Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Cells involved in the regulation...
Mechanisms by which plaque...
Vital roles of ncRNAs in plaque...
Clinical applications
Conclusion and outlook
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(13):3413-3427. doi:10.7150/ijbs.62506 This issue Cite
Review
Targeting non-coding RNAs in unstable atherosclerotic plaques: Mechanism, regulation, possibilities, and limitations
Xiaoxin Li1#, Yanyan Yang1#, Zhibin Wang2, Shaoyan Jiang3, Yuanyuan Meng2, Xiaoxia Song2, Liang Zhao2, Lu Zou1, Min Li1, Tao Yu1,2 ![]()
1. Institute for translational medicine, The Affiliated Hospital of Qingdao University, No. 38 Dengzhou Road, 266021, People's Republic of China.
2. Department of Cardiac Ultrasound, The Affiliated Hospital of Qingdao University, Qingdao 266000, China.
3. Department of Cardiology, The Affiliated Cardiovascular Hospital of Qingdao University, No. 5 Zhiquan Road, Qingdao 266000, China.
#These authors contributed equally to this paper.
Received 2021-5-9; Accepted 2021-7-23; Published 2021-8-3
Abstract

Cardiovascular diseases (CVDs) caused by arteriosclerosis are the leading cause of death and disability worldwide. In the late stages of atherosclerosis, the atherosclerotic plaque gradually expands in the blood vessels, resulting in vascular stenosis. When the unstable plaque ruptures and falls off, it blocks the vessel causing vascular thrombosis, leading to strokes, myocardial infarctions, and a series of other serious diseases that endanger people's lives. Therefore, regulating plaque stability is the main means used to address the high mortality associated with CVDs. The progression of the atherosclerotic plaque is a complex integration of vascular cell apoptosis, lipid metabolism disorders, inflammatory cell infiltration, vascular smooth muscle cell migration, and neovascular infiltration. More recently, emerging evidence has demonstrated that non-coding RNAs (ncRNAs) play a significant role in regulating the pathophysiological process of atherosclerotic plaque formation by affecting the biological functions of the vasculature and its associated cells. The purpose of this paper is to comprehensively review the regulatory mechanisms involved in the susceptibility of atherosclerotic plaque rupture, discuss the limitations of current approaches to treat plaque instability, and highlight the potential clinical value of ncRNAs as novel diagnostic biomarkers and potential therapeutic strategies to improve plaque stability and reduce the risk of major cardiovascular events.
Keywords: atherosclerotic plaques, plaque instability, plaque rupture, non-coding RNAs
Introduction
Atherosclerosis (AS) is a chronic and complex pathological process that is the major cause of cardiovascular diseases. Genetic susceptibility and environmental factors (e.g. smoking, hypertension, hyperlipidemia, diabetes, family history, and obesity) are the main factors that influence AS, with plaque accumulation in the arterial wall being one of the most prominent features [1]. A stable plaque becomes unstable and vulnerable to rupture as AS progresses. Factors that negatively affect plaque stability include necrosis in the plaque core, the presence of inflammatory cells, and thinning of the fibrous cap structure [2, 3]. Moreover, neovascularization in unstable plaques is more likely to exacerbate the rupture process compared to that in stable plaques [4]. While an increase in vessel permeability is conducive to the infiltration of inflammatory cells, the increase in vessel fragility results in easy bleeding and the formation of hematomas in plaques. In the late stages of atherosclerosis, the thin and fragile fiber cap with a large necrotic core, a large number of infiltrated inflammatory cells, intra plaque hematomas, and secondary thrombosis all aggravate the development of AS resulting in serious cardiovascular diseases such as coronary artery disease (CAD), acute coronary syndrome (ACS), myocardial infarction, and stroke [1, 5, 6]. Therefore, maintaining plaque stability and preventing plaque rupture are widely considered to be the principle goals in the clinical treatment of patients with cardiovascular diseases [7].
As discussed above, atherosclerotic plaques are divided into stable plaques and unstable plaques. A stable plaque is characterized by a small swollen and a thick fibrous cap [8]; there is also an absence of clinical symptoms. Conversely, the characteristics of the rupture-prone plaques are (a) a thin fibrous cap with a large lipid core; (b) infiltration of monocytes and macrophages; (c) aggregation of endothelial exfoliation and the presence of surface platelets; (d) deposition of extracellular dense lipid droplets and formation of large lipid droplets around the nucleus; and (e) fibrous connective tissue that forms part of the plaque [9]. The formation of ulcers or fissures in unstable plaques can release or activate transmitters, such as thrombin, adenosine diphosphate (ADP), platelet activating factor (PAF), tissue factor, and oxygen free radicals, which promote platelet aggregation and aggravate arterial mechanical obstruction. Plaque instability is therefore an important factor affecting the occurrence and development of AS, but the mechanisms leading to the instability of atherosclerotic plaques remain to be determined.
Recent studies have shown that non-coding RNAs (ncRNAs) are critically involved in the regulation of plaque instability [10-12]. NcRNAs can not only participate in regulating the expression of lipoproteins and impact the proliferation and differentiation of vascular smooth muscle cells (VSMCs) to affect plaque stability but also regulate the phenotypic transition of immune cells to affect the development of inflammation around plaques. Encouragingly, several microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and small interfering RNAs (siRNAs) have been found to be differentially expressed in the serum from patients with AS and have shown remarkable capabilities in regulating plaque stability, indicating their potential to be used as diagnostic biomarkers or as therapeutic targets [13-18].
Studies examining the relationship between plaque stability and extracellular vesicles and exosomes have also made great progress. For example, it has been shown that the accumulation of extracellular vesicles can aggravate plaque calcification and promote a vasoactive response [19, 20]; changes in calcification morphology and collagen content in plaques are also associated with exosomes [21]. Furthermore, exosomes are thought to be promising carriers for nuclear drugs [22]. Several studies have also presented evidence that exosome delivery systems can control plaque instability [23].
In this review, we introduce the cellular and molecular mechanisms of atherosclerotic plaque stability and highlight the role of ncRNAs in plaque vulnerability. We also summarize new progress in the use of exosomes to treat plaque instability. Finally, we discuss the limitations of the current studies and provide emerging insights into the role of ncRNAs in the regulation of atherosclerotic plaque stability and their potential as targets for novel therapeutic paradigms.
Cells involved in the regulation of plaque instability
Endothelial cells
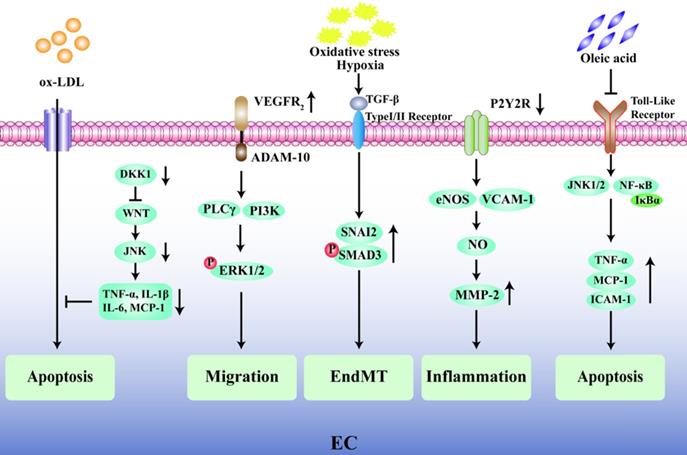
The endothelium plays a pivotal role in the progression of AS and its complications, and endothelial dysfunction is widely recognized as one of the early alterations in the vessel wall preceding the development of plaques [24, 25]. Emerging evidence has shown that the degree of endothelial cell (EC) apoptosis may be a key factor in the transition of a plaque from a stable state to a fragile state [26, 27]. The glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB3) is highly expressed in the ECs present in vulnerable human carotid atherosclerotic plaques, and inhibition of PFKFB3 activity reduces cell apoptosis in plaques and promotes plaque stability [28]. Knockout of the Dickkopf1 (DKK1) gene in apolipoprotein E-deficient (ApoE-/-) mice inhibits the classical Wingless-Related Integration Site (WNT) signal by activating the JNK1 signal transduction pathway and reduces the vulnerability for apoptosis in human umbilical vein endothelial cell (HUVEC) in the presence of oxidized low density lipoprotein (ox-LDL) during the AS process [29]. Moreover, the apoptosis of ECs also leads to local lipid deposition in blood vessels [30], aggravates plaque rupture, and may predispose individuals to arterial thrombosis. In addition, the migration of ECs and the increase in vascular permeability are closely related to the progression and instability of atherosclerotic plaques. Vascular endothelial growth factor receptor 2 (VEGFR2) can induce the expression of disintegrin and metalloprotease 10 (ADAM10) in ECs, and the combination of VEGFR2 and ADAM10 can promote the migration of ECs and accelerate the progression of atherosclerotic plaques [31]. Furthermore, the endothelial to mesenchymal transition (EndMT) plays a key role in cardiovascular disease and the rupture of unstable plaques because EndMT-derived cells are found in oxidative stress and hypoxia-induced atherosclerotic plaques [32]. Chen et al. found that in 25-week-old ApoE-/- mice fed a standard diet, an EC-specific nucleotide P2Y2 receptor (P2Y2R) deficiency prevented vascular adhesion molecule-1 (VCAM-1) in ECs from participating in vascular inflammation and reduced nitric oxide (NO) synthase and matrix metalloproteinase-2 (MMP-2) activity owing to a decrease in macrophage infiltration [33]. A recent intriguing finding has described the protective effect of oleic acid on cardiovascular cells through an inhibition of tumor necrosis factor-α (TNF-α) and a decrease in the expression levels of monocyte chemoattractant protein-1 (MCP-1) and ICAM-1, thereby improving endothelial dysfunction [34, 35]. In general, the key factor affecting the stability of atherosclerotic plaques is endothelial dysfunction, which is mainly manifested by impaired endothelial barrier function, increased vascular permeability, and EC apoptosis (Fig. 2).
