Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(4):198-204. doi:10.7150/ijbs.3.198 This issue Cite
Research Paper
Overexpression of Selenoprotein H Reduces Ht22 Neuronal Cell Death after UVB Irradiation by Preventing Superoxide Formation
Kamel E. Ben Jilani, Jun Panee, Qingping He, Marla J. Berry, Ping-An Li ![]()
Department of Cell Molecular Biology, John A. Burns School of Medicine, University of Hawaii, Honolulu, HI 96813, USA
Received 2006-11-27; Accepted 2007-2-5; Published 2007-2-11
Abstract
Selenoproteins have been shown to exhibit a variety of biological functions, including antioxidant functions, maintaining cellular redox balance, and heavy metal detoxification. UV irradiation-induced damage is partially mediated by increased oxygen radical production. The present study is designed to examine the antioxidative effects of human selenoprotein H (hSelH) after brief period of UVB irradiation on the murine hippocampal neuronal cell line Ht22. Ht22 cells were stably transfected with the hSelH gene or with MSCV empty vector and exposed to UVB irradiation with or without the presence of serum. The results showed that cell viability was significantly higher in hSelH-transfected cells compared to the MSCV vector-transfected cells after 24 h of recovery with or without the presence of serum in the media. Further studies revealed that while the number of superoxide anion (O2˙-) positive cells was increased following a 7 mJ/cm2 of UVB irradiation and 5 h of recovery, overexpression of hSelH significantly reduced superoxide production. These results suggest that hSelH overexpression protects cells from UVB irradiation-induced cell death by reducing the O2˙- formation.
Keywords: selenoprotein H, ultraviolet B irradiation, reactive oxygen species, superoxide anion, cell viability.
1. Introduction
Ultraviolet (UV) irradiation, especially UVB irradiation (290-320 nm), leads to premature skin aging, suppression of immunity, and skin cancer [1]. In neuronal cells, UV irradiation induces apoptosis; therefore, many research groups have used this model as a tool to study the intracellular mechanisms of apoptosis [2-7]. UV irradiation induces cell death via a variety of cellular mechanisms. These include induction of nuclear DNA damage, activation of cell surface death receptors such as the tumor necrosis factor (TNF) superfamily, and formation of reactive oxygen species (ROS) accompanied by mitochondrial dysfunction and release of pro-apoptotic factors [8, 9]. UV irradiation has been demonstrated to produce reactive oxygen species (ROS) such as superoxide anion (•O2) radical, hydroxyl radical, hydrogen peroxide and the highly reactive and destructive oxidant, peroxynitrite (ONOO-) [8-10].
To date, 25 selenoprotein genes have been identified in human. Known biological roles of selenoproteins include antioxidant enzymes, enzymes involved in thyroid hormone metabolism, and proteins functioning in selenium transport and heavy metal detoxification [11-16]. These include several well-known peroxidases and reductases such as glutathione peroxidases (GPxs), phospholipid hydroperoxide glutathione peroxidases (PHGPXs) and thioredoxin reductases (TRs), as well as other lesser characterized selenoproteins such as selenoprotein H (SelH) [17, 18]. Furthermore, selenoprotein P (SelP) may also function as a neural survival factor [19, 20]. Emerging evidence suggests that SelP may also possess antioxidant activity [21, 22]. For example, SelP has been shown to function as a protectant against diquat-induced liver necrosis and lipid peroxidation in the rat [23, 24]. SelP is also protective against peroxynitrite-mediated oxidation and nitration [25]. The antioxidative effects of several other selenoproteins such as Gpx, PHGPXs and TRs have been well characterized [26, 27].
The function of SelH is largely unknown. Recently, it was demonstrated that overexpression of Drosophila melanogaster SelH (dSelH) significantly increased viability in embryos by decreasing lipid peroxidation [28]. The objectives of this study were to investigate whether human Selenoprotein H (hSelH) protects neuronal cells against UVB irradiation-induced cell death and if so, whether the protective effect may associate with ROS production prior to observable cell death. We first determined cell viability following a 7 mJ/cm2 of UVB irradiation injury and 24 h of recovery in the murine hippoccampal neuronal cell line Ht22, transfected with the hSelH gene in Murine Stem Cell Virus (MSCV) or the empty retroviral vector. We then measured O2˙-production after 5 h of recovery in hSelH or empty vector transfected cells using the oxidized dihydroethidine detection method [29, 30]. Our results are consistent with the concept that the neuroprotective effect of SelH is associated with its ability to reduce ROS production.
