Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(7):1671-1681. doi:10.7150/ijbs.57964 This issue Cite
Research Paper
MUC3A induces PD-L1 and reduces tyrosine kinase inhibitors effects in EGFR-mutant non-small cell lung cancer
Yuan Luo1*, Shijing Ma1,2*, Yingming Sun1*, Shan Peng1, Zihang Zeng1, Linzhi Han1, Shuying Li1, Wenjie Sun1, Jieyu Xu1, Xiaoli Tian1, Feng Wang1, Qiuji Wu1, Yu Xiao3, Junhong Zhang1,4,5, Yan Gong3 ![]() , Conghua Xie1,4,5
, Conghua Xie1,4,5 ![]()
1. Department of Radiation and Medical Oncology, Zhongnan Hospital of Wuhan University, Wuhan, China.
2. Department of Geriatrics, Renmin Hospital of Wuhan University, Wuhan, China.
3. Department of Biological Repositories, Zhongnan Hospital of Wuhan University, Wuhan, China.
4. Hubei Key Laboratory of Tumor Biological Behaviors, Zhongnan Hospital of Wuhan University, Wuhan, China.
5. Hubei Cancer Clinical Study Center, Zhongnan Hospital of Wuhan University, Wuhan, China.
* These authors contributed equally to this work.
Received 2021-1-8; Accepted 2021-3-30; Published 2021-4-12
Abstract

The immune checkpoint ligand programmed death-ligand 1 (PD-L1) and the transmembrane mucin (MUC) 3A are upregulated in non-small cell lung cancer (NSCLC), contributing to the aggressive pathogenesis and poor prognosis. Here, we report that knocking down the oncogenic MUC3A suppresses the PD-L1 expression in NSCLC cells. MUC3A is a potent regulator of epidermal growth factor receptor (EGFR) stability, and MUC3A deficiency downregulates the activation of the PI3K/Akt and MAPK pathways, which subsequently reduces the expression of PD-L1. Furthermore, knockdown of MUC3A and tyrosine kinase inhibitors (TKIs) in EGFR-mutant NSCLC cells play a synergistic effect on inhibited proliferation and promoted apoptosis in vitro. In the BALB/c nude mice xenograft model, MUC3A deficiency enhances EGFR-mutated NSCLC sensitivity to TKIs. Our study shows that transmembrane mucin MUC3A induces PD-L1, thereby promoting immune escape in NSCLC, while downregulation of MUC3A enhances TKIs effects in EGFR-mutant NSCLC. These findings offer insights into the design of novel combination treatment for NSCLC.
Keywords: MUC3A, non-small cell lung cancer, EGFR, PD-L1.
Introduction
Lung cancer remains the leading cause of cancer-related mortality worldwide. Non-small cell lung cancer (NSCLC) accounts for approximately 85% lung cancer cases. Therapeutic antibodies blocking programmed death-ligand 1 (PD-L1) brought striking regression of NSCLC. However, only 20% patients responded to the anti-PD-L1 therapy [1]. The expression levels of PD-L1 on the surface of tumor cells were positively correlated with the induction of the inflammatory cytokine interferon-γ [2]. Previous studies suggested that stable expression of mutated epidermal growth factor receptor (EGFR) in immortalized bronchial epithelial cells promoted PD-L1 expression, and that EGFR tyrosine kinase inhibitors (TKIs) inhibited PD-L1 expression in EGFR-mutant NSCLC cells [3]. In addition, epidermal growth factor (EGF) was reported to induce PD-L1 expression through the IL‐6/JAK/STAT3 signaling pathway in EGFR-mutant NSCLC cells [4].
The mucin (MUC) family proteins contain tandem repeat structures, in which the proportions of proline, threonine, and serine are high. The human MUC family consists of members from MUC1 to MUC21, which are sub-classified into the secreted (such as MUC2, MUC5AC, MUC5B, and MUC6) and transmembrane forms (such as MUC1, MUC4, MUC3, and MUC16) [5]. MUCs are large glycoproteins and actively participate in tumor proliferation and metastasis [6]. MUC1 was reported to be upregulated in triple negative breast cancers (TNBCs) [7]. It induced PD-L1 transcription through MYC and NF-κB/p65 pathways in basal B TNBC cells. Targeting MUC1 C-terminal with genetic and pharmacologic approaches suppressed PD-L1 expression [8]. Moreover, MUC4 was highly glycosylated, and the expression of EGFR was modulated by MUC4 [9].
MUC3A is also highly glycosylated. It contains a sperm protein, enterokinase, agrin (SEA) domain, and an EGF domain in its extracellular segment [10]. MUC3A is highly expressed in NSCLC cells and rarely expressed in normal pulmonary epithelial cells, making it a promising tumor biomarker for lung cancer [11]. In gastric, pancreatic, breast, colorectal, renal, and prostate cancers, high expression of MUC3A is an independent factor for poor prognosis [12-17].
MUC family members regulate PD-L1 expression [18], which is associated with the prognosis of immunotherapy for lung cancer. MUC proteins also modulate EGFR [19], whose mutation is popular in NSCLC. EGFR was reported to modulate PD-L1 expression via the PI3K/Akt and MAPK pathway [20]. Therefore, we speculate that MUC3A, containing the EGF domain, may induce PD-L1 expression through PI3K/Akt and MAPK pathway. In the present study, we demonstrated that MUC3A could increase EGFR stability and reduce the effects of TKIs on NSCLC cells. MUC3A was positively correlated with PD-L1 in NSCLC, and the increased MUC3A and PD-L1 levels both indicated poor prognosis of NSCLC patients. Knockdown of MUC3A decreased EGF-induced PD-L1 in EGFR-mutated NSCLC via blocking PI3K/Akt and MAPK pathways. MUC3A deficiency also enhanced TKIs-induced proliferation inhibition and apoptosis promotion in NSCLC cells in vitro and in vivo. Our studies indicated that MUC3A might be a potential target in lung cancer treatment strategies.
