Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. Vascular calcification in...
2. Roles of the pro-inflammatory...
3. Signaling pathways involved...
4. Stimulators that regulate...
5. Discussion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(15):4353-4364. doi:10.7150/ijbs.66537 This issue Cite
Review
IL-1β in atherosclerotic vascular calcification: From bench to bedside
Jialing Shen1*, Ming Zhao2*, Chunxiang Zhang3,6 ![]() , Xiaolei Sun1,2,3,4,5,6,7,8
, Xiaolei Sun1,2,3,4,5,6,7,8 ![]()
1. Department of General Surgery (Vascular Surgery), the Affiliated Hospital of Southwest Medical University, Luzhou 646000, China.
2. Department of Interventional Medicine, the Affiliated Hospital of Southwest Medical University, Luzhou 646000, China.
3. Laboratory of Nucleic Acids in Medicine for National high-level talents, Southwest Medical University, Luzhou 646000, China.
4. School of Cardiovascular Medicine and Sciences, King's College London British Heart Foundation Centre of Research Excellence, Faculty of Life Science and Medicine, King's College London, London SE5 9NU, United Kingdom.
5. Centre for Clinical Pharmacology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London EC1M 6BQ, United Kingdom.
6. Key Laboratory of Medical Electrophysiology, Ministry of Education & Medical Electrophysiological Key Laboratory of Sichuan Province, Collaborative Innovation Center for Prevention and Treatment of Cardiovascular Disease of Sichuan Province, Institute of Cardiovascular Research, Southwest Medical University, Luzhou 646000, China.
7. Cardiovascular and Metabolic Diseases Key Laboratory of Luzhou, Luzhou, 646000, China.
8. Nuclear Medicine and Molecular Imaging Key Laboratory of Sichuan Province, Luzhou 646000, China.
* These authors have contributed equally to this study.
Received 2021-8-27; Accepted 2021-10-11; Published 2021-10-22
Abstract

Atherosclerotic vascular calcification contributes to increased risk of death in patients with cardiovascular diseases. Assessing the type and severity of inflammation is crucial in the treatment of numerous cardiovascular conditions. IL-1β, a potent proinflammatory cytokine, plays diverse roles in the pathogenesis of atherosclerotic vascular calcification. Several large-scale, population cohort trials have shown that the incidence of cardiovascular events is clinically reduced by the administration of anti-IL-1β therapy. Anti-IL-1β therapy might reduce the incidence of cardiovascular events by affecting atherosclerotic vascular calcification, but the mechanism underlying this effect remains unclear. In this review, we summarize current knowledge on the role of IL-1β in atherosclerotic vascular calcification, and describe the latest results reported in clinical trials evaluating anti-IL-1β therapies for the treatment of cardiovascular diseases. This review will aid in improving current understanding of the pathophysiological roles of IL-1β and mechanisms underlying its activity.
Keywords: IL-1β, vascular calcification, cardiovascular events, signaling pathways
Introduction
Atherosclerosis is initiated by disturbances in blood flow and numerous systemic factors including history of smoking, hypertension, diabetes, or hyperlipidemia [1-3]. Atherosclerosis is part of the common pathophysiological basis for most cerebrovascular and cardiovascular diseases [4].
Vascular calcification is a ubiquitous pathological process in atherosclerosis [5, 6]. The annual incidence of atherosclerotic vascular calcification in the general population ranges from less than 5% in individuals under 50 years of age to greater than 12% in individuals over 80 years of age [7]. Imbalance in the calcium dynamics of atherosclerotic vessels can lead to reduced arterial compliance and impaired vascular hemodynamic responses [8, 9]. Clinically, atherosclerotic vascular calcification (Figure 1) is implicated in aortic valve stenosis, congestive heart failure, myocardial infarction, peripheral arterial occlusion, and arterial hypertension [10], which lead to high rates of morbidity and mortality [11, 12].
Vascular calcification in cardiovascular vessels and valves. (A) and (B) show vascular calcification in a 74-year-old woman with a history of rheumatic heart disease and hyperuricemia. (A) Representative CT image of vascular calcification in the left anterior descending (LAD, yellow arrow) and left circumflex artery (LCX, blue arrow) of the left coronary artery (LCA). (B) Representative CT image of vascular calcification in the mitral valve. Images (C), (D), and (E) show vascular calcification in a 56-year-old man with a history of arteriosclerosis obliterans (ASO), severe chronic limb ischemia accompanied by intractable and infectious ulcers, and type 2 diabetes mellitus (T2DM). (C) Representative infrarenal CT angiography (CTA) image shows multiple vascular calcifications. (D) Severe vascular calcification in the infrarenal aorta. (E) Severe vascular calcification in the distal popliteal artery and branches with stenosis and occlusion.
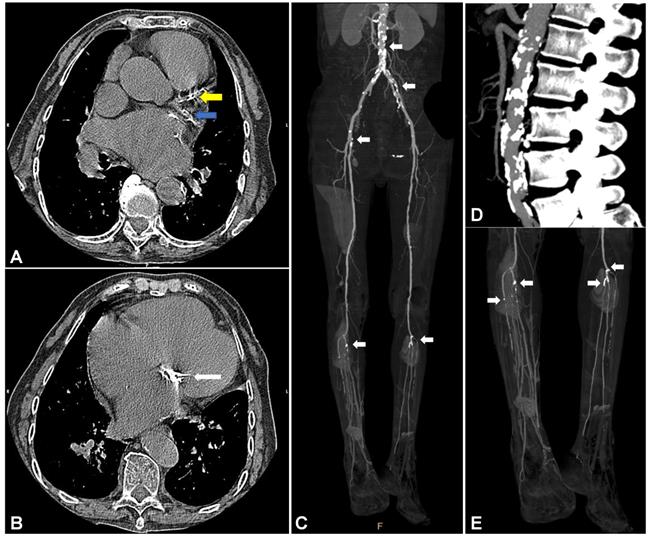
Calcification of the aortic arch was first reported in relation to the risk of coronary heart disease in a cohort study examining 116,309 individuals with a median follow-up of 28-years [12]. That study was also used to assess calcification of the coronary artery in asymptomatic individuals who experienced coronary events such as myocardial infarction or coronary death [11], and calcification of the abdominal aorta in individuals with increased risk for coronary heart disease [13]. Under conditions of atherosclerosis, vascular calcification progresses, and is associated with cardiovascular events and increased mortality [7, 13-15]. A meta-analysis examining 218,080 individuals with a mean follow-up of 10.1-years indicated that vascular calcification results in 4.63-fold higher risk for all-cause mortality, a 3.94-fold higher risk for cardiovascular mortality, and a 3.74-fold higher risk for any coronary events [15].
Although vascular calcification was previously considered passive and degenerative, it is currently recognized as an active and regulated pathobiological process. Vascular calcification, which shares many features with inflammatory atherosclerosis, may be treatable and preventable. The initial feature of calcified atherosclerotic vessels is activation of inflammation [16, 17]. Cytokines secreted by inflammatory cells result in smooth muscle cell (SMC) apoptosis or SMC trans-differentiation into an osteochondrogenic cellular phenotype. Both of these events may contribute to mineral deposition in the plaque [18, 19]. Among the numerous currently known inflammatory signaling pathways, those involving IL-1β are particularly implicated in atherosclerotic vascular calcification. In this review, we summarize the specific roles of IL-1β in atherosclerotic vascular calcification. This information will help delineate the pathogenesis of atherosclerosis, and will help uncover other, currently unknown, mechanisms involving IL-1β, in order to develop anti-IL-1β therapeutics for reducing the incidence of cardiovascular events [20].
1. Vascular calcification in atherosclerosis
Vascular calcification was previously believed to result from passive deposition of calcium and phosphorus on blood-vessel wall. Recent studies have shown, however, that vascular calcification is an active, reversible, highly regulated, and preventable process that is similar to physiological bone development [21]. The two pathobiological mechanisms currently known to underlie vascular calcification are induction of osteogenesis and loss of inhibition of mineralization [22]. Abnormalities in Ca2+ and phosphate metabolism [23], combined with increased oxidative stress [24], stimulation of inflammatory factors [25], dysregulation of certain miRNAs such as miR-34a [26], and disorders of lipid metabolism [27] lead to decreased expression of α-SMA, SM22α, and smooth muscle-myosin heavy chain, which are necessary for the maintenance of vascular function. Meanwhile, the expression of Runx2, SOX9, SP7, MSx2, and OPN, which are factors related to bone formation, is upregulated. This cascade promotes the activity of ALP and expression of BMP-2, resulting in osteogenic or chondral differentiation of VSMCs [8, 28]. Inhibition of autophagy [29], matrix remodeling [30], cellular apoptosis, and matrix vesicles [31] may also accelerate vascular calcification. Recent studies in mouse models and humans have shown that blood vessels can produce and secrete factors, including PPI, OPN, OPG, MGP, FET-A, and Smad 6, which inhibit mineralization or vascular calcification [32-35]. Inhibition of expression in these factors can lead to the initiation of vascular calcification.
