Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(10):1254-1265. doi:10.7150/ijbs.21258 This issue Cite
Research Paper
PTH 1-34 Ameliorates the Osteopenia and Delayed Healing of Stabilized Tibia Fracture in Mice with Achondroplasia Resulting from Gain-Of-Function Mutation of FGFR3
Hangang Chen1, 2, Xianding Sun2, Liangjun Yin1, Shuai Chen1, Ying Zhu2, Junlan Huang2, Wanling Jiang2, Bo Chen2, Ruobin Zhang2, Lin Chen2, Mao Nie1, Yangli Xie2 ![]() , Zhongliang Deng1
, Zhongliang Deng1 ![]()
1. Department of Orthopedic Surgery, the Second Affiliated Hospital, Chongqing Medical University, Chongqing 400010, China;
2. Department of Rehabilitation Medicine, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China.
Received 2017-5-31; Accepted 2017-7-20; Published 2017-9-21
Abstract

Bone fracture healing is processed through multiple stages including the cartilaginous callus formation and its transition to bony callus. FGFR3 negatively regulates chondrogenesis and enhances osteogenesis during skeleton development. We previously found in mice carrying gain-of-function mutation of FGFR3 that FGFR3 delays the healing of un-stabilized fracture that heals mainly through endochondral ossification. Since fracture is regularly treated in clinics with rigid fixation, and stabilized fracture is healed largely through intramembranous ossification, we asked whether FGFR3, a key regulator of osteogenesis, also affect the regeneration of stabilized fracture. We found that gain-of-function mutation of FGFR3 inhibits the initiation of chondrogenesis and the subsequent bone formation. We further studied whether PTH1-34 can improve the osteopenia and delayed healing of the stabilized tibia fracture in mice with achondroplasia. Fracture healing was evaluated by radiography, micro-CT, biomechanical tests, histology, and real-time polymerase chain reaction (RT-PCR) analysis. We found that PTH 1-34 can alleviate the decreased bone mass and compromised architecture in ACH mice. Histological analysis revealed that administration of PTH1-34 increased the size of both the total callus and cartilaginous callus at 14 days after the surgery in ACH mice. RT-PCR data suggested that systemic PTH1-34 accelerated the initiation of chondrogenesis and chondrocyte maturation (earlier and higher levels of expression of chondrogenesis related markers) and enhanced the osteogenic differentiation in the fracture callus in ACH mice. These results indicate that the PTH1-34 administration resulted in an enhanced callus formation during bone fracture healing in ACH mice, which is at least in part mediated by an increase of cartilaginous callus at early stage and the promotion of bone formation in bony callus. In summary, in this study we revealed that FGFR3 delays the regeneration of stabilized fracture by inhibiting both the chondrogenesis and osteogenesis, and PTH1-34 treatment can improve the dysregulated bone metabolism and delayed bone injury healing resulting from gain-of-function mutation of FGFR3.
Introduction
FGFR3 is an essential regulator of skeleton development. In humans, gain of function mutations in FGFR3 cause retarded skeletal development including several types of chondrodysplasia, while mutation leading to decreased FGFR3 activity results in camptodactyly, tall stature, and hearing loss syndrome (CASHL) characterized with overgrowth of axial skeleton [1-3]. Studies using mouse models mimicking these mutations in FGFR3 further support the negative regulatory role of FGFR3 in skeletal development [4, 5].
The role of FGFR3 in the homeostasis and diseases/injuries of skeleton at adult stages that is mainly composed of bone tissue, however, is relatively less studied. We found previously the levels of expression of bone markers including osteocalcin (ocn), osteopotin (op) are enhanced of in the trabecular of mice mimicking human ACH [4]. We further revealed that ACH mice have decreased bone mass resulting from decreased proliferation, enhanced osteogenic differentiation of mesenchymal stromal cells (MSCs), and defective mineralization, as well as enhanced osteoclastogenesis and activity [6]. Since Valverde found that FGFR3 deficient mice also have decreased bone mass and mineralization defect [5], which is similar to that we have found in mice with ACH resulting from gain-of function mutation of FGFR3. Thus, the effects of FGFR3 on osteogenesis and bone homeostasis are assured but complex, more studies are needed to explore its accurate role and mechanisms.
Besides bone homeostasis, fracture is also a very common postnatal disease [7]. Considering the critical role of FGFR3 in chondrogenesis and osteogenesis, it's reasonable to expect the essential role of FGFR3 in fracture healing. The decreased bone mass in ACH patients may lead to increased risk for fracture. Moreover, many ACH patients may undergo orthopedic surgery to lengthen their shortened lower limbs or enlarge the stenosis in spine or foramen magnum (lumbar laminectomy) [8]. Thus, it's important to dissect the role of FGFR3 in fracture healing.
The fracture healing processes are composed of multiple steps, including early bleeding stage, recruitment and proliferation of mesenchymal cells, differentiation of mesenchyme into chondrocytes and osteoblasts, hypertrophic differentiation of chondrocytes, the apoptosis of hypertrophic chondrocytes and finally invasion of blood vessels that brings osteoclasts and osteoprogenitors to replace the cartilaginous callus with bone tissue [9]. There are similar cellular and molecular events between skeletal development and fracture healing, exerting through intramembranous and/or endochondral ossification. The actual healing processes of a fracture are largely dependent on the fixation method. Stably fixed fracture is mainly healed through intramembranous ossification, while un-stabilized fracture is basically healed through endochondral ossification [10].
The essential role of FGFR3 in skeleton development indicates that FGFR3 may be also a key regulator of fracture healing. We have investigated the role of FGFR3 in fracture healing using unstabilized fracture model of mice carrying gain-of-function mutation of FGFR3, and found that FGFR3 delays the fracture healing by inhibiting the initiation of chondrogenesis and hypertrophic differentiation during cartilage formation in the soft callus [11]. There is disadvantage of this study, since rigid fixation is the standard fixing method used to treat fractures and for limb lengthening, an artificial bone fracture commonly used in patients including achondroplasia to length their limbs. Investigating the role of FGFR3 in fracture healing using stabilized fracture model that healed mainly through intramembranous ossification is important.
