Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(7):1037-1044. doi:10.7150/ijbs.7.1037 This issue Cite
Research Paper
Development and Initial Characterization of a HAPPY Panel for Mapping the X. Tropicalis Genome
Zhihua Jiang1, ![]() , Jennifer J. Michal1, Kenneth B. Beckman2, Jessica B. Lyons3, Ming Zhang1, Zengxiang Pan1, Daniel S. Rokhsar3, Richard M. Harland3,
, Jennifer J. Michal1, Kenneth B. Beckman2, Jessica B. Lyons3, Ming Zhang1, Zengxiang Pan1, Daniel S. Rokhsar3, Richard M. Harland3, ![]()
1. Department of Animal Sciences, Washington State University, Pullman, WA 99164-6351, USA;
2. Biomedical Genomics Center, University of Minnesota, Minneapolis, MN 55455, USA;
3. Department of Molecular & Cell Biology, University of California Berkeley, Berkeley, CA 94720-3200, USA.
Received 2011-6-20; Accepted 2011-8-13; Published 2011-8-24
Abstract
HAPPY mapping was designed to pursue the analysis of approximately random HAPloid DNA breakage samples using the PolYmerase chain reaction for mapping genomes. In the present study, we improved the method and integrated two other molecular techniques into the process: whole genome amplification and the Sequenom SNP (single nucleotide polymorphism) genotyping assay in order to facilitate whole genome mapping of X. tropicalis. The former technique amplified enough DNA materials to genotype a large number of markers, while the latter allowed for relatively high throughput marker genotyping with multiplex assays on the HAPPY lines. A total of 58 X. tropicalis genes were genotyped on an initial panel of 383 HAPPY lines, which contributed to formation of a working panel of 146 lines. Further genotyping of 29 markers on the working panel led to construction of a HAPPY map for the X. tropicalis genome. We believe that our improved HAPPY method described in the present study has paved the way for the community to map different genomes with a simple, but powerful approach.
Keywords: HAPPY mapping, whole genome amplification, multiplex genotyping assay, mapping X. tropicalis genome.
Introduction
HAPPY mapping, the analysis of approximately HAPloid DNA samples using the PolYmerase chain reaction, is a genome mapping method based on random DNA breakage and determination of linkage [1-2]. This approach is essentially analogous to classical linkage mapping, but the chromosome breakage and segregation are generated by in vitro analogues with gamma-irradiation or shearing. Genes/markers are then segregated by diluting the resulting fragments to give aliquots that contain approximately 1 haploid genome equivalent. Compared to other conventional genome mapping methods, this HAPPY approach possesses several advantages. First, it allows construction of gene/marker maps without cloning, thus avoiding many potential errors and artifacts [3]. Second, the approach can also be easily adapted to any desired level of resolution, in particular, to a high resolution of genome maps [4]. Third, unlike the radiation hybrid mapping approach, a HAPPY panel contains no carrier DNA, which eases specific PCR amplification of markers and makes multiplexing more amenable. Lastly, HAPPY mapping does not require any polymorphic markers so any piece of DNA can be mapped to a genome region. Therefore, the HAPPY mapping approach is applicable to all species, from human [5], to plant [6] and even to unicellular eukaryotes [7].
However, such a simple and powerful HAPPY mapping method has not yet come into general use even though it was developed by Dear and Cook as early as 1989. Up to date, only eight maps have been generated using the HAPPY approach and all of them were contributed by the inventors' group [1-8]. The bottleneck of the method as far as we can see, is the lack of faithful amplification of the whole DNA that provides enough material for genotyping a large number of markers. It seems that this problem should be now overcome by using a well-developed whole genome amplification method, termed multiple displacement amplification (MDA) [9]. MDA can yield about 20 - 30 ug of product from as few as 1 - 10 copies of genomic DNA. In comparison to other whole genome amplification methods, MDA provides the most reliable genotypes, highest call rates, best genomic coverage, and lowest amplification bias [10]. Therefore, improving the HAPPY approach by solving the bottleneck will help promote its application in genome mapping of many species.