Vascular smooth muscle cells
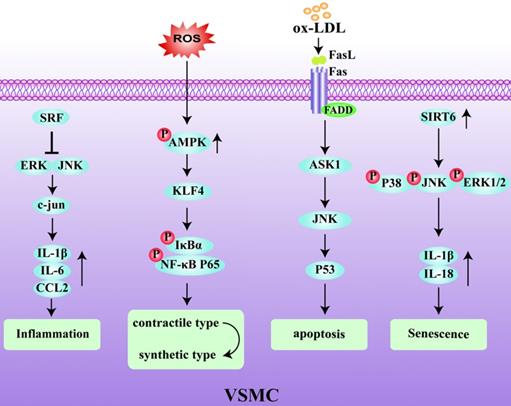
The majority of VSMCs in plaques are derived from the medial layer of the blood vessel. The plasticity of VSMCs play an important role in the occurrence and development of AS and can protect the fibrous cap from rupture and participate in the synthesis of extracellular matrix components [36]. VSMC phenotypic remodeling plays an important role in the early stage of AS [37, 38]. Under pathological inflammatory conditions, VSMCs initially with a stable and normal vasoconstriction phenotype undergo remodeling to a synthetic phenotype. In this latter state, VSMCs express immunoregulatory cytokines, such as IL-6 and CC chemokine ligand (CCL)2, and secrete chemokines to activate the inflammatory state of macrophages, thereby affecting the local stability of the plaques [39, 40]. Serum response factor (SRF), as a key regulator of vascular inflammation, is an important player that regulates the phenotype of VSMCs. Increased expression of SRF reduced the accumulation of macrophages in ApoE-/- mice and inhibited VSMC phenotype changes and the activation of inflammation, thereby enhancing plaque stability [41, 42]. Conditional knockout of the KLF4 gene has been shown to reduce the number of VSMC-derived macrophages and mesenchymal stem cells, resulting in an increase in fibrous cap thickness and a decrease in lesion size [43]. Therefore, VSMC contributes to the stability of atherosclerotic plaques through a KLF4-dependent phenotypic transformation mechanism. In keeping with this, it has also been shown that deletion of AMPKα2 promotes the phenotypic conversion of contractile VSMCs to synthetic VSMCs by increasing KLF4 expression [44]. VSMCs are the main source of collagen in the fiber cap (FC) which is responsible for its tensile strength. A reduction in the number of VSMCs due to the death of initiating cells leads to FC thinning, necrotic nucleus formation, and calcification [1]. Additionally, the Fas receptor/Fas ligand pathway is involved in ox-LDL-induced apoptosis of VSMCs. Interestingly, the activation of p53 makes the VSMCs more sensitive to Fas-mediated apoptosis by transiently increasing Fas expression and translocation from the Golgi [45, 46]. In addition, DNA damage in VSMCs has been shown to be involved in human atherosclerotic plaques both in vitro and in vivo [47, 48], which are manifested by double strand breaks (DSBs) and the increased expression of multiple DNA damage response proteins. Among these, the nuclear deacetylase sirtuin6 (SIRT6) has been reported to participate in the DNA damage response. The overexpression of SIRT6 in VSMCs reduced the activity of nuclear factor-kappa B (NF-κB)-dependent inflammatory factors, inhibited cell senescence, and protected atherosclerotic plaques in ApoE-/- mice [49]. Therefore, inhibiting DNA damage in VSMCs by reducing the relative FC area of late-stage plaques could be a promising target for creating clinically stable plaques [50]. The decreased activity of MMP-2, and the increased migration ability of VSMCs, can increase plaque stability [51]. Early aging of VSMCs and an increased sensitivity to apoptosis in atherosclerotic plaques reduces the ability to repair vulnerable plaques. In addition, the abnormal proliferation of VSMCs after phenotypic transformation also accelerates the process of plaque rupture. Therefore, identifying the factors and mechanisms that can promote the phenotypic transformation of VSMCs and improve plaque stability is an important goal to prevent plaque rupture in the future (Fig. 3).
Immune cells
In the development of AS, phenotypic changes and cytokines secreted by immune cells such as monocytes, macrophages, dendritic cells, and mast cells can stimulate inflammation and affect the stability of plaques. Therefore, understanding the mechanisms by which immune cells promote inflammation and how changes in cell function arise owing to alterations in immune cell surface receptors is likely to be very important in learning how to stabilize plaques and ameliorate the progression of AS.
Monocytes and Macrophages
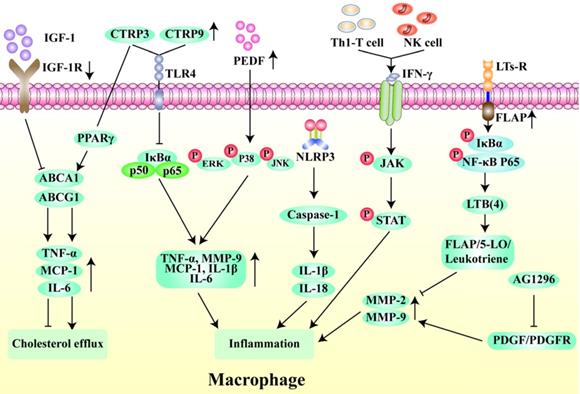
Chemokines mobilize monocytes to migrate and adhere to ECs, which is a key process in macrophage aggregation [52]. Interestingly, the Src family-associated kinases HCK and FGR, mediate the interaction between EC adhesion molecules and β-2 integrins and participate in a series of AS-related processes through activation of Rac/Cdc42, Syk, and Pyk7 effectors. They also lead to dynamic instability of the extracellular matrix by causing an imbalance in monocyte subsets, making the plaque more prone to rupture [53]. Monocyte to macrophage differentiation plays an important role in the early stages of AS [54], and the accumulation of macrophage-derived foam cells in the arterial wall promotes monocyte adhesion and infiltration [55, 56]. Ma et al. found that formononetin can inhibit monocyte adhesion and enhance plaque stability by reducing the expression of SRA in monocytes [57]. Insulin-like growth factor-1 (IGF-1) is highly expressed in monocytes/macrophages in ApoE-/- mice. IGF1R-deficient macrophages inhibit the expression of ABCA1 and ABCG1 and reduce lipid efflux and plaque vulnerability [58]. Interestingly, the activation of MC1-R can not only prevent macrophages from accumulating lipid but also promote the reverse transport of cholesterol by up-regulating the levels of ABCA1 and ABCG1 and reducing the expression of CD36 on the cell surface [59, 60]. Li et al. found that overexpression of C1q/TNF-related protein 9 (CTRP9) can reduce the levels of pro-inflammatory factors such as TNF-α and MCP-1 in macrophages [61]. Wen et al. showed that the levels of phosphorylated ERK-MAPK, p38-MAPK, and JNK-MAPK were significantly reduced by pigment epithelium-derived factor (PEDF) through the regulation of the PPAR-γ and downstream MAPK inflammatory pathways, so as to reduce macrophage inflammation and increase plaque stability [62]. Additionally, several studies have shown that late plaques contain more apoptotic cells than early plaques [58, 63], and the occurrence of modified LDL in the process of early plaque efferocytosis can induce macrophage apoptosis. However, defects will occur with the further development of plaque efferocytosis, resulting in plaque rupture and remodeling of the plaque structure [64]. Therefore, macrophage apoptosis is another key factor in plaque instability (Fig. 4).
Dendritic cells
The high cholesterol environment in patients with atherosclerosis leads to a decrease in the migration of DCs because of the engulfment of the walls of blood vessels with their own antigens, such as ox-LDL. The number of tolerogenic DCs increased when ox-LDL -induced apoptotic DCs (apopox-DCs) were injected intravenously into LDL-/- mice. Interestingly, the collagen levels in mice treated with apopox-DCs were increased by 45% compared with untreated apoptotic dendritic cells, significantly reducing the progression of lesions, which is suggestive of increased plaque stability [65]. In vulnerable plaques, the number of mature DCs is significantly increased and these mature DCs play an important role in the inflammatory processes in atherosclerotic lesions by stimulating effector T cells. Because of the interaction between DCs and regulatory T cells (Treg), there is a direct inhibition of DC migration and adhesion to ECs, leading to the development of plaque instability [66]. In atherosclerotic tissues, the myeloid cell receptor referred to as triggering receptor in myeloid cells (TREM)-1, is a key factor in inflammation, and that the number of DCs increases gradually with the progression of the disease. The expression of TREM-1 in DCs is significantly increased in plaques of patients with symptomatic carotid stenosis, indicating that both DCs and TREM-1 may play an important role in plaque stability [67].
Mast cells
Mast cells are a type of multifunctional white blood cells, which are mainly found in mucosal tissues and connective tissues. An abnormal increase in mast cell number is often accompanied by the occurrence of cardiovascular disease. It is thought that several mast cells and their resultant activation affects the stability of plaques. To further understand the role of the vascular network as a transport channel for several angiogenic and plaque forming factors, Joosp et al. Used indocyanine green video angiography (ICG-VA) during a carotid endarterectomy (CEA) to investigate the correlation between the change in state of the carotid artery and the vulnerability of a carotid plaque and showed that mast cells stained with CD117 were more frequently found in unstable plaques than in stable ones [68]. Mast cells are activated by the binding and cross-linking of antigens to IgE which is itself bound to the Fcε-receptor (FcεR). This results in the release of cytoplasmic granules which contain proinflammatory factors, histamine, and several neutral proteinases. Kritikou et al. used improved flow cytometry to identify specific mast cells that expressed both CD117 and FcεR at the same time and observed that the activation of mast cells in most plaques depends on the expression of the CD63 protein and the presence of IgE fragments on their surfaces [69]. This strongly suggests that the development of AS is related to the number of mast cells and the activation of their surface receptors and, therefore, provides a new strategy for the clinical treatment of AS.
Lymphocytes
At present, while there is no direct research that supports the involvement of lymphocytes in the formation and development of plaques, the possibility has been suggested. For example, Tsaousi et al. suggested that knockout of the T-bet gene inhibits Th1 lymphocyte differentiation to reduce the expression of the M1 macrophage marker NOS-2 and, consequently, the size of atherosclerotic plaques [70]. These phenomena suggest that lymphocytes may be associated with plaque instability, but this needs to be further explored.
Mechanisms by which plaque instability is regulated
Inflammation
Inflammation is widely considered to play a critical role in the formation of atherosclerotic plaques and plaque rupture [71-74]. Histopathology of lateSvulnerable plaques has shown that there are obvious signs of inflammation. In addition, the action of excessive proteolytic enzymes, which stimulate macrophages to participate in the immune response, inhibits the formation of the fibrous cap and degrades the fiber composition of the cap [5]. The accumulation of activated macrophages, T cells, and necrotic core lipids in vulnerable plaques triggers a self-persistent, vicious cycle of inflammatory responses [75, 76]. In patients with AS, activated NLRP3 inflammasomes produce IL-1β and IL-18 [77, 78], which up-regulate VCAM to induce T-cell differentiation and promote a downstream inflammatory response, thus leading to plaque progression [79, 80]. Interferon-γ, a pro-inflammatory cytokine produced by Th1 T cells and natural killer (NK) cells, makes plaques more vulnerable by inhibiting smooth muscle cell differentiation [81] and interstitial collagen gene expression [82]. Likewise, the production of TGF-β can inhibit the activity of Th1 cells and macrophages and reduce plaque inflammation [25, 83]. The cytokine IL-17A alleviates the deleterious mechanical effects of hemodynamics on plaques by promoting collagen gene expression [84, 85]. In addition, the plaque expression levels of the pro-inflammatory factor leukotriene B (LTB) (4) are up-regulated by the BLT1 receptor, which could be a target for treating plaque instability [86]. Leukotriene receptors and their important cofactor 5-lipoxygenase-activating protein (FLAP) are highly expressed in atherosclerotic plaques and promote the production of LTB4 [87]. The levels of the formyl peptide receptor (FPR) subtype FPR2/ALX are significantly increased in atherosclerotic lesions. FPR2/ALX has pro-inflammatory and plaque destabilizing effects on myeloid-derived cells but has the opposite effect on VSMCs [88]. The tyrosine kinase inhibitor AG1296 which inhibits the inflammatory response by blocking the platelet-derived growth factor/platelet-derived growth factor receptor (PDGF/PDGFR) signaling pathway can reduce the expression levels of MMP-2 and MMP-9 to enhance plaque stability [88, 89]. In recent years, the Colchicine Cardiovascular Outcomes (COLCOT) and the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) trials targeting NLRP3/IL-1β pathway have provided potential evidence to treat AS [90], but it is still unknown whether promoting the immune response in patients with AS affects the normal immune homeostasis. Therefore, how to specifically identify vulnerable plaques and target the immune response in the lesions of patients with AS is the next problem that has to be solved (Fig. 4).
