Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(2):209-220. doi:10.7150/ijbs.7.209 This issue Cite
Research Paper
Diversity of Microorganisms Isolated from the Soil Sample surround Chroogomphus rutilus in the Beijing Region
Peng Wang1, 2, Yu Liu1, Yonggang Yin1, 3, Haojie Jin4, Shouxian Wang1, Feng Xu1, ![]() , Shuang Zhao1, Xiaoli Geng1
, Shuang Zhao1, Xiaoli Geng1
1. Institute of Plant and Environmental Protection, Beijing Academy of Agriculture and Forestry Science, Beijing 100097, China;
2. College of Life Sciences, Henan Agricultural University, Zhengzhou 450002, Henan, China;
3. College of Horticulture, Agricultural University of Hebei, Baoding 071001, Hebei, China;
4. Department of Molecular Biology, University of Aarhus, Aarhus C DK-8000, Denmark
Received 2011-1-16; Accepted 2011-2-27; Published 2011-3-2
Abstract
Artificially cultivating Chroogomphus rutilus is too inefficient to be commercially feasible. Furthermore, isolating C. rutilus mycelia in the wild is difficult. Thus, it is important to determine the natural habitat of its fruiting body. This study focused on the ecology of the C. rutilus habitat to isolate and classify beneficial microorganisms that could affect its growth, which could be used in future research on artificial cultivation. In total, 342 isolates were isolated from soil samples collected around a C. rutilus colony in the Beijing region. Of these, 22 bacterial and 14 fungal isolates were selected for sequencing and phylogenetic analysis, based on their growth characteristics and colony morphology. Using 16S rRNA gene sequence analysis, the bacterial isolates were divided into two monophyletic clusters which had significant hits to the genera Bacillus and Pseudomonas, respectively. Using internal transcribed spacer (ITS) sequence analysis, fungal isolates were divided into four monophyletic clusters: Penicillium, Trichoderma, Mortierella, and Bionectria. Moreover, the phylogenetic diversity of these isolates was analysed. The results indicated that numerous microorganisms were present in C. rutilus habitat. This was the first reported examination of the microbiological ecology of C. rutilus.
Keywords: Chroogomphus rutilus, isolation, phylogenetic diversity, 16S rRNA, ITS
Introduction
Chroogomphus rutilus (Schaeff.) O.K. Mill. is a type species in the genus Chroogomphus. The common names pine-spike and spike-cap are based on its shape and close growth association with pine trees. Based on records and our recent investigation, Chroogomphus spp. inhabit the northern hemisphere, including Asia, North America, and Europe [1]. C. rutilus is edible and especially flavourful when dried. Furthermore, it has been investigated as a source of antibiotics and other potentially useful secondary compounds. The fruiting body is economically important for food or herbal medicine use [2].
Since artificially cultivating C. rutilus is difficult, making it too expensive to be a common commodity. There have been many attempts to find an efficient method of cultivating C. rutilus. Researchers have attempted to imitate its natural habitat to promote mycelia growth, such as using soil collected near wild C. rutilus colonies and using pine tree tissue culture medium [3]. Pure mycelia cultures have been isolated; however, artificial cultivation is still inefficient and the fruiting body is difficult to obtain under artificial conditions [4]. Given the short time frame of research and lack of knowledge concerning the niche of C. rutilus in its ecosystem, it is not surprising that cultivating C. rutilus artificially is difficult, even outdoors.
Artificial cultivation may have failed due to the complex habitat C. rutilus requires. C. rutilus may require specific soil chemical composition and the presents of certain plants and microbes in order to grow. Some researchers have examined the plants living in C. rutilus habitats [5]; however, little research has been done to investigate the microorganisms present. Because no organism lives entirely isolated, soil microorganisms could directly or indirectly affect spore germination and fruiting body formation [6]. Furthermore, organisms often establish biological interactions, such as symbiosis, competition, and autoecism. C. rutilus interacts with other fungi and bacteria in its habitat, and may affect each other's growth or life cycles [7].
