Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Elevated Blood levels of Gastrin...
Re-expression of CCK and gastrin...
Cholecystokinin (CCK) Receptors...
Intracellular Signaling via the...
CCK receptors and other cancers
Therapeutic Strategies to...
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(3):283-291. doi:10.7150/ijbs.14952 This issue Cite
Review
The Role of Gastrin and CCK Receptors in Pancreatic Cancer and other Malignancies
Jill P. Smith1 ![]() , Lionel K. Fonkoua2, Terry W. Moody3
, Lionel K. Fonkoua2, Terry W. Moody3
1. Department of Medicine, Georgetown University, Washington, DC, USA
2. Pennsylvania State University, College of Medicine, Hershey, PA, USA
3. National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
Published 2016-1-28
Abstract
The gastrointestinal (GI) peptide gastrin is an important regulator of the release of gastric acid from the stomach parietal cells and it also plays an important role in growth of the gastrointestinal tract. It has become apparent that gastrin and its related peptide cholecystokinin (CCK) are also significantly involved with growth of GI cancers as well as other malignancies through activation of the cholecystokinin-B (CCK-B) receptor. Of interest, gastrin is expressed in the embryologic pancreas but not in the adult pancreas; however, gastrin becomes re-expressed in pancreatic cancer where it stimulates growth of this malignancy by an autocrine mechanism. Strategies to down-regulate gastrin or interfere with its interface with the CCK receptor with selective antibodies or receptor antagonists hold promise for the treatment of pancreatic cancer and other gastrin - responsive tumors.
Keywords: peptide gastrin
Introduction
Advanced pancreatic cancer has a dismal prognosis [1, 2] and the current chemotherapeutic regimens have not significantly improved survival [3]. With the current rise in incidence of pancreatic cancer it is anticipated that this malignancy will surpass colon and breast cancer in the next decade to become the second leading cause of cancer-related deaths in the USA [4]. In this new era of precision medicine, genomic profiling and target-specific therapies to cancer specific receptors has improved the outcome of many recalcitrant cancers [5, 6]. Immune-based therapies have also led to improvements in cancer survival in the past decade [7], especially for the treatment of advanced melanoma [8]. One reason the survival and response to therapies has been so poor for pancreatic cancer is because the current chemotherapeutic regimens are not target specific.
Similar to other malignancies, pancreatic cancer possesses unique growth-related receptors that when activated stimulate tumor proliferation. One such receptor that has been identified and characterized on pancreatic cancer is the cholecystokinin (CCK) receptor, and this receptor is markedly over-expressed in pancreatic cancer [9, 10]. Activation of the CCK receptor in pancreatic cancer by its ligands gastrin [11] or CCK [12, 13] induces signaling through the AKT pathway [14] resulting in cell proliferation. Gastrin is expressed in the developing fetal pancreas where it is thought to play an important role in pancreatic growth and differentiation [15]; however, gastrin expression is turned off at week 14 in the embryo and becomes re-expressed in precancerous lesions of the pancreas, i.e., pancreatic intraepithelial neoplasias (PanINs) [16]. Furthermore, gastrin is markedly over-expressed in human pancreatic cancer where it has been found to stimulate growth by an autocrine mechanism [17]. In this review, we will present the evidence that the CCK-B receptor pathway is a key driver of pancreatic carcinogenesis and pancreatic cancer growth. The role of the gastrin /CCK and the CCK-B receptor pathway in other malignancies will also be discussed.
Elevated Blood levels of Gastrin and CCK and Cancer risk
Both gastrin [18] and CCK [19, 20] are trophic hormones and have been recognized for years as important regulators of growth of the GI tract and pancreas, respectively. When gastrin [11] or CCK [12, 13] were applied to pancreatic cancer cells in tissue culture, these peptides were found to significantly increase proliferation of the cancer cells. With the era of gastric acid suppressing proton pump inhibitors (PPIs), a concern has been raised as to whether the elevation of serum gastrin that occurs with these medications [21-24] could indeed increase the risk of pancreatic cancer and other cancers arising from tissues with CCK receptors. A large study of nearly 130,000 patients with Helicobacter pylori infections reported an approximate 4-fold increase in colorectal cancers in those with elevated gastrin levels [25]. Numerous studies have now been conducted in subjects such as those with atrophic gastritis, pernicious anemia [26] and Zollinger-Ellison syndrome [27], conditions all associated with markedly elevated gastrin blood levels, and it seems that although hypergastrinemia can increase the risk for enterochromaffin tumors and gastric carcinoids, elevated blood gastrin levels do not appear to act as a carcinogen in and by itself [28] for gastrointestinal adenocarcinomas. However, hypergastrinemia has been shown to increase cancer risk in animal models with precancerous colonic polyps and mutations of the APC gene [29]. Likewise, CCK can increase pancreatic cancer risk if an underlying precancerous lesion exists such as a pancreatic intraepithelial neoplasia (PanIN lesion) [30] and a KRAS mutation [30]. And high CCK blood levels from dietary fat have been shown to promote growth of an established pancreatic cancer in animal models [31, 32]. Subjects with chronic pancreatitis, a chronic inflammatory condition, have been shown to have elevated CCK blood levels [33], and these subjects also have an increased risk for the development of pancreatic cancer. These studies in both animals and humans subjects suggest that gastrin and CCK are not mutagenic by themselves; however, these trophic peptides may increase cancer risk when peptide blood levels are elevated in a subject with a precancerous state (i.e., H. pylori, PanINs, colonic polyps) [34] and also stimulate growth of established cancers that possess CCK receptors.
