Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Discovery, Structure,...
Function of Amot in normal...
Amot-p80 regulates tube...
Amot-p130 plays a major role in...
Amotl1 controls cell polarity...
Amotl2 regulates the EMT of...
Role of Amot in cancer
Amot regulates Hippo signaling...
Other signaling pathways
Conclusion and future directions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(6):772-781. doi:10.7150/ijbs.19603 This issue Cite
Review
Angiomotin Family Members: Oncogenes or Tumor Suppressors?
Meng Lv1, Yanwei Shen1, Jiao Yang1, Shuting Li1, Biyuan Wang1, Zheling Chen1, Pan Li1, Peijun Liu2, Jin Yang1 ![]()
1. Department of Oncology, the First Affiliated Hospital of Xian Jiaotong University, Xi'an, Shaanxi Province ,710061, P.R. China;
2. Center for Translational Medicine, the First Affiliated Hospital of Xian Jiaotong University, Xi'an, Shaanxi 710061, P.R. China.
Received 2017-2-10; Accepted 2017-4-9; Published 2017-6-1
Abstract

Angiomotin (Amot) family contains three members: Amot (p80 and p130 isoforms), Amot-like protein 1 (Amotl1), and Amot-like protein 2 (Amotl2). Amot proteins play an important role in tube formation and migration of endothelial cells and the regulation of tight junctions, polarity, and epithelial-mesenchymal transition in epithelial cells. Moreover, these proteins regulate the proliferation and migration of cancer cells. In most cancers, Amot family members promote the proliferation and invasion of cancer cells, including breast cancer, osteosarcoma, colon cancer, prostate cancer, head and neck squamous cell carcinoma, cervical cancer, liver cancer, and renal cell cancer. However, in glioblastoma, ovarian cancer, and lung cancer, Amot inhibits the growth of cancer cells. In addition, there are controversies on the regulation of Yes-associated protein (YAP) by Amot. Amot promotes either the internalization of YAP into the nucleus or the retention of YAP in the cytoplasm of different cell types. Moreover, Amot regulates the AMPK, mTOR, Wnt, and MAPK signaling pathways. However, it is unclear whether Amot is an oncogene or a tumor suppressor gene in different cellular processes. This review focuses on the multifunctional roles of Amot in cancers.
Keywords: Angiomotin, cancer, oncogene, tumor suppressor, YAP.
Introduction
Angiogenesis, the process by which new blood vessels are formed from existing vessels, is considered crucial for the growth of malignant tumors [1, 2]. Since the 1990s, several studies have shown that angiostatin, an angiogenesis inhibitor, potently inhibits angiogenesis and the proliferation and migration of endothelial cells in vitro and in vivo. For this reason, a large number of angiogenesis inhibitors have entered clinical trials [3, 4].
Angiomotin (Amot) belongs to the motin family of angiostatin-binding proteins and is characterized by conserved coiled-coil domains and C-terminal PDZ-binding motifs. Alternative splicing allows the encoding of different Amot isoforms by multiple transcript variants. The Amot family contains Amot (p80 and p130 isoforms), Amot-like protein 1 (Amotl1), and Amot-like protein 2 (Amotl2). The expression of Amot differs spatially and temporally, and has a tissue- and cell-specific pattern. The level of Amot changes from embryogenesis to adulthood. The protein is expressed predominantly in endothelial cells of capillaries and in larger vessels of the placenta, where it may mediate the inhibitory effect of angiostatin on tube formation and migration of endothelial cells toward growth factors during the formation of new blood vessels. Therefore, it is commonly accepted that Amot genes are tumor suppressors. However, recent studies suggest that Amot is actually an oncogene that can regulate many tumor cell processes [5, 6]. There is conflicting evidence and a growing debate on the function of Amot in tumors. This review investigates the role of Amot in cancers, with an emphasis on the discovery, structure, expression, location, and function of these molecules.
Discovery, Structure, Expression, and Location of Amot
In 2001, Troyanovsky used a construct encoding kringle domains 1-4 of angiostatin to screen for angiostatin-binding peptides using a placenta yeast two-hybrid cDNA library. The cDNA contained a 2,025-bp open reading frame predicted to encode a 675-amino acid protein with an estimated molecular mass of 72 kD, and this protein was designated angiomotin [7]. Subsequent analysis of GenBank databases identified two additional polypeptides that had a high sequence homology to Amot: Angiomotin-like 1 (Amotl1, a.k.a. JEAP) and Angiomotin-like 2 (Amotl2, a.k.a. MASCOT) [4, 5]. In 2006, Ernkvist identified a 130-kDa splice isoform of Amot that was expressed in different cell types, including vascular endothelial cells and placental cytotrophoblasts [1, 8, 9].
Three members of the Amot family have been identified to date: Amot (p80 and p130 isoforms), Amotl1, and Amotl2. Amot-p80 (NCBI Accession Code NM_133265) has 675 amino acids and an estimated molecular mass of 80 kDa. Amot-p130 (NCBI Accession Code NM_001113490) has 1084 amino acids and an estimated molecular mass of 130 kDa. Amotl1 (NCBI Accession Code NM_130847) has 956 amino acids and a predicted molecular mass of 106 kDa. Amotl2 (NCBI Accession NM_016201) has 780 amino acids and a predicted molecular mass of 86 kDa. Each of these proteins contains conservative coiled-coil domains and one C-terminal PDZ domain. In addition, three L/P-PXY motifs (106-109 aa in LPTY; 239-242 aa in PPEY, and 284-287 aa in PPEY) are located in the N-terminus of Amot-p130. The L/P-P-X-Y motifs are present in Amotl1 and Amotl2 and are highly conserved, except for the third L/P-P-X-Y motif, which is not conserved in Amotl2. However, these motifs are not present in the short isoform of Amot-p80 (Fig. 1) [10].
Domain structures of proteins from the Amot family (Amot-p80, Amot-p130, Amotl1, and Amotl2).
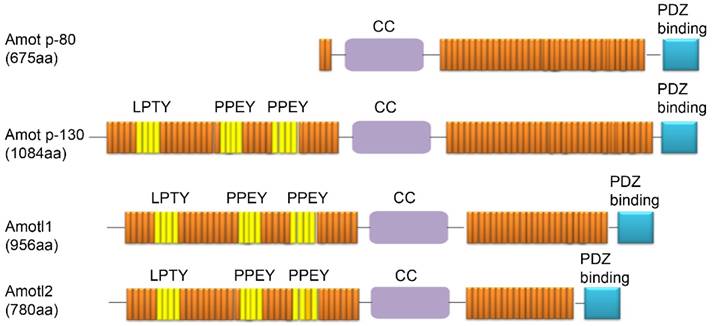
Several studies have evaluated the expression of endogenous Amot family members in many cell lines (Table 1). Endogenous Amot proteins are primarily expressed in endothelial cells (MAE, MS-1, PmT-EC, and HUVEC cells), epithelial cells (HEK293, HEK293T, MDCK, MCF-10A, and HK-2 cells), and some cancer cells (Hela, MCF-7, MDA-MB-468, BC52, MG-63, C4-2B4, 786-o, ACHN, and LCC cells). However, the Amot isoforms identified in the above cell lines are distinct.