Based on the known requirements of casing-soil for the half artificial cultivation of Morchella, Shen et al. (2008) examined the bacterial community contained in the fruiting body of Morchella using a denaturing gradient gel electrophoresis (DGGE) technology. To some extent, the results of sequencing the 16S rRNA genes of 7 kinds of bacteria reflect the bacterial community structure inside Morchella [8]. Shen et al. (2009) also reported that the investigation of fungal communities inhabiting Morchella growth soil using DGGE. Sequence analysis of 12 recovered dominant bands revealed the presence of 10 fungi whose growth may have been inhibited by the dominant Morchella [6]. All of the above studies suggest that co-occurring microbes play an important role in fungal growth.
This study was initiated to investigate the diversity of microorganisms isolated from soil near a C. rutilus colony in the Beijing region. Bacteria and fungi were obtained selectively from soil samples collected around C. rutilus growing areas in the Beijing region based on their growth characteristics and colonial morphology. The 16S rRNA gene sequences of the bacteria isolates and the ITS gene sequences of the fungi isolates were amplified and sequenced respectively. The phylogenetic diversity of the isolated bacteria and fungi was determined based on the 16S rRNA genes and ITS sequences, respectively. This was the first demonstration of the microbe surrounding C. rutilus.
Materials and Methods
Soil sample collection and microbe isolation
Soil samples about 10 cm under the fruiting body of a C. rutilus colony in the Beijing region of the People's Republic of China were collected at an attitude of 790 m. The GPS coordinate was N40 57.186 E116 29.923.The samples were immediately transported to the laboratory for gradient dilution using autoclaved water and aliquots were transferred to appropriate growth medium. Three consecutive propagations using beef extract-peptone agar slants for bacteria and PDA media for fungi were conducted. After a 2-3-day incubation period at 28°C, single colony of per propagation was selected for observation. Out of the colonies owing the same growth characteristics and morphology, only one colony of each morpho-species was selected for further genetic sequencing. In this study, growth characteristics mainly included growth rate and colony morphologies of the isolated strains included size, color, diaphaneity of the colony, the degree of smoothness and so on. All strains in this study (Table 1) were preserved at ‑80°C in their respective liquid media with glycerol added to a final concentration of 15% (v/v).
Strains isolated in this study
| Strain number | Genus | 16S rRNA sequences GenBank accession number | ITS sequences GenBank accession number |
|---|---|---|---|
| I2X | Bacillus sp. | HQ727951 | - |
| I3X | Bacillus sp. | HQ727952 | - |
| I4X | Bacillus sp. | HQ727953 | - |
| II2X | Bacillus sp. | HQ727954 | - |
| II3X | Bacillus sp. | HQ844041 | - |
| III1X | Bacillus sp. | HQ727955 | - |
| III3X | Bacillus sp. | HQ727956 | - |
| IV1X | Bacillus sp. | HQ727957 | - |
| IV3X | Bacillus sp. | HQ727958 | - |
| V1X | Bacillus sp. | HQ727959 | - |
| V2X | Bacillus sp. | HQ727960 | - |
| V3X | Bacillus sp. | HQ727961 | - |
| V4X | Bacillus sp. | HQ727962 | - |
| VI2X | Bacillus sp. | HQ727963 | - |
| VII1X | Bacillus sp. | HQ727964 | - |
| VII2X | Bacillus sp. | HQ727965 | - |
| VII3X | Bacillus sp. | HQ727966 | - |
| II4X | Pseudomonas sp. | HQ727967 | - |
| III4X | Pseudomonas sp. | HQ727968 | - |
| IV2X | Pseudomonas sp. | HQ727969 | - |
| VI3X | Pseudomonas sp. | HQ727970 | - |
| I1X | Pseudomonas sp. | HQ844040 | - |
| I1Z | Penicillium sp. | - | HQ730121 |
| I2Z | Trichoderma sp. | - | HQ730122 |
| II1Z | Penicillium sp. | - | HQ730123 |
| II2Z | Bionectria sp. | - | HQ730124 |
| II3Z | Trichoderma sp. | - | HQ730125 |
| III1Z | Mortierella sp. | - | HQ730126 |
| III2Z | Penicillium sp. | - | HQ730127 |
| V2Z | Penicillium sp. | - | HQ730128 |
| VI1Z | Mortierella sp. | - | HQ730129 |
| VI2Z | Trichoderma sp. | - | HQ730130 |
| VII1Z | Trichoderma sp. | - | HQ730131 |
| VII2Z | Mortierella sp. | - | HQ730132 |
| IV2Z | Trichoderma sp. | - | HQ844042 |
| V3Z | Mortierella sp. | - | HQ844043 |
Genomic DNA extraction from the isolates
Bacterial genomic DNA was extracted according to the methods described by Sambrook (1989) [9] for further 16S rRNA amplification. Fungal genomic DNA was extracted following the methods described by Doyle and Doyle (1987) [10], with some modifications. These pure DNAs extracted from the isolates were then sequenced for phylogenetic analyses.