Materials and Methods
Tissue microarray and bioinformatics analysis
The NSCLC tissue microarray was purchased from Outdo (Shanghai, China), including 92 lung adenocarcinoma (LUAD) and their paired para-carcinoma tissues. The samples come from National Human Genetic Resources Sharing Service Platform (2005DKA21300). Both the intensity and positive percentages of immunohistochemistry (IHC) were used to examine the MUC3A and PD-L1 expression: the IHC H-score (values 0-400) = the scores for intensity of positive staining (less than 5% scored “0”; 5-24% scored “1”; 25-49% scored “2”; 50-74% scored “3”; and more than 74% scored “4”) × the percentage of positive-stained cells × 100 [21]. In the cancer tissues of all the 92 cases, the median MUC3A H-score was 140.
Animals
Five-week-old female BALB/c nude mice were purchased from Charles River Laboratory Animal Technology Co., Ltd, Beijing, China. Mice were housed and handled according to the guidelines of Wuhan University Animal Care Facility and National Institutes of Health. The H1975 cells (5 × 106 cells/mouse) were subcutaneously injected into the right armpit. The mice were randomized into 4 groups: control, MUC3A deficiency, AZD-9291 (5 mg/1 kg/day), and MUC3A deficiency with AZD-9291. The sizes of the subcutaneous tumors were recorded every day. Tumor volume (V) was calculated using the formula: V = π/6 × (major axis) × (minor axis)2.
Cells
Human NSCLC cells (H1975, H1299 and PC9), large cell lung cancer cells (H460), lung mucosal epithelial cells (H292), normal lung epithelial cells (BEAS-2B) were purchased from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. Lung cancer cells were maintained in RPMI-1640 medium, and BEAS-2B cells were maintained in DMEM medium. Mediums were supplied with 10% fetal bovine serum, 100 mg/mL streptomycin and 100 units/mL penicillin. Cells were cultured in a 37 °C incubator (Sanyo, Japan) with 5% CO2. All cells passed the short tandem repeat (STR) analysis of Guangzhou Cellcook Biotech Co., Ltd, China.
Cell viability assay
Cells were seeded into 96-well plate and treated with AZD-9291 and Gefitinib (MCE Ltd., China) at different doses for 48 h. Cell viability was detected by the CCK-8 kit (Dojindo Ltd., Japan) according to the manufacturer's instructions. The optical density was measured at 450 nm through a microplate reader (Rayto Ltd., China).
Flow cytometry
Cells were incubated with Gefitinib (10 μM) and AZD-9291 (0.1 μM) for 48 h. Suspended and adherent cells were both collected. Cell apoptosis was detected by Annexin VFITC/PI Apoptosis Kit (BestBio Ltd., China) according to the manufacturer's instructions and analyzed by flow cytometry (FACS Aria III, BD, USA).
To detect cell membrane expression of PD-L1, cells were digested and incubated in 100 μL PBS containing 2% goat serum at room temperature for 20 min. After washed with PBS for 3 times, cells were incubated with the anti-PD-L1 antibody (listed in Supplemental Table S1) in the dark at 4°C for 30 min. After washing with PBS, the stained cells were resuspended in 400 μL PBS. The samples were then analyzed by flow cytometry (FACS Aria III, BD, USA).
RNA isolation, RT-PCR and qRT-PCR
TRIzol (Sangon Ltd., China) was used to extract total RNA. RNA concentration was detected by a Nanodrop spectrophotometer (Thermo Scientific Ltd., USA). Total RNA (1 μg) was used for the synthesis of first-strand cDNA and reverse transcription reactions were conducted using HiScript® II Q RT SuperMix for qPCR kit (Vazyme Ltd., China). The following primers were used: PD-L1 (forward 5'- GCTGCACTAATTGTCTATTGGGA -3' and reverse 5'- AATTCGCTTGTAGTCGGCACC -3'); GAPDH (forward 5'- GGAGCGAGATCCCTCCAAAAT -3' and reverse 5'- GGCTGTTGTCATACTTCTCATGG -3'). The qRT-PCR reactions were performed using a CFX96 qRT-PCR system (Applied Biosystems Ltd., USA). We used the 2-ΔΔCT method to calculate the fold changes. Data were normalized to GAPDH levels.
Immunoblotting
RIPA lysis buffer (Beyotime Ltd., China) containing protease and phosphatase inhibitor mixture (Sigma Chemical Ltd., USA) was used to extract the whole cells lysis on ice for 30 min. The supernatant was collected after the cell lysates centrifugation at 13,000 g for 20 min at 4 °C. The protein concentration was determined by BCA assay (Beyotime Ltd., China). Protein samples were separated by 7.5-12.5% SDS-PAGE, and the proteins were then transferred to polyvinylidene fluoride membranes. Non-fat milk (5%) was used to block non-specific binding sites. Primary and secondary antibodies used for detection were listed in Supplemental Table S1. Then, the specific bands were visualized with an enhanced chemiluminescence kit (Bio-Rad Ltd., USA) and exposed to the ChemiDoc XRS + system (Bio-Rad Ltd., USA). The Image J program was used to quantify the protein levels.
Histology and IHC
The tumor tissues were fixed with 10% formalin and embedded in paraffin. Tumor tissue sections were used for hematoxylin and eosin (H&E) staining and IHC. IHC was used to detect MUC3A and PD-L1 expression in tumor tissues. Anti-MUC3A antibody and Anti-PD-L1 antibody were listed in Supplemental Table S1.
Statistical analysis
Each experiment was performed for at least 3 times, and data were presented with a representation of at least 3 individual experiments. A two-tailed Student's t-test and one-way analysis of variance (ANOVA) were used to evaluate the statistical significance of different groups. Data were analysed with GraphPad Prism. P values < 0.05 were considered as statistical significance.