In this study, by using stabilized fracture model in mice mimicking human achondroplasia caused by gain-of-function mutation of FGFR3 (FGFR3G369C/+ mice), we explored the effect of FGFR3 on the healing of stabilized fracture. We found that mice mimicking achondroplasia still have delayed healing of the stabilized fracture with both decreased chondrogenesis and osteogenesis, indicating that measures including biological therapies are needed to improve the bad fracture healing in ACH patients. PTH1-34 (teriparatide) is a FDA approved drug to effectively treat osteoporosis by increasing bone formation through the promotion of osteoblastic proliferation, differentiation and the prevention of osteoblast apoptosis [12, 13]. More importantly, PTH plays important roles in regulating both chondrogenesis and bone remodeling and we have found previously that PTHrP and PTH1-34 can alleviate the retarded skeleton development of ACH mice [14, 15]. We thus tested whether PTH1-34 can improve the osteopenia and healing of the stabilized fracture in ACH mice. We found that exogenous PTH1-34 promotes the healing of stabilized fracture of ACH mice by increasing callus areas and accelerating chondrogenesis and osteogenesis. Our results will deepen our understanding about the role of FGFR3 in osteogenesis, fracture healing, subsequently provide an experiment basis for the application of PTH1-34 in the treatment of dysregulated bone metabolism and delayed bone injury (such as fracture and limb lengthening) healing in ACH patients.
Materials and Methods
Mice
FGFR3G369C/+ mice (hereafter referred as ACH mice) were maintained on C57 background and the genotyping was conducted as described previously [4]. All experiments were performed in accordance with protocols approved by the Institutional Animals Care and Use Committee of Daping Hospital (Chongqing, China).
Tibial Shaft Fracture Surgery
8 week old male mice were undergone a standardized, unilateral tibial shaft fracture, adapted from that described by Bonnarens [16], with intramedullary fixation. Briefly, a 5-mm vertical incision in the skin was made to expose the proximal 1/2 of tibia, then a 30-gauge needle was inserted vertically to the intramedullary canal. After blunt dissection of the soft tissue, a fracture was induced by microscissors, then skin was sutured. Buprenorphine was used for control of post-operative pain. Animals were monitored for general postsurgical health and function of the fractured limb.
Administration of PTH 1-34 to mice
Recombinant human PTH 1-34 was purchased from the Anaspec Institute (Fremont, Canada) and dissolved in vehicle (sterile water for injection). ACH mice and their wild-type (WT) littermates were randomly divided into treatment group and control group. PTH1-34 was administered intraperitoneally at the dose of 80 μg/kg body weight per day until the end of the observation period while the control mice were given sterile water.
Measurement of bone mineral density
Right femora were dissected free of soft tissues and fixed. BMD was measured by dual energy X-ray absorptiometry (DEXA; PIXimus Mouse 11 densitometer, GE Medical System, Madison, WI).
X-ray imaging and Micro-computed tomography
Tibias were subjected to high-resolution X-ray examination using Faxitron MX20 (Faxitron X-Ray, Tucson, AZ, USA). After X-ray detection, the bone structure and volume of the femoral distal metaphysis and fracture region were scanned with micro-computed tomography (micro-CT) (viva CT-40, Scanco Medical AG, Switzerland). Image acquisition was performed at 70 kV and 114 μA. Every measurement used the same filtering and segmentation values. Bone structural parameters including BV/TV, Tb.Th and Tb.N were automatically determined after manually selecting trabecular bone and callus areas. Within the callus, newly formed bone was defined by excluding original cortical bone and contouring the edge of the callus on each 10.5 μm thickness 2D slice.
Histology and analysis
A CO2 overdose was used for the euthanasia for all mice. The fractured tibias were dissected and collected on post fracture days (PFD) 3, 5, 7, 10, 14, 21 and 28 with excess muscle and soft tissue excised. The intramedullary needle was removed carefully from the fractured tibia. For histological analysis, the tibias were fixed in 4% paraformaldehyde in 0.01 M PBS (pH 7.4), decalcified in 15% EDTA (pH 7.4) and embedded in paraffin. Five micrometer-thick sections were sliced and staining with Safranin-O, Fast Green, Hematoxylin and Eosin (H&E). The sections were stained using Safranin O/Fast Green and Tartrate Resistant Acid Phosphatase (TRAP, Sigma) staining. Total callus area, total cartilage area, and total woven bone area were quantified. Mice were weighed and injected with calcein (30 mg/kg body weight) at 10 and 3 days prior to sacrifice. The femurs were embedded in a mixture of methyl methacrylate and dibutyl phthalate for analysis of parameters of bone formation. Ten-micron-thick sections were used for fluorescence observation. Histomorphometric analyses were performed with the OsteoMeasure (OsteoMetrics, Atlanta, GA, USA).
RNA isolation and Real-time PCR
Total RNA was isolated using Trizol reagent (Invitrogen Life Technologies, Invitrogen, Carlsbad, USA), which was used to assess the effects of PTH1-34 treatment on mRNA expression of genes of interest. All reactions were performed in Mx3000P PCR machine (Stratagene, Santa Clara, CA, USA) using the Two-Step QuantiTect SYBR Green RT-PCR Kit (Takara) and reaction conditions were optimized for each of the genes by changing the annealing temperature. Each run was replicated three times. Expression levels for each gene of interest were normalized to their corresponding values of endogenous control gene cyclophilin A. PCR data is expressed as fold change in relation to WT control. Each run was replicated three times. The primers for the genes of interest are as follows (in 5'-3' direction): cyclophilin A, GCATACAGGTCCTGGCATCT and TCTTGCTGGTCTTGCCATTC; Sox 9, GGGCTCTACTCCACCTTCACT and AAGATCAGCTCGGTCACCATA; col2, CTGGTGGAGCAGCAAGAGCAA and CAGTGGACAGTAGACGGAGGAAAG; col 10, GCAGCATTACGACCCAAGAT and CATGATTGCACTCCCTGAAG; pthrp, CATCAGCTACTGCATGACAAGG and GGTGGTTTTTGGTGTTGGGAG; cbfa1, CCTGAACTCTGCACCAAGTC and GAGGTGGCAGTGTCATCATC; ocn, TCTGACAAAGCCTTCATGTCC and AATAGTGATACCGTAGATGCG; alp, TGTCTGGAACCGCACTGAACT and CAGTCAGGTTGTTCCGATTCAA; col 1, AATGGTGAGACGTGGAAACCCGAG and CGACTCCTACATCTTCTGAGTTTGG; op, TGCACCCAGATCCTATAGCC and TGTGGTCATGGCTTTCATTG.
