Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Summary
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(10):1242-1253. doi:10.7150/ijbs.21475 This issue Cite
Research Paper
Distinct Effects of Ca2+ Sparks on Cerebral Artery and Airway Smooth Muscle Cell Tone in Mice and Humans
Qing-Yang Zhao1*, Yong-Bo Peng1*, Xiao-Jing Luo1*#, Xi Luo1*, Hao Xu1*, Ming-Yu Wei1*, Qiu-Ju Jiang1, Wen-Er Li1#, Li-Qun Ma1, Jin-Chao Xu1, Xiao-Cao Liu1, Dun-An Zang1, Yu-San She1, He Zhu1, Jinhua Shen1, Ping Zhao1, Lu Xue1, Meng-Fei Yu1, Weiwei Chen1, Ping Zhang2, Xiangning Fu3, Jingyu Chen4, Xiaowei Nie4, Chenyou Shen4, Shu Chen5, Shanshan Chen5, Jingcao Chen6, Sheng Hu7, Chunbin Zou8, Gangjian Qin9, Ying Fang10, Jiuping Ding10, Guangju Ji11, Yun-Min Zheng12, Tengyao Song12, Yong-Xiao Wang12, Qing-Hua Liu1 ![]()
1. Institute for Medical Biology and Hubei Provincial Key Laboratory for Protection and Application of Special Plants in Wuling Area of China, College of Life Sciences, South-Central University for Nationalities, Wuhan 430074, China;
2. Department of Cerebral Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430032, Hubei, China;
3. Department of Thoracic Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430032, Hubei, China;
4. Wuxi &Jiangsu Key Laboratory of Organ Transplantation, Department of Cardiothoracic Surgery, Lung Transplant Group, Wuxi People's Hospital, Nanjing Medical University, Jiangsu, China;
5. Department of Cardiovascular Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430032, Hubei, China;
6. Department of Cerebral Surgery, Zhongnan Hospital, Wuhan University Medical College, Wuhan, 430071, Hubei, China;
7. Department of Medical Oncology, Hubei Cancer Hospital, Wuhan, 430079, Hubei, China;
8. Acute Lung Injury Center of Excellence, Division of Pulmonary, Allergy, and Critical Care Medicine, Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, 15213, USA;
9. Department of Biomedical Engineering, School of Medicine & School of Engineering, University of Alabama Birmingham, AL, 35294, USA;
10. Key Laboratory of Molecular Biophysics of the Ministry of Education, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, China;
11. National Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing, 100101, China;
12. Department of Molecular and Cellular Physiology, Albany Medical College, Albany, NY, 12208, USA.
* Q-Y Z., Y-B P., X-J L., X L., H X., M-Y W. contributed equally to this work.
# Current address: Institute of Pharmacology, University of Technology Dresden, Dresden 01307, Germany.
Received 2017-6-14; Accepted 2017-8-10; Published 2017-9-21
Abstract

The effects of Ca2+ sparks on cerebral artery smooth muscle cells (CASMCs) and airway smooth muscle cells (ASMCs) tone, as well as the underlying mechanisms, are not clear. In this investigation, we elucidated the underlying mechanisms of the distinct effects of Ca2+ sparks on cerebral artery smooth muscle cells (CASMCs) and airway smooth muscle cells (ASMCs) tone. In CASMCs, owing to the functional loss of Ca2+-activated Cl- (Clca) channels, Ca2+ sparks activated large-conductance Ca2+-activated K+ channels (BKs), resulting in a decreases in tone against a spontaneous depolarization-caused high tone in the resting state. In ASMCs, Ca2+ sparks induced relaxation through BKs and contraction via Clca channels. However, the integrated result was contraction because Ca2+ sparks activated BKs prior to Clca channels and Clca channels-induced depolarization was larger than BKs-caused hyperpolarization. However, the effects of Ca2+ sparks on both cell types were determined by L-type voltage-dependent Ca2+ channels (LVDCCs). In addition, compared with ASMCs, CASMCs had great and higher amplitude Ca2+ sparks, a higher density of BKs, and higher Ca2+ and voltage sensitivity of BKs. These differences enhanced the ability of Ca2+ sparks to decrease CASMC and to increase ASMC tone. The higher Ca2+ and voltage sensitivity of BKs in CASMCs than ASMCs were determined by the β1 subunits. Moreover, Ca2+ sparks showed the similar effects on human CASMC and ASMC tone. In conclusions, Ca2+ sparks decrease CASMC tone and increase ASMC tone, mediated by BKs and Clca channels, respectively, and finally determined by LVDCCs.
Keywords: Smooth muscle cells, Ca2+ sparks, BK channels, Ca2+-activated Cl- channels, L-type voltage-dependent Ca2+ channels.
Introduction
The tone of cerebral artery smooth muscle cells (CASMCs) and airway smooth muscle cells (ASMCs) is important for blood perfusion of the brain and air supply to the lung, respectively. Spontaneous, transient local Ca2+ elevations (Ca2+ sparks) [1] play a pivotal role in tone setting. However, their effects and underlying mechanism in CASMCs and ASMCs are still largely uncertain.
Ca2+ sparks decreased the tone in rat and rabbit CASMCs through the activation of BKs [2-7]. However, TMEM16A, a Clca channel [8], has been found to be expressed in rat CASMCs [9] and mediates rat cerebral artery contraction [10]. Therefore, the effects of Ca2+ sparks on CASMC tone and the underlying mechanisms need to be clarified.
Ca2+ release from intracellular stores induced by agonists can activate Cl- and K+ conductance and can cause contraction in guinea-pig tracheal smooth muscle cells [8]. Spontaneous Ca2+ events, Ca2+ sparks, can also occur in airway smooth muscle cells from guinea pig [11], pig [12] and mouse [13] ASMCs. They simultaneously induced relaxation via activating BKs and contraction by triggering Clca channels [11, 14, 15]. However, the integrated effect was either contraction based on Ca2+ sparks being increased and mouse ASMC becoming shortened [16, 17] or relaxation, suggested by the blockade of RyRs with ryanodine mouse ASMC becoming shortened [18]. This discrepancy needs to be elucidated.
In the present study, we comparatively defined the effects of Ca2+ sparks on CASMC and ASMC tone and investigated the underlying mechanisms.
Materials and Methods
For full details of the methods, please see the supporting information.
Study approval
All experiments on animal and human tissues were approved by the Institutional Animal Care and Use Committee and the Ethics Committee of the South-Central University for Nationalities, respectively.
Ionic and mechanical measurements in single CASMCs and ASMCs
Single CASMCs and ASMCs were enzymatically isolated from cerebral arteries and airways [16, 19-21]. Ca2+ sparks [13, 17, 22], cell length simultaneously with [17] and without [16] Ca2+ sparks, STICs, STOCs, and caffeine-induced currents [19, 20], LVDCC currents [23, 24], single BK currents [20] and Vm [25] were then measured.