2. Materials and Methods
hSelH transfection procedures
The plasmid containing the gene encoding hSelH was purchased from Invitrogen (Carlsbad, CA, USA). The target gene was subcloned into Murine Stem Cell retroviral vector pMSCVpuro, purchased from BD Biosciences (Palo Alto, CA, USA). The packaging cells RetroPack PT67 (BD Biosciences) were plated in a 60 mm plate at 70% optical confluency 12 h before transfection. Cells were then transfected with 2 μg of hSelH-carrying retroviral vector or empty vector using Fugene method (Roche, Indianapolis, IN, USA). Culture medium was aspirated from cultures after 4 h of transfection. PT 67 cells were washed twice with PBS, and grown for 24 h in 3 ml complete medium. The cells were then selected by puromycin. The concentration of puromycin was optimized to kill the untransfected control PT67 cells in 7-10 d. The transfected PT67 cells that survived the puromycin selection stably produced virus. For viral infection of the target cells, Ht22 cells were plated 12 h before infection at ~70% optical confluency. The medium from transfected PT67 cells containing virus were collected, filtered through a 0.45 µm filter, and added to the Ht22 cells in the presence of 4 μg/ml Polybrene. The virus-containing medium was replaced after 24 h of incubation. After infection for 48 h, the target cells were subjected to puromycin selection. The selection lasted for ~10 d until the uninfected control cells were killed. The cells that survived the selection made up the hSelH-transfected Ht22 cell line (hSelH-Ht22) and MSCV-transfectant (MSCV-Ht22) cell lines.
Detection of mRNA overexpression on hSelH by Real time PCR
Total RNA was extracted from hSelH-Ht22 and MSCV-Ht22 cells using RNeasy Midi Kit (Qiagen, Valencia, CA, USA). cDNAs were synthesized from total RNA using the SuperScript III First-Strand Synthesis System (Invitrogen), and PCR amplification of target genes were carried out using the Platinum SYBR Green qPCR SuperMix-UDG kit (Invitrogen). The following specific primers were used to amplify hSelH, mSelH, and mGAPDH (house keeping gene) cDNAs:
hSelH forward: (5'-GCTTCCAGTAAAGGTGAACCCG-3')
hSelH reverse: (5'-ACCCAAATCTCCCTACGACAGG-3')
mSelH forward: (5'-GGAAGAAAGCGTAAGGCGGG-3')
mSelH reverse: (5'-GGTTTGGACGGGTTCACTTGC-3')
mGAPDH forward: (5'-TGACATCAAGAAGGTGGTGAAGC-3')
and mGAPDH reverse: (5'-CCCTGTTGCTGTAGCCGTATTC-3')
All PCR reactions were performed in duplicate 20 µl reaction volumes and detection was carried out with a LightCycler 2 real-time PCR machine (Biorad, Hercules, CA, USA).
Cell maintenance and treatment
Murine hippocampal Ht22 neuronal cells were infected either with the MSCV empty vector or with hSelH in MSCV expression vector. For propagation, cells were fed with Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 2 mM glutamine, and 200 mM streptomycin/penicillin (Invitrogen) and then cultured at 70%-75% relative humidity in 5% CO2 at 37˚C. Media was renewed every 3 d. For cell viability assays, cells were seeded in 48-well cell culture plates (Corning, Aton, MA, USA) 48 h before treatment. The cells were allowed to reach 80 % optical confluency before UVB treatment.
UVB irradiation
Cells were seeded in 96 or 24 wells plate and propagated to reach 80% cell confluency. Prior to UVB irradiation, cells were washed twice with cold PBS to remove residual serum and dead cells. Cells were placed in serum-containing media (10%) media or without serum for 1 h of incubation in normoxic incubator at 37˚C. Cells were then, submitted to different doses of UVB irradiation from a 20 watt MEABS-25 bulb (Ultra-Lum, Inc., Claremont, CA, USA), which emits most of its energy (60 %) with the UVB range (290-320 nm) with an emission peak at 310 nm band. The irradiation dosages given were 7 mJ/cm2, 11.5 mJ/cm2, and 23 mJ/cm2. After UVB irradiation, cells were returned to the incubator for 5 h or 24 h of recovery at 37˚C.
