Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(6):748-758. doi:10.7150/ijbs.18472 This issue Cite
Research Paper
Oncogenic Protein Kinase D3 Regulating Networks in Invasive Breast Cancer
Yan Liu1, 2*, Jian Li1*, Jun Zhang1, 2*, Zhenghong Yu3*, Shiyi Yu1, 2*, Lele Wu1, 2, Yuzhi Wang1, 2, Xue Gong1, 2, Chenxi Wu1, 2, Xiuxiu Cai1, 2, Lin Mo3, Mingya Wang1, Jun Gu3, Liming Chen1, 2 ![]()
1. The Key Laboratory of Developmental Genes and Human Disease, Ministry of Education, Institute of Life Science, Southeast University, Nanjing 210096, PR China;
2. Jiangsu Key Laboratory for Molecular and Medical Biotechnology, College of Life Science, Nanjing Normal University, Nanjing 210046, China;
3. Department of General Surgery, Jinling Hospital, Medical School of Nanjing University, NanJing 210002, P. R. China.
* These authors contributed equally to this work.
Received 2016-11-24; Accepted 2017-2-24; Published 2017-5-16
Abstract

Protein Kinase D3 (PRKD3) functions as an important oncogenic driver in invasive breast cancer, which is the leading cause of women mortality. However, PRKD3 regulating network is largely unknown. In this study, we systematically explored PRKD3 regulating networks via investigating phosphoproteome, interactome and transcriptome to uncover the molecular mechanism of PRKD3 in invasive breast cancer. Using iTRAQ, 270 proteins were identified as PRKD3 regulated phosphoproteins from 4619 phosphosites matching 3666 phosphopeptides from 2016 phosphoproteins with p-value <0.005. Transcriptome analysis using affymetrix microarray identified 45 PRKD3 regulated genes, in which 20 genes were upregulated and 25 genes were downregulated with p-value <0.005 upon silencing PRKD3. Using Co-IP in combination of MS identification, 606 proteins were identified to be PRKD3 interacting proteins from 2659 peptides. Further network analysis of PRKD3 regulated phosphoproteins, interacting proteins and regulated genes, reveals 19 hub nodes, including ELAVL1, UBC and BRCA1. UBC was recognized as the most common hub node in PRKD3 regulating networks. The enriched pathway analysis reveals that PRKD3 regulates pathways contributing to multiple cancer related events, including cell cycle, migration and others. Enrichment of cell cycle and cell mobility related pathways across PRKD3 networks, explained the observations that depletion of oncogenic PRKD3 led to alternation of cell cycle and decrease of cell migration ability. Taken together, our current study provided valuable information on the roles as well as the molecular mechanisms of PRKD3 in invasive breast cancer.
Keywords: Invasive breast cancer, PRKD3, PRKD3 regulating networks.
Introduction
Invasive breast cancer is the major cancer type in women. Only in United States, approximately 231,840 new cases of invasive breast cancer and 40,290 breast cancer death were expected in 2015 [1]. The Protein Kinase D (PRKD) family of serine/threonine kinases comprises three members in human, PRKD1, PRKD2 and PRKD3. All PRKD isoforms contain a tandem repeat of zinc finger-like cysteine-rich motifs at its N terminus, which is responsible for diacylglycerol or phorbol ester binding, a pleckstrin homology domain, and a catalytic domain at the C terminus that shares homology with the calmodulin-dependent kinases [2, 3]. Although PRKDs share a homologous catalytic domain, they show structural differences in the N-terminal region and in the regions flanked by the C1 and pleckstrin domains [4, 5]. PRKD3 lacks the alanine- and proline-rich region at its N terminus and an autophosphorylation site at its C terminus. In terms of cancer, PRKDs play important roles in tumor-promoting processes including proliferation, angiogenesis, migration, and invasion via linking to a diverse set of cancer regulated signal transduction pathways, such as the AKT, ERK1/2, JNK, CREB, HDAC and RAS/MAPK pathway[6-8]. In breast cancer, PRKD3 is involved in different aspects of oncogenic signaling, such as mTORC1, PAK4/LIMK and GIT1 [9-11]. PRKD3 mediates the activation of S6K1 through mTORC1 to promote cell proliferation [9]. Phosphorylation of GIT1 by PRKD3 regulates Paxillin trafficking and contributes to cell spreading [11]. Furthermore, endogenous high PRKD3 activity contributes to PAK4 activation to facilitate F-actin re-organization and regulated cell migration [10]. Although diacylglycerol (DAG) is found to bind to the cysteine-rich domain of PRKDs and is crucial for PRKD activation through PKC [2]. To our knowledge, the function, especially the mechanical sides of PRKD3 were not well investigated, and the limited studies on the functions and mechanisms of PRKDs are mainly focused on PRKD1. Besides the not-well understood functions of the PRKD3 in invasive breast cancer, the networks regulated by PRKD3 are totally in blank.
To explore the functions and molecular mechanisms of PRKD3 in invasive breast cancer, in current study, via using in vitro and in vivo assays with combination analysis of comparative phosphoproteome, interactome and transcriptome, we revealed important roles and mechanisms of PRKD3 in promoting invasive breast cancer growth and progression.
