Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(7):927-936. doi:10.7150/ijbs.7.927 This issue Cite
Research Paper
Vitamin E Modulates Cigarette Smoke Extract-induced Cell Apoptosis in Mouse Embryonic Cells
Zhao-Li Chen1*, Jian Tao2*, Jie Yang3*, Zhen-Li Yuan4, Xing-Hua Liu2, Min Jin1, Zhi-Qiang Shen1, Lu Wang3, Hai-Feng Li1, Zhi-Gang Qiu1, Jing-Feng Wang1, Xin-Wei Wang1, Jun-Wen Li1 ![]()
1. Department of Health and Environment, Institute of Health and Environmental Medicine; Key Laboratory of Risk Assessment and Control for Environment & Food Safety, No.1, Dali Road, Tianjin, 300050, P. R. China;
2. Food Sci-Eng College, Northwest A & F University, Yangling, Shanxi Province, 712100, P. R. China;
3. Department of Chronic Disease, Chinese Center for Disease Control and Prevention, Beijing, 100050, P. R. China;
4. School of Public Health, Jilin University, Changchun, 130021, P. R. China.
* The same contribution to the paper.
Received 2011-3-17; Accepted 2011-7-24; Published 2011-7-31
Abstract
Vitamin E (VE) can effectively prevent occurrence of lung cancer caused by passive smoking in mice. However, whether VE prevents smoking-induced cytotoxicity remains unclear. In this study, a primary culture of embryonic lung cells (ELCs) was used to observe the cytotoxic effects of cigarette smoke extract (CSE), including its influence on cell survival, cell cycle, apoptosis, and DNA damage, and also to examine the effects of VE intervention on CSE-induced cytotoxicity. Our results showed that CSE could significantly inhibit the survival of ELCs with dose- and time-dependent effects. Furthermore, CSE clearly disturbed the cell cycle of ELCs by decreasing the proportion of cells at the S and G2/M phases and increasing the proportion of cells at the G0/G1 phase. CSE promoted cell apoptosis, with the highest apoptosis rate reaching more than 40%. CSE also significantly caused DNA damage of ELCs. VE supplementation could evidently inhibit or reverse the cytotoxic effects of CSE in a dose- and time-dependent manner. The mechanism of CSE effects on ELCs and that of VE intervention might involve the mitochondrial pathway of cytochrome c-mediated caspase activation. Our study validate that VE plays a clearly protective effect against CSE-induced cytotoxicity in mouse embryonic lung cells.
Keywords: vitamin E, intervention, cytotoxicity, cigarette smoke extract, embryonic lung cell
Introduction
Cigarette smoking is the main cause of lung cancer and pulmonary emphysema [1, 2]. There were a number of passive smoking populations in China [3], the investigation results indicated that sidestream smoke which contains higher concentrations of hazardous chemicals [4] might be more harmful [5, 6] than mainstream smoke. The harmful effects of smoking and passive smoking are mainly correlated with oxidative stress damage caused by the oxidative substances in cigarette smoke [7, 8]. Although epidemiological studies present contradictory views regarding the preventive effects of antioxidant vitamin E (VE) supplements on smoking-induced lung cancer [9, 10], animal experiments findings have demonstrated that VE can effectively prevent passive smoking-induced lung cancer in mice [11, 12]. Although the pathogenesis of lung cancer caused by cigarette smoke appears to be complex, it is the major pathway that exposure to cigarette smoke causes lung cells DNA damage or apoptosis [13-15]. Recent researches indicated that cigarette smoke extract (CSE) could produce a clear cytotoxic effect [13-19]; however, whether VE prevents CSE-induced cytotoxicity and its mechanisms remain unclear.
In this study, an embryonic lung cells (ELCs) model was used to observe the influence of CSE on cell survival, cell cycle, apoptosis, and DNA damage and VE intervention in CSE-induced cytotoxicity; we also explored the mechanisms of CSE-induced cytotoxicity and VE intervention. We believe that this study will contribute to investigate the underlying mechanism of the preventive effect of VE on smoking-induced lung cancer.
Materials and Methods
Reagents
VE (DL-alpha-tocopherol) was obtained from Sigma (St. Louis, MO, USA). Cigarettes were purchased from the local supermarket (Hongmei Brand; tar content, 15 mg; and nicotine content, 1.2 mg per cigarette). The nicotine standard was obtained from Merck Biosciences (Hohenbrunn, Germany). RPMI-1640 medium, dimethylsulfoxide (DMSO), ribonuclease A (Rnase A), and trypsin were obtained from Invitrogen Corporation (N. Y. USA).
Mouse ELCs preparation and culture
ELCs were isolated from Swiss mouse embryos and cultured as described previously [20, 21]. Briefly, the embryos lungs were removed on the 13th day of gestation. The tissue pieces were shaken in phosphate-buffered saline (PBS) containing 0.25% trypsin on a rotary shaker at 37°C for 20 min. Pipette tissue pieces several times until a single cell suspension was obtained. After standing for 1 min, the cell suspension was centrifuged at 1000 revolutions per minute for 5 min. The cell pellets were resuspended in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/ml), and streptomycin (100 μg/ml) to obtain ELCs suspension. The ELCs were plated at a concentration of 2 × 106 cells/ml and incubated at 37°C in 5% CO2, and were observed and photographed at the logarithmic growth phase under inverted microscope (Olympus) as described previously [20, 21]. The ELCs could grow for more than 10 consecutive passages.