Biomechanical Testing
The biomechanical properties of the fracture calluses were examined by a three-point bending test according to the protocol described previously [17]. Briefly, a downward bending load was applied to the shaft of the posterior aspect of the fractured tibia with the loading nose directly over the fracture site (Instron, Norwood, MA, USA). A load-displacement curve was generated to determine stiffness (N/mm), ultimate force (N) and work to failure (mJ).
Statistical analysis
The data were presented as Means ± SD. Statistical significance was ascertained by two-way ANOVA. When significant levels (P<0.05) were achieved, Tukey's Post Hoc test was performed using PASW 18.0 (IBM, Armonk, USA). The results were considered significantly different at P<0.05.
Results
PTH1-34 alleviates the osteopenia in ACH mice
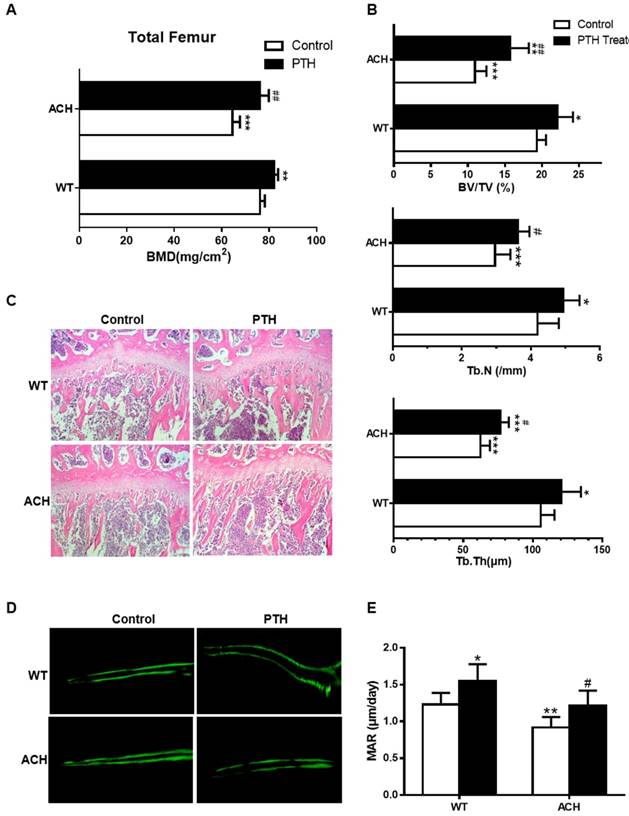
Both ACH patients and mice showed osteopenia, which is a risk factor of fracture. As a FDA-approved drug to treat osteoporosis in humans by promoting bone formation, we first detected the effect of PTH1-34 on bone homeostasis of ACH mice. After being intraperitoneal injected with PTH1-34 (80 μg/kg) or equal volumes of sterile water daily from 8 weeks to 12 weeks, bone mass of femurs was increased in mice of both genotypes. Quantification data of BMD by DEXA showed that PTH1-34 treatment increased total femoral BMD of ACH mice significantly by 18.1% (Fig 1A). Micro-CT analysis of the distal metaphyses of femurs was used to assess the structural parameters including bone volume/tissue volume (%, BV/TV), trabecular number (Tb.N) and trabecular thickness (Tb.Th) in 12-week-old mice. ACH control group displayed a decrease of BV/TV, Tb.N, and Tb.Th, which indicates osteopenia as previously reported. In ACH mice, PTH 1-34 treatment increased BV/TV, Tb.N and Tb.Th markedly by 43.8%, 22.3% and 21.7%, respectively indicating improvement of osteopenia in ACH mice after PTH 1-34 treatment. While in WT littermates, PTH 1-34 administration increased the Tb.N and Tb.Th by 17.9% and 14.5%, respectively (Fig 1B). H&E staining of tibiae revealed that 12-week-old ACH mice exhibited sparser trabecular bone in proximal tibiae of compared with WT mice and PTH 1-34 treatment partially but significantly improved the osteopenia of ACH tibiae (Fig 1C). The reduced mineral appositional rates (MAR) were also improved by PTH 1-34-treatment in ACH mice (Fig 1D and E). These observations indicate that the decreased bone mass and compromised architecture resulting from activated mutation in FGFR3 in adult mice can be ameliorated by PTH 1-34 treatment.
PTH1-34 alleviates the osteopenia in ACH mice. (A) BMD of total femurs from ACH and WT mice measured by DEXA (n=8). (B) Quantitative micro-CT analyses of distal femoral metaphysis (BV/TV, Tb.N and Tb.Th) at 12 weeks (n=6-8). (C) Histology (H&E staining) of the metaphysis of proximal tibiae in wild-type mice and ACH mice treated by vehicle and PTH 1-34 at 12 weeks. (magnification ×200). (D) Double calcein labeling of the undecalcified femurs. (E) Quantitative data of MAR. Graphs show mean value ± SD (*, P <0.05; **, P <0.01; ***, P <0.001; compared with WT control mice. #, P <0.05; ##, P <0.01; compared with ACH control mice).