Measurements of TMEM16A and BK mRNA
Total RNA was extracted from mouse CASMCs and ASMCs and was reverse transcribed to synthesize cDNA. Samples were analyzed by semi-quantitative RT-PCR and quantitative RT-PCR, respectively [26].
Immuno-fluorescence staining
TMEM16A [9, 27] and BK expression [28] in single mouse cells were investigated using immuno-fluorescence staining.
Data analysis
All results are expressed as the means ± SD. Comparisons between two and multiple groups were performed using Student's t-test and one-way ANOVA. Differences with P< 0.05 were considered statistically significant.
Results
Ca2+ sparks decrease mouse CASMC tone and increase ASMC tone
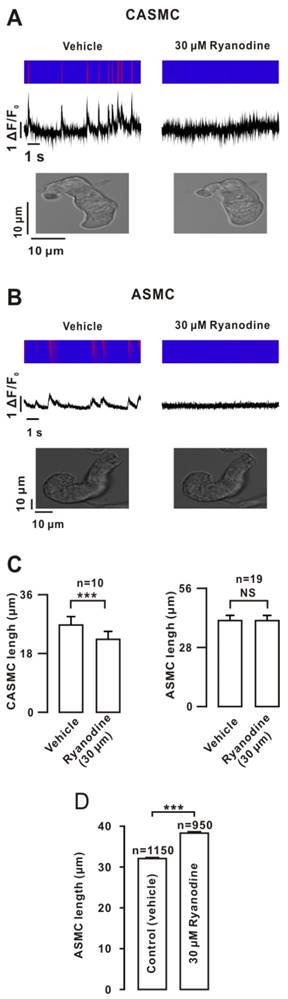
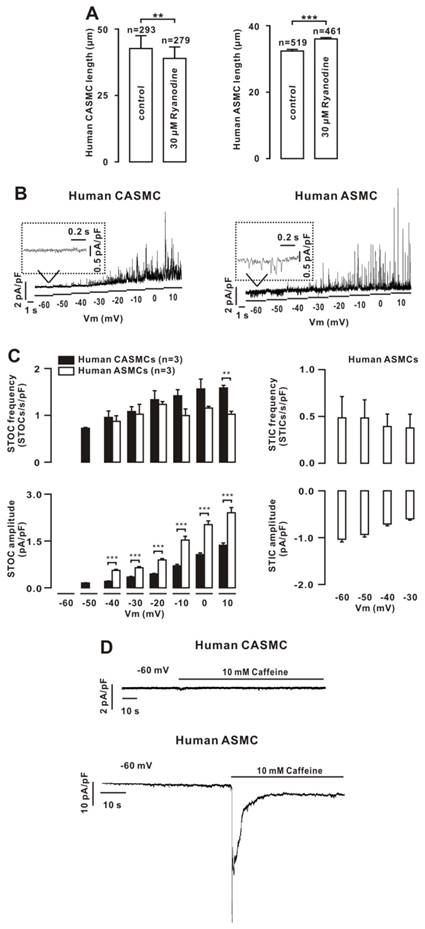
Ryanodine (a selective blocker of RyRs) abrogated Ca2+ sparks in CASMC and caused their shortening. However, the abolishment of Ca2+ sparks did not induce shortening in ASMC (Fig. 1A, B). The summary results suggested that Ca2+ sparks decreased CASMC tone but did not affect ASMC tone (Fig. 1C). However, single ASMCs were elongated after the incubation of ryanodine under suspension conditions (Fig. 1D), suggesting that Ca2+ sparks increased ASMC tone and the cells adhered to the cover slip were prevented from extending (Fig. 1B).Therefore, we used suspended cells to measure the cell length in subsequent experiments. This result was further confirmed by ryanodine and the known bronchodilator isoproterenol (ISO) inducing ASMC relaxation, resulting in increases in the airway lumen area (ALA) (Fig. S1, the detailed explanation for this method in the supplemental material).
Effects of ryanodine on Ca2+ sparks and cell length. (A) Simultaneous recordings of Ca2+ sparks and cell length were performed in mouse CASMCs using an LMS 700 confocal microscope. Ca2+ sparks were abolished by ryanodine, and the cell was shortened. (B) The same experiments were conducted in mouse ASMCs. The cell length did not change. (C) Summary of the results. (D) Single mouse ASMCs were incubated with ryanodine or vehicle for 15 min in the suspended state. Their lengths were increased compared with those of the controls. NS: P> 0.05; ***: P< 0.001. These results indicate that Ca2+ sparks decreased CASMC tone and increased ASMC tone.
The efficiency of Ca2+ sparks is related to their frequencies and amplitudes. We found that Ca2+ sparks had a 2.4-fold higher frequency and a 1.7-fold higher amplitude in CASMCs than those in ASMCs (Fig. S2).
Ca2+ sparks fail to activate STICs
Ca2+ sparks activate Clca channels and BKs to generate STICs and STOCs, respectively. STOCs were observed in CASMCs, and both STICs and STOCs were noted in ASMCs (Fig. 2A, B, C). The frequency of STOCs in CASMCs was higher than that in ASMCs. These data indicate that CASMCs might only have BKs, and ASMCs may have both Clca channels and BKs.
Ca2+ sparks-activated currents in mouse cells. (A) Ca2+ sparks-activated currents at potentials from -60 mV to +10 mV were recorded in CASMC. Only STOCs were observed. (B) The same recordings were performed in an ASMC, showing that Ca2+ sparks simultaneously triggered STOCs and STICs; however, the former occurred prior to the latter shown in the inset box. (C) STOC and STIC frequency and amplitude. (D) Whole-cell ramp currents induced by 10 mM caffeine were recorded in CASMCs and ASMCs. The paxilline-sensitive currents were observed only in CASMCs and these currents with NA-sensitive currents were noted in ASMCs. The paxilline-sensitive currents were larger in CASMCs than in ASMCs. *: P< 0.05. These data indicate that CASMCs had no functional Clca channels; however, they had more STOCs and caffeine-induced inward currents than ASMCs.