CSE preparation and nicotine concentration determination
The CSE was prepared as described previously [17, 22-24]. Briefly, the smoke of four lit cigarettes was collected by a peristaltic pump apparatus into 15 ml of RPMI-1640 medium. By bubbling the smoke in the medium, CSE stock solution was obtained, concentration of nicotine in the CSE stock solution was measured by high-performance liquid chromatography (HPLC) method at the Central Lab, Nankai University, and the concentration of nicotine was about 70000 ng/L in CSE stock solution, and then CSE stock solution was filtered through a 0.22-μm-pore size filter and diluted to the desired concentration with cell medium.
Effects of CSE on survival of ELCs and VE intervention
The ELCs at the logarithmic growth phase were treated with 0.25% trypsin to prepare a single cell suspension. The cells were adjusted to a concentration of 1 × 105 cell/ml and seeded 200 µl cell suspension to each well in a 96-well plate, and 3 parallel wells were set for each group treated with various concentrations of CSE [70000 ng/L (CSE stock solution), 400 ng/L, 200 ng/L, 100 ng/L, and 12.5 ng/L as the equivalent to nicotine]. There was no nicotine in the control group. The cells were incubated for 24 h, 48 h, and 72 h at 37°C under an atmosphere consisting of 5% CO2 and 95% air. Four hours before the end of the incubation, 20 µl of 5 mg/ml MTT was added to each well, and the cells were solubilized with 100 µl DMSO.
The optical density (OD) of each well was measured using ELISA plate reader (Wellscan MK3, Labsystems Dragon) with a test wavelength of 492 nm. The cell survival rate (SR) was determined by the following equation:
SR = (OD value in treated groups/OD value in control) × 100%.
For VE intervention, VE treatment was prepared as described previously [25, 26], VE stock solutions were prepared in ethanol, various concentrations of VE (1 μmol/L, 25 μmol/L, 50 μmol/L, 100 μmol/L and 200 μmol/L) were added to the cell medium 2 h before CSE (200 ng/L as the equivalent to nicotine) treatment. The survival rate was determined using the MTT method as described above.
Effects of CSE on cell cycle and apoptosis of ELCs and VE intervention
The ELCs at the logarithmic growth phase were seeded in a 6-well plate and incubated for 24 h at 37°C under an atmosphere of 5% CO2. Then, the cells were treated with various concentrations of CSE [70000 ng/L(CSE stock solution), 400 ng/L, 200 ng/L, 100 ng/L, and 12.5 ng/L as the equivalent to nicotine] and incubated for a further 48 h. The morphological changes were recorded with an invert microscope equipped with a charge-coupled device (CCD) camera. The CSE-treated cells were trypsinized and adjusted to a concentration of 1×106 cells/ml. The cells were washed with PBS, resuspended in 100 µl of 1 mg/ml Rnase A, and incubated at 37°C for 30 min for RNA digestion, followed by staining with 400 µl of 50 µg/ml propidium iodide ( PI ) for 10 min in the dark for the cell cycle and apoptosis assays. Flow cytometry was performed using a fluorescence-activated cell-sorting (FACS) Calibur flow cytometer (Becton Dickinson). Data were analyzed using ModiFit LT for Mac V 3.0 software (Becton Dickinson), and the percentage of G0/G1, S, G2/M, and apoptotic cells were determined. For VE intervention, various concentrations of VE (1 μmol/L, 25 μmol/L, 50 μmol/L, 100 μmol/L and 200 μmol/L) were added to the medium 2 h before CSE (200 ng/L as the equivalent to nicotine) treatment and analysis was performed using FACS.
DNA damage caused by CSE and VE intervention
The single-cell gel electrophoresis (or comet assay) was used to detect DNA damage [27]. Briefly, after treatment respectively with or without CSE (200 ng/L as the equivalent to nicotine) in presence or absence of VE (100 μmol/L) in 6-well plates for 30h, cells were harvested and washed twice with PBS. An aliquot of 8μl of the cells suspension (2×105 cells/ml) was mixed with 80 μl of 0.75% low melting-point agarose at 37°C and rapidly spreaded on microscope slides pre-coated with 0.5% normal melting-point agarose, the slides were covered with cover slips respectively and allowed to solidify on ice. After 10 min, the slides were placed in a freshly made cold lysing solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10, which 10% DMSO and 1% Triton X-100 had been added immediately prior to use) for 3 h at 4°C. After a ice-cold PBS washing for 3 times, the slides were placed in electrophoresis buffer (300 mM NaOH and 1 mM EDTA, pH 13) for 30 min to allow unwinding of the DNA and DNA breakage at alkali-labile sites. Electrophoresis was conducted in the same buffer (300 mA, 25V) for 20 min. The slides were washed in neutralization buffer (0.4 M Tris, pH 7.5) 3 times for 5 min each, fixation in ethanol for 40 min was followed and finally samples were stained with PI (5 μg/ml) for 15 min. The comets were analyzed at 10×40 magnifications with a fluorescence microscope (Olympus BX51) attached to a CCD video camera equipped with a G excitation filter and connected to a personal computer-based image analysis system, Metaph series 4.5. Fifty cells were randomly selected from each sample and the fraction of DNA tail was measured. DNA tail length was determined to assess the extent of DNA damage.
