Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Regulatory role and mechanisms...
Regulatory role and mechanisms...
Regulatory role and mechanisms...
Regulation of Smad3-dependent...
Therapeutic potential for renal...
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(7):2795-2806. doi:10.7150/ijbs.71595 This issue Cite
Review
Smad3 Signatures in Renal Inflammation and Fibrosis
Wenjing Wu1,2,3,4, Xiaoqin Wang3, Xueqing Yu1 ![]() , Hui-Yao Lan2,5
, Hui-Yao Lan2,5 ![]()
1. Guangdong-Hong Kong Joint Laboratory for Immunological and Genetic Kidney Disease, Department of Pathology, and Guangdong Cardiovascular Institute, Guangdong Academy of Medical Sciences, Guangdong Provincial People's Hospital, Guangzhou, China.
2. Departments of Medicine & Therapeutics, Li Ka Shing Institute of Health Sciences, and Lui Che Woo Institute of Innovative Medicine, The Chinese University of Hong Kong, Hong Kong, China.
3. Hubei University of Chinese Medicine, Wuhan, China.
4. Department of Nephrology, Hubei Provincial Hospital of Traditional Chinese Medicine, Hubei Province Academy of Traditional Chinese Medicine, Wuhan, China.
5. The Chinese University of Hong Kong-Guangdong Academy of Sciences/ Guangdong Provincial People's Hospital Joint Research Laboratory on Immunological and Genetic Kidney Diseases, The Chinese University of Hong Kong, Hong Kong, China.
Received 2022-1-30; Accepted 2022-3-18; Published 2022-3-28
Abstract

Renal inflammation and fibrosis are key pathological features of acute kidney injury (AKI) and chronic kidney disease (CKD). Smad3 is a critical mediator of TGF-β signaling and plays a pathogenic role in both renal inflammation and fibrosis. Smad3 can be activated not only by TGF-β1 but also by many stress molecules including angiotensin II (Ang II), advanced end products (AGEs), and C-reactive protein (CRP) under disease conditions. In addition, Smad3 can interact with other signaling pathways, such as the ERK/p38 MAPK and NF-κB pathways, to mediate renal inflammation and fibrosis. Mechanistically, Smad3 transcriptionally regulates many downstream target genes including microRNAs and long non-coding RNAs to cause cell death, inflammation, and fibrosis. Thus, targeting Smad3 or its downstream genes specifically related to renal inflammation and fibrosis should provide a novel therapeutic strategy to combat kidney diseases.
Keywords: Smad3, renal inflammation and fibrosis, miRNAs, lncRNAs
Introduction
Kidney disease has become a major public health problem worldwide. Chronic kidney disease (CKD) affects more than 10% of the population worldwide [1]. Renal inflammation is a common manifestation of acute kidney injury (AKI) and CKD [2, 3] and may be a driving force from AKI to CKD progression. Fibrosis is a common pathway of progressive CKD that finally leads to the end-stage renal disease. Thus, renal fibrosis accompanied by active inflammation is a major pathological feature in progressive kidney disease [3]. Like Chinese Yin and Yang, transforming growth factor-β1 (TGF-β1) signaling plays a diverse role in renal inflammation and fibrosis [4-7]. It is well documented that TGF-β is a potent anti-inflammatory cytokine and immune regulator that play a protective role in renal inflammation. In the other hand, TGF-β also exerts its pathogenic role in renal fibrosis [4-7]. Upon TGF-β binds to its receptors, it triggers activation of downstream signaling pathways including Smad and non-Smad-dependent pathways. Of them, the canonical Smad pathway is a key regulatory pathway in the pathogenesis of renal inflammation and fibrosis. Of the Smad signaling molecules, Smad3, together with Smad2, is the major receptor-associated Smads. Smad3 has a highly conserved region at the N-terminal and C-terminal, termed mad-homology domain1 (MH1) region and MH2 region. MH1 is mainly associated with DNA binding, while the MH2 region has phosphorylation sites activated by TGF-β1 and has specific sequences that determine their binding to TGF-β1 signal receptors [8, 9].
Smad3 is confirmed to be a major downstream signaling molecule of TGF-β1 in mediating organ inflammation and fibrosis [4]. Smad3 can be activated by many other stress molecules including angiotensin II (Ang II), advanced end products (AGE), and C-reactive protein (CRP). In this review, we focus on the molecular mechanisms of Smad3 in regulating renal inflammation and fibrosis. We also describe the downstream Smad3 signature genes including Smad3-dependent microRNAs and long non-coding RNAs, which regulate renal inflammation and fibrosis. In addition, the new therapeutic approaches for kidney disease by targeting Smad3 signaling, as well as Smad3-dependent microRNAs and lncRNAs are also described.
Regulatory role and mechanisms of Smad3 in renal fibrosis
TGF-β1 has long been known as a key mediator in the pathogenesis of renal fibrosis by activating the downstream Smad proteins, especially Smad3. Once Smad3 becomes activated in response to TGF-β1 and other stress molecules such as Ang II, AGEs, and CRP, it can translocate to the nucleus to directly bind to DNA sequences and regulate the target genes (Figure 1). It is known that many fibrogenic genes responsible for the fibrogenesis including collagen synthesis and epithelial-mesenchymal transition (EMT) are Smad3-dependent [10, 11]. Thus, Smad3 plays a critical role in the development of renal fibrosis in many kidney diseases. An essential role for Smad3 in fibrogenesis is confirmed by the findings that deletion of Smad3 from mice can suppress renal fibrosis in a number of rodent models, including diabetic nephropathy [12], obstructive kidney diseases [13], hypertensive nephropathy [14], and drug-associated nephropathy [15].
Smad3 signaling and crosstalk pathways in renal fibrosis. After binding to TβRII, TGF-β1 activates the TβRI-kinase which phosphorylates Smad3. The phosphorylated Smad3 translocates into the nucleus and regulates the target gene transcription. Smad7 is an inhibitory Smad that functions to block Smad3 activation by degrading the TβRI and preventing phosphorylation of Smad3.Ang II, AGEs and CRP can activate TGF-β1-independent signaling via the ERK/p38/ MAPK crosstalk pathway. Red arrows/ symbols represent pathogenic or positive regulation pathway, while blue lines/ symbols indicate protective or negative regulation pathways in fibrosis.
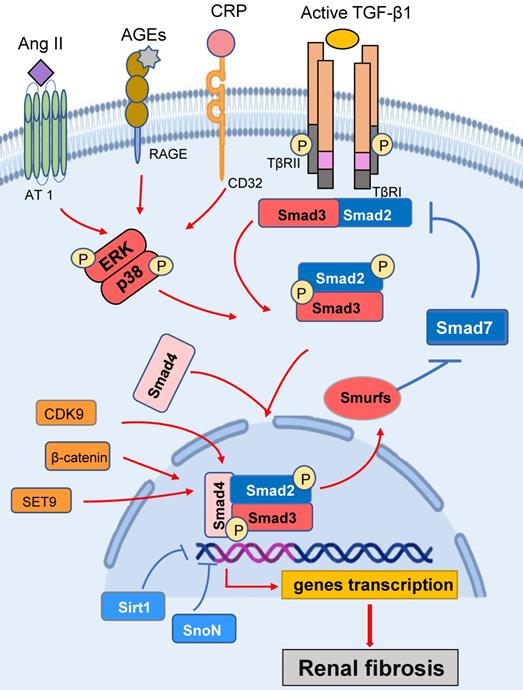
Smad3 can also regulate Smad7 to play a role in renal fibrosis. It is well established that Smad7 is an inhibitory Smad that is induced by Smad3 transcriptionally but exerts its negative feedback mechanism to maintain the homeostasis of TGF-β/Smad signaling [5, 16-19]. In normal situations, renal Smad7 is abundant and exerts its negative feedback mechanism by causing degradation of Type I TGF-β receptor (TβRI) via an ubiquitin proteasome degradation mechanism, thereby preventing the recruitment and phosphorylation of Smad3 [4]. Under disease conditions, Smad3 is overreactive and can also induce a number of E3 ubiquitin ligases such as the Smad ubiquitination regulatory factor 1 (Smurf1), Smad ubiquitination regulatory factor2 (Smurf2), and arkadia, which physically interact with Smad7 and cause an ubiquitin-dependent degradation of renal Smad7 protein [20, 21], resulting in enhanced TGF-β/Smad3 signaling and progressive renal fibrosis [20]. This is further supported by the findings that mice lacking Smad7 largely promote activation of Smad3 signaling and progressive renal fibrosis in both obstructive nephropathy and diabetic kidney disease [22, 23].