Materials and methods
Cell culture
MDA-MB-231 and MDA-MB-468 cellss were cultured in Dulbecco's modified essential medium (DMEM) (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (HyClone; NY, USA) and 1% penicillin-streptomycin solution (Life Technologies). T47D cells were maintained in RPMI 1640 medium (Corning Cellgro) supplemented with 10% FBS and 1% penicillin-streptomycin solution. MCF10A cells were cultured in DMEM/F12 media with 10% horse serum, 20 ng/ml epidermal growth factor (EGF), 100 ng/ml cholera toxin, 0.01 mg/ml insulin, 500 ng/ml hydrocortisone and 1% penicillin-streptomycin solution. Cell lines used in current study were all origin from ATCC and used within 6 months after authenticated via the short tandem repeat (STR) typing.
RNA interference
ON-TARGET plus siRNA targeting PRKD3 (Dharmacon; Lafayette, CO) or the non-targeting control siRNA (Dharmacon) was transfected into breast cancer cells using Lipofectamine RNAi MAX(Invitrogen; Carlsbad, CA) according to the manufacturer's instruction. In brief, cells were plated in 6-well plate with 2 ml growth medium without antibiotics per well and grew to 70-80% confluent for siRNA transfection. SiRNAs were transfected into celss via prepare RNAi duplex-Lipofectamine™ RNAiMAX complexes as follows. First, .30 pmol RNAi duplexes were diluted in 100 µl Opti-MEM®I Reduced Serum Medium without serum. Then, 5 µl Lipofectamine™ RNAiMAX was diluted in 100 µl Opti-MEM® I Reduced Serum Medium. After that the diluted RNAi duplexes were added into the diluted Lipofectamine™ RNAiMAXto make RNAi duplex-Lipofectamine™ RNAiMAX complexes and incubated for 10-20 minutes at room temperature. The RNAi duplex-Lipofectamine™ RNAiMAX complexes were added into cells in the well of plates with final siRNA concentration of 10 nM and mixed gently by rocking the plates back and forth. The cells were incubated at 37°C in a CO2 incubator for 6 hours before changing the medium.
Generation of stable cell lines
MDA-MB-231 cell lines with stable depletion of PRKD3 were generated using lentivirus shRNA system. PL-CMV-GFP-WPRE-U3-NHE1 was used to generate PRKD3 lentiviral shRNA constructs with following primers: 5'CTAGCCAAAAAGCTCCTACTTTCTGTGATTACTCTCTTGAAGTAATCACAGAAAGTAGGAGC3' and 5'CTAGCGCTCCTACTTTCTGTGATTACTTCAAGAGAGTAATCACAGAAAGTAGGAGCTTTTTG3'. Production of lentiviral particles go as follws. First, 3.5x106 293T/17 cells were seeded per 10 cm2 cell culture dish and cultured in low antibiotic growth medium (DMEM + 10% FBS + 0.1x Pen/Strep) for 24 hours in a CO2 incubator (37 °C, 5% CO2)to reach ~70% cell confluence. Then, lentiviral particles were produced via transfecting a plasmid mix (10 µg of lentivector, 6.5 µg of pMDL, 2.5 µg of pVSV-G, 3.5 µg of pREV for a 10 cm2 cell culture dish) using the PEI method. In brief, for a 10 cm2 cell culture dish one dish, 70 ul of 1mg/ml PEI was mixed with the plasmid mix and added into cells. The cells were incubated for 18 hours (37 °C, 5% CO2) before changing the media with growth medium (DMEM + 10% FBS + 1x Pen/Strep). The lentiviral particles were produced and added into target cells to generate stable cell lines.
Western blot
Rabbit antibody against PRKD3 was purchased from Cell Signaling Technology (Beverly, MA). β-actin antibody, anti-rabbit secondary antibody , anti-mouse secondary antibody were purchased from Kangchen. HA antibody was purchased from Santa Cruz Biotechnology. Western blot was carried out following the standard procedure. Briefly, protein lysates were separated by SDS-PAGE, transferred to PVDF membranes, and immunoblotted with the respective antibodies as indicated above and in the figures. Blots were developed with Super Signal West Femto Maximum Sensitivity Substrate (Pierce/Thermo Scientific, Rockford, IL).
Cell proliferation, apoptosis, cell cycle assay and cell migration assay
Cell proliferation was measured with the CCK-8 kit (Dojindo Laboratories, Kumamoto, Japan) according to the protocol recommended by the manufacturer. For the cellular apoptosis assay, cells were stained using Annexin-V/Dead Cell Apoptosis Kit (Invitrogen) and analyzed on a BD FACSCalibur flow cytometry (BD Biosciences, Franklin Lakes, NJ). For cell cycle analysis, unsynchronized cells were harvested by trypsinization and fixed with 70% ethanol. Cells were then stained with propidium iodide for total DNA content and the cell cycle distribution was then analyzed using a BD FACSCalibur flow cytometry (Becton Dickinson). For cell migration assay, monolayers of cells were grown on 6 well plates before a cell-free region was created using a 10 μl pipette tip. Scratch wound width was measured using a graticule at 0 h and 12 h post treatment. Representative images were taken at these time points using a Carl Zeiss Microscopy at ×5 magnification.
PRKD3 regulated phosphoproteome analysis using iTRAQ
Cells were prepared using Ready Prep Protein Extraction kit (Bio-RAD). Extracted protein concentration was determined by BioSpec-nano (Shimadzu Biotech, Kyoto). Approximately 4 mg of protein/sample was used for quantitative phosphoproteomic profiling.
Each protein sample was reduced and alkylated and digested with trypsin (Promega). Following tryptic digestion, peptide samples were desalted using MonoTip C18 (Shimadzu Biotech, Kyoto). The eluted peptides were dried in a SpeedVac and then labeled with iTRAQ8-plex reagents according to the manufacturer's instructions.
