Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(10):1341-1350. doi:10.7150/ijbs.17948 This issue Cite
Research Paper
Clenbuterol Induces Cell Cycle Arrest in C2C12 Myoblasts by Delaying p27 Degradation through β-arrestin 2 Signaling
Min Chen1,3*, Chuncheng Liu1*, Meng Wang1, Hong Wang1, Kuo Zhang1, Yu Zheng1, Zhengquan Yu1, Xiangdong Li1, Wei Guo4, Ning Li1, Qingyong Meng1,2 ![]()
1. State Key Laboratory for Agrobiotechnology, China Agricultural University, Beijing 100193, China
2. Beijing Advanced Innovation Center for Food Nutrition and Human Health, College of Biological Science, China Agricultural University
3. Guangxi Province Center for Disease Control and Prevention, Nanning 530028, China
4. Animal Science/Molecular Biology Bldg, University of Wyoming, Laramie WY82071, USA
* These authors contributed equally to this work
Received 2016-10-17; Accepted 2017-6-16; Published 2017-10-17
Abstract

β2-Adrenoceptor (β2-AR) agonists promote muscle growth. The aim of this study was to elucidate some effects of the selective β2-adrenoceptor agonist clenbuterol (CLB) on myoblast proliferation. We found that CLB induces cell cycle arrest in C2C12 myoblasts. This effect is partly due to the enhanced stability of p27, rather than the increased gene transcription via cAMP response element-binding protein (CREB). Specifically, CLB treatment enhanced the accumulation of p27 in the nucleus while depleting it from the cytosol via a mechanism that requires β2-AR. Surprisingly, p27 accumulation was not reversed by the protein kinase A (PKA) inhibitor H-89, but interestingly, was alleviated by the knockdown of β-arrestin 2. Thus, our work provides a basis for β2-AR agonists inhibit myoblasts proliferation through signaling via β2-AR, β-arrestin 2, and p27.
Keywords: clenbuterol, p27, cell cycle, β2-AR, β-arrestin 2.
Introduction
The balance between protein synthesis and degradation is precisely regulated in muscle cells [1-4]. Hypertrophy is the cumulative effect of increased protein synthesis, suppressed protein degradation, or both [5-7]. CLB, a well-known synthetic agonist of the β2-AR, has been shown previously to induce hypertrophy in both skeletal and cardiac muscles [8-11]. Thus, CLB has been investigated in various rodent models of dystrophy [12, 13], denervation [14, 15] and aging [16, 17], and its anabolic effects have been exploited to improve muscle function and reverse atrophy.
Genetic and pharmacological studies confirmed that these anabolic effects are mediated by β2-AR [11, 18]. Canonically, downstream signaling is triggered when activated β2-AR binds to G-proteins. In turn, the Gα subunit (Gs) enhances the synthesis of cyclic AMP (cAMP) and activates PKA and CREB. When excessive cAMP is produced, further signaling by Gβγ subunits maybe triggered, thereby activating the cascade controlled by phosphoinositol 3-kinase (PI3K) and protein kinase B (AKT) [5, 19]. Additionally, activated β2-AR can also trigger the MAPK pathway [20-22]. In summary, β-agonists can trigger signaling via many pathways that regulate protein metabolism [23-25].
Indeed, although the anabolic functions of CLB are well characterized, its potential role in muscle regeneration has not been fully explored. For example, the net effect of CLB on cell proliferation is unknown, because cAMP and extracellar signal-regulated protein (ERK) signaling have some opposite reports on cell proliferation [26-29]. Thus, it is of great value to investigate the effect of CLB on myoblast proliferation, a critical step in muscle regeneration, which progresses from activation of satellite cells to proliferation, differentiation, and fusion with each other or with existing fibers [30]. C2C12 are murine myoblast derived from satellite cells and cycling myoblasts are comparable to activated satellite cells in muscle fiber [31]. Here, we examine proliferation of C2C12 myoblasts in response to CLB treatment. We show, for the first time, that CLB induces cell cycle arrest by repressing p27 degradation, via a pathway that depends on β2-AR and β-arrestin 2.
Materials and Methods
Cell culture, cell synchronization and transfections
C2C12 and A204 cells were purchased from Cell Resource Center, China. C2C12 and A204 cells were cultured in Dulbecco's modified Eagle's Medium (DMEM) and McCoy's 5A (Modified) medium, respectively, each with 10% fetal bovine serum (FBS). Media were supplemented with 1% penicillin and streptomycin. Both cell lines were cultured at 37oC in a humidified atmosphere containing 5% CO2. C2C12 cells were synchronized in G0/G1 by serum starvation for 24h-48h and then stimulated to enter the cell cycle by re-addition of serum. Synchronization was monitored by flow cytometry.
C2C12 cells were transfected with 3 µg plasmids or 50 pmol siRNA using Nucleofector 2b device (Lonza,Germany) according to the manufactures' instructions.
Materials
CLB (Sigma-Aldrich, C5423); H-89 (Beyotime, S1643); cycloheximide (Beyotime, S1560); β2-AR siRNA (Santa Cruz, sc-39867); β-arrestin 1 shRNA (Origene, TG510701); β-arrestin 2 shRNA (Origene, TF517061). p27 siRNA (si-P, si-8, and si-10) and control siRNA (siNC) were synthesized by Shanghai Genepharma Co., Ltd. with the following respective sequences: 5' ACGTAAACAGCTCCGAATTAA 3', 5' GGUGCCUUUAAUUGGGUCUTT 3', 5' GGCCAACAGAACAGAAGAATT 3', 5' UUCUCCGAACGUGUCACGUTT 3'.