Re-expression of CCK and gastrin and autocrine pancreatic cancer growth
Both gastrin and CCK mRNA and protein expression have been associated with pancreatic cancer. Normal human and porcine pancreas express bioactive amidated gastrins in the embryonic pancreas [15, 35] however, after birth, gastrin immunoreactivity is found only in the G-cells of the gastric antrum [36] and not in the adult pancreas [37]. Although gastrin is not normally expressed in the adult human pancreas [37], it becomes re-expressed in precancerous PanIN lesions [16] and is commonly expressed in human pancreatic cancers where it has been shown to stimulate growth of GI malignancies by an autocrine mechanism [17]. The autocrine mechanism of gastrin stimulating its own growth is substantiated by the finding that endogenous gastrin from cancer cells has been shown to induce its own transcription by activating the CCK- receptor [38]. Thus, pancreatic cells that produce gastrin embryologically become 'silenced' in the normal adult pancreas until a change occurs during carcinogenesis to reactivate its expression. The mechanism involved with the reactivation of gastrin expression is unknown, although there is some evidence that the re-expression may be regulated by microRNAs which are small noncoding RNAs that modulate the expression of mRNA and proteins. Sp-1 or specific protein-1 is a zinc finger transcription factor that binds to GC rich motifs and is often over expressed in cancers including gastrointestinal cancers [39]. A particular miRNA, miRNA-27a, is upregulated in pancreatic cancer [40] and its role has been linked to the down regulation of ZBTB10/RINZF expression: a novel zinc finger protein that inhibits Sp1-dependent activation of the gastrin gene promoter [41]. Gastrin peptide expression is a ubiquitous and important occurrence in pancreatic cancer. When gastrin expression is stably down-regulated with RNA interference in human pancreatic cancer cells, growth of the primary cancer is inhibited and metastases do not occur [42].
CCK is normally produced in the I-cells of the duodenum and not expressed in the pancreas [43]. Gastrin and CCK peptide and their respective mRNA expressions were examined in human pancreatic cancer surgical specimens by radioimmunoassay and RT-PCR [44]. Although high levels of α-amidated gastrins and its precursor were found 74% of the pancreatic tumor specimens, CCK was not detected [44]. Other investigators have, however, reported CCK immunoreactivity in some pancreatic cancer surgical specimens [45]. The role of endogenous CCK peptide expression in pancreatic cancer was examined, and it was found that tumor production of CCK does not influence growth of pancreatic cancer [46] because down-regulation of cancer CCK mRNA rendered effect compared to controls. Hence, both CCK and gastrin peptides may be present in malignant tissue but only the re-expression of endogenous gastrin stimulates pancreatic cancer growth by an autocrine mechanism.
Cholecystokinin (CCK) Receptors and Pancreatic Cancer
Two classic types of CCK receptors have been cloned and characterized [47, 48] and these include the CCK-A receptor (previously referred to as the CCK-1R) and the CCK-B receptor (previously referred to as the CCK-2R). The predominant receptor type in the normal murine pancreas is the CCK-A type, while the predominant form in the normal human pancreas is that of the CCK-B variety [49]. In cancerous tissue, however, both CCK-A and CCK-B receptor phenotypes have been described. For example, in a carcinogen induced pancreatic cancer model using azaserine, de novo CCK-B receptors become expressed in the animals that develop cancers [50]. Likewise in the engineered KRAS mutant mouse model that spontaneously develops pancreatic cancer through a step-wise progression of dedifferentiated PanIN lesions [51], both CCK-A receptors and the de novo expression of CCK-B receptors are described [52]. Weinburg et al [53] studied the gene expression of CCK receptors in both normal human pancreas tissue and pancreatic cancers. He described only the CCK-B phenotype in normal tissue but expression of both CCK-A and CCK-B receptors in pancreatic cancer [53]. Since some pancreatic cancers possess both types of the CCK receptor, it was unclear if both were functional in mediating the growth stimulatory effects of CCK and gastrin. Experiments demonstrated that in one particular human pancreatic cancer cell line (PANC-1) that expresses both the CCK-A and CCK-B receptors [9, 46], that gastrin or CCK-stimulated cancer growth was mediated only through the CCK-B receptor not the CCK-A receptor [11]. The CCK-B receptor phenotype is the major receptor type in normal human pancreas tissue and present with low abundance; however, The CCK-B receptor increases in number significantly with development of cancer [37].
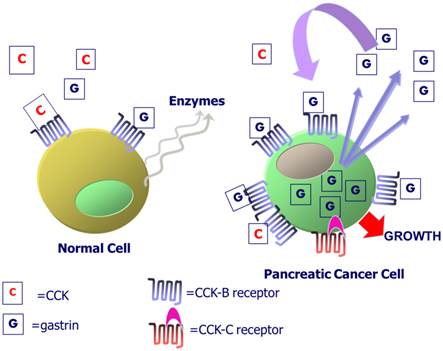
Several mutations and variations of the CCK-B receptor have been described. Song and colleagues [54] described an area in exon 4 that is alternatively spliced resulting in either a “short or long form” of the receptor with a difference of five amino acids in the 3rd intracellular loop. Smith et al described a novel splice variant of this CCK-B receptor in humans (not murine) with alternative splicing of the entire 4th intron resulting in an additional 69 amino acids to the 3rd intracellular loop [55-57], the portion of the receptor involved in GTP-signal transduction and cell proliferation. This alternative splicing results from a single nucleotide polymorphism (SNP) in position 32 of the 4th intron [58] and occurs only in human cancer cells not in normal tissues; hence the mutated CCK-B receptor containing the translated 4th intron has been termed the “CCK-C” or “CCK-cancer” receptor. Transfection of wild type pancreatic cancer cells with the CCK-C receptor accelerates cell growth in culture [58], whereas, down-regulating this 'mutant' receptor, inhibits growth [59]. In fact, the presence of the SNP for the CCK-C receptor has been reported to occur in up to 40% of patients with pancreatic cancer and its presence was associated with a more aggressive course with shortened survival [60]. Some have suggested that the additional 69 amino acids of the CCK-C receptor render the receptor constitutively active where it induces proliferation in even in the absence of gastrin [61]. This splice variant mRNA expression has been detected by RT-PCR in other cancers including insulinomas, bronchial carcinoids, GIST (gastrointestinal stromal tumors) and small cell lung cancer (SCLC) as well as normal tissues [62]. However, it appears that the genetic SNP may be associated with other cancers since it is a germline mutation expressed in about 11-15% of the normal population, however, expression of the CCK-C receptor protein has only been described thus far in those with pancreatic cancer [58]. A diagram of the CCK receptors and autocrine activation of gastrin is shown in Figure 1.
