Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Acknowledgements
Author Contributions
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(2):174-189. doi:10.7150/ijbs.5115 This issue Cite
Research Paper
A Novel Brain Neurovascular Unit Model with Neurons, Astrocytes and Microvascular Endothelial Cells of Rat
Qiang Xue1, 2, 3, Yang Liu1, 2, 3, Hongyi Qi1, 2, 3, Qiang Ma1,2, 3, Ling Xu1, 2, 3, Weihai Chen4, Gang Chen5, Xiaoyu Xu1, 2, 3, ![]()
1. College of Pharmaceutical Sciences & College of Chinese Medicine, Southwest University, Chongqing 400715, China;
2. Chongqing Engineering Research Center for Pharmacological Evaluation, Chongqing 400715, China;
3. Institute of Chinese Medicine, Southwest University, Chongqing 400715, China;
4. College of Psychology, Southwest University, Chongqing 400715, China;
5. Research Center of Medical Chemistry and Chemical Biology, Chongqing Technology and Business University, Chongqing 400067, China.
Received 2012-8-27; Accepted 2013-1-14; Published 2013-2-7
Abstract
A novel triple cell neurovascular unit (NVU) model co-culturing with neurons, brain microvascular endothelial cells (BMECs) and astrocytes was established in this study for investigating the cerebral diseases and screening the candidates of therapeutic drug. We have first performed the cell identification and morphological characterization, analyzed the specific protein expression and determined the blood-brain barrier (BBB) function of the co-culture model under normal condition. Then, we further determined the BBB function, inflammation, cell injury and the variation of neuroprotective factor in this model after anoxia-reoxygenation. The results suggest that this model exhibited a better BBB function and significantly increased expression of P-glycoprotein (Pg-P) and ZO-1 compared with BMECs only or co-culture with astrocytes or neurons. After anoxia-reoxygenation, the pathological changes of this model were basically resemblance to the pathological changes of brain cells and BBB in vivo. And nimodipine, an antagonist of calcium, could reverse those changes as well. According to our observations, we deduce that this triple cell co-culture model exhibits the basic structure, function and cell-cell interaction of NVU, which may offer a more proper in vitro system of NVU for the further investigation of cerebral diseases and drug screening.
Keywords: neurovascular unit, triple cell co-culture model, anoxia-reoxygenation, brain microvascular endothelial cells, astrocytes, neurons.
Introduction
The brain vascular diseases, such as stroke, are characterized by high incidence, high mortality and high morbidity. Over the past decades, a large number of clinical trials of neuroprotective agents have been carried out with various drugs alone or in combination. However, all the trails have been terminated finally as no significant improvement in clinical symptoms and life quality [1]. Further analysis revealed that the key point for these failures may be due to only a single therapeutic target focused by researchers [2-3] and particularly ignoring the interaction between the various components of neurovascular unit (NVU) [4-5].
Due to the facts mentioned above, researchers have to find a solution in view of multi-target, multi-level and combination therapy. In 2003, LO EH and his colleagues have proposed a concept of NVU [6], which refers to an overall structure and function unit consisting of brain neurons, BMECs, glial cells, even with pericytes, basement membrane, microglia cells and extracellular matrix [3,5-6]. This concept emphasizes the dynamic interactions among all of the components and their important influence on the pathophysiology of stroke [7]. This change and mechanism were stressed in the conference of American National Institutes of Neurological Disorders and Stroke (NINDS). Thereafter, simultaneous protection of different cell types in NVU was highlighted in the treatment of stroke [7-8].
Currently, however, there is no proper in vitro NVU model available for investigating the brain vascular diseases and subsequently screening therapeutic drug candidates, In this study, we drawn our inspiration from the previous reports concerning the triple cell co-culture for mimicking the blood brain barrier (BBB). We first established a triple primary cell co-culture model with rat BMECs, neurons and astrocytes as the NVU model in vitro. The morphology and function of this model were also characterized in our study.
Materials and methods
Animals
Newborn Sprague-Dawley (SD) rats of indicated days were obtained from the Experimental Animal Center, Chongqing Medical University (Chongqing, China) and housed in the Experimental Animal Center, College of Pharmaceutical Sciences & College of Chinese Medicine, Southwest University (Chongqing, China). All the experiments were performed in accordance with China's Guidelines for Care and Use of Laboratory Animals.
Materials
DMEM-F12 medium was from GIBCO (Invitrogen Corporation, USA). Trypsin (1:250) and L-glutamine were from Amresco (USA). Fetal bovine serum (FBS) was from Hyclone (Thermo scientific, USA). EDTA and gelatin were from Genview (Gen-view scientific Inc., USA). Collagenase type II, bovine serum albumin (BSA), cytosine arabinoside (Ara-C), poly-L-lysine (MW: 70,000- 150,000), basic fibroblast growth factor (bFGF), sodium fluorescein (SF) and horseradish peroxidase (HRP) were form Sigma (Sigma-Aldrich Co. LLC., USA). All the antibodies used in this study were from Santa Cruz (USA) except anti-GAPDH monoclonal antibody which was from ZSGB-BIO (Beijing, China). All the kits in this study were from ZSGB-BIO (Beijing, China).
Isolation and purification of three kinds of rat cerebral cells
Primary rat BMECs were prepared from the brain of 2-3 weeks old rats as described in previous reports [9-14]. Briefly, meninges, large vessels and white matter were removed carefully and the grey matter was minced into small pieces of approximately 1.0 mm3 in ice-cold D-Hanks. After centrifuged at 150(×g) for 3min, the precipitation layer was added with trypsin (2.5 mg/ml) and digested at 37 °C for 1.5 h. Cold DMEM-F12 (1:1) medium with 10% FBS was added to terminate the digestion and then it was centrifuged at 150(×g) for 5min. The precipitate was re-suspended in 25% BSA and centrifuged at 600 (×g) for 15 min. The microvessels obtained were then digested in collagenase type II (1.0 mg/ml) and DNase (1.5 mg/ml) at 37°C for 1 h and the medium with 10% FBS was added to suspend it. This new suspension was filtered through a 10-mm-pore-size nylon mesh and washed with medium containing 10% FBS. After the filtrate was centrifuged at 150(×g) for 5min, the precipitate layer was re-suspended with a medium with 20% FBS, basic fibroblast growth factor (bFGF, 1.0 ng/ml), heparin (100.0 mg/ml), penicillin (100.0 U/ml), L-glutamine (0.6mg/ml) and streptomycin (100.0 mg/ml). After the density adjustment, the microvessel endothelial cells were seeded on 25cm2 plastic dishes pre-coated with gelatin (15.0 mg/ml) and incubated at 37°C and 5% CO2. The culture medium was changed every 2 days. When the confluence reached 80% (the 6th day), the endothelial cells were further purified with trypsin (2.5 mg/ml) and EDTA (0.2 mg/ml) solution. The purified endothelial cells were used to establish the in vivo NVU model.