Renal fibrosis is characterized by a loss of renal tubules and the accumulation of extracellular matrix (ECM). Myofibroblasts are an active form of fibroblasts that are generally considered to be the main source of ECM production during renal fibrosis [24, 25]. Many studies show that Smad3 has an important role in the transformation of bone marrow-derived fibroblasts in the kidney as genetic disruption of Smad3 inhibits the activation of bone marrow-derived fibroblasts in the kidney in response to obstructive kidney injury in vivo and suppresses monocyte-to-fibroblast transition in vitro [26]. Excitingly, our recent studies have also demonstrated that macrophage-myofibroblast transition (MMT) is a major source of myofibroblast origin (> 60%) that occurs locally within the fibrosing kidney and is regulated by Smad3 [27-29]. Smad3 is required for the efficient transition of recruited macrophages to become collagen I-producing α-SMA+ myofibroblasts within the injured kidney. In addition, the protection from obstructive kidney fibrosis seen in Smad3-/- chimeric mice provides further evidence that bone marrow-derived macrophages make a substantial contribution to the development of renal fibrosis via the Smad3-dependent MMT process [28, 30]. Indeed, bone marrow-derived M2-type pro-fibrotic macrophages are highly proliferative and contribute to renal fibrosis in the UUO kidney [31]. It is proposed that bone marrow-derived M2 macrophages can enter the injured kidney and then transdifferentiate into collagen-producing α-SMA+ myofibroblasts which is under tight control of TGF-β/Smad3 signaling [32]. By using single cell RNA sequence analysis, we reveal that TGF-β1 induces MMT in bone marrow-derive macrophages via the Smad3-Src-POU4F1 pathway as Smad3 can bind Src and POU4F1 promoters to induce MMT and targeting this pathway can block MMT and renal fibrosis in vitro and in vivo [30, 33].
Ang II and AGEs are also able to induce renal fibrosis by activating Smad3 signaling via TGF-β-dependent and-independent pathways (Figure 1). Ang II is able to induce TGF-β1 expression and then activate TGF-β1/Smad3 signaling, which can lead to fibrosis. In addition, a significant finding shows that Ang II can also directly activate Smad3 to induce expression of connective tissue growth factor (CTGF) and collagen I through the AT1-ERK/p38 MAPK crosstalk pathway [34]. This is supported by the findings that addition of Ang II is able to induce Smad3 phosphorylation in tubular epithelial cells (TECs) lacking the TGF-β1 gene, and that blockade of the AT1 receptor, ERK1/2, and p38, is capable of inhibiting Ang II-induced activation of Smad3 and CTGF [34]. This notion is further supported by evidence of knockdown Smad3 to block Ang II-mediated EMT [34]. Ang II-induced overactivation of TGF-β1/Smad3 signaling is also associated with the loss of renal Smad7, which is mediated by a Smurf2-dependent ubiquitin degradation mechanism [35].
It is now well accepted that AGEs are key mediators in diabetic nephropathy. Accumulation of AGEs closely correlates to CTGF expression and EMT. Indeed, the Smad-binding elements are found in the CTGF promoters [36], suggesting that Smad3 may mediate fibrosis by inducing CTGF expression. Because AGEs are able to activate Smad3 via TGF-β1-dependent and independent mechanisms [4, 5] (Figure 1), it is generally believable that AGE-induced CTGF expression via the TGF-β1-independent Smad3 signaling pathway as addition of AGEs is able to stimulate a rapid phosphorylation of Smad2/3, ERK1/2, and p38 and CTGF expression in TECs lacking TGF-β1 gene [37, 38].
C-reactive protein (CRP) acts as one of the most essential inflammatory marker and mediator in various chronic diseases [39, 40] (Figure 1). It mediates renal fibrosis by inducing the early (15 mins) and the late phase (24 hrs) of phosphorylation of Smad3 in HK-2 cells [41, 42]. CRP can activate Smad3 to mediate renal fibrosis directly via the CD32b-ERK/p38 MAP kinase-crosstalk pathway and indirectly through the TGF-β1-dependent mechanism [41]. This is also confirmed by deleting Smad3 to inhibit UUO-induced renal fibrosis in CRP transgenic mice [43].
Smad3 is also activated by other molecules (Figure 1). Of them, cyclin dependent kinases (CDKs), such as CDK9, can promote Smad3-regulating collagen I promoter activity [44]. Ski-related novel protein (SnoN) is a nuclear protein that functions as a negative regulator of TGF-β1/Smad3 signaling [45]. Activation of Smad3 can up-regulate Smurf2 and thus enhances the ubiquitin degradation of SnoN to exert the fibrogenic effects of TGF-β1/Smad3 signaling. Further study shows that Smad3 can repress SnoN transcription by binding to SIE sequences (triple CGACGG box) in the SnoN promoter [46, 47]. β-catenin acts as a co-factor for Smad3 transcriptional activity. Targeted degradation of cytosolic β-catenin or inhibition of β-catenin binding to Smad3 blocks TGF-β1-induced EMT in murine renal tubular epithelial cells [48]. Sirt1 belongs to a highly conserved family of NAD+‐dependent deacetylase and has been reported to deacetylate the lysine residues of a number of nuclear proteins. It has been reported that Sirt1 can bind to Smad3 to reduce the acetylation levels of Smad3 and inhibits renal fibrosis [49]. It is also reported that the methyltransferase SET9 (also known as SETD7) can interact with the Smad3 N-terminal MH1 domain to increase Smad3 activity and upregulate α-SMA expression during renal fibrosis [50]. CREB binding protein (CBP) is also a Smad3 coactivators, it can bind to the Smad complexes to transcriptionally regulate the downstream genes [51]. CREB can competitively inhibit the binding of CBP to Smad3 [52]. Glycogen synthase kinase 3β (GSK3β) is a serine/threonine-protein kinase that inhibits the CREB activities while promoting CBP binding to Smad3 to facilitate renal fibrosis. In TGF-β1-treated renal TECs, the inhibition of GSK3β can enhance the activity of CBP recruitment to CREB and thus ameliorates renal fibrosis [52].
Regulatory role and mechanisms of Smad3 in renal inflammation
Infiltration of immune cells into tissues is a key pathogenetic event in many inflammatory diseases which can be diversely regulated by Smad3. As shown in Figure 2, activation of Smad3 plays a diverse role in immune cell activation and differentiation during renal inflammatory responses. Smad3 is a critical effector molecule of TGF-β1-mediated inhibition of macrophage activation as Smad3 is capable of inhibiting the promoter activities of iNOS and MMP-12 on macrophages [53]. In addition, Smad3 also mediates the TGF-β-dependent inhibition of CD4 T-cell proliferation. Smad3 phosphorylation decreases T-cell receptor (TCR) activation, as well as impeding the effects of CD28 co-stimulation [54]. It is now well defined that Smad3 is a downstream key regulator of TGF-β signaling in T cell immunity [55-57]. It is well documented that Smad3 can bind and regulate expression of Foxp3 to promote Treg cell differentiation and functions in many immunologically-mediated kidney diseases including crescentic glomerulonephritis [58, 59]. In addition, Smad3 is found to be part of a protein complex with RORt, leading to the inhibition of RORt transcriptional activity and then decline Th17 cell generation [60]. Thus, activation of TGF-β/Smad3 signaling exerts its inhibitory effect of on immunologically-mediated diseases by promoting Treg while inhibiting Th17 responses. However, together with IL-6, activation of TGF-β/Smad3 plays an important role in the generation of Th17 cells [61]. Thus, Smad3 is an important regulator in maintaining the balance between Treg and Th17 immune responses, which is supported by the findings that Smad3 deficiency resulted in defective Foxp3 induction but enhanced Th17 cell generation in vitro and in vivo [60].
