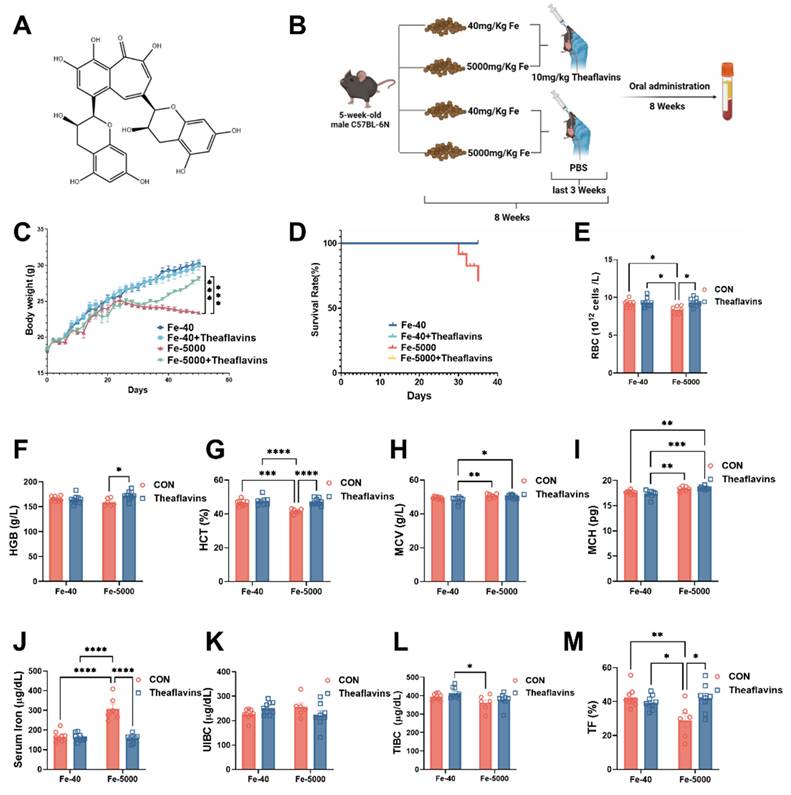
2.2. Animal experiments
The animal experimental protocols were authorized by the Institutional Animal Care and Use Committee of the Laboratory Animal Center at Sichuan Agricultural University (SICAU-2015-033). For this study, 5-week-old male C57BL/6N mice were used and maintained in a controlled setting with a temperature of (21 ± 2) °C, humidity between 45-55%, and a 12-hour light/dark cycle. The mice were randomly divided into a 2*2 factorial arrangement and were provided diets containing either 40 mg/kg or 5000 mg/kg FeSO4 [18, 19]. Additionally, they were administered either 0 or 10 mg /kg theaflavins via gavage during the final three weeks of the experiment. The duration of the experiment was 8 weeks, after which all mice were euthanized (see Fig. 1B).
2.3. Isolation and culture of mouse primary hepatocytes
Mouse primary hepatocytes were isolated and cultured using the following method: liver tissue was digested with collagenase type II (Gibco) to separate the hepatocytes [20, 21]. Live hepatocytes were then isolated by centrifugation using Percoll (Sigma-Aldrich). These hepatocytes were plated onto 6-well or 12-well plates that had been pre-coated with collagen. The primary hepatocytes were initially cultured in DMEM medium for 24 hours, after which the wells were washed once with PBS. Fresh DMEM medium was then added, and the cells were exposed to various reagents. Cells were harvested at specific time points for analysis.
2.4. Determination of hematological parameters
Whole blood samples were collected into EDTA-coated tubes, and hematological parameters, including red blood cell count (RBC), hemoglobin (HGB), hematocrit (HCT), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH) and mean corpuscular hemoglobin concentration (MCHC) were measured using the automatic biochemical analyzer (Shenzhen Myriad Biomedical Electronics Co, China).
2.5. Assessment of iron status
Serum iron levels and tissue non-heme iron concentrations were determined. The quantification of non-heme iron content in liver and spleen was conducted using a previously described method [22]. Blood samples were analyzed to determine serum iron levels, total iron-binding capacity (TIBC), unsaturated iron-binding capacity (UIBC), and serum transferrin (TF) concentrations using a standardized procedure [1].
2.6. ALT, AST and enzyme activity assay
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were measured using an automatic biochemical analyzer to assess liver injury. To assess oxidative stress, malondialdehyde (MDA) and glutathione (GSH) levels were measured using commercially available assay kits from Jiancheng Bioengineering Institute. The assays were performed according to the manufacturer's protocols.
2.7. Real-time quantitative PCR (qPCR) and western blot
Hepatic total RNA extraction was carried out by Trizol reagent (Invitrogen) according to the manufacturer 's specifications, and the mRNA diluted to 1 μg/μl. The working solution was prepared following the protocols provided in the Reverse Transcription Kit (Takara). Relative gene expression levels were determined using the ΔΔCT method, calculating cycle threshold (Ct) values and normalizing them to the housekeeping gene Hprt. Total protein was extracted from the liver using RIPA buffer supplemented with phenylmethanesulfonylfluoride (PMSF, 1 mM). Western blotting was performed according to a previously established protocol. Primary antibodies were diluted 1:1000, and secondary goat anti-mouse and anti-rabbit antibodies conjugated with horseradish peroxidase (HRP) (Santa Cruz) were diluted 1:3000.
2.8. Histological analysis
Liver tissue samples were collected and preserved in 4% paraformaldehyde solution in PBS overnight. After fixation, the tissues were embedded in paraffin. Subsequently, 5-μm thick sections were prepared and stained with hematoxylin and eosin (H&E) as well as Masson's trichrome stain (Masson) for microscopic examination and histological analysis.
2.9. Measurement of lipid ROS and total ROS
To assess cellular levels of reactive oxygen species (ROS) and lipid ROS, primary hepatocytes were treated with 50 μM H2DCFDA (Sigma-Aldrich) and 5 μM CD11-BODIPY (Invitrogen), respectively, for 30 minutes at 37°C. Following incubation, the cells underwent three washes with PBS. Subsequently, the fluorescence intensity and analysis of cells were performed using flow cytometry.
2.10. Determination of labile iron pool (LIP)
FerroFarRedTM (Goryo Chemical) was employed to test the intracellular labile iron (Fe2+) of primary hepatocytes following the manufacturer's provided guidelines. Cells were incubated with 1 μM FerroOrange for 30 min at 37°C, washed with PBS, and analyzed by flow cytometry.