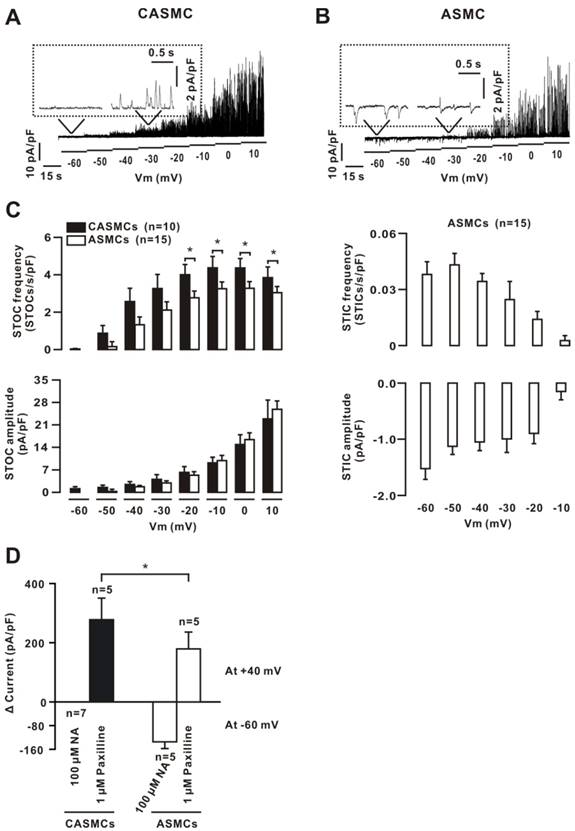
We used caffeine to further confirm these results. The caffeine-induced currents were recorded using a voltage ramp protocol. After washing out, the cells were incubated with NA (a blocker of Clca channels) or paxilline (a blocker of BKs) for 15 min, and then caffeine-induced currents were recorded again. The two peak currents at -60 mV (to represent Clca currents) or 40 mV (to reflect BK currents) were measured and the net values were calculated (Fig. 2D), suggesting that CASMCs only had BKs, and ASMCs had both Clca channels and BKs.
Rat CASMCs express TMEM16A, a Clca channel [9]. We observed large inward currents in rat CASMCs (Fig. S3). Thus, we then measured TMEM16A mRNA (Fig. S4) and protein (Fig. S5) in mouse CASMCs and ASMCs. The results suggest that TMEM16A was expressed in both mouse cell types but did not function as a Clca channel in CASMCs.
To know whether TMEM16A functions in mouse ASMCs, the STICs and caffeine-induced currents were measured. The results show that anti-TMEM16A antibodies declined the amplitude of STICs and caffeine-induced inward currents (not significantly) (Fig. S6), suggesting that other genes may play a role in Clca channels in mouse ASMCs.
Ca2+ sparks result in Vm changes and LVDCC activation
STICs and STOCs affect Vm and then LVDCC activation and inactivation. LVDCC currents were blocked by nifedipine (a selective blocker of LVDCCs [29]). The current-voltage (I-V) curves (Fig. S7) indicate that LVDCCs were activated at approximately -40 mV in CASMCs and ASMCs, however, the currents were larger in the former than in the latter.
Vm in CASMCs exhibited oscillations (Fig. 3), which were abolished, and the baseline was elevated by TEA (a blocker of BKs [2]). NA (an inhibitor of Clca channels [30]) did not induce Vm change. Based on -40 mV of the LVDCC threshold, we constructed distributions of Vm values. The area to the right of -40 mV (S40) represents the LVDCC activation time, showing that the LVDCC opening time was short at the resting state (right) and was longer after TEA treatment (middle) and did not change after NA treatment (left). We further calculated the ratio of P0, S40 divided by the total area (ST), representing the open probability of LVDCCs (Fig. 3B). These results demonstrate that CASMCs had no functional Clca channels, and that, at the resting state, LVDCCs opened (P0= 0.13 ± 0.04, n = 6) by spontaneous depolarization that was potently inhibited by BKs-induced hyperpolarization.
BKs and Clca channels mediate Vm changes in mouse cells. (A) Vm in a CASMC displayed smaller oscillations and TEA abolished oscillations and elevated the baseline. NA did not induce changes. All-point amplitude histograms were constructed, and S40 (the area to the right of -40 mV) and ST (the total area) were marked. (B) The ratios of S40/ST are summarized. (C-D) The same experiments and analyses were performed in ASMCs. *: P< 0.05; ***: P < 0.001. These results indicate that CASMCs had no functional Clca channels; in the resting state, the Po of LVDCCs was lower in CASMCs than in ASMCs, Clca channels-induced depolarization and LVDCC activation were higher than BKs-caused hyperpolarization and LVDCC inactivation in ASMCs, and that BKs-caused hyperpolarization and LVDCC inactivation were higher in CASMCs compared to ASMCs.
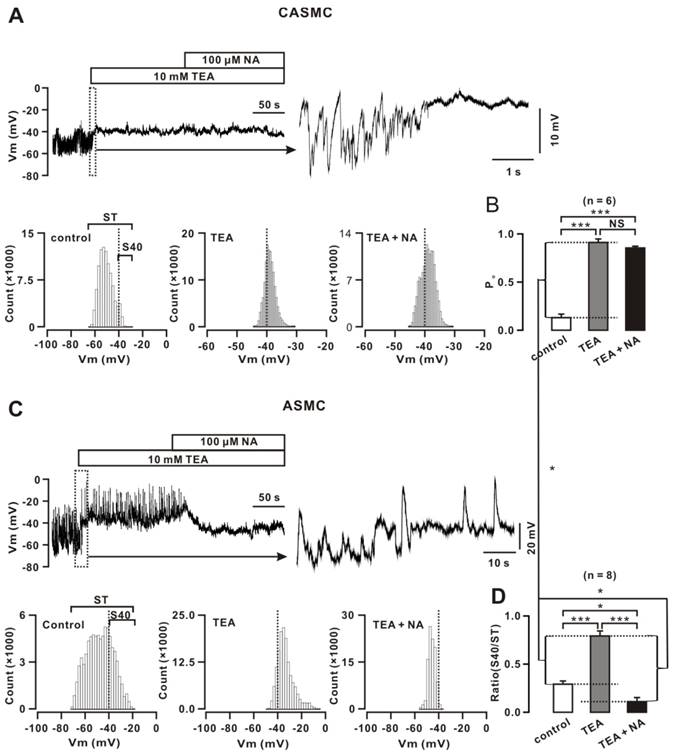
In ASMCs, we performed the same experiments and analyses (Fig. 3C, D). Vm exhibited large oscillations and frequently reached -40 mV (P0= 0.29 ± 0.03, n = 8; P < 0.05 versus that in CASMCs). TEA did not affect the large upward spikes (i.e., depolarization spikes); however, it blocked the small downward spikes (i.e., hyperpolarization spikes) and resulted in the baseline elevating to ~-40 mV. NA abolished the large depolarization spikes, leading the baseline to decline below -40 mV. These results indicate that ASMCs had Clca channels and BKs. The former-induced LVDCC activation degree was larger than the latter-caused LVDCC inactivation degree; therefore, LVDCCs were still opened and mediated contraction. In addition, BKs-induced LVDCC activation degree was larger in CASMCs than ASMCs.