Smad3 signaling and crosstalk pathways in renal inflammation. Smad3 is a key regulator that diversely regulates renal inflammation by either inhibiting or promoting macrophage and T cell. Red arrows/symbols represent pathogenic or positive regulation pathway, while blue lines/symbols indicate protective or negative regulation pathways.
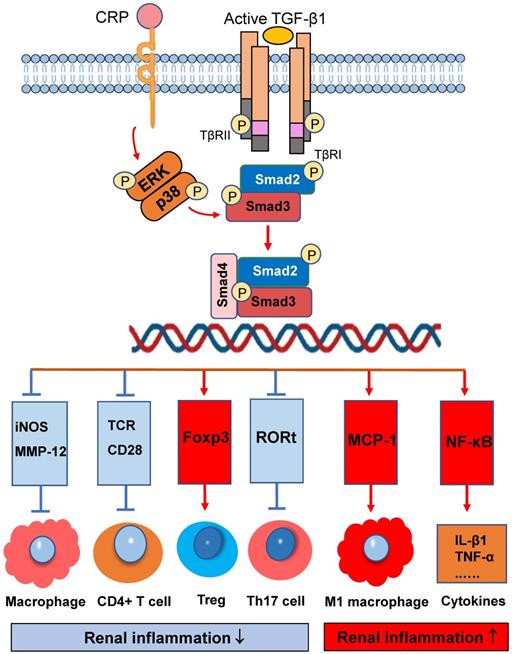
On the other hand, activation of TGF-β/Smad3 signaling may also promote renal inflammation. There are two possible mechanisms responsible for this pro-inflammatory effect of TGF-β/Smad3 signaling. First, Smad3 may exert its chemotactic effect on the macrophage recruitment during renal inflammation as Smad3 can interact with macrophage chemotactic protein-1 (MCP-1) to promote macrophage-dependent renal inflammation [62]. This is supported by the findings that mice lacking Smad3 remarkably suppress renal inflammation by reducing F4/80+ macrophages, together with CD4 and CD8 T cells in the diseased kidneys of obstructive nephropathy [26, 28, 30, 63], Ang II-induced hypertensive nephropathy [14], and ischemic-reperfusion AKI [64]. Interestingly, recent studies also show that deletion of Smad3 from db/db and human CRP transgenic mice can inhibit renal inflammation by blocking MCP-1-dependent macrophage infiltration [65]. Furthermore, deficiency of Smad3 shows to exert its inhibitory effect on NF-κB-driven renal inflammation as seen in many mouse models of kidney disorders [41, 43]. It is likely that deletion of Smad3 may suppress expression of E3 ubiquitin-protein ligases such as Smurf1/Smurf2 that target Smad2, Smad7, and TβRI for degradation. Thus, protection of renal Smad7 from the E3-ligase-dependent ubiquitin degradation in Smad3 KO mice may result in upregulation of IκBα, an inhibitor of NF-κB signaling, thereby inhibiting NF-κB-driven renal inflammation [66, 67].
Regulatory role and mechanisms of Smad3 in cell death during acute kidney injury
Increasing evidence shows that AKI is a major cause of CKD. Smad3 also plays a driving role in AKI by triggering the cell death pathways (Figure 3). Recent studies identified that Smad3 can bind and induce expression of cyclin-dependent kinase inhibitors (CDKIs) including p21/p27 to cause tubular epithelial cell death via the G1 cell-cycle arrest mechanism [68, 69]. It has been reported that CRP can induce the early activation of Smad3 signaling via the CD32-ERK1/2 and p38-dependent mechanism [41]. Thus, mice overexpressing the human CRP develop more severe AKI by activating Smad3-dependent cell death pathway [68, 70], which is reversed by targeting this pathway with a pharmacological Smad3 inhibitor [68]. In addition, activation of TGF-β/Smad3 signaling also plays a key role in the cell senescence during the development of aging kidney, which is mediated via the p16/p21-dependent mechanism [71]. In podocyte-specific TGF-β overexpressing mice, over-activation of TGF-β/Smad3 signaling is involved in the cell senescence via p16 translocation and p21 induction [72]. All these studies suggest that activation of TGF-β/Smad3-p16/p21pathway may not only cause cell death in AKI and but also involves in renal aging and fibrosis in CKD.
Smad3 triggers the cell death pathways in acute kidney injury. Smad3 can bind and activate the p21/p27 to cause tubular epithelial cell death via the G1 cell-cycle arrest mechanism in response to TGF-β1 and CRP under various kidney disease conditions. Red arrows/symbols represent pathogenic or positive regulation pathway, while blue lines/symbols indicate protective or negative regulation pathways.
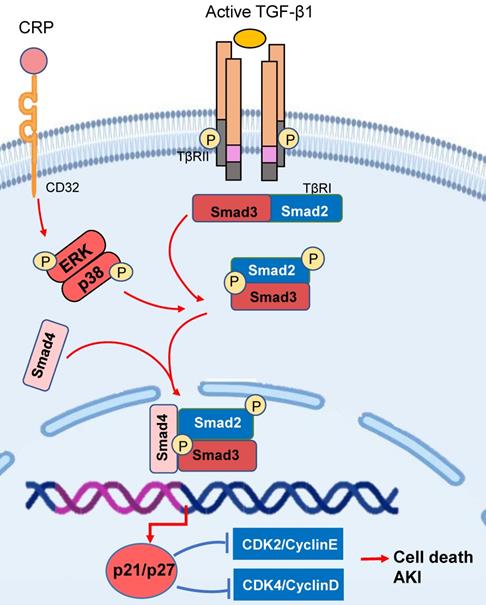
Necroptosis is another cell death pathway leading to AKI [73]. Necroptotic cells can release the components such as high mobility group protein to induce severe necroinflammation [74]. Emerging evidence shows that RIPK1, RIPK3, and MLKL are central regulators in the necroptotic pathway [75, 76]. It has been shown that Smad3 can interact with RIPK and thus loss of Smad3 significantly blocks RIPK-mediated programmed cell death and inflammation [77].
AKI is also a common clinical feature in critically ill patients with COVID-19, particularly in those with inflammatory stress and underlying disease conditions [78]. Strikingly, our most recent study discovered that among SARS-CoV-2 proteins, SARS-CoV-2 N protein is pathogenic for AKI as kidney-specifically overexpressing SARS-CoV-2 N protein can directly induce AKI and promote severe AKI under ischemic conditions [79]. Mechanically, we uncover that SARS-CoV-2 N protein can bind and activate Smad3 signaling to trigger the p21-dependent cell death pathway via the G1 cell cycle arrest mechanism [79, 80]. Thus, targeting Smad3 by genic deletion of Smad3 or pharmacological inhibition of Smad3 signaling can protect against SARS-CoV-2 N-induced AKI [79]. As Smad3 is a key mediator of renal fibrosis [51], it is highly possible that after SARS-CoV-2 infection, intracellular release of SARS-CoV-2 N protein can bind and activate TGF-β/Smad3 signaling to induce the cell death pathway to trigger renal inflammation and “cytokine storm”, resulting in AKI. It is also possible that activation of renal TGF-β/Smad3 signaling results in COVID-19 associated fibrosis including lung and renal fibrosis [81]. Thus, targeting Smad3 may represent as a novel and promising therapeutic strategy for critically ill COVID-19 patients.
