Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(4):298-303. doi:10.7150/ijbs.5.298 This issue Cite
This article has been corrected. See Int J Biol Sci. 2009; 5(6): 621.
Review
Old can be new again: HAPPY whole genome sequencing, mapping and assembly
Zhihua Jiang1 ![]() , Daniel S. Rokhsar2, Richard M. Harland2
, Daniel S. Rokhsar2, Richard M. Harland2
1. Department of Animal Sciences and Center for Reproductive Biology, Washington State University, Pullman, WA 99164-6351, USA
2. Department of Molecular & Cell Biology, University of California Berkeley, Berkeley, CA 94720-3200, USA
Received 2009-3-13; Accepted 2009-4-12; Published 2009-4-15
Abstract
During the last three decades, both genome mapping and sequencing methods have advanced significantly to provide a foundation for scientists to understand genome structures and functions in many species. Generally speaking, genome mapping relies on genome sequencing to provide basic materials, such as DNA probes and markers for their localizations, thus constructing the maps. On the other hand, genome sequencing often requires a high-resolution map as a skeleton for whole genome assembly. However, both genome mapping and sequencing have never come together in one pipeline. After reviewing mapping and next-generation sequencing methods, we would like to share our thoughts with the genome community on how to combine the HAPPY mapping technique with the new-generation sequencing, thus integrating two systems into one pipeline, called HAPPY pipeline. The pipeline starts with preparation of a HAPPY panel, followed by multiple displacement amplification for producing a relatively large quantity of DNA. Instead of conventional marker genotyping, the amplified panel DNA samples are subject to new-generation sequencing with barcode method, which allows us to determine the presence/absence of a sequence contig as a traditional marker in the HAPPY panel. Statistical analysis will then be performed to infer how close or how far away from each other these contigs are within a genome and order the whole genome sequence assembly as well. We believe that such a universal approach will play an important role in genome sequencing, mapping, and assembly of many species; thus advancing genome science and its applications in biomedicine and agriculture.
Keywords: Genome mapping, next-generation sequencing, HAPPY pipeline, genome assembly.
Introduction
Gene map construction can be dated back to the early 19th century when a gene responsible for red-green color blindness was assigned to the human X chromosome [1]. The idea of mapping a genome became a reality in the mid-1970s, due to recombinant DNA technology that helped develop a variety of strategies to facilitate mapping. In situ hybridization allowed scientists to construct a genome-wide physical map by using DNA probes to directly localize, orientate, and order genes [2]. The utilization of DNA variations as markers resulted in a flood of new markers and an explosion in the knowledge of genes' chromosomal whereabouts [3]. The radiation hybrid technique further advanced physical mapping by providing orders and physical locations of markers with high resolution, but without limitation to polymorphic loci [4]. The HAPPY (HAPloid DNA samples using the PolYmerase chain reaction) technique was developed in the late 1980s [5]. Optical mapping could be considered the latest physical mapping method developed in the late 1990s. It is based on restriction digestion patterns on ensembles of individual DNA molecules derived from a variety of clone types [6]. This method has been employed in mapping of relatively small genomes, up to 400 Mb so far [7].
DNA sequencing methods were also developed in the early 1970s, including Maxam-Gilbert sequencing and Sanger termination sequencing. The former DNA sequencing method is based on chemical modification of DNA and subsequent cleavage at specific bases [8], while the latter method makes use of the 2',3'-dideoxy and arabinonucleoside analogues of the normal deoxynucleoside triphosphates, which act as specific chain-terminating inhibitors of DNA polymerase [9]. The Sanger method appears technically simpler and uses fewer toxic chemicals and lower amounts of radioactivity than the method of Maxam and Gilbert. Such advantages made “Sanger sequencing” the only method of choice for the next 30 years [10].
Generally speaking, whole genome mapping and sequencing hold the same goal; to provide a blueprint for understanding genome structure and function. However, these two systems have never worked together in one pipeline although the former is often used as a “skeleton” for whole genome assembly while the latter provides genetic markers, the basic materials for whole genome mapping. Here we share our thoughts with the genome community on how to combine a mapping technique, HAPPY mapping, with the new-generation of sequencing, thus integrating two systems into one pipeline.
