Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(2):154-167. doi:10.7150/ijbs.7.154 This issue Cite
Research Paper
Olmesartan, an AT1 Antagonist, Attenuates Oxidative Stress, Endoplasmic Reticulum Stress and Cardiac Inflammatory Mediators in Rats with Heart Failure Induced by Experimental Autoimmune Myocarditis
Vijayakumar Sukumaran1, Kenichi Watanabe1, ![]() , Punniyakoti T. Veeraveedu1,2, Narasimman Gurusamy3, Meilei Ma1, Rajarajan A. Thandavarayan1, Arun Prasath Lakshmanan1, Ken'ichi Yamaguchi4, Kenji Suzuki5, Makoto Kodama6
, Punniyakoti T. Veeraveedu1,2, Narasimman Gurusamy3, Meilei Ma1, Rajarajan A. Thandavarayan1, Arun Prasath Lakshmanan1, Ken'ichi Yamaguchi4, Kenji Suzuki5, Makoto Kodama6
1. Department of Clinical Pharmacology, Faculty of Pharmaceutical Sciences, Niigata University of Pharmacy and Applied Life Sciences, Niigata, Japan.
2. WPI Immunology Frontier Research Center, Osaka University, Osaka, Japan.
3. Department of Anesthesiology and Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA.
4. Department of Homeostatic Regulation and Development, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan.
5. Department of Gastroenterology and Hepatology, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan.
6. First Department of Medicine, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan.
Received 2010-9-20; Accepted 2011-2-7; Published 2011-2-11
Abstract
Studies have demonstrated that angiotensin II has been involved in immune and inflammatory responses which might contribute to the pathogenesis of immune-mediated diseases. Recent evidence suggests that oxidative stress may play a role in myocarditis. Here, we investigated whether olmesartan, an AT1R antagonist protects against experimental autoimmune myocarditis (EAM) by suppression of oxidative stress, endoplasmic reticulum (ER) stress and inflammatory cytokines. EAM was induced in Lewis rats by immunization with porcine cardiac myosin, were divided into two groups and treated with either olmesartan (10 mg/kg/day) or vehicle for a period of 21 days. Myocardial functional parameters measured by hemodynamic and echocardiographic analyses were significantly improved by the treatment with olmesartan compared with those of vehicle-treated rats. Treatment with olmesartan attenuated the myocardial mRNA expressions of proinflammatory cytokines, [Interleukin (IL)-1β, monocyte chemoattractant protein-1, tumor necrosis factor-α and interferon-γ)] and the protein expression of tumor necrosis factor-α compared with that of vehicle-treated rats. Myocardial protein expressions of AT1R, NADPH oxidase subunits (p47phox, p67phox, gp91phox) and the expression of markers of oxidative stress (3-nitrotyrosine and 4-hydroxy-2-nonenal), and the cardiac apoptosis were also significantly decreased by the treatment with olmesartan compared with those of vehicle-treated rats. Furthermore, olmesartan treatment down-regulated the myocardial expressions of glucose regulated protein-78, growth arrest and DNA damage-inducible gene, caspase-12, phospho-p38 mitogen-activated protein kinase (MAPK) and phospho-JNK. These findings suggest that olmesartan protects against EAM in rats, at least in part via suppression of oxidative stress, ER stress and inflammatory cytokines.
Keywords: Experimental autoimmune myocarditis, oxidative stress, endoplasmic reticulum stress, inflammation, olmesartan
Introduction
Inflammation and autoimmunity are involved in many cardiac diseases. Myocarditis is an inflammatory heart disease, and causes both acute and chronic heart failure as a result [1]. Some patients with myocarditis show a fulminant course and die of intractable cardiogenic shock, and the treatment strategy for the disease is still unresolved [2]. One of the fatal subtypes of myocarditis is giant cell myocarditis, and its etiology is considered to be related to autoimmunity [3, 4]. A model of rat experimental autoimmune myocarditis (EAM) resembles human giant cell myocarditis, and the recurrent form of EAM leads to dilated cardiomyopathy [3, 4]. EAM is induced by T cell activation and the peak of inflammation is observed in the heart around day 21 after immunization [5, 6]. Some evidence supports a role of cellular immune mechanisms in the pathogenesis of myocarditis and subsequent DCM [7].
The renin-angiotensin-aldosterone system (RAAS) plays an important role in the pathogenesis of a variety of clinical conditions, including atherosclerosis, hypertension, left ventricular (LV) hypertrophy, myocardial infarction, and heart failure [8, 9]. As a result, the RAAS represents a logical therapeutic target in the management of hypertension, renal disease, and cardiovascular disease. Inhibition of the RAAS with either angiotensin-converting enzyme inhibitors which block the formation of angiotensin II (Ang-II), the principal effector peptide of the RAAS or Ang-II receptor blockers (ARBs) block the deleterious effects of Ang-II at the Ang-II type 1 receptor (AT1R) has been shown to be effective in lowering blood pressure and reducing cardiovascular mortality and morbidity in various at-risk patient populations. The effects of ARBs in the treatment of hypertension, congestive heart failure and myocardial fibrosis have been well analyzed in human trials, as well as animal models, and the focus of interest is now directed to its pleiotropic effects, especially on inflammatory disorders. ARBs have also been reported to suppress atherosclerotic lesions in animal models and patients with coronary artery disease or hypertension by modulating inflammatory responses [10-12].
Within the ARB class, olmesartan medoxomil is a long acting AT1R antagonist approved for the treatment of mild to severe hypertension, alone or in combination with other agents. Olmesartan reduced the production of reactive oxygen species (ROS), suppressed tissue infiltration of macrophages, and prevented ventricular hypertrophy and fibrosis in hypertensive Dhal salt-sensitive rats with advanced heart failure [13]. Tsuda et al [14] reported that olmesartan synergistically attenuate atherosclerosis at least in part via inhibition of oxidative stress. Moreover, olmesartan is therapeutically effective for the treatment of patients with heart failure by reducing cytokines and oxidative stress through its anti-inflammatory effects [12-14]. Thus the blockade of AT1 receptors is another way to interrupt the renin-angiotensin system (RAS). Recently olmesartan is shown to ameliorate EAM by the suppression of myocardial damage and inflammatory events in the myocardium in addition to hemodynamic modifications [15, 16].
Despite many reports show the beneficial effect of olmesartan in cardiovascular diseases [12-18], its effect on oxidative stress and endoplasmic reticulum (ER) stress is not known in rats with heart failure after EAM. Moreover no study has reported the effects of olmesartan on myocardial function by hemodynamic study, ER stress and MAPK/JNK signaling associated with EAM. Recently we reported that olmesartan improved cardiac function and attenuated cardiac remodeling (fibrosis and hypertrophy) and inflammatory mediators in rats with dilated cardiomyopathy after EAM [19]. In the current study, we have examined the effects of olmesartan on cardiac function, oxidative stress, ER stress, inflammatory cytokines and MAPK/JNK signaling pathways associated with EAM in rats. Our results strongly suggest that olmesartan protects against EAM in rats, at least in part via suppression of oxidative stress, ER stress and inflammatory cytokines.
Materials and Methods
Materials
Olmesartan was generously provided by Daichi-Sankyo Pharmaceutical (Tokyo, Japan), and Lewis rats (male, 8 weeks old) were purchased from Charles River Japan Inc., Kanagawa, Japan.
Experimental design
All experiments were carried out using 8-week-old male Lewis rats and were performed in accordance with the guidelines of our institute [3, 20]. Lewis rats were injected in the footpads with antigen-adjuvant emulsion in accordance with a procedure described previously. In brief, porcine cardiac myosin was dissolved in phosphate-buffered saline at 5 mg/ml and emulsified with an equal volume of complete freund's adjuvant with 11 mg/ml Mycobacterium tuberculosis H37RA (Difco Lab., Detroit, MI, USA). EAM in rats was induced by immunization with 0.1 ml of emulsion once by subcutaneous injection into their rear footpads (0.1 ml to each footpad). The morbidity of EAM was 100% in rats immunized by this procedure [3, 20]. After immunization, the Lewis rats were divided into two groups and received oral administration of olmesartan (10 mg/kg/day; Group-Olm-10) or vehicle (Group-V) for 21 days. Age matched Lewis rats without immunization was used as normal controls (Group-N). Since fibrosis and inflammation plays an important role in myocardial remodeling in our EAM model, we have chosen the antifibrotic, anti-inflammatory and maximal hypotensive dose of olmesartan as previously reported [15, 16, 19, 21]. Moreover, we reported that olmesartan (10 mg/kg/day) improved cardiac function and attenuated cardiac remodeling (fibrosis and hypertrophy) and inflammatory mediators in rats with dilated cardiomyopathy after EAM [19].