The expression and location of Amot family members
| Cell lines | Isoform | Location | Ref. |
|---|---|---|---|
| Endothelial | |||
| MAE | Amot-p80(Ex) | Cytoskeleton | [8,16] |
| Amot-p130(Ex) | Cytoskeleton | [8,16] | |
| Amotl1(En) | Cytoskeleton | [15] | |
| MS-1 | Amot(En) | Cytoskeleton | [15] |
| Amotl1(En) | Cytoskeleton | [15] | |
| Amotl2*En) | TJs | [18] | |
| PmT-EC | Amot-P130(En) | Cytoskeleton | [8] |
| HUVEC | Amotl1(En) | Unknown | [15] |
| Amotl2(En) | Unknown | [29] | |
| Epithelial | |||
| HEK293 | Amot-p80(En) | Nucleus,cytoplasm | [5] |
| Amot-p130(En) | Nucleus,cytoplasm | [5] | |
| Amotl1(En) | Unknown | [59] | |
| HEK293T | Amot-p80(En) | Cytoskeleton | [8,40] |
| Amot-p130(En) | Cytoskeleton | [8,40] | |
| MDCK | Amot(En) | Cytoskeleton | [6] |
| Amot-p80(Ex) | TJs | [20] | |
| Amot-p130(Ex) | TJs ,cytoskeleton, | [20,58] | |
| Amotl2(En) | Cytoskeleton | [58] | |
| MCF 10A | Amot-p130(Ex) | Cytoskeleton | [58] |
| Amotl1(En) | Unknown | [31,41] | |
| Amol2(En) | Unknown | [31] | |
| L | Amot-p80(En) | Cytoskeleton | [46] |
| HK-2 | Amot(En) | Cytoplasm | [6] |
| Cancer | |||
| HeLa | Amot-p130(Ex) | Cytoskeleton | [58] |
| Amotl1(Ex) | Cytoplasm | [31] | |
| Amotl2(Ex) | Cytoplasm | [31] | |
| MCF-7 | Amot(En) | Nucleus | [19] |
| Amot-p130(En) | Unknown | [40] | |
| Amot-p80(Ex) | Cytoplasm | [40] | |
| MDA-MB-468 | Amotl1(En) | Cytoskeleton | [41] |
| BC52 | Amotl1(Ex) | Cytoskeleton | [41] |
| MG-63 | Amot(En) | Unknown | [44] |
| C4-2B4 | Amot-p80(En) | Nucleus,cytoplasm | [46] |
| 786-o | Amot(En) | Nucleus | [6] |
| ACHN | Amot-p80(En) | Unknown | [6] |
| OVCAR3 | Amot-p80(Ex) | Cytoplasm | [50] |
| Amot-p130(Ex) | Cell membrane | [50] | |
| OAW28 | Amot-p80(Ex) | Cytoplasm | [50] |
| Amot-p130(Ex) | Cell membrane | [50] | |
| LCC | Amot(En) | Cytoplasm | [51] |
| H441 | Amotl2(Ex) | Cytoplasm | [62] |
| Other | |||
| SC4 | Amot (En) | Nucleus | [5] |
En: Endogenous; Ex: Exogenous
Moreover, in epithelial and endothelial cells, all Amot isoforms are localized to cell tight junctions (TJs) and play an important role in the apical-basal polarity and stability of the cytoskeleton. Apical-basal polarity involves the interaction of various cell junction complexes, including adherens junctions (AJs), gap junctions, desmosomes, basal lamina hemidesmosomes, and apical TJs [11, 12]. Some studies have shown that all Amot family proteins can interact with Patj at the TJs [13, 14]. Specifically, in epithelial cells (including endothelial cells, HEK 293T cells, L cells, and MDCK cells), either endogenous or exogenous Amot-p80, Amot-p130, Amotl1, and Amotl2 are located to F-actin at the TJs [8, 15-18] (Table 1). However, in HEK 293 cells, SC4 cells, and some cancer cells, endogenous Amot-p130 and Amot-p80 are found in the nucleus of cells, whereas exogenous Amot is expressed in the cytoskeleton and cytoplasm (Table 1) [5, 6, 19].
In summary, in epithelial and endothelial cells, Amot is localized primarily to the cytoskeleton. In most cancer cells, endogenous Amot is localized to the nucleus and exogenous Amot is expressed in the cytoskeleton and cytoplasm. The reasons may be that: 1. The three Amot proteins could not be detected using available antibodies at the time; 2. The type, state, and environment of cells are different, including their process and density; and 3. Endogenous and exogenous Amot are located in various cell regions. However, it is not known whether the different expression patterns and cell-specific features lead to multifunctional roles.
Function of Amot in normal epithelial cells
Previous studies reported that Amot mediated the inhibition of migration and tube formation of endothelial cells by angiostatin [7]. Amot family proteins share similar functions because of their structural similarities. However, some studies have shown that different Amot isoforms play specific roles in different cell types. In some cases, studies from different research groups reported contradictory findings in the same cell line, suggesting that isoforms, cell types, and experimental conditions could significantly affect the function of Amot. Table 2 summarizes the functions of Amot family members in normal cells.
The Function of Amot in normal cells or tissues
| Isoform | Cell or Tissue types | Function | Ref. |
|---|---|---|---|
| Amot-p80 | MAE | Increasing the migration and the tube formation | [7,8,20] |
| MDCK | Inhibiting the function of Amot-p130 | [20] | |
| Placenta, healthy rats | Angiogenesis | [20,21] | |
| Amot-p130 | Placenta | Stabilization and maturation of the blood vessels | [20] |
| MAE | Regulating cytoskeleton organization and cell shape | [8,20] | |
| HEK293,HEK293T, MDCK, MCF10A | Activating Hippo signaling | [17,56-61] | |
| Amotl1 | MAE | Regulating the endothelial migration and tight junction formation | [9,23] |
| Zebrafish | Controling cell polarity and paracellular permeability | [15] | |
| MAE | Increasing the velocity of migration | [15,23] | |
| Retina | Angiogenesis | [25] | |
| HEK293T | Inhibiting the activation of YAP | [17] | |
| Amotl2 | Zebrafish | Regulating cytoskeletal organization | [18] |
| Umbilical vein endothelial cells, MAE cells | Promoting cell proliferation and migration and Regulating cell polarity | [29] | |
| Embryos | Affecting the final pattern and size of sensory organs | [30] | |
| MCF10A | Inhibiting a change of cell morphology, epithelial-mesenchymal transition and the AKT and ERK pathways | [31,32] | |
| Muscle cell | Supporting the involvement of podosomes | [33,34] | |
| HEK293T | Inhibiting the activation of YAP | [17,56] | |
| Zebrafish | Attenuating Wnt/β-catenin signaling | [30,68,69] | |
| Angiogenic endothelial cells | Promoting MAPK/ERK activation | [29] |
Amot-p80 regulates tube formation and migration of endothelial cells
The analysis of in vivo data indicates that Amot isoforms are differentially expressed during angiogenesis. Amot-p80 is expressed in the placenta from embryonic day (E) 11 to the end of gestation, whereas Amot-p130 is expressed in the placenta from E13 to E16 [8]. The analysis of Amot expression in the retina at different postnatal stages indicated that Amot-p80 was highly expressed between postnatal day (P) 3 and P5. However, Amot-p130 was expressed after P7. This result suggests that Amot-p80 is primarily expressed in the early stages of blood vessel formation, during which the major function of endothelial cells is to migrate, whereas Amot-p130 is expressed during the stages of stabilization and maturation of blood vessels [20]. In addition, Amot-p80 regulates the function of Amot-p130. In MDCK cells, Amot-p80 expression results in the translocation of Amot-p130 from TJs, thereby inhibiting the stabilizing function of Amot-p130 [20].