2.11. Hmox1 knockdown
Short hairpin RNAs (shRNAs) targeting mouse Hmox1 (Ho-1-shRNA: AGCCACACAGCACTATGT AAA) were incorporated into the pAdEasy-U6-CMV-mCherry adenovirus vector (HanBio) following the manufacturer's protocol. Recombinant adenovirus amplification was carried out using HEK 293A cells. Primary hepatocytes were seeded in 12-well plates and transduced with Hmox1-shRNA adenoviruses for 24 h before any additional treatment.
2.12. Adenovirus overexpression of Hmox1
Adenoviruses expressing Hmox1 (Ad-Hmox1) were purchased from HanBio (4.0×1010 PFU/ml). Primary hepatocytes were seeded in 12-well plates and transduced with Ad-Hmox1 for 24 h before any additional treatment.
2.13. Statistical analysis
Statistical analyses were performed using GraphPad Prism. All data are presented as means ± standard errors of the means (SEM). Statistical tests comprised unpaired two-tailed Student's t-test and one-way or two-way analysis of variance (ANOVA), as appropriate, followed by Bonferroni post hoc tests. Distribution comparisons were performed using a Kolmogorov-Smirnov test. A p-value less than 0.05 was considered statistically significant. All results were visualized using GraphPad Prism 9.5.0 software.
3. Results
3.1. Theaflavins supplementation ameliorates growth performance decline and inflammatory anemia in mice fed high-iron diets
The experimental procedure and feeding protocol are shown in Fig. 1B. We found that theaflavins played a crucial role in mitigating the decline in growth performance and inflammatory anemia induced by iron overload. Mice in the iron-overloaded group began to lose weight around the fourth week, and by the eighth week, a significant difference in body weight compared to the control group mice was evident. However, oral administration of 200 µL of 10 mg/kg/day theaflavins significantly restored the weight loss caused by iron overload (Fig. 1C). Notably, the iron overload group had a 30% mortality rate at the end of the experiment, a phenomenon not observed in the other treatment groups (Fig. 1D). Iron overload led to a significant decrease in the levels of RBC, HGB, and HCT, all of which were restored normal levels following theaflavins supplementation. Additionally, the levels of MCV, MCH, and HCT levels showed a significant decrease after theaflavins intervention, while the levels of MCV and MCH did not change significantly (Fig. 1E to I). Iron overload consistently increased serum iron and decreased TIBC and TF% compared to the control group, with these serum iron-related indices significantly returning to normal levels following theaflavins administration (p < 0.01). However, UIBC did not exhibit significant changes (Fig. 1J to M).
3.2. Theaflavins administration reduces inflammatory response and attenuates iron overload-induced liver injury
Iron overload typically results in liver injury [23]. To evaluate the impact of theaflavins on the iron overload-induced liver injury, serum samples and liver tissues were collected for analysis. Mice fed high iron (Fe-5000) diets exhibited a significant increase in liver color, liver index and iron content (Fig. 2A, C and D), but without impact on liver weight (Fig. 2B). Meanwhile, the flow cytometry results revealed a dramatic increase in the levels of C11-BODIPY (a marker of ferroptosis), ROS, and Fe2+ in iron-overloaded primary hepatocytes (Fig. 2E-J). However, theaflavins supplementation significantly decreased C11-BODIPY, ROS, and Fe2+ levels, which further confirmed that theaflavins is an inhibitor of ferroptosis (Fig. 2E-J). Iron overload robustly induced hepatic tissue inflammation by upregulating IL-6, IL-1β, and TNF-α mRNA expression, while theaflavins supplementation remarkably attenuated iron overload induced hepatic tissue inflammation by downregulating IL-6, IL-1β, and TNF-α mRNA expression (Fig. S1A-C). Similar results were observed in the protein expression levels of IL-6, IL-1β, and TNF-α (Fig. 2K Fig. S1D-F). Furthermore, hematoxylin and eosin (H&E) staining Perls staining and Masson's trichrome staining of the liver tissues revealed that iron overload induced liver injury and fibrosis (Fig. 2L and M Fig. S1G), findings further supported by elevated serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (Fig. 2N and O). Interestingly, theaflavins administration significantly mitigated iron overload-induced liver injury (Fig. 2L-O). Briefly, high-dose iron leads to hepatic tissue inflammation and injury in mice, while theaflavins addition effectively alleviates iron overload-induced liver inflammation and injury (Fig. 2P).