Measurement of the cytotoxicity
Cell viability was assessed using the Trypan blue exclusion assay as described previously [25]. Briefly, viable and dead cells were discriminated and counted using 0.4% Trypan blue staining with a hemacytometer (Sigma, Saint-Louis, MO, USA). Dead cells were counted as those stained blue by the Trypan blue stain, whereas live and apoptotic cells were those that had excluded the stain.
Detection of superoxide production
MSCV-Ht22 and hSelH-Ht22 cells were exposed to a 7 mJ/cm2 of UVB irradiation in the absence of serum. Production of superoxide anion was assessed using dihydroethidine (HEt) method [31-33] after 5 h of recovery following 7 mJ/cm2 of UVB irradiation, because no frank cell death is seen at this time. HEt was purchased from Molecular Probes (Eugene, OR, USA). Stock solution of HEt (1 mg/ml) was prepared by dissolving in DMSO and stored, protected from light at -20˚C. One µl (1 μg) of HEt was added to 500 μl of cultures 15 min before UVB irradiation. HEt is a cell-permeable fluorescent probe widely used to detect intracellular superoxide anion. It is proposed that the reaction between superoxide and HEt results in the formation of a two-electron oxidized product, which binds to DNA and leads to the enhancement of red fluorescence after being oxidized by superoxide [31, 33, 34]. Twelve microscopic images at the magnification of 20X were captured and the number of oxidized HEt cells, which represent superoxide production, were counted and presented as percentage of total counted cells. Because HEt gives a light background staining, only cells stained with strong bright red color were counted as oxidized HEt-positive cells.
Statistical analysis
All experiments were performed at least in triplicate, repeated two to three times and presented as mean ± SD. ANOVA followed by Scheffe's test was employed for statistical analysis (GraphPad Prism software, SAS, San Diego, CA, USA). A p-value of < 0.05 was considered significant.
3. Results
hSelH infection
hSelH was stably infected into Ht22 cells using MSCV vector. The success of this infection was determined by detection of hSelH mRNA levels using quantitative real-time PCR. The mSelH was a low copy number gene in Ht22 cells. The hSelH mRNA level was increased by about 30 folds compared to endogenous mSelH mRNA level in hSelH-Ht22 cells (Table 1). In control MSCV-Ht22 cells, the murine SelH (mSelH) mRNA level, is comparable to that in SelH-Ht22 but the hSelH mRNA is undetectable.
Detection of mRNA overexpression on hSelH by Real time PCR. Both human SelH (hselh) and mouse SelH (mselh) genes were measured in hSelH-Ht22 and MSCV-Ht22 cells. Each target gene was amplified in duplicates. The table shows a significant drastic increased level of hSelH in Ht22-hSelH cells compared to controls (means ± SD, P < 0.0001).
| Target genes | MSCV-Ht22 | SelH-Ht22 |
|---|---|---|
| hSelH | Non-Detectable | 0.557 ± 0.017* |
| mSelH | 0.0149 ± 0.0019 | 0.0163 ± 0.0005 |
Effects of variable UVB irradiation duration on cell viability
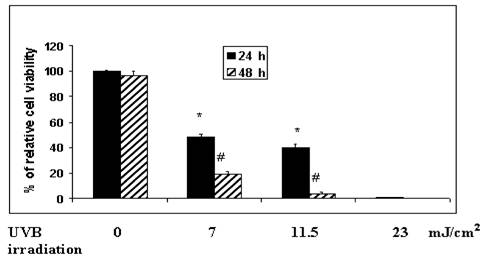
In order to identify an optimal time of UVB exposure, we exposed MSCV-Ht22-transfected and hSelH-Ht22-transfected cells to 0 mJ/cm2, 7 mJ/cm2, 11.5 mJ/cm2, and 23 mJ/cm2 of UVB irradiation. Cell viabilities were measured after 24 and 48 h of recovery. Cell viability after 24 h of recovery, was reduced from 100% in non-radiated control to an average of 50%, 40% and 6% in 7 mJ/cm2, 11.5 mJ/cm2, and 23 mJ/cm2 of UVB irradiation, respectively (Figure 1). Cell viabilities were further decreased after 48 h of recovery. Thus, the average survival rate dropped to 19%, 4% and 0% following 7 mJ/cm2, 11.5 mJ/cm2, and 23 mJ/cm2 of UVB irradiation, respectively (Figure 1). This experiment showed that 7 mJ/cm2 of UVB irradiation resulted in approximately 50% cell death at 24 h of recovery, which was determined to be the optimal length of injury. Therefore, a 7 mJ/cm2 of UVB irradiation was used for the remaining studies.
