Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(4):403-411. doi:10.7150/ijbs.5806 This issue Cite
Research Paper
A Novel Aurora-A Inhibitor, BPR1K0609S1, Sensitizes Colorectal Tumor cells to 5-Fluorofracil (5-FU) Treatment
Yoshimi Shionome1, Wen-Hsing Lin2, Hui-Yi Shiao2, Hsing-Pang Hsieh2, John Tsu-An Hsu2, Toru Ouchi1, 3, ![]()
1. Department of Medicine, NUHS, Pritzker School of Medicine, The University of Chicago, Evanston, USA.
2. Institute of Biotechnology and Pharmaceutical Research National Health Research Institutes, Miaoli, Taiwan 305.
3. Department of Cancer Genetics, Roswell Park Cancer Institute, Buffalo, USA.
Received 2013-1-4; Accepted 2013-3-22; Published 2013-4-29
Abstract
Small synthetic compounds have been implicated in treatment of human cancers. We have synthesized a small compound, BPR1K0609S1 (hereafter, BP), which inhibits Aurora-A kinase. In the present study, we studied the mechanism of BP suppression of tumorigenesis induced by Aurora-A. Given our previous results that inactivation of p53 accelerates MMTV-Aurora-A-mediated tumorigenesis in vivo, we studied the roles of p53 pathway using the isogenic human colon carcinoma cell lines of HCT116, in which p53, Puma, Bax, p21 or Chk2 is deleted. When these isogenic cell lines are treated with BP for 48 h, accumulation of G2M phase and aneuploidy are commonly observed, and HCT116 p21(-) cells show increase in apoptosis. In xenograft assay, s.c. injection of BP efficiently inhibits tumorigenesis of HCT116 deficient for Chk2 or p21. Re-transplantation of BP-resistant tumors indicates that these resistant cells do not acquire advanced tumor growth. Significantly, 5-FU (5-fluorouracil) treatment further induces apoptosis of BP-resistant HCT116 deficient for Chk2 or Puma. These results demonstrate that p21 deficiency enhances BP-mediated suppression of tumor growth, and that BP and 5-FU can collaborate for tumor regression.
Keywords: Aurora-A, kinase inhibitors, mice xenograft, cell cycle, apoptosis
Introduction
Aurora A kinase is frequently amplified in many epithelial tumors, cancers of solid organs and hematological malignancies. Aurora A kinase has been implicated in causing and/or maintaining the malignant phenotype and resistance to microtubule-targeted chemotherapy, such as paclitaxel [1-4]. This kinase regulates many steps of mitosis, such as mitotic entry and exit and bipolar spindle assembly, becoming localized on the centrosome during early G2 phase [1,5]. Previous results have illustrated that phosphorylation of BRCA1 by Aurora-A is crucial for the initiation of the mitosis [6]. As such, inhibition of aurora A kinase activity has been shown to cause centrosome separation and maturation defects, spindle aberrations, cell cycle arrest, and apoptosis [7]. We have generated mice model of Aurora-A tumors, in which Aurora-A cDNA is expressed under control of the MMTV promoter [8]. mTOR and Akt pathways are activated in developed tumors in these mice [8], and combined treatment of tumor cells with inhibitors of Aurora-A, mTOR and Akt induced apoptosis [9], suggesting that network of oncogenic pathways synergize to develop Aurora-A tumors. In that mice model, we have also shown that tumor incidence and malignancy are accelerated when p53 is simultaneously deleted [8]. Extensive studies of functions of p53 have demonstrated that this protein plays a pivotal role in determining cell fate in the process of cell transformation [10-12]. Taken together, these results indicate that Aurora-A functionally interacts with p53 pathway and suggest that integrity of p53 pathways could determine tumor suppression when these cells are exposed to anti-proliferative stimuli.
Through substructure searching for kinase-priviledged fragments in an in-house compound library, we found a lead compound that has a furanopyrimidine scaffold [13]. Aurora-A inhibitor, BP (BPR1K0609S1), was synthesized from this lead compound and tested for its interaction with Aurora-A and epidermal growth factor receptor (EGFR) kinase activity [13]. It has been shown that BP is highly specific for Aurora-A, suggesting that it could be utilized as a lead compound to target Aurora-A tumors in vivo.