Prophylactic effect of theaflavins on high iron-induced liver injury. (A) Gross morphology of intact liver from different treatment groups. (B) Liver weight in different treatment groups. (C) Liver index (liver weight/body weight ratio). (D) Hepatic iron content. (E) Flow cytometry results histogram of total ROS in hepatocytes. (F) Positive rate of total ROS in hepatocytes. (G) Flow cytometry images of lipid peroxidation in hepatocytes. (H) Positive rate of lipid peroxidation in hepatocytes. (I) Flow cytometry histogram of labile iron pool (LIP) in HO-1 knockout hepatocytes. (J) Positive rate of LIP in hepatocytes. (K) Statistical Analysis Western blotting was performed to detect IL-6, IL-1β, and TNF-α protein expression. (L-M) Representative images of hematoxylin and eosin (H&E) staining and Perls staining of mouse liver sections. (N-O) Serum levels of ALT and AST. (P) Overview of Liver injury and inflammatory response. Statistical significance is indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
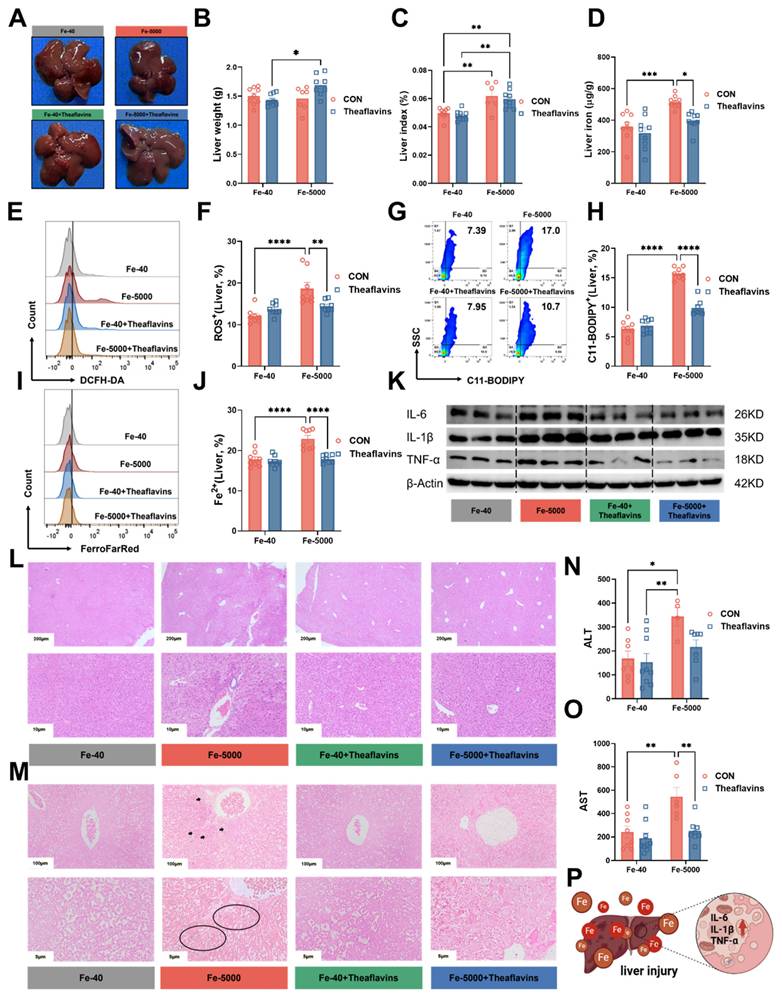
3.3. Theaflavins supplementation inhibits hepatic ferroptosis induced by iron overload in vivo
Ferroptosis is a complex process of cell death triggered by iron-dependent lipid peroxidation [6, 23]. The consequences of this process are profound, leading to changes in cellular biochemistry measurable through indicators like glutathione (GSH) and malondialdehyde (MDA) levels [24]. In this study, iron overload markedly reduced GSH contents while elevating MDA levels in mice liver. However, both indicators were significantly restored after theaflavins treatment (p < 0.01), suggesting theaflavins as a potential an inhibitor of ferroptosis (Fig. 3A-B). Simultaneously, an increase in Ptgs2 (a marker gene for ferroptosis) gene expression was observed in the livers of mice in the iron overload group, which was significantly reduced by the intervention of theaflavins (Fig. 3C). To further investigate this phenomenon, the expression of key genes and pathways involved in ferroptosis was analyzed in the livers of mice with iron overload, with or without theaflavins treatment. The results showed that iron overload increased the mRNA and protein expression of HO-1. Notably, this increase was normalized after theaflavins intervention (Fig. 3D, E and M). A similar result was observed in the mRNA and protein expression of acyl-CoA synthetase long-chain family member 4 (ACSL4), a ferroptosis activator that shapes cellular lipid composition (Fig. 3D and F). Iron overload also significantly increased the protein and gene expression of FTH/L and SLC40A1 in the iron metabolic pathway, which was rescued by theaflavins, indicating theaflavins' ability to inhibit ferroptosis by restoring iron metabolism (Fig. 3D, H-I, M). Iron overload significantly down-regulated the gene expression of TfR1, while the addition of theaflavins restored the expression of TfR1 without significant changes in protein levels (Fig. 3D, G, M). Meanwhile, while the protein levels of SLC7A11 remained unchanged, the expression of SLC3A2 and GPX4 was inhibited by iron overload, with theaflavins showing therapeutic potential (Fig. 3D, J-L, M). Furthermore, the expression of TIMP1 genes was elevated under iron overload, further suggesting that the cells may undergo iron metamorphosis (Fig. 3M). Interestingly, the levels of NCOA4 and LC3II proteins were higher in the iron overload group than in the control group, suggesting that ferritinophagy may play an important role in hepatocytes ferroptosis mediated by iron overload. However, the protein expression of NCOA4 and LC3II return to normal after theaflavins administration (Fig. S2A-C).