Amphibians have been used since the 19th century as vertebrate models for investigating many important aspects of biological sciences [11]. In particular, the study of amphibian embryogenesis has provided important insight into the mechanisms of vertebrate development [12]. In order to meet the Xenopus research community's needs, the National Institutes of Health Xenopus Initiative is supporting the development of genetic and genomic resources, such as (1) complementary deoxyribonucleic acid (cDNA) libraries and expressed sequence tag (EST) sequences, (2) UniGene clusters, (3) full-insert cDNA sequences, (4) a genetic map, (5) genomic libraries, (6) a physical map, (7) genome sequence, (8) microarrays, (9) mutagenesis and phenotyping, and (10) bioinformatics [13-14]. While genomic resources for X. tropicalis have advanced significantly in recent years as described above, tough challenges lie ahead, especially with respect to high-quality assembly of the whole genome for the species. The Department of Energy's Joint Genome Institute produced about 1.33 Gbp of high quality DNA sequences using a seventh-generation inbred Nigerian female [15], but these sequences need to be accurately ordered on 10 chromosomes. Here we present our pilot study to demonstrate the feasibility of the HAPPY approach for construction of whole genome maps as reference for a high quality chromosome-based long-range assembly in X. tropicalis.
Materials and Methods
Animals, Blood Sampling and Cell Preparation. Development of a HAPPY panel for genome mapping of X. tropicalis was carried out by following a protocol developed by Dear and colleagues [5] for construction of a high resolution metric HAPPY map of human chromosome 14, but with modifications. A blood sample from an inbred F10 Nigerian X. tropicalis animal was collected into 0.9X SSC (sodium chloride-sodium citrate buffer) on ice. After collection, the blood cells were resuspended by inverting the tube, and counted using a "Bright Line" Hemocytometer (Hausser Scientific, Horsham, PA). The suspension was then centrifuged at 453 x g for 3 minutes at 4ºC in a tabletop centrifuge, the supernatant was poured off, and the cells were resuspended in PBSG (Phosphate-Buffered Saline + 1% glucose) in aliquots at 6 x 105 cells/mL and 5 x 106 cells/mL, the latter being for high-density controls. Each cell suspension was mixed 1:1 with PBSG + 2% agarose (kept at 37ºC in a water bath), for final concentrations of 3 x 105 cells/mL and 2.5 x 106 cells/mL, respectively, in PBSG + 1% agarose. These mixtures were taken up into approximately 46 or 20 (high density controls) 100 μL calibrated glass pipets (VWR International) and cooled to 4ºC. Each set of “agarose strings” was allowed to fall by gravity into 150 mL of lysis solution (10 mM Tris-Cl, 1mM EDTA, 1% lithium dodecyl sulfate, pH 7.5) and incubated on a rotator at 4ºC. The lysis solution was replaced after intervals of 15 minutes, 30 minutes, 3 hourly intervals, and overnight, and the strings were stored in lysis buffer at 4ºC until use.
Pulsed Field Electrophoresis and DNA Fragmentation. The DNA contained in the cell-agarose strings was separated in a 0.8% chromosomal grade agarose gel under 1X TAE buffer. The cell-agarose strings were cut to fit the length of an electrophoresis well with a razor blade, placed into the well with a spatula and pressed to front of the well such that less than 90% of the height of the well was occupied. The sample plugs were finally sealed into the well with 0.8% low melt agarose in 1X TAE. The electrophoresis was performed using CHEF-DR III Pulsed Field Electrophoresis System (Bio-Rad, Hercules, CA) using S. pombe chromosomes (New England Biolabs, Ipswich, MA) as molecular ladder. The gel was run at 14oC and 3V cm-1 in three blocks: Block 1) 24h with a 96o reorientation angle and 1200 sec switch time; Block 2) 24h with a 100o reorientation angle and 1500 sec switch time; and Block 3) 24h with a 106o reorientation angle and 1800 sec switch time. After electrophoresis was completed, the sides of gel containing yeast standards and high concentration of DNA fragments were excised and stained with ethidium bromide and visualized with UV light. The gel was then placed in a DNA-free environment. A total of 383 plugs were collected with capillary tubes across the gel lane at sizes ranging from ~0.5 Mb to ~6 Mb. In addition, 7 plugs of agarose were also collected from locations outside of the running lane and were used as controls.