Phosphopeptide enrichment was performed using TitanspherePhos-TiO kit (Shimadzu Biotech, Kyoto) according to the manufacturer's instructions. Elution of phosphopeptides were combined, acidified with 100mL of 2.5% trifluoroacetic acid, desalted with the MonoTip C18 (Shimadzu Biotech, Kyoto) and resuspended in 0.1% formic acid. Samples were analyzed using Prominence nanoflow LC system (Shimadzu Biotech, Kyoto) connected to an LCMS-IT-TOF mass spectrometer (Shimadzu Biotech, Kyoto). The detected fragments were searched with ProteinLayer software using Swiss-Prot human database and the phosphorylation sites were determined using a PTM Finder Software (Shimadzu Biotech, Kyoto).
Abundance ratios between samples were quantified by LabSolution Software (Shimadzu Biotech, Kyoto) via the quantification of iTRAQ labeled peptides with a synthetic peptide corresponding to the residues between 14 and 38 (“TQCPDDSTCCELPTGK”) of mouse Granulin3 labeled with [d0]/ [d6]-DMPITC used as an internal standard for quantification (BioworldInc, Minnesota). To minimize contaminating near isobaric ions, only the peptides with isolation specificity more than 75% were quantified.
Transcriptome investigation using Affymetrix microarray
Total RNA from cells was isolated using the RNeasy kit (Qiagen, Hilden, Germany) according to the manufacturer's instruction. Total RNA was eluted in a final volume of 30μl (ddH2O) and stored at -80°C until further processing. The GeneChip 3'IVT Express Kit (Cat#901229, Affymetrix, Santa Clara, CA, US) was used to synthesize double-stranded cDNA and produce biotin-labeled cRNA from 500 ng of total RNA. After fragmentation, 10 μg of cRNA were hybridized at 45°C for 16 h to Affymetrix HG_U133 Plus 2.0 oligonucleotide arrays containing probes to more than 47,000 transcripts. Array hybridization and wash was performed using GeneChip® Hybridization, Wash and Stain Kit (Cat#900720, Affymetrix, Santa Clara, CA, US) in Hybridization Oven 645(Cat#00-0331-220V, Affymetrix, Santa Clara, CA, US) and Fluidics Station 450 (Cat#00-0079, Affymetrix, Santa Clara, CA, US) followed the manufacturer's instructions. Slides were scanned by GeneChip® Scanner 3000 (Cat#00-00212, Affymetrix, Santa Clara, CA, US) and Command Console Software 3.1 (Affymetrix, Santa Clara, CA, US) with default settings.
Data preprocessing of Affymetrix microarray gene expression
The raw data of the expression array, CEL files, were input for a series of analyses including quality control, data pre-processing, and identifying differential expressed genes. Briefly, the array was annotated with hgu133plus2 Homo sapiens ensemble genes [12]. The GCRMA [13] was applied for normalization. Then, a t-test based approach [14] was used for calling differentially expressed genes between sample pair-comparisons.
Real-time RT-PCR
Total RNA was extracted using the RNeasy kit (Qiagen, Hilden, Germany) and reverse transcription of RNA was performed using PrimeScript RT reagent kit (TaKaRa, Otsu, Shiga, Japan) according to the manufacturer's instructions. Real-time RT-PCR reactions were performed with SYBRPremix Ex Taq (TaKaRa) in a Bio- Rad CFX96 Real-Time PCR System (Bio-rad, Hercules, CA). β-actin was used as an internal control. The real-time PCR primer sequences are listed in Supplementary Table 12.
Co-immunoprecipitation and MS identification
Co-immunoprecipitation (Co-IP) was performed using special antibody and control HA-antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and Dynabeads Protein G (Invitrogen, Carlsbad, CA) according to manufacturer's instruction. In brief, cell lysates were incubated with anti- antibody-conjugated beads at 4 °C for 2 h. Then, the beads were washed extensively and boiled in SDS loading buffer. MS analysis and western blot were used to study the immnuoprecipitated proteins.
Experiments were performed on a Q Exactive mass spectrometer that was coupled to Easy nLC (ProxeonBiosystems, now Thermo Fisher Scientific). Six microliter of each fraction was injected for nanoLC-MS/MS analysis. The peptide mixture (5 μg) was loaded onto a the C18-reversed phase column (Thermo Scientific Easy Column, 10 cm long, 75 μm inner diameter,3μm resin) in buffer A (0.1% Formic acid) and separated with a linear gradient of buffer B (80% acetonitrile and 0.1% Formic acid) at a flow rate of 250 nl/min controlled by IntelliFlow technology over 140 min. MS data was acquired using a data-dependent top10 method dynamically choosing the most abundant precursor ions from the survey scan (300-1800 m/z) for HCD fragmentation. Determination of the target value is based on predictive Automatic Gain Control (pAGC). Dynamic exclusion duration was 60 s. Survey scans were acquired at a resolution of 70,000 at m/z 200 and resolution for HCD spectra was set to 17,500 at m/z 200. Normalized collision energy was 30 eV and the underfill ratio, which specifies the minimum percentage of the target value likely to be reached at maximum fill time, was defined as 0.1%. The instrument was run with peptide recognition mode enabled.
