Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(7):775-783. doi:10.7150/ijbs.26077 This issue Cite
Research Paper
Atg7 inhibits Warburg effect by suppressing PKM2 phosphorylation resulting reduced epithelial-mesenchymal transition
Yanling Feng1, Jingwei Liu1, Wendong Guo1, Yi Guan1, Hongde Xu1, Qiqiang Guo1, Xiaoyu Song1, Fei Yi1, Ting Liu1, Wenyu Zhang1, Xiang Dong1, Longyue L. Cao2, Brian P. O'Rourke3, Liu Cao1 ![]()
1. Key Laboratory of Medical Cell Biology, Ministry of Education; Institute of Translational Medicine, China Medical University; Liaoning Province Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, Shenyang, Liaoning Province, China
2. Wilf Family Cardiovascular Research Institute, Department of Medicine (Cardiology), Albert Einstein College of Medicine, Bronx, New York 10461, USA
3. Department of Physiology and Biophysics, Albert Einstein College of Medicine, Bronx, New York 10461, USA
Received 2018-3-15; Accepted 2018-4-16; Published 2018-5-12
Abstract

Metabolic reprogramming is a distinct hallmark in tumorigenesis. Autophagy can rewire cell metabolism by regulating intracellular homeostasis. Warburg effect is a specific energy metabolic process that allows tumor cells to metabolize glucose via glycolysis into lactate even in the presence of oxygen. Although both autophagy and Warburg effect are involved in the stress response to energy crisis in tumor cells, their molecular relationship has remained largely elusive. We found that Atg7, a key molecule involved in autophagy, inhibits the Warburg effect. Mechanistically, Atg7 binds PKM2 and prevents its Tyr-105 phosphorylation by FGFR1. Furthermore, the hyperphosphorylation of PKM2 and its induced Warburg effect due to Atg7 deficiency promote epithelial-mesenchymal transition (EMT). Conversely, overexpression of Atg7 inhibits PKM2 phosphorylation and the Warburg effect, thereby inhibiting EMT of tumor cells. Our work reveals a molecular link between Atg7 and the Warburg effect, which may provide insight into novel strategies for cancer treatment.
Keywords: Autophagy, Warburg effect, Atg7, PKM2, EMT, Metabolic reprogramming
Introduction
Metabolic reprogramming is a distinct hallmark of cancer[1-3]. Under well-oxygenated conditions, most differentiated cells rely primarily on mitochondrial oxidative phosphorylation for energy production [4]. In the 1920s, however, Otto Warburg found that cancer cells undergo metabolic reprogrammming. Even in the presence of oxygen, cancer cells generate energy by metabolizing glucose to lactate through a process known as aerobic glycolysis, or the Warburg effect[5]. Although the Warburg effect is less energetically efficient, a large number of glycolytic intermediates (synthesized proteins, nucleic acids, lipids and other biological macromolecules) can be used by tumor cells for growth and survival[6, 7].
Autophagy is an evolutionarily conserved lysosomal degradation process of cytoplasmic constituents, which can regulate cell energy metabolism and self-update by maintaining intracellular homeostasis[8, 9]. As a process first identified to confer stress tolerance, limit damage, and sustain viability, autophagy has recently been found to be associated with a variety of human diseases including cancer[10, 11]. Autophagy is now emerging as a potent regulator of tumorigenesis, but the exact role of autophagy in cancer development remains contradictory[12]. On one hand, autophagy protects cells from metabolic stress and oxidative damage, thus suppressing tumorigenesis[13-15]. On the other hand, stress-induced autophagy can facilitate tumorgenesis by helping tumor cells overcome nutritional deficiencies or adverse hypoxic environment, thereby promoting more favorable conditions for cell growth[16, 17].
Among multiple factors involved in autophagy signalling pathway, the autophagy-related protein Atg7 is a key factor indispensable for successful autophagic process. Loss of Atg7 impairs the formation of autophagosome, resulting in a deficiency of or even block of autophagy[18, 19]. Except the function in autophagy, Atg7 is involving in many other biological behaviors which are autophagy-independent. We previously found that Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress[20]. And we observed that knockout of Atg7 significantly increased basal glycolytic rates[21], but the underlying molecular mechanism still remains undetermined.
PKM2 is the key mediator for the Warburg effect, and the phosphorylation of PKM2 Tyr105 site leads to less pyruvate kinase activity and promotes the Warburg effect[22]. Here, we found that Atg7 inhibits the Warburg effect by suppressing the phosphorylation of PKM2 Tyr105 site through blocking the binding of PKM2 with its upstream kinase FGFR1, resulting in reduced EMT in tumor cells. The new function of Atg7 in regulating the Warburg effect we unraveled in this study might shed light on novel mechanism of tumorigenesis and provide promising targets for therapy.
