Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(10):1266-1275. doi:10.7150/ijbs.20485 This issue Cite
Research Paper
Triptolide Suppresses Glomerular Mesangial Cell Proliferation in Diabetic Nephropathy Is Associated with Inhibition of PDK1/Akt/mTOR Pathway
Fei Han, Mei Xue, Yunpeng Chang, Xiaoyu Li, Yang Yang, Bei Sun ![]() , Liming Chen
, Liming Chen ![]()
Key Laboratory of Hormones and Development (Ministry of Health), Tianjin Key Laboratory of Metabolic Diseases, Tianjin Metabolic Diseases Hospital & Tianjin Institute of Endocrinology, Tianjin Medical University
Received 2017-4-9; Accepted 2017-8-28; Published 2017-9-21
Abstract

Mesangial cell proliferation has been identified as a mainly contributing factor to glomerulosclerosis, which is typical of diabetic nephropathy. However, the specific mechanisms and therapies remain unclear. PDK1 is a critical regulator of cell proliferation, but the specific role of PDK1 in diabetic nephropathy has not been fully illuminated. In the current study, we demonstrated that triptolide (TP) ameliorated albuminuria in the high fat diet/STZ-induced diabetic rats. TP also suppressed the increased proliferating cell markers Ki-67 and PCNA in the kidney tissues. Our results of MTT and cell cycle analysis further confirmed that TP significantly inhibited mesangial cell proliferation, and the inhibition of PDK1/Akt/mTOR pathway might be the underlying mechanisms. In addition, we also found that the PDK1 activator (PS48) could reverse the cell proliferation inhibition role of TP. These data suggest that TP may be useful in prevention of diabetic glomerulosclerosis and that PDK1/Akt/mTOR pathway might be the underlying mechanism.
Keywords: Diabetic nephropathy, Mesangial cell, Proliferation, PDK1, Triptolide.
Introduction
Diabetic nephropathy (DN) is one of the most serious microvascular complications of diabetes and the leading cause of end-stage renal disease (ESRD) worldwide [1]. DN is clinically characterized by the development of proteinuria followed by decreased glomerular filtration [2]. Pathological features of DN include mesangial expansion, caused by the proliferation of mesangial cells and the excessive accumulation of extracellular matrix, which will lead to kidney fibrosis [3, 4]. Therefore, inhibiting proliferation of mesangial cells is regarded as an effective strategy to ameliorate DN. Despite substantial progress made in recent decades, the mechanism involved in mesangial dysfunction is not fully understood. Therefore, effective agents for prevention and treatment of DN are still in need.
Triptolide (TP) is a major active component of Tripterygium wilfordii Hook F (TWHF), which exerts immunosuppressive and anti-inflammatory therapeutic effects [5-7]. In addition, it was also found to decrease proteinuria in polycystic kidney disease [8, 9], experimental membranous nephropathy [10] and IgA nephropathy [11]. The improvements of inflammation [12], oxidative stress [13] and immunity [14] are all involved in the protective roles of TP in different kinds of kidney diseases. But the effect and mechanisms of TP on DN are still not very clear.
Glomerular mesangial cells play important roles in mesangial matrix homeostasis, regulation of glomerular filtration rate, and phagocytosis of apoptotic cells in the glomerulus [15]. As over- proliferation is one of the typical characters of diabetic mesangial cell dysfunction, we wondered that TP might exert a protective role through this mechanism. It has been reported that TP might prevent diabetic glomerular fibrosis by inhibiting the expression of extracellular matrix proteins and the proliferation of glomerular mesangial cells, which might in relationship with the suppression of TGF-β1 [16]. Despite these studies, the protective mechanisms of TP in DN remain largely unknown.
3-Phosphoinositide dependent protein kinase-1 (PDK1), an ancient serine-threonine kinase belonging to AGC kinase family, is a key element of signaling transduction activated by extracellular ligands, such as growth factors [17, 18]. PDK1 has the ability to promote cell proliferation by activating the Akt pathway [19, 20]. In addition, PDK1 also has tight contact with diabetes [21-23]. Few studies reported that PDK1 might participate in the progression of DN through regulating the apoptosis of podocytes [24, 25]. However, it is not clarified that whether PDK1 mediates the glomerular mesangial cell proliferation in DN. In this study, we examined the effect of TP on DN and mesangial cell proliferation. Besides, we also discussed whether PDK1/Akt pathway mediated the protection role of TP in DN.
Results
TP improves urine microalbumin and glomerulosclerosis in the high fat diet (HFD)/ streptozocin (STZ) induced diabetic rats
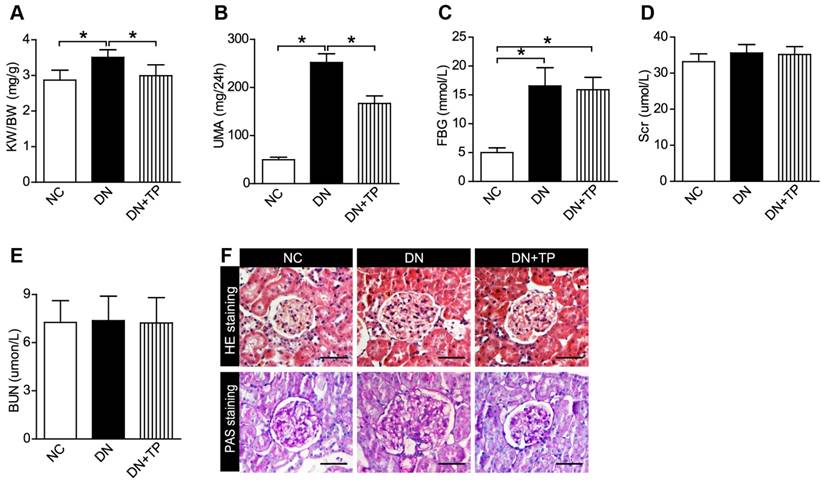
Firstly, we examined that whether TP has a protective effect on DN by using the HFD/STZ induced diabetic rats. We found that TP significantly decreased the relative kidney weight, which reflects kidney hypertrophy, and the 24 h urine microalbumin (UMA) with the blood glucose unaltered (Figure 1A-C). Serum creatinine (Scr) and blood urea nitrogen (BUN) did not differ among the three groups (Figure 1D and E). Hematoxylin and eosin (HE) and periodic acid-Schiff (PAS) staining were performed to examine the kidney pathologic changes. The images in Figure 1F revealed that deposited mesangial matrix, mesangial expansion and the fractional mesangial area were significantly higher in the DN group compared with the NC group, while they were significantly improved by TP treatment.
