Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(2):234-243. doi:10.7150/ijbs.7.234 This issue Cite
Research Paper
TDP-43 Potentiates Alpha-synuclein Toxicity to Dopaminergic Neurons in Transgenic Mice
Tian Tian1, 2*, Cao Huang1*, Jianbin Tong1, Ming Yang1, Hongxia Zhou1 ![]() , Xu-Gang Xia1
, Xu-Gang Xia1 ![]()
1. Department of Pathology, Anatomy & Cell Biology, Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA
2. Department of Neurology, Xiangya Hospital, Central South University, Changsha, Hunan Province 410008, China
*These authors contributed equally to this work.
Received 2011-2-22; Accepted 2011-3-3; Published 2011-3-4
Abstract
TDP-43 and α-synuclein are two disease proteins involved in a wide range of neurodegenerative diseases. While TDP-43 proteinopathy is considered a pathologic hallmark of sporadic amyotrophic lateral sclerosis and frontotemporal lobe degeneration, α-synuclein is a major component of Lewy body characteristic of Parkinson's disease. Intriguingly, TDP-43 proteinopathy also coexists with Lewy body and with synucleinopathy in certain disease conditions. Here we reported the effects of TDP-43 on α-synuclein neurotoxicity in transgenic mice. Overexpression of mutant TDP-43 (M337V substitution) in mice caused early death in transgenic founders, but overexpression of normal TDP-43 only induced a moderate loss of cortical neurons in the transgenic mice at advanced ages. Interestingly, concomitant overexpression of normal TDP-43 and mutant α-synuclein caused a more severe loss of dopaminergic neurons in the double transgenic mice as compared to single-gene transgenic mice. TDP-43 potentiated α-synuclein toxicity to dopaminergic neurons in living animals. Our finding provides in vivo evidence suggesting that disease proteins such as TDP-43 and α-synuclein may play a synergistic role in disease induction in neurodegenerative diseases.
Keywords: TDP-43, alpha-synuclein, mice, dopaminergic neuron, Parkinson's disease, frontotemporal lobe degeneration, genetic model
1. Introduction
TAR DNA-binding protein 43 (TDP-43) is a conserved ribonucleoprotein with diverse functions as exemplified by the ability to regulate gene transcription, mRNA splicing, and RNA stability (1-6). While its physiological functions remain to be elucidated, TDP-43 translocates to the cytoplasm and forms ubiquitinated aggregates in neurodegenerative diseases including amyotrophic lateral sclerosis (ALS) and frontotemporal lobe degeneration (FTLD) (7-10). TDP-43 proteinopathy, a hallmark of sporadic ALS and FTLD, is also observed in the other neurodegenerative diseases such as Parkinson's disease (PD) and Alzheimer's disease (11-13). Neurodegenerative diseases share some features in the pathogenesis and pathology. Most cases of the diseases are sporadic and only a small proportion of them have a clear genetic cause. A neurodegenerative disease is characterized by a selected group of neurons preferably affected at early disease stages, but it often affects a wide range of different neurons at advanced disease stages. For example, heterogeneous neuropathology is observed in sporadic ALS at advanced disease stages (14-16), although motor neurons are preferably affected in the disease at early disease stages. TDP-43 proteinopathy is direct evidence supporting that neurodegenerative diseases may be pathogenically interrelated.
While mutation in TDP-43 is linked to ALS and FTLD (17-21), mutation of the α-synuclein gene is associated with PD (22-26). Overexpression of TDP-43 with or without pathogenic mutation causes a broad neurodegeneration in rodents (27-32), suggesting that normal TDP-43 of elevated expression is neurotoxic. Overexpression of TDP-43 in the substantia nigra induces dopaminergic neuron death (33), suggesting that TDP-43 neurotoxicity is not restricted to the neurons preferably affected in ALS and FTLD. Overexpression of pathogenically mutated α-synuclein causes dopaminergic neuron death in transgenic mice at advanced ages (34). Neurodegenerative diseases are commonly thought of multifactorial pathogenesis. To test this hypothesis, we examined whether TDP-43 potentiates α-synuclein toxicity to dopaminergic neurons in transgenic mice. Transgenic overexpression of the normal TDP-43 caused a significant loss of neurons in the frontal cortex, but not in the substantia nigra pars compacta (SNpc). When normal TDP-43 and mutant α-synuclein were overexpressed simultaneously in the dopaminergic neurons, a dramatic loss of the neurons was detected in the SNpc of transgenic mice. Our results suggest that neurotoxic factors such as TDP-43 and α-synuclein may play a synergistic role in the pathogenesis of neurodegenerative diseases.