Cerebral astrocytes were obtained from brain cortices of 2 day old rats, as described in previous reports [9,15-18]. Meninges, large vessels and white matter were removed carefully and grey matter pieces were dissociated mechanically into small pieces in ice-cold D-Hanks. The cortical pieces were disaggregated in trypsin (2.5 mg/ml) diluted with Ca2+/Mg2+ free PBS at 37°C for 20 min. After centrifuged at 150(×g) for 5 min, the precipitate was re-suspended in the medium with 10% FBS. The suspension was filtered through a 10-mm-pore-size nylon mesh and washed by medium with 10% FBS. Finally, the filtrate was centrifuged at 150(×g) for 5 min and then re-suspended with culture medium containing 10% FBS, penicillin(100.0 U/ml), streptomycin(100.0 mg/ml) and plated on 25cm2 plastic dishes pre-coated with poly-L-lysine (0.1 mg/ml) at 37°C and 5% CO2. On the second day, it was changed with new medium. Then the culture medium was changed every 2 days. When the confluence reached 80% (the 4 -5th day), the plastic dishe was shaken at 220 rpm for 18h at 37 °C to purify the astrocytes. The purified astrocytes were passaged by a brief treatment with trypsin (2.5 mg/ml)-EDTA (0.2 mg/ml) solution and the second passage was used to establish the in vivo NVU model.
Rat cerebral neurons were obtained from the brain cortices of 1 day old rats according to previous researches [19-20]. The cortex was dissected in ice-cold D-Hanks and then digested at 37°C for 30 min in enzymatic solution (2.5 mg/ml). The cortical tissues were homogenated to single cell suspension with medium containing 10% FBS. Then, the suspension was filtered through 10-mm-pore- size nylon mesh and centrifuged at 150(×g) for 5 min. The precipitate was re-suspended with the medium containing 10% FBS, B27(1×), penicillin(100.0 U/ml), streptomycin(100.0 mg/ml) and seeded in plates or plastic culture flask pre-coated with poly-L-lysine (0.1 mg/ml) at 37°C and 5% CO2. On the third day, medium was replaced with new medium supplemented with Ara-C (5.0mg/ml) to inhibit the non-neuronal cell growth for 24 h. Then, the culture medium was changed every 2 days.
Establishment of in vitro NVU model
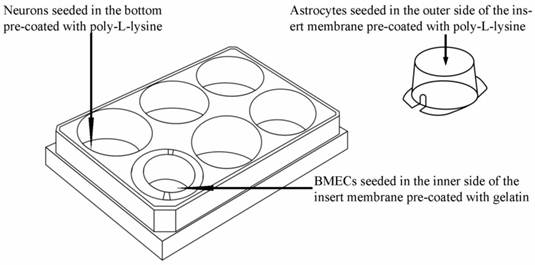
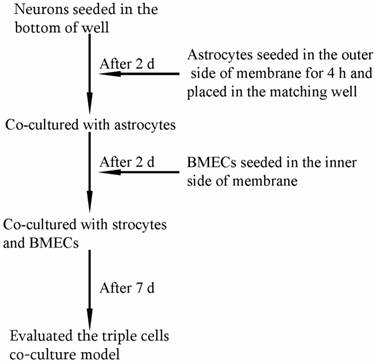
As shown in Fig. 1, the triple cell co-culture system of was established by referring to previous reports [9,21-23] with a modification. Before starting the co-culture, all cells were adapted to the same medium DMEM-F12 (1:1) containing 20% FBS, L-glutamine (0.6mg/ml), penicillin (100.0 U/ml) and streptomycin (100 mg/ml). When the confluence gradually increased up to 90%, the three types of cells were used to establish the model. Astrocytes (1.5×105 cells/cm2) were seeded into the matching well under the insert membrane. Then, the Transwell was placed upside down in the incubator. Depending on the surface tension, the medium couldn't flow out and the astrocytes gradually adhered to the outer side of poly-L-lysine -coated (10.0 mg/ml) insert membrane. After 4 h, the insert was placed in the well where neurons had already grown for 48 h with a seeding density of 0.5×105 cells/cm2. After another 48 h, BMECs (1.0 ×105 cells/cm2) were seeded in the inner side of the insert membrane coated with gelatin (30.0 mg/ml). As controls, BMECs cells were also cultured with neurons alone, with astrocytes alone or without other two primary cells. The day when the endothelial cells were plated was defined as day zero in vitro (day 0). All cells were cultured for 3-5 days with daily change of medium before analysis. The procedure for establishing the model was shown as Fig. 2.
Schematic drawing of triple cell co-culture system.
The procedure for establishing in vitro triple cell co-culture model.
HE staining
Neurons were directly stained in their growing wells of Transwell. Astrocytes were stained after removal of BMECs in the inner side of the insert and BMECs were stained after removal of astrocytes in the outer side of the insert. The staining process was described briefly as followings: cells were washed with PBS and fixed by 4% paraformaldehyde. After washed with PBS again, cells were stained with hematoxylin. After rinsed in running tap water, cells were immerged in 1% acid alcohol and then blued in 0.2% ammonia water after rinsed in running tap water. Finally, cells were stained with eosin after rinsed in tap water. The pictures were taken after they were dehydrated.
Immunocytochemical staining
Different types of primary cells were characterized by specific markers as previous reported [24]. Briefly, cells were first washed with PBS and fixed with 4% paraformaldehyde for 30 min at room temperature. After the removal of excessive paraformaldehyde, the fixed cells were incubated in a fresh blocking buffer (0.5% Triton X-100 in PBS, pH 7.4, containing 10% normal goat serum) for 1 h at room temperature. The triple cells were separately probed overnight at 4 °C with different primary antibodies (anti-factor VIII for BMECs, anti-glial fibrillary acidic protein (GFAP) for astrocytes and anti-neuron-specific enolase (NSE) for neurons), which were diluted 1:100 in PBS with 3% bovine serum albumin. The primary antibodies were detected by incubation with FITC-goat anti-rabbit IgG (1:1000) in PBS containing 3% bovine serum albumin. The cell nucleus was stained with 4ˊ, 6-diamidino-2-phenylindole (DAPI). Images were captured on a Zeiss fluorescence microscope (Carl Zeiss, Germany).
P-glycoprotein and ZO-1 in BMECs were determined as previous reported [25-26]. The astrocytes were scraped from the outer side of the insert membrane. The BMECs on the inner side of insert membrane were kept for the immunocytochemical staining of P-glycoprotein and ZO-1. Anti-P-glycoprotein and anti-ZO-1 were used as the primary antibodies, which were then probed by biotin-conjugated mouse anti-rat secondary antibody for 60 min followed by HRP-labeled streptavidin for 20 min. As a control for nonspecific binding, the above-mentioned procedure was also carried out with omission of the primary antibody.