Variable UVB irradiation exposure parameters on cell viability in MSCV (empty vector) transfected Ht22 cells. The irradiation doses given to the cultures were 0 mJ/cm2, 7 mJ/cm2, 11.5 mJ/cm2, and 23 mJ/cm2, in DMEM medium with serum, respectively. The percentage of cell viability was determined after 24 and 48 h of recovery using the Trypan blue exclusion assay. About 100%, 50%, 40% and 6% of cell viability were observed at 24 h of recovery 0 mJ/cm2, 7 mJ/cm2, 11.5 mJ/cm2, and 23 mJ/cm2 of UVB irradiation, respectively. Cell viabilities were further significantly decreased to 21 %, 4% and 0% at 48 h of recovery at the indicated UVB doses in the Figure. Experiments were performed in triplicate and presented as means ± SD. *Significantly different from non-radiated control at 24 h (P < 0.05) at the indicated UVB dose. #Significantly different from 24 h of recovery at 48 h of recovery (P < 0.05).
Effects of hSelH overexpression on cell viability after UVB irradiation
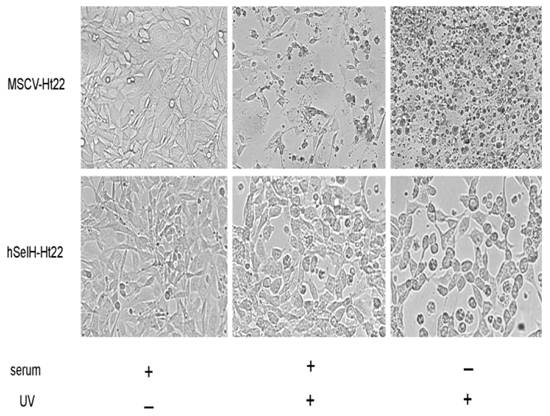
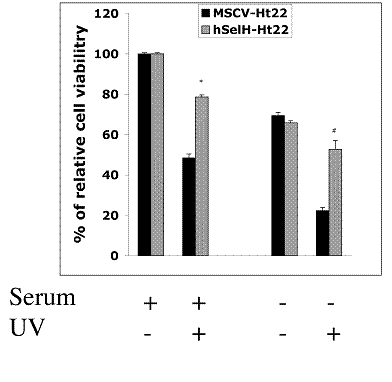
Both MSCV-Ht22 and hSelH-Ht22 cells were exposed to a 7 mJ/cm2 of UVB irradiation in the presence or absence of serum. The cell viabilities were examined at 24 h of recovery and the results were compared to MSCV and hSelH transfectants without UVB irradiation. As shown in Figures 2 and 3, MSCV-Ht22 cells exposed to 7 mJ/cm2 of UVB irradiation in the presence of serum exhibited 48% cell viability compared to controls, as expected. However, the cell viability was significantly increased in cells overexpressing hSelH. That is, hSelH overexpression resulted in appreciatively 80% cell survival compared to controls and this was found to be significantly different than that observed with vector control (P<0.0001). In the absence of serum, cell survival rate decreased to about 70% even without UVB irradiation, with no major difference between the MSCV-Ht22 and hSelH-Ht22 cells. Cell survival rate was further decreased to 23% in MSCV-Ht22 cells after a 7 mJ/cm2 of UVB irradiation. However, with the overexpression of hSelH, cell survival only decreased to 52% (P = 0.0004) when cells were exposed to 7 mJ/cm2 of UVB irradiation without supplementation of serum. Figure 2 shows representative photomicrographs taken from MSCV-Ht22 and hSelH-Ht22 cells treated with or without UVB irradiation in the presence or absence of serum. Figure 3 summarizes the cell viability data collected from three independent experiments.