CCK-B receptors in normal and malignant pancreas cells. Exogenous CCK or gastrin from the peripheral blood binds to the CCK-B receptor in normal human pancreas to activate intracellular signaling and the release of digestive pancreatic enzymes. In pancreatic cancers, the CCK-B receptor is over-expressed and gastrin mRNA and peptide are produced within the cancer cell. Gastrin-17 is released from the cancer cell and binds to the CCK-B receptor to stimulate cell growth by an autocrine mechanism. Some patients with the SNP express a CCK-B spliced variant receptor called the CCK-C receptor that is present only in human pancreatic cancer cells.
The role of the CCK-B receptor in regulating EGFR transactivation in pancreatic cancer cells less well understood. The EGFR was detected on 69% of pancreatic adenocarcinoma tumors examined and 39% of tissues derived from patients with chronic pancreatitis [63]. High glucose promotes pancreatic cancer cellular proliferation by inducing EGF expression and transactivation of the EGFR [64]. In a phase II clinical trial, erlotinib (TKI) with gemcitabine relative to gemcitabine alone significantly decreased the hazard ratio for death and increased the one year survival rate of pancreatic cancer patients [65]. The combination of erlotinib and gemcitabine is approved in Europe for first-line treatment of patients with advanced pancreatic cancer [66]. Erlotinib sensitizes pancreatic cancer cells to AZD8055, a mTOR inhibitor [67]. It remains to be determined if CCK-B receptor antagonists will sensitize pancreatic cancer cells to EGFR TKI.
Intracellular Signaling via the CCK-B receptor
G-protein coupled receptors (GPCRs) like the CCK receptors have been demonstrated to modulate growth of gastrointestinal cancers [68, 69]. In a large pancreatic cancer GWAS analysis, Wei and coworkers [70] screened a database of over 3,000 pancreatic cancer patients and found that the GPCR receptor signaling pathway was the most significant pathway predicting pancreatic cancer risk. When the ligands CCK or gastrin bind and/or activate the CCK-B receptor or the splice variant CCK-C receptor, the receptor undergoes a conformational change that allows exchange of GDP for GTP on the Gα subunit of the heterotrimeric G protein complex. Binding of GTP leads to dissociation of the G protein complex into the Gα subunit and the Gβγ dimer [71]. The GTP-bound Gα subunit then interacts with downstream signaling effectors, leading to activation of various second messenger molecules responsible for eliciting cellular responses including growth, proliferation, differentiation, migration and invasion, angiogenesis, and survival. The Gβγ dimer is also capable of activating PI3K and PLCβ, and acting upon ion channels. When activated by ligand, the CCK receptor signals through Gαq of which there are several subtypes.
We previously demonstrated that down regulation of the CCK-B receptor in pancreatic cancer cells results in apoptosis and halts cell proliferation [72] by interference with intracellular signaling. Gastrin stimulation activates Akt phosphorylation through the CCK-B receptor, [73] and down-regulation of the CCK-B receptor in human pancreatic cancer cells inhibits phosphorylation of Akt [72]. Down regulation of the CCK-B receptor in human PANC-1 pancreatic cancer cells [72] also reduces expression of the X-linked inhibitor of apoptosis protein (XIAP). This factor is typically up-regulated in pancreatic cancer [74, 75, 75] where it binds to active caspase-3 [76, 77] and blocks the increased capase-3 activity induced during apoptosis.
Gastrin activation of the CCK-B receptor, can act in a pro-proliferative manner through the activation of mitogen-activated protein kinases (MAPKs) [including the four sub-groups ERK1/2, JNKs, ERK5, and p38-MAPKs] and cyclins. The ERK1/2 pathway mediates several cellular processes regulated by the CCK-B receptor in enterochromaffin cells, which include proliferation and transcription of gastrin-dependent genes [78]. In CCK-B receptor overexpressing CHO cells or AR42J (rat tumor-derived pancreatic acinar cell line), which endogenously express the CCK-B receptor; gastrin stimulates the ERK1/2 pathway in a protein kinase C dependent manner by tyrosine phosphorylation of Shc, which allow interaction with the Grb2/Sos complex [79-81]. Activation of ERK1/2 in response to gastrin stimulation leads to cellular proliferation and activation of the early response gene c-fos. In gastrointestinal epithelial cells, ERK1/2 activation can involve transactivation of the epidermal growth factor receptor (EGFR), where gastrin induces expression and processing of pro-HB-EGF to EGF and leads to the phosphorylation of EGF receptors and activation of ERK1/2 downstream [82, 83].
CCK receptors and other cancers
CCK receptors have been described and characterized in other GI malignancies including gastric cancer [84, 85] and colon cancers [85-87]. Outside the gastrointestinal tract, both CCK-A and CCK-B receptors are abundantly expressed in lung cancer and binding of CCK or gastrin to small cell lung cancer (SCLC) cells causes elevation of cytosolic Ca2+ [88-90]. Although both receptor types are present (as with some pancreatic cancers) it appears that the only the CCK-B receptor is the receptor type involved in malignant cell proliferation since a CCK-B receptor antagonist, CI-988, inhibits SCLC cell growth in vitro and in vivo [91] as well as the ability of CCK-8 to increase cytosolic Ca2+, focal adhesion kinase (FAK), c-fos mRNA, vascular endothelial cell growth factor (VEGF) mRNA.