PTH1-34 treatment alleviates the impaired fracture healing of ACH mice after tibial shaft fracture with intramedullary fixation
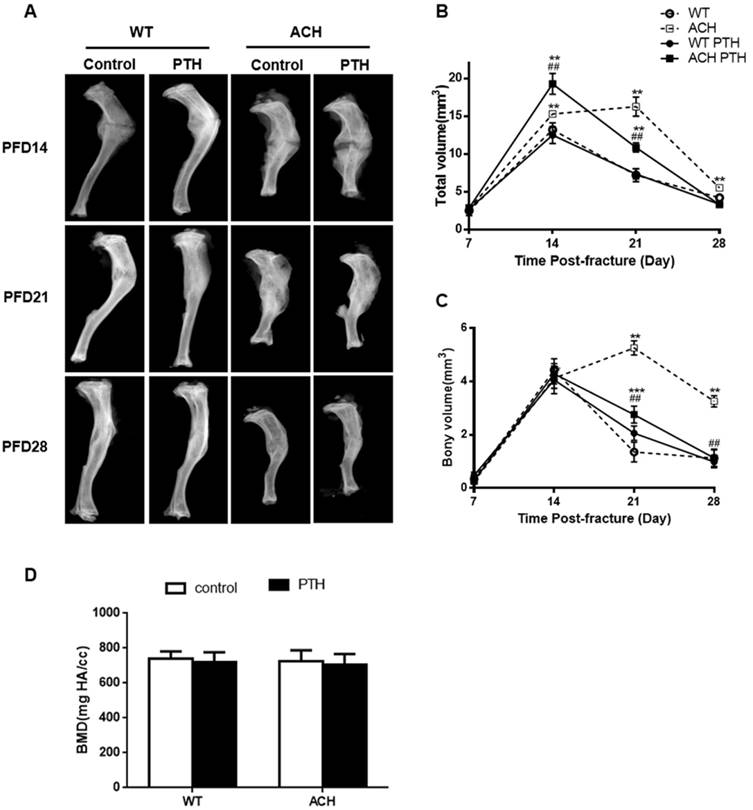
Previous studies showed that gain-of-function mutation of FGFR3 resulted in delayed fracture healing by inhibiting chondrocyte differentiation and resorption in closed non-stabilized fractures [11]. Here, we generated closed stabilized tibial fractures in WT and ACH mice and examined the healing process at different stages. Radiographic evaluation of the fractures over a course of 28 days revealed callus formation in fractured bones. There was no callus formation at PFD3 (Data not shown). By PFD14, a much larger callus with more radiolucency was observed at fracture site in ACH mice. At PFD 28, ACH tibias had more radiolucent fracture callus compared to WT indicating that ACH mice still showed delayed fracture healing in a relatively stable fracture model, which showed less cartilaginous callus than non-stabilized fractures.
Since PTH1-34 can effectively treat osteoporosis and osteoporotic fracture, we asked whether PTH1-34 is an effective biological therapeutic measure for the delayed fracture repair in ACH. PTH 1-34-treated ACH mice showed lager callus than control ACH mice at PFD14 but less radiolucent fracture callus at PFD 28 (Fig 2A). Micro-CT analysis was used to examine the mineralized calluses at the center of calluses. Total callus volume was greatest at 14-day post-fracture in WT mice, and then continuously decreased as resorption of the callus progressed throughout the observation period of study. The total callus volume was significantly larger in the ACH mice (16.25 ± 1.27 mm3) at PFD 21 compared to WT mice (7.20 ± 0.87 mm3) (Fig 2B), bony volume of ACH calluses remained increased at days 21(5.25 ± 0.27 mm3) and deceased at PFD28 (3.35 ± 0.31 mm3) (Fig 2C). After PTH 1-34 treatment, ACH mice showed lager callus in PFD14 and fewer calluses at PFD28 than that of control ACH mice. The similar trend was found in the ratio of bony callus volume to total callus volume (Data not shown). The mineral density had no significant difference at PFD 28 among four groups (Fig 2D). Together, these results indicate that ACH mice showed delayed healing of fracture and this delay can be improved significantly by PTH1-34 treatment.
PTH1-34 treatment alleviates the impaired fracture healing of ACH mice. (A) Representative radiographs of fractured tibias. (B) The quantitative analysis for total callus volume. (C) The quantitative analysis of bony callus volume. (D) The quantitative analysis of BMD. Data are presented as Means ± SD (N=6-8/group; *, P <0.05; **, P <0.01; ***, P <0.001; compared with WT control mice at the same group. ##, P <0.01; compared with ACH control mice at the same group).
Biomechanical properties of the fracture calluses of ACH were improved by PTH 1-34 treatment. (A) Stiffness, (B) ultimate force to failure, and (C) work to failure were assessed by three-point bending in tibias at 28 days post-fracture in four groups. Data are presented as Means±SD (N=6/group, *p<0.05; **p<0.01; ***p<0.001, compared with WT control mice. #, P <0.05; ##, P <0.01; compared with ACH control mice).
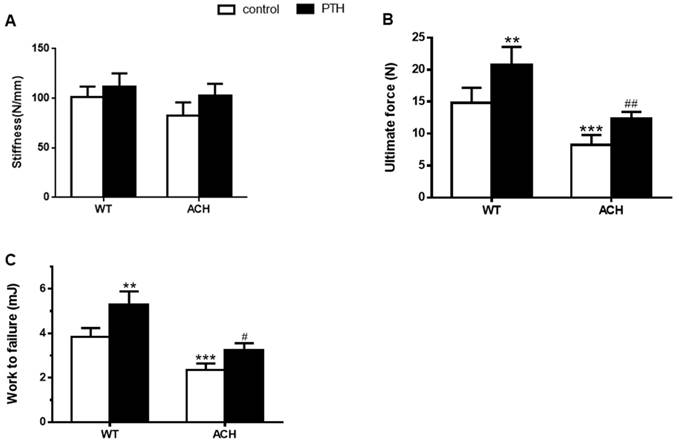
PTH 1-34 enhanced the bone mechanical properties of fractured tibia in ACH mice
Bone mass and architecture determined the bone mechanical properties. To assess the effects of enhanced FGFR3 activity on the mechanical properties of fractured tibia, biomechanical properties were examined by three-point bending in tibias at 4 weeks after fracture in WT and ACH mice. There were a lower work to failure (2.35 ± 0.29 vs 3.84 ± 0.39 mJ, P<0.001) and ultimate force (8.25 ± 1.56 vs 12.35 ± 1.05 N, P<0.001) in the vehicle-treated tibias from ACH mice compared to the vehicle-treated WT controls. After PTH 1-34 treatment, there were significantly increased work to failure and ultimate force in both WT and ACH bone. The work to failure and ultimate force were comparable between PTH 1-34-treated ACH mice and WT mice. These results demonstrated that exogenous PTH administration improved bone strength in the fracture site of WT and ACH mice.
