Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(9):3714-3730. doi:10.7150/ijbs.69989 This issue Cite
Research Paper
Isovalerylspiramycin I suppresses non-small cell lung carcinoma growth through ROS-mediated inhibition of PI3K/AKT signaling pathway
Zeyu Liu1, Moli Huang2, Yue Hong3, Shaoyang Wang1, Yongle Xu1, Cheng Zhong1, Jingyuan Zhang1, Zhengping Zhuang4,5, ![]() , Shan Shan1,
, Shan Shan1, ![]() , Tao Ren1,6,
, Tao Ren1,6, ![]()
1. Department of Respiratory and Clinical Care Medicine, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, 200233, China.
2. Department of Bioinformatics, School of Biological and Basic Medical Sciences, Soochow University, Suzhou, 215123, China.
3. Stem Cell Center, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, 200233, China.
4. Neuro-Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA.
5. Surgical Neurology Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA.
6. Shanghai Key Laboratory of Sleep Disordered Breathing, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, 200233, China.
Received 2021-12-12; Accepted 2022-5-10; Published 2022-5-21
Abstract

Novel drugs are required for non-small cell lung cancer (NSCLC) treatment urgently. Repurposing old drugs as new treatments is a practicable approach with time and cost savings. Some studies have shown that carrimycin, a Chinese Food and Drug Administration (CFDA)-approved macrolide antibiotic, possesses potent anti-tumor effects against oral squamous cell carcinoma. However, its detailed component and underlying mechanisms in anti-NSCLC remain unknown. In our study, isovalerylspiramycin I (ISP-I) was isolated from carrimycin and demonstrated a remarkable anti-NSCLC efficacy in vitro and in vivo with a favorable safety profile. It has been proven that in NSCLC cell lines H460 and A549, ISP-I could induce G2/M arrest and apoptosis, which was mainly attributed to ROS accumulation and subsequently PI3K/AKT signaling pathway inhibition. Numerous downstream genes including mTOR and FOXOs were also changed correspondingly. An observation of NAC-induced reverse effect on ISP-I-leading cell death and PI3K/AKT pathway inhibition, emphasized the necessity of ROS signaling in this event. Moreover, we identified ROS accumulation and PI3K/AKT pathway inhibition in tumor xenograft models in vivo as well. Taken together, our study firstly reveals that ISP-I is a novel ROS inducer and may act as a promising candidate with multi-target and low biological toxicity for anti-NSCLC treatment.
Keywords: Isovalerylspiramycin I (ISP-I), Non-small cell lung cancer (NSCLC), apoptosis, G2/M arrest, ROS, PI3K/AKT signaling pathway
Introduction
Lung cancer is the second most commonly diagnosed cancer worldwide, causing over 1.8 million deaths in 2020 [1]. Although diagnostic and therapeutic methods have markedly improved, the prognosis of lung cancer continues to be poor with a 5-year survival rate of only 23% [2]. NSCLC is the most prevalent histological subtype of lung cancer, accounting for 85-90% of all cases [3]. The first choice of treatment for NSCLC is surgical removal, however, high risk of postoperative recurrence is still the biggest challenge and associates with significantly reduced survival [4]. Besides, a considerable number of patients with advanced NSCLC are inoperable [5]. Chemotherapy and radiotherapy are still the basic anticancer strategies in the treatment of lung cancer [6], yet its efficacy and adverse side effects issues occur frequently and result in cancer recurrence and lethal outcomes [7]. In recent years, the emerging immunotherapy and molecular targeted therapy have made great contributions to the precision NSCLC treatment. Nevertheless, only a small fraction of NSCLC patients can benefit from these two approaches due to its exactly genetic mutation matching and high prices [8-11]. In addition, emergence of drug resistance has limited the benefit of immunotherapy and molecular targeted therapy, especially single-target inhibitors; thus, they cannot provide long-term survival for most patients [10, 11]. Consequently, novel anticancer agents to inhibit NSCLC with low biological toxicity and multi-target are urgently needed.