MS/MS spectra were searched using MASCOT engine (Matrix Science, London, UK; version 2.2) against the UniProtKB human database (149773 entries, downloaded 12/31/15). For protein identification, the following options were used. Peptide mass tolerance=20 ppm, MS/MS tolerance=0.1 Da, Enzyme=Trypsin, Missed cleavage=2, Fixed modification: Carbamidomethyl (C), Variable modification:Oxidation (M)
In vivo mouse model
Female athymic STOCK-Foxn1nu/Nju 4-week-old mice were obtained from Model Animal Research Center of Nanjing University. The stable cell lines (5×106cells) were subcutaneously injected into the armpit of mice respectively. The tumor size was measured for 4 weeks, using a caliper and tumor volume was calculated by the following formula: Volume=0.5×Length×Wideth2. All of the experiments were conducted in accordance with the instructional standard guideline of Southeast University for animal experiments.
Results
PRKD3 promotes breast cancer cell proliferation and breast tumor growth
The expression of PRKD3 was elevated in invasive breast cancer cell lines (MDA-MB-468, MDA-MB-231 and T47D) compared to non-cancerous cell line (MCF10A) (Figure 1A). PRKD3 could be successfully knocked down by anti-PRKD3 siRNAs (Figure 1B). To investigate the functions of PRKD3 in invasive breast cancer, first, we studied the effects of PRKD3 in regulating proliferation of invasive breast cancer cells and found that silencing PRKD3 led to a decrease of cell proliferation of MDA-MB-468, MDA-MB-231 and T47D (Figure 1C).
Oncogenic function of PRKD3 in invasive breast cancer. (A) Elevated expression of PRKD3 in investigated invasive breast cancer cells. (B) Anti-PRKD3 siRNAs silenced PRKD3 in MDA-MB-231. (C) Silencing PRKD3 inhibited proliferations of invasive breast cancer cells. (D) Silencing PRKD3 in MDA-MB-231 reduced xenografts breast tumor growth.
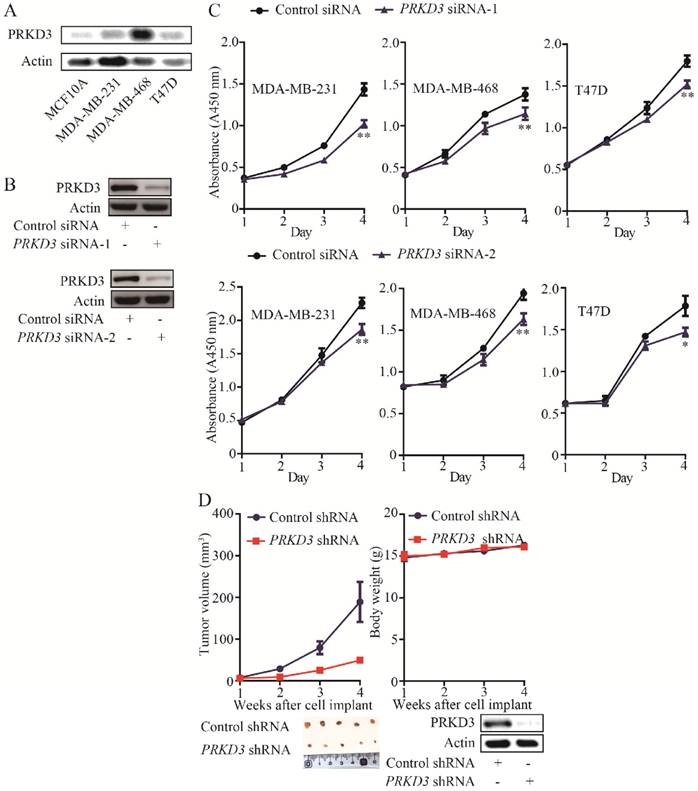
The above findings suggested that PRKD3 promotes breast cancer cell proliferation. Consistently, silencing PRKD3 led to significant reduction of tumor volume in vivo xenograph mouse models (Figure 1D). Taken together, PRKD3 functions as an oncogenic factor in invasive breast cancer.
Since PRKD3 is Serine/Therine kinase, in order to reveal phosphorylation events regulated by PRKD3 in breast cancer cells, comparative iTRAQ labeling phosphoproteome was used (Figure 2A). In total, 3666 phosphopeptides matching 4619 phosphosites from 2016 phosphoproteins were identified with p-value <0.005 (Supplementary Table 1). In detail, 2572 phosphopeptides matching 3057 phosphosites from 1632 phosphoproteins were identified in the PRKD3 phospho-dataset (Figure 2B and Supplementary Table 2).
Phosphoproteome of PRKD3. (A) Flowchart for identification of phosphoproteome of PRKD3. (B) Numbers of identified phosphoproteins, phosphopeptides and phosphosites upon silencing PRKD3. (C) Number of PRKD3 regulated phosphopeptides, phosphosites and phosphoproteins.
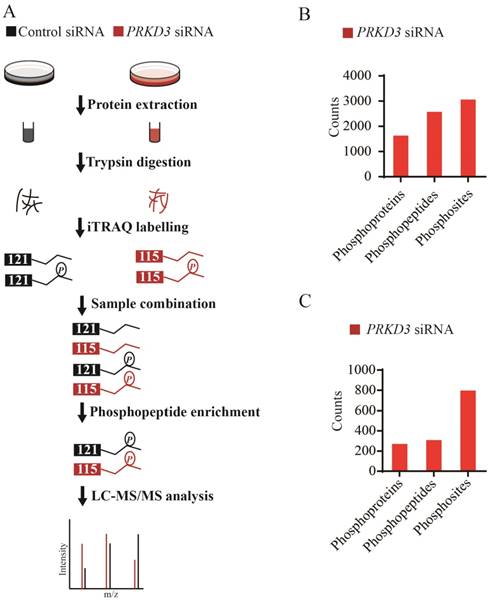
A phosphosite in a phosphopetide of a certain phosphoprotein was regarded to be regulated by PRKD3, if its fold change between the sample and control reached 1.5 fold. In total, 308 phosphopeptides matching 798 phosphosites from 270 phosphoproteins were identified to be PRKD3 regulated (Figure 2C and Supplementary Table 3).