Atherosclerosis usually leads to vascular calcification [36]. In early stages of atherosclerosis, bone-related proteins can be detected histologically, and both the occurrence and development of vascular calcification are associated with the process of atherosclerosis [37]. In the past decade, studies on coronary atherosclerotic calcification were mainly focused on lumen stenosis and plaque vulnerability. While calcification of atherosclerotic plaque core does not increase the vulnerability of the plaque [38], microcalcification of the fibrous caps on atherosclerotic plaques increases circumferential stress, which can, indeed, increase the vulnerability of the plaque. Overall, the size, shape, and location of the microcalcifications can directly define the vulnerability of the plaques [39]. Electron beam computed tomography (CT) and intravascular ultrasound are currently used to detect calcifications in the arteries of 90% of patients with coronary atherosclerotic heart disease. Thus, the degree of vascular calcification may be directly related to the degree of vascular stenosis and risk for cardiovascular events in patients with atherosclerotic diseases [40].
Hence, an accurate, safe, and reproducible clinical detection technique is extremely important for the diagnosis, prevention, and treatment of vascular-calcification-related diseases. In the early stages of vascular calcification, molecular imaging technology, such as optical near-infrared fluorescence imaging [41], can be used to detect osteogenesis early at the (sub)cellular levels [42]. The detection and quantification of advanced vascular calcification can be performed by CT, intravascular ultrasound (IVUS), transthoracic echocardiography, pulse wave velocity measurement, planar radiographs, and magnetic resonance imaging (MRI) [43]. However, there are currently no satisfactory therapeutic approaches to vascular calcification in clinical practice. Even statins, which have been shown to decrease osteogenesis in vivo and vitro [44, 45], have failed to prove beneficial in clinical trials [46]. Preventive measures are critical to decreasing the occurrence of vascular calcification and delaying the progression of vascular calcification. Although the modification of risk conditions (hyperglycemia, uremia, hypertension, hyperlipidemia, secondary hyperparathyroidism, and metabolic syndrome) and other factors (dietary phosphorous, oral activated charcoal, vitamins K and D, magnesium oxide, warfarin, bisphosphonates, antioxidants, estrogen, fetuin, osteopontin, anti-inflammatory agents, mineralocorticoids) might be possible [47-52], the details of any underlying mechanisms are not fully understood, and large-scale clinical trials have been limited.
2. Roles of the pro-inflammatory cytokine IL-1β
IL-1β is implicated in numerous inflammation-related diseases including rheumatoid arthritis, inflammatory bowel disease, osteoarthritis, type 2 diabetes, gout, multiple sclerosis, and Alzheimer's disease [53-55]. As a potent, pro-inflammatory cytokine with a wide range of biological effects, IL-1β is synthesized and secreted by various cells including macrophages, fibroblasts, B lymphocytes, natural killer cells, and smooth muscle cells [56]. As canonical negative feedback regulation to control IL-1β expression and secretion. IL-10 suppresses IL-1β and IL-1β-induced IL-1Ra [57, 58], the natural antagonist of IL-1β. There is also another mechanism underlying IL-1β-TGF-β1-related feedback to decrease the production of IL-1β [59, 60]. One mechanism by which downstream and upstream negative feedback modulate IL-1β via paracrine secretion of interferons has been reported [61]. Of which, IFN-II/IFNγ mediated the downstream regulation via its inhibition of the nitrosylation of NLRP3 inflammasome [62]. While IFN-I/IFNβ mediated the upstream regulation via an IL-10 and STAT-3 dependent manner. The IFN-I/IFNβ secreted predominantly by fibroblasts responded to IL-1β elevation is a strong switch to attenuate the IL-1β induced inflammation at later stages [63]. Recently, the secretion of IL-1β was revealed to be modulated by a negative feedback loop including IL-1β/NF-κB/TIR8/IL-1β during IL‐1β‐induced epithelial‐myofibroblast trans-differentiation [64].
IL-1β also plays an important role in the development of cardiovascular diseases [65]. Various factors can activate IL-1β production and pyrolysis, enabling the participation of IL-1β in the pathophysiological process of cardiovascular diseases [66]. Avolio et al. [67] and Qi et al. [68] discovered excessive activation of IL-1β in the hypothalamic paraventricular nucleus under conditions of hypertension, and found that inhibition of IL-1β can alleviate hypertension by reducing the activity of the sympathetic nervous system. Under conditions of hypertension, IL-1β also participates in the remodeling of aortic blood vessels by activating the renin-angiotensin-aldosterone system [69]. Coronary artery thrombosis and blockade of coronary blood flow resulting from ruptured coronary artery plaques are the main causes of acute myocardial infarction. The ischemic or necrotic myocardium can generate increased levels of ATP and oxidative stress products, which then stimulate the expression of IL-1β [70, 71].
Atrial fibrillation is a common type of arrhythmia possessing a complex mechanism [72]. Increased levels of IL-1β result in atrial myoelectrical and structural remodeling, and induction of atrial fibrillation [73]. Activated Macrophage‐inducible C‐type lectin (Mincle) causes increased expression of IL-1β in the microglial cells residing in the paraventricular nucleus. At 24 hours after myocardial infarction, increased levels of IL-1β are increased further, and are accompanied by excessive activation of the sympathetic nerves. This cascade can result in the occurrence of a malignant ventricular arrhythmia [74].
Recent studies have shown that therapeutic targeting of inflammatory factors can improve cardiovascular outcomes in patients with a history of myocardial infarction [75]. In the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS), administration of the monoclonal IL-1β-neutralizing antibody canakinumab successfully reduced the rate of recurrent cardiovascular events by 17% [20]. However, not all anti-inflammatory therapies benefit in the protection from cardiovascular events [75, 76], the detailed mechanism of anti-IL-1β-mediated activity remains unclear.
3. Signaling pathways involved in IL-1β-mediated regulation of atherosclerotic vascular calcification
3.1. IL-1β induces endothelial-to-mesenchymal transition and promotes atherosclerotic vascular calcification
Endothelial-to-mesenchymal transition (EndMT), a specific form of epithelial-to-mesenchymal transition (EMT), is characterized by the loss of endothelial features and acquisition of specific mesenchymal markers in endothelial cells [77, 78]. EndMT is known to participate in the pathogenesis of atherosclerosis [79-82] and also occurs in atherosclerotic vascular calcification [83, 84].
IL-1β-mediated EndMT contributes to the pathogenesis of various diseases. Lee et al. have shown that injury-induced IL-1β expression induces EndMT in corneal fibrosis by upregulating the expression of FGF-2 [85]. Recombinant IL-1β induces EndMT in human esophageal microvascular endothelial cells, highlighting the important role of IL-1β in early-stage esophageal adenocarcinoma [86]. IL-1β-induced EndMT also impairs the angiogenic potential of human umbilical vein endothelial cells (HUVECs) [87].
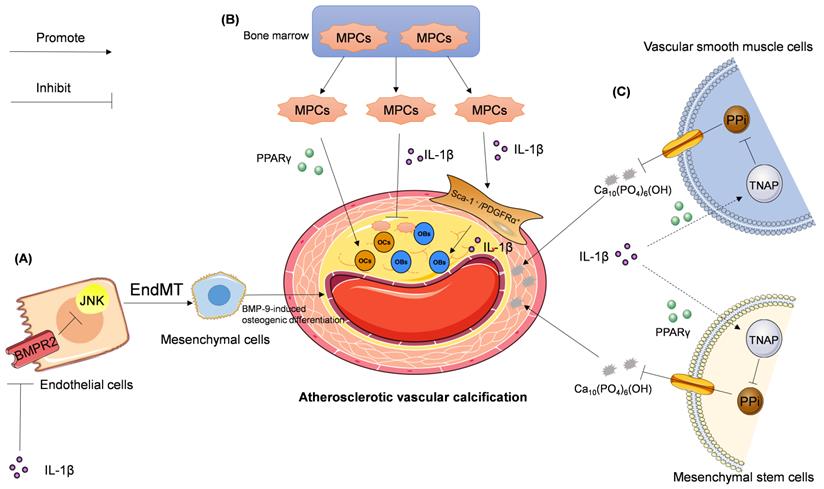
The relationship between IL-1β and EndMT in atherosclerotic vascular calcification is still poorly understood. Sanchez-Duffhues et al. reported that IL-1β-sensitized bone morphogenetic protein-9 (BMP-9)-induces osteogenic differentiation via induction of EndMT. This process is mediated by the downregulation of bone morphogenetic protein receptor type II (BMPR2) expression and subsequent inactivation of the c-Jun N-terminal kinase (JNK) signaling pathway in human primary aortic endothelial cells [88]. This hypothesis was further corroborated in vivo and in patient-derived atherosclerotic tissues [88]. These findings suggest that IL-1β may induce EndMT and promote atherosclerotic vascular calcification (Figure 2A).