Recent studies have shown that tumor cells possess a higher level of ROS and are easier to reach the toxic threshold, thus, further ROS accumulation can act as an effective means of tumor inhibition while normal cells are less or not affected [12]. The overproduction of ROS in NSCLC is served as an initiator and can trigger multiple pathways alterations related to apoptosis, cell cycle and protein synthesis. One prototypical example is the PI3K/AKT signaling pathway [13]. Numerous genes (PIK3CB, AKT1, MTOR, FOXOs, CDKN1A etc.) is involved in PI3K/AKT signaling pathway and can induce expression and activation of multiple receptor tyrosine kinases (RTKs) [14]. Each step in the RTK cascade can be a potential therapeutic target and multi-targeted RTK inhibitors are clinically effective against tumors [15]. Taken together, novel ROS inducer may act as a promising attempt in the development of anti-tumor agent with multi-target and low biological toxicity.
Developing new drugs is a very expensive and time-consuming process [16]. In recent years, “new uses of old drugs” gets great attentions and provides an abundant source for novel anti-tumor agents [17]. Carrimycin is a CFDA-approved newly synthesized macrolide antibiotic, which possesses excellent bioavailability and biosafety in phase I to III clinical trials. Previous studies have revealed that carrimycin can regulate cell physiology, proliferation and immunity through inhibition of protein synthesis, and exhibits a potential anti-tumor activity against oral squamous cell carcinoma [18]. Accordingly, we hypothesize that carrimycin may have the properties of anti-NSCLC, which needs further explore.
To exactly clarify the pharmacological mechanism of carrimycin, we isolated and purified the active component of carrimycin, Isovalerylspiramycin I (ISP-I). The efficacy of ISP-I on the inhibition of NSCLC and its underlying mechanisms were evaluated in the current research. We revealed that ISP-I significantly inhibited NSCLC growth both in vitro and in vivo via excessive ROS accumulation, which mediated PI3K/AKT signaling pathway inhibition. In summary, our study reveals that ISP-I is a novel ROS inducer and may be a promising candidate with low biological toxicity and multi-target ability for anti-NSCLC treatment.
Materials and methods
Reagents and antibodies
ISP-I (95.9% purity) was procured from Shanghai Tonglian Pharmaceutical Co., Ltd. (China). For cell experiments, ISP-I was dissolved to 100mM stock concentration in DMSO (Sigma) and diluted to appropriate concentrations with cell culture medium (final DMSO concentration in culture medium was no greater than 0.015%). For animal experiments, ISP-I was dissolved in Polyethylene glycol (PEG; Sigma), vegetable oil and Tween-80 (Sigma) mixed solvents (v:v:v = 9.5:9.5:1) at 24mg/ml, and then diluted to the final concentration with sterilized water.
The CCK-8 solution, DCFH-DA and crystal violet solution were all procured from Beyotime (Shanghai, China). Dihydroethidium (DHE, #D7008) and N-acetyl-l-cysteine (NAC, A9165) were provided by Sigma (MO, USA). The antibodies used in our research were provided in Supplementary Table S1.
Cell lines and culture
Human NSCLC cell lines (H460 and A549) as well as human lung epithelial cells (Beas2B) were acquired from Chinese Academy of Sciences cell bank. Beas2B was grown in DMEM (Gibco, Grand Island, NY, USA) while H460 and A549 were grown in RPMI-1640 (HyClone, Logan, UT, USA) at 37 °C in a humidified 5% CO2 environment. Media were both supplemented with 10% fetal bovine serum (FBS, HyClone, Logan, UT, USA) and streptomycin/penicillin (1%, Gibco, Grand Island, NY, USA). A Nikon Eclipse Ts2 inverted microscope (100× magnification) was used to observe and image cell morphology and numbers.
CCK-8 assay
Cells were seeded in 96-well plates at 4 × 103 cells/well followed by overnight incubation. Next, they were treated with different doses of ISP-I or DMSO (control) for 24, 48 and 72 h. NSCLC cells after the different treatments were incubated with CCK-8 solution (1:10 dilution) for 1h at 37°C. Fluorescence for every plate was measured by SpectraMax® absorbance reader (Molecular Devices) at 450 nm. The half-maximal inhibitory concentrations (IC50) were evaluated by GraphPad Prism version 8.3.0 software.