Caspase-3 activity assay
Caspase-3 activity was determined using a commercially available caspase-3 assay kit (Keygen); the manufacturer's instructions and the methods described in a previous report [28] were followed. This assay is based on the ability of the activated caspase-3 enzyme to cleave the chromophore from its substrate. After treatment respectively with or without CSE (200 ng/L as the equivalent to nicotine) in presence or absence of VE (100 μmol/L) in dishes for 30 h, cells were harvested and washed twice with PBS, mixed with 50 μl ice-cold lysis buffers, and placed on ice for 20 min. Lysates were then centrifuged at 10,000 × g for 1 min at 4°C. The supernatants were collected, and the concentration of total proteins was adjusted to 2 μg/μl. Next, 50 μl of the supernatants were transferred to microplates and mixed with 50 μl 2× reaction buffers. 5 μl colorimetric caspase-3 substrates were added into each well of microplates, then the plates were incubated for 4 h at 37°C. Cleavage of the peptide substrate was monitored at 405 nm using an ELISA plate reader (Labsystems Dragon). Cell enzymatic activity of caspase-3 was expressed as the OD405.
Western blotting
After treatment respectively with or without CSE (200 ng/L as the equivalent to nicotine) in presence or absence of VE (100 μmol/L) in 6-well plates for 30 h, cells were harvested and washed twice with PBS. For Bax and Bcl-2 detection, total cell proteins were recovered according to a previous method [29]. Cells were lysed in RIPA buffer (50 mM Hepes, 150 mM NaCl, 1% Triton X-100, 5 mM EGTA, 50 mM β-glycerophosphate, 20 mM NaF, 1 mM Na3VO4, 2 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml aprotinin, pH 7.5). After incubation on ice for 30 min, extracts were clarified by centrifugation at 10,000 × g for 30 min at 4°C; the supernatants were subjected to western blot analysis for Bax and Bcl-2. For cytochrome c detection, cytosol proteins were recovered according to a previous method [30]. Cells were lysed in a buffer described previously (250 mM sucrose; 20 mM Hepes-KOH, pH 7.5; 10 mM KCl; 1.5 mM MgCl2; 1 mM EDTA; 1 mM EGTA; 1 mM dithiothreitol; and 0.1 mM phenylmethylsulfonyl fluoride). The lysates were then homogenized and centrifuged 3 times at 3000 × g for 10 min to separate cellular debris and nuclei; the supernatants were subjected to western blot analysis for cytochrome c. The protein content of each supernatant was determined using the Bradford assay. The supernatant solution was mixed with 5× SDS-loading buffer, following treatment in a water bath at 100°C for 10 min. For western blot analysis, equal amounts of total proteins (20 μg) were separated by 12% SDS-PAGE and then transferred and blotted onto a PVDF membrane (Millipore Corporation, Massachusetts, U.S.A.). Immunoblots were analyzed using the following specific primary antibodies (Santa Cruz Biotechnology, Inc., California, U.S.A.): anti-Bax (sc-7480), anti-Bcl-2 (sc-7382), anti-cytochrome c (sc-13156), and anti-actin (sc-1616-R). After exposure to horseradish peroxidase-conjugated secondary antibody (ZB-2301, Zhongshan Goldenbridge Biotechnology, Co., LTD., Beijing, China) for 1 h, protein bands were visualized using Western Blotting Luminol Reagent (sc-2048, Santa Cruz Biotechnology, Inc., California, U.S.A.).
Statistical analysis
All data were presented as mean ± SD. Statistical analysis was performed using one-way ANOVA (LSD test). We used the SPSS 11.5 program for the statistical analysis. A P-value of < 0.05 was considered statistically significant.
Results
Effects of CSE on survival, cell cycle, and apoptosis of ELCs
Effects of CSE on survival of ELCs
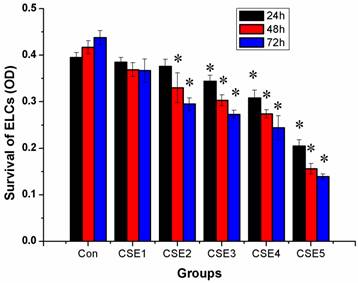
The inhibitory effects on ELCs increased with an increase in the CSE concentration and treatment time, indicating dose- and time-dependency of the inhibitory effects. The CSE with a nicotine concentration of 200 ng/L or higher produced clear inhibitory effects on the growth of ELCs at the 24 h, 48 h, and 72 h time points (Figure 1).
Effects of CSE on survival of ELCs (OD, mean ±S, and n = 3). The CSE could inhibit the cell survival and proliferation of the ELCs, indicating dose- and time-dependency of the inhibitory effect. * P < 0.05 vs control group. R=-0.930, -0.977, -0.986 for 24 h, 48 h and 72 h respectively, P <0.05 (dose-response). Con: control, CSE1: 12.5 ng/L, CSE2: 100 ng/L, CSE3: 200 ng/L, CSE4: 400 ng/L, CSE5: 70,000 ng/L as the equivalent to nicotine.
Effects of CSE on cell cycle of ELCs
The proportion of ELCs in the S and G2/M phases decreased significantly in the groups with CSE containing 200 ng/L or higher concentrations of nicotine treatments compared to that in the control group (P < 0.05, P < 0.01). There was a significant increase in the proportion of ELCs in the G0/G1 phase in the groups treated with 200 ng/L or higher concentration of nicotine compared to that in the control group (P < 0.01). The above results suggested that the CSE decreased the proportion of cells in the S and G2/M phases and mainly arrested the cells in the G0/G1 phase (Figure 2).