The voltage and Ca2+ sensitivity of BKs are different in both cell types
Ca2+ sparks cause local Ca2+ increases and Vm changes and then affect BKs activation. Therefore, we compared the BKs voltage and Ca2+ sensitivity between two cell types. Single BK currents were recorded using the inside-out technique on an excised membrane from a CASMC at 0, 20, 40, and 60 mV with 1 μM (left), 3 μM (middle) and 10 μM free Ca2+ (right, Fig. 4A). The Po values were calculated and presented above the traces. The histograms were constructed and the peak values (i.e., single-channel amplitudes) were obtained. These recordings and analyses were performed in a patch from an ASMC (Fig. 4B). The I-V curves were linear and the conductance values were calculated as 265.7 ± 0.01 pS for CASMCs and 272.6 ± 0.01 pS (P > 0.05) for ASMCs (Fig. 4C), suggesting that the BK conductance did not contribute to the different effects of Ca2+ sparks on tone in the two cell types. The Po-voltage curves (Fig. 4D) showed that, in 1 μM free Ca2+, Po values between both cell types were not different at each of the voltages tested; however, in 3 and 10 μM free Ca2+, Po values at 20, 40 and 60 mV were significantly higher in CASMCs than those in ASMCs. These results suggest that Po value at the same voltage was higher in CASMCs than in ASMCs, indicating that BKs had higher voltage sensitivity and higher Ca2+ sensitivity in the former than the latter, and that BKs were fully activated in both cell types with 10 μM Ca2+ and at a voltage of 60 mV.
Voltage and Ca2+ sensitivity of BKs in mouse CASMCs and ASMCs. (A) Single BK currents were recorded using the inside-out technique at 0, 20, 40, and 60 mV under 1, 3, and 10 μM free Ca2+ conditions in an excised patch from a CASMC. The Po values are shown above each trace. The corresponding all-point amplitude histograms were constructed and fitted, and the single-channel currents were obtained. (B) The same recordings and analyses from an excised patch from an ASMC. (C) I-V curves were constructed, and the conductance values were calculated for CASMCs and ASMCs. (D) Po-voltage curves show that the Po was larger in CASMCs than in ASMCs following the increases in voltage and free Ca2+, indicating that BKs in the former had a higher voltage and Ca2+ sensitivity than in the latter. *: P < 0.05.
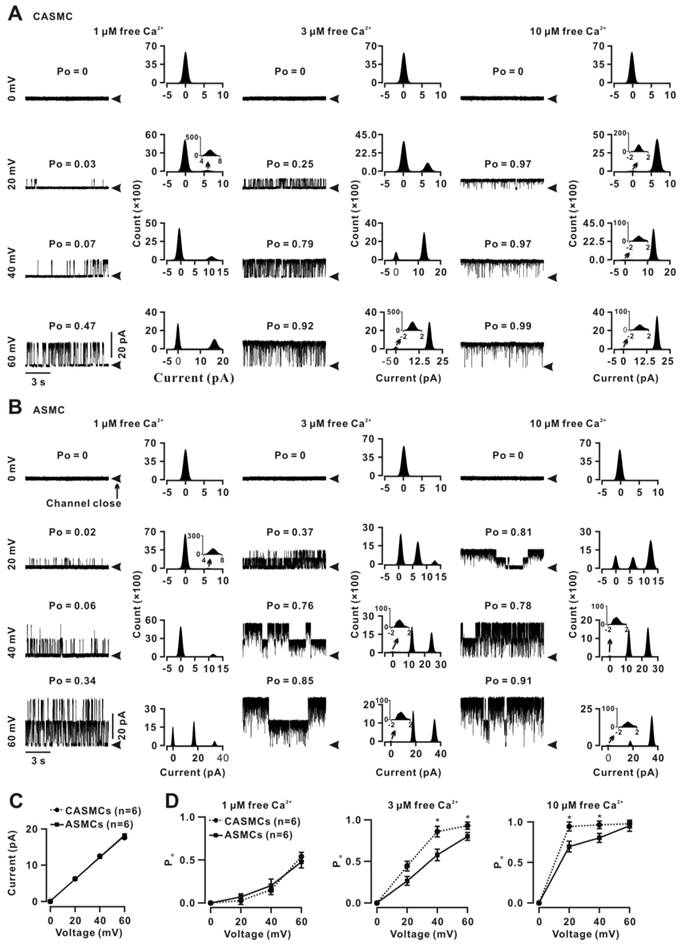
Based on the single and whole-cell BK current amplitudes at 40 mV, the density of BKs was calculated as 31.0 ± 5.0 channels/pF (n = 10) in CASMCs and 16.4 ± 5.2 channels/pF (n = 5, P < 0.05) in ASMCs, suggesting that CASMCs had a 1.9-fold higher BK density than ASMCs. This was supported by staining with anti-BK-FITC antibodies, for which the intensity of FITC fluorescence was higher in CASMCs than in ASMCs (Fig. S8).
Mechanism of the differences in voltage and Ca2+ sensitivity of BKs between the two cell types
Both the voltage and Ca2+ sensitivities of BKs are determined by five subunits α, β1, β2, β3 and β4 [31, 32]. We found that α, β1 and β4 mRNA was expressed in both cell types, and the level of α was 0.54-fold lower, that of β1 was 0.29-fold lower and β4 was 3.8-fold higher in CASMCs than in ASMCs (Fig. S9). The mRNA levels of β2 and β3 were not detected. These results indicate that α, β1 and/or β4 are responsible for the differences in the voltage and Ca2+ sensitivity.
We next defined which subunit plays a role using the antibody neutralization approach [33]. Single BK currents at 0, 20, 40, and 60 mV were recorded using the inside-out configuration in an excised patch from a CASMC and an ASMC in bath solutions with 1 μM intracellular free Ca2+ (Fig. S10). The currents were abolished following the addition of anti-BK α subunit antibodies in the bath. The denatured anti-BK α subunit antibodies had no effect (data not shown). These results were consistent with the α subunit being the pore-forming subunit of BKs [34].
We next studied the role of the β1 subunit. The single BK currents at 0, 20, 40, and 60 mV in bath solutions with 1 and 3 µM free Ca2+ were similarly recorded using the inside-out technique in the absence and presence of anti-β1 subunit antibodies. The Po-voltage curves show that the Po values were decreased by the antibodies, and the decreases were larger in CASMCs than in ASMCs (Fig. 5A, B). These data indicate that β1 is the subunit responsible for the voltage and Ca2+ sensitivity in each cell type, as well as for the higher voltage and Ca2+ sensitivity of BKs in CASMCs than in ASMCs. The denatured anti-β1 subunit antibodies had no effect (data not shown).