Lipid metabolism
Another main reason for the development of AS is the elevated levels of LDL, leading to cholesterol accumulation in the intima, which eventually attracts monocytes. Macrophage phagocytosis of ox-LDL resulting in the creation of foam cells can accelerate the progression of AS [91]. Rinne et al. found that activation of MC1-R in macrophages in atherosclerotic ApoE-/- mice increased the outflow of cholesterol to that of ApoA1 and high-density lipoprotein (HDL), because of the increased reverse transport of cholesterol mediated by ATP-binding cassette transporters ABCA1 and ABCG1 to make the plaque stability signs more obvious [92]. There is evidence that there is an increase in protein deacetylation in macrophages from ApoE-/- mice treated with hydrogen sulfide (H2S), leading to increased deacetylation of several proteins including p53, p65, and sterol response element binding protein. As a result, there is an inhibition of both the synthesis of liver cholesterol and the uptake of cholesterol by macrophages, thereby increasing plaque stability and reducing plaque formation [93]. ApoA-I is the main structural protein component of HDL particles and has been shown to play a key role in reverse cholesterol transport (RCT) [94]; thus, stimulating an increase in endogenous ApoA-I synthesis may be a promising treatment for plaque instability.
Oxidative stress
The excessive production of reactive oxygen species (ROS) is another reason for the development of AS and the formation of unstable plaques. Evidence supports that a dysfunction in endothelial NO synthase (NOS) and an up-regulation of vascular NADPH oxidase in AS are closely related to plaque instability [95]. Ox-LDL activates NOS1 and leads to the expression of CD40 ligand in macrophages. Inhibition of NOS1-derived NO may therefore be an effective strategy to reduce foam cell formation and limit the extent of atherosclerotic plaque expansion [96]. There is evidence that NOX2, a specific inhibitor of NADPH oxidase, can inhibit superoxide dismutase and significantly reduce the expression of hypoxia-inducible factor-1 α, MMP-9, endothelin, and vascular endothelial growth factor in a ApoE-/- mouse model fed a high-fat diet. Therefore, NOX2 can stabilize atherosclerotic plaques and reverse atherosclerotic vascular lesions [97]. In addition, the loss of the transcriptional activator Arnt-like protein-1 (BMAL1) in human aortic endothelium inhibits the intracellular ROS accumulation induced by ox-LDL through BMP-mediated signal transduction, which further aggravates EndMT and negatively affects plaque stability [98]. Deletion of the transcription factor NF-E2 related factor 2 (Nrf2) reduced the extent of atherosclerotic lesion formation in LDLR-/- mice but also led to an increase in the plaque instability index by increasing plaque calcification and oxidative stress in 12-month-old LDLR-/-ApoB100/100 female mice [99-101]. It has further been confirmed that Nrf2 plays a completely different role in different animal models as well as in early and late plaques. In addition, in mouse models of AS, it is clear that there is gender dimorphism in the development of AS. These findings have provided us with new ideas for development of drugs to treat plaque instability in the future.
Exercise training
In patients with stable coronary heart disease (CHD), high degrees of physical activity are associated with low mortality [102, 103]. Additionally, exercise training has been shown to prevent AS and angiotensin (Ang)-induced vulnerable plaque formation in ApoE-/- mice by reducing systemic inflammation [104]. Moderate aerobic exercise can convert calcium deposits in blood vessels into a more stable form [105] and also stabilize plaques by increasing their collagen content [106, 107]. More interestingly, recent studies have shown that exercise may reduce macrophage activity by down-regulating neuropeptide Y (NPY) receptor expression in ApoE-/- mice and increase plaque collagen levels and smooth muscle cell numbers that play a stabilizing role in plaques [108].
Vital roles of ncRNAs in plaque instability
Ample studies are available demonstrating the association between ncRNAs and plaque stability, which provide a rationale for the development of ncRNA-targeted therapeutic strategies in AS (Table 1).
Regulation of ncRNAs in the stability of atherosclerotic plaques
| NcRNAs | Expression | Phenotype | Effect on plaque stability | Reference |
|---|---|---|---|---|
| LncR- TCONS_00024652 | up regulation | promotes ECs proliferation and angiogenesis | increase | [118] |
| LncR-LINC00657 | up regulation | promotes ECs angiogenesis | decrease | [163] |
| LncR-UC.98 | down regulation | promotes ECs proliferation and adhesion | increase | [115] |
| miR-21 | up regulation | regulates macrophage migration and adhesion | decrease | [164-166] |
| miR-200C | up regulation | induces ECs dysfunction to produce ROS | increase | [167] |
| miR-23a-5p | up regulation | cholesterol efflux reduces the formation of foam cells | decrease | [114] |
| miR-124-3p | down regulation | inhibits VSMCs collagen synthesis | decrease | [121, 168] |
| miR-10b | up regulation | induces apoptosis of macrophages | decrease | [127] |
| miR-124 | up regulation | collagen synthesis disorder | decrease | [169] |
| miR-150 | down regulation | increases VSMCs and collagen content, reduce macrophage infiltration and lipid accumulation | increase | [128] |
| miR-19b | up regulation | inhibits STAT3 transcriptional activity affects ECs proliferation, migration and angiogenesis | increase | [170] |
| miR-195 | up regulation | inhibits the TLR2 inflammatory pathway | increase | [129] |
| miR-495 | down regulation | increases of neovascularization after ischemia | increase | [171] |
| miR-455-3p | up regulation | regulates eNOS protein stability and NO production | decrease | [112] |
| miR-133a | up regulation | targets LDLRAP1 reduces lipid accumulation in VSMCs | increase | [119] |
| miR-210 | up regulation | targets tumor suppressor gene APC regulation of VSMCs survival | increase | [122] |
| miR-181b | down regulation | regulates tissue inhibitor of metalloproteinase-3 expression | increase | [172] |
| miR-27b | down regulation | targets Naa15 regulates the activity of CCL20/CCR6 axis regulates ECs angiogenesis | increase | [116] |
| miR-494 | down regulation | cholesterol levels and very low-density lipoprotein (VLDL) components fell | increase | [171, 173] |
| miR-24 | down regulation | increases the expression of MMP-14 in macrophages | increase | [130] |
| miR-145 | up regulation | adjusts the plasticity of VSMCs | increase | [120] |
| miR-33 | down regulation | promotes the expression of ABCA1 and the clearance of cholesterol | increase | [174] |
ncRNAs in ECs
The effects of endothelial dysfunction, inflammation, ROS, and NO production on plaque stability are obvious. The blood flow in atherosclerotic lesions is characterized by turbulent dynamics, and these hemodynamics have a far-reaching impact on the expression of miRNAs, with differentially regulated miRNAs modulating shear stress-mediated transcriptional procedures [109-111]. It has been found that the levels of miR-200C are significantly higher in unstable plaques than in stable plaques. There is also a positive correlation between miR-200C levels and markers of plaque instability. Surprisingly, blood miR-200C expression levels decreased in patients with stable plaques one month after CEA. In addition, it has been found that H2S regulates the stability of NO synthase in ECs by up-regulating miR-455-3p to promote the migration of HUVECs. These data suggest that miR-455-3p is closely related to plaque instability and atherosclerotic progression [112]. Recent studies have shown that hsa_circ_0030042 can ameliorated plaque stability and regulates abnormal autophagy by targeting eukaryotic initiation factor 4A-III (eIF4A3) [113]. The overexpression of miR-19b can inhibit the transcriptional activity of the transcriptional activator 3 (STAT3) [114]. Another study has shown that decreased lncRNAUC.98 expression can stabilize the progression of atherosclerotic plaque by inhibiting the proliferation and migration of ECs [115]. A reduction in miRNA-27b levels has been shown to regulate the activity of the chemokine (C-C motif) ligand 20/C-C chemokine receptor type 6 (CCL20/CCR6) axis by targeting N-alpha-acetyltransferase 15 (Naa15) to promote the stability of atherosclerotic plaques [116]. LncRNA AK136714 silencing inhibits endothelial cell inflammation and protects plaque stability [117]. The lncRNA TCONS_00024652 acts as miR-21 sponge to regulate vascular endothelial cell proliferation and angiogenesis and may be used as a potential method to reduce vascular endothelial dysfunction and treat plaque instability [118].
ncRNAs in VSMCs
The increase in plaque size and decrease in plaque stability caused by the transformation of VSMCs into foam cells is a key step in the formation of AS plaques [43]. Gabunia K et al. reported that interleukin-19 (IL-19), a new type of anti-inflammatory cytokine, has been shown to reduce lipid accumulation in VSMCs. MiR-133a can target and decrease the mRNA levels, stability, and protein expression levels of the LDL receptor adapter protein 1 (LDLRAP1). Mutations in miR-133a lead to LDL receptor dysfunction resulting in human autosomal recessive hypercholesterolemia (ARH). Therefore, miR-133a is a new target to reduce plaque size and rupture vulnerability by reducing Ox-LDL uptake in VSMCs [119]. High expression levels of miR-145 in VSMCs have recently been found to regulate AS and plaque stability. Targeting miR-145 in VSMCs has been shown to increase the area of the fiber cap, collagen content, and plaque stability [120]. Contrary to the effects of miR-145, miR-124-3p inhibits VSMC collagen synthesis by directly targeting prolyl 4-hydroxylase subunit alpha-1 (P4HA1), resulting in atherosclerotic plaque instability [121]. MiR-210 enhances the stability of the fibrous cap in advanced atherosclerotic lesions by targeting the tumor suppressor gene adenomatous polyposis coli (APC), thereby affecting WNT signal transduction, regulating VSMC apoptosis, and preventing plaque rupture [122]. On the one hand, miR-21 can affect the formation of foam cells and the development of local lipid cores by regulating the activity of the NF-κB signal pathway. On the other hand, miR-21 can inhibit VSMC apoptosis by increasing the rate of proliferation of VSMCs in mouse carotid arteries and jointly protecting fibrous cap stability in atherosclerotic plaques [123]. It has been shown that hyperglycemia and streptozotocin-induced type 1 diabetes can reduce the synthesis of collagen and lead to the formation of unstable atherosclerotic plaques in ApoE-/- mice [124]. Metformin can increase the level of activator protein 2 alpha (AP-2α) in carotid atherosclerotic plaques in diabetic ApoE-/- mice, decrease the expression levels of miR-124, and increase the levels of prolyl-4-hydroxylase alpha 1 (P4Hα1) and collagen in VSMCs. Therefore, targeting miR-124 can increase plaque stability by regulating the activity of the AMPKα/AP-2α/miRNA-124/P4Hα1 axis, which provides a new idea for the clinical treatment of AS [125].