Photomicrographs showing effects of hSelH overexpression cell viability in Ht22 cells treated with 7 mJ/cm2 of UVB irradiation followed by 24 h of recovery. Increased cell debris was present in UVB radiated MSCV-Ht22 cell compared to non-treatment control cells. More viable cells were observed when hSelH transfected Ht22 cells were exposed to UVB irradiation. Images were taken at 20X magnification with a Zeiss LSM5 laser-scanning confocal microscope. Bar = 10 μm.
Effects of hSelH overexpression on Ht22 cells exposed to 7 mJ/cm2 of UVB irradiation with or without serum in media. Cell viability was examined after 24 h of recovery and presented as percentage (mean ± SD). Overexpression of hSelH protected cells from UVB-induced damage. *Significantly different from MSCV control, P < 0.0001 and #Significantly different from MSCV transfectant, P = 0.0004, in the presence or in the absence of serum, respectively.
Overexpression of hSelH reduces superoxide formation
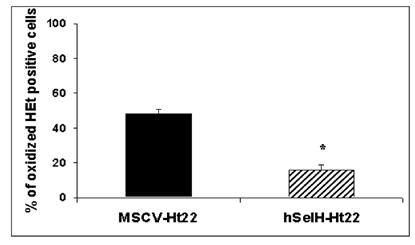
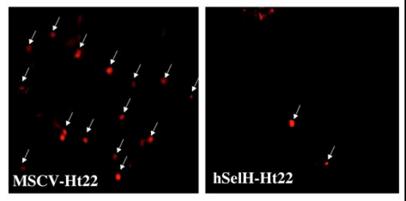
In order to determine the effects of UVB irradiation and overexpression of hSelH on free radical production, we measured superoxide production using the oxidized HEt method. Since a majority of MSCV-transfected cells die after 24 h of recovery, we selected an early recovery time point (5 h) for detection of superoxide formation. A 7 mJ/cm2 of UVB irradiation resulted in 48% of oxidized HEt positive stained cells in MSC-Ht22 after 5 h of recovery. In contrast, the percentage was decreased to 16% (P = 0.0017) in hSelH-Ht22 cells (Figure 4). A set of fluorescent photomicrographs from MSCV-Ht22 and hSelH-Ht22 cells that were exposed to 7 mJ/cm2 of UVB irradiation is shown in Figure 5.
Percentage of oxidized HEt positive cells in MSCV-Ht22 and hSelH-Ht22 cells after 5 h of recovery follow a 7 mJ/cm2 of UVB irradiation. hSelH overexpression significantly reduced oxidized HEt positive cells compared with MSCV-Ht22 cells. *Significantly different from MSCV-Ht22, P = 0.0017.
Representative photomicrographs showing oxidized HEt positive staining in MSCV-Ht22 (A) and hSelH-Ht22 cells (B) after 5 h of recovery follow 7 mJ/cm2 of UVB irradiation. Arrows denote oxidized HEt cells which indicating the formation of superoxide. Images were captured at 20X magnification with a Zeiss LSM5 laser-scanning confocal microscope. Bar = 10 μm.
4. Discussion
Previous published studies on neuronal cells have shown that UV irradiation induced apoptosis is associated with activations of capspase, calpain, JNK, and the mitochondrial apoptosis pathway [4-7]. UV irradiation provokes oxidative stress through the generation of ROS and to induce apoptosis by activation of both receptor-mediated and mitochondrion-initiated cell death pathways [8, 9, 35, 38]. In this study, O2- production increased significantly after 5 h of recovery following a 7 mJ/cm2 of UVB irradiation. This is consistent with the results published by Maglio and colleagues who observed a 5-fold increase in superoxide production in the skin after UV irradiation [39]. Under physiological conditions, superoxide dismutase (SOD) reduces O2˙- to H2O2 that is further reduced to H2O by catalase or glutathione peroxidase. However, excess production of O2˙- leads to formation of the highly toxic hydroxyl radical, •OH, in the presence of metal or peroxynitrite (ONOO-) in the presence of nitric oxide (NO). Hydroxyl radicals and ONOO- are potent oxidants of proteins and DNA, and alter the function of key enzymes through nitration and nitrosylation. Increased ROS in the mitochondrion induces formation of a mitochondrial permeability transition (MPT) pore. The MPT allows release of proapoptotic proteins that subsequently activate a cascade of proteases, ultimately leading to cell death [40, 41].