Materials and Methods
Chemicals and reagents
DMEM medium, penicillin/streptomycin, FBS, and trypsin were purchased from Invitrogen. Puromycin was purchased from Sigma. PKM2(4053s), pPKM2 (Tyr-105)(3827s), Myc(2276s), Flag(2368s) and HRP-conjugated secondary antibodies were purchased from Cell Signaling Technology. Antibodies specific to Atg7(A2856), tubulin(T6199) and β-actin(A1978) were purchased from Sigma. FGFR1(sc-121) antibodies were purchased from Santa Cruz Biotechnology. Unless otherwise stated, chemicals were purchased from Sigma.
Plasmid construction
Expression of the Atg7 shRNA plasmid was previously described[20]. Expression of the Myc-Atg7 plasmid was previously described[23]. The Atg7 shRNA nontargetable mutant Myc-Atg7 plasmid were constructed by PCR amplification and subcloned into pCMV-myc vector (Clontech, 631604). The PKM2 expression plasmid of was bought from OriGene (SC315792). The Flag-PKM2 Y105 point mutation plasmid Y105-E was constructed with the Quickchange Site-Directed Mutagenesis Kit (Stratagene, 200519). GST-tagged Atg7 and PKM2 plasmids were constructed by PCR amplification and subcloned into pGEX-5x-1 (GE Healthcare, 28-9545-53) vectors. PKM2 knockdown lentivirus was bought from Genechem.
Cell Culture and Transfection
HeLa and HEK293T cells were cultured in high-glucose Dulbecco's modified medium (DMEM, Gibco, 12100046), supplemented with 10% fetal bovine serum (FBS, Gibco, 10099141) and 100 units/ml of penicillin/streptomycin at 37oC in a 5%CO2 incubator. Wild-type and atg7-/-MEF cells were obtained as followed: pregnant mice were euthanized to obtain embryos (day e13 to e15). The limb, head, tail and viscera were removed, and subsequently the remaining tissues were minced into smaller pieces and digested in 0.25% trypsin/0.01%EDTA with pipetting. After 10 min of digesting, the mixture was inactivated with fetal bovine serum, centrifuged at 1000 rpm for five minutes, and the cell pellets were resuspended with 15%FBS DMEM. The cells were plated in culture dishes at 37oC in a 5%CO2 incubator.
The cells were plated one day before transfection and reached 70% confluence at the time of transfection. The transfection process was performed using Lipofectamine 2000 (Thermo Fisher Scientific, 11668019) in accordance with the manufacturer's instructions. Cells were harvested 48 h after transfection.
Western Blot Analysis
Cells were lysed for 30 min on ice with RIPA buffer (50 mM Tris/HCl pH 7.5, 200 mM NaCl,12.5% glycerol, 1% NP40, 1 mM EDTA, 1 mM EGTA, and 1 mM DTT) supplemented with protease and phosphatase inhibitor cocktails, and the total protein was harvested by cryogenic centrifugation at 15,000 rpm for 20 min. Protein concentration was measured by G250 and 30 μg of cell lysate were used in samples.
Protein was also isolated from in vivo sources. The fresh tissues were dissected from mice soon after birth. Tissues were immediately frozen in liquid nitrogen, then incubated with RIPA buffer with protease and phosphatase inhibitor cocktails to prepare tissue homogenate. Samples were placed on ice for 30 min with shaking to solubilize the tissue homogenate. The homogenate was centrifuged at 15,000 rpm for 30 min at 4°C. The supernatant was transferred to a new tube and protein concentration was measured by G250. 40 μg of tissue lysate was prepared for separation and blotting.
Samples were separated on 8% SDS PAGE and transferred to PVDF membrane (Millipore, IPVH00010) for 2 h at 80V. The membranes were blocked in 5% BSA in TBST for 1 h at room temperature and probed with primary antibodies overnight at 4°C. Membranes were then washed with TBST three times and incubated with HRP-conjugated secondary antibody for 2 h at room temperature. After three washes, bands were detected by enhanced chemi-luminescence detection kit (ThermoFisher Scientific, 32106) and visualized via the DNR western blot detection system.
Co-immunoprecipitation and GST-pull down Analysis
HeLa or transfected cells were lysed with IP lysis buffer (25 mM Tris, pH 7.6, 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA), and 1 mg protein was incubated with antibody and protein A/G-Sepharose (Santa Cruz, sc-2003) on a rocking platform overnight at 4°C. The beads were then harvested by centrifugation for 5 min at 500 g at 4°C and the supernatant was removed. The beads were then resuspended in IP lysis buffer and repeatedly inverted on a rocking platform for 10 min. This wash was repeated three times to remove the nonspecific binding protein and obtain purified protein complexes that are bound to the antibody-coated beads. The beads were resuspended with loading buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 12.5 mM EDTA, and 0.02% bromphenol blue) for Western blot analysis.