In the current studies, we investigated the roles of p53 pathways in tumor suppression when BP inhibits Aurora-A activity in vitro and in vivo. It has been well demonstrated that p53-mediated cell cycle checkpoint is regulated by its target proteins including p21, Puma, Bax, or by Chk2 kinase [10-12]. On the basis of that, in these studies, we used HCT116 human colon carcinoma cell line and its isogenic variants in which p53, Puma, Bax, p21 or Chk2 is deleted [14-17], and explored whether these proteins are involved in BP-mediated tumor suppression. We further investigated BP-resistant tumors cells recovered from xenograft undergo apoptosis when subsequently treated with 5-FU.
These results indicate that p21 and Chk2 are modifiers of BP-induced tumor suppression, and that combination therapy with 5-FU may efficiently reduce tumor progression.
Materials and Methods
Ethics Statement
We certify that mice were treated in accordance with the guidelines of University of Chicago (Evanston, USA). Protocols of mice studies were approved by Northshore University Health System IACUC. When tumor size reaches 1.5cm, tumors were removed and mice were euthanized by CO2 asphyxiation followed by cervical dislocation.
Cell culture
HCT116 was purchased from ATCC and isogenic HCT116 variants deficient for p53, Puma, Bax, Chk2 or p21 were kindly obtained from Dr. Bert Vogelstein (Johns Hopkins University, Ref. 14-17). They were grown in McCoy's 5A medium supplemented with 10% fetal bovine serum and 100U of penicillin-streptomycin/ml (Invitrogen). HCT116 variants recovered from xenograft were also maintained in the same condition.
Cell cycle analysis of isogenic HCT116 variants when treated with kinase inhibitors
BPR1K0609S1 (BP) was isolated from screen of a library of furanopyrimidine [13]. 5-FU (5-fluorouracil) was purchased from Sigma. Cells were treated with BP (800nM) or 5-FU (375µM) for 48 h followed by FACS analysis with propidium iodide (FACSCalibur and CellQuest Pro software). Results were obtained from two independent experiments using total five mice per one isogenic HCT116 variant.
Xenograft assay
All animal studies were performed by following protocol approved by Institutional Animal Care and Use Committee (IACUC). Female athymic mice at the age of 4 to 5 weeks were purchased from Jackson Laboratory. For xenograft assay, HCT116 isogenic cells (5x106 cells/100 µl of PBS) were transplanted into both flanks of mice. When tumor size reached at 200~400mm3, one side of tumor was s.c. injected with BP (800nM). Size of tumors was measured every other day after injection. When tumor size reached to 2,000mm3, mice were euthanized. Results were obtained from two independent experiments using total five mice per one isogenic HCT116 variant. Using SAS 9.3, two way ANOVA analysis has been conducted for the post hoc analysis on the tumor size changes, using Tukey's studentized range test (HSD) on all main effect mean.
Removal and re-xenograft of drug resistant tumors
After administration of BP compound to tumors developed in nude mice, chemoresistant tumors were excised and placed on cell culture plate in McCoy's 5A medium supplemented with 10% fetal bovine serum and 100U of penicillin-streptomycin/ml, as described above. On the next day, BP was added to cell culture media (400nM or 800nM) of resistant cells, and the cell culture were maintained for at least 2 weeks until cell colonies were isolated. We could only establish BP-resistant HCT116 p53(-), Puma(-), Chk2(-) and Bax(-) cells in our hand. Isolated cells were maintained in the presence of 100nM of BP, and 5x106 cells were transplanted again into nude mice as described above.
Western blot analysis
Cells were lysed with EBC buffer (50mM Tris-HCl, pH8.0, 120mM NaCl, 0.5% NP-40, 100mM NaF, 200nM sodium orthovanadate, 100µg/ml phenylmethysulfonyl fluoride, 2µg/ml leupeptin, 2µg/ml aprotinin). Protein concentration was determined by Bradford method, followed by separation in 5% or 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to Immobilon-P membrane (Milipore) using a semidry transfer method (Trans-Blot, Bio-Rad). Membranes were blocked in 1% nonfat dried milk and 0.05% Tween 20 in PBS. Primary antibodies are: anti Aurora-A, Aurora-A phospho-Thr288, Aurora-B phosphor-Thr232, Aurora-C phosphor-Thr198 (Cell Signaling) and tubulin (Santa Cruz). Secondary antibodies are horseradish peroxidase-conjugated anti mouse IgG or rabbit IgG (Jackson Laboratory).