The selenoprotein family contains 25 members in the human proteome [13]. A majority of published studies have been focused on the expression and functions of GPxs, TRs, iodothyronine deiodinases, and SelP. Studies have shown that these enzymes possess antioxidative effects, in addition to their roles in male fertility, thyroid hormone metabolism, heavy metal detoxification and neurological function and development [20, 26, 42]. For example, glutathione peroxidase acts as peroxynitrite reductase, preventing oxidation and nitration reactions resulting from peroxynitrate [17, 18]. Similarly, SelP protects low-density lipoproteins (LDL) against oxidation (traps ONOO- and protect astrocytes from various injuries including irradiation and tert-butyl hydroperoxide-induced oxidative stress (10, 21, 22, 27]. The functions of most of the recently identified selenoproteins, including SelH, have not been elucidated.
To study whether overexpression of hSelH is capable of protecting cells against UVB-induced oxidative stress, we first transfected the murine hippocampal Ht22 neuron cell line with the hSelH or the empty vector MSCV. As shown in Table 1, the hSelH mRNA level was 30 fold higher than the endogenous mSelH mRNA level in hSelH-Ht22, suggesting a high transcriptional rate of the transfected hSelH gene. We then exposed the cells to a 7 mJ/cm2 of UVB irradiation. Overexpression of hSelH reduced cell death caused by UVB irradiation. This result is consistent with a previous publication showing that overexpression of a selenoprotein homologous to glutathione peroxidase blocks UV-induced cell death [43]. We finally measured O2˙- production in cultured cells at 5 h after a 7 mJ/cm2 of UVB irradiation. The results showed that overexpression of hSelH inhibited O2˙- formation in neurons in the early recovery phase after irradiation. Concurrently, the percent of oxidized HEt-positive cells detected at 5 h of recovery was correlated with the percent of cell death observed at 24 h of recovery, suggesting increased superoxide production might be the underlying cause for the observed cell death. Our findings are in accordance with a previous study reported by Morozova and colleagues [22]. In that study, it was demonstrated that silencing the expression of Drosophila melanogaster SelH (dSelH) gene significantly reduced the total antioxidant capacity and embryo viability. In contrast, overexpression of dSelH aided cell survival by reducing lipid peroxidation in vivo [28].
It is not clear through what mechanism(s) hSelH reduces ROS production. A parallel study carried by Panee and colleagues has shown that overexpression of SelH sustains the total glutathione (GSH) level in Ht22 cells during oxidative stress (Panee et al., manuscript in preparation). GSH plays multiple roles in the protection of cells from ROS, electrophiles and xenobiotics [44, 45]. More interestingly, SelH overexpression upregulated the expression of genes involved in antioxidant defense (Panee et al., manuscript in preparation) and phase II detoxification [44]. Therefore, it is likely that SelH overexpression decreases the ROS level and sustains cell viability in Ht22 cells after UVB irradiation via these pathways.
Taken together, our study shows that overexpression of hSelH protects cells against UVB irradiation-induced cell death by ameliorating ROS formation in the early recovery period. The results are in line with previous publications showing that selenoproteins possess antioxidative capacity.
Acknowledgements
This work was supported in part by NIH R01NS40302, NIH/G12RR03061, NIH/RR16453, and Hawaii Community Foundation. We thank Sarah Timtim and Andrew Sue-Ako for their active participation in this work during their summer research training, Wanyu Liu for technical assistance, and Drs. Rick Bellinger, Peter Hoffman and Steve Seifried for helpful discussions.