In vitro transcription and translation of PKM2 or Atg7 proteins were performed by T7-TNT Kits (Promega, L1170) in accordance with the manufacturer's instructions. Bacterial-expressed GST-PKM2 or GST-Atg7 and control GST proteins were purified with GST sepharose beads (GE Healthcare, 17-0756-01). The GST pull-down assays were performed by incubating equal amounts of GST, GST-fusion proteins bound to GST sepharose beads and in vitro-translated protein for 4 h at 4°C. Then the complexes of proteins were washed three times with binding buffer (20 mM Tris, pH 7.5, 50 mM NaCl, 10% Glycerol, 1% NP-40). The sediments were eluted by 2×loading buffer, and separated by SDS-PAGE for Western blot analysis.
Analysis of glucose consumption and lactate production
Cells were seeded in six-well culture plate. After 6 h adherence, the medium was changed to DMEM media without phenol red. After an additional 48 h of incubation, the media was harvested for measurement of glucose and lactate concentration. Glucose levels were detected with assay kits (Sigma, GAHK20) according to the manufacturer's instructions. Glucose consumption was measured as the difference in glucose levels in the media before and after the 48 h incubation period. The extracellular lactate levels were measured by lactate assay kits (Sigma, MAK065) according to the manufacturer's instruction. The values were normalized to the protein concentration.
Measurement of extracellular acidification rate (ECAR)
ECAR were measured using a Seahorse XFp analyzer (Seahorse Bioscience, 103020-100). In brief, cells were plated on Seahorse XFp plates for 12-15 h at a concentration of 6×103 cells/well. Cells were washed and incubated with assay medium at 37°C for 1 h in a non-CO2 incubator. Glucose, oligomycin and 2-deoxyglucose were injected into the medium at final concentrations of 10 mM, 1 μM, 50 mM, respectively. The ECAR were automatically recorded and calculated by the Seahorse XFp software as per manufacture's recommendation.
Transwell migration assay
To measure cellular capacity for migration, cells were seeded into the upper chamber with the serum-free media in 8 um Boyden Chamber (Corning, 3422). Following 24 h incubation with different treatments, non-invaded cells were removed. Invasive cells on the underside of chambers were fixed in methanol for 5 min. Then, the invaded cells were stained with Giemsa for 30 min and imaged using an inverted microscope. Five individual fields were counted for each sample, and three independent experiments were performed for statistical analysis.
Statistical analysis
Data were statistically analyzed by paired t test or One Way ANOVA using the software GraphPad Prism version 5.0 (GraphPad Software Inc, San Diego, CA, USA). The statistical significance between the experimental groups and their respective control groups were expressed as p values, and p<0.05 was considered statistically different. All experiments were independently repeated at least three times.
Results
Atg7 interacts with PKM2 directly and inhibits PKM2 phosphorylation
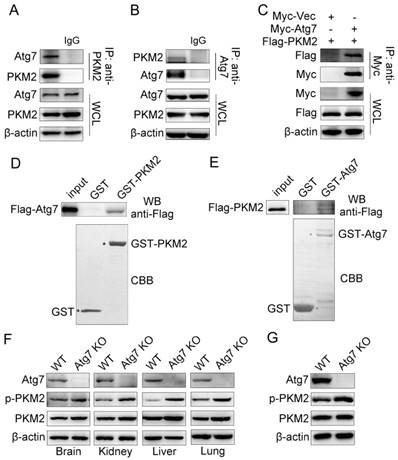
We previously observed that knockout of Atg7 significantly increased basal glycolytic rates[21]. To understand how Atg7 inhibits the Warburg effect, we tested whether Atg7 interacts with PKM2, the key mediator of the Warburg effect. Endogenous Atg7 interacted with endogenous PKM2 in HeLa cells (Figure 1A, 1B). Similarly, Myc-tagged Atg7 co-immunoprecipitated with Flag-tagged PKM2 in 293T cells (Figure 1C). GST pull-down assays using purified glutathione S-transferase (GST)-PKM2 fusion protein and Flag-Atg7 mammalian fusion protein showed that the binding of Atg7 to PKM2 is direct (Figure 1D). The reciprocal pull-down assay using GST-Atg7 and Flag-PKM2 mammalian fusion proteins confirmed this finding (Figure 1E). In order to understand the mechanisms underlying the binding between Atg7 and PKM2, we identified a possible relationship between Atg7 and PKM2. First, we examined the expression of total and Tyr-105 phosphorylated PKM2 in vivo. We found that the phosphorylation of PKM2(Tyr-105) was significantly elevated in Atg7-/- mice tissues, including brain, liver, kidney and lung compared with wild type tissues, whereas total PKM2 expression demonstrated no significant changes (Figure 1F). Cultures of Atg7-/- mouse embryonic fibroblast cells (MEFs) and wild-type MEFs showed no significant differences in total PKM2 but significantly increased in phosphorylation of PKM2(Tyr-105) (Figure 1G). These results suggest that Atg7 interacts with PKM2 directly and inhibits PKM2 phosphorylation.