HAPPY Mapping
HAPPY mapping is a genome mapping method based on random DNA breakage and determination of linkage [5,11]. This approach is analogous to classical linkage mapping, except the chromosome breakage and segregation are generated by in vitro analogues. DNA is broken randomly by gamma-irradiation or shearing. Markers are then segregated by diluting the resulting fragments to give aliquots containing approximately one haploid genome equivalent. PCR reactions determine the presence or absence of markers in each aliquot. For each pair of markers, the number of aliquots containing one, both, or neither of the markers is scored based on gel electrophoresis. Lod and θ values between the pair of markers are then determined assuming a Poisson distribution of fragments among aliquots [11]. Readers can visit Dr. Dear's laboratory website at http://www.mrc-lmb.cam.ac.uk/happy/HappyGroup/Methods.html and learn more about the basics of HAPPY mapping; such as how HAPPY works, making a HAPPY panel, calculating the links, making a map for the links, and limits of the method.
HAPPY mapping allows construction of a map of a piece of DNA with no need to clone it, thus avoiding many potential errors and artifacts [12]. Furthermore, the approach can be easily adapted to any desired levels of resolution, in particular, to high resolution genome maps. For example, Konfortov and colleagues [13] used HAPPY mapping to construct a physical map of Dictyostelium chromosome 6 and mapped 300 sequence-tagged sites to the 4-Mb chromosome, giving an average marker spacing of 14 kb. Unlike the radiation hybrid mapping approach, a HAPPY panel contains no carrier DNA, which eases specific PCR amplification of markers and makes multiplexing more amenable. Additionally, HAPPY mapping does not require any polymorphic markers so any piece of DNA can be mapped to a genome region. These advantages make the HAPPY mapping approach applicable to all species, from human [11] to plant, [14] and even to unicellular eukaryotes [15].
Each aliquot in a HAPPY panel essentially contains very dilute genomic DNA. In theory, the amount of DNA is only good for one marker genotyping. Therefore, one needs to find some unlimited way of amplifying the desired content of each aliquot so that it can be re-typed for several thousand markers. Dear and colleagues [5, 11-15] have used three techniques for this purpose: 1) repeat PCR, 2) primer extension preamplification amplification and 3) restriction fragment whole genome PCR. The first method utilizes primers directed to repeat sequence elements, such as human AluI-repeats, while the second method employs a random primer mix (usually 15-mer) in an attempt to amplify the entire DNA content. The restriction fragment whole genome PCR relies on first generating fragments of the genomic DNA by restriction enzyme cleavage followed by ligation of adaptors, which allow amplification using universal PCR primers. As these methods are based on cyclic amplification with Taq polymerase, several drawbacks exist, such as amplification of relatively small fragments (<3 kb in size), high error rate (3 × 10−5) and uneven amplification of the genomic loci (103-106-fold amplification biases) [16]. As stated by the inventor (http://www.mrc-lmb.cam.ac.uk/happy/HappyGroup/methods/panel/amplify.html), the problem “amplification of each aliquot for unlimited use” still remains; making it a bottleneck for 20 years. This is why such a simple and powerful HAPPY mapping method has not yet come into general use since it was invented 20 years ago.
Next-Generation Sequencing
Between 1995 and 2005, two general strategies were developed for sequencing a complete genome: the BAC by BAC sequencing [17] and shotgun sequencing methods [18]. In the former approach, genomic DNA is cut into pieces of about 150 Mb, inserted into BAC vectors and transformed into E. coli for replication. The BAC inserts are then isolated and mapped to determine the order of each cloned 150 Mb fragment using the latter strategy. Shotgun sequencing randomly shears genomic DNA into small pieces, which are then cloned into plasmids and sequenced on both strands, thus eliminating the BAC step from the former approach. Nevertheless, both strategies use the chain-terminator (or Sanger) method for sequencing, which is costly, time consuming, and labor intensive [19]. Therefore, the high demand for low-cost sequencing has driven the development of high-throughput sequencing technologies, called next-generation sequencing, which can generate many hundreds of thousands or even millions of reads in a relatively short time. There are three such technologies that have been commercialized and recently reviewed by Voelkerding and colleagues. [20]: Roche/454 life science (http://www.454.com), Illumina/Solexa (http://www.Illumina.com) and Applied Biosystem/SOLiD (http://solid.appliedbiosystems.com).