Hemodynamic and echocardiographic studies
To obtain hemodynamic data, rats were anesthetized with 2% halothane in oxygen during the surgical procedures. A catheter-tip transducer (Miller SPR 249; Miller Instruments, Houston, TX) was inserted into the left ventricle through the right carotid artery for the determination of peak left ventricular pressure (LVP) and left ventricular end-diastolic pressure (LVEDP), and the rates of intraventricular pressure rise (+ dP/dt) and decline ( ̶ dP/dt) were recorded as described previously [20]. After instrumentation, the concentration of halothane was reduced to 0.5% to minimize the effects of anesthesia on hemodynamic parameters. In addition, systolic blood pressure (SBP) and diastolic blood pressure (DBP) was measured in conscious rats by using the tail-cuff plethysmographic method (Softron BP-98A, Tokyo, Japan). Echocardiographic studies were carried out with a 7.5-MHz transducer (Aloka Inc., Tokyo, Japan). The left ventricular dimensions in diastole (LVDd) and systole (LVDs) and percentage fractional shortening (FS) were estimated using M-mode measurements.
Cardiac morphometric parameters
The body weight (BW) of rats was noted just before the surgical procedure. After the hemodynamic and echocardiographic analyses, the rats were sacrificed, and the whole myocardium was isolated and weighed to calculate the ratio of heart weight to body weight (HW/BW).
Histopathology
The excised wet myocardium was kept in 10% formalin and the midventricle sections were then embedded with paraffin. Inflammatory cell infiltrations were identified using hematoxylin and eosin (H&E)-stained sections at 200-fold magnification by light microscopy. Several sections of each heart were scored blindly by 2 observers. The scores assigned to these specific sections were averaged as described previously [22]. The extent of cellular infiltration was graded and scored as follows: 0 (normal), 1 (lesion extent between 10-25% of a transverse section), 2 (between 25-50%), 3 (between 50-75%), and 4 (exceeding 75%). In addition, the area of myocardial fibrosis in the midventricle tissue sections stained with Azan-Mallory was quantified using a color image analyzer (CIA-102, Olympus, Tokyo, Japan) and measuring the blue fibrotic areas as opposed to the red myocardium at 200X magnification. The results were presented as the ratio of the fibrotic area to the whole area of the myocardium [20].
Analysis of mRNA levels of inflammatory cytokines
RNA Extraction
Heart tissues were preserved by immersion in RNAlater (Ambion Inc., Austin, TX) immediately after sampling. The extraction of total RNA was performed after homogenization by using Ultra TurraxT8 (IKA Labortechinik, Staufen, Germany) in TRIzol reagent (invitrogen Corp., Carlsbad, CA) in accordance with the standard protocol. Synthesis of cDNA was performed by reverse transcription using total RNA (2 µg) as a template (Super Script II; Invitrogen Corporation, Carlsbad, CA).
Gene expression analysis by real time RT-PCR
Gene expression analysis was performed by real time reverse transcription polymerase chain reaction (RT-PCR) (Smart Cycler; Cepheid, Sunnyvale, CA) using cDNA synthesized from the CHF specimens. Primer sequences were as follows: Interleukin (IL)-1β (forward), CTTCAATCTCACAGCAGCACATCTCG, (reverse), TCCACGGGCAAGACATAGGTAGC; MCP-1 (forward), CTCACCTGCTGCTACTCATTCACT, (reverse), TGCTGCTGGTGATTCTCTTGTAGT; interferon (IFN)-γ (forward), GCTTTGCAGCTCTTCCTCAT, (reverse), GTCACCATCCTTTTGCCAGT; TNF-α (forward), CCCCAAAGGGATGAGAAGTT, (reverse), CACTTGGTGGTTTGCTACGA; GAPDH (forward), GCTCATTTCCTGGTATGACAACG, (reverse), AGGGGTCTACATGGCAACTG. Real time RT-PCR by monitoring with TaqMan probe (TaqMan Gene expression assays; Applied Biosystems, Foster City, CA) was performed in accordance with the following protocol: 600 seconds at 95 ºC, followed by thermal cycles of 15 seconds at 95 ºC, and 60 seconds at 60 ºC for extension. Relative standard curves representing several 10 fold dilutions (1:10:100:1000:10,000:100,000) of cDNA from heart tissue samples were used for linear regression analysis of other samples. Results were normalized to GAPDH mRNA as an internal control and are thus shown as relative mRNA levels.
Immunohistochemistry
Formalin-fixed, paraffin-embedded cardiac tissue sections were used for immunohistochemical staining. After deparaffinization and hydration, the slides were washed in Tris-buffered saline (TBS; 10 mM/l Tris HCl, 0.85% NaCl, pH 7.5) containing 0.1% bovine serum albumin (BSA). Endogenous peroxidase activity was quenched by incubating the slides in methanol and 0.6% H2O2 in methanol. To perform antigen retrieval, the sections were pretreated with trypsin for 15 min at 37 °C. After overnight incubation with the primary antibody, namely mouse polyclonal anti-3-nitrotyrosine (3-NT) (Abcam Inc, MA, USA), mouse monoclonal anti-4-Hydroxynonenal (4-HNE) (Oxis International, Portland, OR) and rabbit polyclonal anti-Ang-II antibody (Peninsula Laboratories Inc, San Carlos, California) (diluted 1:100) at 4 °C, the slides were washed in TBS and horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was then added and the slides were further incubated at room temperature for 45 min. The slides were washed in TBS and incubated with diaminobenzidine tetrahydrochloride as the substrate, and counterstained with hematoxylin. A negative control without primary antibody was included in the experiment to verify the antibody specificity. Measurement of myocardial immunoreactivity for Ang-II was performed in 25 randomly selected fields in heart sections in 400-fold magnification by light microscopy. Ten color images of 4-HNE staining were selected from four sections of the heart and viewed at 400-fold magnifications. The area and intensity of 4-HNE staining were quantified by scoring in a blinded manner. The scoring range was the following: where 0, no visible staining; 1, faint staining; 2 moderate staining; and 3, strong staining [23].
Western Immunoblotting
The myocardial tissue samples obtained from different groups were homogenized with lysis buffer. Protein concentrations in these homogenized samples were measured by the bicinchoninic acid method. For Western blots, proteins were separated by SDS-PAGE and identified with the following antibodies to quantify the myocardial levels of proteins: goat polyclonal anti-p67phox, anti-gp91phox, anti-glucose regulated protein 78 (GRP78), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH), anti-tumor necrosis factor-α antibodies; rabbit polyclonal anti-AT1R, anti-caspase-12, anti-p47phox, nuclear factor kappa B (NF-κB), p38 mitogen-activated protein kinase (p38 MAPK), phospho-p38 MAPK, c-Jun N-terminal Kinase (JNK), phospho-JNK antibody and mouse polyclonal anti-C/EBP homologous protein (CHOP), anti-osteopontin (OPN) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). We used 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Bio-Rad, CA, USA), and electrophoretically transferred to nitrocellulose membranes. Membranes were blocked with 1% nonfat dry milk and 1% BSA (Sigma, St Louis, USA) in TBS-T (20 mM/l Tris, pH 7.6, 137 mM/l NaCl, and 0.05% Tween). After incubation with primary antibody, the bound antibody was visualized with respective HRP-coupled secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and chemiluminescence developing agents (Amersham Biosciences, Buckinghamshire, UK). The level of GAPDH was estimated in every sample to check the equal loading of sample. MAPK and JNK activation was quantified by normalizing phospho-MAPK and phospho-JNK expression level with MAPK and JNK expression in the same sample respectively. Films were scanned, and band densities were quantified with densitometric analysis using Scion Image program (GT-X700, Epson, Tokyo, Japan).