Whole Genome Amplification (WGA) and DNA Measurement. Each plug was then transferred to an individual PCR tube and the first round of WGA was performed using the illustraTM GenomiPhi V2 DNA Amplification Kit (GE Healthcare Lifesciences, Piscataway, NJ). Briefly, 9 µL of sample buffer was added to each agarose plug and DNA was denatured by heating to 95oC for 3 min, followed by cooling to 4oC on ice. Next, 9 µL of reaction buffer was mixed with 1 µL of enzyme mix on ice and added to the cooled sample. Amplification was performed by incubating the sample at 30oC for 2 h, after which time the reaction was terminated by heating to 65oC for 10 min, followed by cooling to 4oC. The second round of WGA was accomplished by taking 1 µl of amplified DNA from the first WGA round and repeating each step as described above. Samples from the second round of WGA were purified by ethanol precipitation with 1.5 M sodium acetate (pH >8)/250 mM EDTA buffer. Purified DNA pellets were resuspended in TE and quantity and quality determined with a Nanodrop spectrophotometer.
HAPPY Panel Characterization. These amplified DNA HAPPY lines were tested for a total of 58 X. tropicalis genes, which have the human orthologs representing all autosomes and X chromosome (see Supplementary Material: Table S1). Putative single nucleotide polymorphisms (SNPs) were identified for each frog gene based on comparison between cDNA and genomic DNA sequences and only one of these sequences were selected to genotype these 383 HAPPY lines using the Sequenom genotyping assay (see Supplementary Material: Table S2). Four frog original whole genome DNA samples served as positive controls and four blanks were used as negative controls. The genotype scores were converted to 1 when there was a call for a genotype or 0 when there was no call for a genotype and the data were then used for a pair-wise similarity analysis. The genotyping data of 58 markers were considered as character string variables, so there are 58 characters for each HAPPY line. The similarity between two character strings was calculated using a VBA (visual basic for application) program by considering both character and the order of the characters in strings. We calculated the similarity in a 383×383 matrix for all these samples. Only the HAPPY lines that were <0.85 similar to others were selected to form a 146-sample working panel.
HAPPY Map Construction: a Pilot Study. A megaBLAST program was used to identify ultraconserved elements (UCEs) between frog scaffolds (v5.1) and the human chromosomes. We selected a region from 0 Mb to ~10 Mb on human chromosome 1 as our target to test the feasibility of the HAPPY panel for map construction. Several pseudo-SNPs were randomly assigned to each of 48 selected UCEs and the Sequenom assay designed picked 29 of them for multiplex genotyping (see Supplementary Material: Table S2). Like the conventional radiation hybrid (RH) mapping, we genotyped the same set of markers twice on the 146-sample working frog HAPPY panel with 5 ng of DNA as template. We scored the genotypes by two different methods. In a simple approach, we assigned “1” or “0” to cases where the call rate of replicates was either = 100% or 0%, while “?” was assigned to cases where discordance of call/no call occurred. The heuristic approach was based on call rate, mean yield and penalty. If call rate = 100% and penalty/yield/skew were acceptable, then “1” was assigned as genotype present. If call rate ≥ 50% and best penalty/best yield were acceptable, then “?” was assigned. The rest of the cases were assigned “0” as absent. The RHMAP 3.0 program was used to construct maps as described previously [16]. Information on markers used in the map construction is presented in Table 1.
UCE marker ID, retention rate (RR) and their locations on human chromosome 1 (HSA1) and X. tropicalis scaffolds (XTS).