These new generation sequencing methods no longer use the Sanger method for sequencing. The 454 technology is based on pyrosequencing and emulsion PCR. Pyrosequencing involves release of a pyrophosphate molecule during incorporation of a nucleotide into a nucleotide acid chain, which is further incorporated into ATP and detected by the production of light [21]. The Solexa technology utilizes a sequencing-by-synthesis approach for sequencing single DNA molecules attached to microspheres. Each sequencing cycle occurs in the presence of all four nucleotides that are incorporated into the oligo-primed cluster fragments by DNA polymerase. These four nucleotides possess reversible fluorophore and termination properties [22]. The SOLiD (supported oligonucleotide ligation and detection) technology is a short-read sequencing method based on ligation [23]. Like other next-generation sequencing methods, DNA fragments for SOLiD sequencing are ligated to oligonucleotide adapters, attached to beads, and clonally amplified by emulsion PCR. Unlike the other platforms, SOLiD then utilizes DNA ligases and a unique approach to sequence the amplified fragments. These next-generation sequencing methods can produce a large amount of sequences in a relatively short time: for example, 500 Mb within 10 hours for Roche 454 GS FLX system, 1.5 Gb within 2.5 days for Illumina Genome Analyzer, and 4 Gb within 6 days for Applied Biosystems SOLiD system [20].
HAPPY Pipeline
Now we propose to combine HAPPY mapping with the new generation sequencing for whole genome sequencing, mapping, and assembly (Figure 1). The process starts with preparation of a HAPPY panel. A HAPPY mapping panel is simply a collection (usually 96) of samplings of genomic DNA, each representing a random subset (less than a complete set) of DNA fragments from a genome [5, 11]. The DNA (the grey cylinders) can be broken into random fragments, using either radiation or mechanical shearing. Generally speaking, it is very likely for genes/markers that sit side by side (illustrated in red and yellow) to remain together on the same random fragments. A random sampling of a subset of these fragments then contributes to form different samples of a HAPPY panel. As discussed above, the panel contains essentially very dilute genomic DNA so that it is insufficient in almost every case for good use. Therefore, here we propose to use a whole genome amplification method, termed multiple displacement amplification (MDA) [24] to overcome the bottleneck. MDA can yield about 20 - 30 µg of products from as few as 1 - 10 copies of genomic DNA. In comparison to other whole genome amplification methods, MDA can provide the most reliable genotypes, highest call rates, best genomic coverage, and lowest amplification bias [25]. Such MDA amplification makes the panel ready to be genotyped on various markers. However, in our newly proposed technique, the amplified DNA is subject to sequencing.
A flow chart demonstration of HAPPY whole genome sequencing, mapping and assembly.
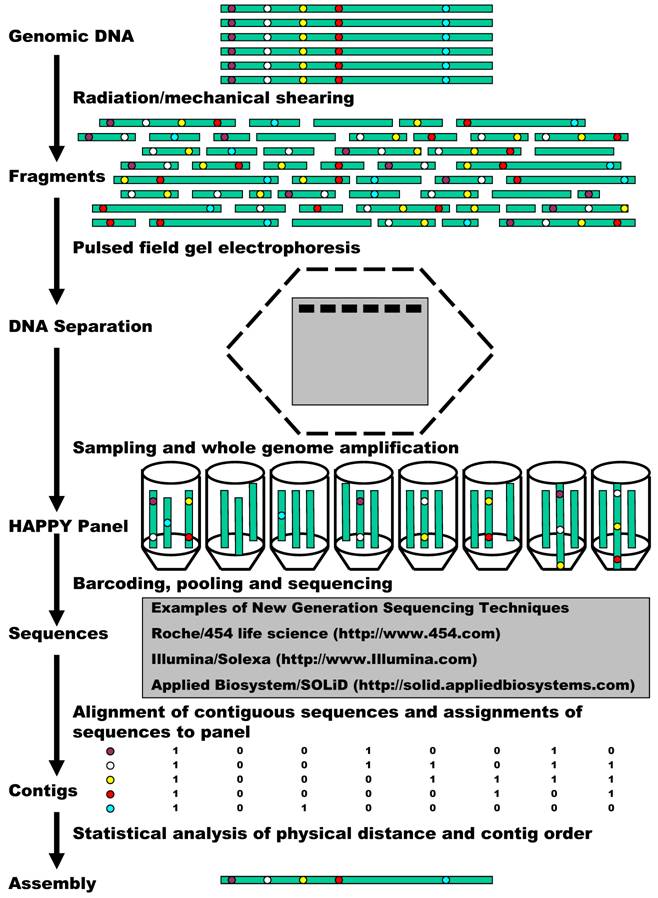