Atg7 interacts with PKM2 directly and inhibits PKM2 phosphorylation. Atg7 interacts with PKM2. (A, B) Coimmunoprecipitation (IP) of endogenous Atg7 with PKM2 in HeLa cells. Cell lysates were subjected to IP using anti-PKM2(rabbit) or anti-Atg7(rabbit) and unrelated rabbit IgG as a control. The resulting precipitates were subjected to Western blot (WB) analysis with anti-Atg7 or anti-PKM2. A portion of the whole-cell lysate (WCL) of the input for IP was also subjected to IB analysis. (C) HEK293T cells were co-transfected with expression plasmids encoding Myc-tag or Myc-tagged Atg7 and Flag-tagged PKM2 as indicated. Cells were lysed and subjected to IP with an anti-Myc antibody. The resulting precipitates were subjected to WB analysis with anti-Flag antibody. A portion of the whole-cell lysate of the input for IP was also subjected to IB analysis. (D, E) GST pull-down assay was performed with glutathione S-transferase (GST) or GST fused PKM2 protein and in vitro translated Flag-Atg7 (D) or GST fused Atg7 protein and in vitro translated Flag-PKM2 (E). Input and pull-down fractions of Flag-Atg7 (D) or Flag-PKM2 (E) were detected by immunoblots using anti-Flag antibodies. The corresponding SDS-PAGE gels show the amount of GST or GST-PKM2/Atg7 immobilized in each assay. Atg7 inhibitsTyr-105 phosphorylation of PKM2. (F) Tissues (brain, kidney, liver and lung) from Atg7 knockout mice and wide type mice were gained soon after born and tissue lysates was used for Western blot assay to detect total and Tyr-105 phosphorylation of PKM2. (G) Western blot analysis of total and Tyr-105 phosphorylation of PKM2 in wide type and Atg7 knockout MEF cells.
Atg7 inhibits PKM2 phosphorylation via blocking the interaction of FGFR1 and PKM2
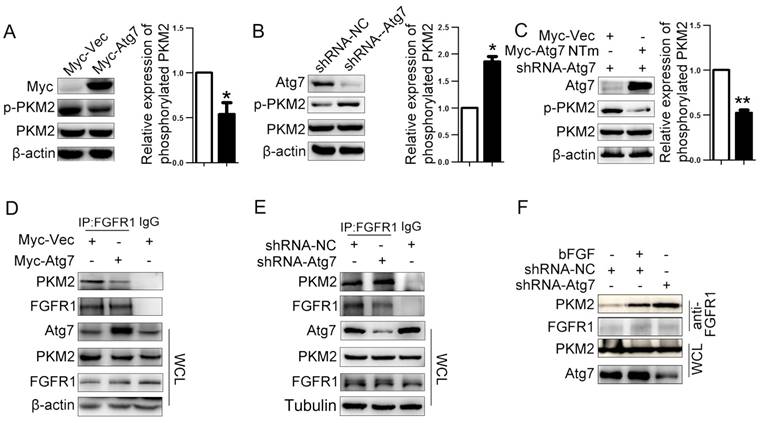
We next explored the molecular mechanism of how Atg7 inhibits PKM2 phosphorylation through both the overexpression Atg7 in HeLa cells using pCMV-myc-Atg7 plasmid and the knockdown of Atg7 in HeLa cells using Atg7-specific short hairpin (sh)RNA lentiviruses. As controls, we used lentiviruses expressing nonspecific shRNA (shNS). Efficient Atg7 knockdown and its influence on autophagy were confirmed by WB (Figure S1A). We found that total PKM2 expression was unchanged with Atg7 overexpression or knockdown, but phosphorylation of PKM2 at the Tyr-105 site was significantly decreased in Atg7 overexpressed cells (Figure 2A) and increased in Atg7 knockdown cells (Figure 2B). Restoration of Atg7 expression in Atg7 knockdown cells rescued the reduction of PKM2 phosphorylation at Tyr-105 (Figure 2C). As fibroblast growth factor receptor 1 (FGFR1) directly phosphorylates PKM2(Tyr-105)[22], we investigated the effect of Atg7 on the binding of FGFR1 with PKM2. We found in HeLa cells that while Atg7 overexpression decreased the binding between FGFR1 and PKM2 when compared with control cells (Figure 2D), Atg7 knockdown significantly enhanced FGFR1 and PKM2 binding (Figure 2E). Restoring Atg7 expression in Atg7 knockdown cells decreased the binding between FGFR1 and PKM2(Figure S1B). Adding bFGF, which activates FGFR1, enhanced binding between FGFR1 and PKM2 similar to Atg7 knockdown cells (Figure 2F). These results showed that Atg7 inhibits PKM2 phosphorylation by blocking the interaction between FGFR1 and PKM2.
