Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(7):1045-1055. doi:10.7150/ijbs.7.1045 This issue Cite
Research Paper
Repertoire of Porcine MicroRNAs in Adult Ovary and Testis by Deep Sequencing
Mingzhou Li1*, Yingkai Liu1*, Tao Wang1, Jiuqiang Guan1, Zonggang Luo1, Haosi Chen2, Xin Wang2, Lei Chen3, Jideng Ma1, Zhiping Mu1, An-an Jiang1, Li Zhu1, Qiulei Lang4, Xiaochuan Zhou4, Jinyong Wang3, Wenxian Zeng5, Ning Li6, Kui Li7, Xiaolian Gao2 ![]() , Xuewei Li1
, Xuewei Li1 ![]()
1. Institute of Animal Genetics & Breeding, College of Animal Science & Technology, Sichuan Agricultural University, Ya'an, Sichuan, China;
2. Department of Biology & Biochemistry, University of Houston, Houston, Texas, USA;
3. Chongqing Academy of Animal Science, Chongqing, China;
4. LC Sciences, Houston, Texas, USA;
5. College of Animal Science & Technology, Northwest A & F University, Yangling, Shaanxi, China;
6. State Key Laboratory of Agrobiotechnology, China Agricultural University, Beijing, China;
7. Institute of Animal Science, Chinese Academy of Agricultural Sciences, Beijing, China.
*These authors contributed equally to this work.
Received 2011-6-3; Accepted 2011-8-9; Published 2011-9-1
Abstract
Background: MicroRNAs (miRNAs), a large family of short endogenous RNAs known to post-transcriptionally repress gene expression, participate in the regulation of almost every cellular process. Changes in miRNA expression are associated with many pathologies. Ovarian folliculogenesis and testicular spermatogenesis are complex and coordinated biological processes, in which tightly regulated expression and interaction of a multitude of genes could be regulated by these miRNAs. Identification and preliminary characterization of gonad-specific miRNAs would be a prerequisite for a thorough understanding of the role that miRNA-mediated posttranscriptional gene regulation plays in mammalian reproduction.
Method: Here, we present the identification of a repertoire of porcine miRNAs in adult ovary and testis using deep sequencing technology. A bioinformatics pipeline was developed to distinguish authentic mature miRNA sequences from other classes of small RNAs represented in the sequencing data.
Results: Using this approach, we detected 582 precursor hairpins (pre-miRNAs) encoding for 732 mature miRNAs, of which 673 are unique. Statistically, 224 unique miRNAs (out of 673, 33.28%) were identified which had significant differential expression (DE) between ovary and testis libraries (P < 0.001). Most of DE miRNAs located on the X chromosome (X-linked miRNAs) (24 out of 34, 70.59%) significantly up-regulated in ovary versus testis (P < 0.001). Predictably, X-linked miRNAs are expressed in a testis-preferential or testis-specific pattern. To explore the potential for co-expression among genomic location clusters of X-linked miRNAs, we surveyed the relationship between the distance separating miRNA loci and the coordinate expression patterns of 32 high confidence X-linked miRNAs in seven normal pig tissues using the real-time quantitative PCR (q-PCR) approach. Our results show that proximal pairs of miRNAs are generally co-expressed implying that miRNAs within 50 kb of genomic bases are typically derived from a common transcript.
Conclusions: The present study characterizes the miRNA transcriptome of adult porcine gonads, with an emphasis on the co-expression patterns of X-linked miRNAs. Our report should facilitate studies of the organ-specific reproductive roles of miRNAs.
Keywords: pig, miRNA, ovary, testis, deep sequencing
Introduction
microRNAs (miRNAs) comprise a large family of ~22 nucleotides (nt) long non-coding small RNAs derived from ~70 nt long stem-loop precursors (pre-miRNAs). miRNAs have emerged as key post-transcriptional regulators of gene expression in eukaryotic organisms [1-3]. In mammals, miRNAs are predicted to regulate the activity of ~50% of the protein-coding genes [4]. The numbers of individual miRNAs expressed in different organisms are comparable to those of transcription factors or RNA-binding proteins. Many miRNAs are expressed in a tissue-specific or developmental stage-specific manner, thereby greatly contributing to cell-type-specific profiles of protein expression [5, 6]. Identifying tissue-specific miRNAs is the first step toward understanding the biological functions of miRNAs, such as the regulation of tissue differentiation and the maintenance of tissue identity.
Folliculogenesis and spermatogenesis are involving a series of complex and precisely regulated processes, which include morphological and functional changes taking place in different types of follicular or spermatogenic cells [7-9]. These activities could be regulated by miRNA-mediated pathways [10, 11]. Previous studies have assessed the expression profiles of miRNAs in different species to decipher the miRNA transcriptome of their reproductive tissues. Ro et al. (2007) identified 122 (15 are novel) and 141 (29 are novel) miRNAs in adult mouse ovary and testis, respectively [12, 13]. Mishima et al. (2008) found that 154 known miRNAs are expressed in the adult ovary and discovered one novel miRNA [14]. Hossain et al. (2009) identified 74 (24 are novel) miRNAs in the bovine ovary [15]. Tripurani et al. (2010) identified 58 (16 are novel) miRNAs in the bovine fetal ovary [16]. More recently, emergence of the deep sequencing approaches and more comprehensive in silico methodology for subsequent data analysis, have stimulated wider spread and more in-depth studies of miRNAs in the reproductive tract. Ahn et al. (2010) identified 516 (118 are novel) miRNAs in mouse newborn ovary using the Illumina Genome Analyzer [17]. In a more comprehensive study, Creighton et al. (2010) identified 404 known and 132 novel miRNAs through deep sequencing (Illumina Genome Analyzer) of small RNA libraries from 103 tissues or cell lines derived from human female reproductive organs including both normal and diseased tissues [18].