16S rRNA gene amplification, cloning, and sequencing
The nearly complete 16S rRNA gene sequence, about 1,500-bp long, was amplified using the universal forward primer P1 and the universal reverse primer P6 [11] (Table 2). The PCR reactions were run using the methods described by Chouari et al. (2003, 2005) [12, 13]. In this study, we used high fidelity Ex taq polymerase (Takara, Japan). The 16S rRNA gene PCR products were purified following the Nucleic Acid Recycling and Purification Kit protocol (Tiangen Biotech, P. R. China). Purified nucleotide sequences and pGEM-T plasmid vectors (Promega, USA) were ligated overnight at 4°C. Recombined plasmids were transformed into Escherichia coli DH10B by electroporation (BTX ECM830, USA) at 500 V for 17 ms and evaluated using blue-white screening. Plasmids containing 16S rRNA gene sequences were extracted and purified according to the method described by Sambrook (1989) [6]. In the present study, three positive clones were selected for sequencing by using the primers RV-M/M13-47 (Table 2) in both directions with an ABI377 automatic sequencer (Applied Biosystems, USA).
Primers used in this study
| Primers | Sequence (5'→3') | Reference |
|---|---|---|
| 16S P1 | AGAGTTTGATCCTGGTCAGAACGCT | Yanagi and Yamasato, 1993 |
| 16S P2 | TACGGCTACCTTGTTACGACTTCACCCC | Yanagi and Yamasato, 1993 |
| ITS1 | TCCGTAGGTGAACCTGCGG | White et al, 1990 |
| ITS4 | TCCTCCGCTTATTGATATGC | White et al, 1990 |
| RV-M | GAGCGGATAACAATTTCACACAGG | Takara, Code No. D3880, Japan. |
| M13-47 | CGCCAGGGTTTTCCCAGTCACGAC | Takara, Code No. D3887, Japan. |
ITS gene amplification, cloning, and sequencing
Recently, the ITS region of the ribosomal operon has become preferentially used for fungi classification instead of 18S ribosome RNA sequences [14]. The entire stretch of ITS1-5.8S-ITS2, approximately 750-bp long, was amplified using the universal forward primer ITS1 and the universal reverse primer ITS4 [15]. Ex taq polymerase (Takara, Japan) was used. The purification and sequencing procedures were the same as those described for the 16S rRNA sequences.
Sequence analysis and phylogenetic tree construction
The 16S rRNA and ITS gene sequences of the isolated strains were compared with sequences in DDBJ/EMBL/GenBank using the basic local alignment search tool (BLAST) [16]. The isolates had significant hits to the genus which owed the lowest e-value in the results of BLAST. When more than one sequence had the same e-value, only the first sequence was selected. Further, sequences were aligned using the Clustal_X software [17]. The distances were calculated according to two-parameter method [18]. The phylogenetic tree was inferred using the neighbour-joining methods [19]. Bootstrap analysis was based on 1,000 re-samplings. The MEGA4.0 package [20] was used for all analyses. All 16S rRNA sequences of the Bacillus and Pseudomonas type strains were obtained from the websites at http://www.bacterio.cict.fr/b/bacillus.html and http://www.bacterio.cict.fr/p/pseudomonas.html, respectively. The ITS sequences of the fungi type strains were obtained from DDBJ/EMBL/GenBank.
Results
Bacteria and fungi isolation from the soil samples
In total, 342 isolates were selectively obtained from soil samples collected near a C. rutilus colony in the Beijing region. Of these, 22 bacterial and 14 fungal isolates were selected out of 342 isolates based on the methods described in Materials and Methods.