Results
BPR1K0609S1 (BP) inhibits phosphorylation of Aurora-A at Thr288
Aurora-A inhibitor, BP, was identified from a library screen of furonapyrimidine [13]. In this study, we investigates anti-tumor activity of BP and the roles of p53 pathway in its tumor suppression activity using isogenic variants of p53-wild type HCT116 human coon cancer cell line, in which p53, p21, Puma, Bax, or Chk2 is deleted.
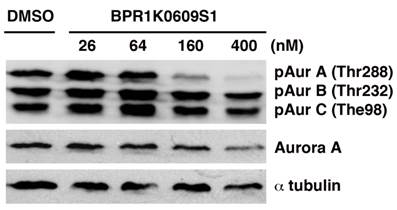
We initiated biochemical characterization of BP using HCT116 human colorectal carcinoma cell line. Cells were treated with BP for 24 h with different concentration of the compound (26, 64, 160 and 400 nM). Phosphorylation of Aurora-A at Thr288, Aurora-B at The232 and Aurora-C at Thr198 has been implicated in their catalytic activation, respectively [18-20]. As shown in Figure 1, BP specifically inhibited phosphorylation of Aurora-A at Thr288 among these phosphorylation sites of Aurora kinases, indicating specificity of this compound to Aurora-A kinase among Aurora family of proteins. The specific activity of BP was similarly observed in HCT116 variants that we used in these studies.
BPR1K0609S1 (BP) specifically inhibits Aurora-A activity. Auto-phosphorylation of Aurora-A at Thr288 is specifically inhibited by BP in concentration dependent manner.
Alteration of cell cycle profiles with BP treatment is determined by integrity of p53-associated proteins
To study the roles of p53 pathway in Aurora-A tumorigenesis and BP-mediated tumor suppression, we used HCT116 human colorectal cancer cell line overexpressing Aurora-A (16), and its isogenic derivatives, in which p53-associated genes (p53, p21, Puma, Bax and Chk2) are stably knocked out in vitro [14-17]. Logarithmically growing cell culture of these variants was treated with BP compound (800nM) for 48 h, and cell cycle profiles were determined by FACS analysis (Figure 2). Among these HCT116 variants, HCT116 p21(-) cells are highly sensitive to BP treatment and they underwent apoptosis (28%). BP-induced apoptosis was much less in parental HCT116 and HCT116 Chk2(-) cells. Interestingly, BP weakly induced HCT116 Bax(-) cells, although pro-apoptotic Bax gene is inactivated in these cells [21,22].
The effects of BPR1K0609S1 (BP) on cell cycle. HCT116 and their isogenic variants, HCT116 p53(-), HCT116 Puma(-), HCT116 Bax(-) HCT116 p21(-) and HCT116 Chk2(-), were treated with BP (800nM) for 48 h, and cell cycle profiles were determined by propidium iodide (PI) staining. Results were obtained from two independent experiments.
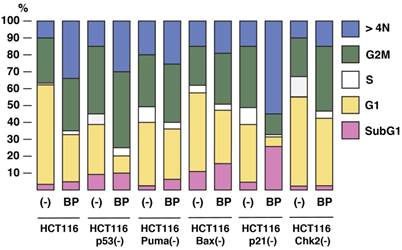
BP potentially induced aneuploidy in all these cell types examined, but significant induction was observed in HCT116 (34%), HCT116 p53(-) (31%), and HCT116 p21(-) (53%). Decrease in G1 phase was commonly observed in all the cell types with BP treatment. Among these cell lines, G1 phase of HCT116 p53(-) and HCT116 p21(-) cells was remarkably decreased from 29% to 11% and 32% to 5%, respectively.
These results indicate that BP compound can efficiently induce apoptosis and aneuploidy in p21-deficient cells, but aneuploidy is well induced in HCT116 and HCT116 p53(-) cell, too.
BP inhibits tumorigenesis of cells deficient for p21 or Chk2
We next explored anti-tumor activity of BP in xenograft model (Figure 3). HCT116 isogenic cell lines were transplanted onto athymic mice, and a size of tumor was measured. Among these cell lines, tumor growth of HCT116 Puma(-) cell was most aggressive. Tumor growth of both HCT116 and HCT116 p21(-) cells were slower than other cell types. When the size became ~200 to 400 mm3, BP (800nM) was directly s.c. injected daily. Of note, tumor growth of HCT116 cells was not significantly reduced with BP treatment. Tumor growth of HCT116 p53(-), HCT116 Bax(-) and HCT116 Puma(-) cells was weakly reduced with BP treatment. Tumorigenesis of HCT116 Chk2(-) and HCT116 p21(-) cells was markedly inhibited with BP injection.