Effects of anti-β1 antibodies on single BK currents in mouse cells. (A) Single BKs-mediated currents were similarly recorded at 0, 20, 40 and 60 mV under 1 and 3 μM free Ca2+ on excised patches from CASMCs and ASMCs using the inside-out technique. After incubation with anti-β1 antibodies, the currents were recorded again. Po values before and after the incubation with antibodies were calculated, and Po-voltage curves were constructed. The results show that the antibodies induced decreases in Po values in both CASMCs and ASMCs, however, the decreases were larger in the former than in the latter. (B) The net decreases in the Po (i.e., ΔPo) values were calculated and used to plot the ΔPo-voltage curves, showing that the ΔPo values were larger following increases in Ca2+ concentration and voltage in CASMCs than in ASMCs. *: P < 0.05. These data demonstrate that β1 subunits mediated the Ca2+ and voltage sensitivity of BKs in both cells and the higher Ca2+ and voltage sensitivity in CASMCs than in ASMCs.
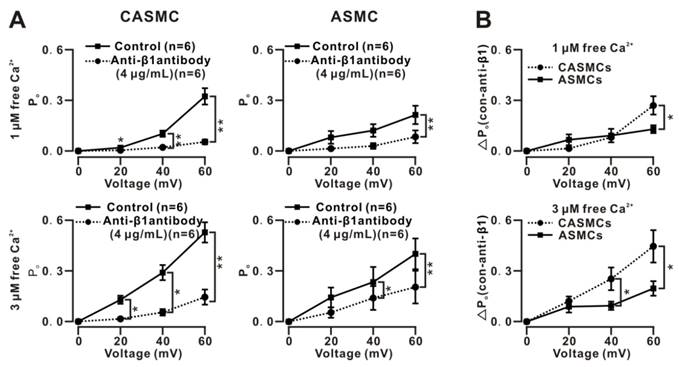
Anti-β4 subunit antibodies failed to affect the Po values (Fig. S11), suggesting that the β4 subunits did not play a role.
Effects of Clca, BK and LVDCC on tone
We further observed the role of Clca, BK and LVDCC. The blocker of BKs (paxilline) induced shortening and the blocker of LVDCCs (nifedipine) induced elongation in CASMCs; in ASMCs, the blocker of Clca channels (NA) and nifedipine induced elongation, and paxilline caused shortening (Fig. S12). These results indicate that Clca channels and BKs mediate tone increases and decreases, respectively. LVDCCs was the final step, based on nifedipine abolishing ryanodine-induced cell length changes (Fig. S13).
Effects and mechanism of Ca2+ sparks on human CASMC and ASMC tone
We next investigated whether Ca2+ sparks exhibit a similar effect on human CAMSC and ASMC tone. Ryanodine induced shortening in human CASMCs and elongation in human ASMCs (Fig. 6A). We hypothesized that these effects would also be mediated by Clca channels, BKs and LVDCCs. Therefore, we recorded STICs and STOCs. Only STOCs were observed in CASMCs, and both were noted in ASMCs (Fig. 6B, C), consistent with the above findings in mouse cells. Moreover, caffeine failed to induce inward currents in human CASMCs; however, it did in human ASMCs (Fig. 6D), further indicating that human CASMCs had no functional Clca channels. In addition, we found that LVDCCs were expressed in both cell types (Fig. S14).
Effects of Ca2+ sparks on human cell tone and the underlying mechanism. (A) Human CASMCs and human ASMCs were incubated with vehicle and ryanodine, and the cell lengths were measured. (B) Only STOCs were recorded in human CASMCs, however, STICs and STOCs were observed in human ASMCs. (C) Average frequency and amplitude of STICs and STOCs, showing that the amplitude of STOCs was lower in CASMCs than in ASMCs. (D) Caffeine failed to induce inward currents in three human CASMCs that occurred in three human ASMCs. **: P < 0.01; ***: P < 0.001. These results indicate that Ca2+ sparks decreased human ASMC tone and increased human CASMC tone and the former had only BKs and the latter had both Clca channels and BKs.
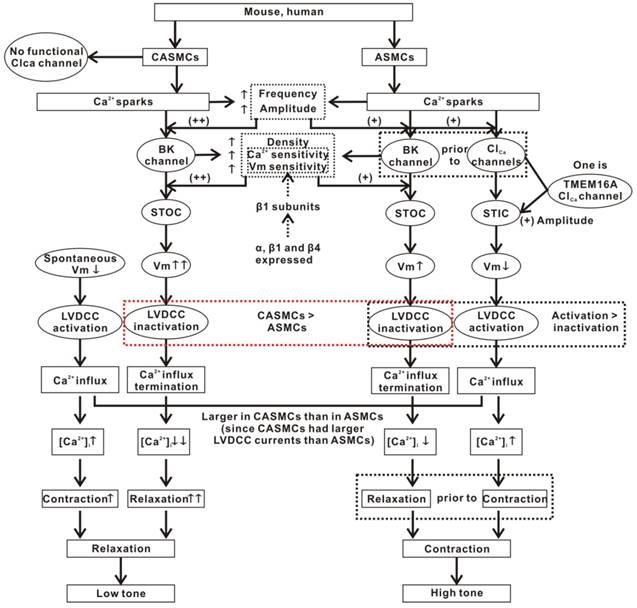
The above results are summarized in Fig. 7.
Mechanisms of Ca2+ sparks-induced decreases in CAMSC tone and increases in ASMC tone. CASMCs lack functional Clca channels. Ca2+ sparks only activate BKs, which mediate relaxation via the STOC-Vm-LVDCC-Ca2+ influx termination pathway. However, this relaxation is partially counteracted by the contraction induced by the spontaneous depolarization of Vm. In ASMCs, except for this BKs-mediated relaxant pathway, Ca2+ sparks also activate Clca channels, which then mediate contraction through the STIC-Vm-LVDCC-Ca2+ influx pathway. However, the integrated result is contraction. The cause may be that Ca2+ sparks activate BKs prior to Clca channels, allowing the contraction is not exactly antagonized by the relaxation. Moreover, Clca channels-induced membrane depolarization and LVDCC activation are larger compared to BKs-caused membrane hyperpolarization and LVDCC inactivation. In addition, the Ca2+ spark frequency and amplitude, BK density, BK currents and β1-determined Ca2+ and voltage sensitivity of BKs were higher in CASMCs than in ASMCS. These allow Ca2+sparks to cause more relaxation in CASMCs than in ASMCs. In addition, TMEM16A will be one type of Clca channels in ASMCs that plays a lesser role in mediating Ca2+sparks' effect on cell tone.