Sequencing and phylogenetic analysis of the 16S rRNA gene sequences
The nearly complete 16S rRNA gene sequences of the 22 bacteria isolates were amplified using the universal primers for the 16S rRNA sequence shown in Table 2 and sequenced. Comparing the 16S rRNA sequences with those in DDBJ/EMBL/GenBank, the strains labeled I2X, I3X, I4X, II2X, II3X, III1X, III3X, IV1X, IV3X, V1X, V2X, V3X, V4X, VI2X, VII1X, VII2X, and VII3X had significant hits to Bacillus spp. and those labeled I1X, II4X, III4X, IV2X, and VI3X had significant hits to Pseudomonas spp.. The 16S rRNA sequence GenBank accession numbers of these strains are indicated after the bacterial names in Table 1.
A phylogenetic tree based on the Bacillus spp. listed in the website http://www.bacterio.cict.fr/b/bacillus.html and the 17 sequenced Bacillus isolates was established (data not shown). Based on genetic distances, Bacillus spp. that had high similarities with the isolates were selected to construct the phylogenetic tree. The 17 Bacillus isolates formed three clusters (Fig. 1). Cluster I included eight isolates. The strains labeled V1X, VII1X, V4X, VII3X, and I3X were clustered with B. simplex, exhibiting 99.9, 99.9, 99.9, 100, and 100% 16S rRNA sequence similarity with B. simplex, respectively. Strains VI2X, V3X, and IV1X clustered with B. sp. LMG 20238, all exhibiting 99.9% 16S rRNA sequence similarity with B. sp. LMG 20238. Cluster II included strains II3X and III1X, which had 99.9% 16S rRNA sequence similarity with B. aryabhattai. Cluster III consisted of seven strains. Strains I2X, V2X, I4X, VII2X, II2X, and III3X were clustered with B. thuringiensis, exhibiting 99.9, 99.9, 100, 99.9, 100, and 100% 16S rRNA sequence similarity, respectively. Strain IV3X showed the highest 16S rRNA sequence similarity, 99.9%, with B. cereus.
Neighbour-joining phylogenetic tree based on 16S rRNA gene sequences showing the distance of isolated strains with the nearest species of the genus Bacillus. Pseudomonas agarici ATCC25941 was used as an out group. Bootstrap percentage values as obtained from 1,000 resamplings of the date set are given at the nodes of the tree. Bar, 0.01 substitutions per nucleotide position.

The phylogenetic diversity of the Pseudomonas isolates was analyzed as described above for Bacillus. A phylogenetic tree was constructed based on the Pseudomonas spp. in the website (http://www.bacterio.cict.fr/p/pseudomonas.html) and the five isolates (data not shown). Using genetic distances, the Pseudomonas spp. which had high similarities with these five isolates were selected to construct the phylogenetic tree. The Pseudomonas isolates formed two clusters (Fig. 2). The Pseudomonas isolates labeled II4X, VI3X, IV2X, and III4X clustered with P. koreensis, exhibiting 99.8, 99.7, 99.8, and 99.8% 16S rRNA sequence similarity, respectively. In the other cluster, strain I1X had 99.8% similarities with P. umsongensis.
Neighbour-joining phylogenetic tree based on 16S rRNA gene sequences showing the distance of isolated strains with the nearest species of the genus Pseudomonas. Bacillus subtilis NCDO 1769T was used as an out group. Bootstrap percentage values as obtained from 1,000 resamplings of the date set are given at the nodes of the tree. Bar, 0.02 substitutions per nucleotide position.

Sequencing and phylogenetic analysis of the ITS sequences
Because ITS sequences can accumulate mutations at a faster rate than the 5.8S, 18S, and 28S rRNA genes, the ITS region is typically most useful for molecular systematics at the species or within species level [21, 22]. The ITS1-5.8S-ITS2 gene sequences of the 14 isolates were compared with sequences listed in DDBJ/EMBL/GenBank, indicating that they had significant hits to the genera Penicillium, Trichoderma, Mortierella and Bionectria. The strains labeled I1Z, II1Z, III2Z, and V2Z had significant hits to Penicillium. Strains I2Z, II3Z, IV2Z, VI2Z and VII1Z had significant hits to Trichoderma. Strains III1Z, V3Z, VI1Z and VII2Z had significant hits to Mortierella. Strain II2Z had significant hits to Bionectria. The ITS gene sequence GenBank accession numbers of the isolated fungi are indicated in Table 1.