Anti-proliferative effects of BPR1K0609S1 (BP) in vivo. Each of the indicated HCT116 variants was transplanted into both flanks of a nude mouse. When tumor size reached at 200-400 mm3, BP (800nM) was injected into one of the developed tumors per mouse. Tumor size was measured every day. Results were obtained from two independent experiments using total five mice per one isogenic HCT116 variant. Using SAS 9.3, two way ANOVA analysis has been conducted for the post hoc analysis on the tumor size changes, using Tukey's studentized range test (HSD) on all main effect mean.
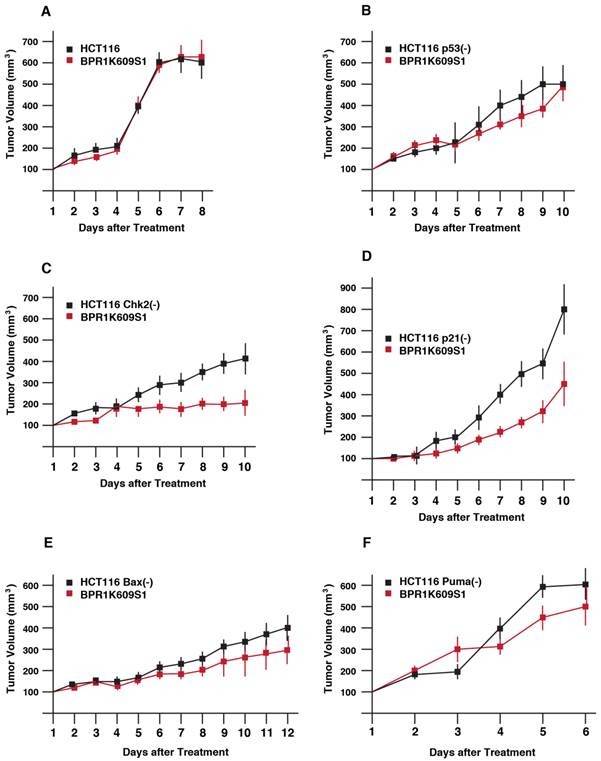
By using SAS 9.3, 2 way ANOVA analysis was conducted for the post hoc analysis on the tumor size fold changes using Tukey's studentized range test (HSD) on all main effect mean. The time points when the 2 groups (untreated, treated with BP) show significant differences is: HCT116 cells; no difference, p53(-) cells; at day 9, Chk2(-) cells; at day 6, p21(-) cells; at day 6, Bax(-) cells; at day 9, Chk2(-) cells; at day 5.
These results demonstrate that Chk2 and p21 can modulate tumor cell's BP sensitivity in xenograft model more significantly than other p53-associated proteins examined.
BP-resistant tumor cells do not show enhanced tumorigenecity
Xenograft experiments indicated that BP show anti-tumor activity, although there is some difference in magnitude of its efficiency. After BP injection, drug-resistant tumors still remained in mice. We characterized whether these drug-resistant cells show more malignant phenotypes than those of the individual original HCT116 variants. After treatment of BP-injection, tumors were removed from xenograft, excised and seeded on cell culture plates. Tumor cells resistant to BP were maintained in the presence of BP (100nM) to establish drug-resistant subclones. Among the removed tumors, HCT116 Puma(-), HCT116 Bax(-), HCT116 Chk2(-) and HCT116 p53(-) cells were successfully established from removed BP-resistant tumors.