Conflict of interests
The authors have declared that no conflict of interest exists.
References
1. Melnikova VO, Ananthaswamy HN. Cellular and molecular events leading to the development of skin cancer. Mutation Res. 2005;571:91-106
2. Licastro F, Sarafian T, Verity AM, Walford RL. Inhibition of polymerases-alpha and -beta completely blocks DNA repair induced by uv irradiation in cultured mouse neuronal cells. Biochem Biophys Res Commun. 1985;132:929-933
3. Subrahmanyam K, Rao KS. Ultraviolet light-induced unscheduled DNA-synthesis in isolated neurons of rat brain of different ages. Mech Ageing Dev. 1991;57:283-291
4. Klums D, Schwartz T. Molecular mechanisms of UV-induced apoptosis. Photodermatol Photoimmunol Photomed. 2000;16:195-201
5. McCollum AT, Nasr P, Estus S. Calpain activates caspase-3 during uv-induced neuronal death but only calpain is necessary for death. J Neurochem. 2002;82:1208-1220
6. McCollum AT, Estus S. Ngf acts via p75 low-affinity neurotrophin receptor and calpain inhibition to reduce uv neurotoxicity. J Neurosci Res. 2004;77:552-564
7. Berglund CM, Radesater AC, Persson MA, Budd Haeberlein SL. UV-induced apoptosis in sh-sy5y cells: Contribution to apoptosis by jnk signaling and cytochrome c. J Neurosci Res. 2004;78:580-589
8. Kulms D, Schwartz T. Independent contribution of three different pathways to ultraviolet-B-induced apoptosis. Biochemical Pharmacol. 2002;64:837-841
9. Kulms D, Schwartz T. Molecular mechanisms involved in UV-induced apoptotic cell death. Skin Pharmacol Appl Skin Physiol. 2002;15:42-47
10. Roussyn I, Briviba K, Masumoto H, Sies H. Selenium-containing compounds protect DNA from single-strand breaks caused by peroxynitrite. Arch Biochem Biophys. 1996;330:216-218
11. Burk RF, Hill KE. Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu Rev Nutr. 2005;25:215-235
12. Kohrle J. Selenium and the control of thyroid hormone metabolism. Thyroid. 2005;15:841-853
13. Kryukov GV, Castellano S, Novoselov SV, Lebanov AV, Zehtab O, Guigo R, Gladyshev VN. Characterization of mammalian Selenoproteomes. Science. 2003;300:1439-1443
14. Rayman MP. The importance of selenium to human health. Lancet. 2000;356:233-241
15. Saito Y, Takahashi K. Characterization of selenoprotein P as a selenium supply protein. Eur J Biochem. 2002;269:5746-5751
16. Saito Y, Sato N, Hirashima M, Takebe G, Nagasawa S, Takahashi K. Domain structure of bi-functional selenoprotein P. Biochem J. 2004;381(Pt 3):841-846
17. Sies H, Sharov VS, Klotz LO, Briviba K. Glutathione peroxidase protects against peroxynitrite-mediated oxidations. A new function for selenoproteins as peroxynitrite reductase. J Biol Chem. 1997;272:27812-27817
18. Sies H, Arteel GE. Interaction of peroxynitrite with selenoproteins and glutathione peroxidase mimics. Free Radic Biol Med. 2000;28:1451-1455
19. Hill KE, Zhou J, McMahan WJ, Motley AK, Atkins JF, Gesteland RF, Burk RF. Deletion of selenoprotein P. alters distribution of selenium in the mouse. J Biol Chem. 2003;278:13640-13646
20. Hirashima M, Naruse T, Maeda H, Nozaki C, Saito Y, Takahashi K. Identification of selenoprotein P fragments as a cell-death inhibitory factor. Biol Pharm Bull. 2003;26:794-798
21. Steinbrenner H, Alili L, Bilgic E, Sies H, Brenneisen P. Involvement of selenoprotein P in protection of human astrocytes from oxidative damage. Free Radic Biol Med. 2006;40:1513-1523
22. Traulsen H, Steinbrenner H, Buchczyk DP, Klotz LO, Sies H. Selenoprotein P protects low-density lipoprotein against oxidation. Free Radic Res. 2004;38:123-128
23. Burk RF, Hill KE, Awad JA, Morrow JD, Kato T. et al. Pathogenesis of diquat-induced liver necrosis in selenium-deficient rats. Assessment of their roles of lipid peroxidation by measurement of F2 isoprostanes. Hepatology. 1995;21:561-569