Effects of CSE on cell cycle of ELCs ( %, mean ±SD, n = 3). CSE decreased the proportion of ELCs in the S and G2/M phases while increased that in the G0/G1 phase of cell cycle. * P < 0.05, # P < 0.01 vs control group. Con: control, CSE1: 12.5 ng/L, CSE2: 100 ng/L, CSE3: 200 ng/L, CSE4: 400 ng/L, CSE5: 70,000 ng/L as the equivalent to nicotine.

Effects of CSE on apoptosis of ELCs
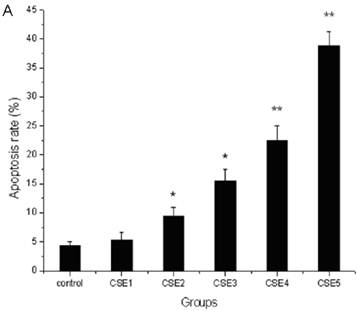
The apoptosis rate of ELCs treated with CSE containing 100 ng/L nicotine or higher concentrations increased significantly compared to that of the control group (P < 0.05, P < 0.01) (Figure 3A). The number of apoptotic and shed cells gradually increased with the increase of CSE concentrations (Figure 3 B, C). The above results illustrated that CSE could promote the apoptosis of ELCs.
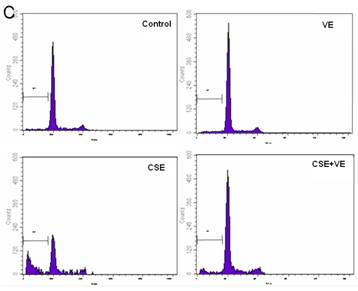
Effects of CSE on ELCs apoptosis. (A) Cell apoptosis rate was determined by the FACS analysis. CSE1: 12.5 ng/L, CSE2: 100 ng/L, CSE3: 200 ng/L, CSE4: 400 ng/L, CSE5: 70,000 ng/L as the equivalent to nicotine. The apoptosis rate of the ELCs treated with CSE with 100 or 200 ng/L nicotine significantly increased compared to that of the control group , furthermore, the apoptosis rate of the ELCs in CSE4 and CSE5 group reached 20% - 40%, showing a significant increase compared to that of the control group. * P < 0.05 vs control group, ** P < 0.01 vs control group. (B) Morphological analysis under a light microscope. The number of shed cells clearly increased in CSE2 group, and almost all cells were shed in CSE5 group. (C) The representative pictures for Cell apoptosis rate determined by the FACS analysis. A hypodiploid peak could be seen in the sub-G0/G1 region, and it reflected an apoptotic state of the cells. VE: 100 μmol/L VE treatment group, CSE: CSE (equivalent to 200 ng/L nicotine) treatment group, CSE+VE: 100 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group.

DNA damage of ELCs caused by CSE
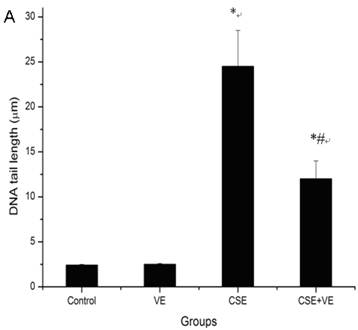
The length of the DNA comet tail in ELCs exposed to CSE (200 ng/L as the equivalent to nicotine) was significantly longer than that in the control group (P<0.05) (Figure 4 A), which was also confirmed by morphological analysis under fluorescence microscope (Figure 4 B). It is suggested that CSE at the experimental concentration was genotoxic and contributed to DNA strand breaks.
CSE-induced ELCs DNA damage and VE intervention. (A) DNA damage expressed as the length of DNA tail (μm) was determined by the comet assay. Data represented the mean ± SD of six independent experiments. * P < 0.05 vs control group, # P < 0.05 vs CSE group. (B) Morphological analysis under a fluorescence microscope (Olympus BX51). VE: 100 μmol/L VE treatment group, CSE: CSE (equivalent to 200 ng/L nicotine) treatment group, CSE+VE: 100 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group.

Preventive effects of VE on CSE-induced ELCs cytotoxicity
Preventive effects of VE on CSE-induced inhibitory effects on ELCs survival
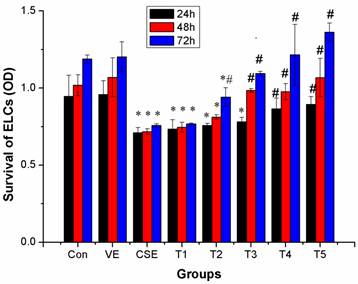
At a dose of 100 μmol/L or higher, VE could clearly prevent the inhibitory effects of CSE treatment at 24 h(P < 0.05); at doses 50 μmol/L and 25 μmol/L, VE clearly eliminated the inhibitory effect of CSE treatments at 48 h and 72 h, respectively(P < 0.05). ELCs survival in the groups treated with 25 μmol/L or higher doses of VE was significantly higher than that in the CSE group at the 72 h time point (P < 0.05). The above results suggested that VE could prevent or reverse CSE-induced inhibition in ELCs survival, indicating dose- and time-dependency of the preventive effect, however, VE alone neither promote nor inhibit ELCs survival (Figure 5).