Signaling pathways involved in IL-1β—mediated regulation of atherosclerotic vascular calcification. (A) IL-1β activates JNK pathway by downregulating BMPR2 expression and subsequent BMPR2-dependent inhibition of JNK, which promotes endothelial to mesenchymal transition (EndMT), leading to BMP-9-induced osteogenic differentiation. (B) IL-1β inhibits the mobilization and infiltration of mesodermal progenitor cells (MPCs), which can bi-directionally differentiate into osteoblasts (OBs) or osteoclasts (OCs). While in hypercholesterolemia IL-1β enhances the mobilization and infiltration of Sca-1+/PDGFRα+ cells, which are differentiated from MPCs and the progenitor cells of OBs. PPARγ promotes the differentiation of MPCs into OCs. (C) IL-1β stimulates tissue-nonspecific alkaline phosphatase (TNAP) expression and activity in both vascular smooth muscle cells (VSMCs) and mesenchymal stem cells followed by pyrophosphate (PPi) degradation. PPi is an effective endogenous inhibitor of Ca10(PO4)6(OH), which is a major component of the calcified aorta. This chain of events accelerates atherosclerotic vascular calcification.
3.2. IL-1β inhibits the mobilization and infiltration of bipotent mesodermal progenitor cells (MPCs), thereby accelerating atherosclerotic vascular calcification
Stem or progenitor cells, and their dynamics, play an important role in cardiovascular diseases [89-92]. Using identification of cell surface markers, such as platelet-derived growth factor receptor alpha (PDGFRα) and stem cell antigen-1 (Sca-1), Cho et al. [93] discovered a cluster of vascular calcifying progenitor cells residing in the arterial adventitia. These cells are derived from the bone marrow and mobilize to the inflamed atherosclerotic lesions. Mesodermal progenitor cells (MPCs), such as Lin-CD29+/Sca-1+/PDGFRα- cells, possess bidirectional differentiation, which enables the development of MPCs into osteoblasts (OBs) or osteoclasts (OCs). MPCs isolated from the adult bone marrow are also progenitors of Sca-1+/PDGFRα+ cells, which can potentially differentiate into OBs [94]. IL-1β, which is elevated in the sera and arteries of hypercholesterolemic ApoE-/- mice, enhances the mobilization and infiltration of Sca-1+/PDGFRα+ cells. Conversely, IL-1β inhibits bipotent MPCs and accelerates atherosclerotic vascular calcification [94]. Thus, current studies suggest that IL-1β is likely a key regulator of bipotent MPCs in vascular calcification.
Activation of peroxisome proliferator-activated receptor γ (PPARγ) promotes the differentiation of bipotent MPCs into OCs; this process alleviates vascular calcification in vitro and in vivo [93]. Collectively, the homeostasis between MPCs and Sca-1+/PDGFRα+ cells may play an important role in vascular calcification under conditions of atherosclerosis (Figure 2B). Induced pluripotent stem (iPS) cells originating from MPC-related cells [95], and monoclonal antibodies specific for IL-1β [96-98], are currently available as sources for the development of new therapeutics for the treatment of patients with atherosclerotic vascular calcification [93].
3.3. IL-1β activates tissue-nonspecific alkaline phosphatase to exacerbate atherosclerotic vascular calcification
Tissue-nonspecific alkaline phosphatase (TNAP) is an enzyme that degrades extracellular pyrophosphate (PPi) and promotes vascular calcification by downregulating the expression of PPi. PPi is a potent endogenous inhibitor of hydroxyapatite [Ca10(PO4)6(OH)] [99-101], which is the main component of the calcified aorta [102]. This decreased level of plasma PPi is associated with genetic or metabolic conditions, such as Hutchinson-Gilford progeria syndrome (HGPS) [99], generalized arterial calcification of infancy (GACI) [103], and advanced chronic kidney disease [104], which predispose vulnerable individuals to vascular calcification [105-107]. Transgenic overexpression of TNAP in VSMCs or in endothelial cells results in pathological calcification in vitro and in vivo [108-110]. Moreover, the upregulation of TNAP expression has also been demonstrated in dialysis-related calcification of human vessels [111].
Ding et al. [112] reported that IL-1β can upregulate TNAP activity and calcification in human mesenchymal stem cells via RUNX2-independent signaling. Lencel et al. [113] utilized VSMCs, stimulated by TNF-α and IL-1β, to demonstrate the cell-specific effects of TNAP, and suggested that PPARγ may mediate these differences in TNAP activity. For these reasons, IL-1β is considered a stimulator of vascular calcification in the context of atherosclerosis. Few studies have examined the roles of IL-1β and TNAP in vascular calcification. However, whether IL-1β can promote vascular calcification via a TNAP-mediated signaling pathway needs further study (Figure 2C).
4. Stimulators that regulate IL-1β expression accelerate atherosclerotic vascular calcification
4.1. Rac2 mediates atherosclerotic vascular calcification via regulation of IL-1β production in macrophages
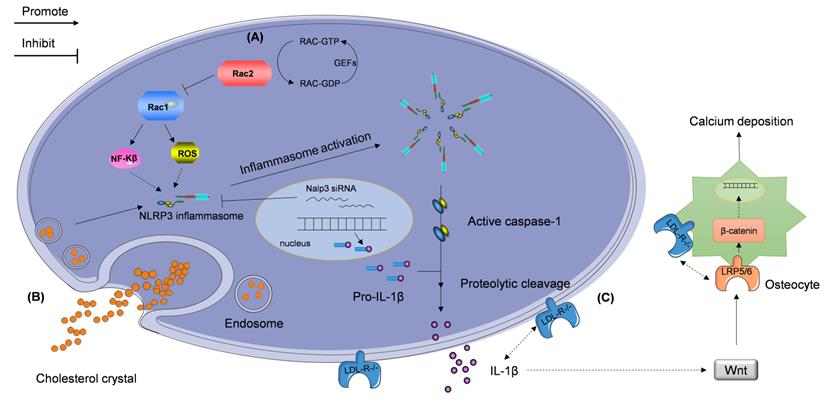
Rac proteins are a subfamily of the Rho family, which consists of small guanosine triphosphate-binding proteins Rac 1, 2, 3, and RhoG [114, 115]. Similar to other small GTPases, Rac switches between a GTP-bound active and GDP-bound inactive state. Stimulus-induced activation of Rac is mediated by guanine nucleotide exchange factors (GEFs) [116-118]; this subfamily of proteins is critical in numerous inflammation-mediating pathological processes. Rac1 and Rac2, which are important signal transducers in inflammatory cells, affect the expression of several cytokines and growth factors [119, 120]. Recently, Ceneri et al. [121] reported a significant decrease in Rac2, and increase in IL-1β, expression, in the aortas of ApoE-/- mice and in calcified plaques in human coronary segments. They also showed that Rac2 deletion can aggravate vascular calcification by increasing macrophage expression of IL-1β, which is associated with NF-κB activation. Rac2 was also found to enhance the production of reactive oxygen species (ROS) elicited by elevation of activated Rac1 expression in Rac2-/-ApoE-/- mice. Thus, Ceneri et al. identified a novel inflammatory signaling pathway that depends on Rac2-mediated regulation of Rac1-dependent IL-1β expression in macrophages (Figure 3A) [121]. This study outlines a pathophysiological signaling mechanism to understand the rationale of the action of IL-1β in the development of atherosclerotic vascular calcification.
Stimulators regulating IL-1β expression accelerate atherosclerotic vascular calcification. The NLRP3 inflammasome participates in the regulation of the progression of atherosclerotic vascular calcification via two signaling pathways described as follows. (A) The reduced Rac2 activity, caused by compromised guanine nucleotide exchange factors (GEFs), elevates the expression of Rac1, which promotes activation of NF-κB pathway and production of reactive oxygen species (ROS). These events lead to the NLRP3 inflammasome production. (B) Extracellular cholesterol crystals are taken up by macrophages and activate the NLRP3 inflammasome. NLRP3 inflammasome-activated caspase-1 cleaves pro-IL-1β and induces the secretion of IL-1β. (C) Deletion of the gene encoding low-density lipoprotein receptor (LDL-R) contributes to elevation in IL-1β expression and subsequent vascular calcification independent of hypercholesterolemia. This cascade may be modulated by the Wnt and β-catenin signaling pathways.