Theaflavins alleviate high-iron induced liver injury by inhibiting the occurrence of ferroptosis in cells. (A) GSH level in the liver. (B) MDA level in the liver. (C) mRNA expression of Ptgs2. (D) Western blot analysis of ferroptosis-related protein expression. (E-L) Quantification of HO-1, ACSL4, TfR1, SLC40A1, FTH/L, SLC3A2, SLC7A11and GPX4 protein expression. (M) Heatmap of correlation analysis between treatment groups and the genes of ferroptosis. Statistical significance is indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
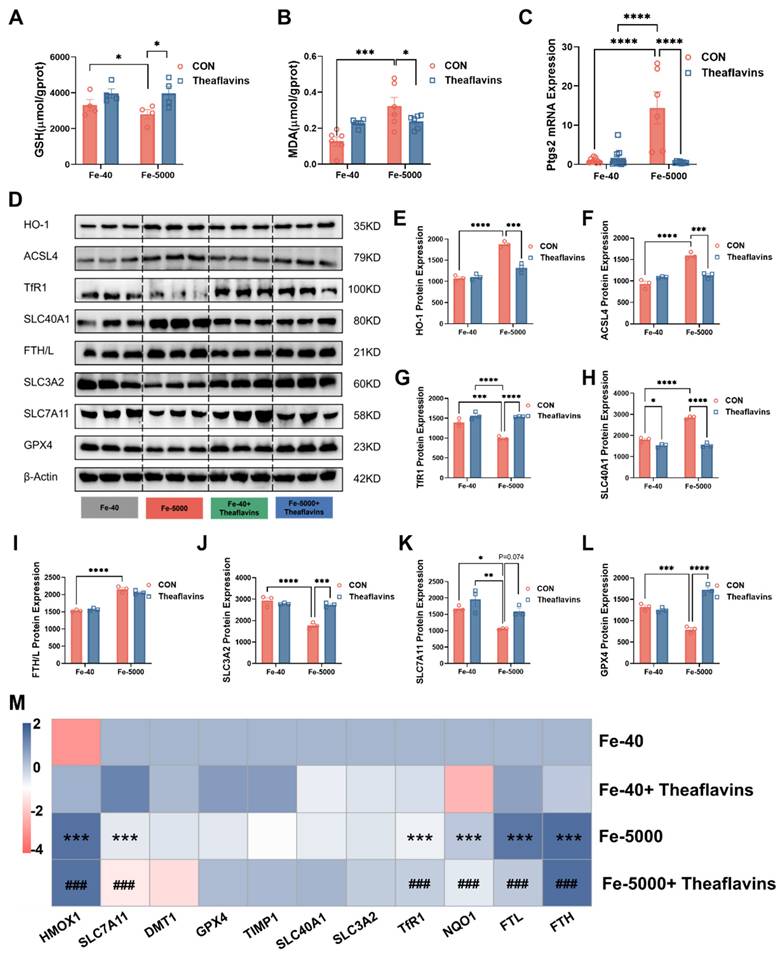
3.4. Theaflavins inhibits iron overload-induced ferroptosis in primary hepatocytes
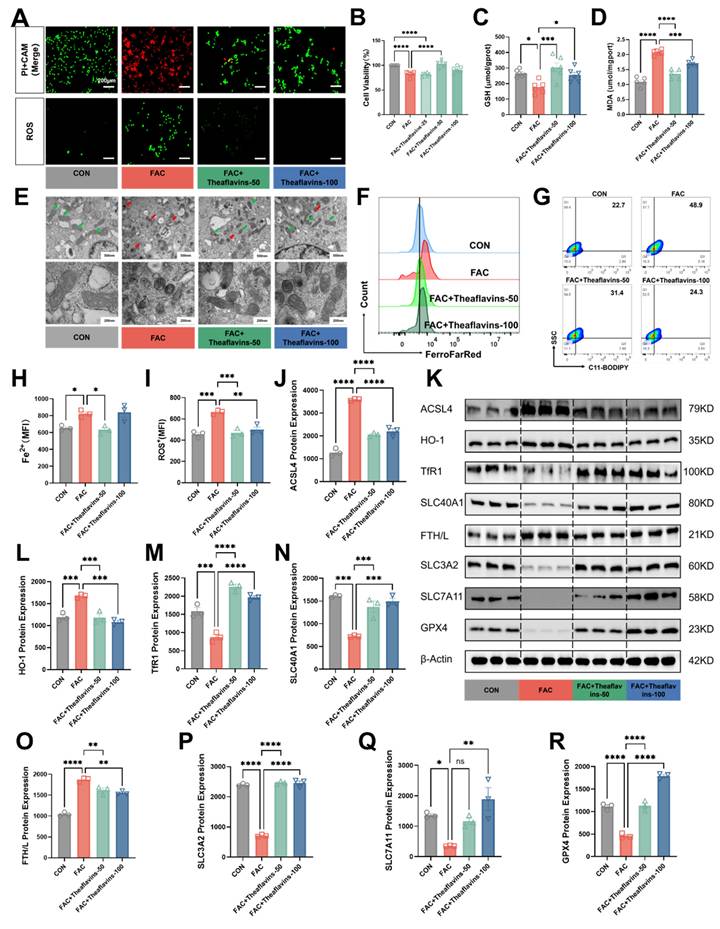
To further clarify the role of theaflavins in ferroptosis induced by iron overload, Ferric ammonium citrate (FAC) at a concentration of 1000 µM was added to the culture medium of primary hepatocytes, followed by the addition of 50 μM and 100 μM theaflavins. Initially, cell viability and cell death of FAC-treated primary hepatocytes were assessed using CAM/PI staining and CCK8 kit. As s shown in Fig. 4A and 4B, treatment with 1000 μΜ FAC markedly decreased the cell viability and increased cell death in primary hepatocytes. However, theaflavins supplementation significantly attenuated high does FAC-induced hepatocyte cell death (Fig. 4A and B). Moreover, FAC treatment decreased GSH content but increased MDA levels in primary hepatocytes, indicating iron overload may induce ferroptosis. Conversely, theaflavins supplementation increased GSH content and decreased MDA levels (Fig. 4C and D). Subsequently, the mitochondrial morphology in the FAC-treated group underwent changes characteristic of ferroptosis, with mitochondria becoming smaller and rounder and cristae beginning to disappear (Fig. 4E). Interestingly, the mitochondrial morphology of the group treated with theaflavins was gradually restored to normal characteristics, further confirming the inhibitory effect of theaflavins on iron overload-induced ferroptosis (Fig. 4E). Meanwhile, the flow cytometry results revealed a dramatic increase in the levels of C11-BODIPY (a maker of ferroptosis), ROS and Fe2+ in FAC-treated primary hepatocytes (Fig. 4A, F-I). However, theaflavins supplementation significantly decreased C11-BODIPY, ROS and Fe2+ level, which further confirmed theaflavins is an inhibitor of ferroptosis (Fig. 4A, F-I). Next, protein expression analysis showed that FAC treatment induced ferroptosis in primary hepatocytes, evidenced by significant changes in the expression of major ferroptosis pathway related proteins (Fig. 4K and S2). Specifically, TfR1, GPX4, SLC7A11, SLC3A2 and SLC40A1 were significantly downregulated, while FTH/L, ACSL4, HO-1, NCOA4 and LC3II were upregulated (Fig. 4J-R, S2D-F). We repeated this experiment using RSL3 as a model of ferroptosis and obtained similar results (Fig. S3). Therefore, these results indicate theaflavins is an inhibitor of ferroptosis.
Inhibition of primary hepatocytes ferroptosis by theaflavins. (A) Fluorescently labeled PI+CAM and ROS (200μm). (B) Cell viability assessed by Cell Counting Kit-8 (CCK-8) assay. (C) GSH levels in hepatocytes. (D) MDA levels in hepatocytes. (E) Transmission electron microscopy results (500nm and 200nm). (F) Flow cytometry histogram of labile iron pool (LIP) in hepatocytes. (G) Flow cytometry results images of lipid peroxidation in hepatocytes. (H) Mean fluorescence intensity (MFI) of LIP in hepatocytes. (I) MFI of total ROS in hepatocyte. (J-R) Western blot analysis and quantification of ferroptosis-related and antioxidant-related protein expression: (J) ACSL4, (K) Western blot analysis of ferroptosis-related protein expression. (L) HO-1, (M) TfR1, (N) SLC3A2, (O) FTH/L, (P) SLC40A1, (Q) SLC7A11, and (R) GPX4. Statistical significance is indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