2. Materials and Methods
Animal experiments
Animal use followed NIH guidelines and the animal use protocol was approved by the Institutional Animal Care and Use Committees at Thomas Jefferson University. As described in our publication (32, 35), the cDNA of normal human TDP-43 and its mutant form (M337V substitution) was created by PCR and was cloned into CAG expression vector. Linearized transgene constructs were injected into the pronuclei of fertilized eggs of C57BL/6J mice. Transgenic mice were identified by PCR amplification of the human TDP-43 transgene with the following primers: 5'-TGCGGGAGTTCTTCTCTCAG-3' (forward) and 5'-AGCCACCTGGATTACCACCA-3' (reverse). Dual-mutant α-synuclein transgenic mice were received from Dr. Richfield and were genotyped by PCR analysis as described in the original publications (34, 36). All transgenic mice were maintained on C57BL6 genomic background.
Mouse psychomotor activity was measured by Rotarod test as described previously (35, 37). Mice were tested on a rotating Rotarod (Med Associates) twice a week since three months of age. On a testing day, a mouse was tested three times separated by 10 minutes of rest and was allowed to run on a rotating Rotarod with accelerated speeds for a maximal period of five minutes. The average time of three tests was calculated as the latency to fall from rotating rotarod.
Immunoblotting
Mouse tissues were homogenized in RAPI buffer supplied with protease inhibitors (Promega). Tissue lysates were cleared by centrifugation and proteins in cleared lysates were measured by BCA assay (Pierce). Total proteins of 25 µg in each lysate were separated on 10% SDS-PAGE and were blotted onto PVDF membrane. The immunoreactivity of both human and mouse TDP-43 was detected with a polyclonal antibody (1: 500; Proteintech) and the immunoreactivity of human TDP-43 was detected with a mouse monoclonal antibody (1: 1000; Abnova, 2E2-D3). Comparable loading of total proteins among samples was estimated by probing the same membranes with a mouse monoclonal antibody recognizing mouse glyceraldehyde-3-phosphatedehydrogenase (1: 2000; Abnova). Immunoreactivity was visualized by ECL reaction (Pierce).
Toluidine blue staining
As described in our publication (32), the ventral and dorsal nerve roots of TDP-43 transgenic mice were examined for axon degeneration by toluidine blue staining. Mice were anesthetized and perfused with a mixture of 4% PFA and 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). The L4 ventral and dorsal roots were removed and post-fixed in the same fixative at 4°C overnight. The roots were further fixed in 1% osmium tetroxide in 0.1 M phosphate buffer (pH 7.4) for 1 h. The well-fixed tissues were dehydrated in graded ethanol and were embedded in Epon 812 (Electron Microscopic Sciences, Fort Washington, PA). Nerve roots were transversely cut into 1 µm-thick sections. Axons in the nerve roots were examined in the semi-thin sections under a light microscope (Nikon 80i).
Immunostaining and histology
Mouse tissues were fixed in 4% PFA and were cryopreserved in 30% sucrose. On a Cryostat, mouse brain was cut into coronal sections of 20 µm and mouse spinal cord was cut into traverse sections of 15 µm. Human TDP-43 immunoreactivity was detected by incubating tissue sections with a monoclonal antibody specific for human TDP-43 (1: 500; Abnova, 2E2-D3) and was visualized using an ABC kit in combination with diaminobenzidine (Vector). Immunostained sections were lightly counterstained with hematoxylin to display the nuclei. Double-labeling immunofluorescence staining was performed as described previously (32). The following antibodies were used for immunofluorescence staining: mouse monoclonal antibody to human TDP-43 (1: 500; Abnova, 2E2-D3), chicken antibody to ubiquitin (1: 1000; Sigma), rabbit antibody to tyrosine hydroxylase (TH; Pel-Freez; 1:1000), and mouse monoclonal antibody to human α-synuclein (1: 1000; Abcam, 4B12). Frozen sections of the gastrocnemius muscle were stained with hematoxylin and eosin (H&E) to display tissue structures.
Stereological cell counting
As described in our publication (38), the number of dopaminergic neurons in the SNpc was estimated by stereological cell counting. PFA-fixed mouse midbrain was cut on a Cryostat into four series of sections and every fourth section was immunostained for TH. The number of TH-positive neurons in the SNpc was estimated with fractionator-based stereology software (Stereologer), which was run on a PC computer attached to a Nikon 80i microscope with a motorized XYZ stage (Prior). The number of cortical neurons in the frontal cortex was also estimated by stereological cell counting as described previously (39).
3. Results
Overexpression of a mutant TDP-43 causes early death in transgenic founders
To develop a genetic animal model for TDP-43-linked ALS and FTLD, we created transgenic mice expressing the human TDP-43 with or without a pathogenic mutation (Fig. 1).