4.2. NLRP3 inflammasome initiates the release of IL-1β by upregulating the expression of caspase-1, leading to atherosclerotic vascular calcification
The NLR family pyrin domain-containing 3 (NLRP3) inflammasome, which is an ROS-sensitive multiprotein complex, accelerates IL-1β maturation via activation of caspase-1 [122, 123]. NLRP3 plays a pivotal role in the pathogenesis of atherosclerosis [124-126]. Numerous studies have reported that NLRP3 is essential for atherogenesis, and that silencing the expression of NLRP3 prevents the rupture of atherosclerotic plaques [127, 128].
Wen et al. [129] found that the expression of NLRP3 inflammasome and IL-1β are upregulated in VSMCs cultured in calcification medium supplemented with β-glycerophosphate (β-GP) and in human calcified popliteal arteries having elevated expression of caspase-1. Downregulation of NLRP3 expression using treatment with NLRP3 siRNA reduces IL-1β secretion, which subsequently reduces vascular calcification, in vitro. However, whether the knockdown of IL-1β can counteract the effect of NLRP3 inflammasome in vascular calcification remains unclear and needs to be studied in vivo. Tangi et al. [130] demonstrated that TNF-α-mediated induction of IL-1β release in aortic smooth muscle cells is also NLRP3-dependent. Additionally, the NLRP3 inflammasome-mediated promotion of IL-1β secretion via upregulation of caspase-1 expression may play an important role in atherosclerotic vascular calcification (Figure 3B). Further investigation is required to uncover the details of this mechanism.
4.3. Hypercholesterolemia promotes the release of IL-1β to induce atherosclerotic vascular calcification
The effect of hypercholesterolemia on vascular calcification was confirmed when Awan et al. [131] reported that patients with familial hypercholesterolemia (FH), who possess mutations in the low-density lipoprotein receptor (LDLR) gene, show severe and extensive vascular calcification in their thoracoabdominal aortas; this process usually commences at 20 years of age in homozygous FH patients. In contrast, vascular calcification in thoracoabdominal aorta is delayed by two decades in individuals affected by heterozygous FH [132]. Although a comparably high level of plasma cholesterol is observed in Ldlr-/-C57BL/6 mice fed a standard-chow diet and wild-type C57BL/6 mice fed a high-cholesterol, high-fat diet, Ldlr-/- mice still develop considerably more extensive aortic vascular calcification compared with that of wild-type mice [133]. None of the currently available therapeutics, including statins, can stop or regress vascular calcification in a clinical setting [134]. Collectively, these findings suggest that vascular calcification in early hypercholesterolemia may involve as of yet undiscovered mechanisms, and that LDLR deficiency may be a key step in vascular calcification.
Duewell et al. [125] and Rajamaki et al. [135] found that crystals of cholesterol are absorbed by macrophages, which then express the activated inflammasome complex. These events result in cleavage and activation of pro-IL-1β and secretion of IL-1β into plasma. Consequently, the binding of plasma IL-1β to IL-1β receptors on vascular endothelial cells promotes the release of various cytokines, SMC proliferation, and macrophage activation. The steps in this cascade contribute to the development of atherosclerosis [136], but the mechanism underlying hypercholesterolemia and IL-1β activity in the context of vascular calcification remains to be uncovered.
The levels of total cholesterol are increased in LDLR-deficient (Ldlr-/-) mice and LDLR-attenuated proprotein convertase subtilisin/kexin type 9 (Pcsk9) transgenic (Tg) mice [137]. However, Awan et al. [138] reported that IL-1β plasma levels in Ldlr-/- mice are twice as high as those in Pcsk9(Tg) mice. They also found that while anti-IL-1β monoclonal antibodies considerably inhibit atherosclerotic vascular calcification in Ldlr-/- mice with hypercholesterolemia, they induce only an insignificant change in the atherosclerotic vascular calcification of Pcsk9(Tg) mice with indiscriminate hypercholesterolemia. This finding suggests a potential mechanism accounting for why Pcsk9(Tg) mice show significantly less calcification compared with that in Ldlr-/- mice. This finding indicates that IL-1β plays a key role in atherosclerotic calcification associated with LDLR deficiency. Although a hypothesis linking IL-1β, LDL-R, and the Wnt/β-catenin signaling pathways to vascular calcification in the setting of hypercholesterolemia has been proposed [133], the detailed mechanism is unknown and further investigation is still needed (Figure 3C).
5. Discussion
Inflammation is proved to be an independent risk factor for cardiovascular disease, even after the lipid-lowering therapies. The SPIRE-1 and SPIRE-2 trials [139] using the statins and PCSK9 antibody bococizumab to reduce atherogenic lipids showed residual inflammation and no improvement for cardiovascular events. Since 2018, several anti-inflammation clinical trials revealed promising and challenging results for the treatment of cardiovascular disease, such as CANTOS [20] and CIRT [76]. The CANTOS trial has shown that treatment with the monoclonal IL-1β-neutralizing antibody canakinumab can reducing the risk of recurrent cardiovascular events in patients with prior heart attack. While the CIRT trial in 5,000 patients with previous coronary disease showed no benefit for the reduction of cardiovascular events with the treatment of methotrexate—an promising anti-inflammatory approach in which once an association with fewer cardiovascular events was observed in patients with rheumatoid. These results provide us with informative implications that more unknown mechanisms exist in the anti-inflammation therapeutics for the prevention of cardiovascular events. A hypothesis of 'innate immune training' of IL-1β in epigenetic reprogramming of myeloid progenitor cells [140] was once proposed to understand the mechanism of the action of IL-1β. However, disputes of this issue exist [75].
Accumulating clinical evidence indicates that vascular calcification is an independent predictor of the occurrence of atherosclerosis-related myocardial infarction and stroke [7, 11-13, 15, 141-143]. Recent studies, including those summarized in this review, have shown that IL-1β plays prominent roles in the development of atherosclerotic vascular calcification. Although the incidence of atherosclerosis-related cardiovascular events has been clinically reduced by the use of anti-IL-1β therapy, as shown in several large-scale population cohort trials (Table 1), the clinical effect of IL-1β inhibition on atherosclerotic vascular calcification remains unclear. It is also unclear whether the beneficial effect induced by anti-IL-1β therapeutics in the setting of atherosclerosis is related to the effect of these therapeutics on atherosclerotic vascular calcification.
Clinical trials examining anti-IL-1β therapy for treatment of patients with cardiovascular diseases
| Trials | Drug type | Dosage and administration | Treatment duration | Outcomes |
|---|---|---|---|---|
| Virginia Commonwealth University-Anakinra Remodeling Trial (VCU-ART) [152] | Recombinant IL-1 receptor antagonist (IL-1Ra) | Anakinra, 100 mg/day, subcutaneous injection | 14 days | Double-blinded, randomized, placebo-controlled; 10 patients with ST-segment elevation of acute myocardial infarction (AMI). Anakinra improved the end-diastolic volume index and left ventricular end-systolic volume index (LVESVi). |
| Virginia Commonwealth University-Anakinra Remodeling Trial 2 (VCU-ART2) [153] | Recombinant IL-1 receptor antagonist (IL-1Ra) | Anakinra, 100 mg/day, subcutaneous injection | 14 days | Double-blinded, randomized, placebo-controlled; 30 patients with stable ST-segment elevation of acute myocardial infarction (AMI). Anakinra had no effect on LVESVi and end-diastolic volume index, but significantly alleviated the upregulation of C-reactive protein and lowered the occurrence of heart failure. Data pooled from the above VCU-ART and VCU-ART2 trials revealed that anakinra reduced the incidence of primary heart failure in contrast with that in the placebo-treated group. |
| MRC-ILA Heart Study [154, 155] | Recombinant IL-1 receptor antagonist (IL-1Ra) | Anakinra, 100 mg/day, subcutaneous injection | 14 days | Phase II, double-blinded, randomized, placebo-controlled; 182 patients diagnosed with non-ST elevation acute coronary syndrome (NSTE-ACS), admitted to hospital <48 h from the development of chest pain. Anakinra reduced the high-sensitivity C-reactive protein levels at 7 days post-treatment. |
| Canakinumab Anti-Inflammatory Thrombosis Outcomes Study [20, 144] | Monoclonal IL-1β-neutralizing antibody | Canakinumab, 50 mg, 150 mg, and 300 mg, subcutaneous injection, every 3 months, respectively | 48 months | A randomized, double-blinded trial enrolling 10,061 patients. Canakinumab, administered at the dose of 150 mg, reduced the rate of recurrent cardiovascular events by 17%. |
Further prospective randomized controlled trials (RCTs) are needed to evaluate the effects of IL-1β-related antibodies and drugs (Table 2) on atherosclerotic vascular calcification and incidence rate of cardiovascular events. Even in the well-known CANTOS trial [20, 144], there is no subgroups treated with anti-IL-1β therapy showing variance in the levels of vascular calcification in the setting of cardiovascular events. Therefore, further clinical trials, especially RCT studies, are needed to explore the roles of IL-1β and related therapeutics in atherosclerotic vascular calcification.