Effect of TP on blood glucose and renal function of HFD/STZ-induced diabetic rats. (A-E) KW/BW (A), UMA (B), FBG (C), Scr (D) and BUN (E) levels of the rats were shown. (F) HE and PAS staining of the kidney sections. The scale bar represents 50 μm. Data were reported as mean ± S.D.. *P < 0.05. KW/BW, kidney weight to body weight ratio; UMA, urinary microalbumin; FBG, fasting blood glucose; Scr, serum creatinine; BUN, blood urea nitrogen.
TP decreases cell proliferation in the kidney of HFD/STZ induced diabetic rats
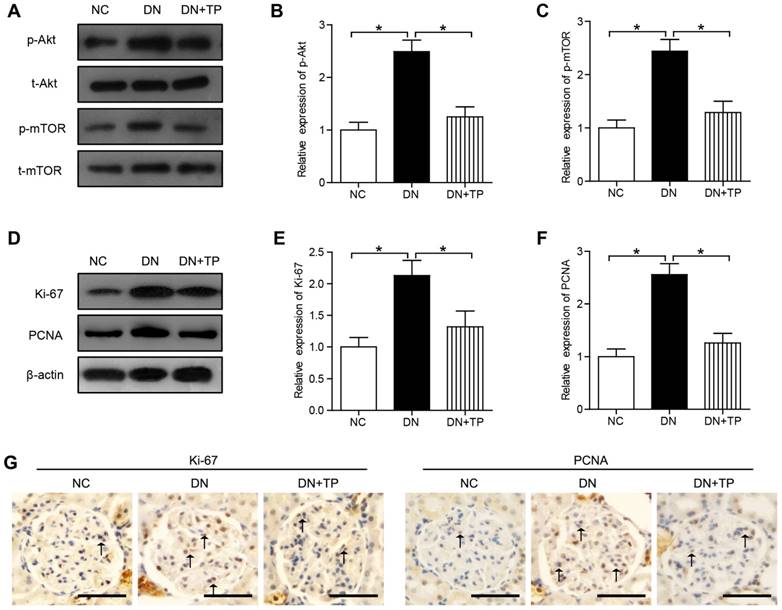
In our current results, the phosphorylation of Akt and mTOR was significantly increased in the diabetic kidney compared with that in the NC group (Figure 2A-C). The protein expression of Ki-67 and proliferating cell nuclear antigen (PCNA) in DN group, markers of proliferating cells, was increased than that in the NC group. Compared with DN group, TP significantly reduced the protein expression of Ki-67 and PCNA (Figure 2D-F). In addition, we also evaluated the cell proliferation in glomerular by immunohistochemistry (IHC) and found that Ki-67-positive cells and PCNA-positive cells were increased in the DN group, but was reduced by TP (Figure 2G).
Cell proliferation in the kidney of HFD/STZ-induced diabetic rats. (A) Protein expression of Akt/mTOR signal pathway in the kidneys of rats. (B and C) Quantification of results in A. (D) Protein expression of Ki-67 and PCNA in the kidneys of rats. (E and F) Quantification of results in D. (G) Immunohistochemistry images of Ki-67 and PCNA. The scale bar represents 50 μm. Data were reported as mean ± S.D.. *P < 0.05
TP suppresses high glucose (HG)-induced proliferation in human renal mesangial cells (HRMCs)
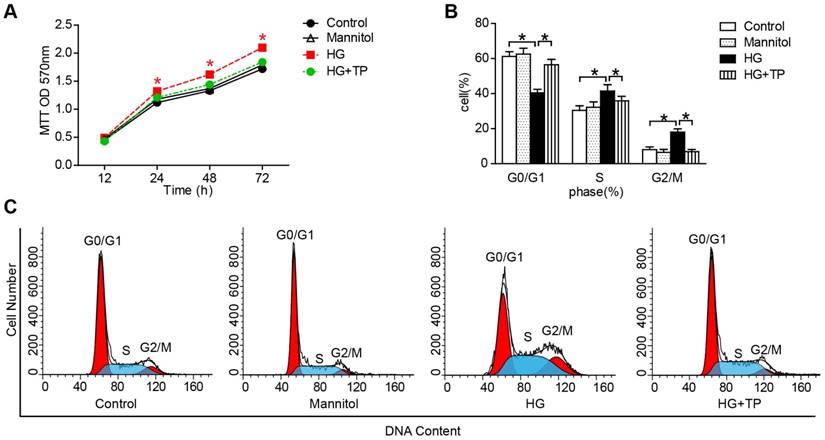
After confirming the protective role of TP in vivo, we further examined the effect of TP on HRMCs proliferation. Our results of 3-(4,5-dimethyl-2-thiazol)-2,5-diphenyl-2H-tetrazolium bromide (MTT) showed that HG markedly increased the cell number, while TP significantly suppressed the cell number (Figure 3A). Then we determined the patterns of cell cycle distribution by flow cytometry. As the data shown, compared with normal control, HG induced a decrement of cell proportion in G0-G1 phase from 61% to 40% (P<0.05), and an increment in S phase from 30% to 41% (P<0.05) and G2-M phase from 8% to 18% (P<0.05). However, TP significantly increased the cell proportion in G0-G1 phase to 55% (P<0.05), decreased the cell proportion in G2-M phase to 38% (P<0.05), and decreased the cell proportion in S phase to 5.7% (Figure 3B and C).