Expression of human TDP-43 in transgenic mice. a, Schematic shows the structure of human TDP-43 transgene. The transgene constructs consist of the CAG promoter, the first noncoding exon and the first intron of the chicken beta actin gene, the human TDP-43 cDNA with or without pathogenic mutation (M337V substitution), and the three repeats of SV40 polyadenylation signal. b, PCR analysis identified transgenic founders carrying the normal or the mutant (M337V) human TDP-43 transgene (hTDP-43). C+: positive control; C-: negative control. c, Immunoblotting detected a robust expression of human TDP-43 in the forebrain of transgenic founder mice. NG: nontransgenic mouse brain. d, Photo showing a transgenic founder mouse and its nontransgenic littermate (NG) at 7 days of age. e, Immunoblotting detected a robust expression of human TDP-43 in the central nerve system of normal TDP-43 transgenic mice. Each lane was loaded with 25 µg of total protein. The same membrane was first probed with a TDP-43 antibody recognizing both human and mouse TDP-43 and was then probed with a GAPDH antibody. Muscle: skeletal muscle; hippo: hippocampus.
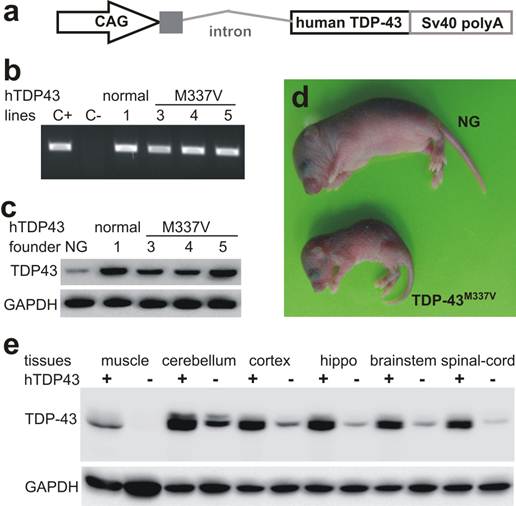
The hybrid promoter CAG is shown to drive transgene expression robustly and ubiquitously in rodents (35, 40, 41), and thus it was used to drive TDP-43 transgenes in mice (Fig. 1a). We chose M337V substitution as an example of TDP-43 mutations because this mutant causes typical ALS with a middle-age onset of symptoms (18). We obtained four transgenic founders expressing mutant TDP-43M337V and one founder expressing normal TDP-43 (Fig. 1). These transgenic founders were born of normal sizes; three TDP-43M337V founders lost mobility soon after birth and died within 10 days of birth (Fig. 1d). One TDP-43M337V female founder lived to 4 months of age, but it was infertile and died shortly after immobility was observed. We examined this mutant founder for neuronal and axonal degeneration in the spinal cord and nerve roots, but did not detect a significant loss of spinal motor neurons or motor axons. Similar to our finding in transgenic rats (32), constitutive overexpression of mutant TDP-43 in mice caused severe phenotypes in transgenic founders, preventing transgenic lines from establishment. By contrast, the transgenic founder overexpressing normal TDP-43 was healthy and fertile.
To expand the normal TDP-43 transgenic mouse colony, we crossed the transgenic founder with C57BL6/J mice newly purchased from Jackson Laboratories. Unexpectedly, some male transgenic mice of the first three generations died within a week after immobility was observed. No detectable phenotype was observed in TDP-43 transgenic mice beyond the fourth generation. We examined the sick mice and did not detect a degeneration of spinal motor neurons or motor axons. The copy number of human TDP-43 transgene was unaltered through mouse generations examined (all 22 copies). We maintained a C57BL6 colony derived from a mating pair that was purchased from the Jackson Laboratories four years ago, and we created TDP-43 transgenic mice on the genomic background of this mouse colony. The genomic background of TDP-43 transgenic mice could thus be considered congenic, but unidentified environmental or genetic factors may alter phenotypic expression in our TDP-43 transgenic mice.