Preventive effects of VE on CSE-induced inhibitory effects on ELCs survival (OD, mean ±SD, and n = 3). VE could prevent or reverse CSE-induced inhibition on ELCs survival, indicating dose- and time-dependency of the preventive effects. * P < 0.05 vs control group. # P < 0.05 vs CSE group. Con: control, VE: 100 μmol/L VE treatment group, CSE: CSE (equivalent to 200 ng/L nicotine) treatment group, T1: 1.0 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group, T2: 25.0 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group, T3: 50 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group, T4: 100 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group, T5: 200 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group.
Intervention of VE on CSE induced changes in cell cycle of ELCs
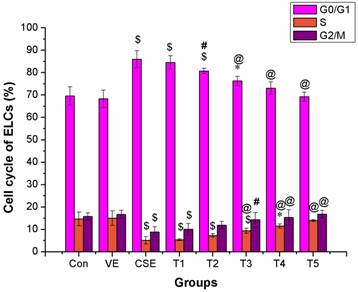
The intervention effect of VE was considerably significant at a dose of 100 μmol/L or above (P < 0.01). The proportion of ELCs in the G0/G1 phase was reversible, and showed no significant difference in 100 μmol/L or above VE treatment groups compared to that of the control group (P > 0.05). The proportion of cells in the S and G2/M phases was clearly higher in 50 μmol/L or higher VE treatment groups than that of the CSE group (P < 0.05), and showed no significant difference compared to that of the control group (P > 0.05). The above results suggested that VE could significantly prevent CSE-induced disturbance of the cell cycle of ELCs, showing a dose-dependent correlation, however, VE alone did not induce any changes in cell cycle of ELCs (Figure 6).
Intervention of VE in CSE-induced change in cell cycle of ELCs (%, mean ±SD, and n = 3). VE could significantly prevent CSE-induced disturbance of the cell cycle of ELCs, showing a dose-dependent correlation, however, VE alone did not induce any change in cell cycle of ELCs. * P < 0.05, $ P < 0.01 vs control group, # P < 0.05, @ P < 0.01 vs CSE group. Con: control, VE: 100 μmol/L VE treatment group, CSE: CSE (equivalent to 200 ng/L nicotine) treatment group, T1: 1.0 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group, T2: 25.0 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group, T3: 50 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group, T4: 100 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group, T5: 200 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group.
Preventive effects of VE on CSE-induced cell apoptosis of ELCs
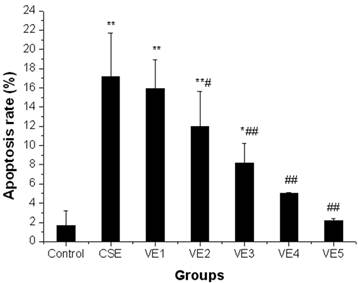
CSE-induced apoptosis of ELCs was markedly reduced with 25 μmol/L or higher doses of VE treatment (P < 0.05, P < 0.01). Moreover, the apoptosis rate of ELCs recovered to normal levels with 100 μmol/L or 200 μmol/L VE treatment (P > 0.05). The above results showed that VE could significantly prevent or reverse CSE-induced cell apoptosis; furthermore, preventive effect improved with an increase of VE dose, indicating a dose-dependent correlation (r=0.937, P<0.05), however, VE alone did not cause any changes to the cell apoptosis of ELCs (Figure 7).
Preventive effects of VE on CSE-induced cell apoptosis of ELCs. VE1, VE2, VE3, VE4 and VE5 refer to VE at the dose of 1 μmol/L, 25 μmol/L, 50 μmol/L, 100 μmol/L, 200 μmol/L, respectively. * P < 0.05, ** P < 0.01 vs control group, # P < 0.05, # # P < 0.01 vs CSE group.
Preventive effects of VE on CSE-induced DNA damage of ELCs
VE (100 μmol/L) could significantly reduce the length of the DNA comet tail of ELCs compared with that in CSE-treated group (P<0.05), suggesting that VE treatment can prevent the CSE-induced DNA damage, but VE alone did not cause any effects to the DNA damage of ELCs (Figure 4 A, B).
Involvement of the mitochondrial pathway in CSE-induced apoptosis through cytochrome c-mediated caspase activation
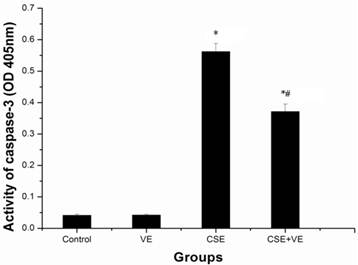
To determine whether CSE induces apoptosis via the activation of caspases, a specific peptide substrate was used to detect caspase-3 proteolytic activity. Marked caspase-3 activation was observed in ELCs after CSE (200 ng/L as the equivalent to nicotine) treatment for 30 h, as shown in Figure 8. This result demonstrated that caspase activation was involved in CSE-induced cell apoptosis. VE (100 μmol/L) intervention significantly reduced caspase-3 activity compared with that in the CSE-treated group (P< 0.05), while VE treatment alone did not cause any effects (P > 0.05).
Activation of caspase-3 by CSE and VE intervention in ELCs. Extracts of whole-cell lysate were analyzed for caspase activity. Specific substrate for caspase-3 was used, and the cleavage of the peptide was monitored at 405 nm. The data represent the mean ± SD from 3 independent experiments. * P < 0.05 vs control group, # P < 0.05 vs CSE group. VE: 100 μmol/L VE treatment group, CSE: CSE (equivalent to 200 ng/L nicotine) treatment group, CSE+VE: 100 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group.