Despite increasing efforts in miRNA identification across various species and diverse tissue types, little is known thus far about porcine gonad-specific miRNAs. The miRBase version 15.0 reports only 175 porcine pre-miRNAs that encoding for 161 distinct miRNAs and 27 miRNA*s, which originates from the hairpin pre-miRNA arm opposite to the annotated miRNA containing arm [19]. In comparison, this number is much less than those pre-miRNAs identified in other mammals, such as human (940), cow (665), chimpanzee (606), mouse (590), macaque (485), horse (343), rat (326), or even dog (325). Identification and preliminary characterization of gonad-specific miRNAs would be the first step toward a thorough understanding of their roles in regulating folliculogenesis and spermatogenesis. In addition to their significance in agriculture, pigs are particularly attractive model animals because of their phylogenetic relation to human beings.
In the present study, we investigated the miRNA transcriptome of adult porcine ovary and testis using deep sequencing technology to elucidate their characteristic organ- and gender-specific expression profiles in terms of their genomic context. Particular emphasis was placed on the features of miRNAs located in X chromosome (X-linked miRNAs).
Materials and Methods
Ethics statement
All research involving animals was conducted according to the Regulations for the Administration of Affairs Concerning Experimental Animals (Ministry of Science and Technology, China, revised in June 2004) and approved by the Institutional Animal Care and Use Committee in the College of Animal Science and Technology, Sichuan Agricultural University, Sichuan, China under permit No. DKY-B20081003.
RNA isolation
Tibetan pigs (a black, Chinese indigenous breed) were used to harvest tissues. Ovaries and testes were taken separately from nine female and nine male Tibetan pigs (210-days-old) respectively, and the tissues were isolated and immediately frozen in liquid nitrogen and stored at -80 oC. Total RNA was extracted using the mirVanaTM miRNA isolation kit (Ambion, Austin, USA) following the manufacturer's procedure. The quantity and purity of total RNA were monitored via analysis by NanoDrop ND-1000 spectrophotometer (Nano Drop, DE, USA) at 260/280 nm (ratio > 2.0). The integrity of total RNA was also tested via analysis by Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA, USA) with RIN number > 6.0.
Small RNA library construction and sequencing
Sequencing of RNA samples was prepared as follows: equal quantities (5 µg) of small RNA isolated from individual female or individual male were pooled. Approximately 45 µg of small RNA representing ovaries or testes was used for library preparation and sequencing. The small RNA fraction between 10-40 nt was isolated by polyacrylamide gel electrophoresis (PAGE) and ligated with proprietary adaptors (Illumina, San Diego, USA). The short RNAs were then converted to cDNA by RT-PCR and the cDNA was sequenced on the Genome Analyzer GA-I (Illumina) following the vendor's recommended protocol for small RNA sequencing.
In silico analysis of the sequence data
Sequenced sequences (Sequ-seqs) were processed using Illumina's Genome Analyzer Pipeline software and then as described by Li et al. with some modification [20]. The sequ-seqs were then subjected to a series of additional filters with acceptance criteria derived from the statistics of mammalian miRNAs in miRBase 15.0: (1) not sequencing adapters; (2) containing no more than 80% A, C, G, or T; (3) containing no more than two N (ambiguious bases); (4) containing not only A and C or G and T, (5) containing no stretches of A7, C8, G6, or T7; (6) being longer than 14 nt or shorter than 27 nt; (7) not containing ≥ 10 repeats of any dimer, ≥ 6 repeats of any trimer, or ≥ 5 repeats of any tetramer; (8) been observed more than two times; and (9) not originating from porcine known classes of RNAs (i.e., mRNA in NCBI database [21]; rRNA, tRNA, small nuclear RNA (snRNA) and small nucleolar RNA (snoRNA) in Rfam database [22]; and repetitive sequence elements in Repbase database [23]). The sequ-seqs that satisfy the acceptance criteria were passed through and called “mappable sequences”.
The mappable sequences were mapped to the pig genome (~2.26 Bbp) (Sscrofa9, ftp://ftp.sanger.ac.uk/pub/S_scrofa/assemblies/) using NCBI Local BLAST. The mapping process included several major steps: (1) map the mappable sequences to the 175 known porcine pre-miRNAs (encoding 189 miRNAs) and then to 5,804 known pre-miRNAs (encoding 6,271 miRNAs) from 22 other mammals in miRBase 15.0; (2) map the mapped sequ-seqs to pig genome to obtain their genomic locations and annotations in Ensembl release 59 (Sscrofa9, April 2009); (3) cluster the unmapped sequences in step 2 (that have been mapped to miRBase sequences, but not to the pig genome) as potential novel miRNAs from gaps in the genome; and (4) predict hairpin RNA structures of the mappable sequences not-mapped to miRBase in step 1 from the adjacent 60 nt sequences in either direction using UNAFold [24]. To avoid ambiguous sequ-seqs that have been assigned to multiple positions in pig genome, only sequ-seqs longer than 18 nt in length were included in step 4.