IL-1β-related antibodies and drugs
| Names | Type | Application |
|---|---|---|
| XOMA052 [156, 157] | Monoclonal anti-IL-1β antibody | In vitro and in vivo |
| Gevokizumab [158] | IL-1β modulator | In vitro and in vivo |
| Anakinra [152-155] | Recombinant IL-1 receptor antagonist (IL-1Ra) | Clinical trial |
| Canakinumab [20, 144] | Monoclonal IL-1β-neutralizing antibody | Clinical trial |
IL1B-/-, IL1R1-/-, IL-1 receptor antagonist-deficient (IL1Ra-/-), and transgenic mice overexpressing either secreted IL-1Ra or intracellular IL-1Ra1, are valuable animal models employed in basic in-vivo and in-vitro research [145-150]. These models are useful to mimic cardiac infarction and ischemic stroke in order to uncover the mechanism linking IL-1β activity, atherosclerotic vascular calcification, and cardiovascular events. New technologic advances, such as using artificial intelligence to conduct big-data analysis, will help us delineate the detailed mechanisms of cardiovascular calcification and design potential therapeutics [151]. These future studies will provide us with a broad understanding of IL-1β roles in atherosclerotic vascular calcification, and will define the value of IL-1β as a potential therapeutic target in the treatment of patients with cardiovascular diseases.
In summary, atherosclerotic vascular calcification is a vascular lesion related to morbidity and mortality worldwide. Currently, no specific and effective treatments are available for this condition because of our insufficient understanding of the detailed molecular mechanisms driving the processes of atherosclerotic vascular calcification. Future research should aim to delineate these molecular mechanisms, including those involving IL-1β-mediated pathways, as related to the development of atherosclerotic vascular calcification.
Abbreviations
ALP: Alkaline phosphatase; AMI: Acute myocardial infarction; ASO: Arteriosclerosis obliterans; BMP-2: Bone morphogenetic protein-2; BMP-9: Bone morphogenetic protein-9; BMPR2: Bone morphogenetic protein receptor type II; CACS: Coronary artery calcium scoring; CANTOS: Canakinumab Anti-Inflammatory Thrombosis Outcomes Study; CTA: Computed tomography angiography; EndMT: Endothelial-to-mesenchymal transition; EMT: Epithelial-to-mesenchymal transition; Fet-A: Fetuin A; FH: Familial hypercholesterolemia; GACI: Generalized arterial calcification of infancy; GEFs: Guanine nucleotide exchange factors; HGPS: Hutchinson-Gilford progeria syndrome; HUVECs: Human umbilical vein endothelial cells; IL-1β: Interleukin-1β; IL-1Ra: IL-1 receptor antagonist; iPS: Induced pluripotent stem; JNK: c-Jun N-terminal kinase; LAD: Left anterior descending; LCA: Left coronary artery; LCX: Left circumflex artery; LDLR: Low-density lipoprotein receptor; Ldlr-/- : LDLR-deficient; LVESVi: Left ventricular end-systolic volume index; MGP: Matrix Gla protein; Mincle: Macrophage‐inducible C‐type lectin; MPCs: Mesodermal progenitor cells; MSx2: Muscle segment homeobox homolog of 2; NLRP3: Nlp family, pyrin domain‐containing 3; NSTE-ACS: Non-ST elevation acute coronary syndrome; OB: Osteoblastic; OC: Osteoclastic; OPN: Osteopontin; OPG: Osteoprotegerin; Pcsk9: Proprotein convertase subtilisin/kexin type 9; PDGFRα: Platelet-derived growth factor receptor alpha; PPARγ: Peroxisome proliferator-activated receptor γ; PPi: Pyrophosphate; RCTs: Randomized controlled trials; Runx 2: Runt-related transcription factor 2; Sca-1: Surface markers stem cell antigen-1; Smad 6: Drosophila mothers against decapentaplegic protein 6; SMC: Smooth muscle cell; Sox 9: SRY-Box transcription factor 9; SP 7: Osterix; T2DM: Type 2 diabetes mellitus; Tg: Transgenic; TNAP: Tissue-nonspecific alkaline phosphatase; TNF-α: Tumor necrosis factor-α; VSMCs: Vascular smooth muscle cells.
Acknowledgements
I (XLS) would like to thank Dr. Ivo Ricardo de Seabra Rodrigues Dias who critically revised the manuscript for grammar and language and Dr. Xiaojun Xie who collected the clinical diagrams used in Figure 1A-B. I (XLS) would like to thank my wife Yarong Ma and my son Andy Chongen Sun. I owe more than I can express to them. Their love and support make everything possible.
Funding
This study was supported by the Collaborative Innovation Center for Prevention and Treatment of Cardiovascular Disease of Sichuan Province Funded Project (Grant code: xtcx2019-20), Key Laboratory of Medical Electrophysiology of Ministry of Education Funded Project (Grant code: KeyME-2018-04), Open Program of Nuclear Medicine and Molecular Imaging Key Laboratory of Sichuan Province (Grant code: HYX19007), and Doctoral Research Initiating Program of Affiliated Hospital of Southwest Medical University (Grant code: 19041).
Author contributions
XLS conceptualized and designed the study, collected the clinical diagrams used in Figure 1C-E, and revised and edited the manuscript. JLS wrote the first draft of the manuscript. MZ critically revised the manuscript. CXZ conceptualized and reviewed the manuscript. All authors read and approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sieve I, Ricke-Hoch M, Kasten M, Battmer K, Stapel B, Falk CS. et al. A positive feedback loop between IL-1β, LPS and NEU1 may promote atherosclerosis by enhancing a pro-inflammatory state in monocytes and macrophages. Vascul Pharmacol. 2018 p: 16-28
2. Pant S, Deshmukh A, Gurumurthy GS, Pothineni NV, Watts TE, Romeo F. et al. Inflammation and atherosclerosis-revisited. J Cardiovasc Pharmacol Ther. 2014;19:170-8
3. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ Res. 2016;118:535-46
4. Torres N, Guevara-Cruz M, Velazquez-Villegas LA, Tovar AR. Nutrition and Atherosclerosis. Arch Med Res. 2015;46:408-26
5. Tolle M, Reshetnik A, Schuchardt M, Hohne M, van der Giet M. Arteriosclerosis and vascular calcification: causes, clinical assessment and therapy. Eur J Clin Invest. 2015;45:976-85
6. Davaine JM, Quillard T, Chatelais M, Guilbaud F, Brion R, Guyomarch B. et al. Bone Like Arterial Calcification in Femoral Atherosclerotic Lesions: Prevalence and Role of Osteoprotegerin and Pericytes. Eur J Vasc Endovasc Surg. 2016;51:259-67
7. McClelland RL, Chung H, Detrano R, Post W, Kronmal RA. Distribution of coronary artery calcium by race, gender, and age: results from the Multi-Ethnic Study of Atherosclerosis (MESA). Circulation. 2006;113:30-7
8. Leopold JA. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med. 2015;25:267-74
9. Peter L, Manfred B, Victor S, Marc T, Jan J, Thomas Z. et al. Medial vascular calcification revisited: review and perspectives. Eur Heart J. 2014;35:1515-25
10. Kaplan NM. Kaplan's clinical hypertension. Philadelphia: Lippincott Williams & Wilkins. 2010
11. Roberto W, Allan Z, Paolo R. High coronary artery calcium scores pose an extremely elevated risk for hard events. J Am Coll Cardiol. 2002;39:225-30
12. Iribarren C, Sidney S, Sternfeld B, Browner WS. Calcification of the aortic arch: risk factors and association with coronary heart disease, stroke, and peripheral vascular disease. JAMA. 2000;283:2810-5
13. Wilson PW, Kauppila LI, O'Donnell CJ, Kiel DP, Hannan M, Polak JM. et al. Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation. 2001;103:1529-34
14. Leopold JA. Vascular calcification: an age-old problem of old age. Circulation. 2013;127:2380-2
15. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5:185-97
16. Manduteanu I, Simionescu M. Inflammation in atherosclerosis: a cause or a result of vascular disorders? J Cell Mol Med. 2012;16:1978-90
17. Nakahara T, Strauss HW. From inflammation to calcification in atherosclerosis. Eur J Nucl Med Mol Imaging. 2017;44:858-60
18. Ikeda K, Souma Y, Akakabe Y, Kitamura Y, Matsuo K, Shimoda Y. et al. Macrophages play a unique role in the plaque calcification by enhancing the osteogenic signals exerted by vascular smooth muscle cells. Biochem Biophys Res Commun. 2012;425:39-44
19. Sallam T, Cheng H, Demer LL, Tintut Y. Regulatory circuits controlling vascular cell calcification. Cell Mol Life Sci. 2013;70:3187-97
20. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C. et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119-31
21. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovascular research. 2018;114:590-600
22. Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. 2006;99:1044-59
23. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109:697-711
24. Luong TTD, Schelski N, Boehme B, Makridakis M, Vlahou A, Lang F. et al. Fibulin-3 Attenuates Phosphate-Induced Vascular Smooth Muscle Cell Calcification by Inhibition of Oxidative Stress. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2018;46:1305-16
25. Bessueille L, Magne D. Inflammation: a culprit for vascular calcification in atherosclerosis and diabetes. Cell Mol Life Sci. 2015;72:2475-89
26. Badi I, Mancinelli L, Polizzotto A, Ferri D, Zeni F, Burba I. et al. miR-34a Promotes Vascular Smooth Muscle Cell Calcification by Downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arteriosclerosis, thrombosis, and vascular biology. 2018;38:2079-90
27. Parhami F, Basseri B, Hwang J, Tintut Y, Demer LL. High-density lipoprotein regulates calcification of vascular cells. Circ Res. 2002;91:570-6
28. Voelkl J, Lang F, Eckardt KU, Amann K, Kuro OM, Pasch A. et al. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci. 2019;76:2077-91
29. Phadwal K, Feng D, Zhu D, MacRae VE. Autophagy as a novel therapeutic target in vascular calcification. Pharmacology & therapeutics. 2020;206:107430
30. Sung DC, Bowen CJ, Vaidya KA, Zhou J, Chapurin N, Recknagel A. et al. Cadherin-11 Overexpression Induces Extracellular Matrix Remodeling and Calcification in Mature Aortic Valves. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:1627-37
31. Yang W, Zou B, Hou Y, Yan W, Chen T, Qu S. Extracellular vesicles in vascular calcification. Clinica chimica acta; international journal of clinical chemistry. 2019;499:118-22
32. Li Y, Sun Z, Zhang L, Yan J, Shao C, Jing L. et al. Role of Macrophages in the Progression and Regression of Vascular Calcification. Frontiers in pharmacology. 2020;11:661
33. Staines KA, MacRae VE, Farquharson C. The importance of the SIBLING family of proteins on skeletal mineralisation and bone remodelling. The Journal of endocrinology. 2012;214:241-55
34. Roumeliotis S, Dounousi E, Eleftheriadis T, Liakopoulos V. Association of the Inactive Circulating Matrix Gla Protein with Vitamin K Intake, Calcification, Mortality, and Cardiovascular Disease: A Review. International journal of molecular sciences. 2019 20
35. Herrmann M, Babler A, Moshkova I, Gremse F, Kiessling F, Kusebauch U. et al. Lumenal calcification and microvasculopathy in fetuin-A-deficient mice lead to multiple organ morbidity. PloS one. 2020;15:e0228503
36. Jansen K, van der Steen AF, van Beusekom HM, Oosterhuis JW, van Soest G. Intravascular photoacoustic imaging of human coronary atherosclerosis. Optics letters. 2011;36:597-9
37. Bobryshev YV. Transdifferentiation of smooth muscle cells into chondrocytes in atherosclerotic arteries in situ: implications for diffuse intimal calcification. The Journal of pathology. 2005;205:641-50
38. Scimeca M, Anemona L, Granaglia A, Bonfiglio R, Urbano N, Toschi N. et al. Plaque calcification is driven by different mechanisms of mineralization associated with specific cardiovascular risk factors. Nutrition, metabolism, and cardiovascular diseases: NMCD. 2019;29:1330-6
39. Tesauro M, Mauriello A, Rovella V, Annicchiarico-Petruzzelli M, Cardillo C, Melino G. et al. Arterial ageing: from endothelial dysfunction to vascular calcification. Journal of internal medicine. 2017;281:471-82
40. Rodondi N, Taylor BC, Bauer DC, Lui LY, Vogt MT, Fink HA. et al. Association between aortic calcification and total and cardiovascular mortality in older women. Journal of internal medicine. 2007;261:238-44
41. Jaffer FA, Libby P, Weissleder R. Optical and multimodality molecular imaging: insights into atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:1017-24
42. New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108:1381-91
43. Rocha-Singh KJ, Zeller T, Jaff MR. Peripheral arterial calcification: prevalence, mechanism, detection, and clinical implications. Catheter Cardiovasc Interv. 2014;83:E212-20
44. Rajamannan NM, Subramaniam M, Caira F, Stock SR, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation. 2005;112:I229-34
45. Monzack EL, Gu X, Masters KS. Efficacy of simvastatin treatment of valvular interstitial cells varies with the extracellular environment. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:246-53
46. Rossebø AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K. et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343-56
47. London GM. Arterial calcification: cardiovascular function and clinical outcome. Nefrologia: publicacion oficial de la Sociedad Espanola Nefrologia. 2011;31:644-7
48. Yutzey KE, Demer LL, Body SC, Huggins GS, Towler DA, Giachelli CM. et al. Calcific aortic valve disease: a consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:2387-93
49. Sheridan K, Logomarsino JV. Effects of serum phosphorus on vascular calcification in a healthy, adult population: A systematic review. Journal of vascular nursing: official publication of the Society for Peripheral Vascular Nursing. 2017;35:157-69
50. Zwakenberg SR, de Jong PA, Bartstra JW, van Asperen R, Westerink J, de Valk H. et al. The effect of menaquinone-7 supplementation on vascular calcification in patients with diabetes: a randomized, double-blind, placebo-controlled trial. The American journal of clinical nutrition. 2019;110:883-90
51. Sakaguchi Y, Hamano T, Obi Y, Monden C, Oka T, Yamaguchi S. et al. A Randomized Trial of Magnesium Oxide and Oral Carbon Adsorbent for Coronary Artery Calcification in Predialysis CKD. Journal of the American Society of Nephrology: JASN. 2019;30:1073-85
52. Gao Y, Wang G, Li Y, Lv C, Wang Z. Effects of oral activated charcoal on hyperphosphatemia and vascular calcification in Chinese patients with stage 3-4 chronic kidney disease. Journal of nephrology. 2019;32:265-72
53. Braddock M, Quinn A. Targeting IL-1 in inflammatory disease: new opportunities for therapeutic intervention. Nature reviews Drug discovery. 2004;3:330-9
54. Dinarello CA. Therapeutic strategies to reduce IL-1 activity in treating local and systemic inflammation. Current opinion in pharmacology. 2004;4:378-85
55. Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095-147
56. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1β secretion. Cytokine & growth factor reviews. 2011;22:189-95
57. Verheyen J, Bonig H, Banning U, Shin DI, Mauz-Körholz C, Körholz D. Co-operation of IL-1 and IL-2 on T-cell activation in mononuclear cell cultures. Immunological investigations. 2001;30:289-302
58. Kopp WC, Urba WJ, Rager HC, Alvord WG, Oppenheim JJ, Smith JW 2nd. et al. Induction of interleukin 1 receptor antagonist after interleukin 1 therapy in patients with cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 1996;2:501-6
59. Cuff CA, Martiney JA, Berman JW, Brosnan CF. Differential effects of transforming growth factor-beta 1 on interleukin-1-induced cellular inflammation and vascular permeability in the rabbit retina. Journal of neuroimmunology. 1996;70:21-8
60. Plata-Salamán CR, Ilyin SE. Interleukin-1beta (IL-1beta)-induced modulation of the hypothalamic IL-1beta system, tumor necrosis factor-alpha, and transforming growth factor-beta1 mRNAs in obese (fa/fa) and lean (Fa/Fa) Zucker rats: implications to IL-1beta feedback systems and cytokine-cytokine interactions. Journal of neuroscience research. 1997;49:541-50
61. Veltman D, Laeremans T, Passante E, Huber HJ. Signal transduction analysis of the NLRP3-inflammasome pathway after cellular damage and its paracrine regulation. Journal of theoretical biology. 2017;415:125-36
62. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA. et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nature immunology. 2013;14:52-60
63. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I. et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213-23