Results
MUC3A expression was positively correlated with PD-L1 expression in NSCLC
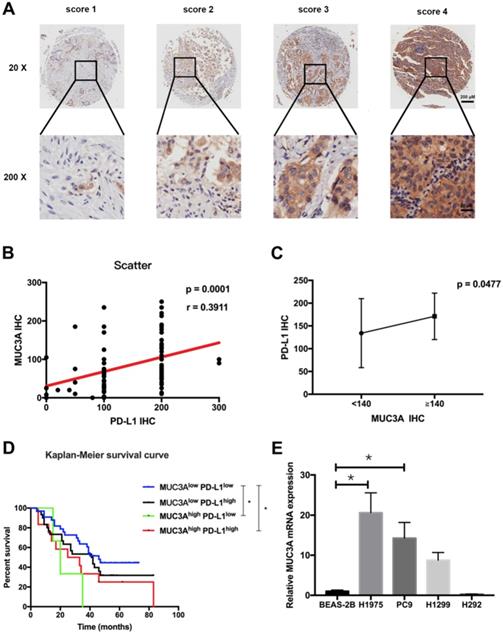
Tissue microarray containing 92 LUAD and paired para-carcinoma tissues with scoring system (Fig. 1A) was used to confirm the increased MUC3A and PD-L1 levels in LUAD tissues compared with normal lung tissues (Fig. S1). The expression levels of MUC3A and PD-L1 were positively correlated (Fig. 1B). Adenocarcinoma with higher levels of MUC3A (IHC > 140) also had higher expression of PD-L1 than the lower ones (Fig. 1C, p < 0.05). Different clinicopathological features of LUAD cases stratified by PD-L1 expression levels were compared, and PD-L1 levels were associated with primary and adjacent carcinoma (p < 0.0001, Table S2). Moreover, LUAD patients were sub-grouped into 4 groups by their PD-L1 and MUC3A expression levels, and the increased MUC3A and PD-L1 levels were correlated with increased lymph node metastasis (p < 0.05, Table 1). Higher expression of both MUC3A and PD-L1 was associated with poorer prognosis (p < 0.05, Kaplan-Meier test, Fig. 1D). To further verify MUC3A expression levels in NSCLC and normal lung epithelial cells, the mRNA levels of MUC3A were measured. MUC3A was highly expressed in LUAD cell lines H1975 and PC9 compared with H1299, and barely expressed in lung mucosal epithelial cells (H292) and normal lung epithelial cells (BEAS-2B) (Fig. 1E). These results suggested that MUC3A expression was upregulated in NSCLC cells and positively correlated with PD-L1 expression.
MUC3A was positively correlated with PD-L1 expression. (A) Representative IHC images of PD-L1 expression. Scoring was measured by the percentage of positive cells with the following staining intensities: less than 5% scored “0”; 5-24% scored “1”; 25-49% scored “2”; 50-74% scored “3”; and more than 74% scored “4”. (B) and (C) The level of MUC3A was positively correlated with PD-L1 expression. (D) High levels of MUC3A and PD-L1 were related to poor clinical outcomes. (E) Relative MUC3A mRNA expression in BEAS-2B, H1975, PC9, H1299 and H292. *, p < 0.05.
Correlation between MUC3A and PD-L1 levels in NSCLC patients and their clinicopathologic characteristics.
| Clinical pathology | MUC3AlowPD-L1low | MUC3Ahigh PD-L1low | MUC3Alow PD-L1high | MUC3Ahigh PD-L1high | N | p value |
|---|---|---|---|---|---|---|
| Gender | ||||||
| Male | 21 | 3 | 17 | 10 | 51 | p=0.1552 |
| Female | 18 | 2 | 17 | 4 | 41 | |
| Age | ||||||
| ≤ 60 | 20 | 0 | 17 | 1 | 38 | p=0.2710 |
| > 60 | 19 | 5 | 17 | 13 | 54 | |
| Tumor size (cm) | ||||||
| < 4 | 19 | 0 | 23 | 3 | 45 | p=0.5735 |
| ≥ 4 | 16 | 3 | 8 | 7 | 34 | |
| None | 4 | 2 | 3 | 4 | 13 | |
| Histological grade | ||||||
| I/I-II | 4 | 0 | 1 | 2 | 7 | p=0.0577 |
| II | 24 | 2 | 19 | 5 | 50 | |
| II-III/III | 9 | 2 | 13 | 7 | 31 | |
| I-III | 2 | 1 | 1 | 0 | 4 | |
| Clinical Stage | ||||||
| I | 11 | 1 | 9 | 4 | 25 | p=0.0917 |
| II | 10 | 1 | 7 | 3 | 21 | |
| III-IV | 10 | 1 | 8 | 2 | 21 | |
| Non | 8 | 2 | 10 | 5 | 25 | |
| Lymph node status | ||||||
| < 4 | 11 | 0 | 12 | 2 | 25 | p < 0.05 |
| ≥ 4 | 27 | 4 | 21 | 8 | 60 | |
| Non | 1 | 1 | 1 | 4 | 7 | |
| Carcinoma | ||||||
| Primary | 39 | 5 | 34 | 14 | 92 | p < 0.05 |
| Adjacent | 86 | 2 | 0 | 0 | 88 |
P value represents the probability from ANOVA for tissue PD-L1 and MUC3A levels between variable subgroups.
Knockdown of MUC3A inhibits EGF-induced PD-L1 expression in EGFR-mutant NSCLC cells
EGF was reported to activate EGFR and then induce PD-L1 expression [22]. To investigate whether EGFR mutations have effects on PD-L1 expression, we searched TCGA database and found that the mRNA levels of PD-L1 were independent of those of EGFR (Fig. 2A), but correlated with EGFR mutations (Fig. 2B). Flow cytometry was used to detect the expression of PD-L1 on the cell membrane surface. EGF with increasing gradient (25, 50, and 75 ng/mL) induced PD-L1 in H1975 cells in a dose-independent way (Fig. 2C). Furthermore, EGF (50 ng/mL) induced the expression of membrane PD-L1 in H1975 and PC9 cells, but not in H1299 and H460 cells (Fig. 2D). The reason should be that H1975 and PC9 are EGFR-mutated cells, while H1299 and H460 are EGFR wild-type cells [4].