Overexpression of the normal TDP-43 causes cortical neuron degeneration in transgenic mice
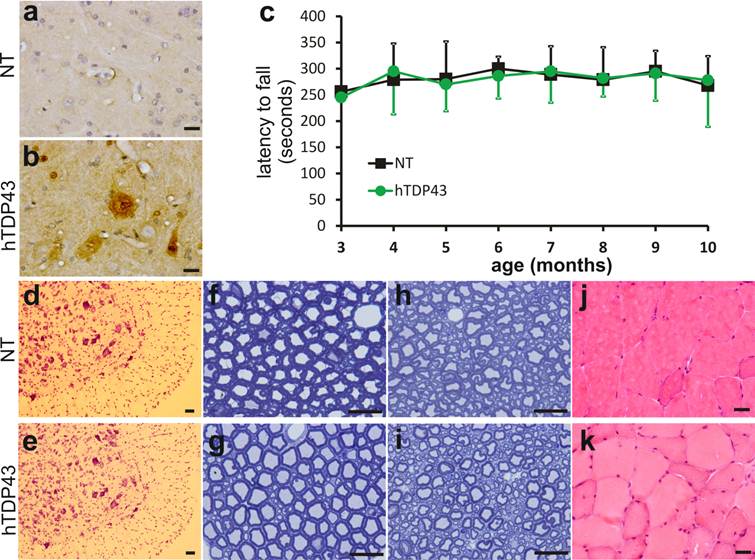
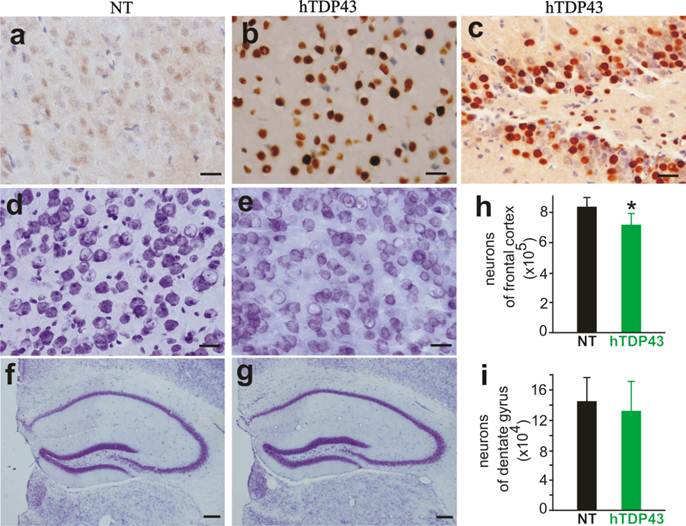
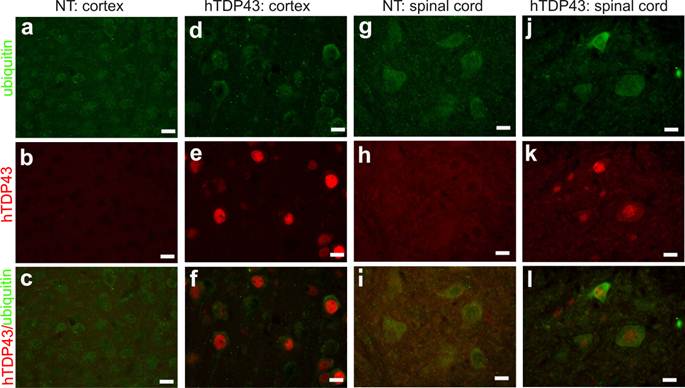
TDP-43-positive inclusion is a hallmark of sporadic ALS and FTLD (42, 43), suggesting that TDP-43 may be involved in the pathogenesis of the sporadic diseases. We used TDP-43 transgenic mice of the sixth generation for the following experiments. A robust expression of human TDP-43 was detected in the brain, spinal cord, and skeletal muscles of TDP-43 transgenic mice (Figs. 1e, 2b, 3b, and 3c), but not in the tissues of nontransgenic controls (Figs. 2a and 3a). On a homogenous genomic background, TDP-43 transgenic mice did not develop abnormality in psychomotor activity by age of one year (Fig. 2c). Accordingly, TDP-43 transgenic mice did not develop detectable pathology in the spinal motor neurons (Fig. 2d-2e), in the dorsal and ventral nerve roots (Fig. 2f-2i), and in the skeletal muscles (Fig. 2j-2k). Mutation of TDP-43 is also linked to FTLD (44). Altered expression of normal TDP-43 may affect cortical neurons in transgenic animals (32). We assessed cortical and hippocampal neurons by an unbiased method—stereological cell counting and detected a significant loss of cortical neurons in the transgenic mice at advanced ages (Fig. 3d-3i). Ubiquitinated TDP-43 aggregates are common to sporadic ALS and FTLD (42, 43). We examined these pathologies in TDP-43 transgenic mice, but did not detect typical TDP-43 or ubiquitin aggregates in the brain and spinal cord of the aged mice (Fig. 4a-4l). Consistent with our findings in TDP-43 transgenic rats (32), ubiquitin and TDP-43 aggregation is not essential to neuronal death.