| UCE ID | RR | UCE size | Identity % | Build 37.1 | V7.1 | V5.1 | |||
|---|---|---|---|---|---|---|---|---|---|
| HSA1 | Location | XTS | Location | XTS | Location | ||||
| Linkage group 1 | |||||||||
| S296794 | 19.4 | 83 | 93 | 1 | 955673 | 7 | 82896424 | 296 | 764590 |
| S57859 | 15.1 | 147 | 80 | 1 | 1248188 | 7 | 89448293 | 57 | 2338758 |
| S57871 | 14.9 | 125 | 80 | 1 | 1479240 | 7 | 83744355 | 57 | 3039740 |
| S160389 | 18.2 | 93 | 83 | 1 | 1956955 | 7 | 84645195 | 160 | 5912 |
| S160377 | 23.5 | 612 | 85 | 1 | 2160467 | 7 | 84947775 | 160 | 316024 |
| S160391 | 12.5 | 67 | 86 | 1 | 2319713 | 7 | 85619891 | 160 | 995945 |
| S342347 | 15 | 63 | 87 | 1 | 2525313 | 7 | 97677337 | 342 | 252045 |
| S342315 | 18.6 | 388 | 80 | 1 | 3028645 | 7 | 97136469 | 342 | 796885 |
| S119425 | 20 | 129 | 84 | 1 | 3563352 | 7 | 96381276 | 119 | 1872405 |
| S266298 | 18.3 | 133 | 81 | 1 | 6196795 | 7 | 93520548 | 266 | 956203 |
| S830158 | 17.2 | 61 | 88 | 1 | 6206330 | 7 | 80088486 | 830 | 178118 |
| S119420 | 15.5 | 62 | 87 | 1 | 6642302 | 7 | 94535051 | 119 | 52808 |
| S119391 | 15.4 | 200 | 82 | 1 | 6681468 | 7 | 94592957 | 119 | 111777 |
| S119371 | 22.9 | 257 | 83 | 1 | 7394747 | 7 | 95439127 | 119 | 917928 |
| S119414 | 14 | 113 | 82 | 1 | 7863756 | 7 | 96208705 | 119 | 1696764 |
| S341348 | 13.5 | 68 | 86 | 1 | 8029469 | 7 | 91619174 | 341 | 758071 |
| S341342 | 14.4 | 58 | 91 | 1 | 8398055 | 7 | 91300336 | 341 | 435537 |
| S689021 | 13.6 | 115 | 86 | 1 | 8845371 | 7 | 81804456 | 689 | 543407 |
| S160396 | 12 | 208 | 85 | 1 | 8926539 | 7 | 81748522 | 160 | 1936586 |
| S160400 | 20.6 | 152 | 82 | 1 | 9416567 | 7 | 86308632 | 160 | 1688790 |
| Linkage group 2 | |||||||||
| S34181 | 15.8 | 118 | 84 | 1 | 3327945 | 5 | 16805411 | 34 | 199580 |
| Not used | |||||||||
| S689042 | 8.3 | 126 | 86 | 1 | 860208 | 7 | 82198263 | 689 | 139505 |
| S182677 | 9 | 108 | 84 | 1 | 935066 | 5 | 11303509 | 182 | 71880 |
| S119431 | 4.1 | 48 | 89 | 1 | 3656842 | 7 | 96262590 | 119 | 1751374 |
| S266312 | 0 | 89 | 83 | 1 | 6100707 | 7 | 93375598 | 266 | 1102680 |
| S694108 | 82.4 | 71 | 87 | 1 | 6206723 | 3a | 4104079 | 694 | 573375 |
| S266308 | 2.1 | 54 | 88 | 1 | 6601968 | 7 | 94427876 | 266 | 58320 |
| S119378 | 6.9 | 132 | 88 | 1 | 7730968 | 7 | 95989531 | 119 | 1475040 |
| S1536311 | 5.5 | 71 | 87 | 1 | 7797086 | 230 | 6128 | 1536 | 14718 |
Results and Discussion
DNA Yields Amplified by WGA. The DNA plugs sampled from the pulsed field electrophoresis gel were the starting materials used in the preparation of the frog HAPPY panel. Based on the S. pombe chromosome standard (New England Biolabs, Ipswich, MA), we were able to collect a total of 383 DNA plugs of various sizes, including 56 plugs ranging from 0.5 Mb to 1 Mb, 71 from 1 Mb to 2 Mb, 48 from 2 Mb to 3 Mb, 71 from 3 Mb to 4 Mb, 32 from 4 Mb to 5 Mb and 8 from 5 Mb to 6 Mb, respectively. In addition, 97 were randomly sampled with sizes of less than 3 Mb. In order to provide enough DNA for the community to map the X. tropicalis genome later, we decided to keep our first round of WGA products as stock. Only 1 µl of the stock was used for a second round of WGA as described above. The second round of WGA was performed on all 383 HAPPY lines, which produced an average of 64.26 ng DNA/µl, varying from 13.21 ng DNA/µl to 133.37 ng DNA/µl. Since the total reaction volume was 20 µl, WGA resulted in a total average yield of 1,285 ng DNA per HAPPY line.