Analysis of cardiac apoptosis by terminal transferase-mediated dUTP nick-end labeling assays
The transferase-mediated dUTP nick-end labeling (TUNEL) assay was performed as specified in the instructions for the in situ apoptosis detection kit (Takara Bio Inc., Shiga, Japan). Sections embedded in paraffin were mounted and examined using light microscopy. Digital photomicrographs were obtained by using a color image analyzer (CAI-102, Olympus) at X400 magnification, and 25 random fields from each heart were chosen and the number of TUNEL positive nuclei was quantified in a blinded manner. For each group, three sections were scored for apoptotic nuclei. Only nuclei that were clearly located in cardiac myocytes were considered.
Statistical analysis
All values are expressed as means ± SEM. Statistical analysis of differences between the groups was performed by one-way ANOVA, followed by Tukey's or Bonferroni's method and two tailed t-test when appropriate. A value of P < 0.05 was considered as statistically significant.
Results
Olmesartan treatment improves myocardial function
Although heart rate was not different among the three groups of rats, central venous pressure (CVP) and LVEDP were significantly higher (6.96±0.8 vs 0.48±0.2 mm Hg P < 0.01; 21.23±2.2 vs 3.18±6.4 mm Hg P < 0.01, respectively) and ±dP/dt were significantly lower in vehicle-treated rats than in group N (2133±219 vs 8004±1462 mm Hg/s P < 0.01; 2003±236 vs 7958±1320 mm Hg/s P < 0.01, respectively), indicating systolic and diastolic dysfunction in vehicle-treated rats (Table 1). CVP and LVEDP were significantly decreased in the olmesartan-treated group compared with those in vehicle-treated group. Myocardial contractility parameters were also improved in EAM rats treated with olmesartan. In addition, SBP and DBP were significantly lowered in vehicle-treated rats than in group N rats and were further lowered by treatment with olmesartan.
Changes in histopathological, hemodynamic and echocardiographic parameters after 3 weeks of treatment with olmesartan in rats with or without EAM
| Group N(n=6) | Group V(n=8) | Group Olm-10 (n=8) | |
|---|---|---|---|
| Histopathology | |||
| BW (g) | 336±6.3 | 253±5.7** | 260±4.1** |
| HW (g) | 1.02±0.01 | 1.47±0.04** | 0.98±0.04## |
| HW/BW (g/kg) | 3.1±0.09 | 5.7±0.44** | 4.3±0.20*## |
| Cardiac histological score (cellular infiltration) | 0 | 2.7±0.60 | 1.0±1.20## |
| Area of fibrosis (%) | 2.5±0.5 | 56.75±2.4** | 32.5±2.1## |
| Hemodynamic data | |||
| CVP (mmHg) | 0.48±0.2 | 6.96±0.8** | 3.67±0.9## |
| SBP (mmHg) | 121 ± 3 | 105 ± 10* | 92 ± 8* |
| DBP (mmHg) | 90 ± 5 | 85 ± 8* | 74 ± 9* |
| LVEDP (mmHg) | 3.18±6.4 | 21.23±2.2** | 7.64±2.5** |
| +dP/dt (mmHg/s) | 8004±1462 | 2133±219** | 2986±316** |
| _dP/dt (mmHg/s) | 7958±1320 | 2003±236** | 2408±224** |
| HR (beats/min) | 352±5.3 | 306±22.6 | 299±19.6 |
| Echocardiographic data | |||
| LVDd (mm) | 7.1±0.6 | 7.6±0.4 | 7.1±0.3 |
| LVDs (mm) | 4.0±0.56 | 6.5±0.51** | 4.4±0.3# |
| FS (%) | 44.7±3.3 | 14.4±3.8** | 39.2±3.3## |
| EF (%) | 80.6±3.3 | 34.2±8.3** | 74.3±3.7## |
Results are presented as the mean ± SEM. n, no. of rats. BW, body weight; HW, heart weight; HW/BW, ratio of heart weight to body weight; CVP, central venous pressure; SBP, systolic blood pressure; DBP, diastolic blood pressure; LVEDP, left ventricular end-diastolic pressure; {±} dP/dt, rate of intra-ventricular pressure rise and decline; HR, heart rate; LVDd, left ventricular dimension in diastole; LVDs, left ventricular dimension in systole; FS, fractional shortening; EF, ejection fraction; group N, aged matched untreated rats; group V, rats with heart failure treated with vehicle; group Olm-10, rats with heart failure treated with olmesartan 10 mg/(kg day), respectively; *P < 0.05 and **P < 0.01 vs. group N; #P < 0.05 and ##P < 0.01 vs. group V.
Echocardiographic studies shown that in group V rats with increased LVDd and LVDs (7.6±0.4 vs 7.1±0.6 mm P < 0.01; 6.5±0.51 vs 4.0±0.56 mm P < 0.01, respectively), and reduced FS (14.4±3.8 vs 44.7±3.3% P < 0.01) and ejection fraction (EF) (34.2±8.3 vs 80.6±3.3% P < 0.01), indicating impaired systolic function compared with that in group N rats (Table 1). Treatment with olmesartan significantly decreased LVDd and increased FS and EF compared with those in vehicle-treated rats (Table 1).
Effects of olmesartan on morphometric parameters
HW and HW/BW were significantly larger in group V than in group N rats (1.47±0.04 vs 1.05±0.01 g P < 0.01; 5.7±0.44 vs 3.1±0.09 g/kg P < 0.01, respectively and Table 1). Olmesartan significantly reduced HW and HW/BW, compared with those in vehicle-treated rats.
Effects of olmesartan on cellular inflammation and inflammatory cytokines
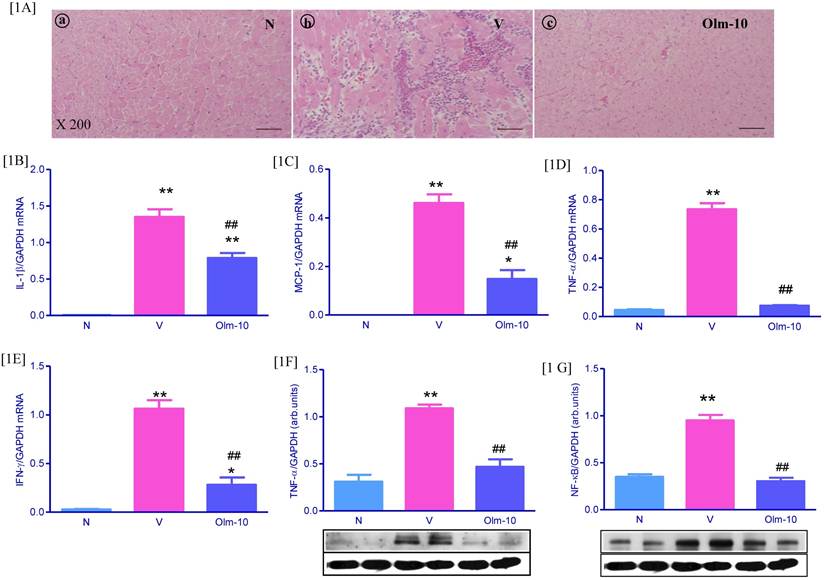
On day 21 at euthanization, severe injuries to myocardium with cellular infiltration were observed in the myocarditis group. The severity of cellular infiltration was significantly reduced in the olmesartan group, indicating a significantly reduced severity of disease (Fig 1A and Table 1). In addition, RT-PCR analyses showed that the expression of IL-1β, MCP-1, TNF-α, and IFN-γ mRNA e were significantly increased in vehicle-treated rats, compared with those in group N rats (Fig 1B-E). In contrast, treatment with olmesartan significantly decreased the myocardial mRNA expressions of IL-1β, MCP-1, TNF-α, and IFN-γ (1.6, 2.8, 1.9 and 3.8-fold, respectively) compared with those in group V rats (Fig 1B-E). In line with RT-PCR data, protein expressions of TNF-α and NF-κB in the myocardial tissue was significantly increased in group V rats compared with those in group N rats (Fig 1F-G), and this was significantly attenuated by olmesartan treatment.