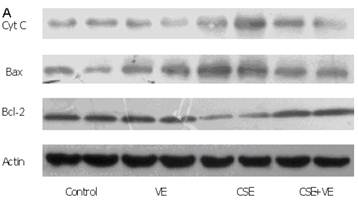
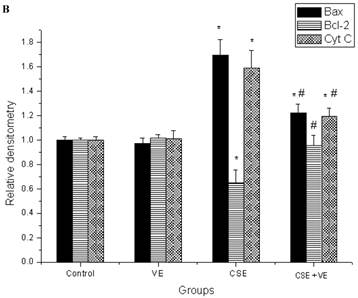
Western blotting analysis revealed an increase in cytosolic cytochrome c, which coincided with both Bax upregulation and Bcl-2 downregulation in ELCs after CSE (200 ng/L as the equivalent to nicotine) treatment for 30 h. Meanwhile, VE (100 μmol/L) intervention decreased cytosolic cytochrome c release, reduced the expression of Bax, and promoted the expression of Bcl-2 compared with that in the CSE-treated group (Figure 9). These observations indicated that the mitochondrial pathway of cytochrome c-mediated caspase activation was involved in the response of ELCs to cigarette smoke exposure.
Release of mitochondrial cytochrome c and expression of Bax and Bcl-2 in ELCs. The proteins from the cytosolic cell lysate (for cytochrome c) and whole-cell lysate (for Bax and Bcl-2) were analyzed by Western blotting. Actin was used as an internal control. The data represent the mean ± SD from 3 independent experiments. (A) The representative pictures for Western blotting in different groups. (B) Expression of proteins of the relevant apoptosis genes in ELCs. * P < 0.05 vs control group, # P < 0.05 vs CSE group. VE: 100 μmol/L VE treatment group, CSE: CSE (equivalent to 200 ng/L nicotine) treatment group, CSE+VE: 100 μmol/L VE + CSE (equivalent to 200 ng/L nicotine) treatment group.
Discussion
Cigarette smoke (CS) contains more than 4,500 chemical compounds, including high concentrations of oxygen free radicals and nitric oxide in the gas phase and organic compounds such as semiquinone radicals in the tar phase. These compounds resulted in the generation of reactive oxygen and nitrogen species [31-35]. It has been recently demonstrated that CSE contains some stable oxidants, i.e., peroxynitrite-like reactants, which cause extensive oxidative stress damage [36, 37]. Since the damage was induced by oxidative stress, antioxidants could play a major role in protection against CS-related lung damage or diseases. VE, a potent peroxyl radical scavenger, is a chain-breaking antioxidant that prevents the propagation of free radical damage in biological membranes [38], and it may have protective effects against CSE-induced cytotoxicity in the lung cells. The significance of VE has been proven as a radical chain breaking antioxidant that can protect the integrity of tissues and play an important role in life process, and it has been found to possess functions that are independent of its antioxidant/radical scavenging ability, such as inhibitory effects on protein kinase C, on the growth of certain cells and on the transcription of some genes, activation on the expression of other genes [39].
Several studies have evaluated the effects of CS on cell proliferation in lung fibroblasts, the results indicated that CSE significantly inhibited fibroblast proliferation and migration, and induced cellular senescence [14, 16]. Our results showed that CSE could produce an obvious inhibitory effect on the survival of ELCs; furthermore, this effect increases with an increase in CSE concentration and treatment time, indicating dose- and time-dependency of the inhibitory effects. VE could remarkably prevent and reverse the CSE-induced inhibitory effects on the growth of ELCs. Furthermore, at doses of 100 μmol/L or above VE treatment, cell growth recovered to normal level and was even better than that in the control group, illustrating that VE could not only prevent CSE-induced inhibitory effects on cell growth but could also significantly promote cell proliferation.
The inhibitory effects of CSE on cell proliferation and the preventive effect of VE on CSE-induced cytotoxicity might be closely correlated with the cell cycle. We found that CSE clearly disturbed the cell cycle of ELCs, produced a significant decrease in the proportion of cells in the S and G2/M phases and an obvious increase in the proportion of cells in the G0/G1 phase. It is suggested that CSE arrested the ELCs at the G0/G1 phase. Nyunoya et al [16] also drew a similar conclusion. CSE severely disturbed the normal progression of the cell cycle, which might be the mechanism underlying CSE-induced inhibition of cell proliferation and CSE-induced apoptosis; also might be the mechanism of tumor formation because it is well kwon that the disorder of cell cycle is one of the mechanisms of tumor formation [14, 16]. We found that VE clearly prevented and improved CSE-induced disturbance of the cell cycle; furthermore, the normal cell cycle was restored with 100 μmol/L VE treatment. The results illustrated that VE exhibits an obvious preventive effect on the CSE-induced disturbance of cell cycle.
Numerous studies have reported that CSE could induce death or apoptosis in different cell lines [17, 18, 40]. Liu et al reported that CSE only induced DNA damage in human bronchial epithelial cells and not cell apoptosis [19].The reason about this different observation might lie in the different CSE doses used in the experiment. Low concentrations of cigarette smoke induced DNA damage and repair without leading to apoptosis in human bronchial epithelial cells, higher concentrations of cigarette smoke, however, could induce either apoptosis or necrosis [41]. Our results showed that CSE clearly induced apoptosis in ELCs, in a dose-dependent manner, i.e., the proportion of cell apoptosis significantly increased with an increase in the CSE concentration. This result confirmed to those of other studies [42, 43]. VE could remarkably prevent and reverse CSE-induced apoptosis in a dose-dependent manner. Apoptosis was restored to normal levels with 200 μmol/L of VE treatment. This result indicated that VE had a preventive effect on CSE-induced apoptosis.