No detectable impairment in the motor system of TDP-43 transgenic mice at advanced ages. a, b, Immunostaining detected human TDP-43 only in the TDP-43 transgenic mouse (hTDP43), but not in its nontransgenic control (NT). Cross sections of lumbar spinal cord were immunostained with a monoclonal antibody recognizing human TDP-43 and the ventral horns of lumbar cords were photographed. c, Rotarod test revealed no difference in psychomotor activity between the TDP-43 transgenic mice (hTDP43) and the nontransgenic littermates (NT). Each group consisted of 8 mice with equal sex composition. d, e, Cresyl violet staining revealed no significant loss of motor neurons in the lumbar cords of TDP-43 transgenic mice. f-i, Toluidine blue staining showed the structure of the L4 ventral roots (f, g) and the L4 dorsal roots (h, i) in TDP-43 transgenic mice and their nontransgenic littermates at the age of 11 months. j, k, H&E staining revealed no atrophy of skeletal muscles in the TDP-43 transgenic mice. Scale bars: a-b and f-k, 20 µm; d-e, 50 µm.
A significant loss of cortical neurons in the frontal cortex of TDP-43 transgenic mice at advanced ages. a-c, Immunostaining revealed a robust expression of human TDP-43 in the cortex and dentate gyrus of TDP-43 transgenic mouse, but not in the cortex of a nontransgenic control (NT). d-g, Cresyl violet staining showed the neurons in the frontal cortex (d, e) and hippocampus (f, g). Scale bars: a-e, 20 µm; and f-g, 50 µm. h, i, Stereological cell counting revealed a significant loss of neurons in the frontal cortex, but not in the dentate gyrus, of TDP-43 transgenic mice. Mice were 10-12 months old. Data are means + SD (n = 5). * indicates p < 0.05.
Ubiquitin inclusion undetectable in TDP-43 transgenic mice at advanced ages. a-l, Double-labeling fluorescence staining revealed no ubiquitin inclusion in TDP-43-overexpressing cells in transgenic mice at 10 months of age. Tissue sections from TDP-43 transgenic mice (hTDP43) or the nontransgenic controls (NT) were immunostained for human TDP-43 and mouse ubiquitin. All scale bars: 20 µm.
Overexpression of normal TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice
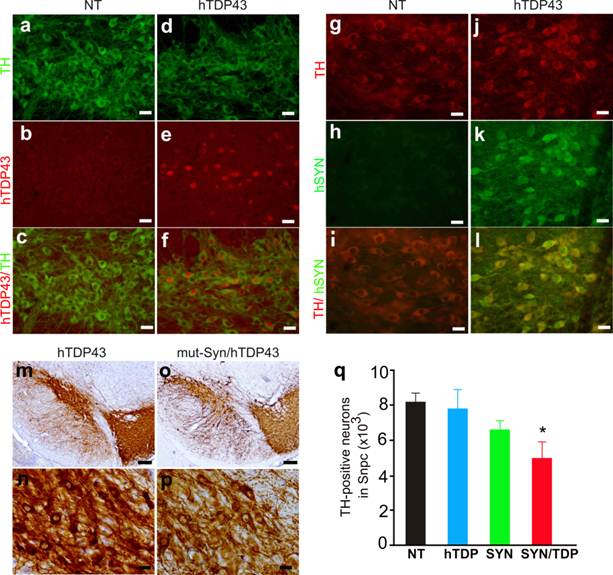
TDP-43 proteinopathy is observed in a wide range of neurodegenerative diseases including PD (12, 13). Transient overexpression of TDP-43 in the substantia nigra causes dopaminergic neuron death (33). Alpha-synuclein is a major component of Lewy body characteristic of PD and pathogenic mutation of α-synuclein is implicated in PD pathogenesis (22-26). We further examined whether TDP-43 modifies α-synuclein toxicity in the double transgenic mice. In TDP-43 transgenic mice, a substantial number of dopaminergic neurons expressed human TDP-43 as shown by double-labeling immunofluorescence staining (Fig. 5a-5f). Overexpression of human TDP-43 alone did not induce a significant loss of dopaminergic neurons in the transgenic mice (Fig. 5q). We then crossed TDP-43 transgene onto a mutant α-synuclein transgenic background (Fig. 5g-5l). Previous study showed that overexpression of mutant α-synuclein causes a significant loss of dopaminergic neurons in male mice at advanced ages (34); however, we did not detect a significant loss of dopaminergic neurons in the mutant α-synuclein transgenic mice though a trend of neuronal loss was observed (Fig. 5q). Our result does not contradict the previous study because we examined three male and three female α-synuclein transgenic mice. Male mice appeared more sensitive to mutant α-synuclein toxicity than female mice. In our study, statistical significance might be obscured by a limited number of animals and a mixture of male and female mice. Interestingly, simultaneous overexpression of normal TDP-43 and mutant α-synuclein caused a significant loss of dopaminergic neurons in the transgenic mice as compared to either single transgenic control (Fig. 5m-5q), suggesting that TDP-43 potentiated α-synuclein neurotoxicity.