Transmission electron microscopy (TEM)
The tight junction (TJ) between BMECs was observed by TEM method as previously described [9,27]. Briefly, the membrane of the Transwell insert in NVU model was removed and fixed with glutaral solution for 30 min at 4°C. Then, the membrane was washed with cacodylate buffer and immerged into the osmium tetroxide solution for 1h. After dehydrated in graded ethanol, cells were stained with uranyl acetate and lead citrate for 1 h and then embedded in embedding medium. The intercellular TJ was examined by a Hitachi 7100 TEM.
Four hours leakage detection
The barrier permeability of BMECs was determined by a 4 h leakage experiment. Briefly, after 5 d co-culture, the insert as the donor pool was filled with medium, while the medium level in the 12-well plates as receptor pool was kept 0.5 cm lower than the insert. Insert with no cell culture was used as control. The medium level in the donor pool was compared before and after 4 h incubation.
Determination of the transendothelial permeability by sodium fluorescein (SF) and horseradish peroxidase (HRP)
The flux of SF and HRP across the endothelial cells was determined as previously described [9,12,27-28]. Briefly, the culture medium in the inserts was replaced with 0.5 ml DMEM-F12 medium containing 100 μg/ml SF. After 2h incubation, the samples were taken from the well of the Transwell, and the absorbances of the samples were measured by fluorospectrophotometer. For HRP, the culture medium in the inserts was replaced with 0.5 ml DMEM-F12 medium containing 50 μg/ml HRP. After 2h incubation, samples were taken from the well of the Transwell and transferred to a microplate. O-phenylenediamine was added to each well and the reaction was terminated by adding H2SO4. The absorbance was determined by a microplate reader.
Determination of gamma-Glutamyl transpeptidase (γ-GT) activity [29-30]
After 7 days co-culture, the BMECs were washed by cold PBS and scraped from the upper side of the membrane and suspended with 10.0 mM TriS-HCI (contain 10.0 mM Tris-HCl solution pH 7.4, 10.0 mM NaCl and l.0 mM MgCl2). All the suspensions were centrifuged at 1000 (×g) for 10 min at 4 °C and then the cells were sonicated (300W, 5min) in the ice bath. The suspension was centrifuged at 13000 (×g) for 30 min at 4 °C. The content of γ-GT in the supernatant was determined by optical density method with a commercial kit (Jiancheng Company, Nanjing, China).
Measurement of transendothelial electrical resistance (TEER) [9,13]
The TEER values of different cells co-culture were measured by an epithelial-volt-ohm resistance meter (ERS-2, Millipore) according to the protocol provided by the company. The background TEER value was measured in the same well under the same condition with no cell seeded. The final result was calculated as the TEER value of different cells subtracted of the corresponding background TEER value and then multiplied by the area of insert membrane. The values are shown as Ω×cm2.
Procedure of anoxia-reoxygenation treatment on triple cell co-culture model
The in vitro anoxia-reoxygenation treatment on triple cell co-culture model was set up as described previously [31-33] with a modification. Briefly, the culture medium was replaced with glucose-free medium without serum (98.5 mM NaCl, 10.0 mM KCl,1.2 mM MgSO4, 0.9 mM Na2HPO4, 6.0 mM NaHCO3, 1.8 mM CaCl2, 40 mM Sodium lactate, 20 mM HEPES with pH6.8). The co-culture system was subsequently transferred into an anaerobic chamber (Fig.3) equilibrated with 95%N2 and 5%CO2 at 37°C for 4 h.
Schematic drawing of the anaerobic device.
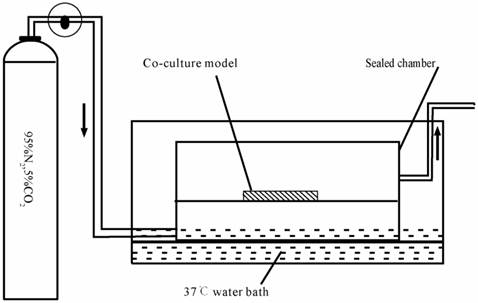
Following the anaerobic treatment, cells were returned to the normoxic incubator with glucose medium without serum(129.5 mM NaCl, 5.0 mM KCl,1.2 mM MgSO4, 0.9 mM Na2HPO4, 20.0 mM NaHCO3, 1.8 mM CaCl2, 55 mM Glucose and 20 mM HEPES with ph7.4) for 2 h. These were randomly divided into 3 groups: (1) normal group; (2) injured group, which was exposed to anoxia- reoxygenation; (3) nimodipine group: 5 μmol.L-1 nimodipine was used during anoxia-reoxygenation.
ELISA analysis
The contents of tumor necrosis factor-α (TNF-α), NO (nitric oxide), lctate dehydrogenase (LDH), malondialdehyde (MDA) and superoxide dismutase (SOD) of the supernatant and the γ-GT of BMECs were detected by sandwich ELISA assay according the manufacturer's instructions, respectively. Absorbances were measured at 495 nm using a microplate ELISA reader (BioTek, USA). Each final value was quantified against a standard curve calibrated with known amounts of protein.
Western blot analysis
BMECs and neurons from the triple cell co-culture model were scraped and collected in 1ml cell lysis buffer (50 mM Tris-HCl PH7.4, 2%SDS, 0.1 M NaCl, 1 mM EDTA, 1%triton X-100, 0.5μg/ml aprotinin, 1 mM sodium rothovanadate, 1 mM PMSF), respectively. Then, each sample was incubated on ice for 20min and centrifugated at 12,000(×g) for 15min. The supernatant of each culture was freeze-dried using vacuum freeze-drying apparatus after added with the protease inhibitors. Then, 10-20μg of total proteins were separated on a 10% Tris/Glycine SDS-PAGE gel and subsequently transferred to PVDF membrane. Following 45 min incubation in a PBST containing 5% non-fat milk the blots were probed with specific antibodies including anti- GAP-43, anti-occludin, anti-IL-6, anti- MMP-9, anti- VEGF, anti- IL-1β, anti-GAPDH. The bound primary antibodies were detected by horseradish peroxidase conjugated sencondary antibodies accordingly. The activity of peroxidase on the blot was visualized by enhanced chemiluminescence (ECL) detection reagents (MILLIPORE). The blots were quantified by Quantity One software (Bio-Rad, USA). The concentration of the loaded cellular proteins was normalized against the internal control GAPDH and then the value was expressed as each normalized data relative to control.
Statistics
All the experiments were repeated at least three times and the data were analyzed by one-way analysis of variance (ANOVA) using SPSS 17.0 and expressed as mean ± S.E.M.