The effect of TP on HG-induced proliferation in HRMCs. (A) MTT assay in the cells treated with HG for different times. *P < 0.05 vs. Control group. (B and C) Flow cytometry analysis of cell cycle in the HRMCs treated for 72 h. Data were reported as mean ± S.D.. *P < 0.05
TP inhibits HG-induced activation of Akt/mTOR pathway in HRMCs
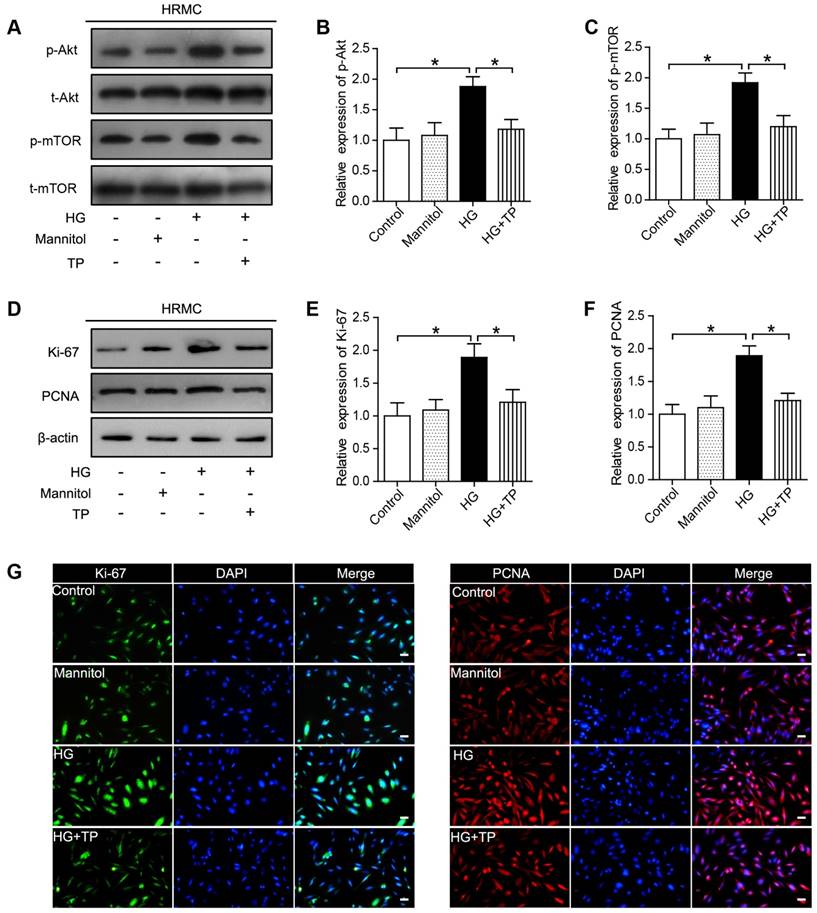
In our current results, Akt/mTOR pathway was significantly activated in the HG-treated HRMCs, while was suppressed by TP (Figure 4A-C). Besides, we also examined the expression of the proliferating cell markers, Ki-67 and PCNA, which were also significantly suppressed by TP, as verified by western blot and immunofluorescence (Figure 4D-G).
The effect of TP on proliferating cell markers in HG-treated HRMCs. (A) Western blot images of Akt/mTOR pathway. (B) Quantification of protein expression of phosphorylation-Akt. (C) Quantification of protein expression of phosphorylation-mTOR. (D) Western blot images of Ki-67 and PCNA. (E) Quantification of Ki-67 protein expression. (F) Quantification of PCNA protein expression. (G) Immunofluorescence images of Ki-67 and PCNA. The scale bar represents 10 μm. Data were reported as mean ± S.D.. *P < 0.05
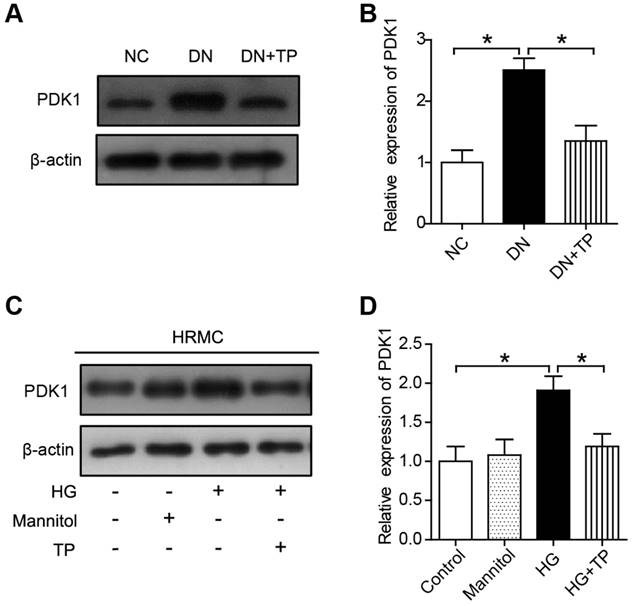
TP inhibits PDK1 activation both in vivo and in vitro
We firstly investigated the protein expression of PDK1 in the kidney of DN group and found it was significantly upregulated, but was downregulated by TP (Figure 5A and B). Moreover, the expression of PDK1 was significantly increased in the cell cultured with HG compared to normal controls. However, TP significantly suppressed the activated PDK1 in the cultured HRMCs (Figure 5C and D).