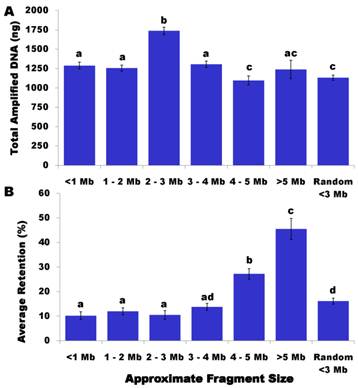
Others have reported higher yields of DNA after WGA. For example, Balogh et al. [17] observed an average yield of 225-350 ng/ µl with 50-500 pg of input DNA after using the GenomiPhi Amplification Kit (Amersham Biosciences). Several reasons might explain the relatively low WGA yield in our present study. First, we did not carry out gamma-irradiation or ultrasonic shearing to break chromosomes into fragments. Chromosome breakage might be only induced during lysis and pulsed field electrophoresis, thus limiting the amount of DNA released from the strings and causing low DNA flows in the gel. This might also provide initial evidence to support that each HAPPY line contains a random subset of frog genome, rather than whole genome content. Second, we used only 1 µl of DNA from the first round of WGA and proceeded to the second round of WGA. As such, we didn't really know how much DNA was input - it could be much lower than 50 pg. Lastly, we used ethanol precipitation to purify WGA products, which may have resulted in loss of some amplified DNA during the purification process. As shown in Figure 1A, HAPPY lines sampled between 2 - 3 Mb in size yielded an average 1,737 ng/line, which was the highest (P<0.05) amount of amplified DNA produced, followed by sample sizes of 3 - 4 Mb (1,307 ng/line), <1 Mb (1,289 ng/line) and 1 - 2 Mb (1,256 ng/line). The HAPPY lines sampled at 4 - 5 Mb and random <3 Mb in size gave the lowest (P<0.05) yields of WGA DNA with 1,097 ng/line and 1,132 ng/line, respectively. Samples collected between 5 - 6 Mb resulted in an average of 1,239 ng WGA DNA per line.
Effects of sampled fragment sizes on WGA yields (A) and marker retention rate (B). Data are presented as least square means ± standard error. The bars without common letters are significantly different (P<0.05).
The WGA procedure was also explored in RH panel amplification [18]. The authors performed two separate WGA amplifications of 10 ng of DNA each of one clone of their canine panel RHDF5000 and analyzed the presence in the amplified DNA of 74 markers known to be present and 18 known to be absent from the original DNA. Among 74 positive markers, the authors found 73 present in one of the amplified DNA samples, while none of the 18 negative markers were detected. In the second amplified DNA sample, the 74 positive markers were all present, whereas all of the 18 negative markers were absent. This indicated that WGA is a reliable method. The team also observed no loss of markers upon two successive amplifications. As such, we feel confident to claim that the second round of WGA on our HAPPY panel would not generate any significant loss or bias from the original round of WGA amplifications.
Marker Retention Rate. A total of 58 gene markers were genotyped on these 383 HAPPY lines using the Sequenom assay in two multiplex sets. As shown in Supplementary Material: Table S1, most of these genes are well distributed among human autosomes and X chromosome and are more than 10 Mb apart, except for two genes on human chromosome 14, three genes on 17 and two genes on X that are less than 6 Mb apart. Fortunately, we were able to find orthologs of all of these human genes in the newest assembly of the X. tropicalis genome (v7.1), including 12 on scaffold 1, 9 on scaffold 2, 7 on scaffold 6, 5 on scaffold 4, 4 each on scaffolds 5, 7, 8 and 9, 3 on scaffold 3, and only one each on scaffolds 10, 35, 83, 181, 532 and 656, respectively. The scaffolds 1 to 10 on the newest assembly are very likely to represent 10 chromosomes in X. tropicalis. Only two genes (PNN and MGA) had a physical distance of less than 0.5 Mb, while the remaining genes are separated by at least 3 Mb up to 74 Mb (Supplementary Material: Table S1).
The HAPPY retention rate was calculated as the percentage of markers per line. Among 383 HAPPY lines genotyped on these 58 markers, only one line had a marker retention rate of 0%. For the remainder of the HAPPY lines, the rate varied from 1.72% to 87.93%, but averaged 14.90%. A total of 157 HAPPY lines had a retention rate of <10% and 20 samples had a retention rate of >40%. As shown in Figure 1B, the sampled fragment size had a significant effect on the marker retention rate. In particular, the HAPPY lines sampled with fragments of 4 - 5 Mb in length had a more than double the marker retention rate (27.2%) compared to lines less than 3 Mb in size (10.2% to 12%) (P<0.05) and nearly twice the figure compared to lines with 3 - 4 Mb in size (13.8%) (P<0.05). The marker retention rate further increased to 45.5% when the HAPPY lines were sampled with fragments ranging from 5Mb to 6 Mb in size (Figure 1B) (P<0.05).