Results
Characterization of cell type and purity by specific markers
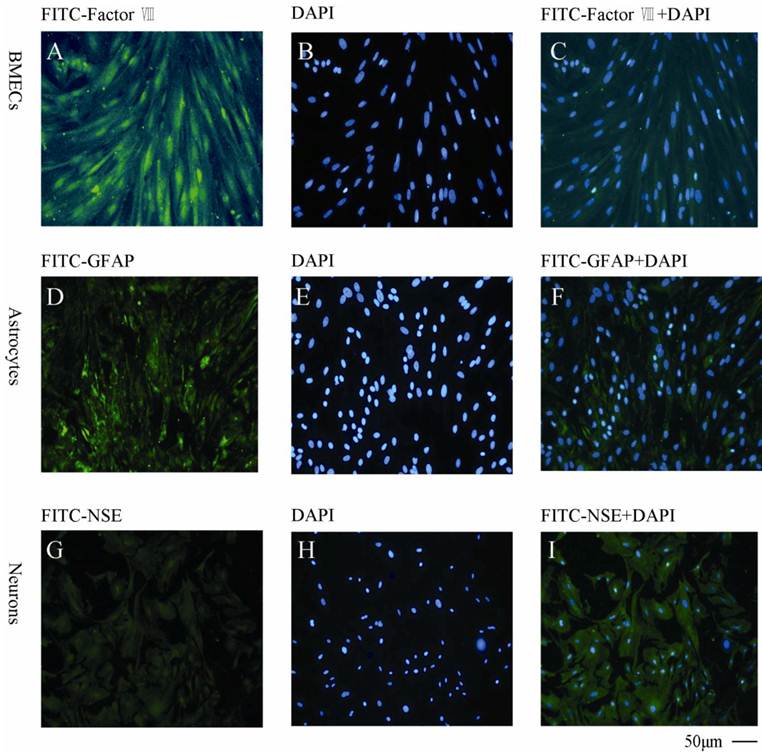
As shown in Fig. 4, the BMECs were detected by the specific anti-factor VIII and the purity was found to be >95% (Fig.4A, B, C). The astrocytes were determined by anti-GFAP (Fig.4D, E, F) with a purity >90%. The neurons was found to be >85% measured by anti-NSE (Fig. 4G, H, I).
Immunocytochemical staining of the primary cells with different cellular markers A, B, C: BMECs with FITC-factor VIII, DAPI and combination of FITC-factor VIII and DAPI, respectively; D, E, F: Astrocytes with FITC-GFAP, DAPI and combination of FITC-GFAP and DAPI, respectively; G, H, I: Neurons with FITC-NSE, DAPI and combination of FITC-NSE and DAPI, respectively. The experiments repeated for three times and 2 wells used in each group.
Morphology of primary cells characterized by HE staining and TEM
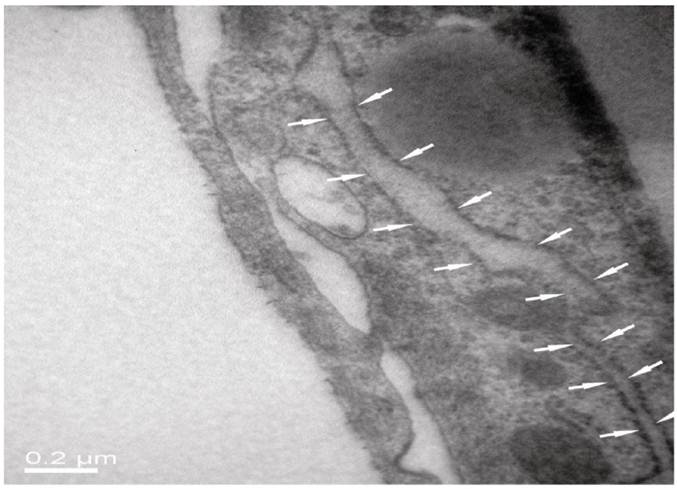
As seen from Fig. 5 of HE staining results, neurons, BMECs and astrocytes in the NVU model formed as a continuous single layer, respectively. Interestingly, numerous eosin staining exists in the micropore of the inner side of the insert membrane, which could be explained that podocytic processes of astrocytes crossed the micropore of the membrane and have a direct contact with endothelial cells. The neurons grown in the bottom of the well have longer axons and more dyed nucleus, which indicated that the cells grew in a preferable condition. The TJ was determined by TEM. As shown in Fig. 6, the presence of electron-dense material in the space between cells reflects the TJ.
HE staining of BMECs (A), astrocytes (B) and neurons (C) in co-culture model.
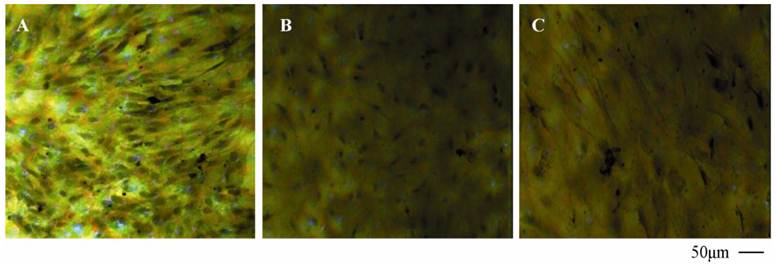
TJ of the neighboring endothelial cells determined by TEM.