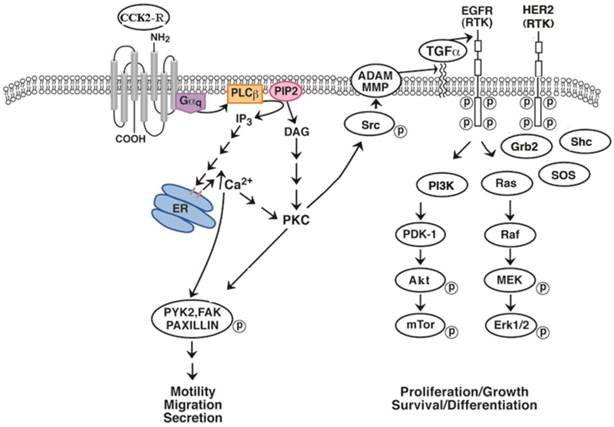
CCK and gastrin may increase the proliferation of some cancers in an EGFR dependent manner. Using NCI-H727 lung carcinoid cells, CCK-8, CCK-8NS and gastrin-17 significantly increased EGFR tyrosine phosphorylation [91]. Figure 2 shows that the activated CCK-B receptor interacts with Gq activating PLCβ and inducing PI turnover. As a result cytosolic Ca2+ is increased and PKC is activated leading to Src phosphorylation. Src phosphorylates numerous proteins such as FAK, paxillin and PYK-2 altering cancer cellular motility and migration. Src activates MMP which cleaves proTGFα to biologically active TGFα which binds to and activates the EGFR. The EGFR can form homodimers with itself or heterodimers with HER2 causing the phosphorylation of protein substrates such as PI3K [92]. PI3K activates PDK-1, Akt and mTOR leading to cancer cellular survival and or differentiation. The EGFR activates the Ras, Raf, MEK and ERK pathway resulting in altered cellular proliferation. The increase in EGFR and ERK tyrosine phosphorylation caused by addition of CCK-8 to lung carcinoid NCI-H727 cells was significantly inhibited by CI-988, PP2, gefitinib and GM6001, a MMP inhibitor [93]. In gastric epithelial cells, gastrin increases MMP9 expression [94]. CCK-8 stimulated secretion of transforming growth factor (TGF) α, an EGFR ligand, from NCI-H727 cells supporting the important role of CCK-8 in MMP activation. An important finding is that CI-988 and gefitinib are synergistic at inhibiting the proliferation of lung carcinoid cells. The results indicate that GPCR antagonists may synergize with TKI at inhibiting the proliferation of epithelial cancers. In lung cancer, resistance to gefitinib develops primarily due to additional EGFR mutations. It remains to be determined if CCK-B receptor antagonists will increase the sensitivity of gefitinib in cancer cells with additional EGFR mutations.
EGFR transactivation. The CCK-B receptor interacts with Gq causing PLC stimulation and PI turnover. The diacylglycerol and IP3 released cause PKC activation and elevation of cytosolic Ca2+ respectively. PKC causes Src phosphorylation leading to FAK, paxillin and PYK2 phosphorylation. Src affects MMP releasing TGFα from its precursor protein causing EGFR tyrosine phosphorylation. The EGFR phosphorylates PI3K activating PDK1, Akt and mTOR. The EGFR interacts with adaptor proteins Grb2 and SOS activating Ras, Raf followed by phosphorylation of MEK and ERK.
Therapeutic Strategies to Interrupt the CCK/ Gastrin: CCK-Receptor Pathway in Cancer
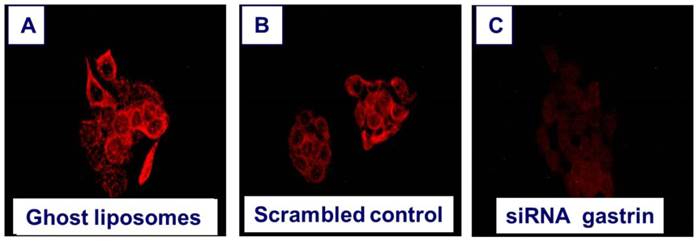
Since gastrin is re-expressed in pancreatic cancer and promotes malignant growth in an autocrine mechanism, obvious strategies to down-regulate or block endogenous production of gastrin have been sought. Treatment of pancreatic cancer cells in culture with antisense oligonucleotides to gastrin inhibited cell growth [95]. When gastrin peptide was stably downregulated by RNAi, the subsequent cell clones failed to grow and metastasize in nude mice [42]. In order to deliver siRNAs to cancer cells in vitro, the siRNAs must be protected from degradation by the nucleases in the peripheral blood; therefore, one strategy that is being developed is the packaging of siRNAs in nanoliposomes or nanoparticles. When nanoliposomes loaded with siRNA to gastrin were applied to gastrin-producing BxPC-3 pancreatic cancer cells in culture, gastrin peptide expression by RT-PCR and immunocytochemistry was significantly decreased compared to the same cells treated with a nanoliposomes containing a nonspecific or scrambled control (Figure 3).
Down regulation of gastrin peptide expression with nanotechnology. Human BxPC-3 pancreatic cancer cells that express high levels of endogenous gastrin were treated for 72 hours with A. nanoliposomes that were empty (ghost) =controls, B. nanoliposomes loaded with a scrambled siRNA =RNA control, or C. nanoliposomes loaded with siRNA specifically directed to gastrin. The cells were washed and reacted with an anti-gastrin antibody (1:500) and secondary goat anti-rabbit AlexaFluor labeled antibody (1:2000), for immunofluorescence analysis of gastrin peptide expression. Immunofluorescent staining was visualized and photographed using Nikon Eclipse upright fluorescent microscope and camera using 40X objective, 2 second exposure and digital zoom of 3.6F. Only the cells treated with nanoliposomes loaded with gastrin siRNA had decreased gastrin immunoreactivity.
Specific gastrin antibodies applied to the culture media of pancreatic cancer cells impairs growth in a dose related fashion [17]. A novel strategy developed by Watson et al [96] involved the raising of an antibody through a vaccine targeted to gastrin called Gastrimmune, or G17DT. This vaccine has a nine amino acid epitope derived from the amino-terminal sequence of gastrin-17 that is conjugated to diphtheria toxoid (DT). The neutralizing ability of rabbit anti-gastrin-17 (G17) antiserum raised by this vaccination was evaluated and found to be effective against colon cancer [96, 97]. G17DT elicited specific and high affinity antibodies that bind gastrin-17, thus preventing its trophic activity. In animals, these antibodies have been shown to reduce the growth and metastatic spread of gastrointestinal tumors [96, 98, 99]. In a randomized placebo controlled clinical study Gilliam and colleagues [100] demonstrated a significant survival benefit in pancreatic cancer patients that elicited neutralizing antibodies toward gastrin in response to vaccination with G17DT. These studies are supportive of the role of gastrin as a key driver in pancreatic cancer growth.