Effects of olmesartan on cellular infiltration and inflammatory cytokines in rats with EAM induced by autoimmune myocarditis. [1A] Histopathology in the heart (H&E-staining) at ×200 fold magnification. (a) Histopathology in a control (group N) (grade 0). (b) Representative histopathology in rat with myocarditis treated with vehicle (group V), the myocardial inflammation was diffused and exceeding 50% of the transverse section (grade 3). (c) Representative histopathology in rat with myocarditis treated with olmesartan (group Olm-10), showed a small focus of cellular infiltration (grade 1). [1B-F] Myocardial messenger RNA expression levels of IL-1β [1B], MCP-1 [1C], TNF-α [1D], IFN-γ [1E] and protein expression of TNF-α [1F], NF-κB [1G] in rats with EAM was determined by quantitative RT-PCR and Western blot. The mRNA expression level of each sample was expressed relative to the expression level of GAPDH gene. [1F-1G] Representative Western blots showing specific bands for TNF-α, NF-κB and GAPDH as an internal control. Equal amounts of protein sample (30 μg) obtained from whole ventricular homogenate were applied in each lane. The mean density value of TNF-α, NF-κB was expressed as a ratio relative to that of GAPDH. Data are mean ± SEM of 4 to 6 rats. Group N, age-matched untreated rats; group V, EAM rats administered with vehicle; group Olm-10, EAM rats treated with olmesartan (10 mg/kg/day). The values are mean ± SEM. *P<0.05, **P<0.01 vs group N; ##P<0.01 vs group V.
Effects of olmesartan on myocardial fibrosis and its marker molecule (OPN)
The hearts from EAM rats showed massive fibrosis and an increased expression of its marker molecule (OPN) compared to those from group N rats (Fig 2 A and A1). Treatment with olmesartan significantly reduced the percent area of fibrosis and OPN expression than in vehicle-treated rats (Fig 2 A and A1).
Effects of olmesartan on cardiac fibrosis and oxidative stress in rats with EAM induced by autoimmune myocarditis. [2A] Azan-Mallory staining for fibrosis of the cross-sectional tissue slices of hearts. Fibrosis is indicated by the blue area as opposed to the red myocardium (×200). [2A1] Myocardial protein expression of osteopontin (OPN). Representative Western blots showing specific bands for OPN and GAPDH as an internal control. Equal amounts of protein sample (30 μg) obtained from whole ventricular homogenate were applied in each lane. The mean density value of OPN was expressed as a ratio relative to that of GAPDH. [2B, C, D] Immunohistochemistry of 4-hydroxy-2-nonenal (4-HNE), 3-nitrotyrosine (3-NT) and angiotensin-II [counterstained with hematoxylin; ×400]. Bar graph shows quantitative analysis of 4-HNE [2B1] and Ang-II positive cardiomyocytes [2D1] in groups N, V and Olm-10. Group N, age-matched untreated rats; group V, EAM rats administered with vehicle; group Olm-10, EAM rats treated with olmesartan (10 mg/kg/day). The bar value indicates 20 μm. The values are mean ± SEM. *P<0.05 and **P<0.01 vs group N; #P<0.05, ##P<0.01 vs group V.
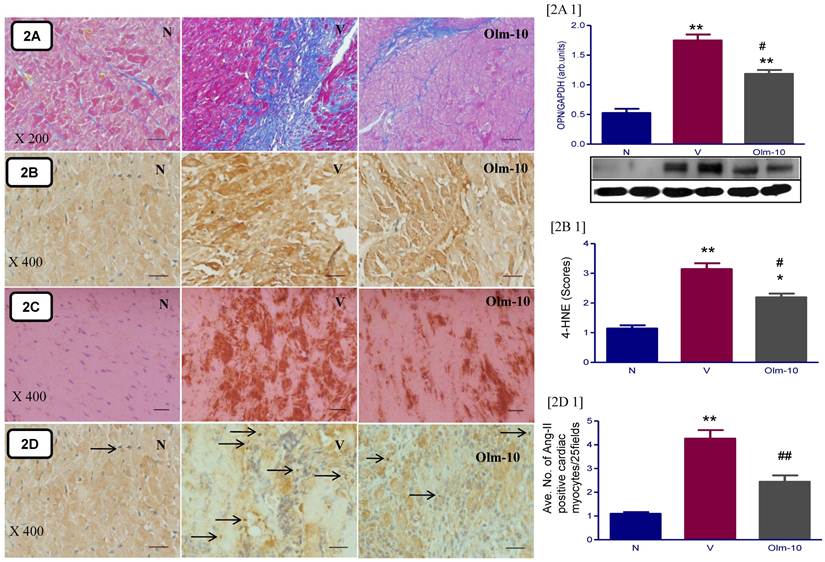
Effects of olmesartan on myocardial 4-HNE and 3-NT content
Myocardial staining intensities of 4-HNE, a byproduct of lipid peroxidation, an index of oxidative stress, were apparently stronger in the vehicle-treated rats compared with that in group N rats. Olmesartan treatment significantly reduced the intensities of 4-HNE in EAM rats (Fig 2B and B1). Semi quantification analysis showed that 4-HNE scores were also increased in the vehicle-treated group and the increase was attenuated by the treatment of olmesartan (Fig 2B and B1). In parallel with increased NADPH oxidase subunits (Fig 3B-D), there was an increase in peroxynitrite formation in vehicle-treated rats, as measured by the content of 3-NT in the cardiac tissue (Fig 2C). In contrast, myocardial 3-NT content was significantly decreased in olmesartan treated rats compared to that in group V.
Effects of olmesartan on myocardial protein expressions of AT1R, p47phox, p67phox, and gp91phox. [3A-D] Representative Western blots showing specific bands for AT1R, p47phox, p67phox, gp91phox and GAPDH as an internal control. Equal amounts of protein sample (30 μg) obtained from whole ventricular homogenate were applied in each lane. These bands are representative of four separate experiments. 3A-D, Densitometric data of protein analysis. The mean density value of AT1R, p47phox, p67phox and gp91phox was expressed as a ratio relative to that of GAPDH. Each bar represents mean ± SEM of 4 to 6 rats. Group N, age-matched untreated rats; group V, EAM rats administered with vehicle; group Olm-10, EAM rats treated with olmesartan (10 mg/kg/day). The values are mean ± SEM. **P<0.01 vs group N; #P<0.05, ##P<0.01 vs group V.
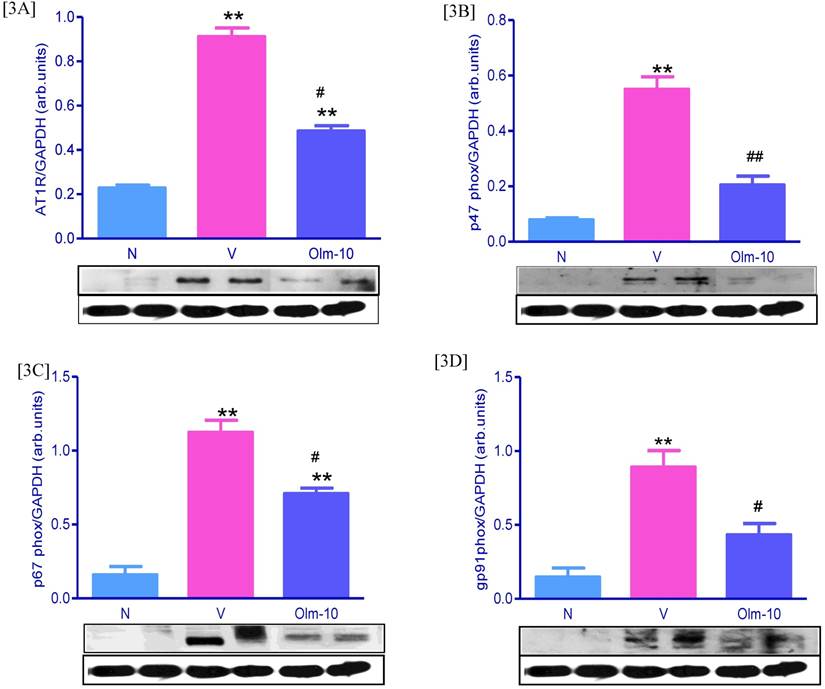
Olmesartan attenuate the expression of cardiac Ang-II, AT1R and NADPH oxidase subunits
Ang-II activates NADPH-dependent oxidases via AT1R stimulation, the most important vascular source of superoxide (O2-). The cardiac expression of Ang-II, AT1R and NADPH oxidase subunits were significantly higher in group V rats compared with that shown in group N rats (Figs 2D, D1 and Fig 3), and these changes were significantly reversed by the treatment with olmesartan (Figs 2D, D1 and Fig 3).