Expression of endothelial P-glycoprotein and ZO-1
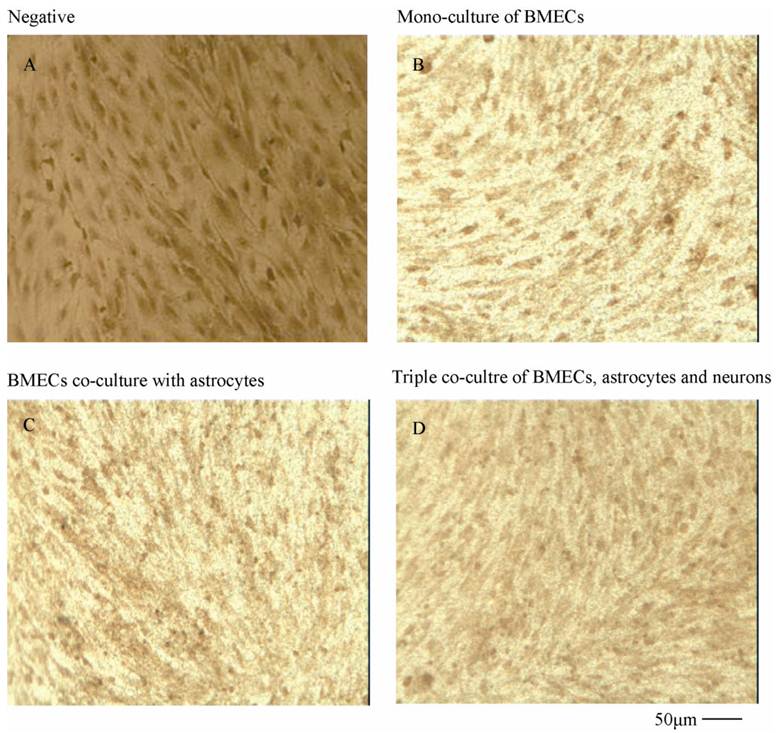
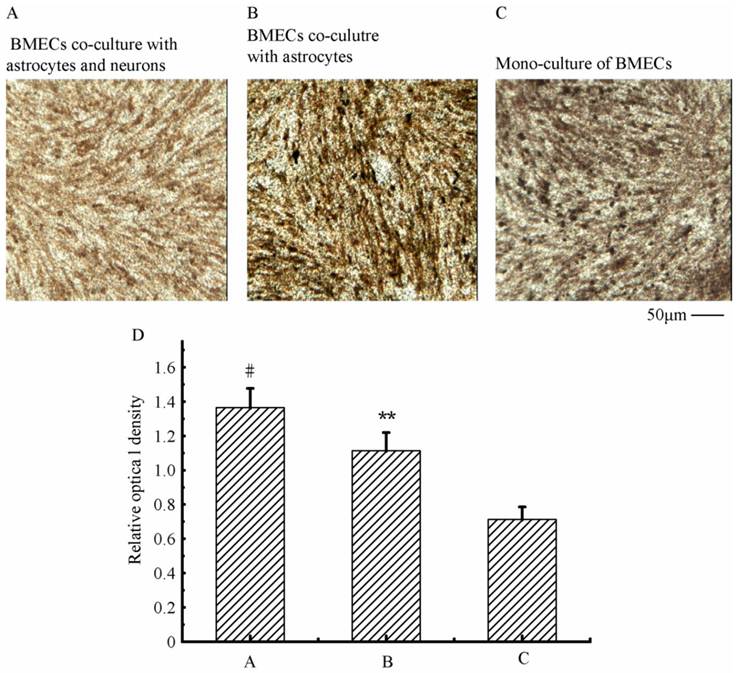
To verify the existing of efflux protein and the barrier integrity of this in vitro NVU model, P-glycoprotein and ZO-1 protein in BMECs were determined, respectively. As shown in Fig.7, the expression of the endothelial P-glycoprotein was increased by participation of astrocytes or neurons and astrocytes. Moreover, P-glycoprotein level of the triple cell co-culture model was highest. These results demonstrated that the existing of neurons and astrocytes could enhance the expression of P-glycoprotein. Similarly, the expression of ZO-1 was relatively low in culture of only BMECs (Fig.8A). And the expression of endothelial ZO-1 significantly increased in the presence of astrocytes (Fig.8B). The highest level of ZO-1 was found in the triple cell co-culture model (Fig.8C). The results demonstrated that endothelial ZO-1 increased after co-culture with neurons and astrocytes, which indicated that the characteristics of this in vitro model are much more in line with the in vivo complex environment.
Immocytochemical staining of P-glycoprotein of BMECs in different models.
Immocytochemical staining of endothelial ZO-1 in different models.
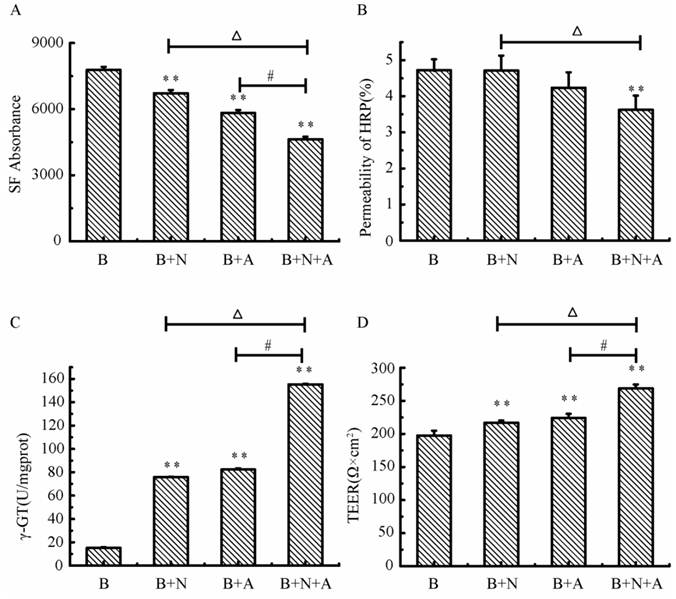
Characterization of the endothelial permeability
As shown in Fig.9, the absorbance of SF and the permeability of HRP in the triple cell co-culture were lowest compared with that in the mono-culture of BMECs or BMECs co-culturing with neurons or astrocytes. The permeability of SF and HRP in triple cell co-culture models was decreased 40.4% and 23.3%, respectively, compared with BMECs culture only. The γ-GT enzyme activity in the triple cell co-culture model reached highest value (155.33 U/mg prot.), which increased 9.2 fold compared to mono-culture of BMECs. TEER reflects the permeability of inorganic ion through the cell model and is the most sensitive indicator of the barrier function of this model. The TEER value of the triple cell co-culture model increased 35.9% compared with BMECs culture only and reached 268.67 Ω×cm2. This indicates that this tripe cells co-culture model exhibits a better TJ contrast with BMECs only or double cells co-culture.
The transendothelial Permeability of SF (A) and HRP (B), γ-GT activity (C) and TEER (D) in different models. A: astrocytes, B: brain microvascular endothelial, N: neurons; *p<0.05, **p<0.01 versus B only, # p<0.01 and △p<0.01. Values represent Mean ± S.E.M (n=4).
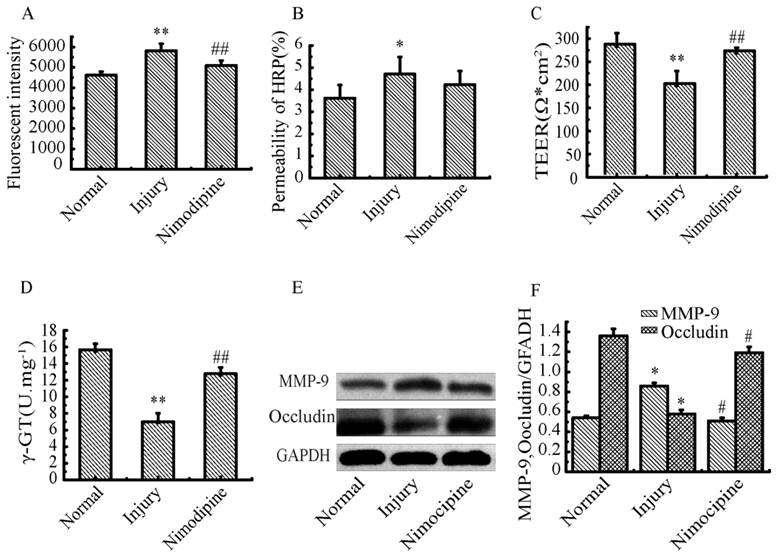
Variations of BBB function in triple cell co-culture model after anoxia-reoxygenation
After anoxia-reoxygenation, compared with the normal group, the permeability of SF (Fig.10A), HRP (Fig.10B) significantly increased, while the TEER value (Fig.10C) and γ-GT activity (Fig.10D) significantly decreased, indicating the whole function of BBB reduced. The expression of MMP-9 (Fig.10E) significantly increased indicating the BBB injury became serious, and the expression of Occludin (Fig.10F) obviously decreased suggesting the endothelial TJ was damaged. Nimodipine treatment significantly reversed the changes caused by anoxia-reoxygenation except the HRP permeability, which decreased in value but wasn't significant. The results demonstrated that nimodipine treatment improved the BBB function remarkably.