Q-PCR
Total RNA was extracted using the mirVanaTM Kit (Ambion) from seven diverse tissue samples, including: hearts, kidneys, livers, lungs, ovaries and spleens, from the nine female Tibetan pigs (210-days-old), and testes from the nine male Tibetan pigs (210-days-old). Removal of genomic DNA from the extracted RNA was accomplished using RNase-free DNase I (TaKaRa, Dalian, China). The forward primers of the 32 selected X-linked miRNAs were identical in sequence and lengths to the miRNA itself (i.e., the most abundant isomiR) based on our sequencing results, and are available in Additional file 2: Supplementary Table 5. EvaGreen-based q-PCR was performed with the High-Specificity miRNA qRT-PCR Detection Kit (Stratagene, La Jolla, USA) on the CFX96™ Real-Time PCR Detection System (Bio-Rad, CA, USA). The ΔΔCt method was used to determine the expression level differences between surveyed samples. All measurements contained a negative control and a no-E. coli poly A polymerase (PAP) control, and each sample of each individual was analyzed in triplicate. Normalized factors (NF) of three internal control genes (U6 snRNA, 18S rRNA and Met-tRNA) [20] and relative quantities of objective miRNAs were analyzed using the qBase software [25].
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE24443.
Results and Discussion
An overview of the sequencing results
All sequ-seqs were counted and the identical sequ-seqs were combined into a single kind. Sequencing of small RNA libraries from porcine adult ovary and testis yielded ~1.37 million (M) and ~2.03M kinds of sequ-seqs representing ~17.62M and ~18.62M counts (i.e., copy numbers) of sequ-seqs, respectively. Additional file 2: Supplementary Table 1 provides the statistics of distribution for small RNAs during a series of filters in order. After removal of 5.61M (ovary: 31.84%) and 8.80M (testis: 47.28%) raw sequ-seqs which did not meet the acceptance criteria of our filters, there were ~12.01M (ovary: 68.16%) and ~9.82M (testis: 52.72%) sequ-seqs considered as “mappable sequences”, which corresponding to 55K (ovary) and 57K (testis) kinds of sequ-seqs.
Interestingly, we found that 2.35M (ovary: 13.35%) and 5.59M (testis: 30.04%) counts of sequ-seqs were out of the boundaries of 15-26 nt in size, and 2.53M (ovary: 14.36%) and 2.41M (testis: 12.94%) counts of sequ-seqs were mapped to certain other known classes of RNA sequences, e.g., mRNA, rRNA, tRNA, snRNA, snoRNA and repetitive sequence elements, which were eliminated from analysis. The proportions of sequ-seqs mapping to known RNA classes are listed in Additional file 2: Supplementary Table 2. It is worth noting that, in this study, we did not filter against the piwi-interacting RNAs (piRNAs), a newly identified class of small regulatory RNAs, abundantly produced in the germline cells of eukaryotes [26-28]. The reasons for this are: (1) no porcine piRNA sequences have been reported or deposited in the public database, such as in the piRNABank (http://pirnabank.ibab.ac.in) [29]; (2) piRNAs sequences are not conserved in mammals [30-32]; and (3) the interaction between PIWI and our small RNAs is not being tested in this study.
The size distribution of mappable sequences was similar in ovary and testis libraries. More than half of the counts of sequ-seqs are 22 nt in length, 6.34M (ovary: 52.83% of mappable sequences) and 4.93M (testis: 50.22%), followed by 23 nt and 21 nt, which is consistent with the typical size of miRNA from Dicer-derived products (Additional file 1: Supplementary Fig. 1). Only the mappable sequences were considered a reliable representation of a miRNA molecule and therefore used for the subsequent analyses, which ensures high reliability of the reported results.
Mapping and cataloging miRNAs expressed in the porcine adult ovary and testis
The statistics of porcine known, novel and candidate miRNAs are summarized in Additional file 2: Supplementary Table 3 based on the counts and kinds of the sequ-seqs. In both libraries, miRNAs frequently exhibited extensive sequence-heterogeneity, producing multiple mature isoforms (named isomiRs) from the miRBase depository as reported in the literature [33-35]. In these cases, as in our previous report [20], the most abundant isomiR was chosen as a reference sequence. This provides the most robust approach for evaluation of differential expression. Measuring the abundance of a given miRNA using the count of the most abundant isomiR correlated well with the expression level of the total counts of all isomiRs (ovary: r = 0.94, Pearson; testis: r = 0.95, Pearson). In general, the following discussions refer to the most abundant isomiR and its counts, when describing a family of sequences that may vary by length and/or vary by one nucleotide.
As shown in the Table 1, the pre-miRNAs and the mature miRNAs identified from the mappable sequences are divided into six groups with high-to-mid confidence in order [36]: (1) 243 miRNAs corresponding to 146 known porcine pre-miRNAs which are also mapped to the pig genome. Specifically, 146 miRNAs and 19 miRNA*s are known in miRBase, and 78 were novel which have not been identified (Additional file 2: Supplementary Tables 4.1); (2) 26 miRNAs correspond to 15 known porcine pre-miRNAs, which cannot be mapped to pig genome. Specifically, 14 miRNAs and three miRNA*s are known in miRBase, and nine were novel (Additional file 2: Supplementary Tables 4.2); (3) 112 miRNAs corresponding to 80 other known miRBase mammalian pre-miRNAs which are mapped to the pig genome. These miRNAs were labeled PN(a) (porcine novel, “a” type) (Additional file 2: Supplementary Tables 4.3); (4) two miRNAs (the most abundant isomiR encoding from the pre-miRNAs in group 1 or 3) corresponding to two novel predicted hairpins in the pig genome. These miRNAs were labeled PN(b) (porcine novel, “b” type) (Additional file 2: Supplementary Tables 4.4); (5) 12 miRNAs (mapped to other known miRBase mammalian pre-miRNAs, which did not map to the pig genome) corresponding to 12 novel predicted hairpins in the pig genome. These were labeled PN(c) (porcine novel, “c” type) (Additional file 2: Supplementary Tables 4.5); and (6) 337 miRNAs (longer than 18 nt and unmapped to any known miRBase mammalian pre-miRNAs) encompassing 327 candidate pre-miRNAs, which were predicted RNA hairpins derived from the pig genome, and were labeled PC (porcine candidate) (Additional file 2: Supplementary Tables 4.6).