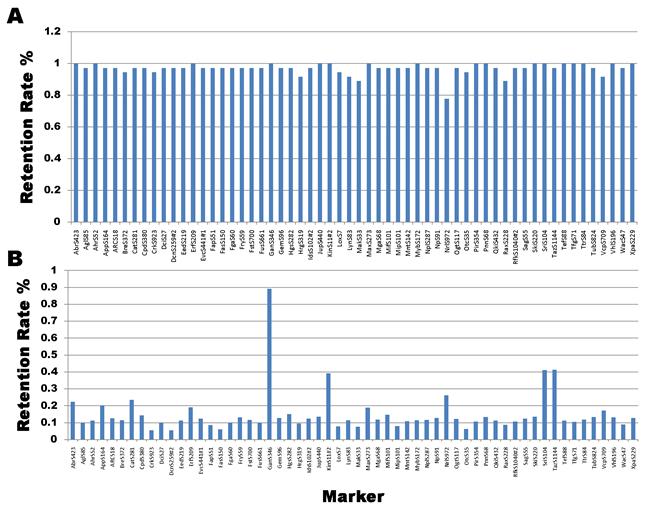
Marker retention rate remained very high (over 96%) for most of the 58 markers genotyped on four frog positive controls, because they represent whole genome DNA (Figure 2A). In contrast, 51 of these 58 genes had a <20% call rate in the HAPPY lines (Figure 2B). The remaining 7 genes include one close to 90% (GanS345) and six between 20% and 45% (Abrs423, CatS372, KinS11#2, NrlS972, Sri104 and TazS1144). The overall retention rate was 21.7% for all 58 genes in our original HAPPY panel. Hukriede and colleagues [19] formed a total of 93 RH lines to map the zebrafish genome. Among them, 81 were derived from a 5,000-rad irradiation dose and 12 from a 4,000-rad dose. After genotyping a total of 1,055 markers on the RH panel, the authors observed an overall retention rate of ≈22%. Therefore, we would speculate that our HAPPY panel would be equivalent to a RH panel with a 5,000-rad irradiation dose.
Marker retention rate on whole genome DNA samples (A) and HAPPY lines (B).
Based on the genotyping characters (1 as presence or 0 as absence) of 58 markers, we calculated the marker similarity among 383 HAPPY lines using a VBA (visual basic for application) program, which considers both characters and their orders between a pair of strings/HAPPY lines. We then selected 146 lines that were <0.85 similar to others and formed a working panel assuming that they randomly represent subsets of the frog whole genome. In order to have enough DNA for mapping a large number of markers on the HAPPY working panel, DNA amplification was achieved again using WGA with the illustraTM GenomiPhi V2 DNA Amplification Kit (GE Healthcare Lifesciences, Piscataway, NJ). Briefly, 2 individual reactions each with 1µl of stock DNA were separately amplified following the manufacturer's instructions. After amplification, products were pooled in order to balance any bias that could have occurred during the amplification process. These pooled samples were subsequently purified with the DNA Clean & Concentrator kit (Zymo Research, Irvine, CA). The frog working panel of 146 lines was then used in construction of a pilot HAPPY map in the present pilot study described below.
HAPPY Mapping Using UCE Markers. Ultraconserved elements (UCEs) are nucleotide sequences that show extreme evolutionary conservation between two or more distinct species. In order to identify UCEs in the current X. tropicalis genome assembly, we used the human genome as our reference. The human build 37.1 contains 219 contigs (including 29 unassigned in the assembly), while frog v5.1 consists of 4,297 assembled scaffolds. As such, a local MegaBLAST was set up to run each of these 4,297 frog scaffolds against all human 22 autosome, X and Y chromosome assemblies. We used the “aligned length x sequence similarity (%) = 25” as an arbitrary cut-off score for collecting the UCEs between human and frog, which requires at least an aligned length of 25 bp with 100% identity. After removal of UCE repeats, 51,498 putative UCE orthologs were established between human and frog (data not shown). We selected 29 UCEs that span a region of 860,330 - 9,416,416 bp on human chromosome 1 as markers for this pilot study to test the feasibility of our working HAPPY panel for map construction of X. tropicalis genome. The locations of UCE markers on human chromosome 1 as well as on frog scaffolds are listed in Table 1.