Discussion
Ca2+ sparks decreased mouse and human CASMC tone. The key reason was the functional loss of Clca channels, although TMEM16A protein, a Clca channel, was expressed in mouse CASMCs. BKs mediated Ca2+ sparks to relax the cells through the pathway of STOC-hyperpolarization-inactivating LVDCC-terminating Ca2+ influx, suppressing spontaneous depolarization-induced contraction to maintain the tone at a low level [35]. However, in ASMCs, Ca2+ sparks activated the pathway of Clca channel (one is TMEM16A)-STIC-depolarization-LVDCC activation-Ca2+ influx, except for the above-described BKs-mediated relaxation. However, the integrated result was contraction. The reasons are that Ca2+ sparks activated BKs prior to Clca channels and the Clca channels-induced LVDCC activation degree was larger than the BKs-caused LVDCC inactivation degree. In addition, compared with ASMCs, CASMCs had great and higher amplitude Ca2+ sparks, a higher BK density and higher voltage and Ca2+ sensitivity of BKs. These allow Ca2+ sparks to induce more relaxation in CASMCs than in ASMCs, additionally supporting Ca2+ sparks in decreasing CASMC tone and increasing ASMC tone. Moreover, β1 subunits determine the higher voltage and Ca2+ sensitivity of BKs in CASMCs than in ASMCs.
Mechanism of Ca2+ sparks decreasing the mouse CASMC tone
Ca2+ sparks induced rat and human CASMC relaxation [2-4, 36]. However, this was not reported in mouse CASMCs [37-39]. We found that Ca2+ spark abolishment induced mouse CASMC elongation (Fig. 1), suggesting that Ca2+ sparks decreased mouse CASMC tone. This was mediated by BKs because paxilline induced cell shortening (Fig. S12). On the other hand, Ca2+ sparks can activate Clca channels to generate STICs and eventually cause contraction. TMEM16A is a Clca channel [40, 41], which was expressed and which mediated a contraction in rat CASMCs ([9, 42], Fig. S3). However, in mouse CASMCs, STICs and caffeine-induced inward currents were not recorded (Fig. 2), Vm was not affected by the Clca channel blocker NA (Fig. 3). These results were consistent with no inward STICs being observed at -40, -50 and -60 mV in mouse CASMCs [37-39]. These results excluded the existence of functional Clca channels, including TMEM16A, although it was expressed (Figs. S4, S5). The underlying reason for this needs to be further defined. Therefore, BKs mediated Ca2+ sparks to decrease the mouse CASMC tone. This relaxant action plays an important role in preventing cerebral artery narrowing; because CASMC keep contracting induced by spontaneous depolarization-activated LVDCCs (Figs. 3, 7). Thus, the key and final step for Ca2+ sparks to decrease CASMC tone is the inactivation of LVDCCs. This was evidenced by the results (Fig. S13).
Mechanism of Ca2+ sparks increasing mouse ASMC tone
In mouse ASMCs, Ca2+ sparks increased, and the cells contracted [16, 17], suggesting that Ca2+ sparks increased tone. In the present study, we abolished Ca2+ sparks, and the cells became elongated (Figs. 1, S1). These data suggest that Ca2+ sparks increased mouse ASMC tone. This was inconsistent with ryanodine inducing mouse ASMC shortening [18].The reason for this discrepancy needs to be further clarified, although it might be due to species differences.
Mouse ASMCs expressed functional BKs and Clca channels (Figs. 2, [18, 43]). These channel-mediated currents resulted in Vm oscillations (Fig. 3, [18]). The upward spikes resulted from Clca channels, and the downward spikes resulted from BKs (Fig. 3), and the former can result in LVDCC activation (Figs. 3, S7; [18]). Therefore, in the physiological state, Ca2+ sparks result in contraction (Fig. S12). However, BKs can also mediateCa2+ sparks to relax the cells (Fig. S12). Why did the two components not completely counteract each other? The cause may be that Ca2+ sparks activated BKs prior to Clca channels (Fig. 1), a phenomenon that was also observed in guinea-pig ASMCs [11], allowing Clca to result in LVDCC activation, and that Clca-induced LVDCC activation was much larger than BK-caused LVDCC inactivation (Fig. 3D). Therefore, the determinant step for Ca2+ sparks to increase ASMC tone is the activation of LVDCCs, an activity that was demonstrated (Fig. S13).
Whether Clca channels in mouse ASMCs were TMEM16A channels [40, 41]? TMEM16A was found to be expressed (Figs. S4, S5), but it might not be the main Clca channel because it did not contribute greatly to STICs and caffeine-induced inward currents (Fig. S6). Previous data have shown that TMEM16A gene deletion in the mouse resulted in an abolishment of STICs in ASMCs and that the inhibition of TMEM16A prevented agonist-induced mouse airway contraction [44]. Such a role was also observed in human [45] and guinea-pig ASMCs [46]. This indicates that the role of TMEM16A needs to be further investigated.
BKs induce larger tone decreases in mouse CASMCs than in mouse ASMCs
CASMCs have more STOCs than ASMCs (Fig. 2), likely due to CASMCs having great and higher amplitude Ca2+ sparks (Fig. S2), more BKs (Figs. 2, 4, S8) and higher voltage and Ca2+ sensitivity of BKs (Fig. 4) than ASMCs. STOCs then induce more hyperpolarization in CASMCs than in ASMCs, resulting in moe inactivation of LVDCCs in the former than in the latter. Indeed, this was confirmed by the results (Fig. 3). However, the blockade of BKs by paxilline failed to induce larger elongation of CASMCs (Fig. S12). The cause may be that CASMCs had larger LVDCC currents (Fig. S7), which resulted in more Ca2+ flux to induce larger contraction than ASMCs, counteracting the BKs-mediated larger relaxation in CASMCs.
BKs are voltage- and Ca2+-dependent channels and each channel includes one pore-forming α and four tissue-specific regulatory β subunits (β1-4) [47, 48]. β1 is widely expressed in smooth muscle [47], consistent with this, its mRNA in both cell types was detected (Fig. S9). β4 was also observed and its level was higher in CASMCs. The level of α was high in both cell types as expected. These mRNA data helped us to narrow the auxiliary subunits to β1 and β4. Our results show that α was the pore-forming subunit (Fig. S10); therefore, its role in voltage and Ca2+ sensitivity of BKs [49] cannot be observed by this approach. Both sensitivities will be determined by β1 (Fig. 5), and not by β4 (Fig. S11), in both cell types, therefore, the higher voltage and Ca2+ sensitivity of BKs in CASMCs than in ASMCs is also attributable to β1 subunit (Fig. 5).
Effects of Ca2+ sparks on human cell tone
In addition, we found that Ca2+ sparks similarly decreased human CASMC tone and increased human ASMC tone, and the underlying mechanism might be the same as that in mouse cells, based on the results (Figs. 6, S14).
Summary
Our results suggest that Ca2+ sparks had distinct effects on the tone of mouse and human CASMCs and ASMCs, mediated by BKs and/or Clca channels and finally determined by LVDCCs (Fig. 7). These findings may provide new interpretations and therapeutic strategies for cerebral ischemia diseases and lung obstructive diseases.