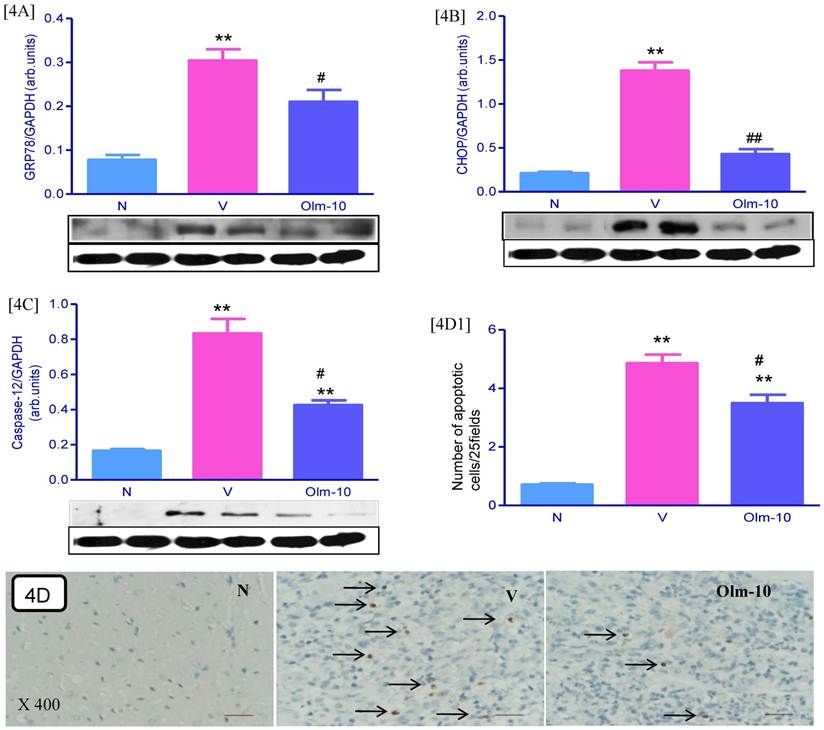
Effects of olmesartan on myocardial protein expressions of GRP78, CHOP and Caspase-12 and apoptosis
The protein levels of GRP78, CHOP and caspase-12 in the myocardium were significantly upregulated in group V rats as compared with those in group N rats. These levels were significantly attenuated in olmesartan treated rats when compared with the vehicle-treated rats (Fig 4A-C). In addition, cardiac apoptosis is further confirmed by TUNEL assay and showed that the numbers of TUNEL positive nuclei were significantly higher in myocardial tissue sections from group V than in group N (Fig 4D and D1). However, treatment with olmesartan significantly decreased the number of TUNEL positive nuclei compared to that in group V (Fig 4D and D1).
Effects of olmesartan on myocardial protein expressions of GRP78, CHOP, caspase-12 and apoptosis. [4A-C], Representative Western blots showing specific bands for GRP78, CHOP, caspase-12 and GAPDH as an internal control. Equal amounts of protein sample (30 μg) obtained from whole ventricular homogenate were applied in each lane. These bands are representative of three separate experiments. 4A-C, Densitometric data of protein analysis. The mean density value of GRP78, CHOP and caspase-12 was expressed as a ratio relative to that of GAPDH. [4D] Myocardial tissue sections stained for TUNEL-positive apoptotic nuclei (indicated by arrows) in the hearts of myosin-immunized rats (×400). [4D1] Bar graph shows quantitative analysis of TUNEL positive cells. Each bar represents mean ± SEM of 4 to 6 rats. Group N, age-matched untreated rats; group V, EAM rats administered with vehicle; group Olm-10, EAM rats treated with olmesartan (10 mg/kg/day). The values are mean ± SEM. **P<0.01 vs group N; #P<0.05, ##P<0.01 vs group V.
Effects of olmesartan on myocardial protein expressions of MAPK and JNK
Myocardial protein expression of phospho-p38 MAPK, phospho-JNK was markedly increased in group V rats compared with that in group N rats (Fig 5A and 5B) and this was significantly attenuated by olmesartan treatment (Fig 5A and 5B).
Myocardial expressions of phospho-p38 MAPK, phospho-JNK family proteins. [5A-B] Representative western blots showing specific bands for phospho-p38 MAPK, phospho-JNK, p38 MAPK and JNK as an internal control. Equal amounts of protein sample (30 μg) obtained from whole ventricular homogenate were applied in each lane. These bands are representative of four separate experiments. 5A-B, Densitometric data of protein analysis. The mean density value of phospho-p38 MAPK and phospho-JNK was expressed as a ratio relative to that of p38 MAPK and JNK. Each bar represents mean ± SEM of 4 to 6 rats. Group N, age-matched untreated rats; group V, EAM rats administered with vehicle; group Olm-10, EAM rats treated with olmesartan (10 mg/kg/day). The values are mean ± SEM. **P<0.01 vs group N; #P<0.05 vs group V.

Discussion
The results of the present study demonstrate that the treatment with oral olmesartan improved both systolic (+dP/dt, % EF and % FS) and diastolic (−dP/dt and LVEDP) functions and reduced the severity of acute EAM in rats, and that the cardioprotection of olmesartan occurs at least in part via the suppression of oxidative stress, endoplasmic reticulum (ER) stress and inflammatory cytokines in addition to hemodynamic modifications.
Proinflammatory cytokines are capable of modulating cardiovascular function by various mechanisms. Several clinical studies have described the participation of proinflammatory cytokines in the pathogenesis of cardiac diseases. The levels of circulating proinflammatory cytokines such as tumor necrosis factor (TNF)-α and IL-1 and IL-6 are elevated in patients with myocarditis [24]. In a murine model of viral myocarditis, the intracardiac expression of TNF-α, IL-1β, interferon-γ, and IL-2 genes were increased [25]. The degree of expression was correlated with the severity of the disease, which suggests that the overproduction of proinflammatory cytokines and chemokines may aggravate the disease. This is supported in part by recent reports that the over expression of TNF-α in heart caused severe myocarditis and cardiomyopathy in transgenic mice [26], and that IL-1β as well as TNF-α promoted the aggravation of viral myocarditis in virus-resistant mice [27]. It has been reported that Ang-II induces inflammation through the production of inflammatory cytokines, reactive oxygen species (ROS), and adhesion molecules [28]. Studies reported that AT1R antagonists are reported to suppress cytokine production and transcription of cytokine genes in vitro and in vivo [13, 15, 16, 19, 29-31]. Interestingly, we could observe an increase in the myocardial mRNA levels of inflammatory cytokines such as TNF-α, MCP-1, IL-1β, and IFN-γ and protein levels of TNF-α, NF-κB and increased inflammatory cell infiltration in rats with EAM, and these changes in mRNA and protein levels were significantly decreased by olmesartan treatment (Fig 1).
Ang-II contributes to cardiac remodeling, hypertrophy, fibrosis and LV dysfunction. Accumulating evidence suggests that many of the adverse cardiac effects of Ang-II are triggered by redox cycling of ROS, generated in part by an NADPH oxidase-dependent pathway [32-35], and NADPH oxidase is one of the major sources of Ang-II-mediated ROS in ventricular myocytes. NADPH oxidase is a multicomponent enzyme complex that is composed of the membrane-bound heterodimer gp91phox (phox indicates phagocytic oxidase; NOX2) and its homologue NOX4; p22phox; the cytosolic regulatory subunits p40phox, p47phox, and p67phox; and the small GTP-binding protein, Rac1 [36, 37]. NADPH oxidase catalyzes the 1-electron reduction of molecular oxygen to superoxide anion, which can react with nitric oxide to form short-lived peroxynitrite. Peroxynitrite then forms stable 3-NT conjugated molecules, which may be used as a marker of oxidative stress [36]. ROS derived from NADPH oxidase activation have been shown to play a critical role in hypertrophy, fibrosis, and remodeling in the heart and vasculature [36-38]. The activation of NADPH oxidase is also documented in pressure overload-induced LV hypertrophy in mice [39]. Further, increase in the expression of NADPH oxidase subunits such as gp91phox and p22phox are found in the infarcted rat and human myocardium [14, 40]. It has been reported that AT1R antagonist synergistically attenuate atherosclerosis, diastolic heart failure, diabetes and murine myocarditis at least partly via inhibition of oxidative stress [14, 30, 31]. In the present study, we confirmed the Ang-II induced ROS activation in remodeling LV myocardium of EAM rats by several independent indices such as immunohistological and western immunoblotting detection of Ang-II expression, protein oxidation (i.e., 4-HNE), superoxide formation measured by 3-NT staining and increased expression of AT1R and NADPH oxidase subunits (Fig 2B-D and Fig 3A-D), respectively. These changes were significantly reversed by treatment with olmesartan in EAM rats. Our present data may link the involvement of Ang-II in LV remodeling process via, at least in part, a pathway of Ang-II induced ROS activation. On the basis of our results, we consider that olmesartan protects the heart from EAM partly through the suppression of inflammatory cytokines and oxidative stress in addition to hemodynamic modifications.