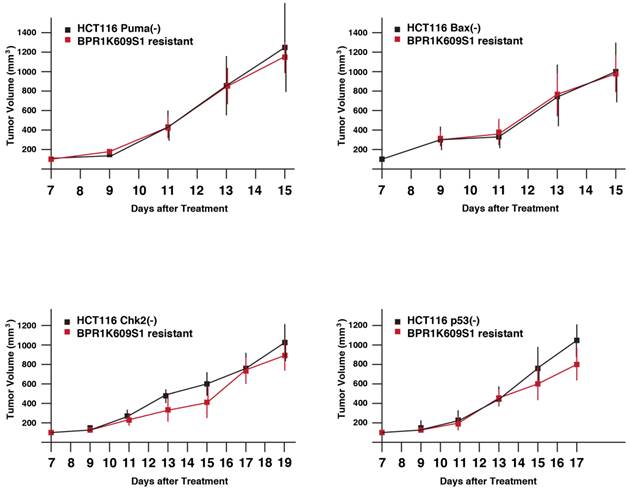
Next, tumor xenograft assay was performed to study whether these recovered cell lines show more malignant phenotypes than each of the original cell lines. Parental and BP-resistant cells (5 x106 cells) were transplanted onto nude mice, and a size of the developed tumors was daily measured from day 7 (Figure 4). Using SAS 9.3, two way ANOVA analysis has been conducted for the post hoc analysis on the tumor size changes, using Tukey's studentized range test (HSD) on all main effect mean. The results indicate the time points when differences between parental and resistant cells are observed are: HCT116 Puma(-) Bax(-) and Chk2(-) cells and their BP-resistant clones; no significant differences, and VX680/MK8745 cells; at day 15, HCT116 Bax(-) and MK8745 cells; at day 13, HCT116 p53(-) and BP cells; at day 15. Of note, these BP-resistant cells did not show faster tumor development compared to their parental cells, respectively. It remains to be elucidated how these cells escaped from apoptosis and aneuploidy after treatment with BP, however, these results indicate that drug-resistant tumors did not acquire accelerated tumorigenesis when xenograft was treated with BP.
Tumor growth of HCT116 variants resistant to BPR1K0609S1 (BP). Parental and BP-resistant HCT116 Puma(-), Bax(-), Chk2(-) and p53(-) cells were transplanted into nude mice. Size of developed tumors was monitored every other day. Results were obtained from two independent experiments using total five mice per one isogenic HCT116 variant. Using SAS 9.3, two way ANOVA analysis has been conducted for the post hoc analysis on the tumor size changes, using Tukey's studentized range test (HSD) on all main effect mean.
5-FU further induces apoptosis of BP-resistant tumor cells
We explored whether BP-resistant tumor cells recovered from xenograft could undergo apoptosis when treated with chemotherapeutic agents. 5-FU is a well-characterized anti-cancer agent and has been used for tumor suppression in vitro and in vivo [29-32]. We studied whether 5-FU could further induce apoptosis of BP-resistant-HCT116 Chk2(-), -HCT116 Bax(-), -HCT116 p53(-) and HCT116 Puma(-) cells (Figure 5). These cells were treated with 5-FU (375µM) for 48 h, and cell cycle profiles were determined by FACS analysis. First, we studied cell cycle profile of these BP-resistant cells without 5-FU treatment and compared them with BP-treated cells that are shown in Figure 2. Population of cells in each phase of the cell cycle was similar in BP-treated and BP-resistant HCT116 Bax(-) cells. No significant difference in cell cycle profile was also observed between BP-treated and BP-resistant HCT116 Puma(-) cells. When HCT116 Chk2(-) cells were treated with BP, ~40% of cells were in G2M phase (Figure 2), however, BP-resistant HCT116 Chk2(-) cells showed decreased G2M phase (27.0%). When HCT116 p53(-) cells were treated with BP, ~9% of cells were in G1 phase (Figure 2). A fraction of BP-resistant HCT116 p53(-) cells in G1 phase was increased to 36.3%. These results indicate that HCT116 Chk2(-) and p53(-) cells became tolerated to BP treatment during xenograft assay.
5-FU treatment further induces cell death of BPR1K0609S1 (BP)-resistant HCT116 isogenic variants. BP-resistant HCT116 Chk2(-), Bax(-), p53(-) and Puma(-) cells were further treated with 5-FU (375µM) for 48 h . Cell cycle profile was determined by FACS analysis of PI-stained samples. Results were obtained from two independent experiments using total five mice per one isogenic HCT116 variant. Using SAS 9.3, two way ANOVA analysis has been conducted for the post hoc analysis on the tumor size changes, using Tukey's studentized range test (HSD) on all main effect mean.