These 29 UCE markers were genotyped on the working panel of 146 HAPPY lines with one multiplex set using the Sequenom assay. Among them, seven markers had a retention rate of <10% and one marker reached a retention rate of > 80% (Table 1). As such, they were excluded from further analysis. Therefore, the remaining 21 UCE markers were used for map construction. In frog assembly v5.1, these 21 UCEs were distributed in scaffolds 34, 57, 119, 160, 266, 296, 341, 342, 689 and 830, respectively (Table 1). Statistical analysis using the RH2PT program of the RHMAP 3.0 package [20] assigned these 21 UCE markers to 2 linkage groups on the basis of a two-locus LOD score of at least 4.0, including 20 markers in group 1 and one marker in group 2. Just recently, we also finished identification of UCEs in the frog assembly v7.1. Interestingly enough, these UCE markers in group 1 were assigned to scaffold 7, while the marker in group 2 was assigned to scaffold 5 (Table 1). The results indicate that our HAPPY assignment of markers supports the newest assembly of X. tropicalis genome v7.1, which integrated 9 scaffolds in v5.1 into a large scaffold in v7.1.
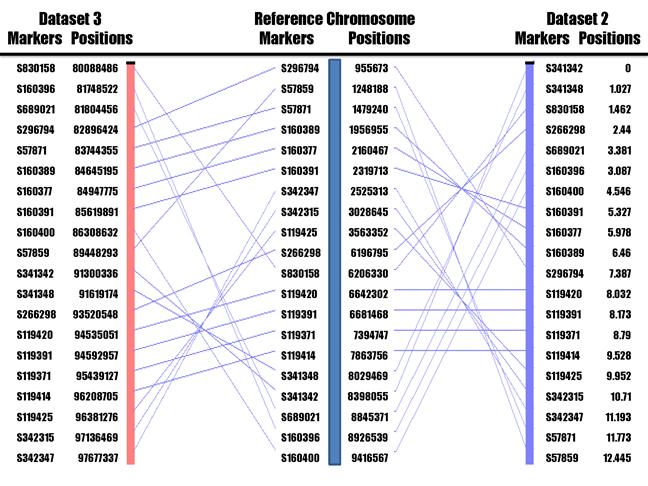
The target region between human chromosome 1 and frog scaffold 7 rearranged during evolution based on these 20 UCE markers linked in group 1 (Figure 3). Genome assembly comparison of marker locations and orders revealed ten putative conserved segments between human (Build 37.1) and frog (v7.1) in the region. In the present study, a conserved segment is defined as a genome region in which UCE content and order are parallel, either in the same or in the opposite orientation between frog and human. Using the RHMAXLIK program of the RHMAP 3.0 package [20], we ordered the same set of 20 UCE markers into a first frog HAPPY map with a total length of 12.45 centihaps (cH) (Figure 3), which should be equivalent to centiRays (cR) in radiation hybrid mapping. Interestingly, alignment of our frog HAPPY map with the human genome assembly found only eight tentative conserved segments between them instead of ten identified above based on the comparison of genome assemblies. We believed that our HAPPY map might have resulted in superior marker order, because they are consistent with the locations on each scaffold assembled in v5.1 (Table 1 and Figure 3). Figure 3 was drawn using the AutoGRAPH program with modification [21].
UCE marker locations and orders on human chromosome 1 (Reference chromosome) as compared to their locations and orders on our frog HAPPY map (Dataset 2) and frog genome assembly v7.1 (Dataset 3).
In summary, the whole genome amplification technique and the Sequenom SNP genotyping assay further improved the conventional HAPPY method and enhanced its capability to map various genomes. The former technique amplified enough DNA materials for genotyping a large number of markers, while the latter allowed for relatively high throughput marker genotyping with multiplex assays on the HAPPY lines. Such an improved HAPPY technique made it possible for us to develop a first HAPPY panel of the X. tropicalis genome and establish a pilot HAPPY map for the species. In the near future, we anticipate that genotyping of markers on a HAPPY panel should be replaced by next generation sequencing, contributing to formation of HAPPY pipeline for whole genome sequencing, mapping and assembly [22].
Supplementary Material
Table S1: Genes selected for genotyping on the X. tropicalis original HAPPY panel. Table S2: Putative SNPs and their flanking sequences of the gene markers genotyped on the original HAPPY panel of X. tropicalis.Table S3. Pseudo-SNPs and their flanking sequences of UCE markers genotyped on the working HAPPY panels of X. tropicalis.