Accumulating evidence indicated that mitochondria play a pivotal role in the apoptotic process in mammalian cells [44]. Activated effector caspase-3 subsequently executed apoptosis by cleaving various cellular substrates vital for cell survival, ultimately resulting in apoptosis [45]. The caspase activation events could be initiated by the release of cytochrome c from the mitochondria into the cytosol, and this was a critical step in the death-signaling cascade [46]. It was the expression and subcellular translocation of cell death initiators or repressors such as Bax and Bcl-2 that regulated the release of cytochrome c [47, 48]. The overexpression of Bcl-2 blocked cytochrome c release in response to a variety of apoptotic stimuli [49]. In contrast, the upregulation and redistribution of Bax from the cytosol to the mitochondria could promote the release of cytochrome c [50]. The evidence from the present study confirmed that CSE promoted ELCs apoptosis and VE intervention yielded the opposite results in which the cytochrome c-mediated caspase activation mitochondrial signaling pathway was involved.
In conclusion, a primary culture of ELCs was used for the first time to observe the preventive effect of the antioxidant VE on CSE-induced cytotoxicity. CSE could clearly inhibit the survival of ELCs, disturb the cell cycle, promote cell apoptosis, and induced DNA damage. VE possessed a significant antagonistic effect on the above effects of CSE. At a certain dose, VE treatment might inhibit or reverse the cytotoxic effects of CSE on ELCs. The mechanisms underlying CSE-induced cytotoxicity and VE intervention might be associated with the mitochondrial pathway of cytochrome c-mediated caspase activation.
Acknowledgements
We thank Zu-Guo Zhao, Shi-Jie Wang, Xiang-Fei Guo and Jing Lang for assistance with preparation of experiments; Zhao-Yun Yin for assistance with data analysis.
Conflict of Interests
The authors declare no competing financial interests.
References
1. Newcomb PA, Carbone PP. The health consequences of smoking. Cancer. Med Clin North Am. 1992;76:305-31
2. Thun MJ, Henley SJ, Calle EE. Tobacco use and cancer: an epidemiologic perspective for geneticists. Oncogene. 2002;21:7307-25
3. Yang GH, Ma JM, Liu N. et al. [Smoking and passive smoking in Chinese, 2002]. Zhonghua Liu Xing Bing Xue Za Zhi. 2005;26:77-83
4. Husgafvel-Pursiainen K. Genotoxicity of environmental tobacco smoke: a review. Mutat Res. 2004;567:427-45
5. Felber Dietrich D, Schwartz J, Schindler C. et al. Effects of passive smoking on heart rate variability, heart rate and blood pressure: an observational study. Int J Epidemiol. 2007;36:834-40
6. Hawamdeh A, Kasasbeh FA, Ahmad MA. Effects of passive smoking on children's health: a review. East Mediterr Health J. 2003;9:441-7
7. Poulsen HE, Prieme H, Loft S. Role of oxidative DNA damage in cancer initiation and promotion. Eur J Cancer Prev. 1998;7:9-16
8. Prasad KN, Edwards-Prasad J. Vitamin E and cancer prevention: recent advances and future potentials. J Am Coll Nutr. 1992;11:487-500
9. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. The Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. N Engl J Med. 1994;330:1029-35
10. Woodson K, Tangrea JA, Barrett MJ. et al. Serum alpha-tocopherol and subsequent risk of lung cancer among male smokers. J Natl Cancer Inst. 1999;91:1738-43
11. Fiala E, Sohn OS, Wang CX. et al. Induction of preneoplastic lung lesions in guinea pigs by cigarette smoke inhalation and their exacerbation by high dietary levels of vitamins C and E. Carcinogenesis. 2005;26:605-12
12. Yang J, Wang L, Chen Z. et al. Antioxidant Intervention of Smoking-Induced Lung Cancer in Mice by Vitamin E and Quercetin. BMC cancer. 2008;8:383
13. Lannan S, Donaldson K, Brown D. et al. Effect of cigarette smoke and its condensates on alveolar epithelial cell injury in vitro. Am J Physiol. 1994;266:L92-100
14. Nakamura Y, Romberger DJ, Tate L. et al. Cigarette smoke inhibits lung fibroblast proliferation and chemotaxis. Am J Respir Crit Care Med. 1995;151:1497-503
15. Nakayama T, Kaneko M, Kodama M. et al. Cigarette smoke induces DNA single-strand breaks in human cells. Nature. 1985;314:462-4
16. Nyunoya T, Monick MM, Klingelhutz A. et al. Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol. 2006;35:681-8
17. Carnevali S, Petruzzelli S, Longoni B. et al. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2003;284:L955-63
18. Baglole CJ, Bushinsky SM, Garcia TM. et al. Differential induction of apoptosis by cigarette smoke extract in primary human lung fibroblast strains: implications for emphysema. Am J Physiol Lung Cell Mol Physiol. 2006;291:L19-29
19. Liu X, Conner H, Kobayashi T. et al. Cigarette smoke extract induces DNA damage but not apoptosis in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2005;33:121-9