The effect of TP on PDK1 expression in vivo and in vitro. (A) The expression of PDK1 in the kidney of HFD/STZ-induced diabetic rats. (B) Quantification of results in A. (C) The expression of PDK1 in HRMCs treated for 72 h. (D) Quantification of results in C. Data were reported as mean ± S.D.. *P < 0.05
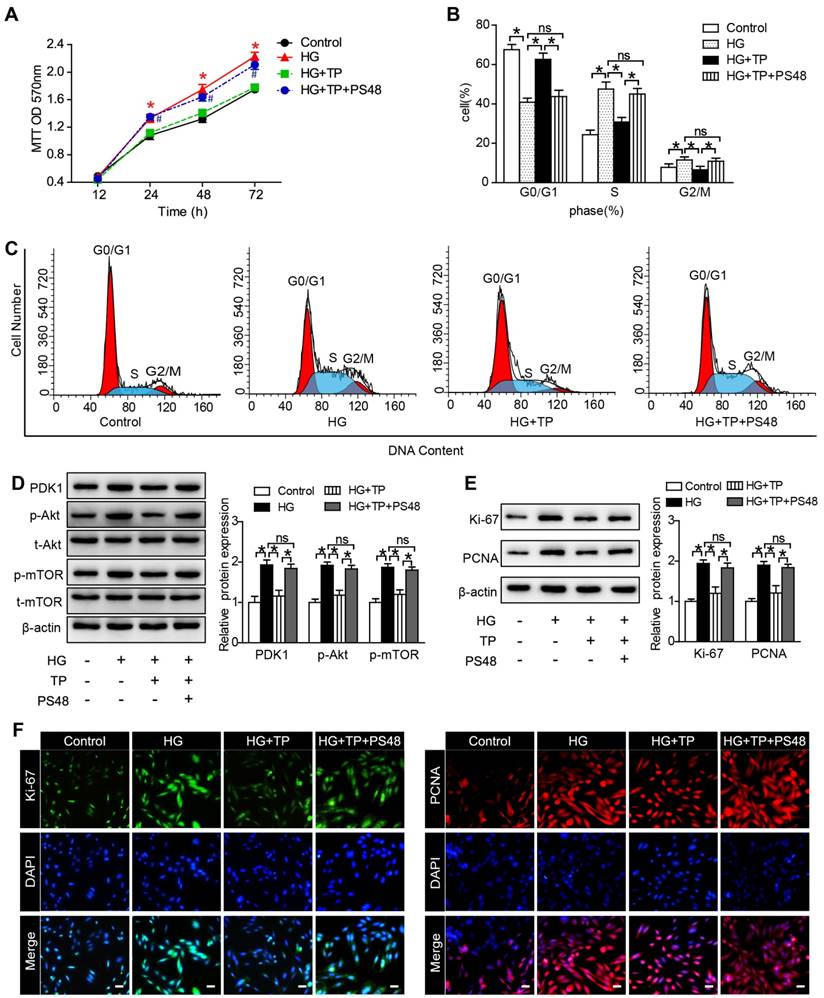
The anti-proliferation role of TP is in association with its suppression of PDK1
To explore the underlying mechanisms, we examined the role of PDK1 in TP-potentiated anti-proliferation by using a PDK1 activator (PS48). As our MTT results shown, the inhibition role of TP in cell number was significantly reversed by PS48 (Figure 6A). We also assessed the effect of PS48 on cell cycle distribution by flow cytometry. TP significantly increased the cell proportion in G0-G1 phase and decreased the cell proportion in G2-M and S phase. However, the effect of TP on cell cycle distribution was all reversed by PS48 (Figure 6B and C). In addition, suppression of PDK1/Akt/mTOR pathway by TP in HG-treated HRMCs was also reversed by PS48 (Figure 6D), as well as the expression of Ki-67 and PCNA (Figure 6E and F). These data suggested that TP might suppress glomerular mesangial cell proliferation through the inhibition of PDK1.
PDK1 activator reversed the effect of TP on mesangial cell proliferation. (A) MTT assay in the cells treated with HG for different times. *P < 0.05 vs. Control group, #P < 0.05 vs. HG+TP group. (B and C) Flow cytometry analysis of cell cycle in the HRMCs treated for 72 h. (D) Protein expression of PDK1/Akt/mTOR pathway in HRMCs treated for 72 h. (E) Protein expression of Ki-67 and PCNA in HRMCs treated for 72 h. (F) Immunofluorescence images of Ki-67 and PCNA. The scale bar represents 10 μm. Data were reported as mean ± S.D.. *P < 0.05; ns represents no significance
Discussion
Mesangial cell proliferation has been identified as main contributing factors to glomerulosclerosis, which is typical of DN [26]. In this study, we demonstrated that TP could decrease the albuminuria in HFD/STZ-induced diabetic rats. Our results suggested that the underlying mechanisms might be the inhibition of mesangial cell proliferation. Importantly, TP can inhibit the activation of PDK1/Akt/mTOR pathway in the kidney of HFD/STZ-induced diabetic rats and in HRMCs cultured with HG. PDK1 activator, PS48, has the ability to reverse the cell proliferation inhibition role of TP in vitro. Thus, we have identified that the activation of PDK1/Akt/mTOR pathway may be a novel player in DN and TP could suppress mesangial cell proliferation via inhibition of PDK1/Akt/mTOR pathway.