In addition to strategies that neutralize or down regulate gastrin; studies have also been conducted to block the interface with the receptor using CCK- receptor antagonists. More than two decades ago, research in this area of pancreatic cancer therapy was temporarily halted when a clinical study in advanced pancreatic cancer patients with a CCK receptor antagonist failed to demonstrate a clinical benefit [101]. The compound used in this early clinical trial however, (MK-329) was a selective CCK-A receptor antagonist [102]. Since it is now well known that the primary type of receptor that mediates cancer growth in pancreatic cancer in humans is the CCK-B receptor isotype [49], research has been reactivated in this area. Over the past decade numerous highly selective CCK-A and CCK-B receptor antagonists have been developed [103]. The CCK-B receptor antagonist netazepide (YF476) was used to treat patients with type 1 gastric carcinoid tumors [104] and further studies are needed using these more potent and selective antagonists in pancreatic cancer.
Recent studies have shown that inflammation [30, 105] and the conditions of the microenvironment are crucial to carcinogenesis progression. Evidence shows that the CCK receptors may play an important role in PanIN progression and perpetuating the inflammatory milieu of chronic and relapsing pancreatitis[49]. Administration of a CCK analogue, cerulein, has been a classic model for experimental pancreatitis in animals[106] and in the KRAS mouse model administration of exogenous CCK stimulates the CCK receptors and significantly accelerates PanIN progression[30]. The nonepithelial component of the microenvironment in the pancreas has also been shown to play an important role carcinogenesis. These nonepithelial components include activated pancreatic stellate cells, neural factors, endothelial elements, collagen, and inflammatory cells[107]. Berna et al recently described the presence of CCK receptors on pancreatic stellate cells [108] and when these receptors are activated, the stellate cells produce collagen and are responsible for the intense desmoplastic reaction associated with pancreatic cancer [109]. When a nonselective CCK receptor antagonist, proglumide, was added to the drinking water of KRASG12D transgenic mice, progression of pancreatic intraepithelial neoplasias (PanINs) was arrested and fibrosis reversed [52]. These results support the important role of the gastrin-CCK receptor pathway in pancreatic carcinogenesis and its microenvironment. Furthermore, these data suggest that the use of selective CCK receptor antagonists may prevent pancreatic cancer development in high risk subjects.
Conclusion
In order to improve the survival of pancreatic cancer, new approaches are needed. Since gastrin is a key driver of pancreatic cancer that stimulates growth through a markedly over-expressed CCK-B receptor, targeting this interface and pathway should have substantial benefits for patient care. Target specific therapies through selective CCK-B receptor antagonist blockade or strategies to down regulate or neutralize the potent trophic effects of gastrin with nanotechnology and immunotherapy show promise in both pancreatic cancers, gastric carcinoid tumors and lung cancers. Lastly a better understanding of the mechanisms involved at the cellular and molecular level of pancreatic cancer will improve our treatment of patients with this lethal disease.
Acknowledgements
Support for this work was provided in part by the William Donner Foundation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605-1617
2. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9-29
3. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039-1049
4. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913-2921
5. Ma WW, Adjei AA. Novel agents on the horizon for cancer therapy. CA Cancer J Clin. 2009;59:111-137
6. Maitland ML, Schilsky RL. Clinical trials in the era of personalized oncology. CA Cancer J Clin. 2011;61:365-381
7. Buonaguro L, Wang E, Tornesello ML, Buonaguro FM, Marincola FM. Systems biology applied to vaccine and immunotherapy development. BMC Syst Biol. 2011;5:146
8. Palathinkal DM, Sharma TR, Koon HB, Bordeaux JS. Current systemic therapies for melanoma. Dermatol Surg. 2014;40:948-963
9. Smith JP, Rickabaugh CA, McLaughlin PJ, Zagon IS. Cholecystokinin receptors and PANC-1 human pancreatic cancer cells. Am J Physiol. 1993;265:G149-G155
10. Smith JP, Liu G, Soundararajan V, McLaughlin PJ, Zagon IS. Identification and characterization of CCK-B/gastrin receptors in human pancreatic cancer cell lines. Am J Physiol. 1994;266:R277-R283
11. Smith JP, Fantaskey AP, Liu G, Zagon IS. Identification of gastrin as a growth peptide in human pancreatic cancer. Am J Physiol. 1995;268:R135-R141
12. Smith JP, Solomon TE, Bagheri S, Kramer S. Cholecystokinin stimulates growth of human pancreatic adenocarcinoma SW-1990. Dig Dis Sci. 1990;35:1377-1384
13. Smith JP, Kramer ST, Solomon TE. CCK stimulates growth of six human pancreatic cancer cell lines in serum-free medium. Regul Pept. 1991;32:341-349
14. Dufresne M, Seva C, Fourmy D. Cholecystokinin and gastrin receptors. Physiol Rev. 2006;86:805-847
15. Brand SJ, Fuller PJ. Differential gastrin gene expression in rat gastrointestinal tract and pancreas during neonatal development. J Biol Chem. 1988;263:5341-5347
16. Prasad NB, Biankin AV, Fukushima N, Maitra A, Dhara S, Elkahloun AG, Hruban RH, Goggins M, Leach SD. Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of Hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 2005;65:1619-1626