Western Blotting
Western Blotting analysis was performed as described previously [32]. Total cell extracts were lysed in cell lysis buffer (Beyotime, P0013), and the lysates were centrifuged at 12,000 rpm for 10 min at 4°C. Samples containing 30 µg protein were loaded on SDS-PAGE gels and transferred to PVDF membranes. Membranes were blocked with 5% nonfat milk or BSA and probed with primary antibodies and horseradish peroxidase-conjugated secondary antibodies. Blots were developed with lumi-light Western blotting substrate detection reagents (Roche, 12015200001). Nuclear- and cytoplasmic-enriched fractions were prepared using Nuclear-Cytosol Extraction Kit (Applygene, P1200), according to manufacturer's instructions. Western Blotting analysis was performed using the anti-p27 (Cell Signaling Technology, 3698S), anti-CREB (Cell Signaling Technology, 9197L), anti-phospho-CREB (Ser133) (Cell Signaling Technology, 9198L), anti-S-phase kinase-associated protein 2 (Skp2) (Cell Signaling Technology, 4313S), anti-CDK2 (Cell Signaling Technology, 2546S), anti-Cyclin E1 (Cell Signaling Technology, 4129S), PARP (Cell Signaling Technology, 9532S), anti-GAPDH (Cell Signaling Technology, 2118L), anti-β-arrestin 1 (Abcam, ab32099), anti-β-arrestin 2 (Abcam, ab54790), anti-tubulin (Beyotime, AT819), anti-cyclin D3 (Beyotime, AC856), anti-p21 (Beyotime, AP021).
Cell Viability
Briefly, 500 C2C12 cells in culture media were plated in 96-well plates, and incubated for 12 h to allow complete attachment. After attachment, CLB prepared in water was added to a final concentration of 0-200 µM. Plates were incubated at 37°C for 2 h, and viability was measured by Caspase-Glo8 Assay (Promega, Cell Titer 96 Aqueous one solution), following manufacturer's protocol.
Flow Cytometry
Cell cycle distribution was performed [33] with slight modification. Cells were harvested, washed with PBS, and fixed with 70% ethanol. Cells were then resuspended in 300 µl PBS, treated with 100 µg/ml RNase A and 0.2% Triton X-100, and incubated at 37°C for 30 min. Finally, samples were treated with 50 µg/ml propidiumiodide (Sigma-Aldrich, P4170) and incubated at room temperature for 30 min in the dark. For each sample, 10,000 cells were analyzed for DNA content in the FL2 channel using FACSCalibur platform (BD Science, USA).
BrdU Incorporation and Immunofluorescence
Cells were synchronized after cultured in serum free medium for at least 24 hours. Then they were released in DMEM with 10% FBS and treated with 10 or 100 µM CLB for 12 h. In the last 2 h of treatment, the cells were labeled with 5-Bromo-2-deoxyUridine (BrdU) (Sigma-Aldrich, B5002) at a final concentration of 10 µM. The experiments were performed [34] with slight modification. Cells were then fixed with 4% paraformaldehyde and permeabilized. Subsequently, samples were treated with 2 M HCL for 30 min, and then with 0.01% trypsin for 12 min at 37°C, washing with PBS after each step. After blocking, the slides were incubated overnight at 4°C with anti-BrdU (Sigma-Aldrich, B2531) diluted 1:1000 in PBS. After washing with PBS, the cells were incubated with fluorescently labeled goat anti-mouse immunoglobulin G (Invitrogen, A-11032) diluted 1:400 in PBS. Finally, the cells were incubated with DAPI for 5 min and then washed with PBS. Control excluding primary antibody was negative. The proliferation rate was calculated as the ratio between cells stained with BrdU and those stained with DAPI.
To label p27, the cells were first fixed with 4% paraformaldehyde for at least 20 min, and then permeabilized for 5 min with a solution containing 0.5% Triton X-100. After blocking, the cells were probed at 4°C over night with 1:400 dilutions of primary antibodies. After washing with PBS three times, the cells were incubated with Alexa 488-labeled goat anti-mouse immunoglobulin G (Invitrogen, A-11034). Nuclei were counterstained with DAPI, and images were taken with AMG EVOS fluorescence microscope.
RNA Analysis
Total RNA was isolated with TRIZOL reagent (Invitrogen, 15596-026) and reverse transcribed into cDNA using oligo-dT primers. Levels of p27, Skp2 and GAPDH mRNA were measured by real-time PCR, using appropriate primers. PCR primers used were the following: 5'-GGGTTAGCGGAGCAGTGTC-3' (forward) and 5'-GGGAACCGTCTGAAACATTTT-3' (reverse) for mouse p27; 5'-GCTAAGCAGCTGCTCCAGACT-3' (forward) and 5'-AGGTTGGGGCATCGTTTAATT-3' (reverse) for mouse Skp2; 5'-AGGTCGGTGTGAACGGATTTG-3' (forward) and 5'-TGTAGACCATGTAGTTGAGGTCA-3' (reverse) for mouse GAPDH.
Statistical analysis
Results are reported as mean ± SD or mean ± SE as indicated. Data were analyzed by Student's t-test using SPSS 16.0, and p < 0.05 was considered statistically significant.