TP, the major active compound extracted from a traditional Chinese medicinal herb TWHF, is found to have effect on many kind of kidney diseases [8-11], as well as DN [14, 27-29]. In this study, we did find that TP decreases albuminuria in HFD/STZ-induced diabetic rats, which are consistent with previously reported [14, 27-29]. Based on the results, we further discussed the related mechanisms underlying the protective role of TP.
Mesangial expansion, caused by cell over-proliferation, is the lesion leading to the loss of glomerular filtration rate and is present in DN patients before the onset of clinical manifestations [30]. Therefore, inhibition of mesangial cell proliferation is now considered one of the targets in the prevention and retardation of DN. Previous study demonstrated that TP was effective in suppressing cancer cell proliferation [31-33]. But it is unknown that whether TP has an effect on mesangial cell proliferation under diabetic conditions. Based on the results of MTT and cell cycle analysis, we confirmed that TP significantly inhibited mesangial cell proliferation, which is consistent with previous report [16]. These data suggested that TP might be useful in preventing the diabetic glomerulosclerosis.
After then, we further discussed the underlying mechanisms. PDK1 can activate Akt kinase on the activation loop at T308, promoting full activity of Akt [20]. In addition, the constitutive activity of PDK1 is a critical regulator of several other important signal transduction pathways that regulate cell proliferation, survival and apoptosis. It has been proved that the activation of PDK1 could upregulate cyclin D1 during cell cycle progression from G0-G1 to S phase [34]. Moreover, PDK1 is also involved in many physiology and pathology changes, such as basilar artery smooth muscle cells proliferation [35], autosomal dominant polycystic kidney disease [36] and insulin resistance [37]. In addition, the role PDK1 plays in chronic kidney disease is drawing more and more attention [38]. Although, there were few studies reported that PDK1 might regulate the apoptosis of podocytes in DN [24, 25]. However, it remains largely unknown that whether PDK1 mediates the glomerular mesangial cell proliferation in DN. In this study, we found that the expression of PDK1 was significantly activated in the glomerular of the diabetic rats and in the HG-treated HRMCs. The results mentioned above suggested that PDK1 might participate in the pathologies of DN. The PDK1 activator PS48 was used to verify the related mechanisms. The results turned out that the inhibition role of TP in mesangial cell proliferation and in Akt/mTOR pathway was both reversed by PS48, suggesting that TP might exert the cell proliferation inhibition role by suppressing the expression of PDK1. Previous studies have shown that TP might exert its protective role through working on microRNA [27], TGF-β1[16], oxidative carbonyl protein [13] and so on. However, our data might provide a new target for TP and for treating DN.
Taken together, our study highlight that TP is effective in decreasing the albuminuria in DN, which might be in relation with the inhibition of mesangial cell proliferation. And the suppression of PDK1/Akt/mTOR pathway may well explain the cell proliferation inhibition role of TP.
Materials and Methods
Animal models
All animal experiments complied with rules of Experimental Animal Care and Use Center in Tianjin Medical University. The protocol was approved by Experimental Animal Ethical Committee of Tianjin Medical University. Thirty-two male Sprague-Dawley (SD) rats of age six weeks and weight 170 ± 10 g were originally obtained from Beijing HFK Bio-Technology Co. Ltd. All rats were first fed with regular rodent chow for one week to adapt and housed in a controlled room at 22 °C with a 12 h day/night cycle. Eight rats were chosen as normal controls (NC group, n = 8) and fed with a standard diet. The rest of them (n = 24) were fed with HFD (Beijing HFK Bio-Technology Co. Ltd) for 6 weeks, which consists of 78.7% standard diet, 10% glucose, 10% animal fat, 1% total cholesterol, and 0.3% sodium cholate. After that, diabetes was induced in the rats fed with HFD by intraperitoneal injection of STZ (Sigma Aldrich, St. Louis, MO, USA) (30 mg/kg) for three consecutive days as previously reported [39]. At the same time, the NC group was injected with citrate-phosphate buffer. The rats were examined 72 h later and those with plasma glucose levels over 16.7 mmol/L were classified as diabetic. Four rats injected with STZ failed to meet the criterion of diabetes and were excluded. Two weeks later, rats with 24 h UMA levels over 30 mg were considered to have DN. The DN rats (n = 20) were randomly divided into diabetic group (DN group, n =10) and TP (Chinese National Institute for the Control of Pharmaceutical and Biological Products)-treated group (DN+TP group, n = 10). The DN+TP group was applied daily with TP (100 ug/kg/d) by oral gavage for 12 weeks [10]. While the DN group was given an oral dose of 1 ml normal saline solution containing 0.4% dimethyl sulfoxide as control. All rats had free access to food and water during the whole experimental time.