Pre-miRNAs and mature miRNAs identified in porcine adult ovary and testis
| Group's description | Pre-miRNA number | Mature miRNA number |
|---|---|---|
| Group 1: known miRNAs (with genome location) | 146 | 243 |
| Group 2: known miRNAs (without genome location) | 15 | 26 |
| Group 3: novel miRNAs, “a” type (PN(a)) | 80 | 112 |
| Group 4: novel miRNAs, “b” type (PN(b)) | 2 | 2 |
| Group 5: novel miRNAs, “c” type (PN(c)) | 12 | 12 |
| Group 6: candidate miRNAs (PC) | 327 | 337 |
| Total | 582 | 732 |
Additional file 2: Supplementary Table 5 provides a complete sequence, name, and relative abundance list for the 732 miRNAs sequences (i.e., the most abundant isomiR) originated from 582 pre-miRNAs detected in this study. Predictably, there exist distinct pre-miRNAs and genomic loci that express identical mature sequences, which resulted in these 732 miRNAs sequences corresponding to 673 unique miRNAs sequences.
A closer look at the mapped known porcine miRNAs from the two libraries in this study found that out of 188 unique miRNAs/miRNA*s deposited in miRBase 15.0, 165 and 156 miRNAs/miRNA*s were detected in ovary and testis, respectively. In both libraries, there is a very high detection rate (169 of 188, 89.89%) of known porcine miRNAs/miRNA*s. This illustrates that the ovary and testis libraries encompass almost the entire repertoire of previously known miRNAs, which are essential for the various biochemistry pathways during folliculogenesis and spermatogenesis [10,11,37,38]. Furthermore, 87 novel miRNA*s have also been identified. Predictably, the majority of counts of mappable sequences, 8.08M (67.25%) and 5.49M (55.98%) in ovary and testis libraries, respectively, can be mapped to the 161 known porcine pre-miRNAs (Additional file 2: Supplementary Table 3). The novel and candidate miRNAs that we identified were typically low abundance as compared to most of the known miRNAs, explaining how they might have eluded previous detection efforts.
miRNAs differentially expressed (DE) between adult testis and ovary
In the groups of cataloged miRNAs, we observed that the majority of miRNAs with abundant counts are represented by a few miRNAs. As shown in Fig. 1, the miRNA transcriptome of porcine gonads consists of unevenly distributed counts of sequences in which the top ten unique miRNAs with the highest expression level account for 24.37% and 26.65% (by counts) versus 30.50% and 31.57% of the total counts of all 451 and 558 unique miRNAs of the mappable sequences in ovary and testis libraries, respectively.
Top ten unique miRNAs with the highest expression level in ovary and testis libraries. Plot of the unique miRNAs versus their % in total counts of the mappable sequences. The dashed vertical lines at 24.37% and 30.50% (above, ovary), and 26.65% and 31.57% (below, testis) represent the accumulative % of the top ten, and total 451 or 558 unique miRNAs in total counts of the mappable sequences, respectively.
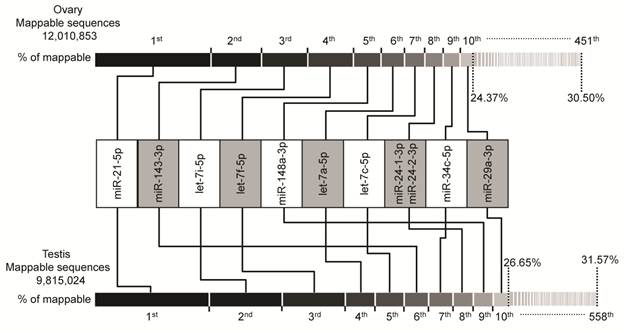
Intriguingly, the plot shows similar results for both ovary and testis libraries. The same unique miRNAs make up the top ten positions, although the rankings are different. The miR-21-5p takes the top ranking in both ovary (0.90M) and testis (0.73M) by counts, these accounts for 7.50% and 7.42% (by counts) of the mappable sequences, respectively. Furthermore, four of top ten miRNAs are from the let-7-family of miRNAs (let-7a-5p, let-7c-5p, let-7i-5p and let-7f-5p). This is consistent with previous reports that let-7-family miRNAs are ubiquitously expressed in various cell and tissues types with a high-expression level and are involved in the cell cycle as master regulators of cell proliferation pathways [39-41]. The relative abundance of these miRNAs in ovary and testis may suggest their housekeeping cellular roles and maybe the most important regulatory miRNA group during ovarian and testicular development. Recent evidence also supports our hypothesis, in which the anomalous expression of these top ten ranked miRNAs may be related to ovarian carcinoma [42-44] and male infertility [37,45,46]. For example, let-7a-5p, let-7c-5p and let-7i-5p were shown to be up-modulated in primary tumors versus normal [47], while let-7f-5p, miR-21-5p [48], miR-143-3p, miR-29a-3p [49], and miR-34c-5p [50] were shown to be down-modulated in ovarian carcinoma cell lines and tissues versus normal, or low-grade cancer versus high-grade cancer. In males, miR-34c-5p plays an essential role in the late steps of spermatogenesis [51], miR-21-5p and let-7a-5p exhibited a high concentration in perinuclear granules in round spermatids (determined by in situ hybridizations) [52], and let-7i-5p, let-7f-5p and miR-29a-3p shown to be down-regulated in testis of non-obstructive azoospermia and normal controls [46]. Taken together, our findings and other evidence support the relevance of these ten miRNAs to ovarian and testicular physiology and indicate that these may be the most important regulatory miRNA group and signature of disease in gonads.