A phylogenetic tree using the Penicillium spp. which had high similarities to the isolated fungi was constructed based on genetic distance (Fig. 3). The four Penicillium spp. isolates formed three clusters (Fig. 3). Cluster I consisted of strain I1Z, which exhibited 100% ITS sequence similarity with P. canescens. Cluster II contained strains II1Z and III2Z, which had the highest ITS sequence similarity to P. restrictum, 99.3% and 99.8%, respectively. Cluster III consisted of V2Z, with 100% similarity to P. aculeatum, and I1Z, with 100% similarity to P. canescens.
Neighbour-joining phylogenetic tree based on ITS sequences showing the distance of isolated strains with the nearest species of the genus Penicillium. Trichoderma viride ATCC20898 was used as an out group. Bootstrap percentage values as obtained from 1,000 resamplings of the date set are given at the nodes of the tree. Bar, 0.005 substitutions per nucleotide position.

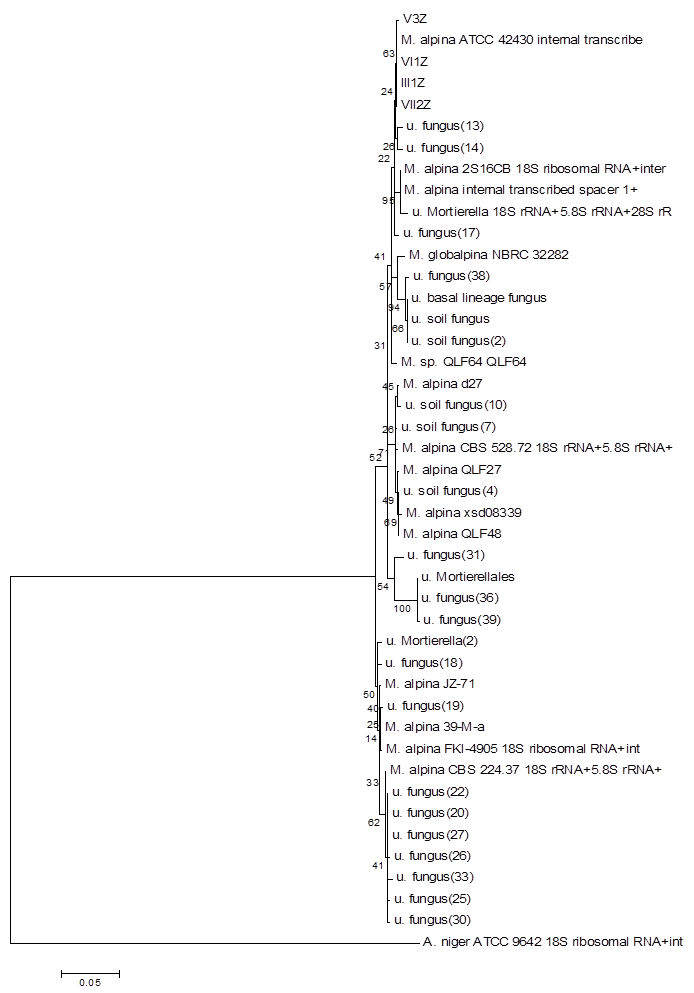
Using the method described for Penicillium, strains I2Z, II3Z, VI2Z, and VII1Z had 100, 99.9, 100, and 100% similarity with Trichoderma koningiopsis, respectively (Fig. 4). Strain IV2Z had 99% similarity with T. atroviride (Fig. 4). The fungi labeled III1Z, VI1Z, VII2Z, and V3Z had 100, 100, 100 and 99.8% similarity with Mortierella alpina, respectively (Fig. 5). The fungus labeled II2Z had 100% similarities with B. ochroleuca (Fig. 6).
Neighbour-joining phylogenetic tree based on 16S rRNA gene sequences showing the distance of isolated strains with the nearest species of the genus Trichoderma. Aspergillus niger ATCC9642 was used as an out group. Bootstrap percentage values as obtained from 1,000 resamplings of the date set are given at the nodes of the tree. Bar, 0.05 substitutions per nucleotide position.