Body weight was monitored daily and blood glucose was tested weekly using the blood drawn from tail vein by a glucose analyzer (Roche, Germany). At the end of study, all rats were placed in individual metabolic cage to collect 24 h urine samples for the measurement of UMA. UMA was determined by the Bradford method. The rats were weighted and anesthetized with intraperitoneal injection of sodium pentobarbital (30 mg/kg body weight). Blood was collected from the inner canthal orbital vein. BUN and Scr were examined by an automatic biochemistry analyzer (CD-1600CS, Abbott Labs, USA). The kidneys were immediately removed and the left one was weighted. One kidney was placed in liquid nitrogen and stored at -80 °C, the other one was fixed with 4% paraformaldehyde.
Cell culture and treatment
Human renal mesangial cells (HRMCs, American Type Culture Collection, Rockville, MD, USA) were cultured with 1640 media, containing 10% fetal bovin serum (Gibco) at 37 °C in 5% CO2. Cells were cultured with D-glucose (Life Technologies) at normal (5.5 mmol/L) or high (25 mmol/L) concentrations in serum-free medium. D-Mannitol (25 mmol/L, Life Technologies) was used for a control of osmolality. TP was reconstituted in 0.01% dimethyl sulfoxide and freshly diluted with culture medium to 10 ug/L before using. To determine the specific role of PDK1 in TP-potentiated anti-proliferation, we applied 5 μmol/L PS48 (MedChem Express, USA) following the treatment of TP.
Cell proliferation assay
MTT assay was used to detect cell proliferation. HRMCs were seeded at a density of 1x105/ml into 96-well plates. After 12, 24, 48 and 72 h incubation with different compounds as described above, 20 ul MTT (5 mg/mL, Invitrogen, USA) was added to each well. Cells were then cultured for an additional 2 h and subsequently lysed using dimethylsulfoxide (150 ul/well; Sigma, USA). When the formazan crystals completely dissolved, the optical density (OD) was measured at 570 nm. The arithmetic mean OD of six wells for each group was calculated.
Cell cycle analysis
Cell cycle analysis was performed using flow cytometry. After 72 hours of treatment, the cells were harvested by trypsinization and washed twice with cold phosphate buffer saline buffer, and then fixed with 75% alcohol for 12 h at 4 °C. After washing, cells were treated with RNase (50 ug/mL) at 37 °C for 30 min. Cells were stained with propidium iodide (50 ug/mL) at 4 °C for 30 min in the dark before being analyzed with a BD FACSCalibur flow cytometer (BD Pharmingen, USA). 1×106 cells were detected for each sample and the cell cycle was analyzed using Modfit software (Verity Software House, USA).
Histology
Kidney tissues were fixed in 4% paraformaldehyde, embedded in paraffin and cut into 4 um thick sections. The kidney sections were stained with a HE staining kit (Baibo Biotechnology Co., Ltd., China) and a PAS staining kit (Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer's protocols. For kidney tissue IHC experiment, kidney slides were blocked for 10 minutes with 3% (v/v) hydrogen peroxide following deparaffinization and rehydration. Sections were then treated with antigen retrieval solution, washed, permeabilized, and blocked. After that, primary antibodies were added overnight at 4 °C as follows: Ki-67 (1:100, Proteintech, USA) and PCNA (1:100, Proteintech, USA). After co-staining for 1 h with an anti-rabbit or mouse, horseradish peroxidase labeled polymer secondary antibody at 37 °C, the stained sections were examined by a blinded manner using light microscopy (Olympus BX-50, Olympus Optical, Tokyo, Japan) in ten randomly selected cortical sections (×400). For cell immunostaining experiment, all procedures were same as IHC analysis. After incubating with primary antibody of Ki-67 or PCNA, the slides were serially incubated with goat anti-rabbit IgG/FITC (1:100, Proteintech, USA) or goat anti-mouse IgG/TRITC (1:100, Proteintech, USA). After that, 4',6-diamidino-2-phenylindole (DAPI) was used to stain the cell nucleus. Slides were captured by a Leica DMI4000 B automated inverted microscope equipped with a Leica DFC300 FX camera.
Western blotting
Total protein in cells and renal cortical tissues was extracted with a protein extraction reagent (Thermo, USA) according to the manufacturer's instructions. After concentrations tested, separation of protein extracts (40 μg/lane) was accomplished with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (10%). The proteins were transferred onto polyvinylidene fluoride membrane (Millipore, USA). The membranes were incubated overnight with primary antibodies. The primary antibodies and their dilutions used were as follows: PDK1 (1:1000, Cell Signaling Technology, USA), total-Akt (1:2000, Cell Signaling Technology, USA), phosphorylation-Akt (1:1000, Cell Signaling Technology, USA), total-mTOR (1:2000, Cell Signaling Technology, USA), phosphorylation-mTOR (1:1000, Cell Signaling Technology, USA), Ki-67 and PCNA (all diluted as 1:1000, Proteintech, USA). After washing, the secondary antibody was used for detection. Proteins were visualized by electrochemiluminescence (Advansta, USA). Intensity of the bands was analyzed with ImageJ software.
Statistical analyses
SPSS 19.0 software (IBM Analytics, USA) was used for statistical analysis. All the values were expressed as mean ± Standard Deviation (S.D.). Data was analyzed using one-way analysis of variance (ANOVA) and post-hoc LSD-test. A value of P < 0.05 was considered statistically significant.