64. Jiang K, Zhang Y, He F, Zhang M, Li T, Tu Z. et al. A negative feedback loop involving NF-κB/TIR8 regulates IL-1β-induced epithelial- myofibroblast transdifferentiation in human tubular cells. J Cell Commun Signal. 2021;15:393-403
65. Haybar H, Shokuhian M, Bagheri M, Davari N, Saki N. Involvement of circulating inflammatory factors in prognosis and risk of cardiovascular disease. Journal of molecular and cellular cardiology. 2019;132:110-9
66. Pedersen BK. Anti-inflammatory effects of exercise: role in diabetes and cardiovascular disease. Eur J Clin Invest. 2017;47:600-11
67. Avolio E, Pasqua T, Di Vito A, Fazzari G, Cardillo G, Alò R. et al. Role of Brain Neuroinflammatory Factors on Hypertension in the Spontaneously Hypertensive Rat. Neuroscience. 2018;375:158-68
68. Qi J, Zhao XF, Yu XJ, Yi QY, Shi XL, Tan H. et al. Targeting Interleukin-1 beta to Suppress Sympathoexcitation in Hypothalamic Paraventricular Nucleus in Dahl Salt-Sensitive Hypertensive Rats. Cardiovascular toxicology. 2016;16:298-306
69. Ren XS, Tong Y, Ling L, Chen D, Sun HJ, Zhou H. et al. NLRP3 Gene Deletion Attenuates Angiotensin II-Induced Phenotypic Transformation of Vascular Smooth Muscle Cells and Vascular Remodeling. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;44:2269-80
70. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H. et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594-604
71. Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV. et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovascular research. 2013;99:164-74
72. Korantzopoulos P, Letsas KP, Tse G, Fragakis N, Goudis CA, Liu T. Inflammation and atrial fibrillation: A comprehensive review. Journal of arrhythmia. 2018;34:394-401
73. Yao C, Veleva T, Scott L Jr, Cao S, Li L, Chen G. et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation. 2018;138:2227-42
74. Wang Y, Yin J, Wang C, Hu H, Li X, Xue M. et al. Microglial Mincle receptor in the PVN contributes to sympathetic hyperactivity in acute myocardial infarction rat. J Cell Mol Med. 2019;23:112-25
75. Moore KJ. Targeting inflammation in CVD: advances and challenges. Nat Rev Cardiol. 2019;16:74-5
76. Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E. et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N Engl J Med. 2019;380:752-62
77. Gong H, Lyu X, Wang Q, Hu M, Zhang X. Endothelial to mesenchymal transition in the cardiovascular system. Life Sci. 2017;184:95-102
78. Cho JG, Lee A, Chang W, Lee M-S, Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol. 2018;9:294
79. Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR. et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. 2016;7:11853
80. Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M. et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. 2015;125:4514-28
81. Souilhol C, Harmsen MC, Evans PC, Krenning G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovascular research. 2018;114:565-77
82. Mudau M, Genis A, Lochner A, Strijdom H. Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc J Afr. 2012;23:222-31
83. Guihard PJ, Yao J, Blazquez-Medela AM, Iruela-Arispe L, Bostrom KI, Yao Y. Endothelial-Mesenchymal Transition in Vascular Calcification of Ins2Akita/+ Mice. PloS one. 2016;11:e0167936
84. Stabley JN, Towler DA. Arterial Calcification in Diabetes Mellitus: Preclinical Models and Translational Implications. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:205-17
85. Lee JG, Ko MHK, Kayab EDP. Endothelial mesenchymal transformation mediated by IL-1β-induced FGF-2 in corneal endothelial cells. Exp Eye Res. 2012;95:35-9
86. Linghui N, Orestis L, Rituparna M, Nebojsa J, Schmidt JL, Otterson MF. et al. Endothelial-mesenchymal transition in normal human esophageal endothelial cells cocultured with esophageal adenocarcinoma cells: role of IL-1β and TGF-β2. Am J Physiol Cell Physiol. 2014;307:859-77
87. Liguori TTA, Liguori GR, Moreira LFP, Harmsen MC. Adipose tissue-derived stromal cells' conditioned medium modulates endothelial-mesenchymal transition induced by IL-1beta/TGF-beta2 but does not restore endothelial function. Cell Prolif. 2019 p: e12629
88. Sanchez-Duffhues G, Garcia de Vinuesa A, van de Pol V, Geerts ME, de Vries MR, Janson SG. et al. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. The Journal of pathology. 2019;247:333-46
89. Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. 2010;7:528-36
90. Gossl M, Modder UI, Atkinson EJ, Lerman A, Khosla S. Osteocalcin expression by circulating endothelial progenitor cells in patients with coronary atherosclerosis. J Am Coll Cardiol. 2008;52:1314-25
91. Hjortnaes J, New SE, Aikawa E. Visualizing novel concepts of cardiovascular calcification. Trends Cardiovasc Med. 2013;23:71-9
92. Zhang L, Issa Bhaloo S, Chen T, Zhou B, Xu Q. Role of Resident Stem Cells in Vessel Formation and Arteriosclerosis. Circ Res. 2018;122:1608-24
93. Cho HJ, Cho HJ, Lee HJ, Song MK, Seo JY, Bae YH. et al. Vascular calcifying progenitor cells possess bidirectional differentiation potentials. PLoS Biol. 2013;11:e1001534
94. Cho HJ, Lee JW, Cho HJ, Lee CS, Kim HS. Identification of Adult Mesodermal Progenitor Cells and Hierarchy in Atherosclerotic Vascular Calcification. Stem Cells. 2018;36:1075-96
95. Sakurai H, Sakaguchi Y, Shoji E, Nishino T, Maki I, Sakai H. et al. In vitro modeling of paraxial mesodermal progenitors derived from induced pluripotent stem cells. PloS one. 2012;7:e47078
96. Alten R, Gram H, Joosten LA, van den Berg WB, Sieper J, Wassenberg S. et al. The human anti-IL-1β monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof-of-concept study in patients with rheumatoid arthritis. Arthritis Res Ther. 2008;10:R67
97. Owyang AM, Maedler K, Gross L, Yin J, Esposito L, Shu L. et al. XOMA 052, an anti-IL-1β monoclonal antibody, improves glucose control and β-cell function in the diet-induced obesity mouse model. Endocrinology. 2010;151:2515-27
98. Chakraborty A, Tannenbaum S, Rordorf C, Lowe PJ, Floch D, Gram H. et al. Pharmacokinetic and pharmacodynamic properties of canakinumab, a human anti-interleukin-1β monoclonal antibody. Clin Pharmacokinet. 2012;51:e1-e18
99. Villa-Bellosta R, Rivera-Torres J, Osorio FG, Acin-Perez R, Enriquez JA, Lopez-Otin C. et al. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation. 2013;127:2442-51
100. Lomashvili KA, Narisawa S, Millan JL, O'Neill WC. Vascular calcification is dependent on plasma levels of pyrophosphate. Kidney Int. 2014;85:1351-6
101. Villa-Bellosta R, Egido J. Phosphate, pyrophosphate, and vascular calcification: a question of balance. Eur Heart J. 2015;38:1801-4
102. Johnsson MS, Nancollas GH. The role of brushite and octacalcium phosphate in apatite formation. Crit Rev Oral Biol Med. 1992;3:61-82
103. Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J. et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90:25-39
104. O'Neill WC, Sigrist MK, McIntyre CW. Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol Dial Transplant. 2010;25:187-91
105. Rutsch F, Nitschke Y, Terkeltaub R. Genetics in arterial calcification: pieces of a puzzle and cogs in a wheel. Circ Res. 2011;109:578-92
106. Jansen RS, Duijst S, Mahakena S, Sommer D, Szeri F, Varadi A. et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:1985-9
107. Orriss IR, Arnett TR, Russell RG. Pyrophosphate: a key inhibitor of mineralisation. Current opinion in pharmacology. 2016;28:57-68
108. Savinov AY, Salehi M, Yadav MC, Radichev I, Millan JL, Savinova OV. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J Am Heart Assoc. 2015;4:e002499
109. Narisawa S, Harmey D, Yadav MC, O'Neill WC, Hoylaerts MF, Millan JL. Novel inhibitors of alkaline phosphatase suppress vascular smooth muscle cell calcification. J Bone Miner Res. 2007;22:1700-10
110. Villa-Bellosta R, Wang X, Millan JL, Dubyak GR, O'Neill WC. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2011;301:H61-H8
111. Shroff RC, McNair R, Figg N, Skepper JN, Schurgers L, Gupta A. et al. Dialysis Accelerates Medial Vascular Calcification in Part by Triggering Smooth Muscle Cell Apoptosis. Circulation. 2008;118:1748-57