3.5. Theaflavins supplementation inhibits hepatocyte ferroptosis by downregulating HO-1
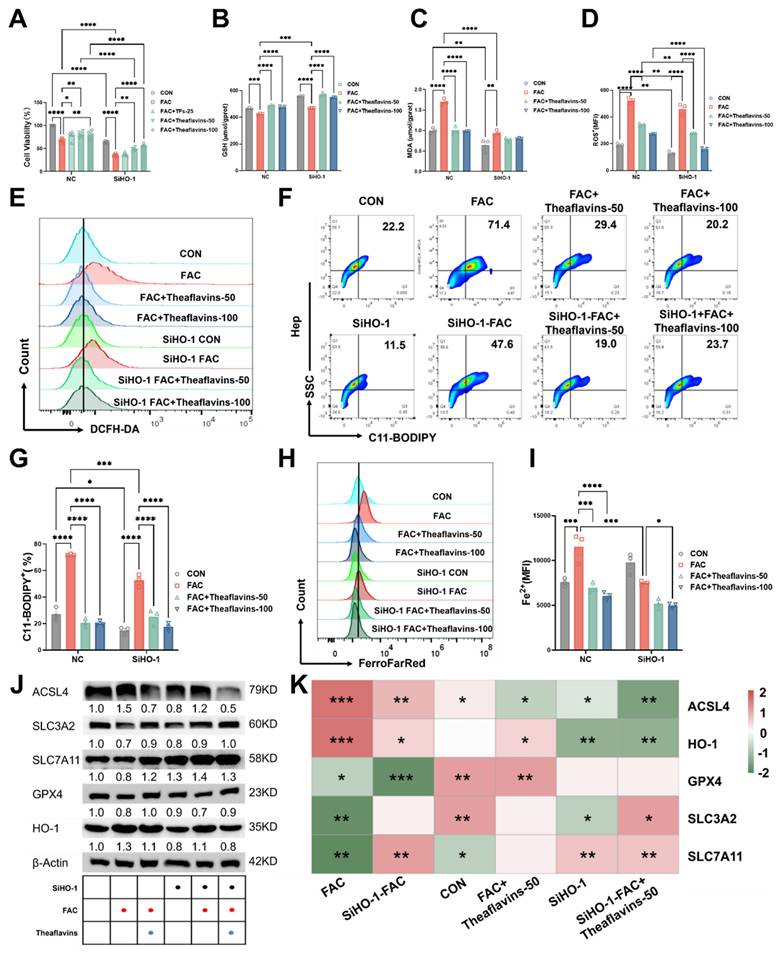
Our prior research revealed that HO-1 exhibits a dual role in ferroptosis and ensuing acute iron overload-induced liver injury resulting from intraperitoneal administration of iron dextran. While it demonstrates certain protective capabilities, its excessive activation can significantly contribute to ferroptosis[1]. This study on significant diet-induced chronic liver damage highlights the need for further exploration of the connection between heme oxygenase-1 (HO-1) and ferroptosis. To further investigate the role of HO-1 in the suppression of ferroptosis mediated by theaflavins, HO-1 was silenced in primary hepatocytes using HO-1 shRNA. In HO-1 knockdown hepatocytes, cell viability was significantly reduced, with a more pronounced decline observed following FAC treatment. However, theaflavin intervention effectively restored cell viability, indicating its protective role (Fig. 5A). Interestingly, knocking down HO-1 resulted in a significant increase in glutathione (GSH) levels, indicating enhanced cellular antioxidant capacity. Conversely, the decrease in malondialdehyde (MDA) levels suggested a reduction in lipid peroxidation (Fig. 5B and C). Notably, FAC treatment, which induces oxidative stress, did not result in significant changes in the HO-1 knockdown cells, indicating that the knockdown HO-1conferred resistance to FAC-induced oxidative damage. This protective effect was further supported by lower ROS, C11-BODIPY, and Fe2+ levels in the FAC-treated HO-1 knockdown cells compared to FAC-treated normal cells (Fig. 5D-I). All the three indexes showed a significant decrease after theaflavins intervention, with the effect of the theaflavins-treated group being more pronounced in the HO-1 knockdown cells compared to normal cells (Fig. 5D-I). Interestingly, knockdown of HO-1 significantly reduced ACSL4 protein expression in the FAC-treated group compared to the normal FAC-treated group, thereby preventing ACSL4 from catalyzing cellular lipid peroxidation to inhibit ferroptosis, and up-regulated the expression of SLC3A2 and SLC7A11 proteins, while GPX4 protein did not change significantly (Fig. 5J-K). Most notably, HO-1 expression was significantly decreased in the FAC-treated group after HO-1 knockdown, with a continued decreasing trend after theaflavins treatment (Fig. 5J-K). Similarly, the same decrease in GSH and increase in MDA after knockdown of HO-1 was observed under the RSL3 ferroptosis model. The promotion of ferroptosis by HO-1 was also further confirmed by flow cytometry results (Fig. S4).
The effects of knockdown of HO-1 on inhibition of ferroptosis by theaflavins. (A) Cell viability assessed by Cell Counting Kit-8 (CCK-8) assay. (B) GSH levels in HO-1 knockout hepatocytes. (C) MDA levels in HO-1 knockout hepatocytes. (D) MFI of total ROS in HO-1 knockout hepatocytes. (E) Flow cytometry results histogram of total ROS in HO-1 knockout hepatocytes. (F) Flow cytometry images of lipid peroxidation in HO-1 knockout hepatocytes. (G) Positive rate of lipid peroxidation in HO-1 knockout hepatocytes. (H) Flow cytometry histogram of labile iron pool (LIP) in HO-1 knockout hepatocytes. (I) MFI of LIP in HO-1 knockout hepatocytes. (J) Western blot analysis of antioxidant pathway-related protein expression in HO-1 knockout hepatocytes. (K) Heatmap of correlation analysis between treatment groups and proteins related to the ferroptosis pathway.
Subsequently, HO-1 was overexpressed in primary hepatocytes using adenovirus-HO-1 (Fig. 6). Primary hepatocytes with HO-1 overexpression were treated with FAC and theaflavins for 6 hours, and ferroptosis was assessed by measuring LIP, ROS, and C11-BODIPY levels. In hepatocytes with HO-1 overexpression, cell viability was markedly impaired, and this effect was further exacerbated by FAC treatment. Theaflavin intervention, however, significantly ameliorated the loss of cell viability (Fig. 6A).