Regulation of Smad3-dependent microRNAs and lncRNA in renal inflammation and fibrosis
Noncoding RNAs, including miRNAs, siRNAs, piwi-interacting RNAs, and various types of lncRNAs, are involved in the progression of kidney diseases. MiRNAs are small noncoding RNAs of approximately 22 nucleotides in length, and they bind to the 30-untranslated region of target genes to regulate gene expression by translational repression or induction of mRNA degradation. MiRNAs are important regulators of cell proliferation, differentiation, and apoptosis [82, 83].
It has been well documented that TGF-β/Smad signaling plays a critical regulatory role in renal inflammation and fibrosis by regulating a number of miRNAs [6, 84] (Figure 4). TGF-β1/Smad3 signaling is capable of inducing miR-21 [85], miR-192 [86] and miR-377 [87], but it reduces the expression of miR-200 [88] and miR-29 families [89]. MiR-21 is reported to play a role in the inflammatory response, immunomodulation and fibrotic disorders [83]. MiR-21 is upregulated in animal models with progressive renal fibrosis and inhibition of miR-21 ameliorates fibrosis in obstructive and diabetic kidney diseases [90-92]. Mechanistically, Smad3 can directly interact with miR-21 and induce the expression of miR-21 in response to TGF-β1 and AGEs [85, 93]. MiR-192 is also a downstream mediator of TGF-β/Smad3 in renal fibrosis. Smad3 could physically interact with the promoter region of miR-192 to induce its expression [94]. Thus, TGF-β-induced tubular miR-192 expression is Smad3-dependent because knockdown of Smad3 can block TGF-β1-induced tubular miR-192 expression and renal fibrosis [94]. This observation is further confirmed in Smad3 KO MEF cells and in the UUO kidney in which deletion of Smad3 inhibits renal miR-192 and progressive renal fibrosis [94]. MiR-433 also acts as a downstream mediator of TGF-β/Smad3-driven renal fibrosis by targeting the antizyme inhibitor Azin1 as silencing miR-433 upregulates Azin1 and inhibits renal fibrosis in a mouse model of UUO [95]. In contrast, miR-29 is protective in renal fibrosis and is negatively regulated by TGF-β via Smad3 as the promoter region of miR-29 contains at least two conserved Smad3 binding-sites and Smad3 could physically interact with the promoter region of miR-29 [89].Thus, Smad3 acts as a suppressor to negatively regulate miR-29 expression during TGF-β-mediated fibrosis, and deletion of Smad3 enhances miR-29b expression, thereby inhibiting collagen matrix expression under high TGF-β1 and diabetic conditions [89, 96, 97].
Smad3-dependent miRNAs and lncRNAs related to renal fibrosis and inflammation and Smad3 targeting therapy for renal fibrosis and inflammation. Smad3 positively regulate pro-fibrogenic or pro-inflammatory miRNAs and lncRNAs, but negatively regulate those to mediate renal fibrosis and inflammation. Specifically targeting Smad3 directly with inhibitors or Smad7 agonists or indirectly to its downstream non-coding RNAs may be the potential therapeutic strategies for renal fibrosis and inflammation. Red arrows/symbols represent pathogenic or positive regulation pathway, while blue lines/symbols indicate protective or negative regulation pathways.
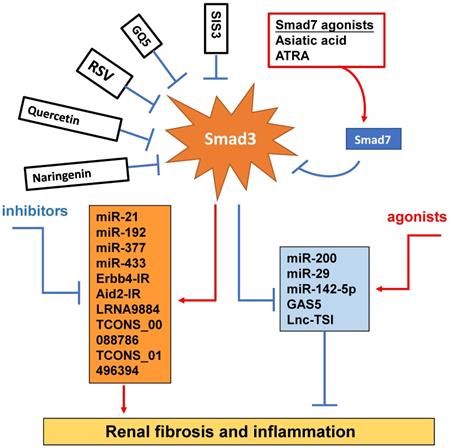
Increasing evidence shows that renal inflammation and fibrosis are also tightly regulated by a few Smad3-dependent long noncoding RNAs (lncRNAs) [98] (Figure 4). Indeed, research into the lncRNAs is more promising for a better understanding of the pathogenic mechanisms of kidney diseases. Compared to miRNAs, lncRNAs are transcripts with lengths exceeding 200 nucleotides without protein-coding functions. LncRNA regulates both target DNAs/RNAs and proteins transcriptionally or post-transcriptionally [99]. By using the high-throughput RNA sequencing, we identify that 413 lncRNAs (plus or minus two-fold to ninefold) are differentially expressed in WT and Smad3 KO kidneys of anti-glomerular basement membranous glomerulonephritis (anti-GBM GN), of them, 21 Smad3-dependent common lncRNAs are altered in both UUO and anti-GBM GN models [100]. Erbb4-IR is a novel Smad3-dependent lncRNA and is highly upregulated in the UUO and diabetic kidneys with progressive renal fibrosis [101, 102]. The functional role of Erbb4-IR in renal fibrosis is demonstrated by silencing this lncRNA to protect kidneys from both UUO and diabetic injury [101, 102]. Mechanistically, Erbb4-IR mediates renal fibrosis in the UUO kidney and diabetic nephropathy by targeting renal Smad7 and miR-29b [101, 102]. The Arid2-IR is also another novel Smad3-related lncRNA. Arid2-IR is one of the most highly upregulated lncRNAs in the UUO kidney with progressive renal inflammation and fibrosis. The promoter region of Arid2-IR contains a Smad3 binding site and thus deletion of Smad3 gene completely blocked upregulation of Arid2-IR in the UUO kidney, suggesting a positive regulatory role for Smad3 in Arid2-IR expression during renal inflammation. Further study reveals that Arid2-IR mediates renal inflammation via the NF-κB-dependent mechanism and thus Arid2-IR may be a downstream mediator of Smad3 and functions to promote NF-κB-driven renal inflammation without effect on TGF-β/Smad3-mediated renal fibrosis in a mouse model of obstructive nephropathy and in vitro [103]. LRNA9884 has been shown to play a proinflammatory role and mediates renal inflammation in db/db mice via a MCP-1-dependent mechanism [104]. Further study also shows that LRNA9884 can induce renal inflammation in AKI mouse model by upregulating macrophage migration inhibitory factor via the NF-κB-dependent mechanism [104, 105]. Lnc-TSI is a lncRNA which is specifically expressed in the fibrotic kidney and negatively correlated with the severity of renal fibrosis. Interestingly, repeated renal biopsy reveals that the lower expression levels of renal Lnc-TSI at the initial kidney biopsy is associated with a more pronounced decline in renal function and fibrosis 4 years later, suggesting that kidney-enriched Lnc-TSI may protect against renal fibrogenesis [106]. It is possible that Lnc-TSI may function as a negative regulator of TGF-β1/Smad3 signaling as overexpression of Lnc-TSI can block Smad3 activation and interaction with TβRI by binding with the MH2 domain of Smad3 [106]. Thus, Smad3 suppresses Lnc-TSI expression through binding with the promoter of Lnc-TSI under TGF-β1 stimulation [106]. GAS5 is also an anti-fibrotic lncRNA and is highly expressed in normal renal tubular epithelial cells but lost in the fibrotic kidney at 7 days after UUO surgery [107]. GAS5 is tightly regulated by Smad3 and thus deletion of Smad3 dramatically inhibits GAS5 in the kidneys of UUO mice in vivo and in TGF-β-stimulated MEFs in vitro [107]. In addition, TCONS_00088786 and TCONS_01496394 are TGF-β/Smad3-associated lncRNAs as they contain potential binding sites for Smad3 and silencing TCONS_00088786 inhibits renal interstitial fibrosis in UUO rat model [108].