Reactome and network analysis of PRKD3 regulated phosphoproteins
To further characterize the functions of PRKD3, the PRKD3 regulated phosphoproteins were undergone reactome analysis [15]. PRKD3 regulated phosphoproteins were found to be enriched in reactomes involved in various biological events, including cell cycle, programmed cell death, DNA replication, chromatin organization, DNA repair, signal transduction, metabolism, organelle biogenesis and maintenance, cellular responses to stress, gene expression, metabolism of proteins, vesicle-mediated transport, cell-cell communication and so on (Figure S1). In addition, 24 enriched pathways with FDR<0.05 were found in the reactomes of PRKD3 regulated phosphoproteins (Supplementary Table 4). In these 24 pathways, 12 pathways are involved in cell cycle, 5 pathways are involved in metabolism of proteins. Other pathways are involved in various biological events, including gene expression (3 pathways), programmed cell death (2 pathways), chromatin organization (1 pathways) and signaling transduction (1 pathways) (Figure 3A and Supplementary Table 4). These results indicated that PRKD3 was likely to play important roles in cancer related events, for example, cell cycle. This claim was further verified by the observations that silencing PRKD3 exhibited obvious effects on cell cycle, while their effects on apoptosis were almost undetectable (Figure 3B-C). These results suggested that PRKD3 is likely to play important roles in cancer related events, at least, in cell cycle regulation.
Reactome and network analysis of PRKD3 regulated phosphoproteins. (A) Enriched pathways in reactome analysis of PRKD3 regulated phosphoproteins. (B-C) Inspection of cell cycle and apoptosis alternation upon silencing PRKD3. (D) PRKD3 regulated phosphoprotein network with labeled hub-nodes. (E) Cancer related network of PRKD3 regulated phosphoproteins.
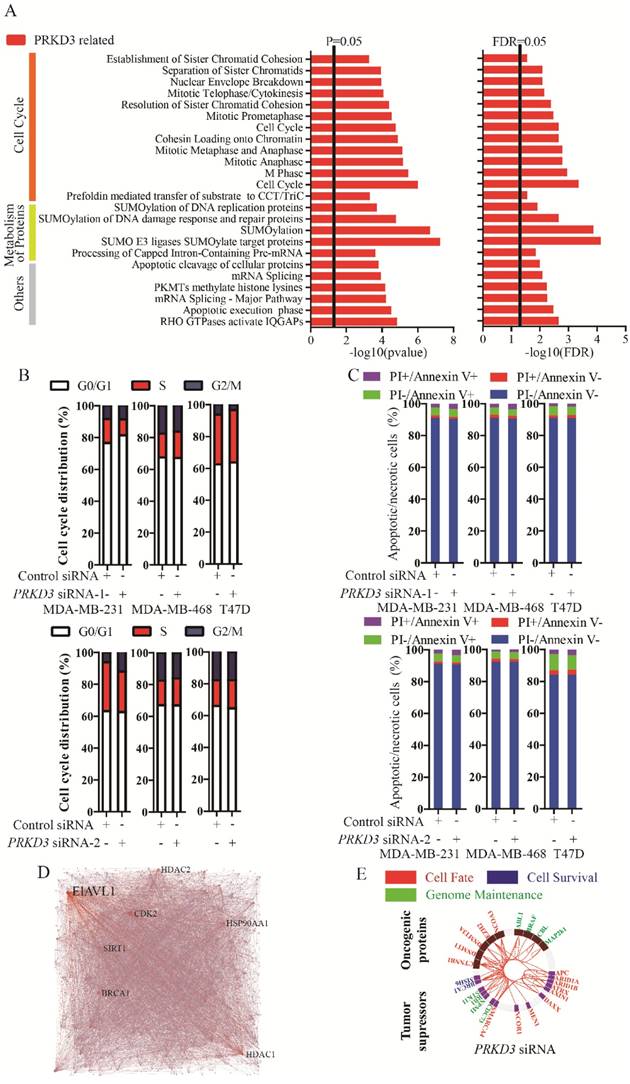
The network of PRKD3 regulated phosphoproteins were further investigated using NetworkAnalyst [16, 17] (Figure 3D, Figure S2 and Supplementary Table 5). The nodes with top 5 largest numbers of degree or betweenness were suggested to be the hub-nodes in the network. In PRKD3 regulated phosphoprotein network, 7 hub-nodes (CDK2, ELAVL1, HDAC1, BRCA1, SIRT1, HSP90AA1 and HDAC2) were identified to be hub-nodes (Figure 3D and Supplementary Table 5). Consistently, cell cycle was found to be one of the most enriched pathways in the enriched pathways in PRKD3 regulated phosphoprotein network (Supplementary Table 5).
When cancer regulated protein regulation networks of PRKD3 were constructed with investigation of known oncogenic proteins and tumor suppressors [18] and only direct interactions were considered, oncogenic proteins and tumor suppressors in the networks contributing to cell fate, cell survival and genomic maintenance were found in PRKD3 regulated phosphoprotein network (Figure 3E and supplementary Table 5).