Neighbour-joining phylogenetic tree based on 16S rRNA gene sequences showing the distance of isolated strains with the nearest species of the genus Mortierella. Aspergillus niger ATCC9642 was used as an out group. Bootstrap percentage values as obtained from 1,000 resamplings of the date set are given at the nodes of the tree. Bar, 0.05 substitutions per nucleotide position.
Neighbour-joining phylogenetic tree based on 16S rRNA gene sequences showing the distance of isolated strains with the nearest species of the genus Bionectria. Aspergillus niger ATCC9642 was used as an out group. Bootstrap percentage values as obtained from 1,000 resamplings of the date set are given at the nodes of the tree. Bar, 0.005 substitutions per nucleotide position.

Discussions
To date, the artificial cultivation of some edible wild fungi, including Chroogomphus spp., Morchella spp., and Matsutake spp., has not been successful. The slow mycelia growth rate on nutrient medium and the difficulty in fruiting body formation is due to the lack of information on the mechanisms of spore germination and fruiting body formation. Fruiting body formation could be affected by factors within the mycelia or externally, in the environment. Many microorganisms secrete beneficial secondary metabolites, which could be important for spore germination or fruiting body formation in wild mushrooms [23]. Rainey et al. (1991) reported that casing was required for the induction of fruiting body formation, which is caused by the presence of saprophytic bacteria in the casing medium [24]. Ntougias et al. (2004) reported that Gram-negative bacteria existed in spent mushroom compost and in mushroom cultivation substrate analyzed immediately after pasteurisation and cooling. The biological properties of compost appear to be important for the induction of fruiting body formation [25]. The microorganisms that exist in proximity to C. rutilus require further detailed examination to determine important interactions, which could advance the artificial cultivation of C. rutilus.
In the present study, 10 gram soil sample were resolved and serial diluted. Only a portion of the solution was spread on the appropriate media. Finally, 36 isolates were obtained for further classification and phylogenetic analysis. Limited by the sample size, there was the chance that some key microbes needed for C. rutilus might be missed. Moreover, traditional cultural method exhibit its limitations in the phylogenetic analysis, since only a small proportion of microbe can be obtained in laboratory condition [26]. Culture-indepentdent methods, such as metagenomic approach, could overcome these difficulties and limitations [27]. To solve these existing problems, metagenomic approach will be applied in the future study. Except of these, several important results from this investigation were summarized below.
First, of the 22 bacteria strains, 17, or about 80%, were Bacillus spp. It was reported that Bacillus spp. are abundant and widely distributed in agro-ecosystem [28, 29]. This could be because these Bacillus strains form a thick outer spore coat increasing resistance to extreme conditions, such as heat, ultraviolet radiation, toxic chemicals, and desiccation. Effects of Bacillus sp. on growth on mushroom was reported by Bis'ko et al. (1995) [30].
Second, four Pseudomonas spp. were isolated from the soil sample. Reports indicated that the presence of saprophytic bacteria, such as Pseudomonas spp., in the casing medium could induce fruiting body formation [31]. Moreover, secondary products excreted by Pseudomonas, such as insecticides and germicides, could affect the growth of C. rutilus mycelia in its natural habitat. Kloepper et al. (2004) reported that Pseudomonas spp. were one of the familiar plant growth-promoting rhizobacteria (PGPR) [32]. This explained the isolation of Pseudomomas spp. in the soil sample in this study. Further research on Pseudomonas spp. and C. rutilus interactions should be conducted, because this interaction might be the key point to artificial cultivation.
Third, we observed that some bacteria isolates could secrete much extracellular polysaccharides in the medium. Extracellular polysaccharides are important in inter-microorganism signaling [33, 34] and are involved in selection recognition, which is important for symbiosis [35]. In this study, the fact that soil samples were collected where C. rutilus grew implied that these extracellular polysaccharides could be important for recognition and signalling between bacteria and C. rutilus. By understanding the molecular mechanisms of these hypothesized interactions, we could enhance the interactions and expand the use of these bacteria.