24. Valentine WM, Hill KE, Austin LM, Valentine HL, Goldowitz D, Burk RF. Brainstem axonal degeneration in mice with deletion of selenoprotein P. Toxicologic Pathology. 2005;33:570-576
25. Arteel GE, Mostert V, Oubrahim H, Briviba K, Abel J, Sies H. Protection by selenoprotein P in human plasma against peroxynitrite-mediated oxidation and nitration. Biol Chem. 1998;379:1201-1205
26. Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathioneperoxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 2003;34:145-169
27. Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, Prolla TA. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496-502
28. Morozova N, Forry EP, Shahid E, Zavacki AM, Harney JW, Kraytsberg Y, Berry MJ. Antioxidant function of a novel selenoprotein in Drosophila melanogaster. Genes Cells. 2003;8:963-971
29. Muranyi M, Li PA. Hyperglycemia increases superoxide production in the CA1 pyramidal neurons after global cerebral ischemia. Neurosci Lett. 2006;393:119-121
30. Muranyi M, Ding C, He Q, Lin Y, Li PA. Streptozotocin-induced diabetes causes astrocyte death after ischemia and reperfusion injury. Diabetes. 2006;55:349-355
31. Bindokas VP, Jordan J, Lee CC, Miller R. Superoxide production in Rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324-1336
32. Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205-213
33. Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vasquez-Vivar J, Kalyanaraman B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic Biol Med. 2003;34:1359-1368
34. Fridovich I. Editorial commentary on "Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide" by H. Zhao et al. Free Radic Biol Med. 2003;34:1357-1358
35. Ho JN, Lee YH, Park JS, Jun WJ, Kim HK, Hong BS, Shin DH, Cho HY. Protective effects of Aucubin isolated from Eucommia ulmoides against UVB-induced oxidative stress in human skin fibroblasts. Biol Pharm Bull. 2005;128:244-1248
36. O'Donovan P, Perrett CM Zhang X, Montaner B Xu Y-Z, Harwood CA McGregor JM, Walker SL Hanaoka F, Karran P . Azathioprine and UVA light generate mutagenic oxidative DNA damage. Science. 2005;306:1871-1874
37. Shiu CT, Lee TM. Ultraviolet-B-induced oxidative stress and responses of the ascorbate-glutathione cycle in a marine macroalga Ulva fasciata. J Exp Bot. 2005;56:2851-2865
38. Valencia A, Moran J. Reactive oxygen species induce different cell death mechanisms in cultured neurons. Free Radic Biol Med. 2004;36:1112-1215
39. Maglio DHG, Paz ML, Ferrari A, Weill FS, Czerniczyniec A, Leoni J, Bustamante J. Skin damage and mitochondrial dysfunction after acute ultraviolet B irradiation: relationship with nitric oxide production. Photodermatol Photoimmunol Photomed. 2005;21:311-317
40. Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309-1312
41. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626-629
42. Chen J, Berry MJ. Selenium and selenoproteins in the brain and brain diseases. J Neurochem. 2003;86:1-12
43. Shisler JL, Senkevich TG, Berry MJ, Moss B. Ultraviolet-induced cell death blocked by a selenoprotein from a human dermatotropic poxvirus. Science. 1998;279:102-151
44. Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 1999;13:1169-1183
45. Maher P. The effects of stress and aging on glutathione metabolism. Aging Res Rev. 2005;4:288-314
Author contact
![]() Correspondence to: Dr. Kamel E. Ben Jilani, Department of Cell Molecular Biology, John A. Burns School of Medicine, 651 Ilalo Street, BSB 217, Honolulu, HI96813, USA; E-mail: kameledu
Correspondence to: Dr. Kamel E. Ben Jilani, Department of Cell Molecular Biology, John A. Burns School of Medicine, 651 Ilalo Street, BSB 217, Honolulu, HI96813, USA; E-mail: kameledu