Results
CLB induces cell cycle arrest in C2C12 cells
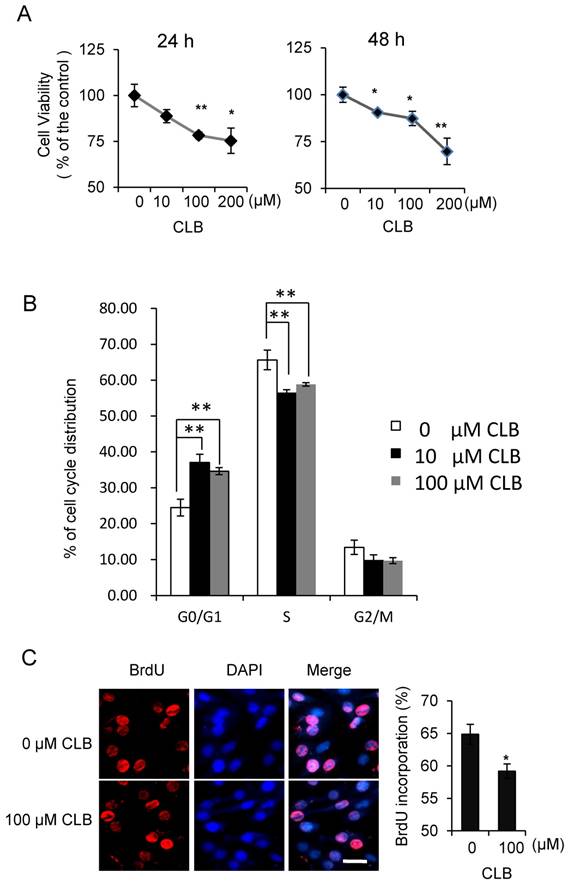
Although isoproterenol, a widely used β2-AR agonist, was reported to induce cell proliferation in HEK293 cells [35], the effect of β2-AR agonist has not been fully studied in the myoblasts. To assess whether CLB interferes with cell proliferation, we first measured its effect on cell viability. We found the viability of C2C12 myoblasts to be decrease when treated with 10-200 µM CLB for 24 or 48 h (Fig. 1A). Furthermore, we monitored cell cycle progression for 24 h after synchronized C2C12 cells were released in 10% fetal bovine serum (data not shown). We found an increase in the proportion of cells in G0/G1 phase after exposure to CLB for 12 h through flow cytometry assay (Fig. 1B). These results suggest that CLB treatment delays cell cycle progression. Moreover, we observed a significant decrease in DNA synthesis after 100 µM CLB treatment, as measured by BrdU incorporation (Fig. 1C). In a similar manner, CLB also inhibited DNA synthesis in skeletal muscle cells derived from human rhabdomyosarcoma A204 (Fig. S1).
CLB induces cell cycle arrest. (A) CLB treatment reduces viability of C2C12 cells treated with 0-200 µM CLB for 24 and 48 h. Results are mean ± SE (n = 5) from three independent experiments. *, p < 0.05; **, p < 0.01. (B) CLB treatment delays cell cycle progression in C2C12 cells. Synchronized cells were treated with 0, 10 or 100 µM CLB for 12 h, and analyzed by flow cytometry. Results are mean ± SD from triplicate experiments. **, p < 0.01. (C) CLB administration reduced DNA synthesis. Synchronized cells were treated with 100 µM CLB for 12 h, labeled with BrdU, and stained with anti-BrdU. DAPI was used to visualize nuclei. Data are mean ± SE (n = 3) from two independent experiments. *, p < 0.05. Scale bar, 20 µm.
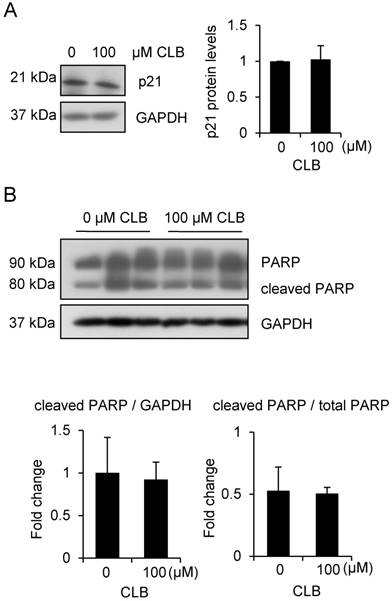
To rule out the possibility that cell apoptosis followed cell cycle arrest, we examined levels of p21 and PARP. There was no significant increase in all these variables (Fig. 2A-B). Thus, CLB induces cell cycle arrest, but not apoptosis, in C2C12 myoblasts.
CLB at a dose of 100 µM does not induce apoptosis. (A) p21 is not significantly changed in cells treated for 1 h with 0 or 100 µM CLB. GAPDH was used as loading control. Results are mean ± SD from three independent experiments. p > 0.05. (B) PARP levels are similar in untreated and CLB-treated cells. Cells were treated for 24 h with 0 or 100 µM CLB, and analyzed by Western blot to measure abundance of cleaved and uncleaved PARP. GAPDH was used to normalize PARP abundance. Results are mean ± SD from three independent experiments. p > 0.05.
CLB-induced cell cycle arrest is due to accumulation of p27
To further understand how CLB modulates cell cycle progression, we examined the expression of proteins that regulate cell cycle progression, including cyclin D and E, CDK2, and CDK inhibitors. We found that levels of CDK2 and cyclin D3, and E1 decreased when synchronized C2C12 cells were treated with CLB for 12 h (Fig. 3A). In contrast, p27, a critical inhibitor of cyclins and CDK2, accumulated (Fig. 3A). These results are hallmarks of an inhibitory effect on cell proliferation.