Colony formation assay
Cells cultured in 6-well plates (103 cells/well) were allowed to attach followed by overnight incubation and treatment with varying of ISP-I or DMSO (control) doses. After different treatments for 24 h, drug-comprising media were exchanged with complete culture media. Medium replacement was done after every 3 days until the surviving cells formed visible colonies 14 days later. Then, we stained the colonies with crystal violet (0.5%, 20 min) and washed thoroughly. Visible colonies with ≥ 50 cells were enumerated.
Cell apoptosis and cell cycle assay
Cells in 6-well plates (2 × 105 cells/well) were incubated overnight, then treated with 5μM, 10μM, 15μM of ISP-I or DMSO (control) for 48h. Apoptosis was estimated by the Annexin V-FITC/PI Apoptosis Kit (AP101, MULTI Sciences), and was detected by flow cytometry (CytoFLEX LX; Beckman Coulter, Inc., USA). Operations were carried out according to kit instructions. Data were processed by CytExpert software (Beckman Coulter, USA). Cell cycles were evaluated by the Cell Cycle Assay Kit (CCS012, MULTI Sciences) and measured using flow cytometry (BD Accuri C6 flow cytometer, BD Biosciences, USA). Results were analyzed by ModFit software.
RNA-sequencing analysis
Total RNA extraction from non-treated as well as ISP-I (10μM) treated NSCLC cells (3 replicates per group) was done by TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and RNeasy Mini Kit (Qiagen, Valencia, CA, USA). The concentrations, quality as well as integrity of extracted RNA were assessed by NanoDrop spectrophotometry (Thermo Scientific). Then, 3 micrograms of RNA (RIN 10.0) from each sample were utilized as input materials for preparation of RNA samples. Samples were sequenced and analyzed by Shanghai Personal Biotechnology Cp. Ltd. Briefly, sequencing libraries were constructed by TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA, USA), quantified using qRTPCR and the Agilent high sensitivity DNA assay on Bioanalyzer 2100 system (Agilent), and then sequenced on Illumina Novaseq 6000 system to generate 150bp pair-end reads. The reads, filtered by FastQC, were mapped to human genome (hg38) using HISAT2 software. Read counts were calculated using HTSeq v0.9.1 and output as FPKM upon normalization. Differentially expressed genes (DEGs) were defined as fold change > 2 and normalized p < 0.05. Heatmap of DEGs was drawn by R. Gene Ontology (GO) (http://www.geneontology.org/) was used for analysis biological processes (BP) while the Kyoto Encyclopedia of Genes and Genomes (KEGG) for pathway analysis. DEGs were subjected to GO_BP enrichment, KEGG analysis as well as Gene Set Enrichment Analysis (GSEA). Circle plots were drawn to depict the related DEGs involved in biological process terms of interest. Raw data and processed data were uploaded to GEO database (accession number: GSE 200370).
Quantitative real-time PCR analysis (qRTPCR)
Total RNA was prepared as mentioned above, after which 1μg of isolated RNA was utilized in reverse transcription assays using a RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) as indicated by the manufacturer.
RT-PCR analysis were performed in a reaction volume of 20μl, including 10ng of cDNA, 200 nM each of forward as well as reverse primer, 1x Power SYBR Green Master Mix (Applied Biosystems) and ddH2O, followed by amplification using the QuantStudio 5 System (Applied Biosystems). Supplementary Table S2 showed the primers used in this study. Final normalized expression values were analyzed using 2-△△CT method relative to the endogenous control GAPDH.
Measurement of ROS
Cells were seeded at 2 × 105 cells/well in 6-well plate and 105 cells/dish in a 35-mm glass bottom dish (Nest, Shanghai). After an overnight culture, 10μM ISP-I or DMSO (control) with or without 2mM NAC (a ROS scavenger) was supplemented to cells and incubated for 48 h [19-21]. Treated cells were then loaded with DCFH-DA (1:1000 dilution) and incubated at 37 °C for 30 min. Removal of excess DCFH-DA was performed by washing with RPMI 1640 medium. Cell esterases could hydrolyze DCFH-DA to form DCFH, which was oxidized to fluorescent DCF by intracellular ROS [22]. Fluorescent intensity of DCF was evaluated by flow cytometry, which was performed on Beckman CytoFLEX LX, followed by analysis using CytExpert software. DCF fluorescent intensity was also visualized by confocal microscopy (Olympus IX83, Tokyo, Japan) with an excitation laser at 473 nm and power at ~0.1 mW level. Image J was used to measure mean fluorescence intensity (MFI).