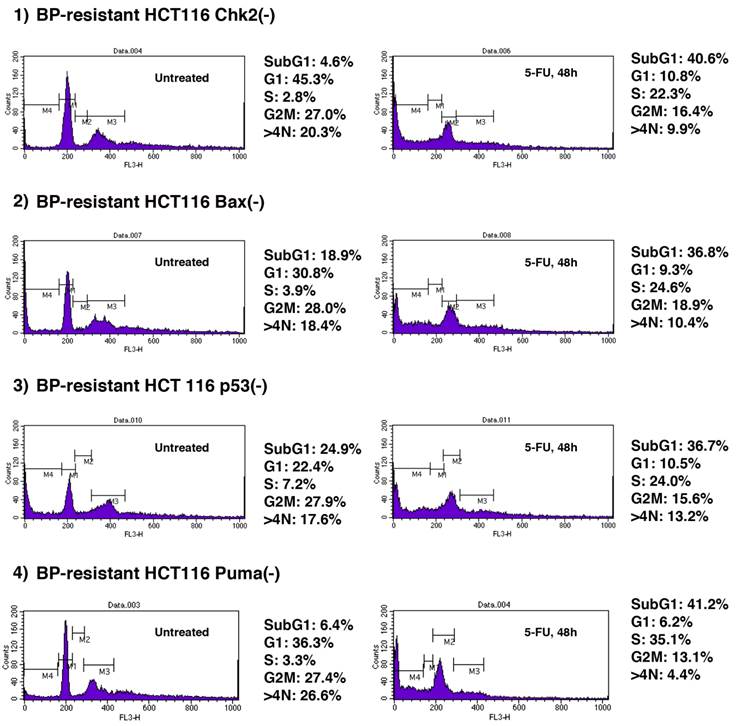
Next, we tested whether these BP-resistant cells undergo apoptosis when treated with 5-FU. 5-FU induced apoptosis of BP-resistant-HCT116 Chk2(-), Bax(-), p53(-) and Puma(-) cells up to 40.6%, 36.8%, 36.7% and 41.2%, respectively. Of note, among these cells, higher increased rate in apoptosis was observed in HCT116 Chk2(-) and Puma(-) cells (from 4.6% to 40.6%, and from 6.4% to 41.2%, respectively). Compared to the parental cell lines, BP resistant cells were more sensitive to 5-FU (Supplementary Table 1). These results indicate that combination therapy with BP and 5-FU may effectively collaborate to induce apoptosis of Aurora-A tumor.
Discussion
In the initial research of translational application of Aurora kinase inhibitors, Aurora-A-specific targeting was considered a more therapeutically viable target given its role in tumorigenesis [24,25]. However, no clinical data in human have shown highly specific inhibitors for Aurora-A to be therapeutically valuable than pan-Aurora inhibitors that target Aurora family of kinases as well as other kinases. Emerging data indicate that combination with spindle reagents, such as taxanes or vinca alkaloids, with Aurora-A kinase inhibitors may prove synergistic [4,26]. Other drawback is that therapeutic dosing of Aurora kinase-specific agents may be difficult to elucidate as higher doses of inhibitors may lead to a pan-Aurora inhibitory effect.
In the present studies, we explored tumor inhibition with a new compound, BP (BPR1K0609S1) that targets Aurora-A kinase. Our preliminary data obtained from MMTV-Aurora-A mice model has shown that inactivation of p53 accelerates tumorigenesis in these mice, suggesting that integrity of p53 pathways is crucial to determine the tumor malignancy. On the basis of these observations, we studied whether p53-associated proteins are involved in BP-induced regression of Aurora-A tumors. HCT116 is a p53 wild type human colon cancer cell line. In vitro knockout of p53 associated genes, such as p53, p21, Bax, Puma, Chk2 etc, in HCT116 cells has been established in previous studies, providing unique and specific systems to investigate the roles of p53 pathways in tumor progression [14-17]. We used these isogenic cell lines in this study, and examined how any of these genes could determine BP-induced tumor suppression in vitro and in vivo. Cell cycle profile of these cell lines after treatment with BP illustrated that BP is potentially inducing accumulation of G2M phase~aneuploidy. This is consistent with the results that Aurora family of kinases are involved in the mitotic (M) phase of the cell cycle, acting to establish the mitotic spindle, bipolar spindle formation, alignment of centrosomes on mitotic spindle, centrosome separation, cytokinesis, and monitoring of the mitotic checkpoint [1,24,25,27]. Thus, Aurora kinases are critical for accurate and organized chromosome division and allocation to each daughter cell. On the basis of these observations, it is conceivable that inhibition of Aurora-A (and other Aurora family kinases) could perturb cell division. HCT116 p21(-) cells showed more accumulation of aneuploidy with BP treatment than, for example, HCT116 p53(-) cells (Figure 2). Although both HCT116 p53(-) and p21(-) cells similarly indicate incomplete cytokinesis and multilobulared nuclei [14], our results suggest that a signaling axis of Aurora-A/p21 regulates mitotic progression.