CLB increases abundance of p27, which is required for cell cycle arrest. (A) CLB exposure downregulates Cdk2 and cyclin D3 and E1, but upregulates p27. Synchronized cells were cultured with 10 and 100 µM CLB for 12 h and harvested. Cell lysates were blotted and probed with antibodies against p27, Cdk2 and cyclin D3 and E1. GAPDH was used as loading control. Fold expression change is indicated below blots. (B) CLB treatment does not increase p27 transcription. C2C12 cells were exposed to 100 µM CLB for indicated times, and p27 expression was measured by real-time PCR. Results are mean ± SD (n = 3). p > 0.05. (C) CLB induces cell cycle arrest under p27 knockdown condition. Synchronized cells were transiently transfected with p27 or control siRNA, treated with 100 µM CLB for 8 h, and analyzed by flow cytometry. Data are mean ± SD (n = 3) from three independent experiments. *, p < 0.05.
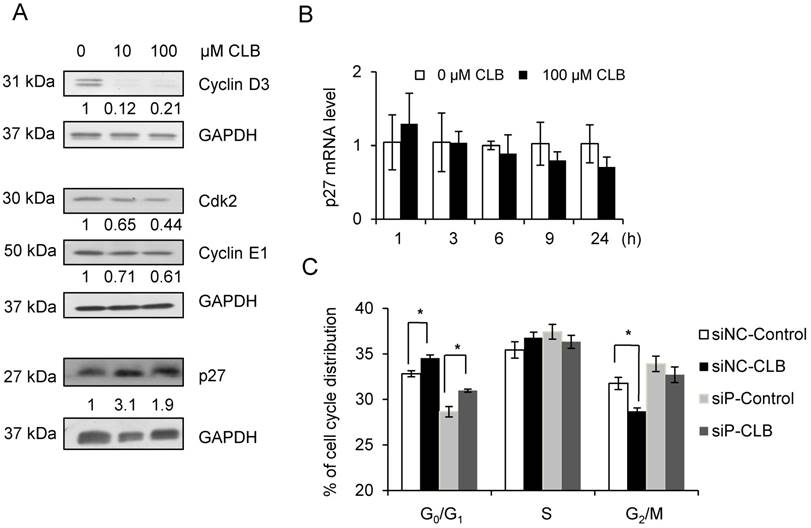
We also checked the mRNA level of p27 by qPCR to test whether p27 accumulation was due to enhanced transcription. We found that p27 mRNA level was not significantly altered in response to treatment with CLB for 1 or 24 h (Fig. 3B).
It is well known that p27 regulates phase transition from G1 to S. We knocked down p27 expression using siRNA. As a result, p27 mRNA was reduced to 40% after transfection with siRNA for 24 and 48 h (Fig. S2). Fig. 3C showed that p27 knockdown induced cell cycle arrest, however, CLB could also induce cell cycle arrest under p27 knockdown condition. These results suggest that the regulation of CLB for p27 may be regulated post-translationally.
CLB modulates p27 localization and degradation
As has been reported [36], p27 could be regulated post-translationally. In addition, the activity and localization of p27 to the nucleus or cytoplasm was tightly associated with post-translational modification. Thus, the cellular distribution of p27 was examined by Western blot. We found that treatment with CLB for 1 h resulted in a remarkable increase in p27 in the nucleus, accompanied by a modest decrease in the cytoplasm (Fig. 4A). Similar results were obtained by immunostaining (Fig. 4B), suggesting that CLB redistributes p27.
Cellular localization and delayed degradation of p27 after CLB treatment. (A-B) CLB increases p27 in the nucleus and depletes it from the cytoplasm, based on Western blotting (A) and immunofluorescence (B). In (A), nuclear and cytosolic p27 were measured in C2C12 cells incubated with 100 µM CLB for 1 h. H3 and GAPDH were used as loading controls for nuclear and cytosolic fractions, respectively. Results are mean ± SD from triplicate experiments. **, p < 0.01; ***, p < 0.001. In (B), synchronized cells were treated with 100 µM CLB for 12 h, probed with anti-p27 antibody, and stained with Alexa 594-conjugated secondary antibody (red). DAPI was used to label the nucleus (blue). Scale bar, 50 µm. Only p27 in the nucleus was quantified. Data are mean ± SD (n = 3). (C) CLB delays p27 degradation. C2C12 cells were incubated with 0 or 100 µM CLB for 1 h and then treated with cycloheximide according to indicated times. p27 stability was examined by quantitative Western blot, results are mean ± SD from two independent experiments. **, p < 0.01.
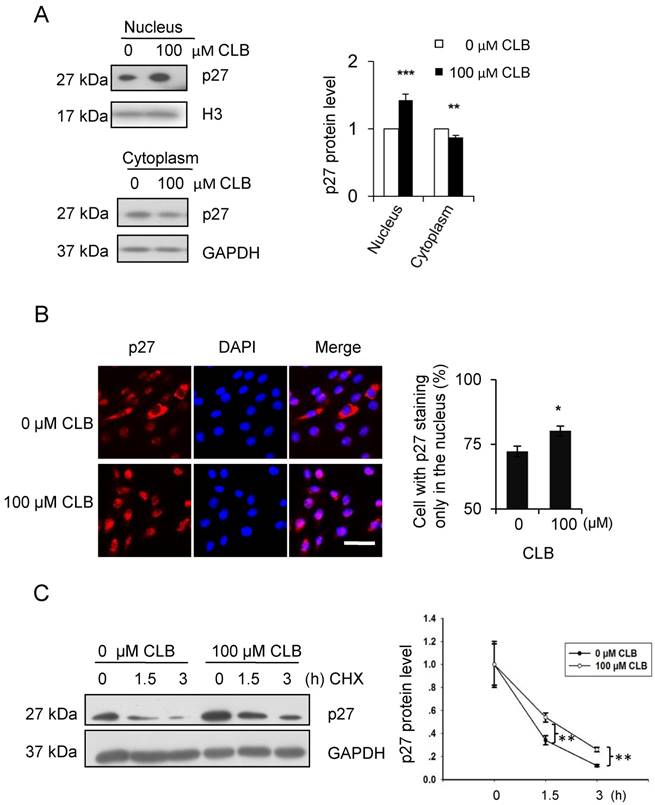
On the other hand, the stability of p27 has also been reported to depend on cellular localization [37]. Hence, we analyzed p27 stability in CLB-treated cells by using cycloheximide chase experiments. As expected, p27 levels decreased more slowly in cells treated with CLB for 1 h (Fig. 4C), clearly indicating that CLB inhibits p27 degradation.