Effect of HO-1 overexpression on the inhibition of cellular ferroptosis by theaflavins. (A) Cell viability assessed by Cell Counting Kit-8 (CCK-8) assay. (B) GSH levels in HO-1 overexpressed hepatocytes. (C) MDA levels in HO-1 overexpressed hepatocytes. (D) Positive rate of total ROS in HO-1 overexpressed hepatocytes. (E) Flow cytometry histogram of total reactive oxygen species (ROS) in HO-1 overexpressed hepatocytes. (F) Flow cytometry images of lipid peroxidation in HO-1 overexpressing hepatocytes. (G) Positive rate of lipid peroxidation in HO-1 overexpressed hepatocytes. (H) Flow cytometry results histogram of labile iron pool (LIP) in HO-1 overexpressed hepatocytes. (I) Positive rate of LIP in HO-1 overexpressed hepatocytes. (J) Expression level of proteins related to antioxidant pathway. (K) Heatmap of correlation analysis between treatment groups and proteins related to the ferroptosis pathway. Statistical significance is indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
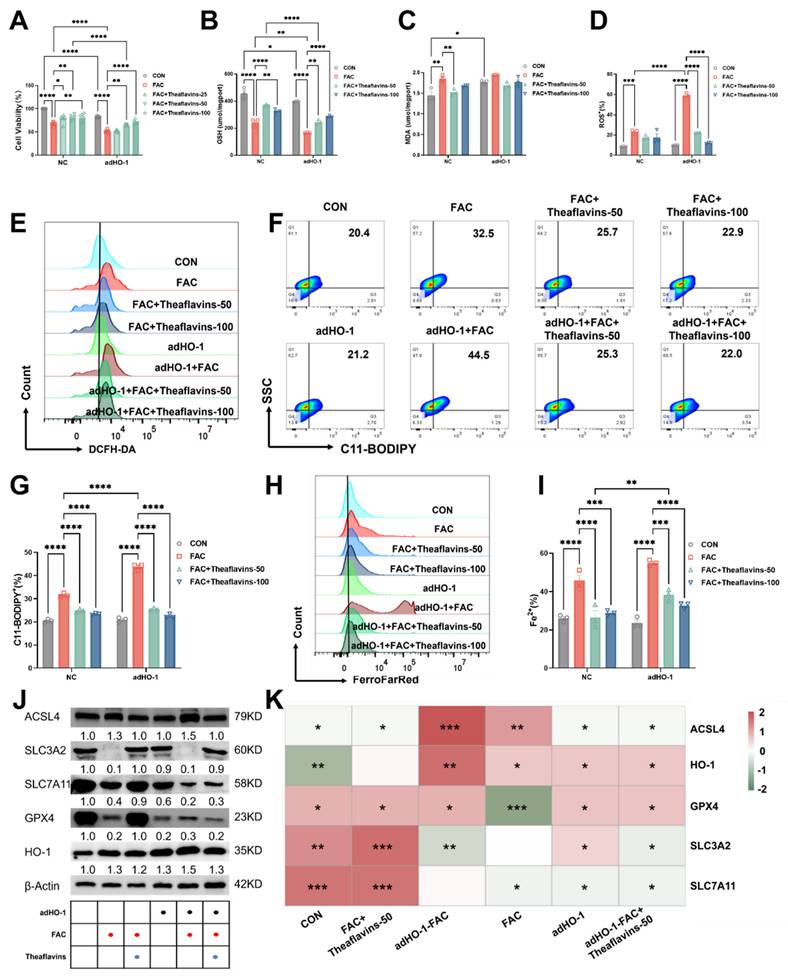
In contrast to HO-1 knockdown, HO-1 overexpression significantly upregulated ferroptosis-related indexes, such as Fe2+, ROS, C11-BODIPY and MDA levels, but decreased GSH content (Fig. 6B-I), indicating HO-1 overexpression further activated ferroptosis in hepatocytes. Interestingly, theaflavins supplementation effectively mitigated ferroptosis induced by HO-1 expression, decreasing the levels of Fe2+, ROS, C11-BODIPY and MDA, but increasing GSH content in primary hepatocytes (Fig. 6B-I). Consistent with our previous findings, theaflavins upregulated the expression of SLC3A2 protein in primary hepatocytes overexpressing HO-1, while downregulating the expression of ACSL4, SLC7A11, and GPX4 (Fig. 6J-K). The same results were corroborated in the RSL3 model, confirming the mechanism by which theaflavins inhibit cellular ferroptosis by inhibiting HO-1 (Fig. S5). Taken together, our findings suggest that theaflavins suppress iron overload-induced ferroptosis in hepatocytes by downregulating heme oxygenase-1 (HO-1) expression.
4. Discussion
Iron, an essential mineral for the human body, plays a crucial role in various biological processes. Unlike other nutrients, the body lacks an active pathway for iron excretion, meaning that any excess iron consumed is stored within the body [25, 26], which can lead to serious diseases, including organ failure [26-28]. The liver and heart are particularly vulnerable to iron overload. The liver is essential for maintaining iron equilibrium within the body. Consequently, any impairment of liver function, particularly in individuals with chronic liver disease, can lead to substantial alterations in iron regulation [3, 29]. Iron-induced damage is mainly caused by its interaction with ROS. This interaction generates hydroxyl radicals, highly reactive species that can induce lipid peroxidation, oxidation of amino acid residues, DNA strand scission, and protein degradation. Recent studies suggest that ferroptosis plays a crucial role in the pathogenesis of acute liver failure induced by acetaminophen (APAP) overdose [30]. Another study mentioned that patients with iron overload-induced liver injury had significantly increased levels of oxidative stress, and that the damage caused by iron overload could be alleviated through the use of ferroptosis inhibitors [31]. These findings suggest that the regulation of cellular ferroptosis is an important target for the treatment of liver diseases, including iron overload-induced liver injury.
Our previous studies and those of others have found that iron overload causes liver injury and liver fibrosis by inducing ROS accumulation and activating ferroptosis in hepatocytes [32]. In the present study, we found that high doses of iron decreased the survival rate of mice after 8 weeks of feeding, which may be related to high-iron induced liver injury as well as other internal organ lesions [33]. Additionally, iron overload was observed to result in a significant increase in IL-6, TNF-α and IL-1β protein expression in mice. These cytokines are key elements of the inflammatory cascade response, and their elevated expression suggests that iron overload may exacerbate the inflammatory response in vivo [33]. Iron metabolism is closely related to inflammation. Hepcidin, a key regulator of systemic iron metabolism, is stimulated by the cytokine IL-6. As a result, hepcidin is classified as an acute-phase protein, produced and released by the liver in response to inflammation. During inflammatory conditions, systemic iron balance is disrupted, typically causing hypoferremia or a reduction in serum iron levels, as our research confirms [34]. Ferroptosis occurs when excess iron accumulates in hepatocytes, leading to a surge in ROS production. These reactive oxygen species, in turn, activate lipid peroxidation, a key step leading to ferroptosis. At the same time, iron overload leads to the upregulation of HO-1 and a massive breakdown of heme (hemoglobin) [35]. The massive breakdown of heme further triggers the release of Fe2+. These Fe2+ accumulate within the cell and cause toxic effects on the cell. Excess Fe2+ lead to a disruption of the redox balance within the cell, which in turn triggers cellular ferroptosis. GSH is a key antioxidant that scavenges intracellular free radicals and maintains cellular redox balance. In the presence of iron overload, the function of system Xc- is inhibited, leading to a decrease in the synthesis of GSH [36, 37]. This decrease in GSH weakens the body's antioxidant capacity, making hepatocytes more susceptible to damage from oxidative stress [38] [1]. This oxidative stress not only exacerbates the cytotoxicity induced by iron overload, but also further increases the sensitivity of hepatocytes to ferroptosis. The present study builds on these findings and explores in greater detail the intricate relationship between iron overload, liver injury and ferroptosis.
Previous studies have explored the mechanisms of iron-induced cell death in mice and found that iron overload can lead to ferroptosis. Iron homeostasis is a delicate balance maintained by several key proteins such as TfR1, SLC40A1 and FTH/L, which are essential for cellular health. Once iron overload disrupts this balance, it triggers a cascade of events leading to cellular dysfunction and death. In this case, the inflammatory response is a key factor, closely linked to the molecular triggers of ferroptosis. GPX4, a glutathione peroxidase, plays a key role in attenuating the inflammatory response and preventing ferroptosis [39]. Its down-regulation in the presence of iron overload indicates a compromised antioxidant defense system. Similarly, SLC7A11 and SLC3A2, which are involved in amino acid transport and glutathione synthesis, respectively, were also down-regulated, further suggesting that the antioxidant mechanism is compromised [40, 41]. Iron overload also upregulated the expression of HO-1, ACSL4, TfR1, PTGS2, and NQO1 proteins. These findings suggest that iron overload promotes iron transport and disrupts the antioxidant system [42, 43].