MUC3A induced PD-L1 in EGFR-mutant NSCLC cell lines. (A) The mRNA levels of PD-L1 were independent of the mRNA levels of EGFR. (B) The mRNA levels of PD-L1 were correlated with EGFR mutation. Data of lung cancer were downloaded from the TCGA database. 544 non-mutation, 32 mutation. (C, D) The PD-L1 mRNA levels were determined by real-time PCR analysis after EGF stimulation for 24 hours. (E, F) The PD-L1 protein levels were determined by flow cytometry analysis after EGF stimulation for 24 hours. (G) The PD-L1 mRNA levels were determined by real-time PCR analysis after EGF stimulation for 24 hours. EGF, 50 ng/mL. *, p < 0.05; **, p < 0.01; ns: not significant. NC: negative control.
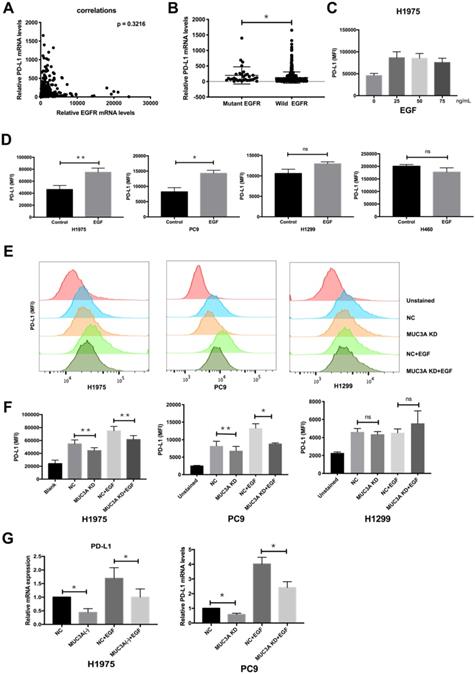
The relationship between MUC3A and EGF-induced PD-L1 in mutated and wild type EGFR cells was then investigated. ShRNAs targeting MUC3A were delivered into the H1975, PC9 and H1299 cells by lentiviruses, and the knockdown efficiency in the stable cell lines was confirmed by immunoblotting (Fig. S2). Flow cytometry results indicated that MUC3A deficiency inhibited EGF-induced PD-L1 expression on the cell membrane surface of H1975 and PC9 cells, but not H1299 cells (Fig. 2E-F). The results of qPCR were consistent. MUC3A deficiency decreased EGF-induced PD-L1 mRNA levels in H1975 and PC9 cells (Fig. 2G). These results indicated that knockdown of MUC3A inhibited EGF-induced PD-L1 expression in EGFR-mutant NSCLC cells, but not in EGFR-wild type cells, suggesting that MUC3A functioned upstream of EGFR.
MUC3A deficiency suppressed PD-L1 expression via blocking PI3K/Akt and MAPK pathways
Activation of PI3K/Akt and MAPK was reported to be associated with upregulated PD-L1 expression in NSCLC cells [20]. One hour after EGF stimulation, reduced PI3K/Akt and MAPK activation was detected in the MUC3A-deficient H1975 and PC9 cells (Fig. 3A-C), which might result from the decreased protein levels of EGFR caused by MUC3A knockdown (Fig. 3A). However, the mRNA levels of EGFR were not affected by the lack of MUC3A (Fig. 3D), suggesting that knockdown of MUC3A downregulated EGFR via reducing its protein translation or stability, instead of mRNA transcription.
MUC3A promoted the activity of PI3K/Akt and MAPK pathway. (A) and (B) MUC3A promoted the activity of PI3K/Akt and MAPK pathway, and knockdown of MUC3A decreased EGFR protein levels. Representative immunoblotting of total and phosphorylated EGFR, AKT, MEK and ERK in H1975 and PC9 cells. (C) Quantifications of phosphorylated AKT, MEK and ERK. (D) MUC3A knockdown had no effect on EGFR mRNA levels, but decreased EGFR protein levels. (E) Immunoblotting of PI3K/Akt and MAPK pathway proteins with MEK and PI3K inhibitor treatment for 1 hour. (F) Flow cytometry of PD-L1 expression on the surface of H1975 cells treated with EGF, MEK and PI3K inhibitor for 24 hours. (G) Immunoblotting of PI3K/Akt and MAPK pathway proteins with AKT and ERK activator treatment for 1 hour. (H) Flow cytometry of PD-L1 expression on the surface of H1975 and PC9 cells treated with SC79 or honokiol for 24 hours. Abbreviations are as follows: MFI, mean fluorescence intensity; EGF, 50 ng/mL; MEK inhibitor GSK1120212, 25 nM; PI3K inhibitor GDC-0941, 500 nM; AKT activator SC79, 5 μg/mL; ERK activator honokiol, 20 μM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns: not significant. NC: negative control.