Therapeutic potential for renal inflammation and fibrosis by targeting Smad3 signaling
Although TGF-β/Smad3 has been considered as a major pathway for fibrogenesis, the diverse roles of this pathway in renal inflammation and fibrosis have hampered the development of anti-TGF-β treatment in general [4-7]. The failure of anti-TGF-β antibodies-based therapy in recent clinical trials has proved that treatment by targeting upstream TGF-β signaling may not be a good strategy for the treatment of kidney diseases [109, 110]. Disappointingly, treatment with a humanized monoclonal neutralizing antibody against TGF-β1 (LY2382770) for patients with diabetic nephropathy shows no efficacy on the improvements of renal dysfunction including serum creatinine, estimated GFR (eGFR), and proteinuria [109]. Similarly, the use of Fresolimumab (another humanized monoclonal antibody) that inhibits all three isoforms of TGF-β also fails to achieve the endpoints of proteinuria reduction in patients with FSGS [110]. It is highly possible that blockade of the entire TGF-β1 signaling may also promote inflammation as TGF-β1 is a potent anti-inflammatory cytokine [4, 111-113]. Thus, targeting the downstream TGF-β signaling molecules specifically related to fibrosis or inflammation could be a better therapeutic approach. Many studies have reported that Smad3 can directly bind to the DNA sequences to regulate expression of several fibrogenic genes and the process of EMT and MMT [51, 84, 114]. Thus, treatment should aim to specifically target Smad3, and its dependent genes directly related to fibrogenesis or inflammation, rather than the entire TGF-β signaling (Figure 4). SIS3 is a small molecule capable of directly suppressing Smad3-mediated expression of collagens matrix [115] and inhibiting the accumulation of α-SMA+ myofibroblasts in the fibrotic kidney by blocking Smad3-dependent myofibroblasts transdifferentiation including MMT [28, 31, 32]. Treatment with SIS3 may also block Smad3-dependent auto-induction of TGF-β1 via a positive feedback loop of TGF-β1/Smad3 signaling [116]. Furthermore, SIS3 can ameliorate renal inflammation and tubular apoptosis in both AKI and UUO kidneys [116, 117]. Excitingly, our recent study also discovered the therapeutic effect of SIS3 on SARS-CoV-2 N-induced AKI by inhibiting Smad3-dependent p21-mediated cell death pathway [79]. Thus, specifically targeting Smad3 may be a novel therapeutic approach for kidney diseases.
In the fibrotic and inflammatory kidney, overactive Smad3 signaling is associated with the loss of renal Smad7 [20, 21, 114]. Thus rebalancing Smad3/Smad7 signaling by either inhibiting Smad3 and/or activating Smad7 may be a better approach for the development of effective and specific therapy for kidney diseases [7]. This is supported by many studies in which overexpressing renal Smad7 can block TGF-β/Smad3-mediated renal fibrosis and NF-κB-driven renal inflammation in diabetic kidney disease [22, 118], crescentic glomerulonephritis [119], UUO [120], AKI [69], and hypertensive nephropathy [22, 120, 121]. Recently, we also identified that naringenin (NG), a flavonoid from grapefruit and citrus fruits [122], functions as a Smad3 inhibitor, whereas asiatic acid (AA), a purified compound from Centella asiatica [123], is a Smad7 agonist. The combination of these two purified traditional Chinese medicine compounds significantly rebalances the Smad3/Smad7 signaling and thus additively enhances the inhibitory effect on TGF-β1/Smad3 signaling and renal fibrosis in vitro and in vivo [124]. Quercetin is also functioning to inhibit Smad3 signaling and has been shown to have therapeutic effect on cisplatin-induced AKI [125, 126]. GQ5 (a small compound isolated from Resina Toxicodendron) can block the interaction of Smad3 with TβRI and attenuates renal fibrosis [127]. Resveratrol (RSV) is a natural plant polyphenol with anti-fibrotic and anti-inflammatory properties. RSV treatment can significantly activate Sirt1 to suppress Smad3 acetylation and the TGF-β1-induced fibrotic response in the remnant kidney of 5/6 nephrectomized rodents, obstructed kidney model or in cultured cells following TGF-β1 treatment [128]. All-trans retinoic acid (ATRA), an active metabolite of vitamin A, belongs to the retinoids family. Treatment with ATRA inactivates Smad3 signaling and protects against the diabetic kidney disease by upregulating renal Smad7 [129].
Increasing evidence also shows that specifically altering the Smad3-dependent microRNAs or lncRNAs related to fibrogenesis or inflammation locally in the diseased kidney could be a better therapeutic approach for combating kidney disorders. As described elsewhere [84, 98] and illustrated in Figure 4, epigenetically targeting miR-21 [85, 91], miR-192 [86, 94], miR-433 [95], miR-29 [96, 97] and miR-200 family [88], Erbb4-IR [102], LRNA9884 [104, 105], Arid2-IR [103], and Lnc-TSI [106] have been shown to be a novel and specific anti-fibrosis and anti-inflammation therapy for kidney diseases. More excitingly, we have developed a kidney-specifically genes delivery system by using non-invasive ultrasound-microbubble-technique, which can effectively transfer the genes or miRNAs/lncRNAs into the kidney to block renal inflammation and fibrosis without detectable side effects [85, 95, 96, 102, 103].
Conclusions
The current advances in research into the regulation of TGF-β signaling and particularly the Smad3-dependent noncoding RNAs have improved our understanding of the molecular mechanisms of renal inflammation and fibrosis in kidney diseases. In term of renal fibrosis, Smad3 is pathogenic and overreactive, whereas Smad7 is protective but lost in the fibrotic kidney. Thus, rebalancing Smad3/Smad7 signaling may be a better therapeutic approach for combating kidney diseases. In addition, epigenetic identification of Smad3-dependent non-coding RANs that specifically regulate renal inflammation and fibrosis may be the key step forwards the development of effective therapy for kidney diseases. It is also highly possible that targeting Smad3 may be a novel therapeutic potential for AKI by protecting kidney cell death from G1 cell cycle arrest.