17. Smith JP, Shih A, Wu Y, McLaughlin PJ, Zagon IS. Gastrin regulates growth of human pancreatic cancer in a tonic and autocrine fashion. Am J Physiol. 1996;270:R1078-R1084
18. Johnson LR, McCormack SA. Regulation of gastrointestinal mucosal growth. 3rd: 611-642. 1994
19. Solomon TE, Petersen H, Elashoff J, Grossman MI. Interaction of caerulein and secretin on pancreatic size and composition in rat. Am J Physiol. 1978;235:E714-E719
20. Solomon TE, Vanier M, Morisset J. Cell site and time course of DNA synthesis in pancreas after caerulein and secretin. Am J Physiol. 1983;245:G99-105
21. Brunner G, Creutzfeldt W. Omeprazole in the long-term management of patients with acid-related diseases resistant to ranitidine. Scand J Gastroenterol Suppl. 1989;166:101-105
22. Chubineh S, Birk J. Proton pump inhibitors: the good, the bad, and the unwanted. South Med J. 2012;105:613-618
23. Koop H, Klein M, Arnold R. Serum gastrin levels during long-term omeprazole treatment. Aliment Pharmacol Ther. 1990;4:131-138
24. Lamberts R, Creutzfeldt W, Struber HG, Brunner G, Solcia E. Long-term omeprazole therapy in peptic ulcer disease: gastrin, endocrine cell growth, and gastritis. Gastroenterology. 1993;104:1356-1370
25. Thorburn CM, Friedman GD, Dickinson CJ, Vogelman JH, Orentreich N, Parsonnet J. Gastrin and colorectal cancer: a prospective study. Gastroenterology. 1998;115:275-280
26. Shah P, Rhim AD, Haynes K, Hwang WT, Yang YX. Diagnosis of pernicious anemia and the risk of pancreatic cancer. Pancreas. 2014;43:422-426
27. Orbuch M, Venzon DJ, Lubensky IA, Weber HC, Gibril F, Jensen RT. Prolonged hypergastrinemia does not increase the frequency of colonic neoplasia in patients with Zollinger-Ellison syndrome. Dig Dis Sci. 1996;41:604-613
28. Maddalo G, Spolverato Y, Rugge M, Farinati F. Gastrin: from pathophysiology to cancer prevention and treatment. Eur J Cancer Prev. 2014;23:258-263
29. Watson SA, Smith AM. Hypergastrinemia promotes adenoma progression in the APC(Min-/+) mouse model of familial adenomatous polyposis. Cancer Res. 2001;61:625-631
30. Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382:561-565
31. Matters GL, Cooper TK, McGovern CO, Gilius EL, Liao J, Barth BM, Kester M, Smith JP. Cholecystokinin mediates progression and metastasis of pancreatic cancer associated with dietary fat. Dig Dis Sci. 2014;59:1180-1191
32. Smith JP, Kramer S, Bagheri S. Effects of a high-fat diet and L364,718 on growth of human pancreas cancer. Dig Dis Sci. 1990;35:726-732
33. Garces MC, Gomez-Cerezo J, Alba D, Codoceo R, Vazquez-Munoz E, Arnalich F, Barbado FJ, Vazquez JJ. Relationship of basal and postprandial intraduodenal bile acid concentrations and plasma cholecystokinin levels with abdominal pain in patients with chronic pancreatitis. Pancreas. 1998;17:397-401
34. Orlando LA, Lenard L, Orlando RC. Chronic hypergastrinemia: causes and consequences. Dig Dis Sci. 2007;52:2482-2489
35. Bardram L, Hilsted L, Rehfeld JF. Progastrin expression in mammalian pancreas. Proc Natl Acad Sci U S A. 1990;87:298-302
36. Forte JG, Wolsin JM. HCl secretion by the gastric oxyntic cell. 853-863. 1987
37. Smith JP, Hamory MW, Verderame MF, Zagon IS. Quantitative analysis of gastrin mRNA and peptide in normal and cancerous human pancreas. Int J Mol Med. 1998;2:309-315
38. Kovac S, Xiao L, Shulkes A, Patel O, Baldwin GS. Gastrin increases its own synthesis in gastrointestinal cancer cells via the CCK2 receptor. FEBS Lett. 2010;584:4413-4418
39. Wang L, Wei D, Huang S, Peng Z, Le X, Wu TT, Yao J, Ajani J, Xie K. Transcription factor Sp1 expression is a significant predictor of survival in human gastric cancer. Clin Cancer Res. 2003;9:6371-6380
40. Ma MZ, Kong X, Weng MZ, Cheng K, Gong W, Quan ZW, Peng CH. Candidate microRNA biomarkers of pancreatic ductal adenocarcinoma: meta-analysis, experimental validation and clinical significance. J Exp Clin Cancer Res. 2013;32:71
41. Tillotson LG. RIN ZF, a novel zinc finger gene, encodes proteins that bind to the CACC element of the gastrin promoter. J Biol Chem. 1999;274:8123-8128
42. Matters GL, Harms JF, McGovern CO, Jayakumar C, Crepin K, Smith ZP, Nelson MC, Stock H, Fenn CW, Kaiser J, Kester M, Smith JP. Growth of human pancreatic cancer is inhibited by down-regulation of gastrin gene expression. Pancreas. 2009;38:e151-e161
43. Dockray GJ. Cholecystokinin. Curr Opin Endocrinol Diabetes Obes. 2012;19:8-12
44. Goetze JP, Nielsen FC, Burcharth F, Rehfeld JF. Closing the gastrin loop in pancreatic carcinoma: coexpression of gastrin and its receptor in solid human pancreatic adenocarcinoma. Cancer. 2000;88:2487-2494
45. Tamiolakis D, Venizelos I, Simopoulos C, Lambropoulou M, Kotini A, Jivannakis T, Alexiadis G, Boglou P, Papadopoulos N. Does neoplastic cholecystokinin expression reflect the embryonal pattern of the protein? A study in human pancreas. Acta Medica (Hradec Kralove). 2004;47:101-105
46. Matters GL, McGovern C, Harms JF, Markovic K, Anson K, Jayakumar C, Martenis M, Awad C, Smith JP. Role of endogenous cholecystokinin on growth of human pancreatic cancer. Int J Oncol. 2011;38:593-601
47. Wank SA, Harkins R, Jensen RT, Shapira H, de Weerth A, Slattery T. Purification, molecular cloning, and functional expression of the cholecystokinin receptor from rat pancreas. Proc Natl Acad Sci U S A. 1992;89:3125-3129
48. Wank SA, Pisegna JR, de Weerth A. Cholecystokinin receptor family. Molecular cloning, structure, and functional expression in rat, guinea pig, and human. Ann N Y Acad Sci. 1994;713:49-66
49. Smith JP, Solomon TE. Cholecystokinin and pancreatic cancer: the chicken or the egg? Am J Physiol Gastrointest Liver Physiol. 2014;306:G91-G101