Finally, it is notable that the fungus strain labeled II2Z had significant hits to the genus Bionectria. Bionectria are endophytic fungi that can colonize living, internal plant tissue without any immediate or overt negative effects [36]. In fact, endophytic fungi were detected and reported in many plants. These fungi showed positive role to help protecting the plants against insects, nematodes, pathogens and so on [37, 38]. In the present study, Bionectria spp. was isolated from the soil sample together with the mycelium of C. rutilus. It could be deduced that Bionectria spp. might interact with the organism nearby. The organism perhaps was the nearby pine tree or the mycelium of C. rutilus. Based on the results obtained in the present study, the detail interaction between C. rutilus and Bionectria sp. II2Z requires more research to determine if this strain played the role on the fruit body formation of C. rutilus.
In this study, microorganisms were isolated and classified in C. rutilus habitat. This laid the foundation of the artificial cultivation of C. rutilus. This report is the first reported examination of the microbiological ecology of C. rutilus. Based on the results of this study, we will examine the exact interactions between C. rutilus and these isolated microorganisms to reveal how these microorganisms assist in the artificial cultivation of C. rutilus. If this research path is successful, the applications of C. rutilus will broaden.
Acknowledgements
We thank the reviewers for the helpful comments and improvement of the text.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Aime M, Miller OK. Proposal to conserve the name Chroogomphus against Brauniellula (Gomphidiaceae). Taxon. 2005;55(1):227-228
2. Li YC, Yang ZL, Tolgor B. Phylogenetic and biogeographic relationships of Chroogomphus species as inferred from molecular and morphological data. Fungal Divers. 2009;38:85-104
3. Yamada A, Ogura T, Ohmasa M. Cultivation of mushrooms of edible ectomycorrhizal fungi associated with Pinus densiflora by in vitro mycorrhizal synthesis. Mycorrhiza. 2001;11(2):59-66
4. Dong HY, Hao C, Niu YR. et al. The ecological elementary research of Chroogomphis rutillus. Chinese Agricultural Science Bulletin. 2010;26(7):191-194
5. Chen QJ, Cheng JH, Hao C. et al. Mycelium growth characters and culture medium formula screening of Comphidius rutilus. Chinese Agricultural Science Bulletin. 2010;26(3):231-233
6. Shen H, Wang H, Zhao YC. et al. Analysis of fungal communities present in Morchella growth soil using denaturing gradient gel electrophoresis. Acta Edulis Fungi. 2009;16(2):10-12
7. Garbaye J, Churin JL, Duponnois R. Effects of substrate sterilization, fungicide treatment, and mycorrhization helper bacteria on ectomycorrhizal formation of pedunculate oak (Quercus robur) inoculated with Laccaria laccata in two peat bare-root nurseries. Biol Fertil Soils. 1992;13:55-57
8. Shen H, Chen MJ, Zhao YC. et al. PCR-DGGE identification of endophytes in Morels. Acta Agriculture Shanghai. 2008;24(2):58-60
9. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. US: Cold Harbor. 1989
10. Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11-15
11. Yanagi M, Yamasato K. Phylogenetic analysis of the family Rhizobiaceae and related bacteria by sequencing of 16S rRNA gene using PCR and DNA sequencer. FEMS Microbiol Lett. 1993;107(1):115-120
12. Chouari R, Le Paslier D, Daegelen P. et al. Molecular evidence for novel planctomycete diversity in a municipal wastewater treatment plant. Appl Environ Microbiol. 2003;69(12):7354- 7363
13. Chouari R, Le Paslie D, Daegelen P. et al. Novel predominant archaeal and bacterial groups revealed by molecular analysis of an anaerobic sludge digester. Environ Microbiol. 2005;7(8):1104- 1115
14. Druzhinina IS, Kopchinskiy AG, Komoń M. et al. An oligonucleotide barcode for species identification in Trichoderma and Hypocrea. Fungal Genet Biol. 2005;42(10):813-828
15. Innis MA, Gelfand DH, Sninsky JJ. et al. PCR Protocols: A Guide to Methods and Applications. New York: Academic Press Inc. 1990:315-322