ncRNAs in macrophages/monocytes
NcRNAs are critically involved in macrophage apoptosis, inflammation, and the phenotypic transformation leading to plaque instability. It has been shown that the serum levels of miR-23a-5p in patients with AS as well as in macrophages in atherosclerotic mice are both significantly increased. A miR-23a-5p inhibitor has been shown to increase cholesterol efflux and reduce the formation of foam cells by up-regulating the expression of ABCA1/G1. A miR-23a-5p anticoagulant therapy has been shown to significantly slow the progression of AS probably by inhibiting an ATP-binding cassette transporter in macrophages that promotes the progression and vulnerability of atherosclerotic plaques [114]. Analogously, the expression levels of miR-10b are increased in the arteries of late atherosclerotic plaques in ApoE-/-mice [126]. In addition, free cholesterol-induced macrophage apoptosis (FC-AM) has been shown to promote the expression of miR-10b in resident peritoneal macrophages (RPM) by up-regulating Mer receptor tyrosine kinase-dependent Twist1/2 [127]. It has been shown that miR-150 significantly enhances the inflammatory response by up-regulating the proliferation, migration, and vascular homeostasis of ECs. It can also reduce infiltration and lipid accumulation in macrophages to promote plaque stabilization [128]. Some studies have found that miR-195 participates in the polarization of macrophages and inhibits mediators of the Toll-like receptor 2 (TLR2) inflammatory pathway. In addition, miR-195 weakens the effect of macrophages on the recruitment and migration of VSMCs [129]. Recent studies have suggested that membrane-1 MMP-14, a selective marker of a subset of invasive macrophages, has been shown to be associated with atherosclerotic plaque progression. The levels of miR-24 in stable plaques are higher than in unstable plaques, and the downregulation of miR-24 promotes the formation of the subset of invasive macrophages, suggesting a new regulatory role for MMP-14 proteolytic activity in plaque stability [130] (Fig. 1).
Non-coding nucleic acids participate the regulation of unstable plaques via mediating functions of endothelial cells, vascular smooth muscle cells and macrophages. As shown, there is a thin fibrous cap in the unstable plaque, and the vulnerable sites are rich in lipid cores, including a large number of apoptotic cells, cholesterol crystals, lipid-rich foam cells, and so on. Among them, in endothelial cells, miR-200C, miR-455-3p, miR-10b and miR-21 were significantly up-regulated, while miR-2b and LncRNA-UC.98 were down-regulated. miR-124-3p was increased and miR-133a, miR-145, miR-210, miR-21, miR-145 were significantly decreased in VSMCs. In addition, miR-23a-5p and miR-10b were up-regulated in macrophages, while miR-150, miR-196 and miR-24 were obviously down-regulated.
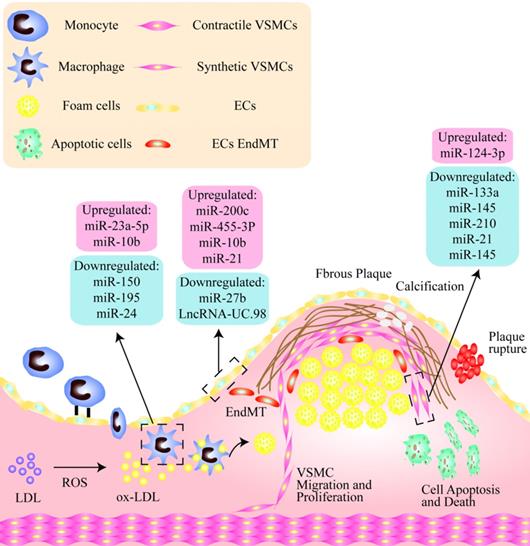
Regulatory mechanism of endothelial cells in unstable plaques. Inhibition of endothelial cell apoptosis induced by ox-LDL was mainly through WNT/JNK pathway after Dickkopf1 (DKK1) silencing. The combination of vascular endothelial growth factor receptor 2 (VEGFR2) and its ligand metalloprotease 10 (ADAM10) induces the phosphorylation of ERK1/2 through PLCγ/PI3K signal transduction and promotes the migration of ECs. Oxidative stress and hypoxia induce transforming growth factor-β (TGF-β) to stimulate the phosphorylation of SNAI2 and SMAD3, resulting in endothelial to mesenchymal transition (EndMT). P2Y2 receptor (P2Y2R) deletion can promote the expression of vascular adhesion molecule-1 (VCAM-1) and endothelial nitric oxide synthase (eNOS) in ECs, thus promote the activity of nitric oxide (NO) and matrix metalloproteinase-2 (MMP-2), then increase endothelial inflammation. Oleic acid suppresses cell apoptosis by inhibiting Toll-like receptor-activated JNK/NF-κB/IκBα pathway and the expression of TNF-α, MCP-1 and ICAM-1.
Regulatory mechanism of VSMC in unstable plaques. Serum response factor (SRF) blocks the inflammatory pathway by inhibiting ERK/JNK/C-Jun pathway and the expression of IL-1β, IL-6 and CCL2. ROS activates the phosphorylation of AMPK and induces phenotypic transformation of VSMCs through the klf4-dependent IκBα/NF-κB p65 pathway. Sirtuin6 (SIRT6) promotes the increase of IL-1β and IL-18 and induces VSMCs senescence by activating the phosphorylated p38/JNK/ERK1-Beat 2 pathway.
Regulatory mechanism of macrophages in unstable plaques. Insulin-like growth factor-1 receptor (IGF-1R) deletion inhibits cholesterol outflow by suppressing the expression of TNF-α, MCP-1 and IL-6 through ABCA1/ABCG1 pathway. In contrast, C1q/tumor necrosis factor-related protein-3 (CTRP3) promotes cholesterol outflow through PPAR-γ. The high expression of CTRP3 and CTRP9 can inhibit the macrophage inflammation through NFκB pathway. Pigment epithelium-derived factor (PEDF) can activate ERK, p38 and JNK phosphorylation to inhibit macrophage inflammation. NLRP3 inflammasomes induces inflammation by activating IL-1β and IL-18. Interferon-γ produced by Th1 T cells and natural killer (NK) cells activates the phosphorylation of JAK and STAT to promote inflammation. Leukotriene receptors (LTs-R) and its cofactor FLAP target FLAP/5-LO/Leukotriene pathway to inhibit inflammation. The tyrosine kinase inhibitor AG1296 suppresses inflammation by reducing the expression of MMP-2 and MMP-9 via PDGF/PDGFR pathway.
Clinical applications
The rupture of unstable atherosclerotic plaques and thrombosis are the most important pathological basis of AS which seriously threatens the life of patients. Therefore, early diagnosis and the identification of unstable plaques are of great value to patients with coronary heart disease [131-134].
Clinically, intravascular ultrasound (IVUS) and optical coherence tomography (OCT) can provide the morphological characteristics of coronary atherosclerotic plaques, which is very helpful for the assessment of plaque stability [135-137]. However, these tools are too invasive and expensive to be widely used in the screening of unstable plaques. Strategies targeting circulating biomarkers, such as ncRNAs, could provide a more convenient method to evaluate plaque stability in patients with coronary heart disease [15, 138, 139]. There are several advantages for ncRNA application in clinical trials. RNA therapy can specially mediate target genes. NcRNAs are associated with complex biological processes such as immune cell development and functions [140-142]. The preclinical development of ncRNA drugs is simpleness in design through gene sequencing. Moreover, the treatment of small nucleic acid drugs at the post-transcriptional level can suppress the activity of special targets whose proteins are difficult to be effective. Besides, RNA therapy can also mediate multiple targets at the same time [143]. For example, a biomimetic exocrine nanocomplex for accurate delivery of miRNA has been developed, which shows excellent targeted delivery and therapeutic effect in mouse myocardial infarction model, which provides a novel idea for the design and development of nucleic acid drug carriers, realizes the accurate delivery of gene drug and microRNA-mediated myocardial repair, and provides a theoretical basis for nucleic acid therapy [144]. Therefore, targeting ncRNAs would be a very promising strategy for the treatment of diseases in future. Panz et al. sequenced the transcripts of blood samples from three patients with stable plaques and three patients with unstable plaques by RNA sequencing, and found 62 species of lncRNAs were differentially expressed in unstable plaques. In particular, lncRNA-snhg7-003 was found to be significantly down-regulated in blood samples from patients with unstable plaques [145]. A microarray analysis of plasma from five patients with AS showed that the expression levels of miR-23a-5p, miR-2110, and miR-320a were all up-regulated compared with those in plasma from healthy controls. In parallel, the levels of miR-4439 and miR-8084 were down-regulated in plasma from patients with vulnerable plaques [114]. The miRNA expression profile of human atherosclerotic plaques has been analyzed using a miRNA chip. It has been found that the expression levels of miR-21, miR-22, miR-210, miR-34a, miR-146a/b, miR-19b, and miR-143 were all clearly up-regulated [146, 147]. Moreover, miR-99b, miR-152, and miR-422a have also been shown to be highly expressed in atherosclerotic plaques but not in healthy blood vessels [148]. Li et al. identified miR-27b as having the higher expression levels in atherosclerotic plaques from high-fat diet-fed ApoE-/-mice [116]. In addition, miR-145 has been shown to be overexpressed in plaques in patients with hypertension [149]. High levels of miR-100 have been shown to be associated with coronary plaques [150], and carotid plaque rupture is accompanied by an increase in the serum circR-284/miR-221 ratio [151]. Therefore, ncRNAs may represent potential diagnostic markers to regulate plaque stability. In the context of human atherosclerotic diseases, the precedent of targeted miRNA therapy has been confirmed. So far, it has been shown that knocking down the expression of miR-27b, miR-210, miR-520c-3p, and lncR-ccl2 protects plaque stability in ApoE-/- mice [116, 122, 152, 153]; therefore, these attractive targets have a huge potential to regulate plaque stability. Regarding plaque stability, the plaques in the widely used ApoE-/- or LDLR-/- mice are usually less prone to rupture as compared to atherosclerotic plaques in humans. Over the past decade, ncRNAs have become the main pathophysiological regulators of atherosclerosis, but most of these studies are based on mice, and conducting extensive studies in humans is an urgent problem yet to be solved. It is worth noting that our candidate target gene has a well-documented role in the regulation of plaque stability (Table 2).