50. Bell RH Jr, Kuhlmann ET, Jensen RT, Longnecker DS. Overexpression of cholecystokinin receptors in azaserine-induced neoplasms of the rat pancreas. Cancer Res. 1992;52:3295-3299
51. Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, Jacks T. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375-387
52. Smith JP, Cooper TK, McGovern CO, Gilius EL, Zhong Q, Liao J, Molinolo AA, Gutkind JS, Matters GL. Cholecystokinin receptor antagonist halts progression of pancreatic cancer precursor lesions and fibrosis in mice. Pancreas. 2014;43:1050-1059
53. Weinberg DS, Ruggeri B, Barber MT, Biswas S, Miknyocki S, Waldman SA. Cholecystokinin A and B receptors are differentially expressed in normal pancreas and pancreatic adenocarcinoma. J Clin Invest. 1997;100:597-603
54. Song I, Brown DR, Wiltshire RN, Gantz I, Trent JM, Yamada T. The human gastrin/cholecystokinin type B receptor gene: alternative splice donor site in exon 4 generates two variant mRNAs. Proc Natl Acad Sci U S A. 1993;90:9085-9089
55. Smith JP, Verderame MF, McLaughlin P, Zagon IS. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Digestion. 1999;60:401-403
56. Smith JP, Verderame MF, McLaughlin PJ, Zagon IS. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Gastroenterology. 1999;116:A1164
57. Smith JP, Verderame MF, McLaughlin P, Martenis M, Ballard E, Zagon IS. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Int J Mol Med. 2002;10:689-694
58. Smith JP, Harms JF, Matters GL, McGovern CO, Ruggiero FM, Liao J, Fino KK, Ortega EE, Gilius EL, Phillips JA III. A single nucleotide polymorphism of the cholecystokinin-B receptor predicts risk for pancreatic cancer. Cancer Biol Ther. 2012;13:164-174
59. Smith JP, Stanley WB, Verderame MF, Zagon IS. Functional significance of the CCK-C receptor in human pancreatic cancer. Pancreas. 2004;29:271-277
60. Smith JP, Whitcomb DC, Matters GL, Brand RE, Liao J, Huang YJ, Frazier ML. Distribution of cholecystokinin-B receptor genotype between patients with pancreatic cancer and controls and its impact on survival. Pancreas. 2015;44:236-242
61. Hellmich MR, Rui XL, Hellmich HL, Fleming RY, Evers BM, Townsend CM Jr. Human colorectal cancers express a constitutively active cholecystokinin-B/gastrin receptor that stimulates cell growth. J Biol Chem. 2000;275:32122-32128
62. Korner M, Waser B, Reubi JC, Miller LJ. CCK(2) receptor splice variant with intron 4 retention in human gastrointestinal and lung tumours. J Cell Mol Med. 2010;14:933-943
63. Bloomston M, Bhardwaj A, Ellison EC, Frankel WL. Epidermal growth factor receptor expression in pancreatic carcinoma using tissue microarray technique. Dig Surg. 2006;23:74-79
64. Han L, Ma Q, Li J, Liu H, Li W, Ma G, Xu Q, Zhou S, Wu E. High glucose promotes pancreatic cancer cell proliferation via the induction of EGF expression and transactivation of EGFR. PLoS One. 2011;6:e27074
65. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960-1966
66. Kundranda M, Kachaamy T. Promising new therapies in advanced pancreatic adenocarcinomas. Future Oncol. 2014;10:2629-2641
67. Wei F, Zhang Y, Geng L, Zhang P, Wang G, Liu Y. mTOR inhibition induces EGFR feedback activation in association with its resistance to human pancreatic cancer. Int J Mol Sci. 2015;16:3267-3282
68. Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79-94
69. Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res. 2010;16:2505-2511
70. Wei P, Tang H, Li D. Insights into pancreatic cancer etiology from pathway analysis of genome-wide association study data. PLoS One. 2012;7:e46887
71. George AJ, Hannan RD, Thomas WG. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 2013;280:5258-5268
72. Fino KK, Matters GL, McGovern CO, Gilius EL, Smith JP. Downregulation of the CCK-B receptor in pancreatic cancer cells blocks proliferation and promotes apoptosis. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1244-G1252
73. Todisco A, Ramamoorthy S, Witham T, Pausawasdi N, Srinivasan S, Dickinson CJ, Askari FK, Krametter D. Molecular mechanisms for the antiapoptotic action of gastrin. Am J Physiol Gastrointest Liver Physiol. 2001;280:G298-G307
74. Lopes RB, Gangeswaran R, McNeish IA, Wang Y, Lemoine NR. Expression of the IAP protein family is dysregulated in pancreatic cancer cells and is important for resistance to chemotherapy. Int J Cancer. 2007;120:2344-2352
75. Satoh K, Kaneko K, Hirota M, Masamune A, Satoh A, Shimosegawa T. Expression of survivin is correlated with cancer cell apoptosis and is involved in the development of human pancreatic duct cell tumors. Cancer. 2001;92:271-278
76. Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300-304
77. Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215-2223
78. Hocker M. Molecular mechanisms of gastrin-dependent gene regulation. Ann N Y Acad Sci. 2004;1014:97-109
79. Seva C, Kowalski-Chauvel A, Blanchet JS, Vaysse N, Pradayrol L. Gastrin induces tyrosine phosphorylation of Shc proteins and their association with the Grb2/Sos complex. FEBS Lett. 1996;378:74-78
80. Daulhac L, Kowalski-Chauvel A, Pradayrol L, Vaysse N, Seva C. Ca2+ and protein kinase C-dependent mechanisms involved in gastrin-induced Shc/Grb2 complex formation and P44-mitogen-activated protein kinase activation. Biochem J. 1997;325( Pt 2):383-389
81. Daulhac L, Kowalski-Chauvel A, Pradayrol L, Vaysse N, Seva C. Src-family tyrosine kinases in activation of ERK-1 and p85/p110-phosphatidylinositol 3-kinase by G/CCKB receptors. J Biol Chem. 1999;274:20657-20663