CLB enhances β2-AR internalization and requires β2-AR for activity
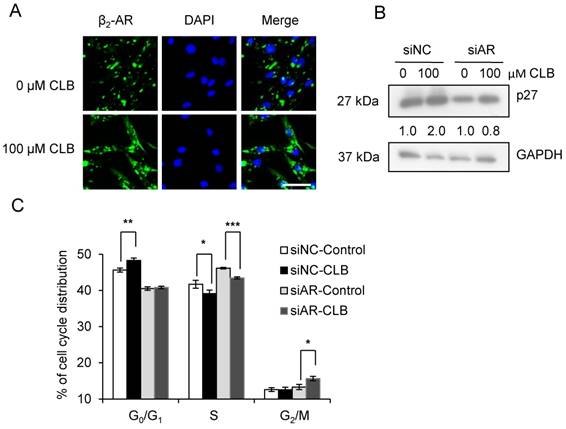
β2-AR is required for the hypertrophy and anti-atrophy properties of CLB [11], and it is activated, internalized, and subsequently recycled after the administration of an agonist [38]. As shown in Fig 5A, we could find that CLB enhanced β2-AR activation and internalization by immunofluorescence assays. To further evaluate the role of β2-AR, we knocked down β2-AR expression using siRNA. Consequently, we observed a reduction in the abundance of monomeric and dimeric β2-AR (Fig. S3). As shown, β2-AR knockdown blocked p27 accumulation (Fig. 5B) and cell cycle arrest (Fig. 5C), suggesting that β2-AR is required for CLB activity.
β2-AR is required for CLB-induced p27 accumulation and cell cycle arrest. (A) CLB treatment relocalizes β2-AR. Synchronized cells were treated with 100 µM CLB for 12 h, and incubated with antibodies against β2-AR, and then with a secondary antibody conjugated to Alexa 488 (green). DAPI was used to label the nucleus (blue). Scale bar, 50 µm. (B) siRNA knockdown of β2-AR reverses p27 accumulation. Synchronized cells were transiently transfected with β2-AR and control siRNA, and then exposed to 100 µM CLB for 60 h. Expression of p27 was measured, using GAPDH as loading control. Fold expression change is indicated below blots. (C) β2-AR knockdown relieves cell cycle arrest. Synchronized cells were transiently transfected with control and β2-AR siRNA, treated with 100 µM CLB for 60 h, and analyzed by flow cytometry. Data are mean ± SD (n = 3). *, p < 0.05; ***, p < 0.001.
CLB stabilizes p27 via β2-AR/β-arrestin 2 instead of Gs/PKA
Several groups have reported that CLB induces muscular hypertrophy and fiber type switching through a mechanism that requires β2-AR, Gs, cAMP, and PKA [18, 39]. We thus asked whether the CLB-induced p27 accumulation was mediated by PKA or Gs using H-89. H-89 is a potent selective inhibitor of PKA.
CREB is a critical transcription factor that is regulated PKA. We found that CREB activation was markedly enhanced by CLB, but inhibited after treated by the PKA inhibitor H-89 (Fig. 6A). To determine whether CLB-mediated regulation of p27 through PKA, C2C12 cells were treated with CLB and H-89. In addition, the suppression of PKA activity by 1-6 µM H-89 also did not alter transcription of p27 (Fig. 6B). Unexpectedly, nuclear p27 still increased remarkably even when cells were exposed to H-89 prior to CLB treatment (Fig. 6C). Thus, accumulation of p27 does not require activation of PKA. Hence, an alternative signaling pathway is involved.
CLB enhances p27 stability via β-arrestin 2 rather than PKA. (A) CLB treatment activates CREB. C2C12 cells were treated with 100 µM CLB for 1 h and nuclear fractions were analyzed for abundance of CREB and p-CREB. Fold expression change is indicated below blots. (B) H-89 does not significantly alter the abundance of p27 mRNA. Cells were treated with different concentrations of H-89 for 18 h and analyzed by real-time PCR. Results are mean ± SD (n = 3), p > 0.05. (C) CLB-induced p27 protein accumulation in the nucleus is independent of PKA. C2C12 cells were pretreated with 5 µM H-89 for 1 h, and then with 0 or 100 µM CLB for 24 h. Abundance of nuclear and cytosolic p27 was analyzed, using H3 and GAPDH as loading controls for nuclear and cytosolic fractions, respectively. Fold expression change is indicated below blots. (D) Knockdown of β-arrestin 2, but not β-arrestin 1, prevents CLB-induced p27 accumulation. Cells were transfected with shRNA for β-arrestin 1 and 2 and then exposed to 0 or 100 µM CLB. Cell lysates were analyzed by Western blot using anti-p27. Tubulin was used as loading control. Fold expression change is indicated below blots. (E) Skp2 protein diminished after CLB treatment. Synchronized cells were treated with 0 or 100 µM CLB for indicated times. Western blotting for Skp2 was performed with GAPDH as normalization control. Fold expression change is indicated below blots.