Atg7 inhibits PKM2 phosphorylation via blocking the interaction of PKM2 and FGFR1. Atg7 inhibitsTyr-105 phosphorylation of PKM2. (A, B, C) Western blot analysis of total and Tyr-105 phosphorylation of PKM2 in HeLa cell expressing Myc-Atg7(A), shRNA-Atg7(B), HeLa cell stable knockdown of endogenous PKM2 and “rescue” expression of Myc-Atg7 NTm (nontargetable mutant). (C) and their corresponding control cells. Right: Quantification of protein expression (Tyr-105 phosphorylation of PKM2) shown in (A, B, C) normalized to β-actin. Data shown are mean±S.E.M. of n≥3 technical replicates and are representative of three independent experiments. P values were calculated by t-test. *P<0.005, **P<0.001. Atg7 blocked the interaction of PKM2 and FGFR1(D, E). Coimmunoprecipitation (IP) of endogenous PKM2 with FGFR1 in HeLa cell transfected with expression plasmids encoding Myc-tag or Myc-tagged Atg7(D), HeLa cell stable knockdown of endogenous PKM2(E). Cell lysates were subjected to IP using anti-FGFR1(rabbit) and unrelated rabbit IgG as a control. The resulting precipitates were subjected to WB analysis with anti-PKM2. A portion of the whole-cell lysate (WCL) of the input for IP was also subjected to IB analysis. (F) HeLa cell stable knockdown of endogenous PKM2 and control cell were treated with bFGF(10 ng/mL), cell lysates were subjected to IP using anti-FGFR1(rabbit). The resulting precipitates were subjected to Immunoblot (IB) analysis with anti-PKM2. A portion of the whole-cell lysate (WCL) of the input for IP was also subjected to WB analysis.
Atg7 inhibits the Warburg effect
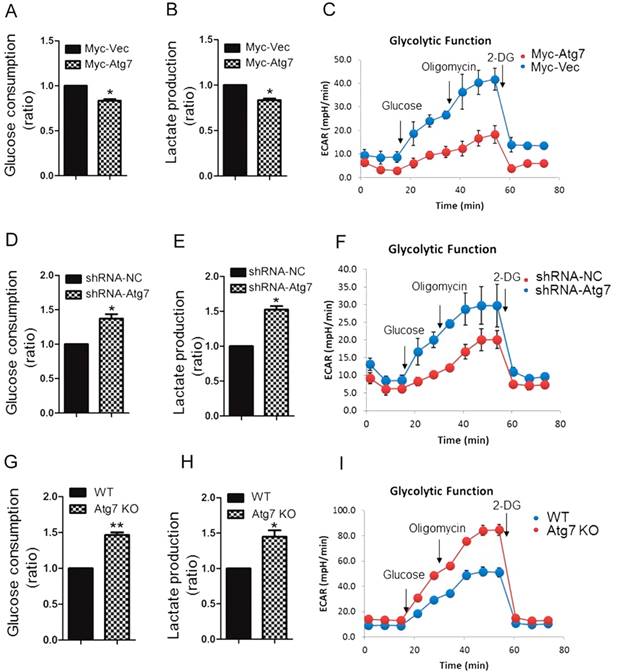
It has been reported that phosphorylation of PKM2 (Tyr-105) promotes the Warburg effect[22]. We have found that Atg7 inhibits PKM2 phosphorylation, so we wanted to investigate the functional role of Atg7 on the Warburg effect. We found that cells overexpressing Atg7 had significantly decreased levels of glucose consumption and lactate production compared with control cells (Figure 3A, 3B). This metabolic change was also accompanied by a decrease in ECAR (Figure 3C) but no obvious change of oxygen consumption rate (OCR). (Figure S1C). Knockdown of Atg7 in HeLa cells increased glucose consumption, lactate production (Figure 3D, 3E), ECAR (Figure 3F) but no obvious change of OCR (Figure S1D) in comparison to control. We found that Atg7-/- MEFs similarly exhibited an increase in glucose consumption and lactate production compared to control cells (Figure 3G, 3H), accompanied by an increase in ECAR (Figure 3I). Taken together, these results suggested that Atg7 inhibits the Warburg effect.
Atg7 inhibits the Warburg effect. Glucose consumption (A, D, G), lactate production (B, E, H) and ECAR (a proxy for the rate of glycolysis) (C, F, I) in HeLa cell expressing Myc-Atg7(A, B, C), HeLa cell knockdown Atg7(D, E, F), Atg7 konckout MEF cell(G, H, I), and their control cells. Data shown are mean±S.E.M. of n≥3 technical replicates and are representative of three independent experiments. P values were calculated by t-test. *P<0.005, **P<0.001.