Western blotting
Total protein extraction from cells was done by the RIPA buffer (#9806, Cell Signaling technology) supplemented with 1× Halt™ protease and protein phosphatase inhibitor cocktail (#78440, Thermo Scientific), and tumor tissue protein was obtained using tissue protein extraction kit (PC201, Epizyme) according to the instruction manuals. The Separation of equivalent amounts of proteins (20µg) was done using different concentrations (6%, 10%, 12.5%) of SDS-PAGE gel (PG110, PG112, Epizyme), and transferred to PVDF membrane, followed by blocking with QuickBlock™ Blocking Buffer (P0252, Beyotime). Then, they were washed thoroughly with TBST and incubated at 4 °C immersed with the primary antibodies overnight. Membranes were washed thrice and then incubated with secondary antibodies (2 h, 25°C). NcmECL Ultra (P10100A, P10100B, New Cell &) were used to visualize immunoreactive proteins via Bio-Rad ChemiDoc Imaging system.
Xenograft tumor model
5×106 H460 and A549 cells were subcutaneously administered into right flanks of male nude mice (5 weeks old, Shanghai South Model Biotechnology Co., Ltd.). When tumors reached approximately 100mm3, tumor-bearing mice were grouped randomly (n = 5/group) for daily oral gavage of ISP-I (30 or 60 mg/kg, experimental groups) or vehicle (water containing PEG, vegetable oil and Tween-80, control group) for 18days. Tumor sizes were determined every three days, and were calculated with the formula V = π/6(length×width2). When the largest tumors approached 2000 mm3 (day18, endpoint), mice were sacrificed, and tumors were harvested and weighed.
Hematoxylin and eosin (HE) and immunohistochemistry (IHC) staining
Tumor tissues and organs of each group were fixed, paraffin-embedded, and sectioned into three-micrometer-thick. Sections were then deparaffinized in xylene after which decreasing ethanol concentrations were used for rehydration. HE staining was conducted following standard protocols for routine analysis. Briefly, staining of deparaffinized sections with hematoxylin was done for 5 min followed by dipping in 1% of acid ethanol and 1% ammonia water. Then, they were eosin-stained for 5 min. For IHC staining, after antigen repair with tetra-acetic acid (EDTA) buffer (PH8.0), sections were incubated in H2O2 solution (3%) to block endogenous peroxidase. Next, they were incubated with primary antibody (Ki-67, 1:200; cleaved caspase-3, 1:100) overnight at 4 °C and then with secondary antibody (HRP-conjugated goat anti-rabbit antibody, 1:100) for 2 h at RT. Immunohistochemical reactions were developed in a substrate solution of DAB and nuclei were counter-stained by hematoxylin. At last, slides were dehydrated with graded alcohol and cleared in xylene. The brown granules in the nucleus or cytoplasm represented positive expression of Ki-67 as well as cleaved caspase-3. The slides were observed and photographed under light microscopy (Olympus IX53, Tokyo, Japan).
Dihydroethidium (DHE) staining
A portion of the tumor tissue from different groups were immediately frozen in OCT embedding agent (Sakura, 4583), and then cut into 6μm thick frozen sections by Thermo FINESSE 325. The frozen tumor tissue sections were next incubated with 20μM DHE (37°C, 30 min) in darkness. Afterwards, sections were thoroughly washed and photographed by a fluorescence microscope (Olympus BX53, Tokyo, Japan).
Blood biochemical analysis
ISP-I (30 or 60 mg/kg, experimental groups) or vehicle (water containing PEG, vegetable oil and Tween-80, control group) were administered daily by oral gavage in nude mice. After 18 days, blood samples were collected from the mice of above groupings by removing eyeballs for serum biochemical analysis. The fresh blood samples were clotted for 2 h at room temperature, and then centrifuged (1000× g, 15 min) at 4°C to obtain serum. 200 μL serum was aliquot and analyzed of liver and kidney function indicators including alanine transaminase (ALT), aspartate transaminase (AST), albumin (ALB), blood urea nitrogen (BUN) and creatinine (Cr) by an automated Chemray 240 clinical analyzer (Rayto, Shenzhen, China).