Identification and analysis of PRKD3 regulated genes
Gene expression is the most fundamental level at which genotype gives rise to the phenotype. However, PRKD3 regulated genes are largely unknown. To further characterize the functions of PRKD3, gene expression profiles upon silencing PRKD3 were investigated using Affymetrix microarrays in duplicates (Supplementary Table 6).
A gene was regarded to be a PRKD3 regulated gene, if its expression fold change between the sample and control reached 1.5 fold with p<0.05 upon silencing PRKD3. In total, 45 genes were identified to be regulated by PRKD3 (Supplementary Table 7). Upon silencing PRKD3, 20 genes were upregulated and 25 genes were downregulated (p<0.05) (Supplementary Table 7).
To validate Affymetrix data of PRKD3, 5 genes were validated using real time RT-PCR and the consistency of these 5 genes expression between real-time RT-PCR and affymetrix experiment were observed (Figure 4B).
Identification and analysis of PRKD3 regulated genes. (A) Heatmap of PRKD3 transcriptome. (B) Real-time RT-PCR validation of PRKD3 regulated genes. (C) Network analysis of PRKD3 regulated genes with labeled hubnodes and differential expression information.
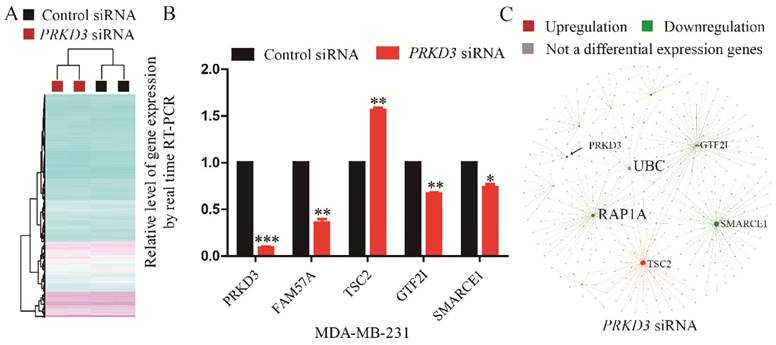
To explore the network of PRKD3 regulated genes, NetworkAnalyst was used and functional hubs in network were identified (Figure 4C, Figure S4 and Supplementary Table 8). Similar to the previous analysis, those nodes which were top 5 of degree or betweenness number were suggested to be the hub-nodes in the network. In PRKD3 regulated gene network, 5 hub-nodes (TSC2, SMARCE1, UBC, RAP1A and GTF2I) were identified (Figure 4C and Supplementary Table 8). In PRKD3 regulated gene network, 3 hub-nodes (SMARCE1, RAP1A and GTF2I) were downregulated, hub-node TSC2 was upregulated and hub-node UBC was not significantly differential regulated upon silencing PRKD3. Be consistent to our previous findings, cell cycle was shown as an enriched pathway in PRKD3 regulated gene network (Supplementary Table 9).
Interactome analysis of PRKD3
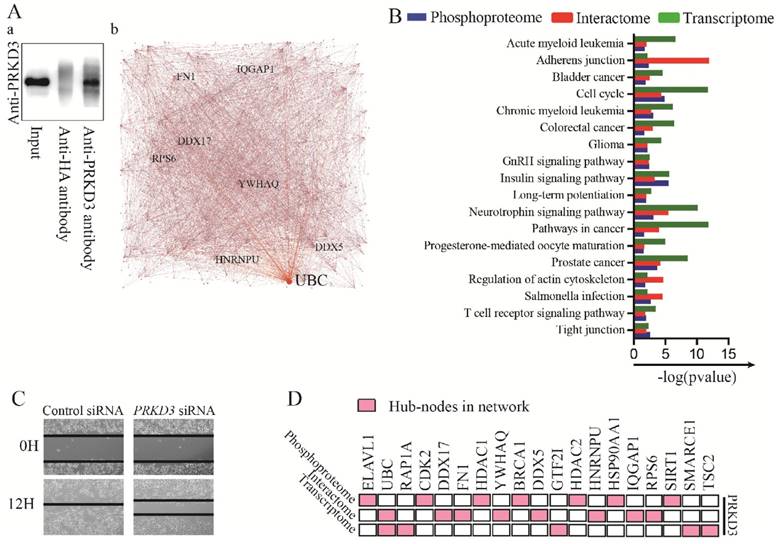
To further explore largely unknown interactome of PRKD3, co-immunoprecipitation (Co-IP) in combination of mass spectrum (MS) identification was used. Those proteins only identified in anti-PRKD3 antibody group, not anti-HA antibody control group were regarded as potential PRKD3 binding proteins (Figure 5A and Supplementary Table 10). In total, we identified 606 proteins from 2659 peptides in PRKD3 interactome. When network of PRKD3 interactome was analyzed and those nodes that were top 5 largest number of degree or betweenness were also suggested to be the hub-nodes in the networks, 8 hub-nodes (UBC, DDX17, IQGAP1, FN1, DDX5, YWHAQ, HNRNPU and RPS6) were identified (Figure 5A-b, Figure S5 and Supplementary Table 10-4). Hub-node UBC was also presented to be a hub-node in PRKD3 interactome network. Cell cycle was also identified to be significantly enriched in PRKD3 interactome (Supplementary Table 10-5).