Amot-p80 primarily regulates the migration of endothelial cells. The expression of Amot-p80 in mouse aortic endothelial (MAE) cells increases cellular migration and induces a migratory phenotype [7, 8]. Moreover, in healthy non-obese rats, exercise-induced angiogenesis may involve an increase in the expression of Amot-p80 [21].
Amot-p130 plays a major role in the control of cell shape
In MAE cells, Amot-p130 is predominantly localized to TJs (E-cadherin and Claudin-1) and regulates cytoskeleton organization and cell shape through the N-terminal region of the protein. However, Amot-p130 does not promote cell migration and does not respond to angiostatin [8, 20]. However, in HEK293 cells, Amot-p130 binds to F-actin at TJs. Phosphorylated Amot-p130 proteins are not localized to TJs and inhibit stress fiber and focal adhesion formation, suggesting that the phosphorylation of Amot inhibits cell migration [22].
Amotl1 controls cell polarity and cell-cell junctions of endothelial cells
Amotl1 and Amot (Amot-p80 and Amot-p130) have similar effects on endothelial migration and TJ formation. Amotl1 shares many features with Amot-p130, including co-localization to F-actin at TJs [9, 23]. Moreover, Amotl1 controls cell polarity and paracellular permeability during zebrafish embryogenesis. The knockdown of Amotl1 causes vascular deficiency during zebrafish embryogenesis [15]. Amotl1 interacts with Amo-p80 through its coiled-coil domain and increases the rate of migration of MAE cells [15, 23]. Furthermore, Amotl1 regulates the function of YAP, pericyte morphology, and coverage of blood vessels [24, 25].
Amotl2 regulates the EMT of epithelial cells and involvement of podosomes in muscle cells
The expression of Amotl2 is cell-type and tissue-specific. Amotl2 is predominantly expressed in the heart and skeletal muscle and was not detected in any cell type of hematopoietic origin investigated (U937, K562, PLB) [26]. Amotl2 regulates cytoskeletal organization and apical TJs [27]. Amotl2 knockdown inhibits cell proliferation and migration and disrupts cell polarity in cultured human umbilical vein endothelial cells and MAE cells [28, 29]. However, some studies show that the knockdown of Amotl2 leads to the over-proliferation and increase in the size of the lateral line primordium (LLP), thus affecting the final pattern and size of sensory organs [30]. Moreover, in mammary epithelial cells (MCF10A), the downregulation of Amotl2 promotes changes in cell morphology and epithelial-mesenchymal transition (EMT) [31, 32]. Amotl2 also plays a crucial role in synaptic maturation by regulating the involvement of podosomes in muscle cells [33, 34]. In addition, under hypoxia conditions, cells preferentially express the Amotl2 isoform. Mojallal et al. found that hypoxia regulated AmotL2 by increasing the binding of c-Fos. C-Fos can bind to the Amotl2 promoter to mediated AmotL2 transcription [27].
The degradation of Amot requires E3 ligases, but different Amot isoforms require different E3 ligases. The WW domains of Nedd4-like ubiquitin E3 ligases (Nedd4, Nedd4-2, and Itch) bind to the L/P-PXY motifs of Amot and subsequently target Amot-p130 and Amotl1 for ubiquitin-dependent degradation. However, Nedd4 does not affect the protein levels of Amotl2 and Amot-p80. Amotl2 has a tyrosine-to-phenylalanine substitution in the third L/P-P-X-Y motif compared to p130-Amot and Amotl1, which suggests that the third L/P-P-X-Y motif in the N-terminal region may be necessary for Nedd4-mediated degradation [10]. Tankyrase binds and targets Amotl2 for ubiquitin-dependent degradation [35]. Therefore, tankyrase inhibitors stabilize Amot proteins [35, 36]. In addition, E3 ligase HECW2 physically interacts with Amotl1 and enhances its stability [37].
In summary, distinct Amot isoforms play specific roles in normal epithelial cells. Amot-p80 regulates tube formation and migration of endothelial cells, whereas Amot-p130 plays an important role in the control of cell shape. Amotl1 controls cell polarity and cell-cell junctions of endothelial cells. Amotl2 regulates the EMT of epithelial cells and the involvement of podosomes in muscle cells.
Role of Amot in cancer
The cytoplasmic and nuclear expression of Amot proteins has been demonstrated in various tumors and cell lines. However, there is a debate about whether Amot is an oncogene or a tumor suppressor gene. Table 3 summarizes the functions of Amot family members in cancers. In this section, we comprehensively review the functional role of Amot in cancers.
The Function of Amot in cancers
| Amot Family | Cancer Types | Function | Possible Mechanism | Ref. |
|---|---|---|---|---|
| Amot | Breast cancer | Oncogene | Increasing the expression of YAP, TAZ and LATS1 and enhancing ERK1/2 pathway | [19,38-40] |
| Osteosarcoma | Oncogene | Unknown | [44,45] | |
| Amot-p80 | Prostate cancer | Oncogene | Unknown | [46] |
| HNSCC | Oncogene | Unknown | [47] | |
| Amot-p130 | Hepatic carcinoma | Oncogene | Augmenting the activity of YAP | [5] |
| Renal cell cancer | Oncogene | Increasing the YAP-related TEAD promoter activity. | [6] | |
| Ovarian cancer | Tumor suppressor | Inhibiting the activation of YAP target genes | [50] | |
| Lung cancer | Tumor suppressor | Sequestering oncogenic YAP/TAZ and decreasing Cyr61 expression | [51] | |
| Amotl1 | Breast cancer | Oncogene | Stimulating Src activity | [41] |
| Cervical cancer | Oncogene | Unkown | [48] | |
| Amotl2 | Colon cancer | Oncogene | Disrupting the apical-basal polarity. | [27] |
| Glioblastoma | Tumor suppressor | Inhibiting the activation of YAP target genes | [49] |
Amot proteins are overexpressed in some tumors and promote tumor cell proliferation and invasion. Below we review the oncogenic roles of Amot in cancers.