As shown in Fig. 2A and Additional file 2: Supplementary Table 5, out of 673 unique miRNAs, 336 (49.93%) unique miRNAs were co-expressed in ovary and testis libraries, 115 (17.09%) and 222 (32.99%) miRNAs appear to be preferentially expressed in ovary and testis libraries, respectively. To determine the significance of differences in miRNA counts between ovary and testis, we utilized the program IDEG6 [53], which performs a normalization calculation to adjust libraries with different counts of mappable sequences. A unique miRNA is said to be differentially expressed when it simultaneously produces P < 0.001 using a Audic-Claverie test, a Fisher exact test and a Chi-squared 2×2 test with the Bonferroni correction to adjust for pair-wise comparison. The analysis of library sequencing data resulted in the identification of 224 unique miRNAs (out of 673, 33.28%) with statistically significant differential expression between ovary and testis libraries. Out of these 224 DE unique miRNAs, 118 (ovary-specific: 23, co-expressed: 95) and 106 (testis-specific: 68, co-expressed: 38) unique miRNAs are down- and up-regulated in ovary versus testis, respectively (Fig. 2A and Additional file 2: Supplementary Table 6).
Characteristics of DE miRNAs between porcine adult testis and ovary. (A) The Venn diagram displays the distribution of 673 unique miRNAs between ovary (left, blue circle) and testis (right, pink circle) libraries. The dashed circles indicate the DE unique miRNAs (P < 0.001, Bonferroni corrected) in ovary versus testis. (B) Chromosomal location of miRNAs based on the number of DE and not DE miRNAs, and (C) the number of significantly up- and down-regulated in ovary versus testis. “ND” means that the genome location of pre-miRNAs has not been determined.

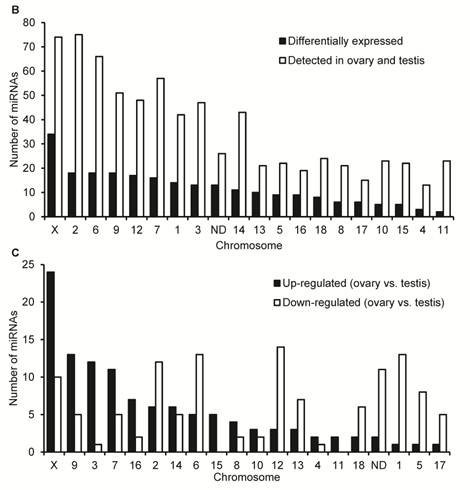
Additional file 2: Supplementary Table 7 lists the detailed sequence, genome location, and context annotation of 582 pre-miRNAs detected in our study, of which 15 pre-miRNAs were registered in miRBase 15.0 which cannot be mapped to the pig genome (Sscrofa9 genome assembly). It is worth noting that the genome locations of mir-28, mir-29c and mir-139 were derived from pig genome assembly (Sscrofa9) in Ensembl release 59 (August, 2010) and absent in miRBase 15.0. The chromosomal distribution of 224 DE unique miRNAs corresponding 235 DE miRNAs are shown in Fig. 2B and 2C. A total of 235 DE miRNAs cover all pig (Sus scrofa) chromosomes (SSC1 to SSC18 and SSCX) except the SSCY for which sequences have not yet been released. Based on the current entries in miRBase for mammals, no miRNAs have been found on the Y chromosome. Intriguingly, similar to the miRNA transcriptome of newborn mouse ovaries [17], most gonad-expressed miRNAs map to chromosome 2 (75 miRNAs), followed by chromosome X (74 miRNAs). These results highlight the homology of gonad-expressed miRNA within mammals.
As shown in Fig. 2B, miRNA expression, as determined by the DE miRNA numbers per chromosome, originated mostly from the SSCX. Out of 74 miRNAs that are located in SSCX and have been detected in ovary and testis, 34 (45.95%) miRNAs have been be defined as DE miRNAs. Secondly, there are 18 DE miRNAs located in each of SSC2 (out of 75, 24.00%), SSC6 (out of 66, 27.27%) and SSC9 (out of 51, 35.29%). As shown in Fig. 2C, most of DE X-linked miRNAs (24 out of 34, 70.59%) significantly up-regulated in ovary versus testis. These results are in accord with previous reports that the X-linked miRNAs were expressed in a testis-preferential or testis-specific pattern [12,14,54]. In silico prediction indicates that the most enriched Gene Ontology (GO) terms found exclusively in mRNA targets of testis X-linked miRNAs were related to the cell cycle process. This corresponds to the massive and continuous cell division, from mitosis of spermatogonia to meiosis of spermatocytes, during the spermatogenesis in testis [55]. Notably, previous reports suggest that Dicer, a crucial enzyme for miRNA biogenesis through processing of pre-miRNAs to a ~20 bp miRNA/miRNA* duplex, is also essential for the development of gonads in mammals [45,56]. In male mice, removal of Dicer from germ cells resulted in infertility in male mouse [57] and in female, the ooplasm of primary oocytes contains Dicer-dependent factors that are crucial for chromosome segregation and meiotic maturation [58].