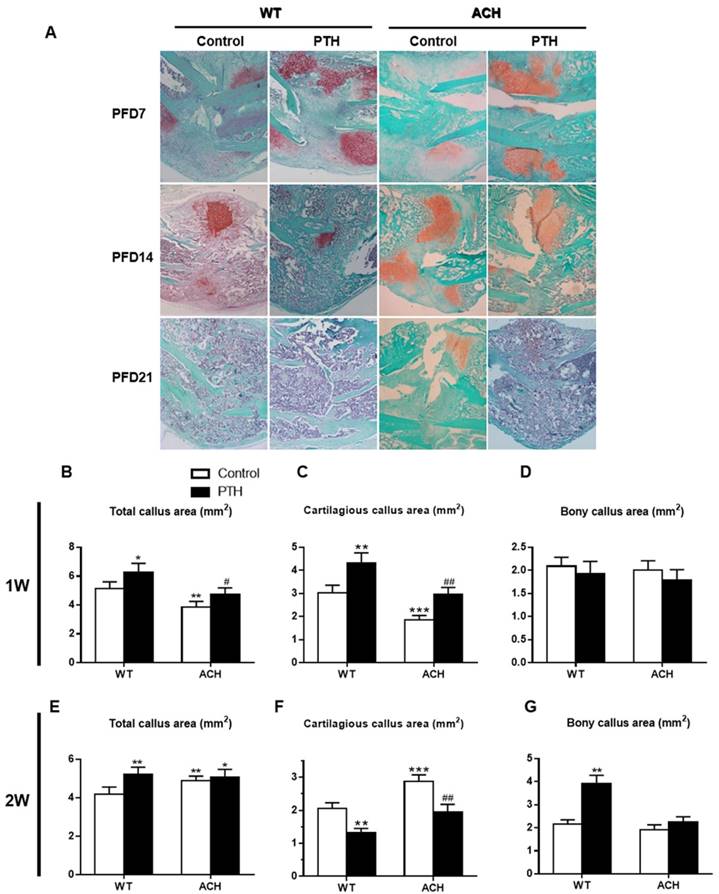
Effects of PTH 1-34 on cartilaginous callus formation and on its transformation into bony callus in ACH mice
PTH/PTHrP/PTHR1 signaling can promote the proliferation of chondrocytes and inhibit the differentiation of chondrocytes through its downstream signaling molecules. While PTHrP/PTHR1 signaling plays a dominant role in regulating the pool of proliferating chondrocytes during limb development, FGFR3 signaling plays a more prominent role in cartilage maturation. Callus tissues from vehicle-treated and PTH 1-34-treated ACH mice were analyzed by histology and computer-assisted image analysis. Compared to those of WT control mice, the total callus areas (ACH 3.85 ± 0.39 vs WT 5.12 ± 0.47 mm2) and cartilaginous callus areas (ACH 1.85 ± 0.22 vs WT 3.02± 0.29 mm2) were reduced in ACH mice at PFD7 (Fig 4B, C). The callus of WT and PTH 1-34-treated ACH mice showed comparable amounts of cartilaginous and bony tissue at PFD7 (Fig 4D). At PFD14, the cartilage tissue in fracture site of WT mice was dramatically reduced, thus ACH mice showed a relative increase of cartilaginous callus compared to WT mice (Fig 4F). The fracture gap was connected by massive accumulation of bony callus, and most of the cartilaginous callus had been resorbed in the WT control and PTH 1-34-treated mice (Fig 4A). In contrast, ACH mice showed apparent cartilaginous callus in the central area of callus at PFD21. The amount of bone tissue was significantly lower where the amounts of remnant cartilaginous and fibrous tissue were significantly higher in ACH mice. These observations indicate that fracture unions are impaired in ACH mice and PTH 1-34-treatment can alleviate this delayed fracture healing.
Effects of exogenous PTH on cartilaginous callus formation and its transformation into bony callus. Representative micrographs of paraffin sections of calluses from mice at 1,2 and 3 weeks post fracture (A). Areas of the total callus (B, E), cartilaginous callus (C, F) and bony callus (D, G) were measured by computer-assisted image analysis. Each value is the Means±SD of determinations in 6-8 animals from each group (*p<0.05; **p<0.01; ***p<0.001, compared with WT control mice. #, P <0.05; ##, P <0.01; compared with ACH control mice).