Abbreviations
ASM: airway smooth muscle; ASMCs: airway smooth muscle cells; CASMCs: cerebral artery smooth muscle cells; Ca2+ sparks: spontaneous, transient local Ca2+ elevations; RyRs: ryanodine receptors; Clca channels: Ca2+-activated Cl- channels; BKs: large-conductance Ca2+-activated K+ channels; STICs: spontaneous transient inward currents; STOCs: spontaneous transient outward currents; LVDCCs: L-type voltage-dependent Ca2+ channels; TMEM16A: transmembrane protein 16A; ALA: airway lumen area; ISO: isoproterenol; Po: probability; Vm: membrane potential; TRs: tracheal rings; TEA: tetraethylammonium chloride; NA: niflumic acid; COPD: chronic obstructive pulmonary disease.
Supplementary Material
Table S1, Figures S1-S14.
Acknowledgements
The authors thank Wen-Bo Sai, Ting Zhang, Li Tan and Hai-Xia Cheng for their technical assistance.
Funding
This research was supported by the National Natural Science Foundation of China (31140087, 30971514 and 31571200 to Q.-H. Liu; 30900816 and 31371307 to Y.-B. Peng) and the Open Foundation of Hubei Provincial Key Laboratory for Protection and Application of Special Plants in Wuling Area of China & the Fundamental Research Funds for the South-Central University for Nationalities (CZW15012, CZW15025, CZP17082).
Author contributions
Qing-Hua Liu,Qing-Yang Zhao, Yong-Bo Peng, Xiao-Jing Luo, Xi Luo, Hao Xu, Ming-Yu Wei designed experiments and wrote the manuscript; Qing-Yang Zhao, Yong-Bo Peng, Xiao-Jing Luo, Xi Luo, Hao Xu, Ming-Yu Wei, Qiu-Ju Jiang, Wen-Er Li, Li-Qun Ma, Jin-Chao Xu, Xiao-Cao Liu, Dun-An Zang, Yu-San She, He Zhu, Ying Fang, Jiuping Ding, Ping Zhang, Xiangning Fu, Jingyu Chen, Xiaowei Nie, Chenyou Shen, Shu Chen, Shanshan Chen, Jingcao Chen, Sheng Hu, Jinhua Shen, Ping Zhao, Lu Xue, Meng-Fei Yu, Weiwei Chen, Ping Zhang, Yun-Min Zheng, Tengyao Song, Yong-Bo Peng, Cunbin Zou, Gangjian Qin, Guangju Ji, Yong-Xiao Wang performed the experiments and analyzed the data; Cunbin Zou, Gangjian Qin, Guangju Ji, Yong-Xiao Wang reviewed the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740-4
2. Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ. et al. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633-7
3. Fay FS. Calcium sparks in vascular smooth muscle: relaxation regulators. Science. 1995;270:588-9
4. Gollasch M, Wellman GC, Knot HJ, Jaggar JH, Damon DH, Bonev AD. et al. Ontogeny of local sarcoplasmic reticulum Ca2+ signals in cerebral arteries: Ca2+ sparks as elementary physiological events. Circ Res. 1998;83:1104-14
5. Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J Physiol. 1998;508( Pt 1):211-21
6. Koide M, Nystoriak MA, Krishnamoorthy G, O'Connor KP, Bonev AD, Nelson MT. et al. Reduced Ca2+ spark activity after subarachnoid hemorrhage disables BK channel control of cerebral artery tone. J Cereb Blood Flow Metab. 2011;31:3-16
7. Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW. et al. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870-6
8. Janssen LJ, Sims SM. Histamine activates Cl- and K+ currents in guinea-pig tracheal myocytes: convergence with muscarinic signalling pathway. The Journal of physiology. 1993;465:661-77
9. Thomas-Gatewood C, Neeb ZP, Bulley S, Adebiyi A, Bannister JP, Leo MD. et al. TMEM16A channels generate Ca(2)(+)-activated Cl(-) currents in cerebral artery smooth muscle cells. American journal of physiology Heart and circulatory physiology. 2011;301:H1819-27
10. 22394518Bulley S, Neeb ZP, Burris SK, Bannister JP, Thomas-Gatewood CM, Jangsangthong W. et al. TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ Res. 2012;111:1027-36
11. ZhuGe R, Sims SM, Tuft RA, Fogarty KE, Walsh JV Jr. Ca2+ sparks activate K+ and Cl- channels, resulting in spontaneous transient currents in guinea-pig tracheal myocytes. J Physiol. 1998;513( Pt 3):711-8
12. Pabelick CM, Prakash YS, Kannan MS, Sieck GC. Spatial and temporal aspects of calcium sparks in porcine tracheal smooth muscle cells. Am J Physiol. 1999;277:L1018-25
13. Liu QH, Zheng YM, Wang YX. Two distinct signaling pathways for regulation of spontaneous local Ca2+ release by phospholipase C in airway smooth muscle cells. Pflugers Arch. 2007;453:531-41
14. Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. American journal of physiology Cell physiology. 2000;278:C235-56
15. Kotlikoff MI, Wang YX. Calcium release and calcium-activated chloride channels in airway smooth muscle cells. American journal of respiratory and critical care medicine. 1998;158:S109-14
16. Liu QH, Zheng YM, Korde AS, Yadav VR, Rathore R, Wess J. et al. Membrane depolarization causes a direct activation of G protein-coupled receptors leading to local Ca2+ release in smooth muscle. Proc Natl Acad Sci U S A. 2009;106:11418-23
17. Tuo QR, Ma YF, Chen W, Luo XJ, Shen J, Guo D. et al. Reactive oxygen species induce a Ca(2+)-spark increase in sensitized murine airway smooth muscle cells. Biochemical and biophysical research communications. 2013;434:498-502
18. Zhuge R, Bao R, Fogarty KE, Lifshitz LM. Ca2+ sparks act as potent regulators of excitation-contraction coupling in airway smooth muscle. The Journal of biological chemistry. 2010;285:2203-10
19. Zhang T, Luo XJ, Sai WB, Yu MF, Li WE, Ma YF. et al. Non-selective cation channels mediate chloroquine-induced relaxation in precontracted mouse airway smooth muscle. PloS one. 2014;9:e101578
20. Wei MY, Xue L, Tan L, Sai WB, Liu XC, Jiang QJ. et al. Involvement of large-conductance Ca2+-activated K+ channels in chloroquine-induced force alterations in pre-contracted airway smooth muscle. PloS one. 2015;10:e0121566
21. Dong L, Zheng YM, Van Riper D, Rathore R, Liu QH, Singer HA. et al. Functional and molecular evidence for impairment of calcium-activated potassium channels in type-1 diabetic cerebral artery smooth muscle cells. J Cereb Blood Flow Metab. 2008;28:377-86