Synergistic effects of TDP-43 and mutant alpha-synuclein on dopaminergic neurons in transgenic mice. a-f, Double-fluorescence labeling revealed a substantial expression of human TDP-43 in the dopaminergic neurons of human TDP-43 transgenic mice (hTDP43), but not in the neurons of nontransgenic mice (NT). Coronal sections of the midbrain were simultaneously immunostained for human TDP-43 and mouse tyrosine hydroxylase (TH, a marker of dopaminergic neurons). g-l, double-labeling fluorescence staining detected a robust expression of a mutant human alpha-synuclein (hSYN) in the dopaminergic neurons of hSYN transgenic mice, but not in the neurons of nontransgenic mice (NT). m-p, Representative photos of low (m, o) or high (n, p) magnification showing dopaminergic neurons in the substantia nigra of TDP-43 single (m, n) and TDP-43/hSYN double (o, p) transgenic mice. Scale bars: a-l, n and p, 20 µm; m, o, 80 µm. q, Stereological cell counting revealed a significant loss of dopaminergic neurons (TH-positive) in the TDP-43 and alpha-synuclein double transgenic mice (SYN/TDP) as compared to nontransgenic (NT) or the single transgenic (hTDP or SYN) mice at 10-12 months of age. Data are means + SD (n = 4-6). The symbol * indicates p < 0.05 as compared to SYN or hTDP transgenic mice.
4. Discussion
Consistent with our findings in transgenic rats (32), pathogenically mutated TDP-43 exhibited higher toxicity than the wildtype form when overexpressed in transgenic mice. Normal TDP-43 of elevated expression was sufficient to induce neurotoxicity in vivo. Moreover, TDP-43 and mutant α-synuclein synergistically induced neurotoxicity when concomitantly overexpressed in the dopaminergic neurons of transgenic mice, suggesting that neuron death is a convergent outcome of the toxicity of disease factors in neurodegenerative diseases such as PD.
While the nature of pathogenic mutation in TDP-43 remains largely unknown, TDP-43 with pathogenic mutation or elevated expression is neurotoxic in vitro and in vivo (28, 45-48). Pathogenic mutation of TDP-43 may cause a gain of function, a loss of function, or a dominant negative effect. The information available is not sufficient to determine the nature of TDP-43 mutation. Silencing of TDP-43 expression by RNAi induces neuronal death in culture (49, 50). Deletion of TDP-43 causes a defect at the neuromuscular junction in Drosophila melanogaster (51). Constitutive or temporal deletion of TDP-43 in mice induces severe phenotypes (52, 53), underscoring the importance of TDP-43 in cellular function. It remains unclear whether neurodegeneration can be induced by cell-specific deletion of TDP-43 in conditional knockout mice. Forced overexpression of human TDP-43 in mice deregulates the expression of mouse endogenous TDP-43 (27). Since it is not known whether human TDP-43 can function properly in mouse tissues, addition of normal human TDP-43 to mouse genome may or may not enhance the functions of TDP-43. In addition, transgenic overexpression of mutant TDP-43 is unable to differentiate a gain of function from a dominant negative effect because both types of mutation can induce disease in transgenic animals (54-56). Although a more sophisticated model such as a TDP-43 knockin mouse is required for determining the natures of TDP-43 mutation, our results, along with previous findings (27, 28, 45-48, 50, 52, 53), suggest a possible gain of function in TDP-43 mutation. We previously showed that overexpression of normal TDP-43 does not induce ALS disease in transgenic rats (32). Consistent with our finding in rats, overexpression of normal TDP-43 in mice failed to induce ALS phenotypes. Interestingly, overexpression of normal TDP-43 caused a significant (though moderate) loss of cortical neurons in asymptomatic mice at advanced ages. It could not be ruled out that overexpression of normal TDP-43 may induce cortical neuron degeneration in aged rats. We will examine cortical neurons in normal TDP-43 transgenic rats at advanced ages in a follow-up study.
TDP-43 and α-synuclein are involved in a wide range of neurodegenerative diseases. While TDP-43 proteinopathy is considered a pathologic hallmark of ALS and FTLD (7-10), it also coexists with Lewy pathology that is a characteristic of sporadic PD (57-59). Alpha-synuclein is a major component of Lewy body and Lewy neurites in PD (57-59). When concomitantly overexpressed in mice, TDP-43 and mutant α-synuclein synergistically induced dopaminergic neurodegeneration. Our finding is not unexpected as TDP-43 and α-synuclein proteinopathy coexist in patients with neurodegenerative diseases (57-59). Clinically, synucleinopathy is more severe in the cases with than in the cases without TDP-43 proteinopathy (60). It remains unclear whether and how disease proteins (such as TDP-43, α-synuclein, β-amyloid, and tau) interact to induce degeneration of selected groups of neurons in patients with neurodegenerative diseases. Our study has provided in vivo evidence suggesting that disease proteins such as TDP-43 and α-synuclein may play a synergistic role in disease induction in neurodegenerative diseases.