Orthotopic lung tumor model
To directly detect the efficacy of ISP-I against NSCLC, we generated orthotopic lung tumors in nude mice by tail vein injection with 1 × 106 H460 or A549 cells. After tumor formation in the lung was ensured using a Skyscan-1176 microCT scanning, we divided the tumor-bearing mice into three groups (4 per group) randomly for daily oral gavage of ISP-I (30 or 60 mg/kg) or vehicle as mentioned above. Mice were sacrificed after 18 days of treatment and lungs from each mouse were taken out for HE staining. The number and size of tumors in the lung were evaluated by gross and histology analysis.
All animal procedures in this study were conducted under IACUC guidelines and approved by the Ethics Committee of Shanghai Jiao Tong University affiliated Sixth People's Hospital.
Statistical analysis
All results were expressed as mean ± standard deviation (S.D.). Mean values were calculated from data obtained from three independent experiments, each performed in triplicate unless otherwise mentioned. Unpaired Student t test (group=2) and one way ANOVA followed by Dunnett's multiple comparisons test (group≥3) were used to compare the differences between groups. All statistical analysis were done by GraphPad Prism software 8.3.0. P < 0.05 was the cut-off for statistically significant.
Results
ISP-I suppressed the proliferation of NSCLC cells
Purity and chemical structure of ISP-I were shown in Fig. 1A. Fig. 1B, C, shows that ISP-I dose- and time-dependently reduced cell viabilities. The IC50 doses of ISP-I for H460 and A549 were 8.648μM and 12.66μM, respectively (Fig. 1C). Then, we treated NSCLC cell lines with ISP-I for 24 h and evaluated colony formation abilities and growth 2 weeks later. It was revealed that colony formation was dose-dependently inhibited by ISP-I (Fig. 1D). Moreover, to evaluate cell toxicity effects of ISP-I in human normal lung epithelial cells, we next treated BEAS-2B with ISP-I. The IC50 doses of ISP-I for BEAS-2B were above 20 μM (Fig. 1E), which was greater than its efficacy doses in NSCLC cells. The cell morphology and number of NSCLC cells (Supplementary Fig. S1) and BEAS-2B cells (Fig. 1F) were observed after treatment with ISP-I at various dosages for 48h. These findings imply that ISP-I toxicity was selective to NSCLC cells, when compared to normal cells.
SP-I suppressed NSCLC cell proliferation. (A) The purity and chemical structure of ISP-I. (B) Cell viabilities of H460 and A549 treated with various doses of ISP-I were assessed by CCK-8 in 96-well plates. (C) IC50 values of ISP-I in H460 and A549 cells were determined after 48 h incubation by GraphPad Prism software. (D) Morphology and clonogenicity of H460 and A549 cells were visualized after crystal violet staining. Colonies were counted. (E) BEAS-2B were treated with varying doses of ISP-I at different durations. CCK-8 assay was performed for cell viability. (F) Morphology and number of BEAS-2B cells were observed under inverted microscope after 48h treatment with various dosages ISP-I. ** p < 0.01 and *** p < 0.001.