Structural alterations, such as progressive myocardial hypertrophy and fibrosis, are considered a pathological basis for the development of diastolic dysfunction [41], and the RAS is suggested to play a pivotal role in the progression of myocardial changes [31, 42] therefore, therapy aiming at reversing myocardial remodeling should be attempted to prevent progression to heart failure. Further, the expression of the inflammatory chemokine osteopontin (OPN) is dramatically increased in cardiomyocytes and inflammatory cells during myocarditis and heart failure in human and animals [43-46] and its high expression levels in acute myocarditis are associated with consecutive development of extensive fibrosis [43]. Studies have demonstrated that Ang-II upregulated cardiac fibrosis and its marker molecule (OPN) expression [43, 47-49] and OPN deficiency could abolish the development of Ang-II-induced cardiac fibrosis [48] suggest that OPN had a pivotal role in the mitogenic effect of Ang-II on rat cardiac fibroblasts. Previous studies have reported that AT1R antagonist is protective against LV fibrosis [13, 19, 31, 43, 46, 49], and decreases OPN expression in non-infarcted myocardium [47] in turn prevents cardiac remodeling. In line with the previous reports [31, 43-48], we could also observe an increased myocardial fibrosis and its marker molecule expression (Fig 2A and A1) in the hearts of EAM rats. The area of fibrosis was significantly lower in the olmesartan-treated rats than in the untreated rats, and the decrease in the area of fibrosis was associated with the decreased OPN expression. Thus, ARB-induced regression of LV fibrosis through the suppression of OPN expression may therefore be another mechanism of the therapeutic effects.
One histological finding characteristic of failing hearts is morphological development of the endoplasmic reticulum (ER), suggesting that ER overload occurs in this condition [50]. Oxidative stress, hypoxia, and enhanced protein synthesis in failing hearts could all potentially enhance ER stress. Emerging data has indicated that excessive and/or prolonged ER stress leads to the initiation of the apoptotic processes promoted by transcriptional induction of CHOP or by the activation of c-Jun-N-terminal kinase (JNK) and/or caspase-12-dependent pathway [51]. Recently, the unfolded protein response (UPR) and/or ER-initiated apoptosis have been implicated in the pathophysiology of various human diseases, including cardiovascular diseases such as cardiac hypertrophy, heart failure, autoimmune cardiomyopathy, atherosclerosis, and ischemic heart disease [52-56]. In addition, authors have shown a marked increased expression of ER chaperones such as GRP78, suggesting that UPR activation is associated with the pathophysiology of heart failure in humans and animals [52, 57]. On the other hand, it has been reported that Ang-II upregulated ER chaperones and induced apoptosis in cultured adult rat cardiac myocytes [52]. Studies have also shown that cardiac myocyte apoptosis itself is one of the mechanisms of the progression of EAM [60, 61]. Apoptotic death of cardiomyocytes and infiltrating T lymphocytes occurs in rat hearts with myosin-induced autoimmune myocarditis, suggesting that apoptosis plays a role in the pathogenesis of myocardial injury in myocarditis [58]. The antagonism of AT1R receptor prevented upregulation of ER chaperones, apoptosis in failing hearts and protects the heart against LV remodeling by inhibiting apoptosis and improves cardiac function [52, 60, 61]. We also investigated ER stress in our model and found that myocardial expression of GRP78, CHOP, caspase-12 and number of TUNEL positive cells were significantly increased in vehicle-treated EAM rats (Fig 4) and these levels were significantly attenuated by treatment with AT1R blocker olmesartan. These findings suggest that cardiac ER stress is induced via an angiotensin II type 1 receptor-dependent pathway. Although it is likely that one of the potential mechanisms by which angiotensin II induced ER stress is enhanced protein synthesis, further investigation about the intracellular signaling pathway by which angiotensin II induces ER stress will be needed.
Excessive ER stress and oxidative stress can trigger the activation of multiple signaling pathways such as phosphorylation of p38 MAPK and JNK, which are closely associated with cell death [62, 63]. Recent studies have demonstrated that the p38 MAPK and JNK pathways are activated in an EAM animal model [64, 65]. Moreover, MAPKs are important mediators of the intracellular signal transduction pathways that are responsible for cell growth and differentiation. Further, the activation of MAPK pathways is a characteristic finding in heart disease and plays a key role in the progression to heart failure which is mediated via Ang-II dependent mechanisms [66-68]. Our data confirms the activation of myocardial MAPK pathways (as assessed by the phosphorylated levels of these kinases) in EAM rats and olmesartan treatment reduces the activation of p38MAPK, and JNK (Fig 5), which suggest that MAPK signaling by stress signaling mediated via Ang II.
In the light of these findings, present study supports the hypothesis that olmesartan improved cardiac function, and attenuated cardiac oxidative stress, ER stress, inflammatory cytokines and MAPK/JNK activation providing further evidence that Ang-II plays a pivotal role in the myocardial remodeling in rats with heart failure after EAM. However, additional experiments will help to reveal the detailed mechanism of action of olmesartan in heart failure after EAM.