Abbreviations
BUN: blood urea nitrogen; DAPI: 4',6-diamidino-2-phenylindole; DN: diabetic nephropathy; ESRD: end-stage renal disease; HE: Hematoxylin and eosin; HFD: high fat diet; HG: high glucose; HRMCs: human renal mesangial cells; IHC: immunohistochemistry; KW/BW: kidney weight to body weight ratio; MTT: 3-(4,5-dimethyl-2-thiazol)-2,5-diphenyl-2H-tetrazolium bromide; OD: optical density; PAS: Periodic acid-Schiff's; PCNA: proliferating cell nuclear antigen; PDK1: 3-Phosphoinositide dependent protein kinase-1; Scr: serum creatinine; SD: Sprague-Dawley; S.D.: Standard Deviation; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; STZ: streptozocin; TP: Triptolide; TWHF: Tripterygium wilfordii Hook F; UMA: urine microalbumin.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no.81273915, 81373864 and 81470187) and Natural Science Foundation of Tianjin (no. 14JCYBJC26200).
Author contributions
Liming Chen and Bei Sun contributed to research design, discussion of result and critical revision of the manuscript. Fei Han contributed to the design of study, conduction of the experiment and the manuscript draft. Mei Xue and Yang Yang performed data analysis. Yunpeng Chang and Xiaoyu Li contributed to discussion of results.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schieppati A, Remuzzi G. Chronic renal diseases as a public health problem: epidemiology, social, and economic implications. Kidney Int Suppl. 2005:S7-S10
2. Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney international. 2008;74:22-36
3. Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. Journal of the American Society of Nephrology: JASN. 2003;14:1358-73
4. Alsaad KO, Herzenberg AM. Distinguishing diabetic nephropathy from other causes of glomerulosclerosis: an update. J Clin Pathol. 2007;60:18-26
5. Wang C, Shan Y, Yang J, Xu X, Zhuang B, Fan Y. et al. Inhibition of Cancer Angiogenesis Using Triptolide Nanoparticles. J Biomed Nanotechnol. 2015;11:805-15
6. Patil S, Lis LG, Schumacher RJ, Norris BJ, Morgan ML, Cuellar RA. et al. Phosphonooxymethyl Prodrug of Triptolide: Synthesis, Physicochemical Characterization, and Efficacy in Human Colon Adenocarcinoma and Ovarian Cancer Xenografts. J Med Chem. 2015;58:9334-44
7. Yi JM, Huan XJ, Song SS, Zhou H, Wang YQ, Miao ZH. Triptolide Induces Cell Killing in Multidrug-Resistant Tumor Cells via CDK7/RPB1 Rather than XPB or p44. Mol Cancer Ther. 2016;15:1495-503
8. Leuenroth SJ, Okuhara D, Shotwell JD, Markowitz GS, Yu Z, Somlo S. et al. Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc Natl Acad Sci U S A. 2007;104:4389-94
9. Leuenroth SJ, Bencivenga N, Igarashi P, Somlo S, Crews CM. Triptolide reduces cystogenesis in a model of ADPKD. Journal of the American Society of Nephrology: JASN. 2008;19:1659-62
10. Chen ZH, Qin WS, Zeng CH, Zheng CX, Hong YM, Lu YZ. et al. Triptolide reduces proteinuria in experimental membranous nephropathy and protects against C5b-9-induced podocyte injury in vitro. Kidney international. 2010;77:974-88
11. He L, Peng X, Liu G, Tang C, Liu H, Liu F. et al. Anti-inflammatory effects of triptolide on IgA nephropathy in rats. Immunopharmacol Immunotoxicol. 2015;37:421-7
12. Zhou Y, Hong Y, Huang H. Triptolide Attenuates Inflammatory Response in Membranous Glomerulo-Nephritis Rat via Downregulation of NF-kappaB Signaling Pathway. Kidney & blood pressure research. 2016;41:901-10
13. Dong XG, An ZM, Guo Y, Zhou JL, Qin T. Effect of triptolide on expression of oxidative carbonyl protein in renal cortex of rats with diabetic nephropathy. Journal of Huazhong University of Science and Technology Medical sciences = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban. 2017;37:25-9
14. Guo H, Pan C, Chang B, Wu X, Guo J, Zhou Y. et al. Triptolide Improves Diabetic Nephropathy by Regulating Th Cell Balance and Macrophage Infiltration in Rat Models of Diabetic Nephropathy. Experimental and clinical endocrinology & diabetes: official journal, German Society of Endocrinology [and] German Diabetes Association. 2016;124:389-98
15. Abboud HE. Mesangial cell biology. Experimental cell research. 2012;318:979-85
16. Cao Y, Huang X, Fan Y, Chen X. Protective Effect of Triptolide against Glomerular Mesangial Cell Proliferation and Glomerular Fibrosis in Rats Involves the TGF- beta 1/Smad Signaling Pathway. Evidence-based complementary and alternative medicine: eCAM. 2015;2015:814089
17. Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG. et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Current biology: CB. 1997;7:776-89
18. Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF. et al. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567-70
19. Primo L, di Blasio L, Roca C, Droetto S, Piva R, Schaffhausen B. et al. Essential role of PDK1 in regulating endothelial cell migration. The Journal of cell biology. 2007;176:1035-47
20. Liu Y, Wang J, Wu M, Wan W, Sun R, Yang D. et al. Down-regulation of 3-phosphoinositide-dependent protein kinase-1 levels inhibits migration and experimental metastasis of human breast cancer cells. Molecular cancer research: MCR. 2009;7:944-54