Technically, it is not a problem at all to sequence every sample of the HAPPY panel described above. However in all practicality, it would still be costly when 96 or more samples of a HAPPY panel are sequenced individually. This problem can be overcome by taking advantage of the barcoding technique, which is becoming a standard approach to increase the numbers of samples run on high throughput instruments [26]. For example, the authors developed a pyrosequencing-tailored barcoding approach that allows for the unambiguous assignment of nucleic acid sequences from a mixture of libraries from up to 48 different samples on a Roche/454 Life Sciences sequencer. In the Applied Biosystem/SOLiD system, 16 barcodes are selected and can be added to the 3' end of the target sequence using a modified version of the P2 adaptor. (http://www3.appliedbiosystems.com/) These barcodes possess uniform melting temperatures, low-error rate, and orthogonal sequences that are unique in color space. In the Illumina/Solexa system, DNA can be randomly sheared and amplified with primers that contain a 3 bp barcode. Using current instruments, reagents, and protocols, one Solexa "lane" generates ~120 Mb in ~3 million reads of ~40 bp. When each Solexa lane is multiplexed with 12 barcodes, for example, it will provide on average, ~10 Mb of sequence in ~250,000 reads for each sample. At this level of multiplexing, one Solexa instrument "run" (7 lanes plus control) would allow tag sequencing of 84 HAPPY samples. This means, one can finish 192 HAPPY samples in a maximum of three runs. New-generation sequencing combined with the barcode technique will produce innumerous amounts of sequences for assembly.
Barcode sequencing will further allow us to determine the presence/absence of a sequence contig as a traditional marker in the HAPPY panel. When two contigs (illustrated in Figure 1 in red and yellow) occur often together, it signifies that they lie next to each other within the genome. However, when contigs occur independently, they must be located apart from each other on the same chromosome or on different chromosomes. An example of this can be seen in Figure 1 by comparing the blue contig to the red or yellow contig. Statistical analysis of the similarities and differences between the patterns of many such sequence contigs will help us infer how close or how far away from each other these contigs are within the genome, and hence their order, to produce a map. Certainly, such a map orders the whole genome sequence assembly as well.
In eukaryotic species, genome size varies quite a lot [27]. For example, genome sizes in plants ranges from 0.01 pg (~10 Mb) in some unicellular algae (e.g. Cyanidium caldarium) to 127.4 pg (~124,600 Mb) in the tetraploid angiosperm Fritillaria assyriaca. The animal genome sizes have been reported to range from ~0.03 pg (~29Mb) in the root-knot nematode Meloidogyne graminicola to ~133 pg (~130,000 Mb) in the marbled lungfish Protopterus aethiopicus. In contrast, fungi usually have relatively small genomes, ranging from 10 Mb to 60 Mb, with an average of ~37 Mb. Therefore, a detailed HAPPY pipeline needs to be justified accordingly, once a given species is chosen. For example, using the SOLiD system to generate whole genome draft sequences as references might be an initial step before the HAPPY pipeline is applied for mapping and assembly of large genomes.
In summary, integration of HAPPY mapping and next-generation sequencing in one pipeline actually involves two replacement events: 1) the initial fragmentation and cloning step in traditional genome sequencing is replaced by a HAPPY panel creation and 2) the genes/markers genotyping in traditional HAPPY mapping is substituted by the sequencing. Such a universal approach will play an important role in genome sequencing, mapping, and assembly of many species; thus advancing genome science and its applications in biomedicine and agriculture.