20. Freshney RI. Culture of animal cells: A manual of basic technique. New York, USA: Wiley-Liss, Inc. 2005
21. Mather JP, Barnes D. Animal Cell Culture Methods. New York, USA: Academic Press Inc. 1998
22. Bernhard D, Huck CW, Jakschitz T. et al. Development and evaluation of an in vitro model for the analysis of cigarette smoke effects on cultured cells and tissues. J Pharmacol Toxicol Methods. 2004;50:45-51
23. Panayiotidis MI, Stabler SP, Allen RH. et al. Cigarette smoke extract increases S-adenosylmethionine and cystathionine in human lung epithelial-like (A549) cells. Chem Biol Interact. 2004;147:87-97
24. Kim H, Liu X, Kobayashi T. et al. Reversible cigarette smoke extract-induced DNA damage in human lung fibroblasts. Am J Respir Cell Mol Biol. 2004;31:483-90
25. Sen CK, Khanna S, Roy S. et al. Molecular basis of vitamin E action. Tocotrienol potently inhibits glutamate-induced pp60(c-Src) kinase activation and death of HT4 neuronal cells. J Biol Chem. 2000;275:13049-55
26. Naziroğlu M, Lückhoff A. Effects of antioxidants on calcium influx through TRPM2 channels in transfected cells activated by hydrogen peroxide. J Neurol Sci. 2008;270:152-8
27. Blasiak J, Kowalik J. A comparison of the in vitro genotoxicity of tri- and hexavalent chromium. Mutat Res. 2000;469:135-45
28. Wickenden JA, Clarke MC, Rossi AG. et al. Cigarette smoke prevents apoptosis through inhibition of caspase activation and induces necrosis. Am J Respir Cell Mol Biol. 2003;29:562-7
29. Xu XL, Wang X, Chen ZL. et al. Overexpression of Grb2-associated binder 2 in human lung cancer. Int J Biol Sci. 2011;7:497-505
30. Chen ZL, Zhang YX, Yang J. et al. Estrogen promotes benzo[a]pyrene-induced lung carcinogenesis through oxidative stress damage and cytochrome c-mediated caspase-3 activation pathways in female mice. Cancer Letters. 2011;308:14-22
31. Kinnula VL, Fattman CL, Tan RJ. et al. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med. 2005;172:417-22
32. DeMeo DL, Zanobetti A, Litonjua AA. et al. Ambient air pollution and oxygen saturation. Am J Respir Crit Care Med. 2004;170:383-7
33. Pryor WA, Prier DG, Church DF. Electron-spin resonance study of mainstream and sidestream cigarette smoke: nature of the free radicals in gas-phase smoke and in cigarette tar. Environ Health Perspect. 1983;47:345-55
34. Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111-26
35. Leanderson P, Tagesson C. Cigarette smoke-induced DNA damage in cultured human lung cells: role of hydroxyl radicals and endonuclease activation. Chem Biol Interact. 1992;81:197-208
36. Ishii T, Matsuse T, Igarashi H. et al. Tobacco smoke reduces viability in human lung fibroblasts: protective effect of glutathione S-transferase P1. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1189-95
37. Yamaguchi Y, Nasu F, Harada A. et al. Oxidants in the gas phase of cigarette smoke pass through the lung alveolar wall and raise systemic oxidative stress. J Pharmacol Sci. 2007;103:275-82
38. Traber MG, Packer L. Vitamin E: beyond antioxidant function. Am J Clin Nutr. 1995;62(Suppl 6):S1501- S1509
39. Azzi A, Stocker A. Vitamin E: non-antioxidant roles. Prog Lipid Res. 2000;39:231-55
40. Baumgartner KB, Samet JM, Coultas DB. et al. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Collaborating Centers. Am J Epidemiol. 2000;152:307-15
41. Togo S, Sugiura H, Nelson A. et al. Hepatic growth factor (HGF) inhibits cigarette smoke extract induced apoptosis in human bronchial epithelial cells. Exp Cell Res. 2010;316:3501-16
42. Bartecchi CE, Mackenzie TD, Schier RW. The human costs of tobacco use. N Engl J Med. 1994;330:907-12
43. Davies KJ, Lin SW, Pacifici RE. Protein damage and degradation by oxygen radicals. IV. Degradation of denatured protein. J Biol Chem. 1987;262:9914-20
44. Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309-12
45. Srinivasula SM, Ahmad M, Fernandes-Alnemri T. et al. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949-57
46. Luo X, Budihardjo I, Zou H. et al. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481-90
47. Pervin S, Singh R, Chaudhuri G. Nitric-oxide-induced Bax integration into the mitochondrial membrane commits MDA-MB-468 cells to apoptosis: essential role of Akt. Cancer Res. 2003;63:5470-9
48. Chinnaiyan AM, Orth K, O'Rourke K. et al. Molecular ordering of the cell death pathway. Bcl-2 and Bcl-xL function upstream of the CED-3-like apoptotic proteases. J Biol Chem. 1996;271:4573-6
49. Budihardjo I, Oliver H, Lutter M. et al. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269-90
50. Esposti MD. The roles of Bid. Apoptosis. 2002;7:433-40
Author contact
![]() Corresponding author: Jun-Wen Li, Tel:+86-22-84655345; fax: +86-22-23328809, E-mail address: junwen9999com.
Corresponding author: Jun-Wen Li, Tel:+86-22-84655345; fax: +86-22-23328809, E-mail address: junwen9999com.