Acknowledgements
This work was supported by NIH/NIGMS grant 1R01GM086321-01 to R.M.H. and D.S.R. with a subaward to Z.J.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Dear PH, Cook PR. Happy mapping: a proposal for linkage mapping the human genome. Nucleic Acids Res. 1989;17:6795-807
2. Dear PH, Cook PR. Happy mapping: linkage mapping using a physical analogue of meiosis. Nucleic Acids Res. 1993;21:13-20
3. Piper MB, Bankier AT, Dear PH. A HAPPY map of Cryptosporidium parvum. Genome Res. 1998;8:1299-1307
4. Konfortov BA, Cohen HM, Bankier AT, Dear PH. A high-resolution HAPPY map of Dictyostelium discoideum chromosome 6. Genome Res. 2000;10:1737-1742
5. Dear PH, Bankier AT, Piper MB. A high-resolution metric HAPPY map of human chromosome 14. Genomics. 1998;48:232-41
6. Thangavelu M, James AB, Bankier A. et al. HAPPY mapping in a plant genome: reconstruction and analysis of a high-resolution physical map of a 1.9 Mbp region of Arabidopsis thaliana chromosome 4. Plant Biotechnol J. 2003;1:23-31
7. Hamilton EP, Dear PH, Rowland T. et al. Use of HAPPY mapping for the higher order assembly of the Tetrahymena genome. Genomics. 2006;88:443-51
8. Walter G, Tomlinson IM, Cook GP. et al. HAPPY mapping of a YAC reveals alternative haplotypes in the human immunoglobulin VH locus. Nucleic Acids Res. 1993;21:4524-9
9. Dean FB, Hosono S, Fang L. et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99:5261-6
10. Lovmar L, Syvänen AC. Multiple displacement amplification to create a long-lasting source of DNA for genetic studies. Hum Mutat. 2006;27:603-614
11. Pollet N, Mazabraud A. Insights from Xenopus genomes. Genome Dyn. 2006;2:138-53
12. Yergeau DA, Mead PE. Manipulating the Xenopus genome with transposable elements. Genome Biol. 2007;8(Suppl 1):S11
13. Klein SL, Strausberg RL, Wagner L. et al. Genetic and genomic tools for Xenopus research: The NIH Xenopus initiative. Dev Dyn. 2002;225:384-91
14. Klein SL, Gerhard DS, Wagner L. et al. Resources for genetic and genomic studies of Xenopus. Methods Mol Biol. 2006;322:1-16
15. Hellsten U, Harland RM, Gilchrist MJ. et al. The genome of the Western clawed frog Xenopus tropicalis. Science. 2010;328:633-6
16. Jiang Z, He H, Hamasima N. et al. Comparative mapping of Homo sapiens chromosome 4 (HSA4) and Sus scrofa chromosome 8 (SSC8) using orthologous genes representing different cytogenetic bands as landmarks. Genome. 2002;45:147-56
17. Balogh MK, Børsting C, Sa´nchez Diz P. et al. Application of whole genome amplification for forensic analysis. International Congress Series. 2006;1288:725- 727
18. Senger F, Priat C, Hitte C. et al. The first radiation hybrid map of a perch-like fish: the gilthead seabream (Sparus aurata L). Genomics. 2006;87:793-800
19. Hukriede NA, Joly L, Tsang M. et al. Radiation hybrid mapping of the zebrafish genome. Proc Natl Acad Sci U S A. 1999;96:9745-50
20. Boehnke M, Lange K, Cox DR. Statistical methods for multipoint radiation hybrid mapping. Am J Hum Genet. 1991;49:1174-88
21. Derrien T, André C, Galibert F, Hitte C. AutoGRAPH: an interactive web server for automating and visualizing comparative genome maps. Bioinformatics. 2007;23:498-499
22. Jiang Z, Rokhsar DS, Harland RM. Old can be new again: HAPPY whole genome sequencing, mapping and assembly. Int J Biol Sci. 2009;5:298-303
Author contact
![]() Corresponding author: Dr. Zhihua Jiang (jiangzedu) or Dr. Richard M. Harland (harlandedu).
Corresponding author: Dr. Zhihua Jiang (jiangzedu) or Dr. Richard M. Harland (harlandedu).