Acknowledgements
This work was supported by NIH/NIGMS grant 1R01GM086321-01 to R.M.H. and D.S.R. with a subaward to Z.J. We thank Ms. Jennifer Michal and Ms. Vanessa Michelizzi for editing the manuscript.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Beverly Mertz. http://www.accessexcellence.org/RC/AB/IE/Short_History_of_Mapping.php
2. Raudsepp T, Chowdhary BP. FISH for mapping single copy genes. Methods Mol Biol. 2008;422:31-49
3. White R, Lalouel JM. Chromosome mapping with DNA markers. Sci Am. 1988;258:40-48
4. Cox DR, Burmeister M, Price ER, Kim S, Myers RM. Radiation hybrid mapping: a somatic cell genetic method for constructing high-resolution maps of mammalian chromosomes. Science. 1990;250:245-250
5. Dear PH, Cook PR. Happy mapping: a proposal for linkage mapping the human genome. Nucleic Acids Res. 1989;17:6795-6807
6. Lai Z, Jing J, Aston C. et al. A shotgun optical map of the entire Plasmodium falciparum genome. Nat Genet. 1999;23:309-313
7. Zhou S, Bechner MC, Place M. et al. Validation of rice genome sequence by optical mapping. BMC Genomics. 2007;8:278
8. Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci U S A. 1977;74:560-564
9. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463-5467
10. Schuster SC. Next-generation sequencing transforms today's biology. Nat Methods. 2008;5:16-18
11. Dear PH, Cook PR. Happy mapping: linkage mapping using a physical analogue of meiosis. Nucleic Acids Res. 1993;21:13-20
12. Piper MB, Bankier AT, Dear PH. A HAPPY map of Cryptosporidium parvum. Genome Res. 1998;8:1299-1307
13. Konfortov BA, Cohen HM, Bankier AT, Dear PH. A high-resolution HAPPY map of Dictyostelium discoideum chromosome 6. Genome Res. 2000;10:1737-1742
14. Thangavelu M, James AB, Bankier A, Bryan GJ, Dear PH, Waugh R. HAPPY mapping in a plant genome: reconstruction and analysis of a high-resolution physical map of a 1.9 Mbp region of Arabidopsis thaliana chromosome 4. Plant Biotechnol J. 2003;1:23-31
15. Hamilton EP, Dear PH, Rowland T, Saks K, Eisen JA, Orias E. Use of HAPPY mapping for the higher order assembly of the Tetrahymena genome. Genomics. 2006;88:443-451
16. Silander K, Saarela J. Whole genome amplification with Phi29 DNA polymerase to enable genetic or genomic analysis of samples of low DNA yield. Methods Mol Biol. 2008;439:1-18
17. Lander ES, Linton LM, Birren B. et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860-921
18. Venter JC, Adams MD, Myers EW. et al. The sequence of the human genome. Science. 2001;291:1304-1351
19. Metzker ML. Emerging technologies in DNA sequencing. Genome Res. 2005;15:1767-1776
20. Voelkerding KV, Dames SA, Durtschi JD. Next-Generation Sequencing: From Basic Research to Diagnostics. Clin Chem. 2009 [Epub ahead of print]
21. Nyrén P, Pettersson B, Uhlén M. Solid phase DNA minisequencing by an enzymatic luminometric inorganic pyrophosphate detection assay. Anal Biochem. 1993;208:171-175
22. Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387-402
23. Shendure J, Porreca GJ, Reppas NB. et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728-1732
24. Dean FB, Hosono S, Fang L. et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99:5261-5266
25. Lovmar L, Syvänen AC. Multiple displacement amplification to create a long-lasting source of DNA for genetic studies. Hum Mutat. 2006;27:603-614
26. Parameswaran P, Jalili R, Tao L. et al. A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res. 2007;35:e130
27. Gregory TR, Nicol JA, Tamm H. et al. Eukaryotic genome size databases. Nucleic Acids Res. 2007;35:D332-338
Author contact
![]() Correspondence to: Zhihua Jiang, Tel: +509 335 8761; Fax: +509 335 4246; E-mail: jiangzedu
Correspondence to: Zhihua Jiang, Tel: +509 335 8761; Fax: +509 335 4246; E-mail: jiangzedu