Acknowledgements
This research was supported by a Yujin Memorial Grant, Ministry of Education, Culture, Sports, Science and Technology, Japan, and by a grant from the promotion and Mutual Aid Corporation for Private Schools, Japan. We also express our sincere gratitude to Dr. Masaki Nagata (Division of Oral and Maxillofacial Surgery, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan) for carrying out the RT-PCR analysis in this study.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death. Circulation. 1999;99:1091-1100
2. Feldman AM, McNamara D. Myocarditis. N Engl J Med. 2000;343:1388-1398
3. Kodama M, Matsumoto Y, Fujiwara M, Masani F, Izumi T, Shibata A. A novel experimental model of giant cell myocarditis induced in rats by immunization with cardiac myosin fraction. Clin Immunol Immunopathol. 1990;57:250-262
4. Kodama M, Matsumoto Y, Fujiwara M, Zhang SS, Hanawa H, Itoh E, Tsuda T, Izumi T, Shibata A. Characteristics of giant cells and factors related to the formation of giant cells in myocarditis. Circ Res. 1991;69:1042-1050
5. Kodama M, Matsumoto Y, Fujiwara M. In vivo lymphocytemediated myocardial injures demonstrated by adoptive transfer of experimental autoimmune myocarditis. Circulation. 1992;85:1918- 1926
6. Fuse K, Kodama M, Ito M, Okura Y, Kato K, Hanawa H, Aoki S, Aizawa Y. Polarity of helper T cell subsets represents disease nature and clinical course of experimental autoimmune myocarditis in rats. Clin Exp Immunol. 2003;134:403-408
7. Binah O. Pharmacologic modulation of the immune interaction between cytotoxic lymphocytes and ventricular myocytes. J Cardiovasc Pharmacol. 2001;38:298-316
8. Ferrario CM, Strawn WB. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol. 2006;98:121-128
9. Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM. Renin-angiotensin system and cardiovascular risk. Lancet. 2007;369:1208-1219
10. Navalkar S, Parthasarathy S, Santanam N, Khan BV. Irbesartan, an angiotensin type 1 receptor inhibitor, regulates markers of inflammation in patients with premature atherosclerosis. J Am Coll Cardiol. 2001;37:440-444
11. Koh KK, Ahn JY, Han SH, Kim DS, Jin DK, Kim HS, Shin MS, Ahn TH, Choi IS, Shin EK. Pleiotropic effects of angiotensin II receptor blocker in hypertensive patients. J Am Coll Cardiol. 2003;42:905-910
12. Fliser D, Buchholz K, Haller H. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation. 2004;110:1103-1107
13. Yoshida J, Yamamoto K, Mano T, Sakata Y, Nishikawa N, Nishio M, Ohtani T, Miwa T, Hori M, Masuyama T. AT1 receptor blocker added to ACE inhibitor provides benefits at advanced stage of hypertensive diastolic heart failure. Hypertension. 2004;43:686-691
14. Tsuda M, Iwai M, Li JM, Li HS, Min LJ, Ide A, Okumura M, Suzuki J, Mogi M, Suzuki H, Horiuchi M. Inhibitory effects of AT1 receptor blocker, olmesartan, and estrogen on atherosclerosis via anti-oxidative stress. Hypertension. 2005;45:545-551
15. Yuan Z, Nimata M, Okabe TA, Shioji K, Hasegawa K, Kita T, Kishimoto C. Olmesartan, a novel AT1 antagonist, suppresses cytotoxic myocardial injury in autoimmune heart failure. Am J Physiol Heart Circ Physiol. 2005;289:H1147-1152
16. Nimata M, Kishimoto C, Yuan Z, Shioji K. Beneficial effects of olmesartan, a novel angiotensin II receptor type 1 antagonist, upon acute autoimmune myocarditis. Molecular and Cellular Biochem. 2004;259:217-222
17. Kim-Mitsuyama S, Izumi Y, Izumiya Y, Yoshida K, Yoshiyama M, Iwao H. Additive beneficial effects of the combination of a calcium channel blocker and an angiotensin blocker on a hypertensive rat-heart failure model. Hypertens Res. 2004;27:771-779
18. Yao L, Kobori H, Rahman M, Seth DM, Shokoji T, Fan Y, Zhang GX, Kimura S, Abe Y, Nishiyama A. Olmesartan improves endothelininduced hypertension and oxidative stress in rats. Hypertens Res. 2004;27:493-500
19. Sukumaran V, Watanabe K, Veeraveedu PT, Thandavarayan RA, Gurusamy N, Ma M, Yamaguchi K, Suzuki K, Kodama M, Aizawa Y. Beneficial effects of olmesartan, an angiotensin II receptor type 1 antagonist, in rats with dilated cardiomyopathy. Exp Biol Med (Maywood). 2010;235:1338-1346
20. Watanabe K, Ohta Y, Nakazawa M, Higuchi H, Hasegawa G, Naito M, Fuse K, Ito M, Hirono S, Tanabe N, Hanawa H, Kato K, Kodama M, Aizawa Y. Low dose carvedilol inhibits progression of heart failure in rats with dilated cardiomyopathy. Br J Pharmacol. 2000;130:1489-1495
21. Jia N, Okamoto H, Shimizu T, Chiba S, Matsui Y, Sugawara T, Akino M, Kitabatake A. A newly developed angiotensin II type 1 receptor antagonist, CS866, promotes regression of cardiac hypertrophy by reducing integrin beta1 expression. Hypertens Res. 2003;26:737-742
22. Yuan Z, Liu Y, Liu Y, Zhang J, Kishimoto C, Ma A, Liu Z. Peroxisome proliferator-activated receptor-gamma ligands ameliorate experimental autoimmune myocarditis associated with inhibition of self-sensitive T cells. J Cardiovasc Pharmacol. 2004;43:868-875
23. Liu YH, Carretero OA, Cingolani OH, Liao TD, Sun Y, Xu J, Li LY, Pagano PJ, Yang JJ, Yang XP. Role of inducible nitric oxide synthase in cardiac function and remodeling in mice with heart failure due to myocardial infarction. Am J Physiol Heart Circ Physiol. 2005;289:H2616-H2623
24. Matsumori A, Yamada T, Suzuki H, Matoba Y, Sasayama S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br Heart J. 1994;72:561-566
25. Nakamura H, Yamamura T, Umemoto S, Fukuta S, Shioi T, Matsumori A, Sasayama S, Matsuzaki M. Autoimmune response in chronic ongoing myocarditis demonstrated by heterotopic cardiac transplantation in mice. Circulation. 1996;94:3348-3354
26. Kubota T, McTiernan CF, Frye CS, Demetris AJ, Feldman AM. Cardiac-specific overexpression of tumor necrosis factor-alpha causes lethal myocarditis in transgenic mice. J Card Fail. 1997;3:117-124
27. Kishimoto C, Takada H, Kawamata H, Umatake M, Ochiai H. Immunoglobulin treatment prevents congestive heart failure in murine encephalomyocarditis viral myocarditis associated with reduction of inflammatory cytokines. J Pharmacol Exp Ther. 2001;299:645-651
28. Willemsen JM, Westerink JW, Dallinga-Thie GM, Van Zonneveld AJ, Gaillard CA, Rabelink TJ, de Koning EJ. Angiotensin-II type 1 receptor blockade improves hyperglycemia-induced endothelial dysfunction and reduces proinflammatory cytokine release from leukocytes. J Cardiovasc Pharmacol. 2007;49:6-12
29. Kramer C, Sunkomat J, Witte J, Luchtefeld M, Walden M, Schmidt B. Angiotensin II receptor-independent anti-inflammatory and anti-aggregatory properties of losartan: role of the active metabolite EXP 3179. Circ Res. 2002;90:770-776
30. Seko Y. Effect of the angiotensin II receptor blocker olmesartan on the development of murine acute myocarditis caused by coxsackievirus B3. Clin Sci. 2006;110:379-386
31. Nishio M, Sakata Y, Mano T, Yoshida J, Ohtani T, Takeda Y, Miwa T, Masuyama T, Yamamoto K, Hori M. Therapeutic effects of angiotensin II type 1 receptor blocker at an advanced stage of hypertensive diastolic heart failure. J Hypertens. 2007;25:455-461
32. Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471-478
33. Harrison DG, Cai H, Landmesser U, Griendling KK. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J Renin Angiotensin Aldosterone Syst. 2003;4:51-61
34. Sowers JR. Hypertension, angiotensin II, and oxidative stress. N Engl J Med. 2002;346:1999-2001
35. Das DK, Maulik N, Engelman RM. Redox regulation of angiotensin II signaling in the heart. J Cell Mol Med. 2004;8:144-152
36. Habibi J, Whaley-Connell A, Qazi MA, Hayden MR, Cooper SA, Tramontano A, Thyfault J, Stump C, Ferrario C, Muniyappa R, Sowers JR. Rosuvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, decreases cardiac oxidative stress and remodeling in Ren2 transgenic rats. Endocrinology. 2007;148:2181-2188
37. Whaley-Connell A, Govindarajan G, Habibi J, Hayden MR, Cooper SA, Wei Y, Ma L, Qazi M, Link D, Karuparthi PR, Stump C, Ferrario C, Sowers JR. Angiotensin II-mediated oxidative stress promotes myocardial tissue remodeling in the transgenic (mRen2) 27 Ren2 rat. Am J Physiol Endocrinol Metab. 2007;293:E355-E363