Breast cancer
Breast cancer tissues express significantly higher levels of Amot transcripts compared with normal mammary tissues. The levels of Amot are significantly higher in grade 2 and grade 3 tumors compared to grade 1 tumors. Furthermore, high levels of Amot transcript are associated with shorter overall survival. The expression levels of Amotl1 and Amotl2 are non-significantly higher in tumors than in normal tissues [38]. Similarly, immunohistochemistry shows that Amot is highly expressed in breast cancer tissues, particularly in the center of these tissues, compared with adjacent non-cancerous tissues. In addition, the expression levels of Amot were increased in specimens from patients with a high level of Ki-67, which is an immunohistochemical proliferation marker in many types of cancer [19]. Furthermore, the downregulation of Amot decreases the growth and invasion of MCF-7 cells in vitro [19, 39]. The expression of YAP, YAP/TAZ, and LATS1 is significantly decreased in MCF-7 cells following the knockdown of Amot. Moreover, the expression of YAP is obviously decreased in the nucleoprotein [19]. The expression of Amot-p80 enhances the ERK1/2-dependent proliferation of MCF7 cells [40]. The results of a large cohort of human breast tumors indicate that Amotl1 protein levels are upregulated during cancer progression and that Amotl1 expression is upregulated in tumors with ER-negative status. Moreover, Amotl1 expression stimulates the proliferation and migration of breast cancer cells (MDA-MB-468 cells) [41]. Amotl1 promotes the proliferation of breast cancer cells in vitro and in vivo by stimulating Src activity, possibly by localizing it to specific domains where it can be activated. The elevated activity of c-Src tyrosine kinase, which is also known as proto-oncogene tyrosine-protein kinase Src, may be linked to cancer progression by stimulating other signaling pathways [41]. In summary, Amot and Amotl1 act as oncogenes in breast cancer and play a major role in the activity of YAP, ERK1/2, and Src.
Moreover, some vaccines against Amot-p80 have been studied for cancer therapy. Holmgren found that Amot administered via DNA vaccination could inhibit pathological angiogenesis and tumor growth in BALB/C mice [42]. The period of onset of breast cancer in BALB-neuT female offspring born from and fed by pAmot-vaccinated mothers was significantly delayed compared to female offspring born from and fed by control vaccinated mothers [43].
Colon cancer
The expression of Amotl2 increases in more than 50% of colon cancers (Dukes B-C). Kaplan-Meier analysis indicates that Amotl2 is associated with shorter overall survival and is a marker of poor prognosis in colon cancer. Furthermore, Amotl2 promotes tumor formation and invasion in vivo [27] and disrupts apical-basal sorting of membrane proteins by mislocalizing the Par3 and Crb3 polarity complexes, promoting cell invasion [27]. In summary, Amotl2 acts as an oncogene in colon cancer by disrupting the apical-basal polarity.
Hepatic carcinoma and renal cell cancer
After liver injury, Amot deletion impedes the proliferation of biliary epithelial cells and tumorigenesis. Amot-p130 promotes the nuclear translocation of YAP and the consequent proliferation of Nf2-deficient cells by inhibiting the interaction between YAP and Lats1/2 [5]. In renal cell cancer (RCC), Amot is expressed predominantly in the nucleus of RCC cells and tissues, and in the cytoplasm and nucleus of renal epithelial cells and paracancerous tissues. Furthermore, the silencing of Amot inhibits the proliferation of non-tumor renal epithelial HK-2 and RCC 786-O cells, whereas the upregulation of Amot triggers the proliferation of ACHN cells [6]. In hepatic carcinoma and RCC cells, the expression of Amot-p130 increases the expression of nuclear YAP, CTGF, and Cyr61, and acts as a YAP cofactor, preventing YAP phosphorylation and augmenting its activity toward a specific set of genes that facilitate tumorigenesis [5, 6]. In summary, Amot-p130 acts as an oncogene in hepatic carcinoma and RCC by augmenting the activity of YAP.
Osteosarcoma, prostate cancer, head and neck squamous cell carcinoma, and cervical cancer
In osteosarcoma, Amot mRNA expression is upregulated compared with normal tissues and cells. The downregulation of Amot suppresses cell proliferation, migration, and invasion [44, 45]. In prostate cancer cells, Amot-p80 has been shown to be a novel component of the Cad11 complex and increases cell migration and tumor growth [46]. In head and neck squamous cell carcinoma (HNSCC) cells, the transient transfection of Amot-p80 induces an increase in the proliferation and migration of HNSCC cells [47]. In Hela and C33A cells, the knockdown of Amotl1 inhibits cell migration, whereas the overexpression of Amotl1 stimulates cell migration [48]. In summary, Amot family members as oncogenes in osteosarcoma, prostate cancer, head and neck squamous cell carcinoma, and cervical cancer, but the mechanism is unclear until now.
On the other hand, in some cancer cell lines, Amot inhibits cell proliferation and invasion. Below we review the tumor suppressor roles of Amot in cancers.
Glioblastoma
In primary glioblastoma cells, the overexpression of Amotl2 in U87 cells inhibits foci formation, growth, and metastasis in vitro and in vivo. In U87 cells, Amotl2 binds to YAP, inhibits nuclear translocation and the subsequent activation of YAP target genes, and reduces cell proliferation and invasion [49]. In summary, Amotl2 acts as a tumor suppressor in glioblastoma cells by inhibiting the activation of YAP target genes.
Ovarian cancer and lung cancer
In ovarian cancer, the overexpression of Amot-p130 inhibits the growth and soft agar colony formation of ovarian cancer cells in vitro [50]. In lung cancer, Amot expression is significantly decreased in clinical lung cancer and highly proliferative and invasive lung cancer cells (LCC). Furthermore, the knockdown of Amot-p130 in lung cancer cell lines promotes cancer proliferation, migration, invasion, and EMT [51]. In ovarian serous cancer cells (OVCAR3 and OAW28) and LCC, the knockdown of Amot-p130 promotes cancer progression by decreasing cytoplasmic sequestration and increasing the nuclear translocation of oncogenic co-activators YAP/TAZ, leading to the increase in the expression of Cyr61, which is a YAP target gene [50, 51]. In summary, Amot acts as a tumor suppressor in ovarian cancer and lung cancer by inhibiting the activation of YAP target genes.
Lymphoma
A somatic variant of Amotl1 has been identified in a pediatric case of nodal marginal zone lymphoma. However, Amotl1 mutations were not observed in pediatric cases of follicular lymphoma [52].
In summary, the knockdown of Amot decreases cellular proliferation and migration in breast cancer, osteosarcoma, colon cancer, prostate cancer, head and neck squamous cell carcinoma, cervical cancer, liver cancer, and RCC. However, in primary glioblastoma, ovarian cancer, and LCC, Amot inhibits cellular growth and migration. The difference may be due to the following reasons: 1. The function of Amot is cell-specific and tissue-specific; 2. The expression pattern of Amot proteins differs in the cell, i.e., most of the endogenous Amot is present in the nucleus [5, 6, 19], whereas exogenous Amot is usually expressed in the cytoplasm [5, 49]; 3. Amot isoforms assume distinct functions. Moreover, several other mechanisms may cause this phenomenon, including Hippo-YAP and ERK signaling pathways. In most reports, Amot acts as an oncogene or a tumor suppressor in tumor cells by augmenting or inhibiting the activity of YAP. Recently more and more studies have been primarily focused on the regulatory role of Amot in Hippo-YAP signaling. Below we review the relationship between Amot and YAP.