Theaflavins interventions are effective in restoring such changes. By scavenging harmful ROS and suppressing inflammatory signaling pathways, theaflavins may help mitigate the damage caused by iron overload [44]. Theaflavins successfully mitigated the negative impact of elevated iron levels on serum iron and erythropoiesis. Studies have shown that although polyphenols reduce iron absorption through their anti-inflammatory properties, they can still indirectly promote erythropoiesis by modulating inflammatory responses and improving the microenvironment, thereby demonstrating a positive effect on the body's hematopoietic function [45]. The research observed that theaflavins bind to iron with high affinity, preventing its uncontrolled accumulation in the bloodstream. This binding not only stabilizes iron levels but also ensures its efficient delivery to erythroid precursor cells for hemoglobin synthesis [46]. Surprisingly, theaflavins appeared to alleviate liver injury due to iron overload by inhibiting cellular ferroptosis through decreasing the expression of HO-1. In this study, we performed HO-1 knockdown and overexpression in hepatocytes to further validate this result. This observation suggests that theaflavins may act by inhibiting HO-1 expression while activating system Xc-, thereby mitigating the deleterious effects of iron overload [47]. Up-regulation of HO-1 levels in the cell is a hallmark of oxidative stress, with downstream effects especially in the pro-oxidative state [48, 49]. By targeting HO-1, theaflavins can modulate iron metabolism and mitigate iron-induced cell death. Future studies will aim to further elucidate the molecular mechanisms underlying these observations and explore the potential clinical applications of theaflavins in the treatment of iron-overload related disorders.
5. Conclusion
This research revealed that excessive iron accumulation in mice disrupts iron balance and the antioxidant defense system. This disturbance led to ferroptosis in the liver, causing oxidative damage and inflammatory anemia. Theaflavins can effectively attenuate iron overload-induced oxidative damage and ferroptosis by suppressing HO-1 expression while activating system Xc-. These results underscore the promising therapeutic potential of theaflavins in alleviating diseases associated with iron metabolism disorders.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the Joint Funds of the National Natural Science Foundation of China (Grant No. U22A20513) and National Natural Science Foundation of Sichuan Province (Grant No. 2024NSFSC0297).
Author contributions
Junzhou Chen., Mingdao Mu., Xin Lai., Ping Zheng., Hui Yan., Quyuan Wang and Huifen Wang: performed experiments. Junzhou Chen., Mingdao Mu., Daiwen Chen and Aimin Wu: designed experiments. Junzhou Chen., Mingdao Mu., Chen Liu., Junqiu Luo and Jie Yu: wrote the paper. Junzhou Chen., Mingdao Mu., Yuheng Luo., Jun He., Bing Yu., Daiwen Chen and Aimin Wu: interpreted data and modified the paper. Daiwen Chen and Aimin Wu: funding acquisition.
Abbreviations
ALT: alanine transaminase
AST: aspartate transaminase
DMT1: divalent metal transporter 1
RBC: red blood cell count
FAC: ferric citrate
FTH/L: ferritin heavy/light chain
Fpn: ferroportin
GSH: glutathione
GSSG: glutathione, Oxydized
GPX4: glutathione peroxidase 4
HGB: hemoglobin
HCT: hematocrit
HO-1: heme oxygenase 1
MCV: mean corpuscular volume
MCH: mean corpuscular hemoglobin
MCHC: mean corpuscular hemoglobin concentration
MDA: malondialdehyde
NOQ1: quinone oxidoreductase 1
Ptgs2: Prostaglandin G/H synthase 2
ROS: reactive oxygen species
SLC3A2: solute carrier family 3 member 2
SLC7A11: solute carrier family 7 member 11
TF: transferrin saturation
TIBC: total iron binding capacity
TfR1: transferrin receptor 1
UIBC: unsaturated iron binding capacity
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wu A, Feng B, Yu J, Yan L, Che L, Zhuo Y. et al. Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021;46:102131
2. Chen J, Li X, Ge C, Min J, Wang F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 2022;29:467-80
3. Milic S, Mikolasevic I, Orlic L, Devcic E, Starcevic-Cizmarevic N, Stimac D. et al. The role of iron and iron overload in chronic liver disease. Med Sci Monit. 2016;22:2144
4. Goldberg D, Ditah IC, Saeian K, Lalehzari M, Aronsohn A, Gorospe EC. et al. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology. 2017;152:1090-9 e1
5. Yiannikourides A, Latunde-Dada GO. A short review of iron metabolism and pathophysiology of iron disorders. Medicines. 2019;6:85
6. Li J, Cao F, Yin H-l, Huang Z-j, Lin Z-t, Mao N. et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88
7. Cheng Z, Li Y. What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: an update. Chem Rev. 2007;107:748-66
8. Stoyanovsky D, Tyurina Y, Shrivastava I, Bahar I, Tyurin V, Protchenko O. et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? FRBM. 2019;133:153-61
9. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693 -+
10. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149:1060-72
11. Kontoghiorghes GJ, Kontoghiorghe CN. Iron and chelation in biochemistry and medicine: new approaches to controlling iron metabolism and treating related diseases. Cells. 2020;9:1456
12. Takemoto M, Takemoto H. Synthesis of theaflavins and their functions. Molecules. 2018;23:918
13. He H-F. Research progress on theaflavins: Efficacy, formation, and preparation. Food Nutr Res. 2017;61:1344521
14. Brune M, Rossander L, Hallberg L. Iron absorption and phenolic compounds: importance of different phenolic structures. EJCN. 1989;43:547-57
15. Das A, Baidya R, Chakraborty T, Samanta AK, Roy S. Pharmacological basis and new insights of taxifolin: A comprehensive review. Biomedicine & Pharmacotherapy. 2021;142:112004
16. Ma N, Wei W, Fan X, Ci X. Farrerol attenuates cisplatin-induced nephrotoxicity by inhibiting the reactive oxygen species-mediated oxidation, inflammation, and apoptotic signaling pathways. FRONT PHYSIOL. 2019;10:485666