Variations of BBB function in co-culture model. (A) Fluorescent intensity of SF. (B) permeability of HRP (%). (D) γ-GT activity. (E, F) Expression of MPP-9 and Occludin detected by western blotting. Values represent Mean ± S.E.M (n=4).*P<0.05 and **P<0.01 versus normal, #P<0.05 and ##P<0.01 versus injury.
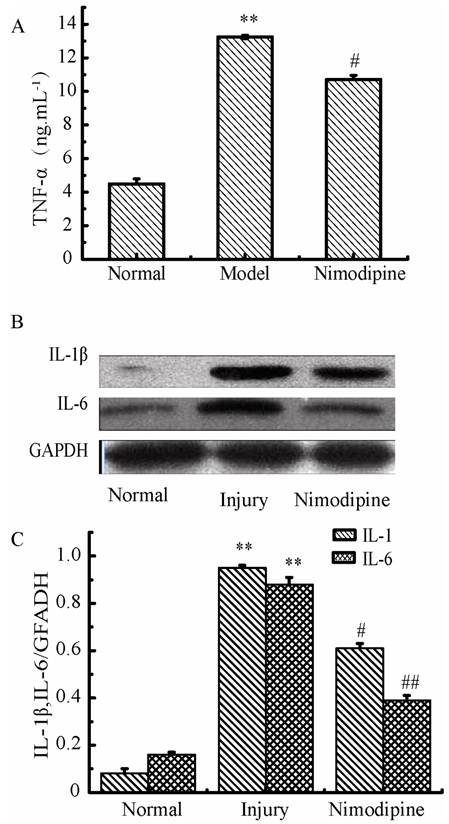
Variations of Inflammation cytokines in triple cell co-culture model after anoxia-reoxygenation
As shown in Fig. 11, the TNF-α, IL-1β and IL-6 were increased significantly in culture supernatants after anoxia-reoxygenation compared with the normal group, which indicated the inflammatory response was activated and involved in cell injury. Nimodipine treatment markedly attenuated the increase of inflammation.
Expressions of inflammation cytokines in triple cell co-culture after anoxia-reoxygenation. (A) TNF-α level in the culture supernatant. (B, C) Expression of IL-1β and IL-6 detected by western blotting, Values represent Mean ± S.E.M (n=4). **P<0.01 versus normal, #P<0.05 and ##P<0.01 versus injury.
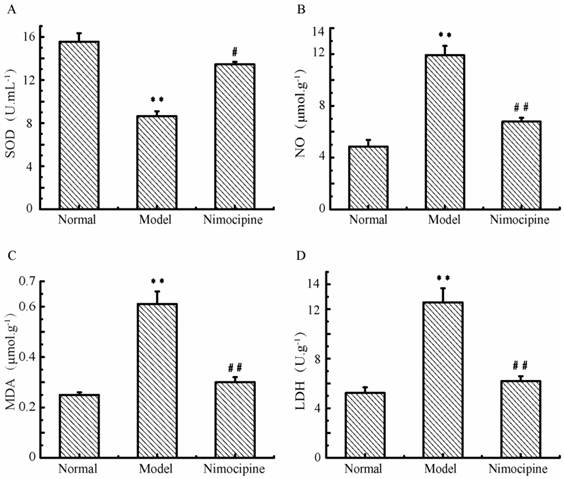
Oxidative damage in triple cell co-culture model after anoxia-reoxygenation
After anoxia-reoxygenation, SOD (Fig.12A) decreased and NO (Fig.12B) increased significantly compared with the normal group, which indicated there were increased free radicals. Meanwhile, LDH (Fig.12C) and MDA levels (Fig.12D) also increased significantly after anoxia-reoxygenation, which indicated there may be cell damage caused by lipid peroxidation and oxidative stress. After nimodipine treatment, these four parameters were improved significantly, indicating an effectiveness of nimodipine on cell injury.
Oxidative damage in triple cell co-culture model after anoxia -reoxygenation. (A) SOD concentration;(B) NO concentration; (C) MDA concentration; (D) LDH concentration; Values represent Mean ± S.E.M (n=4); **P<0.01 versus normal, ##P<0.01 versus injury.
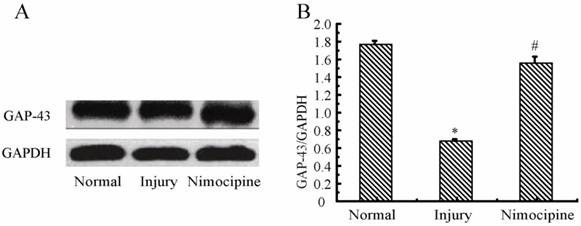
Variations of GAP-43 in triple cell co-culture model after anoxia- reoxygenation
As shown in Fig.13, compared with the normal group, the expressions of GAP-43 after anoxia- reoxygenation decreased obviously, which implied the neuron protection was weakened. After treated with nimodipine, the expression of GAP-43 was increased obviously, which evidenced nimodipine may play a role in promoting the neuron protection.
Expression of GAP-43 and VEGF detected by western blotting in the triple cell co-culture model after anoxia-reoxygenation. Values represent Mean ± S.E.M (n=4). (A) The bands and (B) its relative optical density values of GAP-43 and VEGF, *P<0.05 versus normal, #P<0.05 versus injury.
Discussion
Focusing on neurovascular unit is becoming a prerequisite for investigating the physiology and pathology of brain, screening drug candidates and discovering therapeutic targets. The normal physiological function of brain depends on the interactions among various CNS cells. The neurons and astrocytes depend on the functional integrity of the vascular system to provide oxygen and energy substances [34]. Astrocytes and neurons release a variety of factors such as VEGF, bFGF, EPO, angiopoietin to modulate the migration, proliferation, angiogenesis of endothelial cells and form the sound vascular network [35-37]. Compared with neurons only, neurovascular unit, as a complete structural and functional unit, is more advantaged in representing the integrity of cerebral function and reflecting the intercellular communications of neurons with other type cells under the condition of physiology or pathology, which may be more meaningful in the investigation of cerebral diseases and therapeutic drug screening.