Amot regulates Hippo signaling pathway
The Hippo signaling pathway is involved in an evolutionarily conserved kinase cascade and regulates cell fate determination, including tumorigenesis [53]. The pathway is composed of a core kinase cascade, in which Mst1/2 kinases, upon interaction with scaffold protein WW45, phosphorylate LATS1/2 and Mob1. Phosphorylated LATS1/2 binds to and further activate Mob1 kinase, which in turn, phosphorylates the transcriptional coactivator YAP. The phosphorylation of YAP prevents it from entering into the nucleus, where it can form transcriptionally active complexes with TEAD and other transcription factors to drive the expression of pro-proliferative or anti-apoptotic genes. Amot-p130, Amotl1, and Amotl2—but not Amot-p80—interact with the WW domains of YAP via PPxY motifs [54]. However, there is a debate on the regulation of YAP by Amot (Figure 2).
Schematic models for YAP regulation by Amot. (A). Amot-p130 facilitates the nuclear entry and transcriptional activity of YAP, promoting the transcription of TEAD-target genes. (B) Amot binds to Merlin at the tight junctions and phosphorylates Mst1/2, LATS1/2, and Mob1. Phosphorylated LATS1/2 phosphorylates YAP, which promotes the ubiquitination and degradation of YAP. (C) Amot promotes the phosphorylation of YAP by LATS2 in the cytoplasm, leading to YAP degradation. (D) LATS-mediated phosphorylation of Amot promotes the interaction between Amot and YAP and subsequent degradation of YAP. (E) F-actin and YAP compete for binding to Amot at the tight junctions. Amot binds to YAP and maintains in the cytoplasm. (F) Amot physically interacts with YAP leading to localization to the actin cytoskeleton.
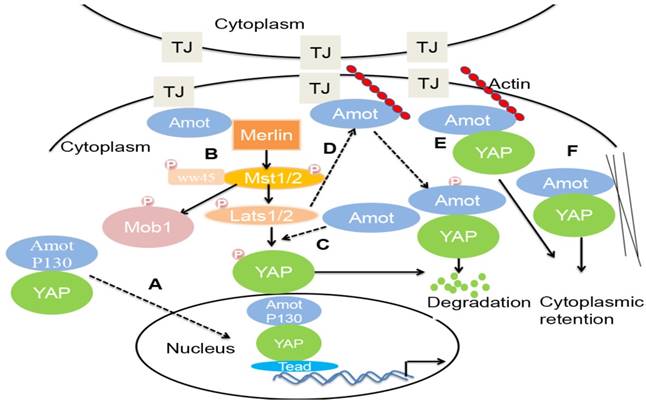
First, some studies reported that Amot acts as a YAP cofactor, preventing YAP phosphorylation and augmenting its activity toward a specific set of genes that facilitate tumorigenesis. Amot facilitates the nuclear entry and transcriptional activity of YAP and is required in the liver for ductal cell proliferation and tumorigenesis in response to liver injury or Nf2 deletion [5]. In addition, Amotl1 translocates to the nucleus, in a complex with YAP, promoting proliferation and hypertrophy of cardiomyocytes [55].
Second, in HEK293 cells, the coiled-coil (CC) domain of Amot interacts with the helical region between the FERM domain and the inhibitory CTD of Merlin. Amot binds to and activates Merlin. Both Merlin and Amot are present at the AJs and allow Hippo signaling to occur [56].
Third, in HEK293T cells, Amotl1, Amotl2, and Amot130 promote LATS2 phosphorylation of YAP in the cytoplasm and inhibit the activity of YAP [17].
Fourth, in HEK 293 cells, LATS can synergize with F-actin perturbations by phosphorylating Amot and preventing it from associating with F-actin. Through phosphorylation by LATS1/2, Amot binds to ubiquitin ligase atrophin-1 interacting protein (AIP)4. The Amot130-AIP4 complex promotes the ubiquitination and degradation of YAP and consequently inhibits YAP-dependent transcription and cell growth [5, 22, 57].
Fifth, in MDCK cells, Amot recruits YAP/TAZ to various compartments, including TJs and F-actin. F-actin and YAP compete for binding to Amot. The knockdown of Amot activates YAP—as indicated by the decreased localization of YAP to TJs—attenuates YAP phosphorylation, increases the amount of YAP in the nucleus, and induces the expression of the YAP target gene [58, 59].
Sixth, in MCF7 and 10A cells, Amot binds YAP to the actin cytoskeleton. The overexpression of Amot and Amotl1 causes the cytoplasmic retention of TAZ and suppresses its transcriptional outcome, including the expression of CTGF and Cyr61 [60, 61]. Similarly, the overexpression of Amotl2 in H441 cells induces a significant translocation of TAZ from the nucleus to the cytoplasm [62].
Other signaling pathways
In addition to the Hippo signaling pathway, other pathways control Amot proteins. Any alterations in signaling can change the function of Amot.
AMPK signaling pathway
AMP-activated protein kinase (AMPK) is a metabolic sensor ubiquitously expressed in various species. Cellular AMPK is phosphorylated and activated under certain stress conditions, such as energy deprivation, and in turn, activates several downstream substrates to modulate adaptive changes and maintain metabolic homeostasis. Some evidence has demonstrated the potential role of AMPK signaling in tumor initiation and progression [63]. In HEK 293A cells, AMPK stabilizes and increases the levels of Amotl1 steady-state proteins by directly phosphorylating Ser793 of Amotl1, inhibiting cellular proliferation, and increasing survival [64].
mTOR pathway
Mammalian/mechanistic target of rapamycin (mTOR) is a serine-threonine kinase that controls several functions in mammalian cells. mTOR affects translation, transcription, protein degradation, cell signaling, metabolism, and cytoskeleton dynamics. mTOR also participates in tumorigenesis [65, 66]. In glioblastoma cells, mTORC2 interacts and phosphorylates Amotl2. Phosphorylated Amotl2 cannot bind to YAP and increases the nuclear entry and transcriptional activity of YAP, leading to enhanced cellular growth and invasiveness [49].
Wnt signaling pathway
Wnt is a key signaling cascade involved in the regulation of cellular development and stemness, and is strongly associated with cancer. The role of Wnt signaling in carcinogenesis has been described for many types of cancer, including colorectal cancer and pancreatic ductal adenocarcinoma [67]. In zebrafish, the transfection of Amotl2 or other motin genes attenuates Wnt/β-catenin signaling, whereas the co-knockdown of three human motin genes upregulates Wnt/β-catenin signaling. Amotl2 blocks the accumulation of β-catenin in the nucleus by maintaining it in cytoplasmic punctate structures via the N-terminal glutamine-rich and middle coiled-coil domains, thus limiting cellular proliferation [30, 68, 69].