Acknowledgements
This study was supported by grants from Research Grants Council of Hong Kong (14117418, 14104019, 14101121); The Guangdong-Hong Kong-Macao-Joint Labs Program from Guangdong Science and Technology Department (2019B121205005); and the Lui Che Woo Institute of Innovative Medicine (CARE program).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hill NR, Fatoba ST, Oke JL, Hirst JA, O'Callaghan CA, Lasserson DS. et al. Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis. PLoS One. 2016;11:e0158765
2. Singbartl K, Formeck CL, Kellum JA. Kidney-Immune System Crosstalk in AKI. Semin Nephrol. 2019;39:96-106
3. Eddy AA, Neilson EG. Chronic kidney disease progression. J Am Soc Nephrol. 2006;17:2964-6
4. Lan HY, Chung AC. TGF-β/Smad signaling in kidney disease. Semin Nephrol. 2012;32:236-43
5. Lan HY. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011;7:1056-67
6. Gu YY, Liu XS, Huang XR, Yu XQ, Lan HY. Diverse Role of TGF- β in Kidney Disease. Front Cell Dev Biol. 2020;8:123
7. Lan HY. The yin and yang role of transforming growth factor-β in kidney disease. Integr Med Nephrol Androl. 2021;8:1
8. Attisano L, Lee-Hoeflich ST. The Smads. Genome Biol. 2001;2:REVIEWS3010
9. Wrana JL, Attisano L. The Smad pathway. Cytokine Growth Factor Rev. 2000;11:5-13
10. Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-β/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. 2001;276:17058-62
11. Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M. et al. Functional characterization of transforming growth factor β signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276:19945-53
12. Fujimoto M, Maezawa Y, Yokote K, Joh K, Kobayashi K, Kawamura H. et al. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem Biophys Res Commun. 2003;305:1002-7
13. Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486-94
14. Liu Z, Huang XR, Lan HY. Smad3 mediates ANG II-induced hypertensive kidney disease in mice. Am J Physiol Renal Physiol. 2012;302:F986-97
15. Zhou L, Fu P, Huang XR, Liu F, Chung AC, Lai KN. et al. Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am J Physiol Renal Physiol. 2010;298:F1006-17
16. von Gersdorff G, Susztak K, Rezvani F, Bitzer M, Liang D, Bottinger EP. Smad3 and Smad4 mediate transcriptional activation of the human Smad7 promoter by transforming growth factor β. J Biol Chem. 2000;275:11320-6
17. Nagarajan RP, Zhang J, Li W, Chen Y. Regulation of Smad7 promoter by direct association with Smad3 and Smad4. J Biol Chem. 1999;274:33412-8
18. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH. et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF β receptor for degradation. Mol Cell. 2000;6:1365-75
19. Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T. et al. Smurf1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J Biol Chem. 2001;276:12477-80
20. Inoue Y, Imamura T. Regulation of TGF-β family signaling by E3 ubiquitin ligases. Cancer Sci. 2008;99:2107-12
21. Lan HY. Smad7 as a therapeutic agent for chronic kidney diseases. Front Biosci. 2008;13:4984-92
22. Chen HY, Huang XR, Wang W, Li JH, Heuchel RL, Chung AC. et al. The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes. 2011;60:590-601
23. Chung AC, Huang XR, Zhou L, Heuchel R, Lai KN, Lan HY. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol Dial Transplant. 2009;24:1443-54
24. Neilson EG. Mechanisms of disease: Fibroblasts-a new look at an old problem. Nat Clin Pract Nephrol. 2006;2:101-8
25. Strutz F, Muller GA. Renal fibrosis and the origin of the renal fibroblast. Nephrol Dial Transplant. 2006;21:3368-70
26. Chen J, Xia Y, Lin X, Feng XH, Wang Y. Smad3 signaling activates bone marrow-derived fibroblasts in renal fibrosis. Lab Invest. 2014;94:545-56
27. Tang PM, Nikolic-Paterson DJ, Lan HY. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. 2019;15:144-58
28. Wang YY, Jiang H, Pan J, Huang XR, Wang YC, Huang HF. et al. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J Am Soc Nephrol. 2017;28:2053-67
29. Nikolic-Paterson DJ, Wang S, Lan HY. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int Suppl (2011). 2014;4:34-8
30. Tang PM, Zhang YY, Xiao J, Tang PC, Chung JY, Li J. et al. Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage-myofibroblast transition. Proc Natl Acad Sci U S A. 2020;117:20741-52
31. Meng XM, Wang S, Huang XR, Yang C, Xiao J, Zhang Y. et al. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016;7:e2495
32. Wang S, Meng XM, Ng YY, Ma FY, Zhou S, Zhang Y. et al. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget. 2016;7:8809-22
33. Tang PM, Zhou S, Li CJ, Liao J, Xiao J, Wang QM. et al. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 2018;93:173-87
34. Yang F, Chung AC, Huang XR, Lan HY. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-β-dependent and -independent Smad pathways: the role of Smad3. Hypertension. 2009;54:877-84
35. Yang F, Huang XR, Chung AC, Hou CC, Lai KN, Lan HY. Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J Pathol. 2010;221:390-401
36. Holmes A, Abraham DJ, Sa S, Shiwen X, Black CM, Leask A. CTGF and SMADs, maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem. 2001;276:10594-601
37. Li JH, Huang XR, Zhu HJ, Oldfield M, Cooper M, Truong LD. et al. Advanced glycation end products activate Smad signaling via TGF-β-dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. 2004;18:176-8
38. Chung AC, Zhang H, Kong YZ, Tan JJ, Huang XR, Kopp JB. et al. Advanced glycation end-products induce tubular CTGF via TGF-β-independent Smad3 signaling. J Am Soc Nephrol. 2010;21:249-60
39. Liu F, Chen HY, Huang XR, Chung AC, Zhou L, Fu P. et al. C-reactive protein promotes diabetic kidney disease in a mouse model of type 1 diabetes. Diabetologia. 2011;54:2713-23
40. Zhang R, Zhang YY, Huang XR, Wu Y, Chung AC, Wu EX. et al. C-reactive protein promotes cardiac fibrosis and inflammation in angiotensin II-induced hypertensive cardiac disease. Hypertension. 2010;55:953-60
41. You YK, Huang XR, Chen HY, Lyu XF, Liu HF, Lan HY. C-Reactive Protein Promotes Diabetic Kidney Disease in db/db Mice via the CD32b-Smad3-mTOR signaling Pathway. Sci Rep. 2016;6:26740
42. Li ZI, Chung AC, Zhou L, Huang XR, Liu F, Fu P. et al. C-reactive protein promotes acute renal inflammation and fibrosis in unilateral ureteral obstructive nephropathy in mice. Lab Invest. 2011;91:837-51
43. You YK, Wu WF, Huang XR, Li HD, Ren YP, Zeng JC. et al. Deletion of Smad3 protects against C-reactive protein-induced renal fibrosis and inflammation in obstructive nephropathy. Int J Biol Sci. 2021;17:3911-22
44. Qu X, Jiang M, Sun YB, Jiang X, Fu P, Ren Y. et al. The Smad3/Smad4/CDK9 complex promotes renal fibrosis in mice with unilateral ureteral obstruction. Kidney Int. 2015;88:1323-35
45. Wegner K, Bachmann A, Schad JU, Lucarelli P, Sahle S, Nickel P. et al. Dynamics and feedback loops in the transforming growth factor β signaling pathway. Biophys Chem. 2012;162:22-34
46. Liu L, Wang Y, Yan R, Li S, Shi M, Xiao Y. et al. Oxymatrine Inhibits Renal Tubular EMT Induced by High Glucose via Upregulation of SnoN and Inhibition of TGF-β1/Smad Signaling Pathway. PLoS One. 2016;11:e0151986
47. Wang Y, Zhang X, Mao Y, Liang L, Liu L, Peng W. et al. Smad2 and Smad3 play antagonistic roles in high glucose-induced renal tubular fibrosis via the regulation of SnoN. Exp Mol Pathol. 2020;113:104375
48. Tian X, Zhang J, Tan TK, Lyons JG, Zhao H, Niu B. et al. Association of β-catenin with P-Smad3 but not LEF-1 dissociates in vitro profibrotic from anti-inflammatory effects of TGF-beta1. J Cell Sci. 2013;126:67-76
49. Huang XZ, Wen D, Zhang M, Xie Q, Ma L, Guan Y. et al. Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J Cell Biochem. 2014;115:996-1005
50. Shuttleworth VG, Gaughan L, Nawafa L, Mooney CA, Cobb SL, Sheerin NS. et al. The methyltransferase SET9 regulates TGFB1 activation of renal fibroblasts via interaction with SMAD3. J Cell Sci. 2018 131
51. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325-38
52. Chen B, Wang P, Liang X, Jiang C, Ge Y, Dworkin LD. et al. Permissive effect of GSK3beta on profibrogenic plasticity of renal tubular cells in progressive chronic kidney disease. Cell Death Dis. 2021;12:432
53. Werner F, Jain MK, Feinberg MW, Sibinga NE, Pellacani A, Wiesel P. et al. Transforming growth factor-β1 inhibition of macrophage activation is mediated via Smad3. J Biol Chem. 2000;275:36653-8
54. Delisle JS, Giroux M, Boucher G, Landry JR, Hardy MP, Lemieux S. et al. The TGF-β-Smad3 pathway inhibits CD28-dependent cell growth and proliferation of CD4 T cells. Genes Immun. 2013;14:115-26
55. Li MO, Flavell RA. TGF-β: a master of all T cell trades. Cell. 2008;134:392-404
56. Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-β regulation of immune responses. Annu Rev Immunol. 2006;24:99-146
57. Sanjabi S, Oh SA, Li MO. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol. 2017 9
58. Yang C, Huang XR, Fung E, Liu HF, Lan HY. The Regulatory T-cell Transcription Factor Foxp3 Protects against Crescentic Glomerulonephritis. Sci Rep. 2017;7:1481
59. Xu L, Kitani A, Strober W. Molecular mechanisms regulating TGF-β-induced Foxp3 expression. Mucosal Immunol. 2010;3:230-8
60. Martinez GJ, Zhang Z, Chung Y, Reynolds JM, Lin X, Jetten AM. et al. Smad3 differentially regulates the induction of regulatory and inflammatory T cell differentiation. J Biol Chem. 2009;284:35283-6
61. Zhang Y, Meng XM, Huang XR, Wang XJ, Yang L, Lan HY. Transforming growth factor-β1 mediates psoriasis-like lesions via a Smad3-dependent mechanism in mice. Clin Exp Pharmacol Physiol. 2014;41:921-32
62. Zhang F, Tsai S, Kato K, Yamanouchi D, Wang C, Rafii S. et al. Transforming growth factor-β promotes recruitment of bone marrow cells and bone marrow-derived mesenchymal stem cells through stimulation of MCP-1 production in vascular smooth muscle cells. J Biol Chem. 2009;284:17564-74
63. Inazaki K, Kanamaru Y, Kojima Y, Sueyoshi N, Okumura K, Kaneko K. et al. Smad3 deficiency attenuates renal fibrosis, inflammation,and apoptosis after unilateral ureteral obstruction. Kidney Int. 2004;66:597-604
64. Nath KA, Croatt AJ, Warner GM, Grande JP. Genetic deficiency of Smad3 protects against murine ischemic acute kidney injury. Am J Physiol Renal Physiol. 2011;301:F436-42
65. Xu BH, Sheng J, You YK, Huang XR, Ma RCW, Wang Q. et al. Deletion of Smad3 prevents renal fibrosis and inflammation in type 2 diabetic nephropathy. Metabolism. 2020;103:154013
66. Wang W, Huang XR, Li AG, Liu F, Li JH, Truong LD. et al. Signaling mechanism of TGF-β1 in prevention of renal inflammation: role of Smad7. J Am Soc Nephrol. 2005;16:1371-83
67. Ng YY, Hou CC, Wang W, Huang XR, Lan HY. Blockade of NFκB activation and renal inflammation by ultrasound-mediated gene transfer of Smad7 in rat remnant kidney. Kidney Int Suppl. 2005:S83-91.