In this study, we have proposed this in vitro neurovascular unit model with a distinct destination compared with the previous multiple cells co-culture models, which only focused on BBB function. In most previous reports, various type cells co-culture were established for brain research, however, mainly limited in two type cells [38-40]. So far, only a few papers have reported the triple cells co-culture used to study the blood brain barrier permeability. Schiera G et al [22-23] established triple cell- co-culture model with immortalized vascular endothelial cells, astrocytes and neurons. Sequentially, Nakagawa S et al [9,21] established primary rat BMECs, astrocytes and pericytes cells co-culture model. These models were mainly applied for studying the BBB permeability and not for studying the intercellular communications during the brain damage and repair within a functional unit. In our work, we have established the triple primary cells co-culture system with primary rat neurons, astrocytes and BMECs. The previous triple cells co-culture model established by Nakagawa S et al [9,21] was absence of neurons. In Schiera G et al's model [22-23], they have utilized the immortalized vascular endothelial cells, which are not much closer to cells in vivo in phenotype than primary cells. Therefore, the tripe primary cell co-culture model established in our study may be more effective on mimicking the in vivo situation and reproducing the cell-cell interactions that control NVU damage and maintenance in the mammalian brain.
NVU is acknowledgedly composed by neurons as the core component together with BMECs, astrocytes, pericyte, basal membrane, microglia and extracellular matrix. Previous investigations have demonstrated that astrocytes play a crucial role between the communications of neurons and BMECs [41-42]. Therefore, neurons, BMECs, and astrocytes are the key components of NVU. Based on these evidences, we have adopted these three types of cells to establish the in vitro NVU model.
The common dysfunctions in CNS contain abnormality of BBB permeability, cellular edema, neuronal degeneration, inflammation, cerebral blood flow and microcirculatory disturbance, and ischemia/reperfusion injury. In our study, we have not only investigated the structure and the most of these functions in vitro NVU model under normal condition, but also compared the changes of this model after anoxia-reoxygenation with the reversing effect of nimodipine as well.
In our experimental results, the alteration of MPP-9, Occludin, SF, HRP, TEER and γ-GT shown in the triple cells model after anoxia-reoxygenation suggested that the damage of BBB structure and function led to significantly increased permeability. The changes of TNF-α, IL-1β and IL-6 implied the activation and participation of inflammation in cell damage. The variations of NO, LDH, MDA and SOD levels indicated the cell damage, especially the cell membrane, may be attributed to effects of free radicals, lipid peroxidation and acidosis induced by lactate accumulation. The decreased expression of GAP-43 after anoxia-reoxygenation indicated the protective effect of neurons became weak, while these changes could be ameliorated by nimodipine. These results demonstrated this triple cell co-culture model showed the basic physiological and pathological processes and characteristics of NVU after anoxia-reoxygenation in the aspects of BBB permeability, ischemia/reperfusion injury and inflammation, etc.
Furthermore, when comparing the protein expression of Pg-P, ZO-1 and values of TEER between the triple cells co-culture model and two types of cells co-culture (astrocytes and BMECs or neurons and BMECs), we found that the existence of astrocytes and neurons could promote the formation of tight junction of BMECs and the increase of TEER. Together with the changes of GAP-43 protein expression after anoxia-reoxygenation, these results may support the presence of communication and interaction among the triple cells in our co-culture model. Therefore, this in vitro NVU model established in our study showed the main characteristics of NVU under both the physiological and pathological condition. However, the investigation in the electrophysiology and neurotransmitter metabolism is not involved in this study. Other factors may also be behind the increase of TEER [43]. All of these questions are needed to be investigated in future.
Conclusion
In this study, we have successfully established the in vitro triple cell- NVU model with the main three types of primary cells of NVU. Furthermore, we have observed the changes of this model after anoxia-reoxygenation injury and also the reversal effect of nimodipine.
Our results demonstrated that this in vitro NVU model has the basic morphological structure and physiological and pathological function. Moreover, it could reflect the integrity of NVU and the interactions among these triple cells. Thus, this model could be employed to assess the physiological and pathological processes of NVU in vitro and investigate the therapeutic targets and agents for several CNS disorders, such as: cerebrovascular disease, Alzheimer's disease, migraine, hypertension and epilepsy.
Acknowledgements
This work was supported by the NSFC Projects (No.81073084; No.81202946), the Projects of Chongqing Science & Technology (CSTC, 2010AC5011, 2012jjA10142), the Key Projects of Chinese Medicine Research of Chongqing Municipal Health Bureau (2010[60] 2010-1-4), the Fundamental Research Funds for Central Universities (No.XDJK2009C081), the Southwest University Doctor Foundation (No.104290-20710906) and the Fundamental Research Funds for the Southwest University Graduate Student (No ky2009017).
Author Contributions
Performed the research: QX, YL, QM, LX; Designed the research study: XX; Contributed essential reagents or tools: GC, WC; Analyzed the data: QM, QX, HQ; Wrote the paper: QX, HQ, QM, XX.
Conflict of Interest
The authors confirm that there are no conflicts of interest.