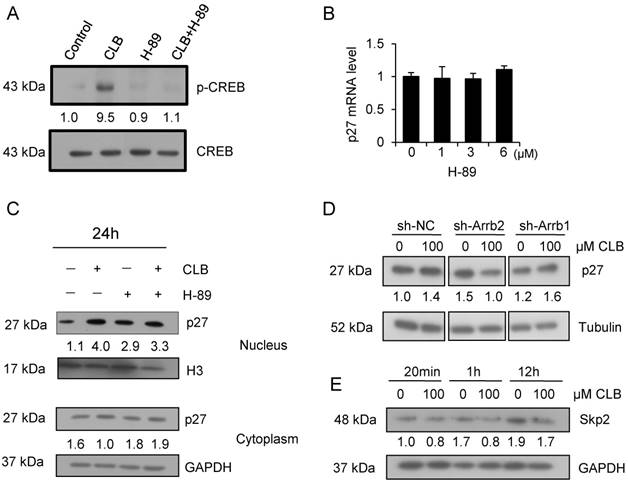
β-arrestins are adaptor proteins with key roles downstream of β2-AR [40, 41]. To verify whether β-arrestins mediate p27 accumulation in response to CLB treatment, we silenced β-arrestin 1 and 2 by RNA interference (Fig. S4). Silencing β-arrestin 2, but not β-arrestin 1, blocked p27 accumulation significantly (Fig. 6D), indicating that β-arrestin 2 is required for this process.
Skp2 is well known as the regulator for p27 stability, which tags p27 for proteasome degradation [42]. We found that there was a significant decrease in the abundance of the protein level of Skp2 (Fig. 6E), suggesting that CLB enhances p27 stability by β-arrestin 2 and Skp2.
Discussion
CLB, a selective β2-adrenoceptor agonist, promotes skeletal muscle growth mainly by modulating protein synthesis and degradation [5]. In addition, fenoterol enhances skeletal muscle regeneration [43], an ordered progression from the activation of satellite cells to proliferation and finally differentiation. We have observed CLB to promote C2C12 myoblast differentiation in vitro (unpublished data), a process that naturally requires exit from the cell cycle. Here, we report that CLB treatment induces cell cycle arrest in C2C12 myoblasts. Thus, CLB appears to promote myoblast differentiation by forcing exit from the cell cycle, and thus stimulate muscle regeneration.
A previous report has demonstrated that the exogenous β2-AR agonist isoproterenol promotes cell proliferation during embryogenesis [44]. However, the effects of CLB on the proliferation of muscle progenitor cells have not been clarified. Previous studies using autoradiographic techniques showed that CLB enhanced the proliferation of satellite cells in transplanted skeletal muscles [45]. However, a different report demonstrated that CLB did not affect proliferation in muscle-derived cells [46]. In our experiments, we found that CLB treatment induced the accumulation of p27 and cell cycle arrest in skeletal myoblasts. Thus, the effects of CLB appear to depend on cell type.
p27 is tightly regulated during proliferation and differentiation in myogenic cells. For example, p27 abundance is correlated inversely with proliferative potential in satellite cells [47-49]. During myogenesis, p27 is upregulated to ensure terminal growth arrest [50] and to promote differentiation [51, 52]. Indeed, our experiments show that p27 accumulation in the nucleus, along with decreased expression of CDK2, and cyclin D and E, contributes to cell cycle arrest.
Nuclear p27 is a critical cell cycle regulator and the G1/S transition in the cell cycle is regulated by the nuclear p27, which can bind to and prevent the activation of the cyclin and CDKs complex. Whereas cytoplasmic p27 regulates cell migration (Besson, 2008). So, the function of p27 associated with its localization. In our research, CLB changed the distribution of p27. In addition, CLB also attenuated the degradation of p27. However, it is unclear whether CLB regulates p27 via cAMP and PKA. Before checking the role of PKA, we found β2-AR is needed to regulate the cell cycle and p27 accumulation. However, the inhibitor of PKA pathway, H89, could not prevent the regulation of p27 in the C2C12. However, we showed that β-arrestin 2 modulated the translocation p27 through RNAi.
It is well known that p27 stability is regulated mainly by phosphorylation, which induces degradation via the ubiquitin-proteasome machinery [36]. When Thr187 in p27 is phosphorylated in late G1 phase, a binding site is formed for the complex that contains Skp2 and E3 ubiquitin ligase [36, 53, 54]. Which tags p27 for proteasome degradation [42]. We found that the expression of Skp2 at the mRNA level was unchanged after CLB treatment (Fig. S4C). However, there was a significant decrease in the abundance of the protein (Fig. 6D), suggesting that CLB enhances p27 stability by reducing Skp2 through β-arrestin 2 pathway. Additional, β-arrestin 2 can participate in the regulation of Akt [55], and Akt could interact with and directly phosphorylate Skp2 [56]. So CLB may regulate the stability of p27 via Akt/Skp2 in C2C12 myoblasts.
Abbreviations
CLB: clenbuterol; CREB: cAMP response element-binding protein; β2-AR: β2-Adrenoceptor; PKA: protein kinase A; cAMP: cyclic AMP; Skp2: S-phase kinase-associated protein 2; BrdU: 5-Bromo-2-deoxyUridine; ERK: extracellar signal-regulated protein.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the National Basic Research Program of China (2015CB943103), the Modern Agro-industry Technology Research Systems of China (CARS-37), National Transgenic Breeding Project of China (2014ZX08010004, 2016ZX08010004-002) and National “863” Project of the China Ministry of Science and Technology (No. 2012AA020601). We'd like to appreciate Bioyong tech (Beijing) Co. for their kind help in data association analysis.