Analysis of PRKD3 interactome and integrated analysis of PRKD3 regulating networks from phosphoproteome, interactome to transcriptome. (A) Identification of PRKD3 interactome: a. Co-IP of PRKD3 interactome, b. Network with labeled hubnodes of PRKD3 interactome. (B) Comparison of phosphoproteome, interactome and transcriptome common enriched pathways in PRKD3 regulating networks. (C) Migration assays of investigated invasive breast cancer cells upon silencing PRKD3. (D) Integrated analysis of hub-nodes from PRKD3 regulating networks.
Integrated analysis PRKD3 phosphoproteome, interactome and transcriptome
When phosphoproteome, interactome and transcriptome of PRKD3 enriched pathways were compared, 18 enriched pathways across phosphoproteome, interactome and transcriptome were identified in PRKD3 regulatory networks (Figure 5B and Supplementary Table 11). Consistent to the results shown in the above sections, cell cycle was one of these 18 pathways, supporting the roles of PRKD3 in modulating cell cycle progression, promoting cell proliferation and breast tumor growth. In addition, the enriched adherens junction and regulation of actin cytoskeleton across PRKD3 imply the important role of PRKD3 in modulating cancer mobility and contributing to cancer metastasis. Indeed, depletion of PRKD3 was found to be able to reduce cancer cell migration ability (Figure 5C).
When hub-nodes of PRKD3 networks from phosphoproteome, interactome to transcriptome were pooled together, in total 19 hub-nodes of PRKD3 were found (Figure 5D and Supplementary Table 11-2). When the network consisting only hub-nodes was constructed using STRING [19], all the hub nodes were connected and form a new network with UBC as the most distinguishable hub followed by CDK2, BRCA1, HDAC1 and HSP90AA1 (Figure S6 and Supplementary Table 11-3).
Discussion
PRKD3 was suggested to carry out oncogenic functions in invasive breast cancer with the findings showing that the silencing PRKD3 lead to decrease of invasive breast cancer proliferation, migration and in vivo tumor growth (Figure 1C-D, Figure 5C). The oncogenic roles of PRKD3 in invasive breast cancers were also further supported with the reports from other groups: PRKD3 depletion strongly inhibited cell proliferation of TNBC cells [9], silencing PRKD3 significantly inhibited HCC1806 cell proliferation [20], knockdown of PRKD3 or treatment with PRKD inhibitor decreases growth of primary tumors and metastasis in vivo [21]. The important oncogenic roles of PRKD3 in invasive breast cancer can be further supported via observation that PRKD3 regulating networks with hub nodes containing known breast cancer drivers, such as BRCA1 and HDAC2, and the enriched pathways involves in multiple cancer related events including cell cycle, apoptosis, migration, cancer metabolism, cancer immunity and so on (Figure 1, Figure 3-5). Consistently identified enriched cell cycle and cell mobility regulated pathways including adherens junction and regulation of actin cytoskeleton from phosphoproteome, interactome to transcriptome across PRKD3 gives mechanical supports to our observations that depletion of PRKD3 significantly affect the cancer cell cycle and migration ability (Figure 3&5 and Supplementary Table 11). Insulin signaling pathway was well known to play a key role in energy metabolism and growth, also play important roles in neoplasia via modulating both cancer growth and metastasis [22]. Although PRKD3 has been linked to energy metabolism events of invasive breast cancer, the consistent enrichment of Insulin signaling pathway from phosphoproteome, interactorme to transcriptome across PRKD3 strongly indicates that PRKD3 is likely to play a role in energy metabolism events in invasive breast cancer (Figure 5B). Pathway of breast cancer was not identified to be enriched pathways across all PRKD3 phosphoproteome, interactome and transcriptome due to limited studies on the mechanical sides of PRKD3 in breast cancer, while the enriched pathways of 4 other cancers, including bladder cancer, chromic myeloid leukemia, glioma and prostate cancer, at least raised an indication that the identified networks and pathways would shed a light on the breast tumorigenesis and breast cancer development. Although pathway of salmonella infection did not directly linked to breast cancer, salmonella infection was suggested to contribute to the tumor cell transformation of colon cancer and modulate cancer related STAT-β-catenin pathway [23]. In addition, pathway of bacterial invasion of epithelial cells, was reported to be strongly implicated in breast cancer [24]. Activation of T cells through the T cell antigen receptor (TCR) is essential for thymocyte development and effector T cell function as key effectors of the adaptive immune response [25]. T cell receptor signaling pathway was observed to be enriched from phosphoproteome, interactome and transcriptome upon depletion of PRKD3, indicating that PRKD3 might be involved in cancer immunity (Figure 5 and Supplementary Table 11). In the PRKD3 regulating networks, UBC was the most common hub-nodes among the 19 hub nodes across PRKD3 networks from phosphoproteome, interactome to transcriptome. UBC, which encoded a polyubiquitin precursor, is associated with protein degradation to regulate cell signaling pathways to modulate multiple cellular processes including cell cycle [26]. The rest hub nodes contains well known cancer divers, such as BRCA1, indicating the important roles of PRKD3 in invasive breast cancer.
Our current pioneer study of PRKD3 regulating networks from phosphoproteome, interactome, to transcriptome shed light on the oncogenenic roles and mechanisms of PRKD3 in invasive breast cancer at systematically level.
Supplementary Material
Additional File 1Supplementary Table 1.
Supplementary Table 2.
Supplementary Table 3.
Supplementary Table 4.