The biomarkers of ncRNAs of the stability plaques detected in human serum of atherosclerosis
| ncRNA | Expression | Fold change (FC) | Sample | Reference |
|---|---|---|---|---|
| miR-2110 | up regulation | 2 | blood samples | [114] |
| miR-4439 | down regulation | 1.4 | blood samples | [114] |
| miR-8084 | down regulation | 1.4 | blood samples | [114] |
| miR-23a-5p | up regulation | 4 | blood samples | [114] |
| miR-320a | up regulation | 4.2 | blood samples | [114, 175] |
| miR-200C | up regulation | 2 | tissue sample | [167] |
| miR-210 | up regulation | 3.12 | blood samples | [146, 147] |
| miR-21 | up regulation | 5.3 | blood samples | [146, 147, 164, 175] |
| miR-34a | up regulation | 2 | tissue sample | [146, 147, 176] |
| miR-146a/b | up regulation | 4.15 | blood samples | [146] |
| miR-19b | up regulation | 4 | blood samples | [147] |
| miR-22 | up regulation | - | blood samples | [147] |
| miR-143 | up regulation | 5 | blood samples | [147] |
| miR-99b | up regulation | - | blood samples | [177] |
| miR-152 | up regulation | 4 | blood samples | [177] |
| miR-422a | up regulation | 3.8 | blood samples | [177] |
| miR-145 | up regulation | 2.2 | tissue sample | [148] |
| miR-100 | up regulation | 1.8 | tissue sample | [148] |
| circR-284 | up regulation | 3 | blood samples | [151] |
Conclusion and outlook
The transition of an atherosclerotic plaque from stable to unstable is a multi-step process involving multiple cells. The purpose of this study was to investigate the regulatory effect of ncRNAs targeting endothelial cells, vascular smooth muscle cells, and immune cells on plaque stability. Inflammation, lipid metabolism, and oxidative stress pathway are also closely related to plaque stability. NcRNAs can stably exist in the plasma and other body fluids, and the use of ncRNA-targeted therapies has been widely recognized. The identification of unstable plaques at an early stage will be helpful for effective intervention in patients with CAD and significantly improve their prognosis [154]. LncRNAs are potential regulators of the inflammatory response [155] and can regulate gene expression through epigenetic regulation, transcriptional regulation, post-transcriptional regulation, molecular sponge effects, and as molecular chaperones, thus playing a key role in the regulation of plaque stability[145, 156]. MiRNAs are expressed in the vascular system, participate in vascular inflammation and smooth muscle cell proliferation, and are now recognized as main regulators in the signaling pathways leading to plaque instability [147, 157].
Although lncRNAs transcripts are more numerous than protein-coding genes, their expression, function, and mechanism of action in the relevant cell types during development of the atherosclerotic plaque are unclear. Stable miRNAs in circulating blood can be used as endogenous disease biomarkers [158, 159]. However, there are still some studies on how the release of miRNAs in the plasma reflects the dynamics and regulates the instability of carotid plaque. Some tools such as Tissue Atlas [157] and IMOTA (https://ccb-web.cs.uni-saarland.de/imota/) provide powerful means for the subsequent detection of the expression of miRNAs and their target genes in specific tissues. Some studies have demonstrated that lncRNAs combined with miRNAs play an important role in plaque stability, but whether this mechanism is universal has not been confirmed, and the relationship between lncRNAs and miRNAs remains to be further studied. To date, most studies have been carried out only in vitro; therefore, the role of ncRNAs and their targets needs to be further studied in vivo. In recent years, exosomes have been widely reported as a medium for extracellular transport. Some studies have shown that ncRNAs can be detected in extracellular transport vesicles in exosomes carrying biological media; therefore, it is thought that ncRNAs secreted to extracellular bodies are loaded by exosomes, and the special structure of the exosome's bilayer lipid membranes protect them from degradation, but the specific loading mechanism is not clear [160, 161]. Whether exocrine ncRNAs can enter the cell cycle and play a role in regulating plaque stability still needs to be further explored. In addition, the development of methods for efficiently producing batches of exosomes and ncRNAs that can act on host cells remains a difficult problem. Importantly, discovering ncRNAs from scratch is very costly and often requires many sequencing efforts, without established standards for data analysis methods. In addition, ncRNAs undergo some modifications in vivo, such as methylation and splicing, making it more difficult to study the mechanism of plaque stability. With continuous improvements in RNA sequencing technology, more efficient RNA analysis methods can be applied to a small number of blood samples from specific patients [162]. We believe that great progress will be made in exploring the utility of ncRNAs in the treatment of plaques in the future. Although our knowledge of the role of ncRNAs in plaque stability remains preliminary, this field is worthy of deeper exploration and greater research efforts.
Abbreviations
CVDs: Cardiovascular diseases; CAD: Coronary artery disease; ACS: Acute coronary syndrome; ADP: Adenosine diphosphate; PAF: Platelet activating factor; NcRNAs: Non-coding RNAs; SiRNAs: Small interfering RNAs; EC: Endothelial cell; VSMCs: Vascular smooth muscle cells; PFKFB3: 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase; WNT: Wingless-Related Integration Site; HUVEC: Human umbilical vein endothelial cell; VEGFR2: Vascular endothelial growth factor receptor 2; MMP-2: Matrix metalloproteinase-2; EndMT: Endothelial to mesenchymal transition; VCAM-1: Vascular adhesion molecule-1; CTRP9: C1q/TNF-related protein 9; PEDF: Pigment epithelium-derived factor; CHD: Coronary heart disease; P4HA1: Prolyl 4-hydroxylase subunit alpha-1; TLR2: Toll-like receptor 2.
Acknowledgements
This work was supported by The National Natural Science Foundation of China (No. 81870331, China), and the Qingdao municipal science and technology bureau project (No. 21-1-4-rkjk-12-nsh, China).
Author contributions
X.L. collected materials and wrote the manuscript. T.Y., Z.W. and Y.Y. provided the idea. X.L., B.B. and M.L. are responsible for the schematic diagram within this article. T.Y., Y.Y., Y.M., X.S. and L.Z. helped with the final revision of the article. All authors reviewed the manuscript and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Barrett TJ, Schlegel M, Zhou F, Gorenchtein M, Bolstorff J, Moore KJ. et al. Platelet regulation of myeloid suppressor of cytokine signaling 3 accelerates atherosclerosis. Sci Transl Med. 2019 11
2. Perrotta P, Emini Veseli B, Van der Veken B, Roth L, Martinet W, De Meyer GRY. Pharmacological strategies to inhibit intra-plaque angiogenesis in atherosclerosis. Vascul Pharmacol. 2019;112:72-8
3. Chistiakov DA, Orekhov AN, Bobryshev YV. Contribution of neovascularization and intraplaque haemorrhage to atherosclerotic plaque progression and instability. Acta Physiol (Oxf). 2015;213:539-53
4. Yang Z, Huang Y, Zhu L, Yang K, Liang K, Tan J. et al. SIRT6 promotes angiogenesis and hemorrhage of carotid plaque via regulating HIF-1alpha and reactive oxygen species. Cell Death Dis. 2021;12:77
5. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317-25
6. Fu X, He X, Yang Y, Jiang S, Wang S, Peng X. et al. Identification of transfer RNA-derived fragments and their potential roles in aortic dissection. Genomics. 2021;113:3039-49
7. Dhawan UK, Singhal A, Subramanian M. Dead cell and debris clearance in the atherosclerotic plaque: Mechanisms and therapeutic opportunities to promote inflammation resolution. Pharmacol Res. 2021;170:105699
8. Alloza I, Goikuria H, Idro JL, Trivino JC, Fernandez Velasco JM, Elizagaray E. et al. RNAseq based transcriptomics study of SMCs from carotid atherosclerotic plaque: BMP2 and IDs proteins are crucial regulators of plaque stability. Sci Rep. 2017;7:3470
9. Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the unstable plaque. Prog Cardiovasc Dis. 2002;44:349-56
10. Li M, Yang Y, Wang Z, Zong T, Fu X, Aung LHH. et al. Piwi-interacting RNAs (piRNAs) as potential biomarkers and therapeutic targets for cardiovascular diseases. Angiogenesis. 2021;24:19-34
11. Cheng M, Yang Y, Xin H, Li M, Zong T, He X. et al. Non-coding RNAs in aortic dissection: From biomarkers to therapeutic targets. J Cell Mol Med. 2020;24:11622-37
12. Wang Q, Liu B, Wang Y, Bai B, Yu T, Chu XM. The biomarkers of key miRNAs and target genes associated with acute myocardial infarction. PeerJ. 2020;8:e9129
13. Huang CK, Kafert-Kasting S, Thum T. Preclinical and Clinical Development of Noncoding RNA Therapeutics for Cardiovascular Disease. Circ Res. 2020;126:663-78
14. Huang Y. The novel regulatory role of lncRNA-miRNA-mRNA axis in cardiovascular diseases. J Cell Mol Med. 2018;22:5768-75
15. Poller W, Dimmeler S, Heymans S, Zeller T, Haas J, Karakas M. et al. Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur Heart J. 2018;39:2704-16
16. Kumar S, Boon RA, Maegdefessel L, Dimmeler S, Jo H. Role of Noncoding RNAs in the Pathogenesis of Abdominal Aortic Aneurysm. Circ Res. 2019;124:619-30
17. Zong T, Yang Y, Zhao H, Li L, Liu M, Fu X. et al. tsRNAs: Novel small molecules from cell function and regulatory mechanism to therapeutic targets. Cell Prolif. 2021;54:e12977
18. Liu Y, Yang Y, Wang Z, Fu X, Chu XM, Li Y. et al. Insights into the regulatory role of circRNA in angiogenesis and clinical implications. Atherosclerosis. 2020;298:14-26
19. New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K. et al. Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res. 2013;113:72-7
20. Yang L, Han D, Zhan Q, Li X, Shan P, Hu Y. et al. Blood TfR+ exosomes separated by a pH-responsive method deliver chemotherapeutics for tumor therapy. Theranostics. 2019;9:7680-96
21. Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W. et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. 2016;15:335-43