Our results demonstrated that HCT116 Chk2(-) cells are more sensitive to BP in xenograft assay among the cell lines examined. Interestingly, these cells indicated marked accumulation of aneuploidy when treated with BP (Figure 2), suggesting that inhibition of tumor development with BP is associated with this phenotype. Recent studies have illustrated that Chk2 deficient cells undergo mitotic catastrophe followed by apoptosis under conditions of cell stress [28]. Molecular mechanism underlying these phenotypes reveals that mitotic catastrophe occurs in a p53-independent manner and involves a primary activation of caspase-2, upstream of cytochrome c release, followed by caspase-3 activation and chromatin condensation. Thus, inhibition of tumor growth of HCT116 Chk2(-) cells with BP might be due to induced apoptosis of this caspase pathway.
We studied in vivo phenotypes of tumor cells recovered from xenograft assays after BP treatment. Among the removed tumors, only four cell types, HCT116 Puma(-), Bax(-), Chk2(-) and p53(-) cells could be re-cultured in our hand. Those cells were used for re-transplantation into nude mice, and their tumor growth assay was performed. Our results indicate that there are no significant differences in tumor growth between each of their parental cells and re-transplanted cells. Mechanisms of how these cells acquired BP-resistant phonotypes remain to be elucidated, and that this drug-resistance pathway is not directly involved in enhanced tumor growth. In our previous studies, we have found that mTOR/Akt is activated in the Aurora-A tumors and this activation occurs at the late stage of cell transformation [9]. Given that, we assume that activation of mTOR/Akt could provide drug-resistant phenotypes (submitted).
Since final goal of cancer therapy is to eliminate developed tumors, we examined whether BP-resistant cells could further undergo apoptosis when treated with other conventional therapeutic agents. 5-Fluorouracil (5-FU) has been the first-choice chemotherapy drug for colorectal cancer for many years. Although 5-FU in combination with other chemotherapeutic agents improves response rates and survival in breast and head and neck cancers, 5-FU has the greatest impact in treatment of colorectal cancer [29]. Nonetheless, response rates for 5-FU-based chemotherapy as a first-line treatment for advanced colorectal cancer are only 10-15% [30]. The combination of 5-FU with newer chemotherapies such as IRINOTECAN and OXALIPLATIN has improved the response rates for advanced colorectal cancer to 40-50% [31,32]. Among the BP-resistant clones described above, HCT116 Chk2(-) and HCT116 Puma(-) cells are more sensitive to subsequent 5-FU treatment compared to HCT116 p53(-) and Bax(-) clones (Figure 5). We do not understand the mechanism of regulation of chemosensitivity when BP-resistant cells are further treated with 5-FU. Of note, cell cycle profile of BP-resistant clones is different from that of parental cells when transiently treated with BP (Figure 2 and Figure 5, left). Thus, BP-resistant cells have overcome aneuploidy, therefore it is possible that DNA synthesis, which is indicated by a target of 5-FU, could be increased in those cells due to disrupted checkpoint. Increase in S phase after 5-FU treatment of BP-resistant clones support this notion.
These results suggest that combined therapy using both BP and 5-FU might effectively inhibit tumor development in vivo. Biological markers that could define the prospective evaluation of tumor's response to either BP or 5-FU need to be determined.
Supplementary Material
Supplementary Table 1Acknowledgements
The authors thank all the members of the Ouchi Laboratory for helpful discussion.
Grant Support
This work is supported by grants, R01CA79892 and R01CA90631, from National Institutes of Health, and a Breast Cancer Research Grant from Susan Komen Foundation (T.O.).
Competing Interests
No potential conflicts of interest were disclosed.