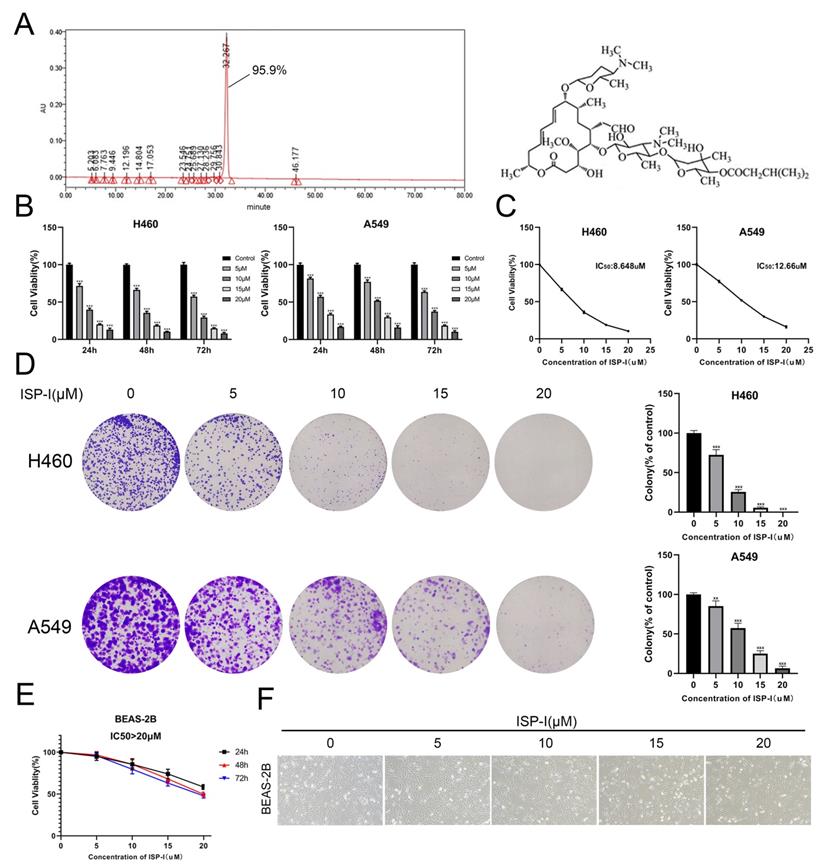
ISP-I induced apoptosis and G2/M arrest in NSCLC cells
We analyzed apoptosis and cell cycle in H460 and A549 to further determine the details of ISP-I induced cytotoxicity in NSCLC. Here, we revealed that 48 h treatment with ISP-I at a gradually increased dose of 5μM, 10μM and 15μM induced 11.22%, 26.95%, 40.55% apoptosis in H460 and induced 9.54%, 19.01%, 27.09% apoptosis in A549 cells (Fig. 2A). To additional validate our cell apoptosis data, we checked mitochondria-induced endogenous apoptosis associated genes BAX, BBC3 (Puma) and BCL2 expression levels [23], and also examined activation of caspase-9 (endogenous apoptosis initiator), caspase-8 (exogenous apoptosis initiator) and caspase-3 (apoptosis executioner) in the treated samples (Fig. 2B). These results implied that ISP-I induced both endogenous apoptosis and exogenous apoptosis meanwhile.
ISP-I induced apoptosis and G2/M arrest in H460 and A549. (A) NSCLC cells were exposed to increasing doses of ISP-I for 48h, and apoptosis was evaluated by flow cytometry analysis after Annexin V and PI double staining. (B) Bax, Bcl-2, Puma and cleaved Caspase 3/ 8/ 9 expression levels were evaluated when H460 and A549 were treated with ISP-I at 0, 5, 10 and 15 μM for 48h. (C) Cell cycle analysis after treatment for 48 h with increasing dosages of ISP-I. (D) Protein levels of p21, phospho-cdc2, cdc2 and cyclin B1 were investigated after treatment with increasing doses of ISP-I for 48 h by western blotting. ns p > 0.05, * p < 0.05, ** p < 0.01 and *** p < 0.001.
With regards to the mechanism of anti-proliferative effects of ISP-I, Fig. 2C showed that ISP-I treatment for 48h obviously ascended the abundance of cells in G2/M phase. The transition from G2 phase to mitosis was initiated by activated cyclin B1/cdc2 complex, which could be inhibited by the increased p21 [24, 25]. As shown in Fig. 2D, ISP-I elevated phospho-cdc2 and p21 whilst suppressing cyclin B1 and cdc2 protein levels. Collectively, the above results indicated that ISP-I caused G2/M arrest by altering vital molecules during G2/M phase transitions.