22. Tian C, Yang Y, Bai B, Wang S, Liu M, Sun RC. et al. Potential of exosomes as diagnostic biomarkers and therapeutic carriers for doxorubicin-induced cardiotoxicity. Int J Biol Sci. 2021;17:1328-38
23. Yang S, Xia YP, Luo XY, Chen SL, Li BW, Ye ZM. et al. Exosomal CagA derived from Helicobacter pylori-infected gastric epithelial cells induces macrophage foam cell formation and promotes atherosclerosis. J Mol Cell Cardiol. 2019;135:40-51
24. Asare Y, Campbell-James TA, Bokov Y, Yu LL, Prestel M, El Bounkari O. et al. Histone Deacetylase 9 Activates IKK to Regulate Atherosclerotic Plaque Vulnerability. Circ Res. 2020;127:811-23
25. Xue Q, He N, Wang Z, Fu X, Aung LHH, Liu Y. et al. Functional roles and mechanisms of ginsenosides from Panax ginseng in atherosclerosis. J Ginseng Res. 2021;45:22-31
26. Grechowa I, Horke S, Wallrath A, Vahl CF, Dorweiler B. Human neutrophil elastase induces endothelial cell apoptosis by activating the PERK-CHOP branch of the unfolded protein response. FASEB J. 2017;31:3868-81
27. Bai B, Yang Y, Ji S, Wang S, Peng X, Tian C. et al. MicroRNA-302c-3p inhibits endothelial cell pyroptosis via directly targeting NOD-, LRR- and pyrin domain-containing protein 3 in atherosclerosis. J Cell Mol Med. 2021
28. Poels K, Schnitzler JG, Waissi F, Levels JHM, Stroes ESG, Daemen M. et al. Inhibition of PFKFB3 Hampers the Progression of Atherosclerosis and Promotes Plaque Stability. Front Cell Dev Biol. 2020;8:581641
29. Di M, Wang L, Li M, Zhang Y, Liu X, Zeng R. et al. Dickkopf1 destabilizes atherosclerotic plaques and promotes plaque formation by inducing apoptosis of endothelial cells through activation of ER stress. Cell Death Dis. 2017;8:e2917
30. Paone S, Baxter AA, Hulett MD, Poon IKH. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell Mol Life Sci. 2019;76:1093-106
31. Donners MM, Wolfs IM, Olieslagers S, Mohammadi-Motahhari Z, Tchaikovski V, Heeneman S. et al. A disintegrin and metalloprotease 10 is a novel mediator of vascular endothelial growth factor-induced endothelial cell function in angiogenesis and is associated with atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:2188-95
32. Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR. et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. 2016;7:11853
33. Chen X, Qian S, Hoggatt A, Tang H, Hacker TA, Obukhov AG. et al. Endothelial Cell-Specific Deletion of P2Y2 Receptor Promotes Plaque Stability in Atherosclerosis-Susceptible ApoE-Null Mice. Arterioscler Thromb Vasc Biol. 2017;37:75-83
34. Perdomo L, Beneit N, Otero YF, Escribano O, Diaz-Castroverde S, Gomez-Hernandez A. et al. Protective role of oleic acid against cardiovascular insulin resistance and in the early and late cellular atherosclerotic process. Cardiovasc Diabetol. 2015;14:75
35. Wang Q, Yang Y, Fu X, Wang Z, Liu Y, Li M. et al. Long noncoding RNA XXYLT1-AS2 regulates proliferation and adhesion by targeting the RNA binding protein FUS in HUVEC. Atherosclerosis. 2020;298:58-69
36. Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118:692-702
37. He X, Lian Z, Yang Y, Wang Z, Fu X, Liu Y. et al. Long Non-coding RNA PEBP1P2 Suppresses Proliferative VSMCs Phenotypic Switching and Proliferation in Atherosclerosis. Mol Ther Nucleic Acids. 2020;22:84-98
38. Han L, Yin Q, Yang L, van Rijn P, Yang Y, Liu Y. et al. Biointerface topography regulates phenotypic switching and cell apoptosis in vascular smooth muscle cells. Biochem Biophys Res Commun. 2020;526:841-7
39. Hasanov Z, Ruckdeschel T, Konig C, Mogler C, Kapel SS, Korn C. et al. Endosialin Promotes Atherosclerosis Through Phenotypic Remodeling of Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol. 2017;37:495-505
40. Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156-64
41. Ackers-Johnson M, Talasila A, Sage AP, Long X, Bot I, Morrell NW. et al. Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arterioscler Thromb Vasc Biol. 2015;35:817-28
42. Li M, Deng WS, Zhang J, Zheng WP, Yu T, Zhou QH. Aligned Electrospun PLLA/Graphene Microfibers with Nanotopographical Surface Modulate the Mitochondrial Responses of Vascular Smooth Muscle Cells. Adv Mater Interfaces. 2021 8
43. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM. et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628-37
44. Ding Y, Zhang M, Zhang W, Lu Q, Cai Z, Song P. et al. AMP-Activated Protein Kinase Alpha 2 Deletion Induces VSMC Phenotypic Switching and Reduces Features of Atherosclerotic Plaque Stability. Circ Res. 2016;119:718-30
45. Zhu XL, Li T, Cao Y, Yao QP, Liu X, Li Y. et al. tRNA-derived fragments tRF(GlnCTG) induced by arterial injury promote vascular smooth muscle cell proliferation. Mol Ther Nucleic Acids. 2021;23:603-13
46. Zou Y, Yang Y, Fu X, He X, Yu TJMT-NA. The regulatory roles of aminoacyl-tRNA synthetase in cardiovascular disease. 2021.
47. Choubey D, Panchanathan R. IFI16, an amplifier of DNA-damage response: Role in cellular senescence and aging-associated inflammatory diseases. Ageing Res Rev. 2016;28:27-36
48. Gray K, Bennett M. Role of DNA damage in atherosclerosis-bystander or participant? Biochem Pharmacol. 2011;82:693-700
49. Grootaert MOJ, Finigan A, Figg NL, Uryga AK, Bennett MR. SIRT6 Protects Smooth Muscle Cells From Senescence and Reduces Atherosclerosis. Circ Res. 2021;128:474-91
50. Gray K, Kumar S, Figg N, Harrison J, Baker L, Mercer J. et al. Effects of DNA damage in smooth muscle cells in atherosclerosis. Circ Res. 2015;116:816-26
51. Liu S, Yang Y, Jiang S, Xu H, Tang N, Lobo A. et al. MiR-378a-5p Regulates Proliferation and Migration in Vascular Smooth Muscle Cell by Targeting CDK1. Front Genet. 2019;10:22
52. Nasser MI, Zhu S, Huang H, Zhao M, Wang B, Ping H. et al. Macrophages: First guards in the prevention of cardiovascular diseases. Life Sci. 2020;250:117559
53. Medina I, Cougoule C, Drechsler M, Bermudez B, Koenen RR, Sluimer J. et al. Hck/Fgr Kinase Deficiency Reduces Plaque Growth and Stability by Blunting Monocyte Recruitment and Intraplaque Motility. Circulation. 2015;132:490-501
54. Kasikara C, Schilperoort M, Gerlach BD, Xue C, Wang X, Zheng Z. et al. Deficiency of macrophage PHACTR1 impairs efferocytosis and promotes atherosclerotic plaque necrosis. J Clin Invest. 2021
55. van Diepen JA, Berbee JF, Havekes LM, Rensen PC. Interactions between inflammation and lipid metabolism: relevance for efficacy of anti-inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis. 2013;228:306-15
56. Catapano AL, Pirillo A, Norata GD. Vascular inflammation and low-density lipoproteins: is cholesterol the link? A lesson from the clinical trials. Br J Pharmacol. 2017;174:3973-85
57. Ma C, Xia R, Yang S, Liu L, Zhang J, Feng K. et al. Formononetin attenuates atherosclerosis via regulating interaction between KLF4 and SRA in apoE(-/-) mice. Theranostics. 2020;10:1090-106
58. Higashi Y, Sukhanov S, Shai SY, Danchuk S, Tang R, Snarski P. et al. Insulin-Like Growth Factor-1 Receptor Deficiency in Macrophages Accelerates Atherosclerosis and Induces an Unstable Plaque Phenotype in Apolipoprotein E-Deficient Mice. Circulation. 2016;133:2263-78
59. Lin J, Liu Q, Zhang H, Huang X, Zhang R, Chen S. et al. C1q/Tumor necrosis factor-related protein-3 protects macrophages against LPS-induced lipid accumulation, inflammation and phenotype transition via PPARgamma and TLR4-mediated pathways. Oncotarget. 2017;8:82541-57
60. Li J, Zhang P, Li T, Liu Y, Zhu Q, Chen T. et al. CTRP9 enhances carotid plaque stability by reducing pro-inflammatory cytokines in macrophages. Biochem Biophys Res Commun. 2015;458:890-5
61. Huang C, Zhang P, Li T, Li J, Liu T, Zuo A. et al. Overexpression of CTRP9 attenuates the development of atherosclerosis in apolipoprotein E-deficient mice. Mol Cell Biochem. 2019;455:99-108
62. Wen H, Liu M, Liu Z, Yang X, Liu X, Ni M. et al. PEDF improves atherosclerotic plaque stability by inhibiting macrophage inflammation response. Int J Cardiol. 2017;235:37-41
63. Otsuka F, Kramer MC, Woudstra P, Yahagi K, Ladich E, Finn AV. et al. Natural progression of atherosclerosis from pathologic intimal thickening to late fibroatheroma in human coronary arteries: A pathology study. Atherosclerosis. 2015;241:772-82
64. Van Vre EA, Ait-Oufella H, Tedgui A, Mallat Z. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:887-93
65. Legein B, Janssen EM, Theelen TL, Gijbels MJ, Walraven J, Klarquist JS. et al. Ablation of CD8alpha(+) dendritic cell mediated cross-presentation does not impact atherosclerosis in hyperlipidemic mice. Sci Rep. 2015;5:15414
66. Dietel B, Cicha I, Voskens CJ, Verhoeven E, Achenbach S, Garlichs CD. Decreased numbers of regulatory T cells are associated with human atherosclerotic lesion vulnerability and inversely correlate with infiltrated mature dendritic cells. Atherosclerosis. 2013;230:92-9
67. Rai V, Rao VH, Shao Z, Agrawal DK. Dendritic Cells Expressing Triggering Receptor Expressed on Myeloid Cells-1 Correlate with Plaque Stability in Symptomatic and Asymptomatic Patients with Carotid Stenosis. PLoS One. 2016;11:e0154802
68. Joo SP, Lee SW, Cho YH, Kim YS, Seo BR, Kim HS. et al. Vasa Vasorum Densities in Human Carotid Atherosclerosis Is Associated with Plaque Development and Vulnerability. J Korean Neurosurg Soc. 2020;63:178-87
69. Kritikou E, Kalogiouri NP, Kolyvira L, Thomaidis NS. Target and Suspect HRMS Metabolomics for the Determination of Functional Ingredients in 13 Varieties of Olive Leaves and Drupes from Greece. Molecules. 2020 25
70. Tsaousi A, Hayes EM, Di Gregoli K, Bond AR, Bevan L, Thomas AC. et al. Plaque Size Is Decreased but M1 Macrophage Polarization and Rupture Related Metalloproteinase Expression Are Maintained after Deleting T-Bet in ApoE Null Mice. PLoS One. 2016;11:e0148873
71. Aukrust P, Sandberg WJ, Otterdal K, Vinge LE, Gullestad L, Yndestad A. et al. Tumor necrosis factor superfamily molecules in acute coronary syndromes. Ann Med. 2011;43:90-103
72. Fu X, Zong T, Yang P, Li L, Wang S, Wang Z. et al. Nicotine: Regulatory roles and mechanisms in atherosclerosis progression. Food Chem Toxicol. 2021;151:112154