The delayed endochondral bone formation and osteoblast differentiation of ACH mice is partially rescued by PTH 1-34 treatment
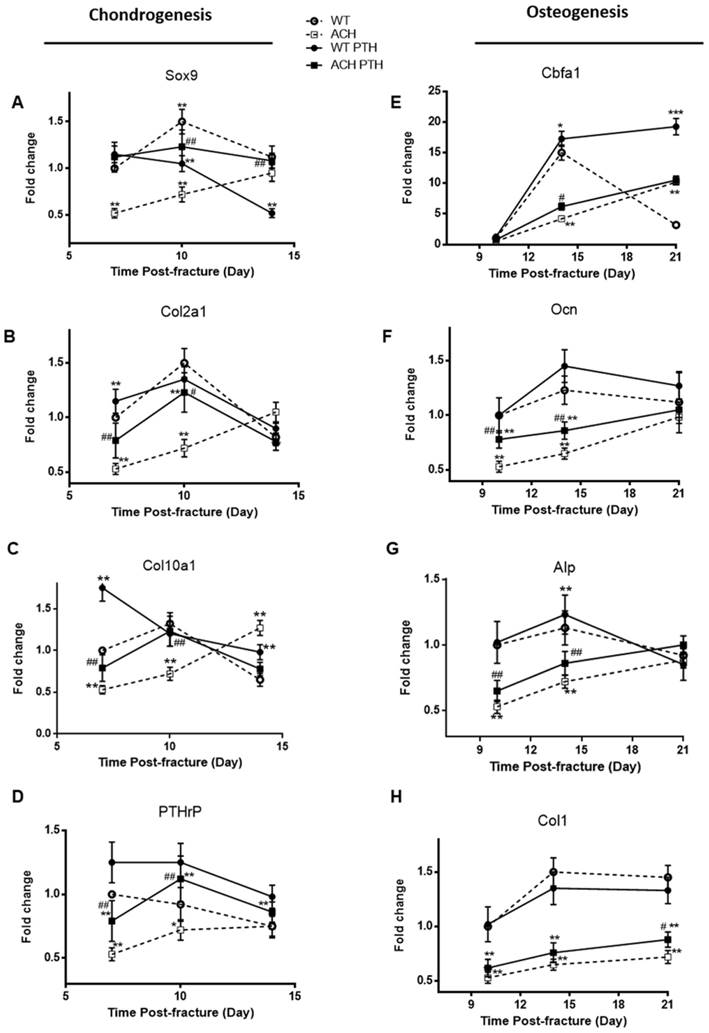
To determine whether the alterations of the callus areas were associated with the changes of the expression of genes associated with endochondral bone formation, quantitative real-time PCR analysis was performed. Sox 9, collagen type II (Col2a1) and collagen type X (Col10a1) expression reached at peak value earlier during fracture repair in the WT and PTH 1-34-treated ACH mice, and the expression levels of these two groups were comparable, which are significantly lower at PFD7 and 10 in the ACH mice (Fig 5A-C). The expressions of Col2a1 and Col10a1 peaked in ACH fractures at PFD14, while they peaked at PFD10 in WT and PTH 1-34-treated ACH fracture. The delayed appearance and disappearance of these chondrogenic genes in the fracture callus are consistent with the delayed endochondral bone formation in the ACH mice and PTH 1-34-treatment can restore this delay in some extent. We also found that PTH 1-34 can increase the expression of PTHrP at PFD10 in both WT and ACH mice (Fig 5D). Despite the improved chondrogenesis, we checked the expression levels of genes involved in osteoblastic differentiation in the callus tissue of fractures site in control and PTH 1-34-treated ACH mice. The expression level of cbfa1 was significantly lower in tissues containing the fracture callus in ACH mice compared with WT control at PFD14 (Fig 5E). The other osteoblast differentiation markers, including collagen type I (Col1a1), alkaline phosphatase (Alp), and Ocn also had delayed maximal expression (at PFD21) in fracture callus from ACH, consistent with the delayed bony tissue formation in fracture site. After PTH 1-34-treatment, the expression levels of Col1a1, Alp and Ocn were increased in ACH mice at the fracture site at both PFD14 and PFD21(Fig 5F-H), while the delayed peak expression of these genes were not ameliorated. These observations demonstrated that systemic PTH 1-34 accelerated chondrocyte maturation and enhanced osteogenic differentiation in the fracture callus in ACH mice.
Analysis of the PTH 1-34 effects on cellular differentiation during fracture healing in WT and ACH mice. Total RNA was extracted from callus in mice at various time points after fracture. Real-time RT-PCR mRNA expression analyses were performed for evaluating expressions of genes involved in chondrogenesis in callus from mice after fracture and results were expressed as fold changes relative to the expression level of WT callus at PFD7(A-D). The following primer sets were used: cbfa1 (E), alp (F), ocn (G) and col1 (H) to evaluate the osteogenic differentiation during fracture healing. Results were expressed as fold changes relative to the expression level of WT callus at PFD7. The analysis was repeated for three times. Data are presented as Means±SD (N=5-6 mice/group; *, P <0.05; **, P <0.01; ***, P <0.001; compared with WT control mice at the same group. #, P <0.05; ##, P <0.01; compared with ACH control mice at the same group).
Discussion
ACH is the most common genetic dwarfism [18]. ACH patients have normal lifespan, and usually will experience the harmful effects of maldeveloped and abnormally maintained skeleton. ACH patients have osteopenia, which may make them more susceptible to fracture. Moreover, many ACH patients will undergo limb lengthening or decompression surgery for their stenosed foramens [19]. Whether the fracture healing processes of ACH patients are different to patients without FGFR3 mutation, especially the underlying mechanisms are not clarified.
We previously studied the role of FGFR3 in fracture healing using un-stablized fracture model, which is not regularly used in clinics, as rigid fixation is routinely used clinically. In this study, we used stabilized fracture model to study the role of FGFR3 in the healing of fracture fixed with clinically regularly used stabilization approach. Compared with unstable fracture model, this stabilized fracture model has less endochondral ossification and more intramembranous ossification [20].
A variety of studies including ours demonstrated that FGFR3 is a negative regulator of skeleton growth mainly by affecting chondrogenesis during endochondral ossification. However, the effect of FGFR3 on bone formation or osteogenesis either in endochondral or intramembranous ossification during skeleton development and fracture is not fully clarified presently.
In this study we found that in stabilized fracture model of ACH mice there was still exhibited delayed chondrogenesis with less cartilaginous callus and postponed replacement of cartilage with bone tissue, although the extent was much less than that in un-stabilized model. More importantly, we revealed that although stabilized fracture model is supposed to have more osteogenesis (intramembranous ossification), the osteogenic differentiation was still inhibited in ACH mice compared with that in WT mice as revealed by delayed and decreased expressions of osteogenic markers including cbfa1. The mechanisms for the inhibited osteogenesis are not fully clarified presently. As described above, the bone formation (osteogenesis) in fracture healing is either secondary to chondrogenesis (as the later part of endochondral ossification) or directly derived from osteogenic differentiation of MSCs in non-cartilaginous region (intramembranous ossification). Thus, the inhibited osteogenesis in ACH mice may be related to the delayed chondrogenesis and smaller cartilage template. Presently, there are inconsistent results about the effects of FGFR3 on osteogenic differentiation of MSCs, ECM synthesis and mineralization. The direct effect of FGFR3 on osteogenesis during fracture healing is not clear now. Our preliminary data from study using tibia cortical defect model in ACH mice suggest that FGFR3 may directly inhibit the osteogenesis during the healing of cortical defect (unpublished data). Moreover, considering the recent progresses about the direct differentiation of chondrocytes to osteoblasts, the delayed (inhibited) osteogenesis may be also related to the inhibited chondrogenesis in ACH mice.