The anti-tumor effect of ISP-1 depended on ROS accumulation
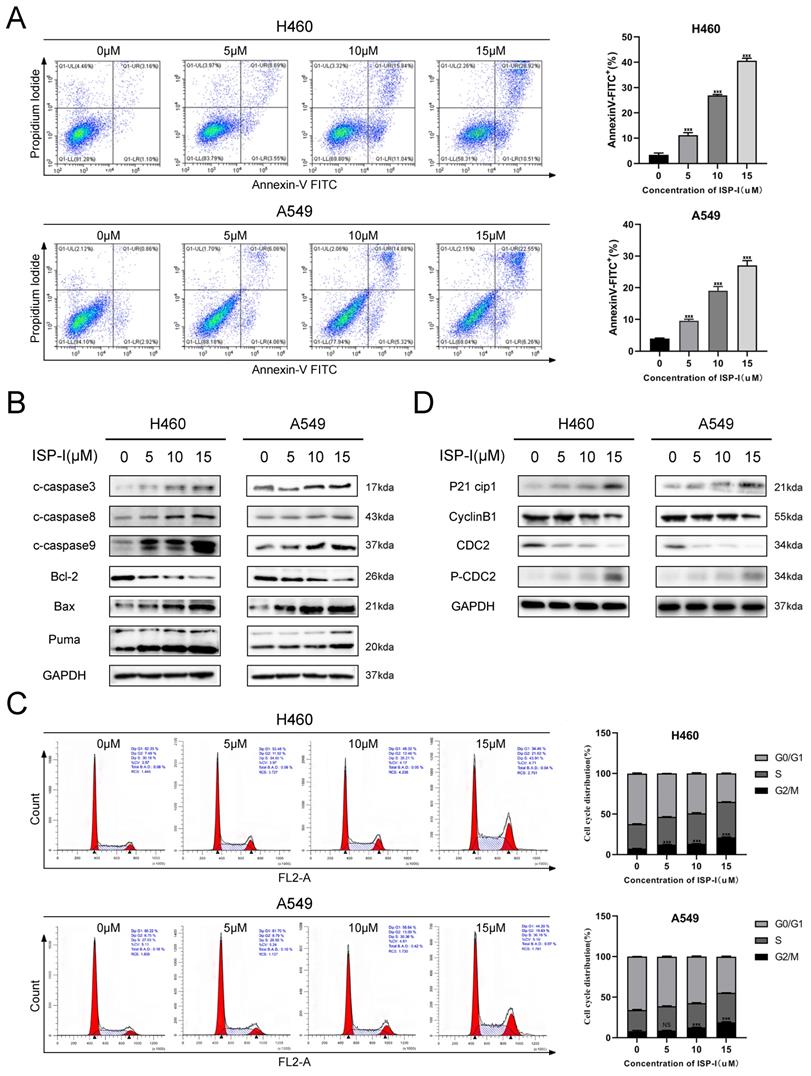
To explore the possible mechanisms of ISP-I-induced apoptosis and G2/M arrest, we performed RNA-seq comparisons of NSCLC cells after ISP-I (10μM) treatment for 48h. The results demonstrated that 3571 genes were up-regulated and 2790 genes were down-regulated after ISP-I treatment in H460, similar results were observed in A549 (Fig. 3A, 3B). We analyzed DEGs by Gene Ontology (GO) enrichment analysis. Here, we focused on the BP. All possibly relevant BP terms were presented in the bubble chart and ROS related terms (e.g., GO: 0006979, GO: 0000302, GO: 0034599) were significantly enriched (Fig. 3C). GSEA was also performed. The results demonstrated that response to ROS was markedly positive regulation in H460 and A549 treated with ISP-I (H460: normalized enrichment score (NES) = 1.35, normalized p-value (P) = 0.000; A549: NES = 1.36, P = 0.000) (Fig. 3D). According to the above analysis, we hypothesized that ROS accumulation was pivotal biological processes triggered by ISP-I. Next, we identified excess intracellular ROS contents through flow cytometry (Fig. 3E) and confocal microscopy (Fig. 3F). The data indicated that treatment with ISP-I (10μM) for 48h significantly increased cellular ROS levels in H460 and A549 cells, and 2 mM ROS scavenger NAC could effectively decrease ROS accumulation.