73. Yu T, Wang Z, Jie W, Fu X, Li B, Xu H. et al. The kinase inhibitor BX795 suppresses the inflammatory response via multiple kinases. Biochem Pharmacol. 2020;174:113797
74. Ren JL, Chen Y, Zhang LS, Zhang YR, Liu SM, Yu YR. et al. Intermedin1-53 attenuates atherosclerotic plaque vulnerability by inhibiting CHOP-mediated apoptosis and inflammasome in macrophages. Cell Death Dis. 2021;12:436
75. Bullenkamp J, Dinkla S, Kaski JC, Dumitriu IE. Targeting T cells to treat atherosclerosis: odyssey from bench to bedside. Eur Heart J Cardiovasc Pharmacother. 2016;2:194-9
76. Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res. 2014;114:1867-79
77. Ridker PM. Anticytokine Agents: Targeting Interleukin Signaling Pathways for the Treatment of Atherothrombosis. Circ Res. 2019;124:437-50
78. Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y. et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020;11:776
79. Khambhati J, Engels M, Allard-Ratick M, Sandesara PB, Quyyumi AA, Sperling L. Immunotherapy for the prevention of atherosclerotic cardiovascular disease: Promise and possibilities. Atherosclerosis. 2018;276:1-9
80. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15:203-14
81. Hansson GK, Hellstrand M, Rymo L, Rubbia L, Gabbiani G. Interferon gamma inhibits both proliferation and expression of differentiation-specific alpha-smooth muscle actin in arterial smooth muscle cells. J Exp Med. 1989;170:1595-608
82. Ovchinnikova O, Robertson AK, Wagsater D, Folco EJ, Hyry M, Myllyharju J. et al. T-cell activation leads to reduced collagen maturation in atherosclerotic plaques of Apoe(-/-) mice. Am J Pathol. 2009;174:693-700
83. Klingenberg R, Gerdes N, Badeau RM, Gistera A, Strodthoff D, Ketelhuth DF. et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J Clin Invest. 2013;123:1323-34
84. MacLeod AS, Hemmers S, Garijo O, Chabod M, Mowen K, Witherden DA. et al. Dendritic epidermal T cells regulate skin antimicrobial barrier function. J Clin Invest. 2013;123:4364-74
85. Gistera A, Robertson AK, Andersson J, Ketelhuth DF, Ovchinnikova O, Nilsson SK. et al. Transforming growth factor-beta signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin-17-dependent pathway. Sci Transl Med. 2013;5:196ra00
86. Back M, Bu DX, Branstrom R, Sheikine Y, Yan ZQ, Hansson GK. Leukotriene B4 signaling through NF-kappaB-dependent BLT1 receptors on vascular smooth muscle cells in atherosclerosis and intimal hyperplasia. Proc Natl Acad Sci U S A. 2005;102:17501-6
87. Back M, Sultan A, Ovchinnikova O, Hansson GK. 5-Lipoxygenase-activating protein: a potential link between innate and adaptive immunity in atherosclerosis and adipose tissue inflammation. Circ Res. 2007;100:946-9
88. Petri MH, Laguna-Fernandez A, Gonzalez-Diez M, Paulsson-Berne G, Hansson GK, Back M. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc Res. 2015;105:65-74
89. Dong M, Zhou C, Ji L, Pan B, Zheng L. AG1296 enhances plaque stability via inhibiting inflammatory responses and decreasing MMP-2 and MMP-9 expression in ApoE-/- mice. Biochem Biophys Res Commun. 2017;489:426-31
90. Ridker PM. From CANTOS to CIRT to COLCOT to Clinic: Will All Atherosclerosis Patients Soon Be Treated With Combination Lipid-Lowering and Inflammation-Inhibiting Agents? Circulation. 2020;141:787-9
91. Yu XH, Zhang DW, Zheng XL, Tang CK. Cholesterol transport system: An integrated cholesterol transport model involved in atherosclerosis. Prog Lipid Res. 2019;73:65-91
92. Rinne P, Rami M, Nuutinen S, Santovito D, van der Vorst EPC, Guillamat-Prats R. et al. Melanocortin 1 Receptor Signaling Regulates Cholesterol Transport in Macrophages. Circulation. 2017;136:83-97
93. Du C, Lin X, Xu W, Zheng F, Cai J, Yang J. et al. Sulfhydrated Sirtuin-1 Increasing Its Deacetylation Activity Is an Essential Epigenetics Mechanism of Anti-Atherogenesis by Hydrogen Sulfide. Antioxid Redox Signal. 2019;30:184-97
94. Holleboom AG, Jakulj L, Franssen R, Decaris J, Vergeer M, Koetsveld J. et al. In vivo tissue cholesterol efflux is reduced in carriers of a mutation in APOA1. J Lipid Res. 2013;54:1964-71
95. Forstermann U, Xia N, Li H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ Res. 2017;120:713-35
96. Roy A, Saqib U, Baig MS. NOS1-mediated macrophage and endothelial cell interaction in the progression of atherosclerosis. Cell Biol Int. 2021
97. Quesada IM, Lucero A, Amaya C, Meijles DN, Cifuentes ME, Pagano PJ. et al. Selective inactivation of NADPH oxidase 2 causes regression of vascularization and the size and stability of atherosclerotic plaques. Atherosclerosis. 2015;242:469-75
98. Zhu M, Tang H, Tang X, Ma X, Guo D, Chen F. BMAL1 suppresses ROS-induced endothelial-to-mesenchymal transition and atherosclerosis plaque progression via BMP signaling. Am J Transl Res. 2018;10:3150-61
99. Ruotsalainen AK, Inkala M, Partanen ME, Lappalainen JP, Kansanen E, Makinen PI. et al. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc Res. 2013;98:107-15
100. Ruotsalainen AK, Lappalainen JP, Heiskanen E, Merentie M, Sihvola V, Napankangas J. et al. Nuclear factor E2-related factor 2 deficiency impairs atherosclerotic lesion development but promotes features of plaque instability in hypercholesterolaemic mice. Cardiovasc Res. 2019;115:243-54
101. Barajas B, Che N, Yin F, Rowshanrad A, Orozco LD, Gong KW. et al. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler Thromb Vasc Biol. 2011;31:58-66
102. Stewart RAH, Held C, Hadziosmanovic N, Armstrong PW, Cannon CP, Granger CB. et al. Physical Activity and Mortality in Patients With Stable Coronary Heart Disease. J Am Coll Cardiol. 2017;70:1689-700
103. Liu S, Yang Y, Jiang S, Tang N, Tian J, Ponnusamy M. et al. Understanding the role of non-coding RNA (ncRNA) in stent restenosis. Atherosclerosis. 2018;272:153-61
104. Pellegrin M, Aubert JF, Bouzourene K, Amstutz C, Mazzolai L. Voluntary Exercise Stabilizes Established Angiotensin II-Dependent Atherosclerosis in Mice through Systemic Anti-Inflammatory Effects. PLoS One. 2015;10:e0143536
105. Hsu JJ, Fong F, Patel R, Qiao R, Lo K, Soundia A. et al. Changes in microarchitecture of atherosclerotic calcification assessed by (18)F-NaF PET and CT after a progressive exercise regimen in hyperlipidemic mice. J Nucl Cardiol. 2020
106. Pellegrin M, Miguet-Alfonsi C, Bouzourene K, Aubert JF, Deckert V, Berthelot A. et al. Long-term exercise stabilizes atherosclerotic plaque in ApoE knockout mice. Med Sci Sports Exerc. 2009;41:2128-35
107. Cardinot TM, Lima TM, Moretti AI, Koike MK, Nunes VS, Cazita PM. et al. Preventive and therapeutic moderate aerobic exercise programs convert atherosclerotic plaques into a more stable phenotype. Life Sci. 2016;153:163-70
108. Wu WQ, Peng S, Wan XQ, Lin S, Li LY, Song ZY. Physical exercise inhibits atherosclerosis development by regulating the expression of neuropeptide Y in apolipoprotein E-deficient mice. Life Sci. 2019;237:116896
109. Araldi E, Suarez Y. MicroRNAs as regulators of endothelial cell functions in cardiometabolic diseases. Biochim Biophys Acta. 2016;1861:2094-103
110. Tang N, Jiang S, Yang Y, Liu S, Ponnusamy M, Xin H. et al. Noncoding RNAs as therapeutic targets in atherosclerosis with diabetes mellitus. Cardiovasc Ther. 2018;36:e12436
111. Yang Y, Yu T, Jiang S, Zhang Y, Li M, Tang N. et al. miRNAs as potential therapeutic targets and diagnostic biomarkers for cardiovascular disease with a particular focus on WO2010091204. Expert Opin Ther Pat. 2017;27:1021-9
112. Li XH, Xue WL, Wang MJ, Zhou Y, Zhang CC, Sun C. et al. H2S regulates endothelial nitric oxide synthase protein stability by promoting microRNA-455-3p expression. Sci Rep. 2017;7:44807
113. Yu F, Zhang Y, Wang Z, Gong W, Zhang C. Hsa_circ_0030042 regulates abnormal autophagy and protects atherosclerotic plaque stability by targeting eIF4A3. Theranostics. 2021;11:5404-17
114. Yang S, Ye ZM, Chen S, Luo XY, Chen SL, Mao L. et al. MicroRNA-23a-5p promotes atherosclerotic plaque progression and vulnerability by repressing ATP-binding cassette transporter A1/G1 in macrophages. J Mol Cell Cardiol. 2018;123:139-49
115. Fan Z, Zhang Y, Xiao D, Ma J, Liu H, Shen L. et al. Long noncoding RNA UC.98 stabilizes atherosclerotic plaques by promoting the proliferation and adhesive capacity in murine aortic endothelial cells. Acta Biochim Biophys Sin (Shanghai). 2020;52:141-9
116. Qun L, Wenda X, Weihong S, Jianyang M, Wei C, Fangzhou L. et al. miRNA-27b modulates endothelial cell angiogenesis by directly targeting Naa15 in atherogenesis. Atherosclerosis. 2016;254:184-92
117. Bai J, Liu J, Fu Z, Feng Y, Wang B, Wu W. et al. Silencing lncRNA AK136714 reduces endothelial cell damage and inhibits atherosclerosis. Aging (Albany NY). 2021;13:14159-69
118. Halimulati M, Duman B, Nijiati J, Aizezi A. Long noncoding RNA TCONS_00024652 regulates vascular endothelial cell proliferation and angiogenesis via microRNA-21. Exp Ther Med. 2018;16:3309-16
119. Gabunia K, Herman AB, Ray M, Kelemen SE, England RN, DeLa Cadena R. et al. Induction of MiR133a expression by IL-19 targets LDLRAP1 and reduces oxLDL uptake in VSMC. J Mol Cell Cardiol. 2017;105:38-48
120. Lovren F, Pan Y, Quan A, Singh KK, Shukla PC, Gupta N. et al. MicroRNA-145 targeted therapy reduces atherosclerosis. Circulation. 2012;126:S81-90
121. Chen W, Yu F, Di M, Li M, Chen Y, Zhang Y. et al. MicroRNA-124-3p inhibits collagen synthesis in atherosclerotic plaques by targeting prolyl 4-hydroxylase subunit alpha-1 (P4HA1) in vascular smooth muscle cells. Atherosclerosis. 2018;277:98-107
122. Eken SM, Jin H, Chernogubova E, Li Y, Simon N, Sun C. et al. MicroRNA-210 Enhances Fibrous Cap Stability in Advanced Atherosclerotic Lesions. Circ Res. 2017;120:633-44
123. Jin H, Li DY, Chernogubova E, Sun C, Busch A, Eken SM. et al. Local Delivery of miR-21 Stabilizes Fibrous Caps in Vulnerable Atherosclerotic Lesions. Mol Ther. 2018;26:1040-55
124. Biscetti F, Tinelli G, Rando MM, Nardella E, Cecchini AL, Angelini F. et al. Association between carotid plaque vulnerability and high mobility group box-1 serum levels in a diabetic population. Cardiovasc Diabetol. 2021;20:114