68. Lai W, Tang Y, Huang XR, Ming-Kuen Tang P, Xu A, Szalai AJ. et al. C-reactive protein promotes acute kidney injury via Smad3-dependent inhibition of CDK2/cyclin E. Kidney Int. 2016;90:610-26
69. Fu S, Tang Y, Huang XR, Feng M, Xu AP, Lan HY. Smad7 protects against acute kidney injury by rescuing tubular epithelial cells from the G1 cell cycle arrest. Clin Sci (Lond). 2017;131:1955-69
70. Tang Y, Huang XR, Lv J, Chung AC, Zhang Y, Chen JZ. et al. C-reactive protein promotes acute kidney injury by impairing G1/S-dependent tubular epithelium cell regeneration. Clin Sci (Lond). 2014;126:645-59
71. Fang Y, Gong AY, Haller ST, Dworkin LD, Liu Z, Gong R. The ageing kidney: Molecular mechanisms and clinical implications. Ageing Res Rev. 2020;63:101151
72. Ueda S, Tominaga T, Ochi A, Sakurai A, Nishimura K, Shibata E. et al. TGF-β1 is involved in senescence-related pathways in glomerular endothelial cells via p16 translocation and p21 induction. Sci Rep. 2021;11:21643
73. Liu W, Chen B, Wang Y, Meng C, Huang H, Huang XR. et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc Natl Acad Sci U S A. 2018;115:E1475-E84
74. Gao L, Liu MM, Zang HM, Ma QY, Yang Q, Jiang L. et al. Restoration of E-cadherin by PPBICA protects against cisplatin-induced acute kidney injury by attenuating inflammation and programmed cell death. Lab Invest. 2018;98:911-23
75. Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z. et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J Am Soc Nephrol. 2015;26:2647-58
76. Jiang L, Liu XQ, Ma Q, Yang Q, Gao L, Li HD. et al. hsa-miR-500a-3P alleviates kidney injury by targeting MLKL-mediated necroptosis in renal epithelial cells. FASEB J. 2019;33:3523-35
77. Yang Q, Gao L, Hu XW, Wang JN, Zhang Y, Dong YH. et al. Smad3-Targeted Therapy Protects against Cisplatin-Induced AKI by Attenuating Programmed Cell Death and Inflammation via a NOX4-Dependent Mechanism. Kidney Dis (Basel). 2021;7:372-90
78. Chen J, Wang W, Tang Y, Huang XR, Yu X, Lan HY. Inflammatory stress in SARS-COV-2 associated Acute Kidney Injury. Int J Biol Sci. 2021;17:1497-506
79. Wang W, Chen J, Hu D, Pan P, Liang L, Wu W. et al. SARS-CoV-2 N Protein Induces Acute Kidney Injury via Smad3-Dependent G1 Cell Cycle Arrest Mechanism. Adv Sci (Weinh). 2021:e2103248.
80. Zhao X, Nicholls JM, Chen YG. Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-β signaling. J Biol Chem. 2008;283:3272-80
81. Chen W. A potential treatment of COVID-19 with TGF-β blockade. Int J Biol Sci. 2020;16:1954-5
82. Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010;11:252-63
83. Lorenzen JM, Haller H, Thum T. MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nat Rev Nephrol. 2011;7:286-94
84. Meng XM, Tang PM, Li J, Lan HY. TGF-β/Smad signaling in renal fibrosis. Front Physiol. 2015;6:82
85. Zhong X, Chung AC, Chen HY, Meng XM, Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol. 2011;22:1668-81
86. Kato M, Zhang J, Wang M, Lanting L, Yuan H, Rossi JJ. et al. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-β-induced collagen expression via inhibition of E-box repressors. Proc Natl Acad Sci U S A. 2007;104:3432-7
87. Wang Q, Wang Y, Minto AW, Wang J, Shi Q, Li X. et al. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J. 2008;22:4126-35
88. Xiong M, Jiang L, Zhou Y, Qiu W, Fang L, Tan R. et al. The miR-200 family regulates TGF-β1-induced renal tubular epithelial to mesenchymal transition through Smad pathway by targeting ZEB1 and ZEB2 expression. Am J Physiol Renal Physiol. 2012;302:F369-79
89. Qin W, Chung AC, Huang XR, Meng XM, Hui DS, Yu CM. et al. TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol. 2011;22:1462-74
90. Ning ZW, Luo XY, Wang GZ, Li Y, Pan MX, Yang RQ. et al. MicroRNA-21 Mediates Angiotensin II-Induced Liver Fibrosis by Activating NLRP3 Inflammasome/IL-1β Axis via Targeting Smad7 and Spry1. Antioxid Redox Signal. 2017;27:1-20
91. Kolling M, Kaucsar T, Schauerte C, Hubner A, Dettling A, Park JK. et al. Therapeutic miR-21 Silencing Ameliorates Diabetic Kidney Disease in Mice. Mol Ther. 2017;25:165-80
92. Yamada M, Kubo H, Ota C, Takahashi T, Tando Y, Suzuki T. et al. The increase of microRNA-21 during lung fibrosis and its contribution to epithelial-mesenchymal transition in pulmonary epithelial cells. Respir Res. 2013;14:95
93. Lyu H, Li X, Wu Q, Hao L. Overexpression of microRNA-21 mediates Ang II-induced renal fibrosis by activating the TGF-β1/Smad3 pathway via suppressing PPARalpha. J Pharmacol Sci. 2019;141:70-8
94. Chung AC, Huang XR, Meng X, Lan HY. miR-192 mediates TGF-β/Smad3-driven renal fibrosis. J Am Soc Nephrol. 2010;21:1317-25
95. Li R, Chung AC, Dong Y, Yang W, Zhong X, Lan HY. The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-β/Smad3-Azin1 pathway. Kidney Int. 2013;84:1129-44
96. Chen HY, Zhong X, Huang XR, Meng XM, You Y, Chung AC. et al. MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol Ther. 2014;22:842-53
97. Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol Ther. 2014;22:974-85
98. Gu YY, Dou JY, Huang XR, Liu XS, Lan HY. Transforming Growth Factor-β and Long Non-coding RNA in Renal Inflammation and Fibrosis. Front Physiol. 2021;12:684236
99. Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7-21
100. Zhou Q, Chung AC, Huang XR, Dong Y, Yu X, Lan HY. Identification of novel long noncoding RNAs associated with TGF-β/Smad3-mediated renal inflammation and fibrosis by RNA sequencing. Am J Pathol. 2014;184:409-17
101. Feng M, Tang PM, Huang XR, Sun SF, You YK, Xiao J. et al. TGF-β Mediates Renal Fibrosis via the Smad3-Erbb4-IR Long Noncoding RNA Axis. Mol Ther. 2018;26:148-61
102. Sun SF, Tang PMK, Feng M, Xiao J, Huang XR, Li P. et al. Novel lncRNA Erbb4-IR Promotes Diabetic Kidney Injury in db/db Mice by Targeting miR-29b. Diabetes. 2018;67:731-44