21. Hashimoto N, Kido Y, Uchida T, Asahara S, Shigeyama Y, Matsuda T. et al. Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nature genetics. 2006;38:589-93
22. Kawano Y, Nakae J, Watanabe N, Fujisaka S, Iskandar K, Sekioka R. et al. Loss of Pdk1-Foxo1 signaling in myeloid cells predisposes to adipose tissue inflammation and insulin resistance. Diabetes. 2012;61:1935-48
23. Wang LY, Hung CL, Chen YR, Yang JC, Wang J, Campbell M. et al. KDM4A Coactivates E2F1 to Regulate the PDK-Dependent Metabolic Switch between Mitochondrial Oxidation and Glycolysis. Cell reports. 2016;16:3016-27
24. Saurus P, Kuusela S, Dumont V, Lehtonen E, Fogarty CL, Lassenius MI. et al. Cyclin-dependent kinase 2 protects podocytes from apoptosis. Scientific reports. 2016;6:21664
25. Saurus P, Kuusela S, Lehtonen E, Hyvonen ME, Ristola M, Fogarty CL. et al. Podocyte apoptosis is prevented by blocking the Toll-like receptor pathway. Cell death & disease. 2015;6:e1752
26. Steffes MW, Osterby R, Chavers B, Mauer SM. Mesangial expansion as a central mechanism for loss of kidney function in diabetic patients. Diabetes. 1989;38:1077-81
27. Han F, Wang S, Chang Y, Li C, Yang J, Han Z. et al. Triptolide prevents extracellular matrix accumulation in experimental diabetic kidney disease by targeting microRNA-137/Notch1 pathway. Journal of cellular physiology. 2017
28. Ma R, Liu L, Liu X, Wang Y, Jiang W, Xu L. Triptolide markedly attenuates albuminuria and podocyte injury in an animal model of diabetic nephropathy. Experimental and therapeutic medicine. 2013;6:649-56
29. Gao Q, Shen W, Qin W, Zheng C, Zhang M, Zeng C. et al. Treatment of db/db diabetic mice with triptolide: a novel therapy for diabetic nephropathy. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2010;25:3539-47
30. Ichinose K, Kawasaki E, Eguchi K. Recent advancement of understanding pathogenesis of type 1 diabetes and potential relevance to diabetic nephropathy. American journal of nephrology. 2007;27:554-64
31. Jiang C, Fang X, Zhang H, Wang X, Li M, Jiang W. et al. AMD3100 combined with triptolide inhibit proliferation, invasion and metastasis and induce apoptosis of human U2OS osteosarcoma cells. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;86:677-85
32. Yuan S, Wang L, Chen X, Fan B, Yuan Q, Zhang H. et al. Triptolide inhibits the migration and invasion of human prostate cancer cells via Caveolin-1/CD147/MMPs pathway. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2016;84:1776-82
33. Xiong J, Su T, Qu Z, Yang Q, Wang Y, Li J. et al. Triptolide has anticancer and chemosensitization effects by down-regulating Akt activation through the MDM2/REST pathway in human breast cancer. Oncotarget. 2016;7:23933-46
34. Gao Y, Wu F, Zhou J, Yan L, Jurczak MJ, Lee HY. et al. The H19/let-7 double-negative feedback loop contributes to glucose metabolism in muscle cells. Nucleic acids research. 2014;42:13799-811
35. Yu ZL, Wang JN, Wu XH, Xie HJ, Han Y, Guan YT. et al. Tanshinone IIA Prevents Rat Basilar Artery Smooth Muscle Cells Proliferation by Inactivation of PDK1 During the Development of Hypertension. Journal of cardiovascular pharmacology and therapeutics. 2015;20:563-71
36. Zhao X, Paterson AD, Zahirieh A, He N, Wang K, Pei Y. Molecular diagnostics in autosomal dominant polycystic kidney disease: utility and limitations. Clinical journal of the American Society of Nephrology: CJASN. 2008;3:146-52
37. Kan J, Velliquette RA, Grann K, Burns CR, Scholten J, Tian F. et al. A novel botanical formula prevents diabetes by improving insulin resistance. BMC complementary and alternative medicine. 2017;17:352
38. Tobisawa T, Yano T, Tanno M, Miki T, Kuno A, Kimura Y. et al. Insufficient activation of Akt upon reperfusion because of its novel modification by reduced PP2A-B55alpha contributes to enlargement of infarct size by chronic kidney disease. Basic research in cardiology. 2017;112:31
39. Wang D, Luo P, Wang Y, Li W, Wang C, Sun D. et al. Glucagon-like peptide-1 protects against cardiac microvascular injury in diabetes via a cAMP/PKA/Rho-dependent mechanism. Diabetes. 2013;62:1697-708
Author contact
![]() Corresponding authors: Liming Chen, MD, PhD. E-mail: xfx22081163.com. Tel: +86-22-23333266; Bei Sun, MD, PhD. E-mail: beisunedu.cn. Tel: +86-22-23542602 Address: Key Laboratory of Hormones and Development (Ministry of Health), Tianjin Key Laboratory of Metabolic Diseases, Tianjin Metabolic Diseases Hospital & Tianjin Institute of Endocrinology, Tianjin Medical University, 300070 Tianjin, China Fax: +86-22-23528460
Corresponding authors: Liming Chen, MD, PhD. E-mail: xfx22081163.com. Tel: +86-22-23333266; Bei Sun, MD, PhD. E-mail: beisunedu.cn. Tel: +86-22-23542602 Address: Key Laboratory of Hormones and Development (Ministry of Health), Tianjin Key Laboratory of Metabolic Diseases, Tianjin Metabolic Diseases Hospital & Tianjin Institute of Endocrinology, Tianjin Medical University, 300070 Tianjin, China Fax: +86-22-23528460