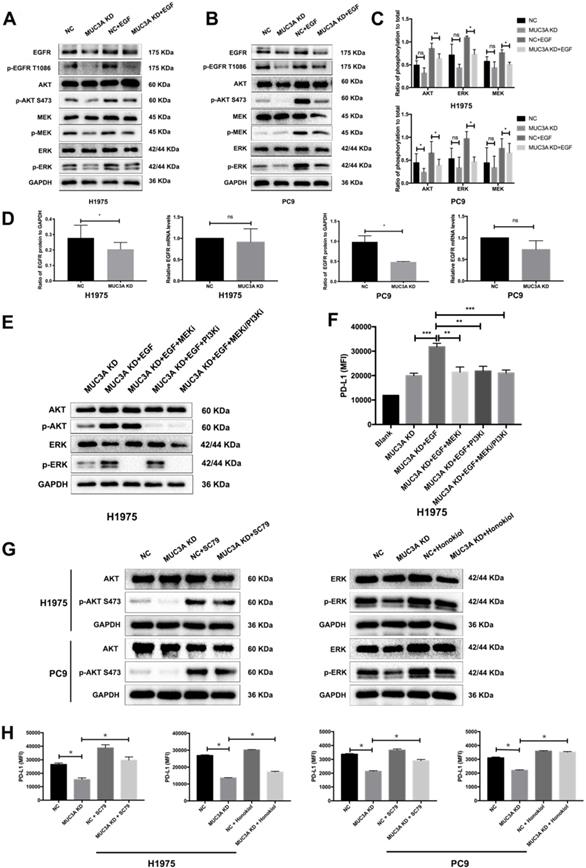
To confirm the involvement of PI3K/Akt and MAPK pathways in MUC3A-mediated PD-L1 modulation, specific inhibitors of MEK, GSK1120212 (trametinib), and pan type I PI3Ks, GDC-0941 (pictilisib), were used to treat NSCLC cells (Fig. 3E). MEK and PI3K inhibitors blocked EGF-induced PD-L1 expression in MUC3A-deficient H1975 cells (Fig. 3F). Activators of AKT, SC79, and activators of ERK, honokiol, were used to treat NSCLC cells (Fig. 3G). It was observed that both the activators could restore Akt and ERK activation attenuated by MUC3A, and restore the expression of PD-L1 (Fig. 3H). These results indicated that MUC3A knockdown inhibited PD-L1 expression induced by EGF through PI3K/Akt and MAPK pathway.
Knockdown of MUC3A improved NSCLC sensitivity to TKIs in vitro
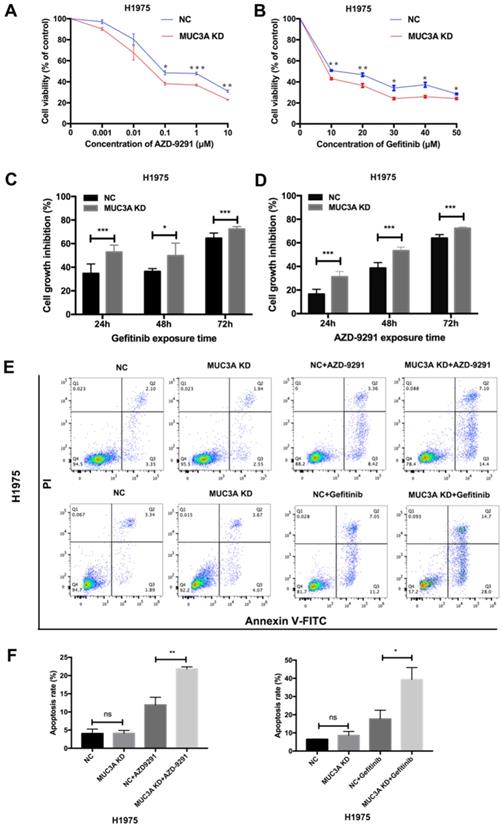
MUC3A deficiency reduced the levels of EGFR proteins, we next explored whether MUC3A affected the in vitro drug sensitivity of EGFR-mutant cells to TKIs. Two TKIs were selected, AZD-9291 (sensitive to H1975) and gefitinib (resistant to H1975). The results of cell viability assay indicated that MUC3A deficiency increased the inhibitory effects of TKIs on H1975 cell growth in a dose-dependent manner (Fig. 4A-B). TKIs also markedly inhibited the cell growth of MUC3A-deficient H1975 cells in a time-dependent manner (Fig. 4C-D). Moreover, knockdown of MUC3A increased TKIs-induced cell apoptosis in H1975 cells (Fig. 4E-F). These results suggested that MUC3A deficiency promoted TKIs sensitivity in NSCLC cells in vitro.
MUC3A deficiency improved NSCLC sensitivity to TKIs in vitro. (A) and (B) MUC3A deficiency potentiated gefitinib and AZD-9291-induced growth inhibition in H1975 cells. (C) and (D) Cell growth inhibition after exposed to AZD-9291 (0.1 μM) and Gefitinib (10 μM) for 24, 48 and 72 hours. (E) and (F) MUC3A knockdown increases NSCLC cell apoptosis induced by Gefitinib (10 μM) and AZD-9291 (0.1 μM). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns: not significant. NC: negative control.
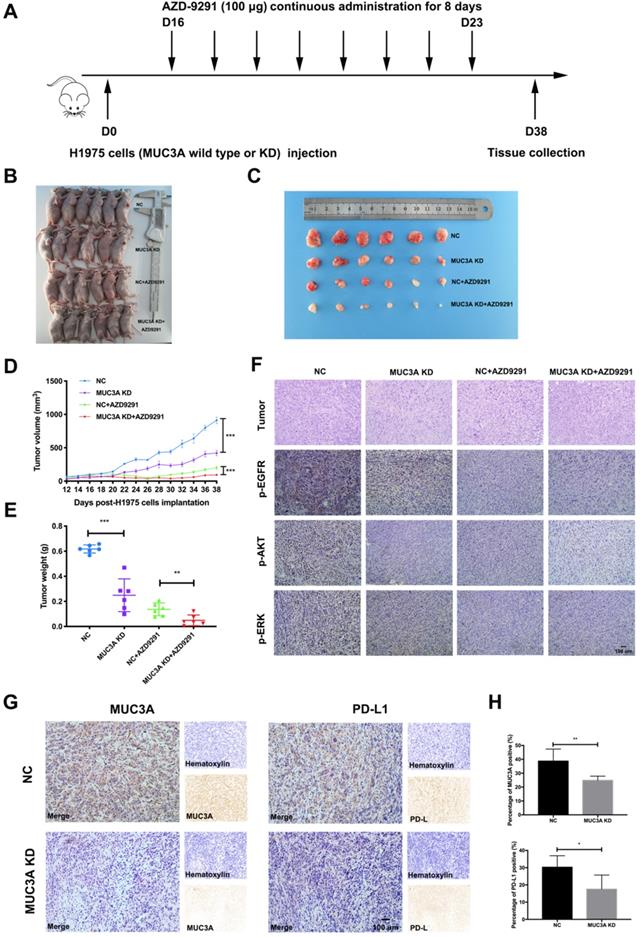
MUC3A deficiency improved NSCLC sensitivity to TKIs in vivo
To investigate the effects of MUC3A on NSCLC cell sensitivity to TKIs in vivo, MUC3A-deficient or wild-type H1975 cells were subcutaneously implanted into the BALB/c nude mice, followed by AZD-9291 treatment (100 μg/mouse/day for 8 days, Fig. 5A). MUC3A deficiency significantly suppressed tumor volume, and the combination with AZD-9291 exerted synergistic effect on the reduction of tumor burden (Fig. 5B-E). H&E staining was performed to confirm the tumor tissues (Fig. 5F). Moreover, p-EGFR, p-AKT and p-ERK were decreased in the MUC3A deficient cells after treatment with AZD-9291. IHC of the tumor sections was used to confirm MUC3A knockdown by shRNA lentiviruses, and the xenografts from the mice injected with MUC3A-deficient cells presented significantly less MUC3A and PD-L1 (Fig. 5G-H). These results suggested that MUC3A deficiency impaired tumor growth and promoted sensitivity to TKIs in vivo.