Exploring the expression patterns of porcine X-linked miRNAs
The XX/XY system is one of the most common sex-determination systems and is found in the vast majority of mammals. Intriguingly, we found that eight and one X-linked pre-miRNAs mapped to two and three genomic loci, respectively (Additional file 2: Supplementary Table 7), which resulting to 52 X-linked pre-miRNAs been mapped to 62 genome loci in SSCX.
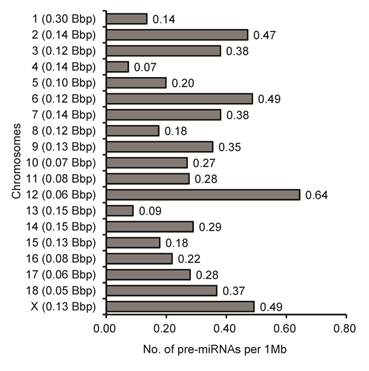
To examine more deeply the differences of miRNA densities between SSCX and autosomes (SSC1 to SSC18), we calculated the genomic density distribution of 567 porcine gonad-expressed pre-miRNAs corresponding to 637 genome loci (number of pre-miRNA loci per Mb of individual chromosome). Similarly to previous reports [20,55], we found a higher density (1.69-fold) of pre-miRNA loci on SSCX (0.49 pre-miRNA loci per Mb of chromosome) than the average of the 18 other autosomes (0.29 pre-miRNA loci per Mb of chromosome) in pig (Fig. 3).
Densities of pre-miRNAs on chromosomses in pigs. Densities were calculated by dividing the number of pre-miRNA loci on the individual by the length of nucleotides on the corresponding chromosome (shown in left brackets), which is shown as the number of pre-miRNA loci per megabase of DNA. Bbp: billion base-pairs.
miRNAs adjacently located on a chromosome (named clustered miRNAs), located in a polycistron, are co-expressed with neighboring miRNAs, implying that they also generally derive from a common transcript [59,60]. We grouped 52 X-linked pre-miRNAs corresponding to 62 genome loci into nine clusters (at least two genome loci in a cluster) based on their inter-distance being less than 50 kb, which resulted in 33 (53.23%) of 62 X-linked pre-miRNA loci likely to cluster (Additional file 2: Supplementary Table 8).
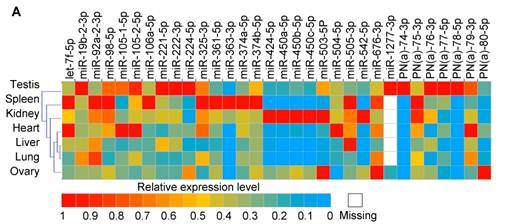
Together with three known porcine X-linked miRNAs (miR-105-1-5p, miR-105-2-5p and miR-450b-5p) that were not detected in our ovary and testis libraries, we measured the tissue-specific expression pattern for 32 high confidence X-linked miRNAs which are the predominantly expressed miRNAs based on the counts of most abundant isomiRs in two libraries in our study (25 are known porcine miRNAs and seven are PN(a) types) using a q-PCR approach (Fig. 4A). Most of them were ubiquitously expressed in almost all tissues analyzed. Nonetheless, miR-1277-3p was detected only in ovary and testis tissues. Predictably, these X-linked miRNAs preferentially exhibited a higher abundance in testes.
Expression patterns of X-linked miRNAs. (A) Q-PCR analysis of expression of 32 selected X-linked miRNAs across seven porcine normal tissues. (B) Relationship between the distance separating X-linked miRNA loci and their coordinate expression in seven normal pig tissues. Each of these miRNAs was paired with each of the others lying in the same orientation on the same chromosome. For each pair, the distance between the two loci was ranked, and the correlation coefficient (r, Pearson) for their expression was plotted according this rank (yellow triangles, left axis). A five-point moving average of r is also shown (blue rhombus), as are the distances between miRNAs in the SSCX (red circles with a line through the middle, right axis), and the average r for all the plotted pairs (dashed line). Turning point discussed in the text are annotated (a and b).
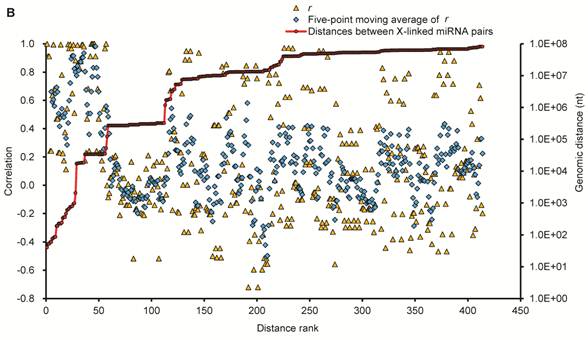
To explore the potential for co-expression among clusters of 32 X-linked miRNAs, we surveyed the relationship between the distance separating miRNA loci as described by Baskerville et al. [61] (Fig. 4B), and their coordinate expression in seven normal pig tissues. The distances separating pairs of miRNAs on the SSCX ranged from 40 nt (from mir-450a to mir-450c) to > 81 Mb (from PN(a)-80 to mir-221). Pairwise comparisons were made between the expression profiles of all miRNAs oriented in the same direction, calculating for each pair a Pearson correlation coefficient (r) ranging between -1 (anti-correlated) and 1 (perfectly correlated). The miRNAs were ranked by the distance separating each pair of miRNAs from each other, and correlations were then plotted based on this ranking, with closer genes having lower rank numbers (Fig. 4B). Similar to the observations made by Baskerville and colleagues in human miRNAs [61], we found that most porcine miRNAs within 50 kb of each other have highly correlated expression patterns. This is consistent with the idea that they are processed from polycistronic primary transcripts. Beyond the 50 kb range, the correlation dropped abruptly to background levels, suggesting that the upper bound for a majority of miRNA polycistronic transcripts is about 50 kb. Intriguingly, the turning point miRNA pairs are mir-676 and itself (r = 1, small points a), since mir-676 (104 nt in length) occurred two times in the SSCX (from 55,113,034 to 55,113,113 and 55,165,137 to 55,165,216 at reverse strand), which are separated by 52,024 bp (empty circles b). The next ranked miRNA pairs are mir-325 / PN(a)-76 (r = 0.00), and mir-450b / mir-106a (r = -0.08), which are separated by 122,184 nt and 267,370 nt respectively.