MAPK/ERK signaling pathway
The generic mitogen-activated protein kinases (MAPK) signaling pathway is shared by four distinct kinase cascades, including the extracellular signal-related kinases (ERK1/2), Jun amino-terminal kinases (JNK1/2/3), p38-MAPK, and ERK5. The MAPK/ERK pathway is associated with cell proliferation, differentiation, migration, senescence, and apoptosis [70]. In endothelial cells, Amotl2 promotes MAPK/ERK activation and plays a pivotal role in cell polarity, migration, and proliferation [29]. In addition, Amot may enhance ERK and AKT signaling and promote the proliferation of renal epithelial, RCC, and colon cancer cells [6, 71]. However, contrary to the results in other cell types, Amotl2 inhibits the AKT and ERK pathways in MCF 10A cells [31].
MicroRNAs
MicroRNAs (miRNAs) can regulate various pathways involved in the development of tumors by targeting oncogenes and tumor suppressor genes. Studies show that miRNAs directly target the 3′ untranslated regions of Amot mRNAs and regulate reporter gene expression, and MiR-497 and MiR-205 regulate Amot proteins in some cancer cell lines, including osteosarcoma and breast cancer cells. MiR-497 and MiR-205 significantly inhibit the proliferation and invasion of cancer cells by decreasing the expression of Amot [39, 45]. Similarly, miR-124 and MiR23b inhibit angiogenesis and tube formation of endothelial cells by decreasing the expression of Amot [48, 72].
Conclusion and future directions
Amot, an angiostatin binding protein, plays a critical role in angiogenesis, proliferation, and migration of cancer cells and endothelial cells in vitro and in vivo. The three members of the Amot family have different tissue- and cell-specific expression patterns and assume distinct functions in different cell types. Amot-p80 regulates tube formation and migration of endothelial cells. Amot-p130 controls cell shape. Amotl1 regulates cell polarity and cell-cell junctions. Amotl2 regulates the EMT and involvement of podosomes. However, there is a debate about whether Amot is an oncogene or a tumor suppressor gene. In most cancers, Amot proteins promote the proliferation and invasion of tumor cells, including breast cancer, osteosarcoma, colon cancer, prostate cancer, HNSCC, cervical cancer, liver cancer, and RCC. However, Amot inhibits cellular growth in lung cancer, glioblastoma, and ovarian cancer.
Moreover, there is strong evidence that Amot regulates YAP, a key downstream transcriptional co-activator of Hippo signaling pathway, which can trigger the proliferation of tumor cells. However, it is unknown whether Amot allows YAP to enter into the nucleus or to remain in the cytoplasm. In addition to the Hippo signaling pathway, Amot regulate the Wnt and MAPK/ERK signaling pathway. One hand, Amot promotes the proliferation and invasion of tumor cells as an oncogene by facilitating the nuclear entry of YAP and MAPK/ERK activation in some cancers. On the other hand, Amot inhibits cellular growth as a tumor suppressor gene by causing the cytoplasmic retention of YAP andβ-catenin in some cancers. In short, in different cell types, Amot play distinct functions by different mechanisms. The reasons may be that: 1. Amot family contains three members and Amot isoforms may assume distinct functions. But in many reports, Amot isoforms were not distinguished. 2. The expression pattern of Amot proteins differs in different cell types, the endogenous Amot is present in the nucleus, whereas exogenous Amot is usually expressed in the cytoplasm. 3. The function of Amot is distinct in different cell types and cellular processes.
Therefore, more studies are required to elucidate the role of Amot in tumors in the future study. On the basis of the above review, new questions arise: Firstly, which Amot family member is predominantly expressed in tumor cells should be identified. Secondly, does Amot play distinct roles in tumor cells and non-tumor epithelial cells? Thirdly, what are the specific mechanisms and conditions under which Amot regulates the dual role of YAP? Fourthly, what is the upstream regulator of Amot? Is Amot involved in the crosstalk between the Hippo, AMPK, Wnt, MAPK/ERK, and mTOR signaling pathways?
Abbreviations
Amot: Angiomotin; Amotl1: Amot-like protein; Amotl2: Amot-like protein 2; YAP: Yes-associated protein; TJs : tight junctions; AJs: adherens junctions; E: embryonic day; P: postnatal day; MAE cells: mouse aortic endothelial cells; EMT: epithelial-mesenchymal transition; HNSCC : head and neck squamous cell carcinoma; RCC : renal cell cancer; LCC: lung cancer cells; AIP: ubiquitin ligase atrophin-1 interacting protein; CC: coiled-coil; AMPK: AMP-activated protein kinase; mTOR: Mammalian/mechanistic target of rapamycin; MAPK: mitogen-activated protein kinases; ERK: extracellular signal-related kinases; JNK: Jun amino-terminal kinases; En: Endogenous; Ex: Exogenous.
Acknowledgements
This work is supported by a grant from the National Natural Science Foundation of China (No. 81572527).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bratt A, Wilson WJ, Troyanovsky B, Aase K, Kessler R, Van Meir EG. et al. Angiomotin belongs to a novel protein family with conserved coiled-coil and PDZ binding domains. Gene. 2002;298:69-77
2. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353-364
3. O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Cao Y. et al. Angiostatin: a circulating endothelial cell inhibitor that suppresses angiogenesis and tumor growth. Cold Spring Harbor symposia on quantitative biology. 1994;59:471-482
4. Ji WR, Castellino FJ, Chang Y, Deford ME, Gray H, Villarreal X. et al. Characterization of kringle domains of angiostatin as antagonists of endothelial cell migration, an important process in angiogenesis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1998;12:1731-1738
5. Yi C, Shen Z, Stemmer-Rachamimov A, Dawany N, Troutman S, Showe LC. et al. The p130 isoform of angiomotin is required for Yap-mediated hepatic epithelial cell proliferation and tumorigenesis. Science signaling. 2013;6:ra77
6. Lv M, Li S, Luo C, Zhang X, Shen Y, Sui YX. et al. Angiomotin promotes renal epithelial and carcinoma cell proliferation by retaining the nuclear YAP. Oncotarget. 2016;7:12393-12403
7. Troyanovsky B, Levchenko T, Mansson G, Matvijenko O, Holmgren L. Angiomotin: an angiostatin binding protein that regulates endothelial cell migration and tube formation. The Journal of cell biology. 2001;152:1247-1254