MUC3A deficiency improved NSCLC sensitivity to TKIs in vivo. (A) The overall scheme of animal experiments. (B) Twenty-four nude mice were sacrificed on day 38. (C) Gross view of tumor. N = 6. (D) Growth curves of tumor volume indicate that the combination therapy significantly inhibits tumor growth in vivo compared with single treatment groups. N = 6. (E) Tumor weight. N = 6. (F) Representative H&E staining images of tumor, and representative IHC images of p-EGFR, p-AKT and p-ERK, scale bar: 100 μm. (G) and (H) Representative IHC images of MUC3A and PD-L1 in tumor tissues. The MUC3A deficiency group had less MUC3A and PD-L1 expression than that of control. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns: not significant. NC: negative control.
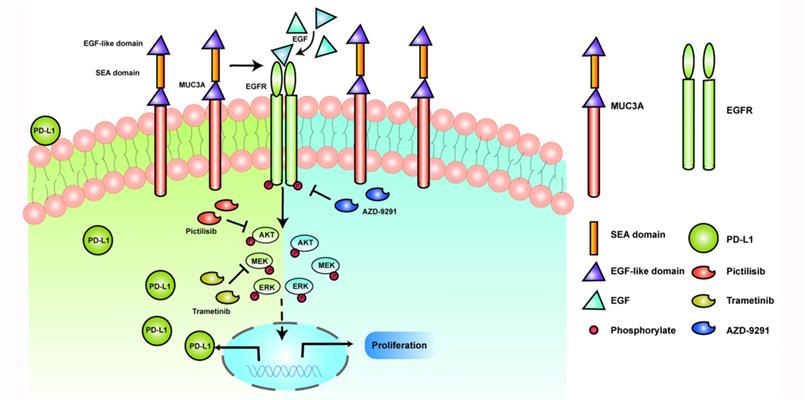
Discussion
MUCs have been identified as adverse prognosis markers and attractive therapeutic targets [5]. In our study, we identified that MUC3A induced PD-L1 and reduced TKIs effects in EGFR-mutant NSCLC (Fig. 6). MUC3A was reported to be highly expressed in human lung cancer [23, 24]. Our tissue microarray showed the same results that MUC3A was highly expressed in non-small cell lung cancer. Moreover, PD-L1 was also upregulated in NSCLC, and positively related with MUC3A. PD-L1 is one of the most important predictors for the efficacy of NSCLC anti-PD-L1 related-immunotherapy [25]. Numerous studies suggested that mutant-EGFR signaling induced PD-L1 expression [4, 22, 26, 27]. In our study, we revealed that MUC3A altered the levels of PD-L1 via affecting the stability of mutant EGFR protein.
Schematic model proposed for the role of MUC3A inducing PD-L1 and reducing TKI effects in EGFR-mutant NSCLC. MUC3A induced PD-L1 via PI3K/AKT and MAPK pathway. MEK and PI3K inhibitors (trametinib and pictilisib) blocked EGF-induced PD-L1 expression. TKIs (AZD-9291) inhibited the cell proliferation.
Transmembrane protein MUC3A contains an extracellular cysteine-rich domain with 2 EGF-like motifs [28]. Theoretical structure-function relationship analysis of the conserved domains indicated that all of the MUCs of this subfamily could interact with ErbB family member [29]. EGFR is a member of the ErbB family and its mutation is one of the most common drivers in NSCLC [30]. Compared with wild-type EGFR, EGFR mutation increases both mRNA and protein levels of PD-L1 [3, 31]. Based on the TCGA database, we found that the expression levels of PD-L1 in EGFR-mutant lung cancer were significantly increased compared with wild-type EGFR one. Previous studies demonstrated that MUC1C induced PD-L1 and immune evasion in TNBC [8]. MUC3A shares the same SEA domain with MUC1, suggesting that this domain is critical for its autoproteolysis to impact EGF-induced functions. Upon EGF stimulation, the PD-L1 expression of EGFR mutant cell lines H1975 and PC9 was significantly upregulated, while the PD-L1 expression in EGFR wild-type cell lines H1299 and H460 did not change significantly in our study. After MUC3A knockdown in EGFR-mutant and wild-type NSCLC cells, the expression of PD-L1 was reduced only in EGFR-mutant cells upon EGF stimulation. However, other studies showed that EGFR activation by EGF stimulation upregulated PD-L1 expression in EGFR wild-type BEAS-2B cells [22]. There may be several reasons for this discrepancy. One possible reason is the difference of PD-L1 expression baselines. Another reason may be the different protocols of EGF stimulation. In brief, our current results indicated that MUC3A induced PD-L1 in EGFR-mutant NSCLC cells.