Above all, although the detailed effects and mechanisms of FGFR3 on osteogenesis during fracture healing are not clarified, our present results support the ideas that FGFR3 directly/indirectly inhibits osteogenesis at least during fracture healing processes. More studies including using inducible activation/inactivation of FGFR3 at specific cell lineages at adult stage are needed to explore the detailed effects and mechanisms of FGFR3 on bone formation during fracture healing.
Since ACH mice have delayed fracture healing either in un-stabilized or stabilized fracture models, we need to find biological measures to improve the poor healing of ACH patients. ACH is a developmental abnormality caused by FGFR3 mutation, which indicates that the abnormal fracture healing, a pathological process that largely recapitulates skeleton development, in ACH may be also related to the similar cellular and molecular mechanisms with skeleton development.
Since direct targeting FGFR3, the casual molecule, is difficult, we thus decided to find molecules involved in the pathogenesis of the maldevelopment of skeleton in ACH. A variety of signaling pathways have been found involved in the regulation of skeleton development by FGFR3 including PTH signaling [21]. PTH signaling is very important for skeleton development and homeostasis. During skeleton development, PTH signaling promotes proliferation and inhibits differentiation of chondrocytes. Mutations leading to constitutive activation or inactivating of PTH signaling cause Jansen metaphyseal chondrodysplasia and Blomstrand's lethal chondrodysplasia, respectively [22]. PTH signaling also plays important roles in bone homeostasis by regulating osteoblastic lineages mediated bone formation and osteoclast-mediated bone resorption [23].
PTHR1, a receptor for PTH /PTHrP, is expressed in multiple cell types during fracture healing, including chondrocytes and osteoblasts. Furthermore, multiple studies have demonstrated that PTH1-34 can improve fracture healing by preferentially enhancing chondrocyte recruitment, and the rate of chondrocyte maturation to stimulate endochondral ossification [24]. PTH 1-34 can also stimulate bone formation by increasing osteoblast number and activity, and delay the transformation of mature osteoblasts into lining cells [25]. These studies demonstrated that PTH /PTHrP signaling has important effects on both chondrogenesis and osteogenesis during skeleton development, bone homeostasis and fracture healing.
Since FGFR3 also plays essential roles in these processes, it is intriguing to speculate that there is interaction between FGFR3 and PTH signaling among those physiology and pathological events during skeleton development, homeostasis and regeneration. Indeed we have previously shown that ACH mice have decreased expression of PTHR1 in their growth plates, and PTHrP treatment can partially rescue the retarded growth of the cultured bone rudiments from ACH mice [14]. Furthermore, we previously found that intermittent subcutaneous injection of PTH 1-34 can rescue the retarded skeletal development and prevent early postnatal lethality of ACH mice by alleviating the inhibited chondrocyte differentiation and proliferation through its up-regulation of PTHrP expression and down-regulation of FGFR3 level [15]. We thus asked whether exogenous PTH 1-34 can affect the delayed fracture healing of ACH mice.
In this study, we found that exogenous PTH 1-34 promotes the healing of stabilized fracture of ACH mice by increasing callus areas and accelerating both chondrogenesis and osteogenesis. Our results using both X-ray and CT imaging showed that PTH 1-34-treated fractures generated a larger total callus volume. Although PTH 1-34 enhanced the callus volume, it did not increase the average mineral density of the callus tissue relative to controls at post-fracture 3 weeks. Both the expression levels of the chondrogenic and osteogenic related genes were increased in bone callus from PTH 1-34-treated ACH mice.
Quantitative comparison of the expression levels of the chondrogenic versus osteogenic extracellular matrix genes in callus tissues across the anabolic phase of fracture repair showed that PTH 1-34 preferentially enhanced chondrogenesis over osteogenesis. In addition to our observations showing that PTH 1-34 increased the volume of cartilage callus, we also observed an increased rate in chondrocyte hypertrophy in fracture tissues treated with PTH 1-34. The earlier induction of chondrocyte hypertrophy in the callus was indicated by an earlier peak in collagen type X expression. Previous studies have shown that endogenous PTH is essential for trabecular bone formation in the fetus and neonate and intermittent administration of PTH1-34 exhibits an anabolic effect primarily on trabecular bone [26, 27]. Our data suggest that intermittent PTH1-34 can also have positive effects on bone formation during fracture healing, which may be associated with the direct promotion of ossification and secondary effect of promoted endochondral bone formation. The combination of an increased callus volume and more rapid mineralization of the cartilaginous callus would presumably produce a more mechanically stable environment around fracture site.
Our results, for the first time demonstrated that FGFR3, a well-known key regulator of chondrogenesis, also inhibits the healing process of stabilized fracture by inhibiting both chondrogenesis and osteogenesis, and PTH 1-34, a clinically available drug, can be used to effectively improve the delayed fracture healing in ACH mice. Our current study will deepen our understanding about the role of FGFR3 in osteogenesis during skeleton development and fracture healing, subsequently provide an experiment basis for application of PTH 1-34 in the treatment of ACH patients that usually have dysregulated bone metabolism, or undergo fracture or corrective surgery for their malformed skeleton. This study also suggests that FGFR3 is an important targeting molecule for the treatment of osteoporosis and fracture/delayed fracture union in patients other than ACH.