8. Ernkvist M, Aase K, Ukomadu C, Wohlschlegel J, Blackman R, Veitonmaki N. et al. p130-angiomotin associates to actin and controls endothelial cell shape. The FEBS journal. 2006;273:2000-2011
9. Nishimura M, Kakizaki M, Ono Y, Morimoto K, Takeuchi M, Inoue Y. et al. JEAP, a novel component of tight junctions in exocrine cells. The Journal of biological chemistry. 2002;277:5583-5587
10. Wang C, An J, Zhang P, Xu C, Gao K, Wu D. et al. The Nedd4-like ubiquitin E3 ligases target angiomotin/p130 to ubiquitin-dependent degradation. The Biochemical journal. 2012;444:279-289
11. Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335-344
12. Miyoshi J, Takai Y. Structural and functional associations of apical junctions with cytoskeleton. Biochimica et biophysica acta. 2008;1778:670-691
13. Wells CD, Fawcett JP, Traweger A, Yamanaka Y, Goudreault M, Elder K. et al. A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell. 2006;125:535-548
14. Sugihara-Mizuno Y, Adachi M, Kobayashi Y, Hamazaki Y, Nishimura M, Imai T. et al. Molecular characterization of angiomotin/JEAP family proteins: interaction with MUPP1/Patj and their endogenous properties. Genes to cells: devoted to molecular & cellular mechanisms. 2007;12:473-486
15. Zheng Y, Vertuani S, Nystrom S, Audebert S, Meijer I, Tegnebratt T. et al. Angiomotin-like protein 1 controls endothelial polarity and junction stability during sprouting angiogenesis. Circulation research. 2009;105:260-270
16. Bratt A, Birot O, Sinha I, Veitonmaki N, Aase K, Ernkvist M. et al. Angiomotin regulates endothelial cell-cell junctions and cell motility. The Journal of biological chemistry. 2005;280:34859-34869
17. Paramasivam M, Sarkeshik A, Yates JR, Fernandes MJ, McCollum D. Angiomotin family proteins are novel activators of the LATS2 kinase tumor suppressor. Molecular biology of the cell. 2011;22:3725-3733
18. Hultin S, Zheng Y, Mojallal M, Vertuani S, Gentili C, Balland M. et al. AmotL2 links VE-cadherin to contractile actin fibres necessary for aortic lumen expansion. Nature communications. 2014;5:3743
19. Lv M, Lv M, Chen L, Qin T, Zhang X, Liu P. et al. Angiomotin promotes breast cancer cell proliferation and invasion. Oncology reports. 2015;33:1938-1946
20. Ernkvist M, Birot O, Sinha I, Veitonmaki N, Nystrom S, Aase K. et al. Differential roles of p80- and p130-angiomotin in the switch between migration and stabilization of endothelial cells. Biochimica et biophysica acta. 2008;1783:429-437
21. Roudier E, Chapados N, Decary S, Gineste C, Le Bel C, Lavoie JM. et al. Angiomotin p80/p130 ratio: a new indicator of exercise-induced angiogenic activity in skeletal muscles from obese and non-obese rats? The Journal of physiology. 2009;587:4105-4119
22. Dai X, She P, Chi F, Feng Y, Liu H, Jin D. et al. Phosphorylation of angiomotin by Lats1/2 kinases inhibits F-actin binding, cell migration, and angiogenesis. The Journal of biological chemistry. 2013;288:34041-34051
23. Gagne V, Moreau J, Plourde M, Lapointe M, Lord M, Gagnon E. et al. Human angiomotin-like 1 associates with an angiomotin protein complex through its coiled-coil domain and induces the remodeling of the actin cytoskeleton. Cell motility and the cytoskeleton. 2009;66:754-768
24. Oka T, Schmitt AP, Sudol M. Opposing roles of angiomotin-like-1 and zona occludens-2 on pro-apoptotic function of YAP. Oncogene. 2012;31:128-134
25. Zheng Y, Zhang Y, Barutello G, Chiu K, Arigoni M, Giampietro C. et al. Angiomotin like-1 is a novel component of the N-cadherin complex affecting endothelial/pericyte interaction in normal and tumor angiogenesis. Scientific reports. 2016;6:30622
26. Moreau J, Lord M, Boucher M, Belleau P, Fernandes MJ. Protein diversity is generated within the motin family of proteins by alternative pre-mRNA splicing. Gene. 2005;350:137-148
27. Mojallal M, Zheng Y, Hultin S, Audebert S, van Harn T, Johnsson P. et al. AmotL2 disrupts apical-basal cell polarity and promotes tumour invasion. Nature communications. 2014;5:4557
28. Huang H, Lu FI, Jia S, Meng S, Cao Y, Wang Y. et al. Amotl2 is essential for cell movements in zebrafish embryo and regulates c-Src translocation. Development. 2007;134:979-988
29. Wang Y, Li Z, Xu P, Huang L, Tong J, Huang H. et al. Angiomotin-like2 gene (amotl2) is required for migration and proliferation of endothelial cells during angiogenesis. The Journal of biological chemistry. 2011;286:41095-41104
30. Agarwala S, Duquesne S, Liu K, Boehm A, Grimm L, Link S. et al. Amotl2a interacts with the Hippo effector Yap1 and the Wnt/beta-catenin effector Lef1 to control tissue size in zebrafish. eLife. 2015;4:e08201
31. Wang W, Huang J, Chen J. Angiomotin-like proteins associate with and negatively regulate YAP1. The Journal of biological chemistry. 2011;286:4364-4370
32. Kim M, Kim M, Park SJ, Lee C, Lim DS. Role of Angiomotin-like 2 mono-ubiquitination on YAP inhibition. EMBO reports. 2015;17:64-78
33. Proszynski TJ, Sanes JR. Amotl2 interacts with LL5beta, localizes to podosomes and regulates postsynaptic differentiation in muscle. Journal of cell science. 2013;126:2225-2235
34. Bernadzki KM, Rojek KO, Proszynski TJ. Podosomes in muscle cells and their role in the remodeling of neuromuscular postsynaptic machinery. European journal of cell biology. 2014;93:478-485
35. Campbell CI, Samavarchi-Tehrani P, Barrios-Rodiles M, Datti A, Gingras AC, Wrana JL. The RNF146 and tankyrase pathway maintains the junctional Crumbs complex through regulation of angiomotin. Journal of cell science. 2016;129:3396-3411
36. Wang W, Li N, Li X, Tran MK, Han X, Chen J. Tankyrase Inhibitors Target YAP by Stabilizing Angiomotin Family Proteins. Cell reports. 2015;13:524-532
37. Choi KS, Choi HJ, Lee JK, Im S, Zhang H, Jeong Y. et al. The endothelial E3 ligase HECW2 promotes endothelial cell junctions by increasing AMOTL1 protein stability via K63-linked ubiquitination. Cellular signalling. 2016;28:1642-1651
38. Jiang WG, Watkins G, Douglas-Jones A, Holmgren L, Mansel RE. Angiomotin and angiomotin like proteins, their expression and correlation with angiogenesis and clinical outcome in human breast cancer. BMC cancer. 2006;6:16
39. Zhang H, Fan Q. MicroRNA-205 inhibits the proliferation and invasion of breast cancer by regulating AMOT expression. Oncology reports. 2015;34:2163-2170
40. Ranahan WP, Han Z, Smith-Kinnaman W, Nabinger SC, Heller B, Herbert BS. et al. The Adaptor Protein AMOT Promotes the Proliferation of Mammary Epithelial Cells via the Prolonged Activation of the Extracellular Signal-regulated Kinases. Cancer Res. 2011;71:2203-11