Supplementary Table 5.
Supplementary Table 6.
Supplementary Table 7.
Supplementary Table 8.
Supplementary Table 9.
Supplementary Table 10.
Supplementary Table 11.
Supplementary Table 12.
Supplementary Figures.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant No.: 81572712 to L. Chen), National Basic Research Program of China (973 Program) (Grant No.: 2015CB965000 to L. Chen), the Fundamental Research Funds for the Central Universities (Grant No.: 3231005410 to L. Chen), grants from Natural Science Foundation of Jiangsu Province (Grant No. : SBK2016030027 to L. Chen), the Six talent peaks project in Jiangsu Province (Grant No.: 2015-JY-002 to L. Chen), Jiangsu Shuangchuang talent program to L. Chen. Natural Science Foundation of Jiangsu Province, China (Grant No.: BK20151403 to J. Li), the Fundamental Research Funds for the Central Universities (Grant No.: 3231006205& 3231006402 to J. Li) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA: a cancer journal for clinicians. 2016;66(1):31-42
2. Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. The Journal of biological chemistry. 2005;280(14):13205-13208
3. Sturany S, Van Lint J, Muller F, Wilda M, Hameister H, Hocker M, Brey A, Gern U, Vandenheede J, Gress T. et al. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J Biol Chem. 2001;276(5):3310-3318
4. Wang QJ. PKD at the crossroads of DAG and PKC signaling. Trends in pharmacological sciences. 2006;27(6):317-323
5. Rykx A, De Kimpe L, Mikhalap S, Vantus T, Seufferlein T, Vandenheede JR, Van Lint J. Protein kinase D: a family affair. FEBS letters. 2003;546(1):81-86
6. Fu Y, Rubin CS. Protein kinase D: coupling extracellular stimuli to the regulation of cell physiology. Embo Rep. 2011;12(8):785-796
7. Chen J, Deng F, Singh SV, Wang QJ. Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCepsilon/PKD3 pathway downstream of Akt and ERK 1/2. Cancer research. 2008;68(10):3844-3853
8. LaValle CR, George KM, Sharlow ER, Lazo JS, Wipf P, Wang QJ. Protein kinase D as a potential new target for cancer therapy. Biochimica et biophysica acta. 2010;1806(2):183-192
9. Huck B, Duss S, Hausser A, Olayioye MA. Elevated protein kinase D3 (PKD3) expression supports proliferation of triple-negative breast cancer cells and contributes to mTORC1-S6K1 pathway activation. The Journal of biological chemistry. 2014;289(6):3138-3147
10. Doppler H, Bastea LI, Borges S, Spratley SJ, Pearce SE, Storz P. Protein kinase d isoforms differentially modulate cofilin-driven directed cell migration. PloS one. 2014;9(5):e98090
11. Huck B, Kemkemer R, Franz-Wachtel M, Macek B, Hausser A, Olayioye MA. GIT1 phosphorylation on serine 46 by PKD3 regulates paxillin trafficking and cellular protrusive activity. J Biol Chem. 2012;287(41):34604-34613
12. Gautier L, Cope L, Bolstad BM, Irizarry RA. affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307-315
13. Lim WK, Wang K, Lefebvre C, Califano A. Comparative analysis of microarray normalization procedures: effects on reverse engineering gene networks. Bioinformatics. 2007;23(13):i282-288
14. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47
15. Stein LD. Using the Reactome database. Curr Protoc Bioinformatics. 2004 Chapter 8:Unit 8 7
16. Xia J, Gill EE, Hancock RE. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat Protoc. 2015;10(6):823-844
17. Xia J, Benner MJ, Hancock RE. NetworkAnalyst-integrative approaches for protein-protein interaction network analysis and visual exploration. Nucleic Acids Res. 2014;42(Web Server issue):W167-174
18. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558
19. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research. 2015;43(Database issue):D447-452
20. Hao Q, McKenzie R, Gan H, Tang H. Protein kinases D2 and D3 are novel growth regulators in HCC1806 triple-negative breast cancer cells. Anticancer research. 2013;33(2):393-399
21. Borges S, Perez EA, Thompson EA, Radisky DC, Geiger XJ, Storz P. Effective Targeting of Estrogen Receptor-Negative Breast Cancers with the Protein Kinase D Inhibitor CRT0066101. Molecular cancer therapeutics. 2015;14(6):1306-1316
22. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nature reviews Cancer. 2008;8(12):915-928
23. Lu R, Wu S, Zhang YG, Xia Y, Zhou Z, Kato I, Dong H, Bissonnette M, Sun J. Salmonella Protein AvrA Activates the STAT3 Signaling Pathway in Colon Cancer. Neoplasia. 2016;18(5):307-316
24. Shigemizu D, Hu Z, Hung JH, Huang CL, Wang Y, DeLisi C. Using functional signatures to identify repositioned drugs for breast, myelogenous leukemia and prostate cancer. PLoS computational biology. 2012;8(2):e1002347
25. Chakraborty AK, Weiss A. Insights into the initiation of TCR signaling. Nat Immunol. 2014;15(9):798-807
26. Sarcevic B, Mawson A, Baker RT, Sutherland RL. Regulation of the ubiquitin-conjugating enzyme hHR6A by CDK-mediated phosphorylation. The EMBO journal. 2002;21(8):2009-2018
Author contact
![]() Corresponding author: Liming Chen Email:chenliming1981edu.cn
Corresponding author: Liming Chen Email:chenliming1981edu.cn