Acknowledgements
Grant sponsors: National Natural Science Foundation of China (81530071, 81672230), Special Funds for Major State Basic Research Program of China (2014CB 942904), Foundation of army (16CXZ016) and Basic and Advanced Research Project in Chongqing (CSTC2013jcyjC0009).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes & development. 2002;16:1446-65
2. Escobar LF, Tucker M, Bamshad M. A second family with CATSHL syndrome: Confirmatory report of another unique FGFR3 syndrome. American journal of medical genetics Part A. 2016;170:1908-11
3. Toydemir RM, Brassington AE, Bayrak-Toydemir P, Krakowiak PA, Jorde LB, Whitby FG. et al. A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. American journal of human genetics. 2006;79:935-41
4. Chen L, Adar R, Yang X, Monsonego EO, Li C, Hauschka PV. et al. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest. 1999;104:1517-25
5. Valverde-Franco G, Liu H, Davidson D, Chai S, Valderrama-Carvajal H, Goltzman D. et al. Defective bone mineralization and osteopenia in young adult FGFR3-/- mice. Human molecular genetics. 2004;13:271-84
6. Su N, Sun Q, Li C, Lu X, Qi H, Chen S. et al. Gain-of-function mutation in FGFR3 in mice leads to decreased bone mass by affecting both osteoblastogenesis and osteoclastogenesis. Human molecular genetics. 2010;19:1199-210
7. Loi F, Cordova LA, Pajarinen J, Lin TH, Yao Z, Goodman SB. Inflammation, fracture and bone repair. Bone. 2016;86:119-30
8. Devmurari KN, Song HR, Modi HN, Venkatesh KP, Ju KS, Song SH. Callus features of regenerate fracture cases in femoral lengthening in achondroplasia. Skeletal radiology. 2010;39:897-903
9. Hadjiargyrou M, O'Keefe RJ. The convergence of fracture repair and stem cells: interplay of genes, aging, environmental factors and disease. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2014;29:2307-22
10. Miclau T, Lu C, Thompson Z, Choi P, Puttlitz C, Marcucio R. et al. Effects of delayed stabilization on fracture healing. J Orthop Res. 2007;25:1552-8
11. Su N, Yang J, Xie Y, Du X, Lu X, Yin Z. et al. Gain-of-function mutation of FGFR3 results in impaired fracture healing due to inhibition of chondrocyte differentiation. Biochemical and biophysical research communications. 2008;376:454-9
12. Dobnig H, Turner RT. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology. 1995;136:3632-8
13. Orwoll ES, Scheele WH, Paul S, Adami S, Syversen U, Diez-Perez A. et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2003;18:9-17
14. Chen L, Li C, Qiao W, Xu X, Deng C. A Ser(365)->Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Human molecular genetics. 2001;10:457-65
15. Xie Y, Su N, Jin M, Qi H, Yang J, Li C. et al. Intermittent PTH (1-34) injection rescues the retarded skeletal development and postnatal lethality of mice mimicking human achondroplasia and thanatophoric dysplasia. Human molecular genetics. 2012;21:3941-55
16. Bonnarens F, Einhorn TA. Production of a standard closed fracture in laboratory animal bone. J Orthop Res. 1984;2:97-101
17. Weng T, Mao F, Wang Y, Sun Q, Li R, Yang G. et al. Osteoblastic molecular scaffold Gab1 is required for maintaining bone homeostasis. Journal of cell science. 2010;123:682-9
18. Kostenuik P, Mirza FM. Fracture healing physiology and the quest for therapies for delayed healing and nonunion. J Orthop Res. 2017;35:213-23
19. Accogli A, Pacetti M, Fiaschi P, Pavanello M, Piatelli G, Nuzzi D. et al. Association of achondroplasia with sagittal synostosis and scaphocephaly in two patients, an underestimated condition? American journal of medical genetics Part A. 2015;167A:646-52
20. Garcia P, Holstein JH, Histing T, Burkhardt M, Culemann U, Pizanis A. et al. A new technique for internal fixation of femoral fractures in mice: impact of stability on fracture healing. Journal of biomechanics. 2008;41:1689-96
21. Xie Y, Zhou S, Chen H, Du X, Chen L. Recent research on the growth plate: Advances in fibroblast growth factor signaling in growth plate development and disorders. Journal of molecular endocrinology. 2014;53:T11-34
22. Schipani E, Provot S. PTHrP, PTH, and the PTH/PTHrP receptor in endochondral bone development. Birth defects research Part C, Embryo today: reviews. 2003;69:352-62
23. Karaplis AC, Goltzman D. PTH and PTHrP effects on the skeleton. Reviews in endocrine & metabolic disorders. 2000;1:331-41
24. Kakar S, Einhorn TA, Vora S, Miara LJ, Hon G, Wigner NA. et al. Enhanced chondrogenesis and Wnt signaling in PTH-treated fractures. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2007;22:1903-12
25. Jang MG, Lee JY, Yang JY, Park H, Kim JH, Kim JE. et al. Intermittent PTH treatment can delay the transformation of mature osteoblasts into lining cells on the periosteal surfaces. J Bone Miner Metab. 2016;34:532-9
26. Jobert AS, Zhang P, Couvineau A, Bonaventure J, Roume J, Le Merrer M. et al. Absence of functional receptors for parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J Clin Invest. 1998;102:34-40
27. Miao D, He B, Karaplis AC, Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. J Clin Invest. 2002;109:1173-82
Author contact
![]() Corresponding authors: Yangli Xie, MD, Department of Rehabilitation Medicine, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: xieyangli 841015com. Zhongliang Deng, Department of Orthopedic Surgery, Second Affiliated Hospital, Chongqing Medical University, Chongqing 400010, China. Tel.: +86-13608367586; Fax: + 86-023-63711527; Email: zhongliang.dengcom
Corresponding authors: Yangli Xie, MD, Department of Rehabilitation Medicine, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: xieyangli 841015com. Zhongliang Deng, Department of Orthopedic Surgery, Second Affiliated Hospital, Chongqing Medical University, Chongqing 400010, China. Tel.: +86-13608367586; Fax: + 86-023-63711527; Email: zhongliang.dengcom