Author contributions
M.C. designed and performed experiments, analyzed data and wrote the paper; C.L performed experiments, analyzed data and revised the paper; M.W., Y.Z. and K.Z helped performed cell culture experiments; H.W. helped analyze the data. Z.Y., X.L. and W.G. discussed data and revised paper. N.L. participated in the design of the study. Q.M. conceived the study, analyzed the data and wrote the paper. All authors contributed to the final version of the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tang H, Inoki K, Lee M, Wright E, Khuong A, Khuong A. et al. mTORC1 Promotes Denervation-Induced Muscle Atrophy Through a Mechanism Involving the Activation of FoxO and E3 Ubiquitin Ligases. Sci Signal. 2014;7:ra18
2. Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E. et al. BMP signaling controls muscle mass. Nature genetics. 2013;45:1309-18
3. Sartorelli V, Fulco M. Molecular and cellular determinants of skeletal muscle atrophy and hypertrophy. Science's STKE: signal transduction knowledge environment. 2004;2004:re11
4. Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol. 2006;174:677-87
5. Lynch GS, Ryall JG. Role of beta-adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiol Rev. 2008;88:729-67
6. Claeys MC, Mulvaney DR, McCarthy FD, Gore MT, Marple DN, Sartin JL. Skeletal muscle protein synthesis and growth hormone secretion in young lambs treated with clenbuterol. J Anim Sci. 1989;67:2245-54
7. Benson DW, Foley-Nelson T, Chance WT, Zhang FS, James JH, Fischer JE. Decreased myofibrillar protein breakdown following treatment with clenbuterol. J Surg Res. 1991;50:1-5
8. Bhavsar PK, Brand NJ, Felkin LE, Luther PK, Cullen ME, Yacoub MH. et al. Clenbuterol Induces Cardiac Myocyte Hypertrophy via Paracrine Signalling and Fibroblast-derived IGF-1. J Cardiovasc Transl Res. 2010
9. Wong K, Boheler KR, Bishop J, Petrou M, Yacoub MH. Clenbuterol induces cardiac hypertrophy with normal functional, morphological and molecular features. Cardiovasc Res. 1998;37:115-22
10. Petrou M, Wynne DG, Boheler KR, Yacoub MH. Clenbuterol induces hypertrophy of the latissimus dorsi muscle and heart in the rat with molecular and phenotypic changes. Circulation. 1995;92:II483-9
11. Hinkle RT, Hodge KM, Cody DB, Sheldon RJ, Kobilka BK, Isfort RJ. Skeletal muscle hypertrophy and anti-atrophy effects of clenbuterol are mediated by the beta2-adrenergic receptor. Muscle Nerve. 2002;25:729-34
12. Hayes A, Williams DA. Examining potential drug therapies for muscular dystrophy utilising the dy/dy mouse: I. Clenbuterol. J Neurol Sci. 1998;157:122-8
13. Dupont-Versteegden EE. Exercise and clenbuterol as strategies to decrease the progression of muscular dystrophy in mdx mice. J Appl Physiol. 1996;80:734-41
14. Cockman MD, Jones MB, Prenger MC, Sheldon RJ. Magnetic resonance imaging of denervation-induced muscle atrophy: effects of clenbuterol in the rat. Muscle Nerve. 2001;24:1647-58
15. Sneddon AA, Delday MI, Maltin CA. Amelioration of denervation-induced atrophy by clenbuterol is associated with increased PKC-alpha activity. Am J Physiol Endocrinol Metab. 2000;279:E188-95
16. Ramos BP, Colgan LA, Nou E, Arnsten AF. Beta2 adrenergic agonist, clenbuterol, enhances working memory performance in aging animals. Neurobiol Aging. 2008;29:1060-9
17. Zeman RJ, Peng H, Danon MJ, Etlinger JD. Clenbuterol reduces degeneration of exercised or aged dystrophic (mdx) muscle. Muscle Nerve. 2000;23:521-8
18. Choo JJ, Horan MA, Little RA, Rothwell NJ. Anabolic effects of clenbuterol on skeletal muscle are mediated by beta 2-adrenoceptor activation. Am J Physiol. 1992;263:E50-6
19. Kline WO, Panaro FJ, Yang H, Bodine SC. Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J Appl Physiol. 2007;102:740-7
20. Hill SJ, Baker JG. The ups and downs of Gs- to Gi-protein switching. Br J Pharmacol. 2003;138:1188-9
21. Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88-91
22. Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gamma. Science. 1997;275:394-7
23. Pearen MA, Ryall JG, Maxwell MA, Ohkura N, Lynch GS, Muscat GE. The orphan nuclear receptor, NOR-1, is a target of beta-adrenergic signaling in skeletal muscle. Endocrinology. 2006;147:5217-27
24. Glass DJ. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nature cell biology. 2003;5:87-90
25. Spanou D, Perimenis P, Mantzouratou P, Gomatos C, Cokkinos D, Mourouzis I. et al. Clenbuterol favorably remodels neonatal cardiac cells via activation of p38 MAPK signalling pathway. The Journal of cardiovascular surgery. 2012;53:789-95
26. Guereschi MG, Araujo LP, Maricato JT, Takenaka MC, Nascimento VM, Vivanco BC. et al. Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur J Immunol. 2013;43:1001-12
27. Rocha AS, Paternot S, Coulonval K, Dumont JE, Soares P, Roger PP. Cyclic AMP inhibits the proliferation of thyroid carcinoma cell lines through regulation of CDK4 phosphorylation. Mol Biol Cell. 2008;19:4814-25
28. Dumaz N, Marais R. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft fur Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J. 2005;272:3491-504
29. Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258-66