References
1. Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer. 2005;5(1):42-50
2. Nishida N, Nagasaka T, Kashiwagi K, Boland CR, Goel A. High copy amplification of the Aurora-A gene is associated with chromosomal instability phenotype in human colorectal cancers. Cancer Biol Ther. 2007;6(4):525-33
3. Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, Fiorica JV, Nicosia SV, Cheng JQ. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003;9(4):1420-6
4. Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3(1):51-62
5. Karthigeyan D, Prasad SB, Shandilya J, Agrawal S, Kundu TK. Biology of Aurora A kinase: Implications in cancer manifestation and therapy. Med Res Rev. 2010 [Epub ahead of print]
6. Ouchi M, Fujiuchi N, Sasai K, Katayama H, Minamishima YA, Ongusaha PP, Deng C, Sen S, Lee SW, Ouchi T. BRCA1 phosphorylation by Aurora-A in the regulation of G2 to M transition. J Biol Chem. 2004;279(19):19643-8
7. Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN Jr, Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14(6):1639-48
8. Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi M, Ouchi T, Deng CX. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene. 2006;25(54):7148-58
9. Taga M, Hirooka E, Ouchi T. Essential roles of mTOR/Akt pathway in Aurora-A cell transformation. Int J Biol Sci. 2009;5(5):444-50
10. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9(10):749-58
11. Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9(10):714-23
12. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9(12):862-73
13. Coumar MS, Chu CY, Lin CW, Shiao HY, Ho YL, Reddy R, Lin WH, Chen CH, Peng YH, Leou JS, Lien TW, Huang CT, Fang MY, Wu SH, Wu JS, Chittimalla SK, Song JS, Hsu JT, Wu SY, Liao CC, Chao YS, Hsieh HP. Fast-forwarding hit to lead: aurora and epidermal growth factor receptor kinase inhibitor lead identification. J Med Chem. 2010;53(13):4980-8
14. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282(5393):1497-501
15. Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100(4):1931-6
16. Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290(5493):989-92
17. Jallepalli PV, Lengauer C, Vogelstein B, Bunz F. The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J Biol Chem. 2003;278(23):20475-9
18. Ferrari S, Marin O, Pagano MA, Meggio F, Hess D, El-Shemerly M, Krystyniak A, Pinna LA. Aurora-A site specificity: a study with synthetic peptide substrates. Biochem J. 2005;390(Pt 1):293-302
19. Yasui Y, Urano T, Kawajiri A, Nagata K, Tatsuka M, Saya H, Furukawa K, Takahashi T, Izawa I, Inagaki M. Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J Biol Chem. 2004;279(13):12997-3003
20. Li XY, Sakashita G, Matsuzaki H. et al. Urano Direct association with inner centromere protein (INCENP) activates the novel chromosomal passenger protein. Aurora C J. Biol. Chem. 2004;279:47201-47211
21. Ishikawa T, Watanabe N, Nagano M, Kawai-Yamada M, Lam E. Bax inhibitor-1: a highly conserved endoplasmic reticulum-resident cell death suppressor. Cell Death Differ. Aug. 2011;18(8):1271-8
22. Renault TT, Manon S. Bax: Addressed to kill. Biochimie. 2011;93(9):1379-91
23. Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330-8
24. Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4(11):842-54
25. Ducat D, Zheng Y. Aurora kinases in spindle assembly and chromosome segregation. Exp Cell Res. 2004;301(1):60-7
26. Mazumdar A, Henderson YC, El-Naggar AK, Sen S, Clayman GL. Aurora kinase A inhibition and paclitaxel as targeted combination therapy for head and neck squamous cell carcinoma. Head Neck. 2009;31(5):625-34
27. Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5(1):1-10
28. Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K, Horne D, Feunteun J, Lenoir G, Medema R, Vainchenker W, Kroemer G. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004May27;23(25):4362-70
29. Efficacy of adjuvant fluorouracil and folinic acid in colon cancer. International Multicentre Pooled Analysis of Colon Cancer Trials (IMPACT) investigators. Lancet. 1995;345(8955):939-44
30. Johnston PG, Kaye S. Capecitabine: a novel agent for the treatment of solid tumors. Anticancer Drugs. 2001;12(8):639-46
31. Giacchetti S, Perpoint B, Zidani R, Le Bail N, Faggiuolo R, Focan C, Chollet P, Llory JF, Letourneau Y, Coudert B, Bertheaut-Cvitkovic F, Larregain-Fournier D, Le Rol A, Walter S, Adam R, Misset JL, Lévi F. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2000;18(1):136-47
32. Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J, Alakl M, Gruia G, Awad L, Rougier P. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355(9209):1041-7
Author contact
![]() Corresponding author: Toru Ouchi, Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlson Streets, Buffalo, NY 14263, USA. Phone: (716) 845-7173. Fax: (716) 845-1698 Email: Toru.Ouchiorg.
Corresponding author: Toru Ouchi, Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlson Streets, Buffalo, NY 14263, USA. Phone: (716) 845-7173. Fax: (716) 845-1698 Email: Toru.Ouchiorg.