125. Liang WJ, Zhou SN, Shan MR, Wang XQ, Zhang M, Chen Y. et al. AMPKα inactivation destabilizes atherosclerotic plaque in streptozotocin-induced diabetic mice through AP-2α/miRNA-124 axis. J Mol Med (Berl). 2018;96:403-12
126. Yu X, Li Z, Chen G, Wu WK. MicroRNA-10b Induces Vascular Muscle Cell Proliferation Through Akt Pathway by Targeting TIP30. Curr Vasc Pharmacol. 2015;13:679-86
127. Wang D, Wang W, Lin W, Yang W, Zhang P, Chen M. et al. Apoptotic cell induction of miR-10b in macrophages contributes to advanced atherosclerosis progression in ApoE-/- mice. Cardiovasc Res. 2018;114:1794-805
128. Gong FH, Cheng WL, Wang H, Gao M, Qin JJ, Zhang Y. et al. Reduced atherosclerosis lesion size, inflammatory response in miR-150 knockout mice via macrophage effects. J Lipid Res. 2018;59:658-69
129. Bras JP, Silva AM, Calin GA, Barbosa MA, Santos SG, Almeida MI. miR-195 inhibits macrophages pro-inflammatory profile and impacts the crosstalk with smooth muscle cells. PLoS One. 2017;12:e0188530
130. Di Gregoli K, Jenkins N, Salter R, White S, Newby AC, Johnson JL. MicroRNA-24 regulates macrophage behavior and retards atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:1990-2000
131. Sanz J, Fayad ZA. Imaging of atherosclerotic cardiovascular disease. Nature. 2008;451:953-7
132. Okura H, Asawa K, Kubo T, Taguchi H, Toda I, Yoshiyama M. et al. Incidence and predictors of plaque rupture in the peripheral arteries. Circ Cardiovasc Interv. 2010;3:63-70
133. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852-66
134. Yu T, Zhang Y, Li PF. Mitochondrial Ubiquitin Ligase in Cardiovascular Disorders. Adv Exp Med Biol. 2017;982:327-33
135. Jacob SS, Hassan M, Yacoub MH. Utility of mass spectrometry for the diagnosis of the unstable coronary plaque. Glob Cardiol Sci Pract. 2015;2015:25
136. Garcia-Garcia HM, Jang IK, Serruys PW, Kovacic JC, Narula J, Fayad ZA. Imaging plaques to predict and better manage patients with acute coronary events. Circ Res. 2014;114:1904-17
137. Fujii K, Hao H, Ohyanagi M, Masuyama T. Intracoronary imaging for detecting vulnerable plaque. Circ J. 2013;77:588-95
138. May JM, Bylicky M, Chopra S, Coleman CN, Aryankalayil MJ. Long and short non-coding RNA and radiation response: a review. Transl Res. 2021
139. Lu H, Shi C, Wang S, Yang C, Wan X, Luo Y. et al. Identification of NCAPH as a biomarker for prognosis of breast cancer. Mol Biol Rep. 2020;47:7831-42
140. Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol. 2016;16:279-94
141. Amit M, Takahashi H, Dragomir MP, Lindemann A, Gleber-Netto FO, Pickering CR. et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature. 2020;578:449-54
142. Tian Z, Liang G, Cui K, Liang Y, Wang Q, Lv S. et al. Insight Into the Prospects for RNAi Therapy of Cancer. Front Pharmacol. 2021;12:644718
143. Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2017;16:167-79
144. Yao C, Wu W, Tang H, Jia X, Tang J, Ruan X. et al. Self-assembly of stem cell membrane-camouflaged nanocomplex for microRNA-mediated repair of myocardial infarction injury. Biomaterials. 2020;257:120256
145. Pan Z, Fan Z, Ma J, Liu H, Shen L, He B. et al. Profiling and functional characterization of circulation LncRNAs that are associated with coronary atherosclerotic plaque stability. Am J Transl Res. 2019;11:3801-15
146. Raitoharju E, Lyytikainen LP, Levula M, Oksala N, Mennander A, Tarkka M. et al. miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis. 2011;219:211-7
147. Markus B, Grote K, Worsch M, Parviz B, Boening A, Schieffer B. et al. Differential Expression of MicroRNAs in Endarterectomy Specimens Taken from Patients with Asymptomatic and Symptomatic Carotid Plaques. PLoS One. 2016;11:e0161632
148. Cipollone F, Felicioni L, Sarzani R, Ucchino S, Spigonardo F, Mandolini C. et al. A unique microRNA signature associated with plaque instability in humans. Stroke. 2011;42:2556-63
149. Santovito D, Mandolini C, Marcantonio P, De Nardis V, Bucci M, Paganelli C. et al. Overexpression of microRNA-145 in atherosclerotic plaques from hypertensive patients. Expert Opin Ther Targets. 2013;17:217-23
150. Soeki T, Yamaguchi K, Niki T, Uematsu E, Bando S, Matsuura T. et al. Plasma microRNA-100 is associated with coronary plaque vulnerability. Circ J. 2015;79:413-8
151. Bazan HA, Hatfield SA, Brug A, Brooks AJ, Lightell DJ Jr, Woods TC. Carotid Plaque Rupture Is Accompanied by an Increase in the Ratio of Serum circR-284 to miR-221 Levels. Circ Cardiovasc Genet. 2017 10
152. Wang J, Hu X, Hu X, Gao F, Li M, Cui Y. et al. MicroRNA-520c-3p targeting of RelA/p65 suppresses atherosclerotic plaque formation. Int J Biochem Cell Biol. 2021;131:105873
153. Khyzha N, Khor M, DiStefano PV, Wang L, Matic L, Hedin U. et al. Regulation of CCL2 expression in human vascular endothelial cells by a neighboring divergently transcribed long noncoding RNA. Proc Natl Acad Sci U S A. 2019;116:16410-9
154. Yang X, Yang Y, Guo J, Meng Y, Li M, Yang P. et al. Targeting the epigenome in in-stent restenosis: from mechanisms to therapy. Mol Ther Nucleic Acids. 2021;23:1136-60
155. Mathy NW, Chen XM. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J Biol Chem. 2017;292:12375-82
156. Zhang K, Shi ZM, Chang YN, Hu ZM, Qi HX, Hong W. The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene. 2014;547:1-9
157. Wang D, Atanasov AG. The microRNAs Regulating Vascular Smooth Muscle Cell Proliferation: A Minireview. Int J Mol Sci. 2019 20
158. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344-52
159. Keller A, Leidinger P, Bauer A, Elsharawy A, Haas J, Backes C. et al. Toward the blood-borne miRNome of human diseases. Nat Methods. 2011;8:841-3
160. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14:195-208
161. Mori MA, Ludwig RG, Garcia-Martin R, Brandao BB, Kahn CR. Extracellular miRNAs: From Biomarkers to Mediators of Physiology and Disease. Cell Metab. 2019;30:656-73
162. He X, Yang Y, Wang Q, Wang J, Li S, Li C. et al. Expression profiles and potential roles of transfer RNA-derived small RNAs in atherosclerosis. J Cell Mol Med. 2021
163. Bao MH, Li GY, Huang XS, Tang L, Dong LP, Li JM. Long Noncoding RNA LINC00657 Acting as a miR-590-3p Sponge to Facilitate Low Concentration Oxidized Low-Density Lipoprotein-Induced Angiogenesis. Mol Pharmacol. 2018;93:368-75
164. He W, Zhu L, Huang Y, Zhang Y, Shen W, Fang L. et al. The relationship of MicroRNA-21 and plaque stability in acute coronary syndrome. Medicine (Baltimore). 2019;98:e18049
165. Cao J, Zhang K, Zheng J, Dong R. MicroRNA-146a and -21 cooperate to regulate vascular smooth muscle cell proliferation via modulation of the Notch signaling pathway. Mol Med Rep. 2015;11:2889-95
166. Gao L, Zeng H, Zhang T, Mao C, Wang Y, Han Z. et al. MicroRNA-21 deficiency attenuated atherogenesis and decreased macrophage infiltration by targeting Dusp-8. Atherosclerosis. 2019;291:78-86
167. Magenta A, Sileno S, D'Agostino M, Persiani F, Beji S, Paolini A. et al. Atherosclerotic plaque instability in carotid arteries: miR-200c as a promising biomarker. Clin Sci (Lond). 2018;132:2423-36
168. Zhai C, Cong H, Hou K, Hu Y, Zhang J, Zhang Y. et al. Effects of miR-124-3p regulation of the p38MAPK signaling pathway via MEKK3 on apoptosis and proliferation of macrophages in mice with coronary atherosclerosis. Adv Clin Exp Med. 2020;29:803-12
169. Liang WJ, Zhou SN, Shan MR, Wang XQ, Zhang M, Chen Y. et al. AMPKalpha inactivation destabilizes atherosclerotic plaque in streptozotocin-induced diabetic mice through AP-2alpha/miRNA-124 axis. J Mol Med (Berl). 2018;96:403-12
170. Li S, Geng Q, Chen H, Zhang J, Cao C, Zhang F. et al. The potential inhibitory effects of miR19b on vulnerable plaque formation via the suppression of STAT3 transcriptional activity. Int J Mol Med. 2018;41:859-67
171. Welten SMJ, de Jong RCM, Wezel A, de Vries MR, Boonstra MC, Parma L. et al. Inhibition of 14q32 microRNA miR-495 reduces lesion formation, intimal hyperplasia and plasma cholesterol levels in experimental restenosis. Atherosclerosis. 2017;261:26-36
172. Di Gregoli K, Mohamad Anuar NN, Bianco R, White SJ, Newby AC, George SJ. et al. MicroRNA-181b Controls Atherosclerosis and Aneurysms Through Regulation of TIMP-3 and Elastin. Circ Res. 2017;120:49-65
173. Wezel A, Welten SM, Razawy W, Lagraauw HM, de Vries MR, Goossens EA. et al. Inhibition of MicroRNA-494 Reduces Carotid Artery Atherosclerotic Lesion Development and Increases Plaque Stability. Ann Surg. 2015;262:841-7 discussion 7-8
174. Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S. et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921-31
175. Chen C, Wang Y, Yang S, Li H, Zhao G, Wang F. et al. MiR-320a contributes to atherogenesis by augmenting multiple risk factors and down-regulating SRF. J Cell Mol Med. 2015;19:970-85
176. Zhong X, Li P, Li J, He R, Cheng G, Li Y. Downregulation of microRNA34a inhibits oxidized lowdensity lipoproteininduced apoptosis and oxidative stress in human umbilical vein endothelial cells. Int J Mol Med. 2018;42:1134-44
177. Bidzhekov K, Gan L, Denecke B, Rostalsky A, Hristov M, Koeppel TA. et al. microRNA expression signatures and parallels between monocyte subsets and atherosclerotic plaque in humans. Thromb Haemost. 2012;107:619-25
Author contact
![]() Corresponding authors: Tao Yu, PhD, Department of Cardiac Ultrasound, The Affiliated hospital of Qingdao University, No. 16 Jiangsu Road, Qingdao 266000, China; Center for Regenerative Medicine, Institute for translational medicine, School of Basic Medicine, Qingdao University, 38 Deng Zhou Road, Qingdao 266021, China. Tel: +86-532-82991791; fax: +86-532-82991791; E-mail: yutao0112edu.cn.
Corresponding authors: Tao Yu, PhD, Department of Cardiac Ultrasound, The Affiliated hospital of Qingdao University, No. 16 Jiangsu Road, Qingdao 266000, China; Center for Regenerative Medicine, Institute for translational medicine, School of Basic Medicine, Qingdao University, 38 Deng Zhou Road, Qingdao 266021, China. Tel: +86-532-82991791; fax: +86-532-82991791; E-mail: yutao0112edu.cn.