41. Couderc C, Boin A, Fuhrmann L, Vincent-Salomon A, Mandati V, Kieffer Y. et al. AMOTL1 Promotes Breast Cancer Progression and Is Antagonized by Merlin. Neoplasia. 2016;18:10-24
42. Holmgren L, Ambrosino E, Birot O, Tullus C, Veitonmaki N, Levchenko T. et al. DNA vaccine targeting angiomotin inhibits angiogenesis and suppresses tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9208-9213
43. Barutello G, Curcio C, Spadaro M, Arigoni M, Trovato R, Bolli E. et al. Antitumor immunization of mothers delays tumor development in cancer-prone offspring. Oncoimmunology. 2015;4:e1005500
44. Ruan W, Wang P, Feng S, Xue Y, Li Y. Long non-coding RNA small nucleolar RNA host gene 12 (SNHG12) promotes cell proliferation and migration by upregulating angiomotin gene expression in human osteosarcoma cells. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37:4065-4073
45. Ruan WD, Wang P, Feng S, Xue Y, Zhang B. MicroRNA-497 inhibits cell proliferation, migration, and invasion by targeting AMOT in human osteosarcoma cells. OncoTargets and therapy. 2016;9:303-313
46. Ortiz A, Lee YC, Yu G, Liu HC, Lin SC, Bilen MA. et al. Angiomotin is a novel component of cadherin-11/ -catenin/p120 complex and is critical for cadherin-11-mediated cell migration. The FASEB Journal. 2014;29:1080-1091
47. Hakami F, Darda L, Stafford P, Woll P, Lambert DW, Hunter KD. The roles of HOXD10 in the development and progression of head and neck squamous cell carcinoma (HNSCC). British journal of cancer. 2014;111:807-816
48. Wan HY, Li QQ, Zhang Y, Tian W, Li YN, Liu M. et al. MiR-124 represses vasculogenic mimicry and cell motility by targeting amotL1 in cervical cancer cells. Cancer letters. 2014;355:148-158
49. Artinian N, Cloninger C, Holmes B, Benavides-Serrato A, Bashir T, Gera J. Phosphorylation of the Hippo Pathway Component AMOTL2 by the mTORC2 Kinase Promotes YAP Signaling, Resulting in Enhanced Glioblastoma Growth and Invasiveness. The Journal of biological chemistry. 2015;290:19387-19401
50. Wang Y, Justilien V, Brennan KI, Jamieson L, Murray NR, Fields AP. PKCiota regulates nuclear YAP1 localization and ovarian cancer tumorigenesis. Oncogene. 2017;36:534-545
51. Hsu YL, Hung JY, Chou SH, Huang MS, Tsai MJ, Lin YS. et al. Angiomotin decreases lung cancer progression by sequestering oncogenic YAP/TAZ and decreasing Cyr61 expression. Oncogene. 2015;34:4056-4068
52. Ozawa MG, Bhaduri A, Chisholm KM, Baker SA, Ma L, Zehnder JL. et al. A study of the mutational landscape of pediatric-type follicular lymphoma and pediatric nodal marginal zone lymphoma. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2016;29:1212-1220
53. Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nature cell biology. 2011;13:877-883
54. Moleirinho S, Guerrant W, Kissil JL. The Angiomotins-from discovery to function. FEBS letters. 2014;588:2693-2703
55. Ragni CV, Diguet N, Le Garrec JF, Novotova M, Resende TP, Pop S, Charon N, Guillemot L, Kitasato L, Badouel C, Dufour A, Olivo-Marin JC, Trouvé A, McNeill H, Meilhac SM. Amotl1 mediates sequestration of the Hippo effector Yap1 downstream of Fat4 to restrict heart growth. Nature communications. 2017;8:14582
56. Li Y, Zhou H, Li F, Chan SW, Lin Z, Wei Z. et al. Angiomotin binding-induced activation of Merlin/NF2 in the Hippo pathway. Cell Research. 2015;25:801-817
57. Mana-Capelli S, Paramasivam M, Dutta S, McCollum D. Angiomotins link F-actin architecture to Hippo pathway signaling. Molecular biology of the cell. 2014;25:1676-1685
58. Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q. et al. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes & development. 2011;25:51-63
59. Chan SW, Lim CJ, Guo F, Tan I, Leung T, Hong W. Actin-binding and cell proliferation activities of angiomotin family members are regulated by Hippo pathway-mediated phosphorylation. The Journal of biological chemistry. 2013;288:37296-37307
60. Chan SW, Lim CJ, Chong YF, Pobbati AV, Huang C, Hong W. Hippo pathway-independent restriction of TAZ and YAP by angiomotin. The Journal of biological chemistry. 2011;286:7018-7026
61. Cox CM, Mandell EK, Stewart L, Lu R, Johnson DL, McCarter SD. et al. Endosomal regulation of contact inhibition through the AMOT:YAP pathway. Molecular biology of the cell. 2015;26:2673-2684
62. Lucci V, Di Palma T, D'Ambrosio C, Scaloni A, Zannini M. AMOTL2 interaction with TAZ causes the inhibition of surfactant proteins expression in lung cells. Gene. 2013;529:300-306
63. Cheng J, Zhang T, Ji H, Tao K, Guo J, Wei W. Functional characterization of AMP-activated protein kinase signaling in tumorigenesis. Biochimica et biophysica acta. 2016;1866:232-251
64. DeRan M, Yang J, Shen CH, Peters EC, Fitamant J, Chan P. et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell reports. 2014;9:495-503
65. Yuan ZX, Mo J, Zhao G, Shu G, Fu HL, Zhao W. Targeting Strategies for Renal Cell Carcinoma: From Renal Cancer Cells to Renal Cancer Stem Cells. Frontiers in pharmacology. 2016;7:423
66. Gu G, Dustin D, Fuqua SA. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Current opinion in pharmacology. 2016;31:97-103
67. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461-1473
68. Li Z, Wang Y, Zhang M, Xu P, Huang H, Wu D. et al. The Amotl2 gene inhibits Wnt/beta-catenin signaling and regulates embryonic development in zebrafish. The Journal of biological chemistry. 2012;287:13005-13015
69. Dalle Nogare D, Chitnis AB. Connecting physical cues and tissue patterning. eLife. 2015;4:e11375
70. Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. Journal of receptor and signal transduction research. 2015;35:600-604
71. Zhang Y, Yuan J, Zhang X, Yan F, Huang M, Wang T. et al. Angiomotin promotes the malignant potential of colon cancer cells by activating the YAP-ERK/PI3K-AKT signaling pathway. Oncology reports. 2016;36:3619-3626
72. Hannafon BN, Carpenter KJ, Berry WL, Janknecht R, Dooley WC, Ding WQ. Exosome-mediated microRNA signaling from breast cancer cells is altered by the anti-angiogenesis agent docosahexaenoic acid (DHA). Molecular cancer. 2015;14:133
Author contact
![]() Corresponding author: Professor Jin Yang, Department of Oncology, the First Affiliated Hospital of Xian Jiaotong University, Xi'an, Shaanxi Province , 710061, P.R. China (E-mail : yangjinxjtu.edu.cn)
Corresponding author: Professor Jin Yang, Department of Oncology, the First Affiliated Hospital of Xian Jiaotong University, Xi'an, Shaanxi Province , 710061, P.R. China (E-mail : yangjinxjtu.edu.cn)