22. Liu QH, Zheng YM, Korde AS, Li XQ, Ma J, Takeshima H. et al. Protein kinase C-epsilon regulates local calcium signaling in airway smooth muscle cells. Am J Respir Cell Mol Biol. 2009;40:663-71
23. Sai WB, Yu MF, Wei MY, Lu Z, Zheng YM, Wang YX. et al. Bitter tastants induce relaxation of rat thoracic aorta precontracted with high K(+). Clin Exp Pharmacol Physiol. 2014;41:301-8
24. Zhang CH, Lifshitz LM, Uy KF, Ikebe M, Fogarty KE, ZhuGe R. The cellular and molecular basis of bitter tastant-induced bronchodilation. PLoS biology. 2013;11:e1001501
25. Xiao JH, Zheng YM, Liao B, Wang YX. Functional role of canonical transient receptor potential 1 and canonical transient receptor potential 3 in normal and asthmatic airway smooth muscle cells. Am J Respir Cell Mol Biol. 2010;43:17-25
26. Cai Y, Bolte C, Le T, Goda C, Xu Y, Kalin TV. et al. FOXF1 maintains endothelial barrier function and prevents edema after lung injury. Science signaling. 2016;9:ra40
27. Caci E, Scudieri P, Di Carlo E, Morelli P, Bruno S, De Fino I. et al. Upregulation of TMEM16A Protein in Bronchial Epithelial Cells by Bacterial Pyocyanin. PLoS One. 2015;10:e0131775
28. Bukiya A, Dopico AM, Leffler CW, Fedinec A. Dietary cholesterol protects against alcohol-induced cerebral artery constriction. Alcohol Clin Exp Res. 2014;38:1216-26
29. Yamamura A, Yamamura H, Guo Q, Zimnicka AM, Wan J, Ko EA. et al. Dihydropyridine Ca(2+) channel blockers increase cytosolic [Ca(2+)] by activating Ca(2+)-sensing receptors in pulmonary arterial smooth muscle cells. Circ Res. 2013;112:640-50
30. Boccaccio A, Menini A. Temporal development of cyclic nucleotide-gated and Ca2+ -activated Cl- currents in isolated mouse olfactory sensory neurons. J Neurophysiol. 2007;98:153-60
31. Balderas E, Zhang J, Stefani E, Toro L. Mitochondrial BKCa channel. Front Physiol. 2015;6:104
32. Evanson KW, Bannister JP, Leo MD, Jaggar JH. LRRC26 is a functional BK channel auxiliary gamma subunit in arterial smooth muscle cells. Circ Res. 2014;115:423-31
33. Shi J, Ju M, Saleh SN, Albert AP, Large WA. TRPC6 channels stimulated by angiotensin II are inhibited by TRPC1/C5 channel activity through a Ca2+- and PKC-dependent mechanism in native vascular myocytes. J Physiol. 2010;588:3671-82
34. Zemen BG, Lai MH, Whitt JP, Khan Z, Zhao G, Meredith AL. Generation of Kcnma1fl-tdTomato, a conditional deletion of the BK channel alpha subunit in mouse. Physiol Rep. 2015:3
35. Kidd MW, Leo MD, Bannister JP, Jaggar JH. Intravascular pressure enhances the abundance of functional Kv1.5 channels at the surface of arterial smooth muscle cells. Science signaling. 2015;8:ra83
36. Wellman GC, Nathan DJ, Saundry CM, Perez G, Bonev AD, Penar PL. et al. Ca2+ sparks and their function in human cerebral arteries. Stroke. 2002;33:802-8
37. Rueda A, Fernandez-Velasco M, Benitah JP, Gomez AM. Abnormal Ca2+ spark/STOC coupling in cerebral artery smooth muscle cells of obese type 2 diabetic mice. PLoS One. 2013;8:e53321
38. Pluger S, Faulhaber J, Furstenau M, Lohn M, Waldschutz R, Gollasch M. et al. Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca(2+) spark/STOC coupling and elevated blood pressure. Circ Res. 2000;87:E53-60
39. Essin K, Welling A, Hofmann F, Luft FC, Gollasch M, Moosmang S. Indirect coupling between Cav1.2 channels and ryanodine receptors to generate Ca2+ sparks in murine arterial smooth muscle cells. J Physiol. 2007;584:205-19
40. Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019-29
41. Yu K, Duran C, Qu Z, Cui YY, Hartzell HC. Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ Res. 2012;110:990-9
42. Bulley S, Neeb ZP, Burris SK, Bannister JP, Thomas-Gatewood CM, Jangsangthong W. et al. TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ Res. 2012;111:1027-36
43. Bao R, Lifshitz LM, Tuft RA, Bellve K, Fogarty KE, ZhuGe R. A close association of RyRs with highly dense clusters of Ca2+-activated Cl- channels underlies the activation of STICs by Ca2+ sparks in mouse airway smooth muscle. J Gen Physiol. 2008;132:145-60
44. Zhang CH, Li Y, Zhao W, Lifshitz LM, Li H, Harfe BD. et al. The transmembrane protein 16A Ca(2+)-activated Cl- channel in airway smooth muscle contributes to airway hyperresponsiveness. Am J Respir Crit Care Med. 2013;187:374-81
45. Gallos G, Remy KE, Danielsson J, Funayama H, Fu XW, Chang HY. et al. Functional expression of the TMEM16 family of calcium-activated chloride channels in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2013;305:L625-34
46. Danielsson J, Perez-Zoghbi J, Bernstein K, Barajas MB, Zhang Y, Kumar S. et al. Antagonists of the TMEM16A calcium-activated chloride channel modulate airway smooth muscle tone and intracellular calcium. Anesthesiology. 2015;123:569-81
47. Contreras GF, Castillo K, Enrique N, Carrasquel-Ursulaez W, Castillo JP, Milesi V. et al. A BK (Slo1) channel journey from molecule to physiology. Channels (Austin). 2013;7:442-58
48. Torres YP, Granados ST, Latorre R. Pharmacological consequences of the coexpression of BK channel alpha and auxiliary beta subunits. Front Physiol. 2014;5:383
49. Yang Y, Sohma Y, Nourian Z, Ella SR, Li M, Stupica A. et al. Mechanisms underlying regional differences in the Ca2+ sensitivity of BK(Ca) current in arteriolar smooth muscle. J Physiol. 2013;591:1277-93
Author contact
![]() Corresponding author: Dr. Qing-Hua Liu, Institute for Medical Biology, College of Life Sciences, South-Central University for Nationalities, 82MinZu Ave., Wuhan 430074, Hubei, People's Republic of China Tel.: 0086-27-67842576, Fax: 0086-27-67841906 liu258qcom; qinghualiuscuec.edu.cn
Corresponding author: Dr. Qing-Hua Liu, Institute for Medical Biology, College of Life Sciences, South-Central University for Nationalities, 82MinZu Ave., Wuhan 430074, Hubei, People's Republic of China Tel.: 0086-27-67842576, Fax: 0086-27-67841906 liu258qcom; qinghualiuscuec.edu.cn