17. Weerawatanakorn M, Lee Y-L, Tsai C-Y, Lai C-S, Wan X, Ho C-T. et al. Protective effect of theaflavin-enriched black tea extracts against dimethylnitrosamine-induced liver fibrosis in rats. F&F. 2015;6:1832-40
18. Yang C, Wu A, Tan L, Tang D, Chen W, Lai X. et al. Epigallocatechin-3-Gallate alleviates liver oxidative damage caused by Iron overload in mice through inhibiting Ferroptosis. Nutrients. 2023;15:1993
19. Ha J-H, Doguer C, Collins JF. Consumption of a high-iron diet disrupts homeostatic regulation of intestinal copper absorption in adolescent mice. Am J Physiol Gastrointest Liver Physiol. 2017;313:G353-G60
20. Zhang Z, Zhang F, Guo X, An P, Tao Y, Wang F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology. 2012;56:961-71
21. Feng B, Jiao P, Helou Y, Li Y, He Q, Walters MS. et al. Mitogen-Activated Protein Kinase Phosphatase 3 (MKP-3)-Deficient Mice Are Resistant to Diet-Induced Obesity. Diabetes. 2014;63:2924-34
22. Wang H, An P, Xie E, Wu Q, Fang X, Gao H. et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66:449-65
23. Mao L, Zhao T, Song Y, Lin L, Fan X, Cui B. et al. The emerging role of ferroptosis in non-cancer liver diseases: hype or increasing hope? Cell Death Dis. 2020;11:518
24. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X. et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369-79
25. Kohgo Y, Ikuta K, Ohtake T, Torimoto Y, Kato J. Body iron metabolism and pathophysiology of iron overload. INT J HEMATOL. 2008;88:7-15
26. Kremastinos DT, Farmakis D. Iron overload cardiomyopathy in clinical practice. Circulation. 2011;124:2253-63
27. Ludwig H, Evstatiev R, Kornek G, Aapro M, Bauernhofer T, Buxhofer-Ausch V. et al. Iron metabolism and iron supplementation in cancer patients. Wien Klin Wochenschr. 2015;127:907-19
28. Garbowski MW, Cabantchik I, Hershko C, Hider R, Porter JB. The clinical relevance of detectable plasma iron species in iron overload states and subsequent to intravenous iron-carbohydrate administration. Am J Hematol. 2023;98:533-40
29. Li L-X, Guo F-F, Liu H, Zeng T. Iron overload in alcoholic liver disease: underlying mechanisms, detrimental effects, and potential therapeutic targets. CMLS. 2022;79:201
30. Wu A, Li M, Chen Y, Zhang W, Li H, Chen J. et al. Multi-enzyme Active Manganese Oxide Alleviates Acute Liver Injury by Mimicking Redox Regulatory System And Inhibiting Ferroptosis. Adv Healthcare Mater. 2024;13:2302556
31. Feng G, Byrne CD, Targher G, Wang F, Zheng MH. Ferroptosis and metabolic dysfunction-associated fatty liver disease: Is there a link? Liver Int. 2022;42:1496-502
32. Lai X, Wu A, Bing Y, Liu Y, Luo J, Yan H. et al. Retinoic acid protects against lipopolysaccharide-induced ferroptotic liver injury and iron disorders by regulating Nrf2/HO-1 and RARβ signaling. FRBM. 2023;205:202-13
33. Wang C, Wang X, Song G, Xing H, Yang L, Han K. et al. A high-fructose diet in rats induces systemic iron deficiency and hepatic iron overload by an inflammation mechanism. J. Food Biochem. 2021;45:e13578
34. Rosenblum SL. Inflammation, dysregulated iron metabolism, and cardiovascular disease. Front Aging. 2023;4:1124178
35. Yang Y, Lin Y, Wang M, Yuan K, Wang Q, Mu P. et al. Targeting ferroptosis suppresses osteocyte glucolipotoxicity and alleviates diabetic osteoporosis. Bone Res. 2022;10:26
36. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32:920-37
37. Sönmez Aydın F, Hukkamlı B, Budak H. Coaction of hepatic thioredoxin and glutathione systems in iron overload-induced oxidative stress. J Biochem Mol Toxicol. 2021;35:e22704
38. Yamada N, Karasawa T, Kimura H, Watanabe S, Komada T, Kamata R. et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020;11:144
39. Zhao T, Yu Z, Zhou L, Wang X, Hui Y, Mao L. et al. Regulating Nrf2-GPx4 axis by bicyclol can prevent ferroptosis in carbon tetrachloride-induced acute liver injury in mice. Cell Death Discov. 2022;8:380
40. Wang Y, Wang T, Xiang Q, Li N, Wang J, Liu J. et al. GPR116 promotes ferroptosis in sepsis-induced liver injury by suppressing system Xc-/GSH/GPX4. Cell Biol Toxicol. 2023;39:3015-30
41. Yang L, Ye F, Liu J, Klionsky DJ, Tang D, Kang R. Extracellular SQSTM1 exacerbates acute pancreatitis by activating autophagy-dependent ferroptosis. Autophagy. 2023;19:1733-44
42. Grube J, Woitok MM, Mohs A, Erschfeld S, Lynen C, Trautwein C. et al. ACSL4-dependent ferroptosis does not represent a tumor-suppressive mechanism but ACSL4 rather promotes liver cancer progression. Cell Death Dis. 2022;13:704
43. Wang C, Liu T, Tong Y, Cui R, Qu K, Liu C. et al. Ulinastatin protects against acetaminophen-induced liver injury by alleviating ferroptosis via the SIRT1/NRF2/HO-1 pathway. AM J TRANSL RES. 2021;13:6031
44. Zhan J, Cao H, Hu T, Shen J, Wang W, Wu P. et al. Efficient preparation of black tea extract (BTE) with the high content of theaflavin mono-and digallates and the protective effects of BTE on CCl4-induced rat liver and renal injury. J Agric Food Chem. 2021;69:5938-47
45. Xu T, Zhang X, Liu Y, Wang H, Luo J, Luo Y. et al. Effects of dietary polyphenol supplementation on iron status and erythropoiesis: a systematic review and meta-analysis of randomized controlled trials. AJCN. 2021;114:780-93
46. Srole DN, Ganz T. Erythroferrone structure, function, and physiology: Iron homeostasis and beyond. J Cell Physiol. 2021;236:4888-901
47. Chen Y, Zhang P, Chen W, Chen G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol Lett. 2020;225:9-15
48. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. CMLS. 2016;73:3221-47
49. Silva-Islas CA, Maldonado PD. Canonical and non-canonical mechanisms of Nrf2 activation. Pharm Res. 2018;134:92-9
Author contact
![]() Corresponding authors: Daiwen Chen and Aimin Wu; E-mail address: dwchenedu.cn; wuaimin0608com; wuaiminedu.cn.
Corresponding authors: Daiwen Chen and Aimin Wu; E-mail address: dwchenedu.cn; wuaimin0608com; wuaiminedu.cn.