Upregulation of PD-L1 was reported to be modulated by activation of MAPK [32, 33] and PI3K/Akt [32, 34, 35], as well as transcriptional factors HIF-1α [36], STAT3 [37] and NF-ĸB pathyway [38]. In our study, knockdown of MUC3A downregulated phosphorylation of MEK, ERK and AKT. Flow cytometry results showed that MEK and PI3K inhibition reduced the expression of membrane PD-L1. Our results suggested that MUC3A induced PD-L1 through MAPK and PI3K/Akt pathways.
MUC1-mediated protection against EGFR degradation can increase total cellular pools of EGFR over time [39, 40]. MUC4 was reported to modulate the expression of EGFR [9]. MUC3A has a domain structure similar to MUC4, with N-terminal tandem repeats, EGF-like sequence and a SEA module [5]. Our results demonstrated that MUC3A deficiency significantly decreased the protein levels of EGFR, instead of mRNA levels. These studies indicated that MUC3A affected the stability of the EGFR proteins.
Tumors with constitutively active EGFR mutations were reported in up to 40% cases of NSCLC in Asian populations [41]. TKIs targeting mutant EGFR reduce the cancer growth successfully, but acquired resistance inevitably occurs [42]. Degradation of endogenous mutant EGFR is a common mechanism for the most clinically relevant TKIs-sensitizing [43]. Our results showed here that MUC3A deficiency increased TKIs-induced NSCLC cells proliferation inhibition and apoptosis. Our studies further indicated that MUC3A reduced TKIs effects in EGFR-mutant NSCLC via increasing the stability of EGFR proteins. Moreover, in vivo studies collectively supported the conclusion that MUC3A deficiency increased TKIs drug sensitivity and downregulated PD-L1 expression.
Mutant EGFR induces PD-L1 via PI3K/Akt and MAPK pathway. We demonstrated that the transmembrane mucin MUC3A increased EGFR protein stability in EGFR-mutant cell lines. MUC3A can also reduce the effect of TKIs through EGFR modulation in NSCLC. Our studies provided rationale to target MUC3A combining with TKIs in EGFR-mutant lung cancers.
Abbreviations
NSCLC: non-small cell lung cancer; TKI: tyrosine kinase inhibitor; MUC: mucin; LUAD: lung adenocarcinoma; EGFR: epidermal growth factor receptor; EGF: epidermal growth factor; TNBCs: triple negative breast cancers; SEA: sperm protein, enterokinase, agrin; IHC: Immunohistochemistry; H&E: hematoxylin and eosin.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Availability of data and materials
The datasets used in current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was approved by the Medical Ethics Committee at Zhongnan Hospital of Wuhan University and the Institutional Animal Care and Use Committee of Wuhan University.
Funding
This study was supported by National Natural Science Foundation of China (81773236, 81800429 and 81972852), Key Research & Development Project of Hubei Province (2020BCA069), Nature Science Foundation of Hubei Province (2020CFB612), Health Commission of Hubei Province Medical Leading Talent Project, Young & Middle-Aged Medical Backbone Talents of Wuhan (WHQG201902), Application Foundation Frontier Project of Wuhan (2020020601012221), Zhongnan Hospital of Wuhan University Science, Technology and Innovation Seed Fund (znpy2019001, znpy2019048 and ZNJC201922), and Chinese Society of Clinical Oncology TopAlliance Tumor Immune Research Fund (Y-JS2019-036).
Author contributions
Yuan Luo, Yan Gong, and Conghua Xie conceived and designed the experiments. Yuan Luo, Shijing Ma, Yingming Sun, Shan Peng, Linzhi Han, Wenjie Sun, Shuying Li, Qiuji Wu, Jieyu Xu, Xiaoli Tian, Feng Wang, Yu Xiao and Junhong Zhang performed part of the experiments. Yuan Luo, Shijing Ma, Yingming Sun, Shan Peng and Zihang Zeng analyzed the data. Yuan Luo contributed to the writing-original draft. Yuan Luo, Yan Gong, and Conghua Xie revised the paper. Conghua Xie and Yan Gong provided financial support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC. et al. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017;18:895-903
2. Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM. et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. 2006;580:755-62
3. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL. et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355-63
4. Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu Z. et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol. 2016;49:1360-8
5. Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9:874-85
6. Chugh S, Gnanapragassam VS, Jain M, Rachagani S, Ponnusamy MP, Batra SK. Pathobiological implications of mucin glycans in cancer: Sweet poison and novel targets. Biochim Biophys Acta. 2015;1856:211-25
7. Kufe DW. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene. 2013;32:1073-81
8. Maeda T, Hiraki M, Jin C, Rajabi H, Tagde A, Alam M. et al. MUC1-C Induces PD-L1 and Immune Evasion in Triple-Negative Breast Cancer. Cancer Res. 2018;78:205-15
9. Li W, Wu C, Yao Y, Dong B, Wei Z, Lv X. et al. MUC4 modulates human glioblastoma cell proliferation and invasion by upregulating EGFR expression. Neurosci Lett. 2014;566:82-7
10. Gum JR Jr, Ho JJ, Pratt WS, Hicks JW, Hill AS, Vinall LE. et al. MUC3 human intestinal mucin. Analysis of gene structure, the carboxyl terminus, and a novel upstream repetitive region. J Biol Chem. 1997;272:26678-86
11. Copin MC, Devisme L, Buisine MP, Marquette CH, Wurtz A, Aubert JP. et al. From normal respiratory mucosa to epidermoid carcinoma: expression of human mucin genes. Int J Cancer. 2000;86:162-8
12. Duncan TJ, Watson NF, Al-Attar AH, Scholefield JH, Durrant LG. The role of MUC1 and MUC3 in the biology and prognosis of colorectal cancer. World J Surg Oncol. 2007;5:31
13. Park HU, Kim JW, Kim GE, Bae HI, Crawley SC, Yang SC. et al. Aberrant expression of MUC3 and MUC4 membrane-associated mucins and sialyl Le(x) antigen in pancreatic intraepithelial neoplasia. Pancreas. 2003;26:e48-54