112. Ding J, Ghali O, Lencel P, Broux O, Chauveau C, Devedjian JC. et al. TNF-alpha and IL-1beta inhibit RUNX2 and collagen expression but increase alkaline phosphatase activity and mineralization in human mesenchymal stem cells. Life Sci. 2009;84:499-504
113. Lencel P, Delplace S, Pilet P, Leterme D, Miellot F, Sourice S. et al. Cell-specific effects of TNF-alpha and IL-1beta on alkaline phosphatase: implication for syndesmophyte formation and vascular calcification. Lab Invest. 2011;91:1434-42
114. Tybulewicz VL, Henderson RB. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol. 2009;9:630-44
115. Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509-14
116. Bokoch GM. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005;15:163-71
117. Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost. 2010;103:40-55
118. Citi S, Spadaro D, Schneider Y, Stutz J, Pulimeno P. Regulation of small GTPases at epithelial cell-cell junctions. Mol Membr Biol. 2011;28:427-44
119. Li B. Role of the Guanosine Triphosphatase Rac2 in T Helper 1 Cell Differentiation. Science. 2000;288:2219-22
120. Morrison AR, Yarovinsky TO, Young BD, Moraes F, Ross TD, Ceneri N. et al. Chemokine-coupled beta2 integrin-induced macrophage Rac2-Myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J Exp Med. 2014;211:1957-68
121. Ceneri N, Zhao L, Young BD, Healy A, Coskun S, Vasavada H. et al. Rac2 Modulates Atherosclerotic Calcification by Regulating Macrophage Interleukin-1beta Production. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:328-40
122. Hua KF, Chou JC, Ka SM, Tasi YL, Chen A, Wu SH. et al. Cyclooxygenase-2 regulates NLRP3 inflammasome-derived IL-1beta production. J Cell Physiol. 2015;230:863-74
123. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677-87
124. Baldrighi M, Mallat Z, Li X. NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis. 2017;267:127-38
125. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357-61
126. Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ Res. 2018;122:1722-40
127. Zheng F, Xing S, Gong Z, Mu W, Xing Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. 2014;2014:507208
128. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J Cell Physiol. 2018;233:2116-32
129. Wen C, Yang X, Yan Z, Zhao M, Yue X, Cheng X. et al. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int J Cardiol. 2013;168:2242-7
130. Tangi TN, Elmabsout AA, Bengtsson T, Sirsjo A, Fransen K. Role of NLRP3 and CARD8 in the regulation of TNF-alpha induced IL-1beta release in vascular smooth muscle cells. Int J Mol Med. 2012;30:697-702
131. Awan Z, Alrasadi K, Francis GA, Hegele RA, McPherson R, Frohlich J. et al. Vascular calcifications in homozygote familial hypercholesterolemia. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:777-85
132. Alrasadi K, Alwaili K, Awan Z, Valenti D, Couture P, Genest J. Aortic calcifications in familial hypercholesterolemia: potential role of the low-density lipoprotein receptor gene. Am Heart J. 2009;157:170-6
133. Fantus D, Awan Z, Seidah NG, Genest J. Aortic calcification: Novel insights from familial hypercholesterolemia and potential role for the low-density lipoprotein receptor. Atherosclerosis. 2013;226:9-15
134. Rossebø AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K. et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343-56
135. Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT. et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PloS one. 2010;5:e11765
136. Fearon WF, Fearon DT. Inflammation and cardiovascular disease: role of the interleukin-1 receptor antagonist. Circulation. 2008;117:2577-9
137. Zaid A, Roubtsova A, Essalmani R, Marcinkiewicz J, Chamberland A, Hamelin J. et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. 2008;48:646-54
138. Awan Z, Denis M, Roubtsova A, Essalmani R, Marcinkiewicz J, Awan A. et al. Reducing Vascular Calcification by Anti-IL-1beta Monoclonal Antibody in a Mouse Model of Familial Hypercholesterolemia. Angiology. 2016;67:157-67
139. Pradhan AD, Aday AW, Rose LM, Ridker PM. Residual Inflammatory Risk on Treatment With PCSK9 Inhibition and Statin Therapy. Circulation. 2018;138:141-9
140. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K. et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell. 2018;172:162-75.e14
141. Yahagi K, Kolodgie FD, Lutter C, Mori H, Romero ME, Finn AV. et al. Pathology of Human Coronary and Carotid Artery Atherosclerosis and Vascular Calcification in Diabetes Mellitus. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:191-204
142. Shaw LJ, Raggi P, Schisterman E, Berman DS, Callister TQ. Prognostic value of cardiac risk factors and coronary artery calcium screening for all-cause mortality. Radiology. 2003;228:826-33
143. Thompson RC, Allam AH, Lombardi GP, Wann LS, Sutherland ML, Sutherland JD. et al. Atherosclerosis across 4000 years of human history: the Horus study of four ancient populations. Lancet. 2013;381:1211-22
144. Everett BM, Donath MY, Pradhan AD, Thuren T, Pais P, Nicolau JC. et al. Anti-Inflammatory Therapy With Canakinumab for the Prevention and Management of Diabetes. J Am Coll Cardiol. 2018;71:2392-401
145. Elhage R, Maret A, Pieraggi MT, Thiers JC, Arnal JF, Bayard F. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation. 1998;97:242-4
146. Kikuo I, Masaru S, Norio I, Taizo M, Reiko H, Kenichirou N. et al. Deficiency of interleukin-1 receptor antagonist promotes neointimal formation after injury. Circulation. 2003;108:516-8
147. Kikuo I, Shojiro S, Norio I, Taizo M, Koji M, Masatoshi K. et al. Lack of interleukin-1 receptor antagonist modulates plaque composition in apolipoprotein E-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1068-73
148. Faten MS, Kwak BR, David M, Christos C, Marina B, Graziano P. et al. Interleukin-1 plays a major role in vascular inflammation and atherosclerosis in male apolipoprotein E-knockout mice. Cardiovascular research. 2017;66:583
149. Chamberlain J, Evans D, King A, Dewberry R, Dower S, Crossman D. et al. Interleukin-1β and signaling of interleukin-1 in vascular wall and circulating cells modulates the extent of neointima formation in mice. Am J Pathol. 2006;168:1396-403
150. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y. et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:656-60
151. Rogers MA, Aikawa E. Cardiovascular calcification: artificial intelligence and big data accelerate mechanistic discovery. Nat Rev Cardiol. 2019;16:261-74
152. Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R. et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am J Cardiol. 2010;105:1371-7 e1
153. Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW. et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am J Cardiol. 2013;111:1394-400
154. Morton AC, Rothman AM, Greenwood JP, Gunn J, Chase A, Clarke B. et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA Heart Study. Eur Heart J. 2015;36:377-84
155. Crossman DC, Morton AC, Gunn JP, Greenwood JP, Hall AS, Fox KA. et al. Investigation of the effect of Interleukin-1 receptor antagonist (IL-1ra) on markers of inflammation in non-ST elevation acute coronary syndromes (The MRC-ILA-HEART Study). Trials. 2008;9:8
156. Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H. et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis. 2011;216:313-20
157. Owyang AM, Issafras H, Corbin J, Ahluwalia K, Larsen P, Pongo E. et al. XOMA 052, a potent, high-affinity monoclonal antibody for the treatment of IL-1β-mediated diseases. mAbs. 2011;3:49-60
158. Roubille F, Busseuil D, Shi Y, Nachar W, Mihalache-Avram T, Mecteau M. et al. The interleukin-1β modulator gevokizumab reduces neointimal proliferation and improves reendothelialization in a rat carotid denudation model. Atherosclerosis. 2014;236:277-85
Author contact
![]() Corresponding authors: Xiaolei Sun, M.D., Ph.D., E-mail: sunxiaoleiedu.cn; xiaolei.sunac.uk. Chunxiang Zhang, M.D., Ph.D., E-mail: zhangchx999com.
Corresponding authors: Xiaolei Sun, M.D., Ph.D., E-mail: sunxiaoleiedu.cn; xiaolei.sunac.uk. Chunxiang Zhang, M.D., Ph.D., E-mail: zhangchx999com.