Inhibition of Atg7 promotes epithelial-mesenchymal transition (EMT) through PKM2 mediated Warburg effect
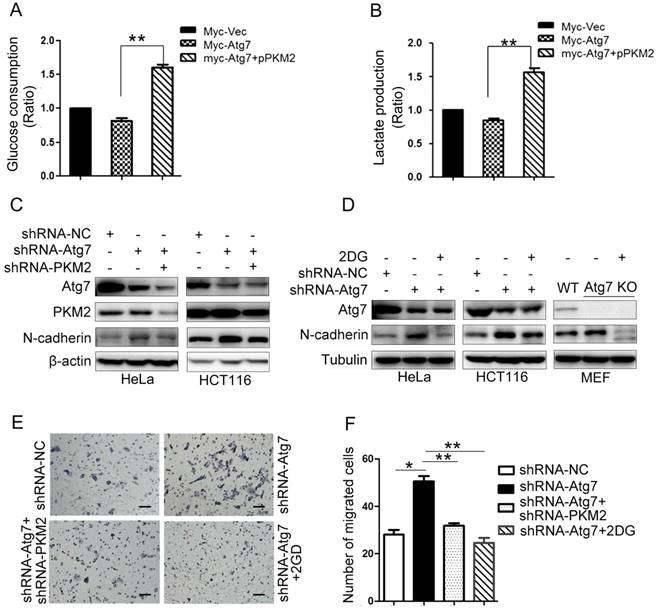
As we confirmed that Atg7 inhibites the Warburg effect (Figure 3), we further explored if Atg7 influences EMT by altering PKM2 mediated Warburg effect. Cells overexpressing Atg7 had a significantly decreased glucose consumption and lactate production previously described (Figure 3A, 3B). We next mutated tyrosine 105 phosphorylation site to artificially induce phosphomimetic mutant (Y105E) and overexpressed PKM2 (Y105E) would recover the effect caused by Atg7 overexpression (Figure 4A, 4B). These results showed that Atg7 inhibits the Warburg effect via PKM2 phosphorylation(Tyr-105). Atg7 knockdown in HeLa and HCT-116 cells activated EMT, as demonstrated by the increase in N-cadherin expression (Figure 4C). Knockdown of PKM2 in Atg7-knockdown HeLa and HCT-116 cells could rescue this effect caused by Atg7 inhibition (Figure 4C). In addition, 2DG (inhibitor of glycolysis) administration in Atg7-knockdown HCT-116, HeLa, and MEF cells consistently rescued the enhancement of EMT observed in these cells (Figure 4D). As shown in Figure 4E, 4F, Atg7 knockdown promotes HCT-116 cell migration, while 2DG administration revered this effect. These results demonstrate that inhibition of Atg7 promotes EMT through PKM2 mediated Warburg effect.
Atg7 inhibition promotes epithelial-mesenchymal transition through enhanced Warburg effect. Mutational analysis revealed that substitution of Y105 to E105 results in a significant increase glucose consumption(A), lactate production(B) in Atg7 knockdown cells. P values were calculated by One Way ANOVA. *P<0.005, **P<0.001. (C) Western blot analysis of EMT markers in knockdown Atg7 and PKM2. (D) 2DG (inhibitor of glycolysis) administration in Atg7-knockdown cell, Western blot analysis of EMT markers in HCT-116, HeLa and MEF cells. (E) Transwell assay in HCT-116 2DG administration. Original magnification, ×200. Scale bar represents 100 μm. (F) Statistical analysis result of transwell assay in (E). P values were calculated by One Way ANOVA. *P<0.005, **P<0.001.
Discussion
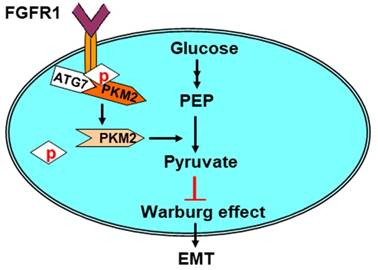
Our key findings described here are summarized in a model presented in Figure 5. Here, we revealed that Atg7 interacts with PKM2, the protein responsible for the Warburg effect during tumorigenesis. Atg7 inhibits the phosphorylation of PKM2 Tyr105 by blocking the binding of PKM2 with its upstream kinase, FGFR1. This in turn inhibits the Warburg effect and reduces EMT.
Model for Atg7 inhibiting Warburg effect by suppressing PKM2 phosphorylation resulting reduced EMT. Atg7 interacts with PKM2 directly and inhibits PKM2 phosphorylation. PKM2 dephosphorylation inhibits the Warburg effect, thereby inhibiting EMT. See text for details.
Metabolic reprogramming is one of the critical hallmarks of cancer cells[1-3]. Cancer cells metabolize glucose via glycolysis to lactate even in the presence of oxygen, a process known as the Warburg effect[5]. This metabolic switch can provide cancer cells with growth and invasion advantages through the biosynthesis of materials needed cells to thrive in an acidic tumor microenvironment[24]. Understanding the regulation and molecular mechanism underlying the Warburg effect could help us find a new way for cancer therapy.