14. Rakha EA, Boyce RW, Abd El-Rehim D, Kurien T, Green AR, Paish EC. et al. Expression of mucins (MUC1, MUC2, MUC3, MUC4, MUC5AC and MUC6) and their prognostic significance in human breast cancer. Mod Pathol. 2005;18:1295-304
15. Wang RQ, Fang DC. Alterations of MUC1 and MUC3 expression in gastric carcinoma: relevance to patient clinicopathological features. J Clin Pathol. 2003;56:378-84
16. Shibahara H, Higashi M, Yokoyama S, Rousseau K, Kitazono I, Osako M. et al. A comprehensive expression analysis of mucins in appendiceal carcinoma in a multicenter study: MUC3 is a novel prognostic factor. PLoS One. 2014;9:e115613
17. Arul GS, Moorghen M, Myerscough N, Alderson DA, Spicer RD, Corfield AP. Mucin gene expression in Barrett's oesophagus: an in situ hybridisation and immunohistochemical study. Gut. 2000;47:753-61
18. Pyzer AR, Stroopinsky D, Rosenblatt J, Anastasiadou E, Rajabi H, Washington A. et al. MUC1 inhibition leads to decrease in PD-L1 levels via upregulation of miRNAs. Leukemia. 2017;31:2780-90
19. Mukhopadhyay P, Lakshmanan I, Ponnusamy MP, Chakraborty S, Jain M, Pai P. et al. MUC4 overexpression augments cell migration and metastasis through EGFR family proteins in triple negative breast cancer cells. PLoS One. 2013;8:e54455
20. Coelho MA, de Carné Trécesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M. et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity. 2017;47:1083-99.e6
21. Li J, Tan Q, Yan M, Liu L, Lin H, Zhao F. et al. miRNA-200c inhibits invasion and metastasis of human non-small cell lung cancer by directly targeting ubiquitin specific peptidase 25. Mol Cancer. 2014;13:166
22. Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S. et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. Journal of Thoracic Oncology. 2015;10:910-23
23. Nguyen PL, Niehans GA, Cherwitz DL, Kim YS, Ho SB. Membrane-bound (MUC1) and secretory (MUC2, MUC3, and MUC4) mucin gene expression in human lung cancer. Tumour Biol. 1996;17:176-92
24. Lakshmanan I, Ponnusamy MP, Macha MA, Haridas D, Majhi PD, Kaur S. et al. Mucins in lung cancer: diagnostic, prognostic, and therapeutic implications. J Thorac Oncol. 2015;10:19-27
25. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A. et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016;375:1823-33
26. Ji M, Liu Y, Li Q, Li X, Ning Z, Zhao W. et al. PD-1/PD-L1 expression in non-small-cell lung cancer and its correlation with EGFR/KRAS mutations. Cancer Biol Ther. 2016;17:407-13
27. Cho JH, Zhou W, Choi YL, Sun JM, Choi H, Kim TE. et al. Retrospective Molecular Epidemiology Study of PD-L1 Expression in Patients with EGFR-Mutant Non-small Cell Lung Cancer. Cancer Res Treat. 2018;50:95-102
28. Ho SB, Dvorak LA, Moor RE, Jacobson AC, Frey MR, Corredor J. et al. Cysteine-Rich Domains of Muc3 Intestinal Mucin Promote Cell Migration, Inhibit Apoptosis, and Accelerate Wound Healing. Gastroenterology. 2006;131:1501-17
29. Liberelle M, Jonckheere N, Melnyk P, Van Seuningen I, Lebegue N. EGF-Containing Membrane-Bound Mucins: A Hidden ErbB2 Targeting Pathway? J Med Chem. 2020;63:5074-88
30. Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R. et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051-8
31. Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S. et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J Thorac Oncol. 2015;10:910-23
32. Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19:598-609
33. Balan M, Mier y Teran E, Waaga-Gasser AM, Gasser M, Choueiri TK, Freeman G. et al. Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J Biol Chem. 2015;290:8110-20
34. Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S. et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2:361-70
35. Song M, Chen D, Lu B, Wang C, Zhang J, Huang L. et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS One. 2013;8:e65821
36. Noman MZ, Chouaib S. Targeting hypoxia at the forefront of anticancer immune responses. Oncoimmunology. 2014;3:e954463
37. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M. et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A. 2008;105:20852-7
38. Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS One. 2015;10:e0123410
39. Pochampalli MR, el Bejjani RM, Schroeder JA. MUC1 is a novel regulator of ErbB1 receptor trafficking. Oncogene. 2007;26:1693-701
40. Hisatsune A, Nakayama H, Kawasaki M, Horie I, Miyata T, Isohama Y. et al. Anti-MUC1 antibody inhibits EGF receptor signaling in cancer cells. Biochem Biophys Res Commun. 2011;405:377-81
41. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760-74
42. Wu SG, Shih JY. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer. 2018;17:38
43. Menard L, Floc'h N, Martin MJ, Cross DAE. Reactivation of Mutant-EGFR Degradation through Clathrin Inhibition Overcomes Resistance to EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2018;78:3267-79
Author contact
![]() Corresponding authors: Dr. Conghua Xie, Department of Radiation and Medical Oncology, Zhongnan Hospital of Wuhan University, 169 Donghu Road, Wuhan, Hubei 430071, China, Phone: +86-27-67812607, Email: chxie_65edu.cn; and Dr. Yan Gong, Department of Biological Repositories, Zhongnan Hospital of Wuhan University, 169 Donghu Road, Wuhan, Hubei 430071, China, Phone: +86-27-67811461, Email: yan.gongedu.cn.
Corresponding authors: Dr. Conghua Xie, Department of Radiation and Medical Oncology, Zhongnan Hospital of Wuhan University, 169 Donghu Road, Wuhan, Hubei 430071, China, Phone: +86-27-67812607, Email: chxie_65edu.cn; and Dr. Yan Gong, Department of Biological Repositories, Zhongnan Hospital of Wuhan University, 169 Donghu Road, Wuhan, Hubei 430071, China, Phone: +86-27-67811461, Email: yan.gongedu.cn.