Taken together, there has been accumulating evidence suggesting that miRNAs closely spaced in the genome are co-regulated, which is essential in regulating a complex cell signaling network, and is more efficient than the regulatory pattern mediated by discrete miRNAs [61-63].
Supplementary Material
Additional File 1Supplementary Figure 1: Size distribution of mappable sequences.
Supplementary Table 1. Statistics of the distribution for small RNAs during a series of filters in order. Supplementary Table 2. Proportion of sequ-seqs mapping to known RNA classes.Supplementary Table 3. Statistics based on the counts and kinds of the mappable sequences.Supplementary Table 4. Profile of the porcine miRNAs.Supplementary Table 5. Porcine unique miRNAs.Supplementary Table 6. miRNAs differentially expressed (DE) between porcine adult ovary and testis.Supplementary Table 7. Porcine pre-miRNA annotations.Supplementary Table 8. Genome location clusters of porcine pre-miRNAs.
Acknowledgements
This work was supported by grants from the National Special Foundation for Transgenic Species of China (2009ZX08009-155B and 2008ZX08006-003), the National Natural Science Foundation of China (30901024), the International Science & Technology Cooperation Program of China (2011DFB30340), the Funds for Distinguished Young Scientists of Chongqing (CSTC2010BA1007) to X.L., M.L., and J.W., and the NIH (R41 DA029063) to X.Z. and X.G. We thank Chris Heble and Dr. G. Kenneth Smith (LC Sciences, Houston) for critically reading of the manuscript.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597-610
2. Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642-55
3. Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228-34
4. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215-33
5. Siomi H, Siomi MC. Posttranscriptional regulation of microRNA biogenesis in animals. Mol Cell. 2010;38:323-32
6. Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010;11:252-63
7. Piprek RP. Molecular and cellular machinery of gonadal differentiation in mammals. Int J Dev Biol. 2010;54:779-86
8. DeFalco T, Capel B. Gonad morphogenesis in vertebrates: divergent means to a convergent end. Annu Rev Cell Dev Biol. 2009;25:457-82
9. Perheentupa A, Huhtaniemi I. Aging of the human ovary and testis. Mol Cell Endocrinol. 2009;299:2-13
10. Carletti MZ, Christenson LK. MicroRNA in the ovary and female reproductive tract. J Anim Sci. 2009;87:E29-38
11. He Z, Kokkinaki M, Pant D, Gallicano GI, Dym M. Small RNA molecules in the regulation of spermatogenesis. Reproduction. 2009;137:901-11
12. Ro S, Park C, Sanders KM, McCarrey JR, Yan W. Cloning and expression profiling of testis-expressed microRNAs. Dev Biol. 2007;311:592-602
13. Ro S, Song R, Park C, Zheng H, Sanders KM. et al. Cloning and expression profiling of small RNAs expressed in the mouse ovary. RNA. 2007;13:2366-80
14. Mishima T, Takizawa T, Luo SS, Ishibashi O, Kawahigashi Y. et al. MicroRNA (miRNA) cloning analysis reveals sex differences in miRNA expression profiles between adult mouse testis and ovary. Reproduction. 2008;136:811-22
15. Hossain MM, Ghanem N, Hoelker M, Rings F, Phatsara C. et al. Identification and characterization of miRNAs expressed in the bovine ovary. BMC Genomics. 2009;10:443
16. Tripurani SK, Xiao C, Salem M, Yao J. Cloning and analysis of fetal ovary microRNAs in cattle. Anim Reprod Sci. 2010;120:16-22
17. Ahn HW, Morin RD, Zhao H, Harris RA, Coarfa C. et al. MicroRNA transcriptome in the newborn mouse ovaries determined by massive parallel sequencing. Mol Hum Reprod. 2010;16:463-71
18. Creighton CJ, Benham AL, Zhu H, Khan MF, Reid JG. et al. Discovery of novel microRNAs in female reproductive tract using next generation sequencing. PLoS One. 2010;5:e9637
19. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154-8
20. Li M, Xia Y, Gu Y, Zhang K, Lang Q. et al. MicroRNAome of porcine pre- and postnatal development. PLoS One. 2010;5:e11541
21. Pruitt KD, Tatusova T, Klimke W, Maglott DR. NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res. 2009;37:D32-6
22. Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL. et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136-40
23. Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474
24. Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol. 2008;453:3-31
25. Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19
26. Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199-202
27. Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P. et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203-7
28. Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T. et al. Characterization of the piRNA complex from rat testes. Science. 2006;313:363-7
29. Sai Lakshmi S, Agrawal S. piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res. 2008;36:D173-7
30. Thomson T, Lin H. The biogenesis and function of PIWI proteins and piRNAs: progress and prospect. Annu Rev Cell Dev Biol. 2009;25:355-76