30. Berdeaux R, Stewart R. cAMP signaling in skeletal muscle adaptation: hypertrophy, metabolism, and regeneration. Am J Physiol Endocrinol Metab. 2012;303:E1-17
31. Burattini S, Ferri P, Battistelli M, Curci R, Luchetti F, Falcieri E. C2C12 murine myoblasts as a model of skeletal muscle development: morpho-functional characterization. Eur J Histochem. 2004;48:223-33
32. Li Y, He J, Sui S, Hu X, Zhao Y, Li N. Clenbuterol upregulates histone demethylase JHDM2a via the beta2-adrenoceptor/cAMP/PKA/p-CREB signaling pathway. Cell Signal. 2012;24:2297-306
33. Lin SL, Chang DC, Ying SY, Leu D, Wu DT. MicroRNA miR-302 inhibits the tumorigenecity of human pluripotent stem cells by coordinate suppression of the CDK2 and CDK4/6 cell cycle pathways. Cancer Res. 2010;70:9473-82
34. Sachidanandan C, Sambasivan R, Dhawan J. Tristetraprolin and LPS-inducible CXC chemokine are rapidly induced in presumptive satellite cells in response to skeletal muscle injury. J Cell Sci. 2002;115:2701-12
35. Schmitt JM, Stork PJ. beta 2-adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein rap1 and the serine/threonine kinase B-Raf. J Biol Chem. 2000;275:25342-50
36. Vervoorts J, Luscher B. Post-translational regulation of the tumor suppressor p27(KIP1). Cell Mol Life Sci. 2008;65:3255-64
37. Susaki E, Nakayama KI. Multiple mechanisms for p27(Kip1) translocation and degradation. Cell Cycle. 2007;6:3015-20
38. Claing A. Regulation of G protein-coupled receptor endocytosis by ARF6 GTP-binding proteins. Biochem Cell Biol. 2004;82:610-7
39. Goncalves DA, Silveira WA, Lira EC, Graca FA, Paula-Gomes S, Zanon NM. et al. Clenbuterol suppresses proteasomal and lysosomal proteolysis and atrophy-related genes in denervated rat soleus muscles independently of Akt. Am J Physiol Endocrinol Metab. 2012;302:E123-33
40. Shiina T, Kawasaki A, Nagao T, Kurose H. Interaction with beta-arrestin determines the difference in internalization behavor between beta1- and beta2-adrenergic receptors. J Biol Chem. 2000;275:29082-90
41. Kobayashi H, Narita Y, Nishida M, Kurose H. Beta-arrestin2 enhances beta2-adrenergic receptor-mediated nuclear translocation of ERK. Cell Signal. 2005;17:1248-53
42. Kossatz U, Dietrich N, Zender L, Buer J, Manns MP, Malek NP. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 2004;18:2602-7
43. Beitzel F, Gregorevic P, Ryall JG, Plant DR, Sillence MN, Lynch GS. Beta2-adrenoceptor agonist fenoterol enhances functional repair of regenerating rat skeletal muscle after injury. J Appl Physiol (1985). 2004;96:1385-92
44. Ryall JG, Lynch GS. The potential and the pitfalls of beta-adrenoceptor agonists for the management of skeletal muscle wasting. Pharmacology & therapeutics. 2008;120:219-32
45. Roberts P, McGeachie JK. The effects of clenbuterol on satellite cell activation and the regeneration of skeletal muscle: an autoradiographic and morphometric study of whole muscle transplants in mice. J Anat. 1992;180( Pt 1):57-65
46. Scheffler JM. Lysophosphatidic Acid, But Neither Clenbuterol Nor Salbutamol, Stimulates Increases in ERK-1/2 Phosphorylation which is Not Associated with an Appreciable Increase in Proliferation. University of Nebraska - Lincoln. 2007
47. Chakravarthy MV, Abraha TW, Schwartz RJ, Fiorotto ML, Booth FW. Insulin-like growth factor-I extends in vitro replicative life span of skeletal muscle satellite cells by enhancing G1/S cell cycle progression via the activation of phosphatidylinositol 3'-kinase/Akt signaling pathway. J Biol Chem. 2000;275:35942-52
48. Allen RE, Boxhorn LK. Regulation of skeletal muscle satellite cell proliferation and differentiation by transforming growth factor-beta, insulin-like growth factor I, and fibroblast growth factor. J Cell Physiol. 1989;138:311-5
49. Spangenburg EE, Chakravarthy MV, Booth FW. p27Kip1: a key regulator of skeletal muscle satellite cell proliferation. Clin Orthop Relat Res. 2002:S221-7
50. Zabludoff SD, Csete M, Wagner R, Yu X, Wold BJ. p27Kip1 is expressed transiently in developing myotomes and enhances myogenesis. Cell Growth Differ. 1998;9:1-11
51. Chu CY, Lim RW. Involvement of p27(kip1) and cyclin D3 in the regulation of cdk2 activity during skeletal muscle differentiation. Biochim Biophys Acta. 2000;1497:175-85
52. Leshem Y, Spicer DB, Gal-Levi R, Halevy O. Hepatocyte growth factor (HGF) inhibits skeletal muscle cell differentiation: a role for the bHLH protein twist and the cdk inhibitor p27. J Cell Physiol. 2000;184:101-9
53. Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9:661-4
54. Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193-9
55. Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261-73
56. Lin HK, Wang G, Chen Z, Teruya-Feldstein J, Liu Y, Chan CH. et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nature cell biology. 2009;11:420-32
Author contact
![]() Corresponding author: Qingyong Meng, State Key Laboratory for Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing 100193, China E-mail addresses: qymengedu.cn
Corresponding author: Qingyong Meng, State Key Laboratory for Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing 100193, China E-mail addresses: qymengedu.cn