Autophagy is an evolutionarily conserved process for lysosomal degradation of cytoplasmic constituents. It can modulate cellular energy metabolism and self-regulate to maintain intracellular homeostasis and cell development[8, 9].
Recently, it was reported that autophagy might be implicated in the Warburg effect. It has been shown that autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia[25]. Other studies have suggested that epithelial cancer cells may induce the Warburg effect in neighboring stromal fibroblasts in which autophagy was activated[26]. The autophagy related protein Atg7 is key component of autophagy as silencing Atg7 expression reduces or even blocks autophagy, while enhanced levels of Atg7 expression were sufficient to induce autophagy[27]. Recently, it has been reported that Atg7 is tightly associated with the Warburg effect, but the role of Atg7 remains controversial. Some studies have shown that monoallelic loss of Atg7 in a mouse AML model was sufficient to increase glycolytic metabolism[25], and that Atg7-/- MEFs exhibited a reduction in basal oxygen consumption and an increase in ECAR[21]. However, other work has found that shATG7-expressing K562 cells are less glycolytic compared with control cells[28]. In addition, paclitaxel-resistant cervical cancer cells with activation of autophagy genes, including Atg7, revealed a significant switch of cellular metabolism from oxidative phosphorylation to glycolysis[29]. However, most of these studies illustrate the relationship between autophagy and Warburg effects from a phenomenological perspective and lacks in-depth study at the molecular level.
Recent studies have demonstrated that PKM2 is highly expressed in cancer cells, where phosphorylation of PKM2 Tyr105 hinders the formation of tetramer and subsequently inhibits PKM2 activity and promotes the Warburg effect[22]. In this study, we identified the molecular connection between Atg7 and the Warburg effect. We found that Atg7 directly interacts with PKM2, a key regulator of the Warburg effect. We then confirmed the influence of Atg7 on the Warburg effect. Cells overexpressing Atg7 had a significantly decreased level of glucose consumption and lactate production, accompanied by a decreased ECAR. This results from the inhibition of the phosphorylation (Tyr-105) and subsequent activation of PKM2 in these cells, as addition of constitutively active PKM2 could rescue this effect caused by Atg7 overexpression. These results showed that Atg7 inhibits the Warburg effect by inhibiting PKM2 phosphorylation(Tyr-105). In accordance with this, Knockdown of Atg7 in HeLa cell lines or knockout of Atg7 in MEF cells increased glucose consumption, lactate production, and ECAR. In Atg7-/- mice tissues, the phosphorylation of PKM2 (Tyr-105) was significantly elevated in many important organs, including brain, liver, kidney and lung. This result was confirmed in Atg7 overexpression, knockdown or knockout cells which indicated that Atg7 inhibited the the Warburg effect was a consequence of PKM2 dephosphorylation.
The phosphorylation of PKM2(Tyr105) is directly regulated by the kinase FGFR1[22]. Therefore, we checked the effect of Atg7 on the binding of FGFR1 to PKM2. We found that Atg7 blocked the interaction between FGFR1 and PKM2 and that knockdown of Atg7 enhanced the interaction between FGFR1 and PKM2. The enhancement of this interaction seen with Atg7 knockdown was similar to that seen after bFGF treatment. Taken together, these studies show that Atg7 inhibites PKM2 phosphorylation by blocking the interaction between FGFR1and PKM2. In further support of this, we found that Atg7 can interact with FGFR1 (Figure S1E) and PTP1B (dephosphorylates PKM2) (Figure S1F). It has been reported that ROS promotes the phosphorylation of PKM2[30] and that defects in autophagy increases intracellular ROS levels[31]. We applied NAC (ROS scavenging agent) to both Atg7 knockdown cells and controls cells to access the influence of ROS, and found that NAC partly influenced the phosphorylation of PKM2 (Figure SG, H).
Epithelial to mesenchymal transition (EMT) is a complex phenomenon by which epithelial cells lose their polarity and cell-cell connections, and gain mesenchymal features such as of movement and invasion[32]. This allows them to detach from their primary site and invade surrounding tissues and blood vessels. EMT plays a fundamental role in embryonic development and tissue repair[33], as well as in tumor progression and metastasis[34]. Induction of autophagy inhibits migration and invasion through degrading EMT transcriptional factors, whereas Knockdown of ATG5 or ATG7 stimulates the expression of the EMT regulators Snail and Slug, and promotes cell invasion[35].
Here we suggest for the first time that the inhibition of Atg7 promotes EMT through PKM2 mediated Warburg effect. Atg7 knockdown cells demonstrated activated EMT, as indicated by an increase in N-cadherin expression. Both Knockdown of PKM2 and addition of a glycolysis inhibitor 2DG could rescue this phenotype, which suggests that the enhancement of EMT seen in these Atg-knockdown cells is dependent on the Warburg effect. Our findings provide a novel insight into the molecular relationships between Atg7 and Warburg effect, which might offer novel therapeutic targets in future.