31. Klattenhoff C, Theurkauf W. Biogenesis and germline functions of piRNAs. Development. 2008;135:3-9
32. Betel D, Sheridan R, Marks DS, Sander C. Computational analysis of mouse piRNA sequence and biogenesis. PLoS Comput Biol. 2007;3:e222
33. Kuchenbauer F, Morin RD, Argiropoulos B, Petriv OI, Griffith M. et al. In-depth characterization of the microRNA transcriptome in a leukemia progression model. Genome Res. 2008;18:1787-97
34. Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A. et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610-21
35. Glazov EA, Cottee PA, Barris WC, Moore RJ, Dalrymple BP. et al. A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. Genome Res. 2008;18:957-64
36. Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC. et al. A uniform system for microRNA annotation. RNA. 2003;9:277-9
37. Calvel P, Rolland AD, Jegou B, Pineau C. Testicular postgenomics: targeting the regulation of spermatogenesis. Philos Trans R Soc Lond B Biol Sci. 2010;365:1481-500
38. McFarlane L, Wilhelm D. Non-coding RNAs in mammalian sexual development. Sex Dev. 2009;3:302-16
39. Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008;18:505-16
40. Barh D, Malhotra R, Ravi B, Sindhurani P. MicroRNA let-7: an emerging next-generation cancer therapeutic. Curr Oncol. 2010;17:70-80
41. Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N. et al. Inhibition of translational initiation by let-7 microRNA in human cells. Science. 2005;309:1573-6
42. Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F. et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699-707
43. Dahiya N, Morin PJ. MicroRNAs in ovarian carcinomas. Endocr Relat Cancer. 2010;17:F77-89
44. Wyman SK, Parkin RK, Mitchell PS, Fritz BR, O'Briant K. et al. Repertoire of microRNAs in epithelial ovarian cancer as determined by next generation sequencing of small RNA cDNA libraries. PLoS One. 2009;4:e5311
45. Papaioannou MD, Nef S. microRNAs in the testis: building up male fertility. J Androl. 2010;31:26-33
46. Lian J, Zhang X, Tian H, Liang N, Wang Y. et al. Altered microRNA expression in patients with non-obstructive azoospermia. Reprod Biol Endocrinol. 2009;7:13
47. Yang N, Kaur S, Volinia S, Greshock J, Lassus H. et al. MicroRNA microarray identifies let-7i as a novel biomarker and therapeutic target in human epithelial ovarian cancer. Cancer Res. 2008;68:10307-14
48. Dahiya N, Sherman-Baust CA, Wang TL, Davidson B, Shih Ie M. et al. MicroRNA expression and identification of putative miRNA targets in ovarian cancer. PLoS One. 2008;3:e2436
49. Nam EJ, Yoon H, Kim SW, Kim H, Kim YT. et al. MicroRNA expression profiles in serous ovarian carcinoma. Clin Cancer Res. 2008;14:2690-5
50. Zhang L, Volinia S, Bonome T, Calin GA, Greshock J. et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2008;105:7004-9
51. Bouhallier F, Allioli N, Lavial F, Chalmel F, Perrard MH. et al. Role of miR-34c microRNA in the late steps of spermatogenesis. RNA. 2010;16:720-31
52. Kotaja N, Bhattacharyya SN, Jaskiewicz L, Kimmins S, Parvinen M. et al. The chromatoid body of male germ cells: similarity with processing bodies and presence of Dicer and microRNA pathway components. Proc Natl Acad Sci U S A. 2006;103:2647-52
53. Romualdi C, Bortoluzzi S, D'Alessi F, Danieli GA. IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol Genomics. 2003;12:159-62
54. Luo L, Ye L, Liu G, Shao G, Zheng R. et al. Microarray-based approach identifies differentially expressed microRNAs in porcine sexually immature and mature testes. PLoS One. 2010;5(8):e11744
55. Guo X, Su B, Zhou Z, Sha J. Rapid evolution of mammalian X-linked testis microRNAs. BMC Genomics. 2009;10:97
56. Gonzalez G, Behringer RR. Dicer is required for female reproductive tract development and fertility in the mouse. Mol Reprod Dev. 2009;76:678-88
57. Maatouk DM, Loveland KL, McManus MT, Moore K, Harfe BD. Dicer1 is required for differentiation of the mouse male germline. Biol Reprod. 2008;79:696-703
58. Mattiske DM, Han L, Mann JR. Meiotic maturation failure induced by DICER1 deficiency is derived from primary oocyte ooplasm. Reproduction. 2009;137:625-32
59. Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S. et al. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005;33:2697-706
60. Zhang Y, Zhang R, Su B. Diversity and evolution of microRNA gene clusters. Sci China C Life Sci. 2009;52:261-6
61. Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241-7
62. Kim YK, Yu J, Han TS, Park SY, Namkoong B. et al. Functional links between clustered microRNAs: suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res. 2009;37:1672-81
63. Xu J, Wong C. A computational screen for mouse signaling pathways targeted by microRNA clusters. RNA. 2008;14:1276-83
Author contact
![]() Corresponding author: X.L.G (E-mail: xgaoedu) or X.W.L (E-mail: lixuewei9125com)
Corresponding author: X.L.G (E-mail: xgaoedu) or X.W.L (E-mail: lixuewei9125com)