38. Wei Y, Whaley-Connell AT, Chen K, Habibi J, Uptergrove GM, Clark SE, Stump CS, Ferrario CM, Sowers JR. NADPH oxidase contributes to vascular inflammation, insulin resistance, and remodeling in the transgenic (mRen2) rat. Hypertension. 2007;50:384-391
39. Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, Shah AM. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. 2003;93:802-805
40. Fukui T, Yoshiyama M, Hanatani A, Omura T, Yoshikawa J, Abe Y. Expression of p22phox and gp91phox, essential components of NADPH oxidase, increases after myocardial infarction. Biochem Biophys Res Commun. 2001;281:1200-1206
41. Aurigemma GP, Gaasch WH. Clinical practice. Diastolic heart failure. N Engl J Med. 2004;351:1097-1105
42. Sakata Y, Masuyama T, Yamamoto K, Doi R, Mano T, Kuzuya T, Miwa T, Takeda H, Hori M. Renin angiotensin system-dependent hypertrophy as a contributor to heart failure in hypertensive rats: different characteristics from renin angiotensin system-independent hypertrophy. J Am Coll Cardiol. 2001;37:293-299
43. Szalay G, Sauter M, Haberland M, Zuegel U, Steinmeyer A, Kandolf R, Klingel K. Osteopontin: a fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host. Circ Res. 2009;104:851-859
44. Hanawa H, Abe S, Hayashi M, Yoshida T, Yoshida K, Shiono T, Fuse K, Ito M, Tachikawa H, Kashimura T, Okura Y, Kato K, Kodama M, Maruyama S, Yamamoto T, Aizawa Y. Time course of gene expression in rat experimental autoimmune myocarditis. Clin Sci (Lond). 2002;103:623-632
45. Shin T, Ahn M, Kim H, Kim HM, Matsumoto Y. Increased expression of osteopontin in the heart tissue of Lewis rats with experimental autoimmune myocarditis. J Vet Med Sci. 2006;68:379-382
46. Tokuda K, Kai H, Kuwahara F, Yasukawa H, Tahara N, Kudo H, Takemiya K, Koga M, Yamamoto T, Imaizumi T. Pressure-independent effects of angiotensin II on hypertensive myocardial fibrosis. Hypertension. 2004;43:499-503
47. Kusuyama T, Yoshiyama M, Omura T, Nishiya D, Enomoto S, Matsumoto R, Izumi Y, Akioka K, Takeuchi K, Iwao H, Yoshikawa J. Angiotensin blockade inhibits osteopontin expression in non-infarcted myocardium after myocardial infarction. J Pharmacol Sci. 2005;98:283-289
48. Matsui Y, Jia N, Okamoto H, Kon S, Onozuka H, Akino M, Liu L, Morimoto J, Rittling SR, Denhardt D, Kitabatake A, Uede T. Role of osteopontin in cardiac fibrosis and remodeling in angiotensin II-induced cardiac hypertrophy. Hypertension. 2004;43:1195-1201
49. Kim S, Yoshiyama M, Izumi Y, Kawano H, Kimoto M, Zhan Y, Iwao H. Effects of combination of ACE inhibitor and angiotensin receptor blocker on cardiac remodeling, cardiac function, and survival in rat heart failure. Circulation. 2001;103:148-154
50. Maron BJ, Ferrans VJ. Intramitochondrial gylcogen deposits in hypertrophied human myocardium. J Mol Cell Cardiol. 1975;7:697-702
51. Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic β-cells. Apoptosis. 2002;7:335-345
52. Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705-712
53. Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y, Suzuki M, Nishino T, Nakaya H, Koseki H, Aoe T. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol. 2004;24:8007-8017
54. Mao W, Fukuoka S, Iwai C, Liu J, Sharma VK, Sheu SS, Fu M, Liang CS. Cardiomyocyte apoptosis in autoimmune cardiomyopathy: mediated via endoplasmic reticulum stress and exaggerated by norepinephrine. Am J Physiol Heart Circ Physiol. 2007;293:H1636-1645
55. Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, Bochaton-Piallat ML, Mochizuki N, Kitakaze M. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226-1233
56. Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, Glembotski CC. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186-1193
57. Sun Y, Liu G, Song T, Liu F, Kang W, Zhang Y, Ge Z. Upregulation of GRP78 and caspase-12 in diastolic failing heart. Acta Biochim Pol. 2008;55:511-516
58. Futamatsu H, Suzuki J, Mizuno S, Koga N, Adachi S, Kosuge H, Maejima Y, Hirao K, Nakamura T, Isobe M. Hepatocyte growth factor ameliorates the progression of experimental autoimmune myocarditis: a potential role for induction of T helper 2 cytokines. Circ Res. 2005;96:823-830
59. Ishiyama S, Hiroe M, Nishikawa T, Shimojo T, Abe S, Fujisaki H, Ito H, Yamakawa K, Kobayashi N, Kasajima T, Marumo F. The Fas/Fas ligand system is involved in the pathogenesis of autoimmune myocarditis in rats. J Immunol. 1998;161:4695-4701
60. Kanamori H, Takemura G, Li Y, Okada H, Maruyama R, Aoyama T, Miyata S, Esaki M, Ogino A, Nakagawa M, Ushikoshi H, Kawasaki M, Minatoguchi S, Fujiwara H. Inhibition of Fas-associated apoptosis in granulation tissue cells accompanies attenuation of postinfarction left ventricular remodeling by olmesartan. Am J Physiol Heart Circ Physiol. 2007;292:H2184-2194
61. Matsusaka H, Kinugawa S, Ide T, Matsushima S, Shiomi T, Kubota T, Sunagawa K, Tsutsui H. Angiotensin II type 1 receptor blocker attenuates exacerbated left ventricular remodeling and failure in diabetes-associated myocardial infarction. J Cardiovasc Pharmacol. 2006;48:95-102
62. Ueda S, Matsutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J. Redoxcontrol of cell death. Antioxid Redox Signal. 2002;4:405-414
63. Okada K, Minamino T, Kitakaze M. Role of endoplasmic reticulum stress in cardiac development and dysfunction. Folia Pharmacolo Jpn. 2005;126:385-389
64. Shimazaki H, Watanabe K, Veeraveedu PT, Harima M, Thandavarayan RA, Arozal W, Tachikawa H, Kodama M, Aizawa Y. The antioxidant edaravone attenuates ER-stress-mediated cardiac apoptosis and dysfunction in rats with autoimmune myocarditis. Free Radic Res. 2010;44:1082-1090
65. Haga T, Suzuki J, Kosuge H, Ogawa M, Saiki H, Haraguchi G, Maejima Y, Isobe M, Ueda T. Attenuation of experimental autoimmune myocarditis by blocking T cell activation through 4-1BB pathway. J Mol Cell Cardiol. 2009;46:719-727
66. Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev. 2000;52:11-34
67. Kyaw M, Yoshizumi M, Tsuchiya K. et al. Antioxidants inhibit JNK and p38 MAPK activation but not ERK 1/2 activation by angiotensin II in rat aortic smooth muscle cells. Hypertens Res. 2001;24:251-261
68. Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589-600
Author Biography
Mr Vijayakumar Sukumaran obtained his master degree in Pharmacy (Pharmaceutical chemistry) in 2002 from K. M. College of Pharmacy (Dr. MGR. Medical University, Tamil Nadu, India) and worked as a lecturer at JSS University (India) as well as INTI International University (Malaysia). Currently, he studies doctoral programme in Department of Clinical Pharmacology, Niigata University of Pharmacy and Applied Life Sciences (NUPALS, Japan). His research interests spreads over the fields of cardiovascular disease, diabetic complications, inflammation, toxicity studies and metabolic syndrome. He has over 10 publications in his credit.
Prof. Kenichi Watanabe obtained his MD in 1974 followed by a Medical PhD from Niigata Medical University in 1985 and a Pharmaceutical PhD from Shizuoka Prefectural University in 1995. Currently, he is head of the Department of Clinical Pharmacology, NUPALS, Niigata City, Japan. Prior to NUPALS, Prof. Kenichi Watanabe had held faculty appointments at Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan. His areas of expertise include heart failure, diabetes, hypertension, inflammation, cardiac imaging, cell signaling and metabolic syndrome. He has over 100 publications in his credit.
![]() Corresponding author: Prof. Kenichi Watanabe, MD, Ph.D., Department of Clinical Pharmacology, Faculty of Pharmaceutical Sciences, Niigata University of Pharmacy and Applied Life Sciences, 265-1 Higashizima, Niigata city, 956-8603, Japan. Tel.: +81 250 255267; fax: +81 250 25 5021. E-mail: watanabeac.jp (Kenichi Watanabe); svkumar1979com (Vijayakumar Sukumaran).
Corresponding author: Prof. Kenichi Watanabe, MD, Ph.D., Department of Clinical Pharmacology, Faculty of Pharmaceutical Sciences, Niigata University of Pharmacy and Applied Life Sciences, 265-1 Higashizima, Niigata city, 956-8603, Japan. Tel.: +81 250 255267; fax: +81 250 25 5021. E-mail: watanabeac.jp (Kenichi Watanabe); svkumar1979com (Vijayakumar Sukumaran).