Conclusion
In summary, our study suggests that Atg7 inhibits PKM2 Tyr105 phosphorylation and suppresses the Warburg effect by blocking the binding of PKM2 with its upstream kinase, FGFR1. Inhibition of Atg7 promotes epithelial-mesenchymal transition (EMT) through enhancing the Warburg effect.
Supplementary Material
Supplementary figure S1.
Acknowledgements
We are grateful to M. Komatsu (Japan) for providing the Atg+/- mice. This work was supported by National Key R&D Program of China (2016YFC1302400). The Ministry of education innovation team development plan to Liu Cao (IRT_17R107&IRT13101). Key project of the National Natural Science Foundation to Liu Cao (81130042, 31171323, 81770001, 2015225003) National Science Foundation of China to Xiaoyu Song (31300963, LFWK201725), Qiqiang Guo (81502400), Yi Guan (81502447) and Fei Yi (81502414). Liaoning education department general project to Hongde Xu (LK201627).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pinheiro C, Garcia EA, Morais-Santos F, Moreira MA, Almeida FM, Jube LF. et al. Reprogramming energy metabolism and inducing angiogenesis: co-expression of monocarboxylate transporters with VEGF family members in cervical adenocarcinomas. BMC cancer. 2015;15:835
2. Saeidi N, Meoli L, Nestoridi E, Gupta NK, Kvas S, Kucharczyk J. et al. Reprogramming of intestinal glucose metabolism and glycemic control in rats after gastric bypass. Science. 2013;341:406-10
3. Soga T. Cancer metabolism: key players in metabolic reprogramming. Cancer science. 2013;104:275-81
4. Ciccarone F, Vegliante R, Di Leo L, Ciriolo MR. The TCA cycle as a bridge between oncometabolism and DNA transactions in cancer. Seminars in cancer biology. 2017;47:50-6
5. Warburg O. On the origin of cancer cells. Science. 1956;123:309-14
6. Dell' Antone P. Energy metabolism in cancer cells: how to explain the Warburg and Crabtree effects? Med Hypotheses. 2012;79:388-92
7. Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol. 2009;92:329-33
8. Kim KH, Lee MS. Autophagy-a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10:322-37
9. Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells. 2013;36:7-16
10. Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G. et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338:956-9
11. Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745-7
12. Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta. 2003;1603:113-28
13. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891-906
14. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY. et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062-75
15. Liu H, He Z, Simon HU. Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy. 2014;10:372-3
16. Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155:1216-9
17. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H. et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717-29
18. Tanida I, Mizushima N, Kiyooka M, Ohsumi M, Ueno T, Ohsumi Y. et al. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol Biol Cell. 1999;10:1367-79
19. Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol. 2004;15:231-6
20. Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N. et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336:225-8
21. Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM. et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY). 2009;1:425-37
22. Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K. et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73
23. Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE. et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374-9
24. Carmona-Fontaine C, Bucci V, Akkari L, Deforet M, Joyce JA, Xavier JB. Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. Proc Natl Acad Sci U S A. 2013;110:19402-7
25. Watson AS, Riffelmacher T, Stranks A, Williams O, De Boer J, Cain K. et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015:1
26. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG. et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984-4001
27. Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109:151-60
28. Karvela M, Baquero P, Kuntz EM, Mukhopadhyay A, Mitchell R, Allan EK. et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy. 2016;12:936-48
29. Peng X, Gong F, Chen Y, Jiang Y, Liu J, Yu M. et al. Autophagy promotes paclitaxel resistance of cervical cancer cells: involvement of Warburg effect activated hypoxia-induced factor 1-alpha-mediated signaling. Cell Death Dis. 2014;5:e1367
30. Guo D, Gu J, Jiang H, Ahmed A, Zhang Z, Gu Y. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to the development of pulmonary arterial hypertension. J Mol Cell Cardiol. 2016;91:179-87
31. Zhao Z, Zhao J, Xue J, Zhao X, Liu P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am J Cancer Res. 2016;6:2162-77
32. Banyard J, Bielenberg DR. The role of EMT and MET in cancer dissemination. Connect Tissue Res. 2015;56:403-13
33. Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A. et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell. 2013;24:466-80
34. Grant CM, Kyprianou N. Epithelial mesenchymal transition (EMT) in prostate growth and tumor progression. Transl Androl Urol. 2013;2:202-11
35. Grassi G, Di Caprio G, Santangelo L, Fimia GM, Cozzolino AM, Komatsu M. et al. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis. 2015;6:e1880
Author contact
![]() Corresponding author: lcaoedu.cn
Corresponding author: lcaoedu.cn