References
1. Birmingham K. Future of neuroprotective drugs in doubt. Nat Med. 2002;8:5
2. Iadecola C, Li J, Ebner TJ. et al. Nitric oxide contributes to functional hyperemia in cerebellar cortex. The American journal of physiology. 1995;268:R1153-62
3. Guo S, Kim WJ, Lok J. et al. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7582-7
4. Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328-35
5. Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347-60
6. Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399-415
7. del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2003;23:879-94
8. Grotta JC, Jacobs TP, Koroshetz WJ. et al. Stroke program review group - An interim report. Stroke. 2008;39:1364-70
9. Nakagawa S, Deli MA, Kawaguchi H. et al. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochemistry International. 2009;54:253-63
10. Abbott NJ, Hughes CC, Revest PA. et al. Development and characterisation of a rat brain capillary endothelial culture: towards an in vitro blood-brain barrier. Journal of cell science. 1992;103( Pt 1):23-37
11. Diglio CA, Grammas P, Giacomelli F. et al. Primary culture of rat cerebral microvascular endothelial cells. Isolation, growth, and characterization. Laboratory investigation, a journal of technical methods and pathology. 1982;46:554-63
12. Kis B, Deli MA, Kobayashi H. et al. Adrenomedullin regulates blood-brain barrier functions in vitro. Neuroreport. 2001;12:4139-42
13. Veszelka S, Pasztoi M, Farkas AE. et al. Pentosan polysulfate protects brain endothelial cells against bacterial lipopolysaccharide-induced damages. Neurochem Int. 2007;50:219-28
14. M.A D, CA z, NTK D. et al. Immunohistochemical and electron microscopy detections on primary cultures of rat cerebral endothelial cells. In: Boer, AG, Sutanto, W (Eds), drug transport across the Blood-Brain Barrier: In Vivo and In Vitro Techniques. Amsterdam:Harwood Academic Publishers. 1997:.23-28
15. Beetsch JW, Olson JE. Taurine transport in rat astrocytes adapted to hyperosmotic conditions. Brain research. 1993;613:10-5
16. Olson J, Holtzman D. Respiration and cell volume of primary cultured cerebral astrocytes in media of various osmolarities. Brain research. 1982;246:273-9
17. McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890-902
18. Noble M, Murray K. Purified astrocytes promote the in vitro division of a bipotential glial progenitor cell. The EMBO journal. 1984;3:2243-7
19. Singer CA, Figueroa-Masot XA, Batchelor RH. et al. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1999;19:2455-63
20. Suvarna NU, O'Donnell JM. Hydrolysis of N-methyl-D-aspartate receptor-stimulated cAMP and cGMP by PDE4 and PDE2 phosphodiesterases in primary neuronal cultures of rat cerebral cortex and hippocampus. J Pharmacol Exp Ther. 2002;302:249-56
21. Nakagawa S, Deli MA, Nakao S. et al. Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cellular and Molecular Neurobiology. 2007;27:687-94
22. Schiera G, Bono E, Raffa MP. et al. Synergistic effects of neurons and astrocytes on the differentiation of brain capillary endothelial cells in culture. J Cell Mol Med. 2003;7:165-70
23. Schiera G, Sala S, Gallo A. et al. Permeability properties of a three-cell type in vitro model of blood-brain barrier. J Cell Mol Med. 2005;9:373-9
24. Silva RF, Falcao AS, Fernandes A. et al. Dissociated primary nerve cell cultures as models for assessment of neurotoxicity. Toxicol Lett. 2006;163:1-9
25. Maines LW, Antonetti DA, Wolpert EB. et al. Evaluation of the role of P-glycoprotein in the uptake of paroxetine, clozapine, phenytoin and carbamazapine by bovine retinal endothelial cells. Neuropharmacology. 2005;49:610-7
26. Vorbrodt AW, Dobrogowska DH. Molecular anatomy of interendothelial junctions in human blood-brain barrier microvessels. Folia Histochem Cytobiol. 2004;42:67-75
27. Hamm S, Dehouck B, Kraus J. et al. Astrocyte mediated modulation of blood-brain barrier permeability does not correlate with a loss of tight junction proteins from the cellular contacts. Cell and Tissue Research. 2004;315:157-66
28. Franke H, Galla HJ, Beuckmann CT. An improved low-permeability in vitro-model of the blood-brain barrier: transport studies on retinoids, sucrose, haloperidol, caffeine and mannitol. Brain research. 1999;818:65-71
29. Roux F, Durieu-Trautmann O, Chaverot N. et al. Regulation of gamma-glutamyl transpeptidase and alkaline phosphatase activities in immortalized rat brain microvessel endothelial cells. Journal of cellular physiology. 1994;159:101-13
30. DeBault LE, Cancilla PA. gamma-Glutamyl transpeptidase in isolated brain endothelial cells: induction by glial cells in vitro. Science (New York, NY). 1980;207:653-5
31. Lièvre V, Becuwe P, Bianchi A. et al. Free radical production and changes in superoxide dismutases associated with hypoxia/reoxygenation-induced apoptosis of embryonic rat forebrain neurons in culture. Free Radical Biology and Medicine. 2000;29:1291-301
32. Kokura S, Yoshida N, Imamoto E. et al. Anoxia/reoxygenation down-regulates the expression of E-cadherin in human colon cancer cell lines. Cancer Letters. 2004;211:79-87
33. He F, Wu L-X, Shu K-X. et al. HGF protects cultured cortical neurons against hypoxia/reoxygenation induced cell injury via ERK1/2 and PI-3K/Akt pathways. Colloids and Surfaces B: Biointerfaces. 2008;61:290-7
34. Deli MA, Ábrahám CS, Kataoka Y. et al. Permeability Studies on In Vitro Blood-Brain Barrier Models: Physiology, Pathology, and Pharmacology. Cellular and Molecular Neurobiology. 2005;25:59-127
35. Haseloff RF, Blasig IE, Bauer HC. et al. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cellular and Molecular Neurobiology. 2005;25:25-39
36. Bauer HC, Bauer H. Neural induction of the blood-brain barrier: still an enigma. Cellular and Molecular Neurobiology. 2000;20:13-28
37. Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. Journal of Anatomy. 2002;200:629-38
38. Kidambi S, Lee I, Chan C. Primary Neuron/Astrocyte Co-Culture on Polyelectrolyte Multilayer Films: A Template for Studying Astrocyte-Mediated Oxidative Stress in Neurons. Advanced Functional Materials. 2008;18:294-301
39. Gaillard PJ, Voorwinden LH, Nielsen JL. et al. Establishment and functional characterization of an in vitro model of the blood-brain barrier, comprising a co-culture of brain capillary endothelial cells and astrocytes. European journal of pharmaceutical sciences: official journal of the European Federation for Pharmaceutical Sciences. 2001;12:215
40. Hayashi K, Nakao S, Nakaoke R. et al. Effects of hypoxia on endothelial/pericytic co-culture model of the blood-brain barrier. Regulatory peptides. 2004;123:77-83
41. Zonta M, Angulo MC, Gobbo S. et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nature Neuroscience. 2003;6:43-50
42. Moretto G, Xu RY, Monaco S. et al. EXPRESSION AND DISTRIBUTION OF GAP-43 IN HUMAN ASTROCYTES IN CULTURE. Neuropathology and Applied Neurobiology. 1995;21:362-7
43. Stanness KA, Neumaier JF, Sexton TJ. et al. A new model of the blood-brain barrier: co-culture of neuronal, endothelial and glial cells under dynamic conditions. Neuroreport. 1999;10:3725-31
Author contact
![]() Corresponding author: Professor Xiaoyu XU, Laboratory of Molecular Pharmacology, College of Pharmaceutical Sciences & College of Chinese Medicine, Southwest University, Chongqing 400715, China. Tel.: +86-23-6825-0761; Fax: +86-23-6825-1225; E-mail: xuxiaoyuedu.cn.
Corresponding author: Professor Xiaoyu XU, Laboratory of Molecular Pharmacology, College of Pharmaceutical Sciences & College of Chinese Medicine, Southwest University, Chongqing 400715, China. Tel.: +86-23-6825-0761; Fax: +86-23-6825-1225; E-mail: xuxiaoyuedu.cn.