103. Zhou Q, Huang XR, Yu J, Yu X, Lan HY. Long Noncoding RNA Arid2-IR Is a Novel Therapeutic Target for Renal Inflammation. Mol Ther. 2015;23:1034-43
104. Zhang YY, Tang PM, Tang PC, Xiao J, Huang XR, Yu C. et al. LRNA9884, a Novel Smad3-Dependent Long Noncoding RNA, Promotes Diabetic Kidney Injury in db/db Mice via Enhancing MCP-1-Dependent Renal Inflammation. Diabetes. 2019;68:1485-98
105. Zhang Y, Tang PM, Niu Y, Garcia Cordoba CA, Huang XR, Yu C. et al. Long Non-coding RNA LRNA9884 Promotes Acute Kidney Injury via Regulating NF-kB-Mediated Transcriptional Activation of MIF. Front Physiol. 2020;11:590027
106. Wang P, Luo ML, Song E, Zhou Z, Ma T, Wang J. et al. Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-β/Smad3 pathway. Sci Transl Med. 2018 10
107. Zhang YY, Tan RZ, Yu Y, Niu YY, Yu C. LncRNA GAS5 protects against TGF-β-induced renal fibrosis via the Smad3/miRNA-142-5p axis. Am J Physiol Renal Physiol. 2021;321:F517-F26
108. Sun J, Zhang S, Shi B, Zheng D, Shi J. Transcriptome Identified lncRNAs Associated with Renal Fibrosis in UUO Rat Model. Front Physiol. 2017;8:658
109. Voelker J, Berg PH, Sheetz M, Duffin K, Shen T, Moser B. et al. Anti-TGF-β1 Antibody Therapy in Patients with Diabetic Nephropathy. J Am Soc Nephrol. 2017;28:953-62
110. Vincenti F, Fervenza FC, Campbell KN, Diaz M, Gesualdo L, Nelson P. et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int Rep. 2017;2:800-10
111. Petersen M, Thorikay M, Deckers M, van Dinther M, Grygielko ET, Gellibert F. et al. Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008;73:705-15
112. Moon JA, Kim HT, Cho IS, Sheen YY, Kim DK. IN-1130, a novel transforming growth factor-β type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 2006;70:1234-43
113. Rodon J, Carducci MA, Sepulveda-Sanchez JM, Azaro A, Calvo E, Seoane J. et al. First-in-human dose study of the novel transforming growth factor-β receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res. 2015;21:553-60
114. Tang PM, Zhang YY, Mak TS, Tang PC, Huang XR, Lan HY. Transforming growth factor-β signalling in renal fibrosis: from Smads to non-coding RNAs. J Physiol. 2018;596:3493-503
115. Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y. et al. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59:2612-24
116. Zhang Y, Meng XM, Huang XR, Lan HY. The preventive and therapeutic implication for renal fibrosis by targetting TGF-β/Smad3 signaling. Clin Sci (Lond). 2018;132:1403-15
117. Ji X, Wang H, Wu Z, Zhong X, Zhu M, Zhang Y. et al. Specific Inhibitor of Smad3 (SIS3) Attenuates Fibrosis, Apoptosis, and Inflammation in Unilateral Ureteral Obstruction Kidneys by Inhibition of Transforming Growth Factor β (TGF-β)/Smad3 Signaling. Med Sci Monit. 2018;24:1633-41
118. Ka SM, Yeh YC, Huang XR, Chao TK, Hung YJ, Yu CP. et al. Kidney-targeting Smad7 gene transfer inhibits renal TGF-β/MAD homologue (SMAD) and nuclear factor κB (NF-κB) signalling pathways, and improves diabetic nephropathy in mice. Diabetologia. 2012;55:509-19
119. Ka SM, Huang XR, Lan HY, Tsai PY, Yang SM, Shui HA. et al. Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J Am Soc Nephrol. 2007;18:1777-88
120. Lan HY, Mu W, Tomita N, Huang XR, Li JH, Zhu HJ. et al. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol. 2003;14:1535-48
121. Liu GX, Li YQ, Huang XR, Wei LH, Zhang Y, Feng M. et al. Smad7 inhibits AngII-mediated hypertensive nephropathy in a mouse model of hypertension. Clin Sci (Lond). 2014;127:195-208
122. Liu X, Wang W, Hu H, Tang N, Zhang C, Liang W. et al. Smad3 specific inhibitor, naringenin, decreases the expression of extracellular matrix induced by TGF-β1 in cultured rat hepatic stellate cells. Pharm Res. 2006;23:82-9
123. Schaneberg BT, Mikell JR, Bedir E, Khan IA. An improved HPLC method for quantitative determination of six triterpenes in Centella asiatica extracts and commercial products. Pharmazie. 2003;58:381-4
124. Meng XM, Zhang Y, Huang XR, Ren GL, Li J, Lan HY. Treatment of renal fibrosis by rebalancing TGF-β/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget. 2015;6:36984-97
125. Tan RZ, Wang C, Deng C, Zhong X, Yan Y, Luo Y. et al. Quercetin protects against cisplatin-induced acute kidney injury by inhibiting Mincle/Syk/NF-κB signaling maintained macrophage inflammation. Phytother Res. 2020;34:139-52
126. Gu YY, Zhang M, Cen H, Wu YF, Lu Z, Lu F. et al. Quercetin as a potential treatment for COVID-19-induced acute kidney injury: Based on network pharmacology and molecular docking study. PLoS One. 2021;16:e0245209
127. Ai J, Nie J, He J, Guo Q, Li M, Lei Y. et al. GQ5 Hinders Renal Fibrosis in Obstructive Nephropathy by Selectively Inhibiting TGF-β-Induced Smad3 Phosphorylation. J Am Soc Nephrol. 2015;26:1827-38
128. Li J, Qu X, Ricardo SD, Bertram JF, Nikolic-Paterson DJ. Resveratrol inhibits renal fibrosis in the obstructed kidney: potential role in deacetylation of Smad3. Am J Pathol. 2010;177:1065-71
129. Sierra-Mondragon E, Rodriguez-Munoz R, Namorado-Tonix C, Molina-Jijon E, Romero-Trejo D, Pedraza-Chaverri J. et al. All-Trans Retinoic Acid Attenuates Fibrotic Processes by Downregulating TGF-β1/Smad3 in Early Diabetic Nephropathy. Biomolecules. 2019 9
Author contact
![]() Corresponding authors: Hui-Yao Lan, Departments of Medicine & Therapeutics, Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong, China. E-mail: hylanedu.hk; and Xueqing Yu, Guangdong Academy of Medical Sciences, Guangdong Provincial People's Hospital, Guangzhou, China, E-mail: yuxqsysu.edu.cn.
Corresponding authors: Hui-Yao Lan, Departments of Medicine & Therapeutics, Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong, China. E-mail: hylanedu.hk; and Xueqing Yu, Guangdong Academy of Medical Sciences, Guangdong Provincial People's Hospital, Guangzhou, China, E-mail: yuxqsysu.edu.cn.