16. Altschul SF, Gish W, Miller W. et al. Basic local alignment search tool. J Mol Biol. 1990;215:403-410
17. Thompson JD, Gibson TJ, Plewniak F. et al. The clustal_X windows interface: flexible strategies for multiple sequence alignement aided by quality analysis tools. Nucleic Acid Res. 1997;25:4876-4882
18. Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111- 120
19. Saitou N, Nei M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406-425
20. Tamura K, Dudley J, Nei M. et al. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0 . Mol Niol Evol. 2007;24( 8):1596-1599
21. Baldwin BG. Phylogenetic utility of the internal transcribed spacers of nuclear ribosomal DNA in plants: An example from the Compositaogy. Mol Phylogenet Evol. 1992;1(1):3-16
22. Schmidt O, Moreth U. Data bank of rDNA-ITS sequences from building-rot fungi for their identification. Wood Sci Technol. 2002;36:429-433
23. Li CG, Li XM, Kong WD. et al. Effect of monoculture soybean on soil microbial community in the Northeast China. Plant Soil. 2010;330(1-2):423-433
24. Rainey PB. Effect of Pseudomonas putida on hyphal growth of Agaricus bisporus. Mycol Res. 1991;95:699-704
25. Ntougias S, Zervakis G, Kavroulakis N. et al. Bacterial diversity in spent mushroom compost assessed by amplified rDNA restriction analysis and sequencing of cultivated isolates. System Appl Microbiol. 2004;27(6):746-754
26. Amann RI, Ludwig W, Schleifer KH. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev. 1995;59:143-169
27. Streit WR, Schmitz RA. Metagenomics - the key to the uncultured microbes. Curr Opin Microbiol. 2004;7(5):492-498
28. Ruussos S, Garcia JL, Rinaudo G. Distribution of heterotrophic aerobic microflora and specially denitrifying and free-living nitrogen-fixing bacteria in the rhizosphere of rice. Ann Microbiol. 1980;131(2):197-207
29. Choudhary DK, Johri BN. Interactions of Bacillus spp. and plants - with special reference to induced systemic resistance (ISR). Microbiol Res. 2009;164(5):493-513
30. Bis'ko NA, Bilay VT. Effects of Bacillus macerans Fr. on growth of Pleurotus ostreatus (Jacq.:Fr.) Kumm. In: (ed.) Elliott T.J. Science and Cultivation of Edible Fungi. Rotterdam: Balkema. 1995:843-846
31. Kim MK, Math RK, Cho KM. et al. Effect of Pseudomonas sp. P7014 on the growth of edible mushroom Pleurotus eryngii in bottle culture for commercial production. Bioresour Technol. 2008;99(8):3306- 3308
32. Kloepper JW, Ryu CM, Zhang S. Induced systemic resistance and promotion of plant growth by Bacillus spp. Phytopathology. 2004;94(11):1259-1266
33. Van Workum WAT, Cremers H, Wijfjes AHM. et al. Role of exopolysaccharides of Rhizobium leguminosarum bv. viciae as host plant-specific molecules required for infection thread formation during nodulation of Vicia sativa. Mol Plant Microbe In. 1998;11(12):1233-1241
34. Djordjevic SP, Chen H, Batley M. Nitrogen fixation ability of exopolysaccharide synthesis mutants of Rhizobium sp. strain NGR234 and Rhizobium trifolii is restored by the addition of homologous exopolysaccharides. J Bacteriol. 1987;169(1):53-60
35. Gaskins MH, Albrecht SL, Hubbell DH. Rhizosphere bacteria and their use to increase plant productivity: A review. Agr Ecosyst Environ. 1985;12(2):99-116
36. Hirsch GU, Braun U. Communities of parasitic microfungi. In: (ed.) Winterhoff W. Handbook of vegetation science: fungi in vegetation science, 19. Dordrecht: Kluwer Academic. 1992:225-250
37. Vega FE, Posada F, Aime MC. et al. Entomopathogenic fungal endophytes. Biol Control. 2008;46:72- 82
38. Zhou XW, Zhu HF, Liu L. et al. A review: recent advances and future prospects of taxol-producing endophytic fungi. Appl Microbiol Biotechnol. 2010;86(6):1707- 1717
Author contact
![]() Corresponding author: Feng Xu. Institute of Plant and Environmental Protection, Beijing Academy of Agriculture and Forestry Science, Beijing 100097, China. Tel: +86 10 51503432, Fax: +86 10 51503899, E-mail: xxu1022com.
Corresponding author: Feng Xu. Institute of Plant and Environmental Protection, Beijing Academy of Agriculture and Forestry Science, Beijing 100097, China. Tel: +86 10 51503432, Fax: +86 10 51503899, E-mail: xxu1022com.