Acknowledgements
We thank Ms. Dian Wang, Ms. Xiaotao Wei, and Ms. Zhen Qu for technical assistance.
This work is supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NS064042 and NS072113 to X.G.X) and by the National Institutes of Health/National Institute of Environmental Health Sciences (ES016760 to X.G.X). The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH institutes.
Conflict of Interests
The authors declare no conflicts of interest.
References
1. Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280(45):37572-37584
2. Abhyankar MM, Urekar C, Reddi PP. A novel CpG-free vertebrate insulator silences the testis-specific SP-10 gene in somatic tissues: role for TDP-43 in insulator function. J Biol Chem. 2007;282(50):36143-36154
3. Bose JK, Wang IF, Hung L, Tarn WY, Shen CK. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem. 2008;283(43):28852-28859
4. Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69(6):3584-3596
5. Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276(39):36337-36343
6. Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, Shoesmith C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35(2):320-327
7. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A. et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668-1672
8. Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R. et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63(4):535-538
9. Banks GT, Kuta A, Isaacs AM, Fisher EM. TDP-43 is a culprit in human neurodegeneration, and not just an innocent bystander. Mamm Genome. 2008;19(5):299-305
10. Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B. et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65(4):470-473
11. Lippa CF, Rosso AL, Stutzbach LD, Neumann M, Lee VM, Trojanowski JQ. Transactive response DNA-binding protein 43 burden in familial Alzheimer disease and Down syndrome. Arch Neurol. 2009;66(12):1483-1488
12. Chanson JB, Echaniz-Laguna A, Vogel T, Mohr M, Benoilid A, Kaltenbach G, Kiesmann M. TDP43-positive intraneuronal inclusions in a patient with motor neuron disease and Parkinson's disease. Neurodegener Dis. 2010;7(4):260-264
13. Markopoulou K, Dickson DW, McComb RD, Wszolek ZK, Katechalidou L, Avery L, Stansbury MS, Chase BA. Clinical, neuropathological and genotypic variability in SNCA A53T familial Parkinson's disease. Variability in familial Parkinson's disease. Acta Neuropathol. 2008;116(1):25-35
14. Nishihira Y, Tan CF, Toyoshima Y, Yonemochi Y, Kondo H, Nakajima T, Takahashi H. Sporadic amyotrophic lateral sclerosis: Widespread multisystem degeneration with TDP-43 pathology in a patient after long-term survival on a respirator. Neuropathology. 2009 epub
15. Machida Y, Tsuchiya K, Anno M, Haga C, Ito T, Shimo Y, Wakeshima T, Iritani S, Ikeda K. Sporadic amyotrophic lateral sclerosis with multiple system degeneration: a report of an autopsy case without respirator administration. Acta Neuropathol. 1999;98(5):512-515
16. Tsuchiya K, Sano M, Shiotsu H, Akiyama H, Watabiki S, Taki K, Kondo H, Nakano I, Ikeda K. Sporadic amyotrophic lateral sclerosis of long duration mimicking spinal progressive muscular atrophy exists: additional autopsy case with a clinical course of 19 years. Neuropathology. 2004;24(3):228-235
17. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572-574
18. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A. et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668-1672
19. Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL. et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409-416
20. Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH. et al. Novel Mutations in TARDBP (TDP-43) in Patients with Familial Amyotrophic Lateral Sclerosis. PLoS Genet. 2008;4(9):e1000193
21. Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B. et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Annals of Neurology. 2009;65:470-474
22. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276(5321):2045-2047
23. Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18(2):106-108
24. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C. et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302(5646):841
25. Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364(9440):1167-1169
26. Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364(9440):1169-1171
27. Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, Malunda J, Xu Y, Winton MJ, Trojanowski JQ, Lee VM. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest. 2011;121(2):726-738
28. Shan X, Chiang PM, Price DL, Wong PC. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci U S A. 2010;107(37):16325-16330
29. Stallings NR, Puttaparthi K, Luther CM, Burns DK, Elliott JL. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol Dis. 2010;40(2):404-414
30. Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106(44):18809-18814
31. Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107(8):3858-3863
32. Zhou H, Huang C, Chen H, Wang D, Landel CP, Xia PY, Bowser R, Liu YJ, Xia XG. transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010;6(3):e1000887
33. Tatom JB, Wang DB, Dayton RD, Skalli O, Hutton ML, Dickson DW, Klein RL. Mimicking Aspects of Frontotemporal Lobar Degeneration and Lou Gehrig's Disease in Rats via TDP-43 Overexpression. Molecular Therapy. 2009;17(4):607-613
34. Thiruchelvam MJ, Powers JM, Cory-Slechta DA, Richfield EK. Risk factors for dopaminergic neuron loss in human alpha-synuclein transgenic mice. Eur J Neurosci. 2004;19(4):845-854
35. Zhou H, Huang C, Yang M, Landel CP, Xia PY, Liu YJ, Xia XG. Developing tTA Transgenic Rats for Inducible and Reversible Gene Expression. Int J Biol Sci. 2009;2(5):171-181
36. Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol. 2002;175(1):35-48
37. Xia XG, Zhou H, Huang Y, Xu Z. Allele-specific RNAi selectively silences mutant SOD1 and achieves significant therapeutic benefit in vivo. Neurobiol Dis. 2006;23(3):578-586
38. Zhou H, Falkenburger BH, Schulz JB, Tieu K, Xu Z, Xia XG. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int J Biol Sci. 2007;3(4):242-250
39. Huang C, Zhou H, Tong J. et al. FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. PLOS Genetics. 2011 in press
40. Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193-199
41. Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28(3-4):147-155
42. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130-133
43. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602-611
44. Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B. et al. TARDBPmutations in motoneuron disease with frontotemporal lobar degeneration. Annals of Neurology. 2009;65(4):470-473
45. Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y, Neumann M, Trojanowski JQ, Lee VM. Expression Of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem. 2009Mar27;284(13):8516-24
46. Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proceedings of the National Academy of Sciences. 2010;107(8):3858-3863
47. Li Y, Ray P, Rao EJ, Shi C, Guo W, Chen X, Woodruff EA, Fushimi K, Wu JY. A Drosophila model for TDP-43 proteinopathy. Proceedings of the National Academy of Sciences. 2010;107(7):3169-3174
48. Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proceedings of the National Academy of Sciences. 2009;106(44):18809-18814
49. Iguchi Y, Katsuno M, Niwa J, Yamada S, Sone J, Waza M, Adachi H, Tanaka F, Nagata K, Arimura N, Watanabe T, Kaibuchi K, Sobue G. TDP-43 depletion induces neuronal cell damage through dysregulation of Rho family GTPases. J Biol Chem. 2009;284(33):22059-22066
50. Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, Rochefort D, Bel Hadj S, Durham HD, Velde CV, Rouleau GA, Drapeau P. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2009;18:18
51. Feiguin F, Godena VK, Romano G, D'Ambrogio A, Klima R, Baralle FE. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009;583(10):1586-1592
52. Chiang PM, Ling J, Jeong YH, Price DL, Aja SM, Wong PC. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci U S A. 2010;107(37):16320-16324
53. Wu LS, Cheng WC, Hou SC, Yan YT, Jiang ST, Shen CK. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;15:15
54. Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, Wu D, Xue R, Andrade M, Tankou S, Mori S, Gallagher M, Ishizuka K, Pletnikov M, Kida S. et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci U S A. 2007;104(36):14501-14506
55. Lai C, Lin X, Chandran J, Shim H, Yang W, Cai H. The G59S mutation in p150(glued) causes dysfunction of dynactin in mice. J Neurosci. 2007;27(51):13982-13990
56. Kudo T, Kure S, Ikeda K, Xia AP, Katori Y, Suzuki M, Kojima K, Ichinohe A, Suzuki Y, Aoki Y, Kobayashi T, Matsubara Y. Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum Mol Genet. 2003;12(9):995-1004
57. Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M. et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171(1):227-240
58. Leverenz JB, Yu CE, Montine TJ, Steinbart E, Bekris LM, Zabetian C, Kwong LK, Lee VM, Schellenberg GD, Bird TD. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain. 2007;130(Pt 5):1360-1374
59. Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR, Uitti RJ, Rademakers R, Adamson J, Baker M, Hutton ML. et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007;66(2):142-151
60. Yokota O, Davidson Y, Arai T, Hasegawa M, Akiyama H, Ishizu H, Terada S, Sikkink S, Pickering-Brown S, Mann DM. Effect of topographical distribution of α-synuclein pathology on TDP-43 accumulation in Lewy body disease. Acta Neuropathol. 2010;120(6):789-801
Author contact
![]() Corresponding author: H.Z. (Hongxia.zhouedu) or X.G.X (xugang.xiaedu)
Corresponding author: H.Z. (Hongxia.zhouedu) or X.G.X (xugang.xiaedu)