82. Miyazaki Y, Shinomura Y, Tsutsui S, Zushi S, Higashimoto Y, Kanayama S, Higashiyama S, Taniguchi N, Matsuzawa Y. Gastrin induces heparin-binding epidermal growth factor-like growth factor in rat gastric epithelial cells transfected with gastrin receptor. Gastroenterology. 1999;116:78-89
83. Sinclair NF, Ai W, Raychowdhury R, Bi M, Wang TC, Koh TJ, McLaughlin JT. Gastrin regulates the heparin-binding epidermal-like growth factor promoter via a PKC/EGFR-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2004;286:G992-G999
84. Smith JP, Shih AH, Wotring MG, McLaughlin PJ, Zagon IS. Characterization of CCK-B/gastrin-like receptors in human gastric carcinoma. Int J Oncol. 1998;12:411-419
85. McWilliams DF, Watson SA, Crosbee DM, Michaeli D, Seth R. Coexpression of gastrin and gastrin receptors (CCK-B and delta CCK-B) in gastrointestinal tumour cell lines. Gut. 1998;42:795-798
86. Singh P, Walker JP, Townsend CM Jr, Thompson JC. Role of gastrin and gastrin receptors on the growth of a transplantable mouse colon carcinoma (MC-26) in BALB/c mice. Cancer Res. 1986;46:1612-1616
87. Smith JP, Stock EA, Wotring MG, McLaughlin PJ, Zagon IS. Characterization of the CCK-B/gastrin-like receptor in human colon cancer. Am J Physiol. 1996;271:R797-R805
88. Staley J, Jensen RT, Moody TW. CCK antagonists interact with CCK-B receptors on human small cell lung cancer cells. Peptides. 1990;11:1033-1036
89. Yoder DG, Moody TW. High affinity binding of cholecystokinin to small cell lung cancer cells. Peptides. 1987;8:103-107
90. Staley J, Fiskum G, Moody TW. Cholecystokinin elevates cytosolic calcium in small cell lung cancer cells. Biochem Biophys Res Commun. 1989;163:605-610
91. Moody TW, Jensen RT. CI-988 inhibits growth of small cell lung cancer cells. J Pharmacol Exp Ther. 2001;299:1154-1160
92. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117-1134
93. Moody TW, Nuche-Berenguer B, Moreno PJRT. CI-988 inhibits EGFR transactivation and proliferation caused by addition of CCK/Gastrin to lung cancer cells. J mole Neuroscience. 2015 in press
94. Wroblewski LE, Pritchard DM, Carter S, Varro A. Gastrin-stimulated gastric epithelial cell invasion: the role and mechanism of increased matrix metalloproteinase 9 expression. Biochem J. 2002;365:873-879
95. Smith JP, Verderame MF, Zagon IS. Antisense oligonucleotides to gastrin inhibit growth of human pancreatic cancer. Cancer Lett. 1999;135:107-112
96. Watson SA, Michaeli D, Grimes S, Morris TM, Crosbee D, Wilkinson M, Robinson G, Robertson JF, Steele RJ, Hardcastle JD. Anti-gastrin antibodies raised by gastrimmune inhibit growth of the human colorectal tumour AP5. Int J Cancer. 1995;61:233-240
97. Watson SA, Michaeli D, Grimes S, Morris TM, Robinson G, Varro A, Justin TA, Hardcastle JD. Gastrimmune raises antibodies that neutralize amidated and glycine-extended gastrin-17 and inhibit the growth of colon cancer. Cancer Res. 1996;56:880-885
98. Watson SA, Michael D, Justin TA, Grimes S, Morris TM, Robinson G, Clarke PA, Hardcastle JD. Pre-clinical evaluation of the Gastrimmune immunogen alone and in combination with 5-fluorouracil/leucovorin in a rat colorectal cancer model. Int J Cancer. 1998;75:873-877
99. Watson SA, Michaeli D, Grimes S, Morris TM, Varro A, Clarke PA, Smith AM, Justin TA, Hardcastle JD. A comparison of an anti-gastrin antibody and cytotoxic drugs in the therapy of human gastric ascites in SCID mice. Int J Cancer. 1999;81:248-254
100. Gilliam AD, Broome P, Topuzov EG, Garin AM, Pulay I, Humphreys J, Whitehead A, Takhar A, Rowlands BJ, Beckingham IJ. An international multicenter randomized controlled trial of G17DT in patients with pancreatic cancer. Pancreas. 2012;41:374-379
101. Abbruzzese JL, Gholson CF, Daugherty K, Larson E, DuBrow R, Berlin R, Levin B. A pilot clinical trial of the cholecystokinin receptor antagonist MK-329 in patients with advanced pancreatic cancer. Pancreas. 1992;7:165-171
102. Chang RS, Lotti VJ, Chen TB, Kunkel KA. Characterization of the binding of [3H]-(+/-)-L-364,718: a new potent, nonpeptide cholecystokinin antagonist radioligand selective for peripheral receptors. Mol Pharmacol. 1986;30:212-217
103. Berna MJ, Jensen RT. Role of CCK/gastrin receptors in gastrointestinal/metabolic diseases and results of human studies using gastrin/CCK receptor agonists/antagonists in these diseases. Curr Top Med Chem. 2007;7:1211-1231
104. Fossmark R, Sordal O, Jianu CS, Qvigstad G, Nordrum IS, Boyce M, Waldum HL. Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide (YF476) results in regression of tumours and normalisation of serum chromogranin A. Aliment Pharmacol Ther. 2012;36:1067-1075
105. Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D, Gomez SB, Ji B, Huang H, Wang H, Fleming JB, Logsdon CD, Cruz-Monserrate Z. A High-Fat Diet Activates Oncogenic Kras and COX2 to Induce Development of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology. 2013;145:1449-1458
106. Lampel M, Kern HF. Acute interstitial pancreatitis in the rat induced by excessive doses of a pancreatic secretagogue. Virchows Arch A Pathol Anat Histol. 1977;373:97-117
107. Apte MV, Wilson JS, Lugea A, Pandol SJ. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology. 2013;144:1210-1219
108. Berna MJ, Seiz O, Nast JF, Benten D, Blaker M, Koch J, Lohse AW, Pace A. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J Biol Chem. 2010;285:38905-38914
109. Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, Ramm GA, Buchler M, Friess H, McCarroll JA, Keogh G, Merrett N, Pirola R, Wilson JS. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas. 2004;29:179-187
Author contact
![]() Corresponding author: Email jps261edu; PH: 1-202-687-2020
Corresponding author: Email jps261edu; PH: 1-202-687-2020