ISP-I significantly enhanced the intracellular ROS levels in NSCLC cells. (A) The cluster heat map of DEGs expression patterns of H460 and A549 after treatment with ISP-I vs. control as detected by RNA-Seq (up-regulated, red; down-regulated, blue). (B) Volcano plots showed DEG number of H460 and A549 after ISP-I treatment vs. control (up-regulated, red; down-regulated, blue; unchanged, black). (C) List of enriched GO_BP terms. (D) GSEA enrichment plots after ISP-I treated, NES and P were shown in each plot. (E) The relative fluorescence intensity for ROS generation by flow cytometry in H460 and A549 after ISP-I (10μM, 48h) with or without NAC (2 mM) treatment for 48h. (F) Confocal microscopy images of intracellular ROS generation in H460 and A549. Treatment protocols were the same as described in Fig. 3E. The mean fluorescence intensity for ROS generation was analyzed by Image J. Scale bars=20μm. ns p > 0.05, * p < 0.05 and *** p < 0.001.
ROS accumulation could trigger cell death in many aspects [26]. We next assessed whether ROS was involved in ISP-I-induced NSCLC growth inhibition. As shown in Fig. 4A and 4B, the changes of cell morphology (Fig. 4A) and viability (Fig. 4B) after ISP-I treatment in H460 and A549 were remarkably reversed by the addition of NAC. Taken together, ROS accumulation is required for ISP-I-mediated tumor growth inhibition in NSCLC cells.
The anti-tumor effect of ISP-1 depended on ROS accumulation. (A) Changes in the morphology and cell numbers of H460 and A549 after indicated treatments as imaged by inverted microscope (magnification, 100×). (B) Viabilities of H460 and A549 cells after ISP-I (10μM, 48h) treatment with or without NAC (2 mM) were assessed by CCK-8 assay. *** p < 0.001.

The ROS-mediated PI3K/AKT signaling pathway was involved in the anti-tumor performance of ISP-I
To find the detailed mechanism underlying ROS-mediated tumor inhibition, we analyzed DEGs by KEGG pathway. Several pathways could be affected by ISP-I and one of the most enriched pathways was the PI3K/AKT signaling pathway (Fig. 5A). This pathway is notably ROS-regulated and performs important functions in cancer cell proliferation and apoptosis [27]. Here, we identified that PI3K/AKT signaling pathway was significantly suppressed by ISP-I. A heat map showed the mRNA level alteration of several related genes, including PIK3CB (PI3K), AKT1 (AKT), MTOR, EIF4EBP1 (4eBP1), EIF4E (eIF-4E), FOXO1, FOXO3, CDKN1A (p21), CDK1 (cdc2), CCNB1 (cyclinB1), GADD45A (GADD45), BBC3 (Puma), BAX, Fas (Apo1, CD95), TNFRSF10A (Apo2, DR4), TNFRSF10B (DR5) and FADD, which were the crucial genes involved in PI3K/AKT signaling pathway and directly led to apoptosis and G2/M arrest (Fig. 5B). Next, we validated the expression of key differential genes mentioned above by qRTPCR, and the results were consistent with the RNA-seq analysis (Fig. 5C). We also used western blotting to verify PI3K/AKT signaling pathway inhibition. In H460 and A549, the expression levels of total PI3K, total AKT, total mTOR, p-PI3K, p-AKT and p-mTOR were obviously decreased after 48h treatment with ISP-I in a dosage-dependent manner, whereas total FOXO1/3a and p-FOXO1/3a were up-regulated (Fig. 5D).
The PI3K/AKT signaling pathway was significantly suppressed. (A) The significantly enriched pathways of DEGs related with NSCLC growth were shown in the bubble chart by KEGG pathway analysis. (B) A heat map from RNA-seq dataset demonstrated the mRNA levels of crucial genes involved in PI3K/AKT signaling pathway in H460 and A549 after ISP-I treatment for 48h vs. control. (high expression, red; low expression, blue). (C) The mRNA expression levels of genes mentioned above was assessed by qRTPCR. (D) The protein expression levels of total PI3K, total AKT, total mTOR, total FOXO3a, total FOXO1, p-PI3K, p-AKT, p-mTOR, p-FOXO3a and p-FOXO1 were examined after ISP-I treatment for 48h in H460 and A549 by western blotting. *** p < 0.001.

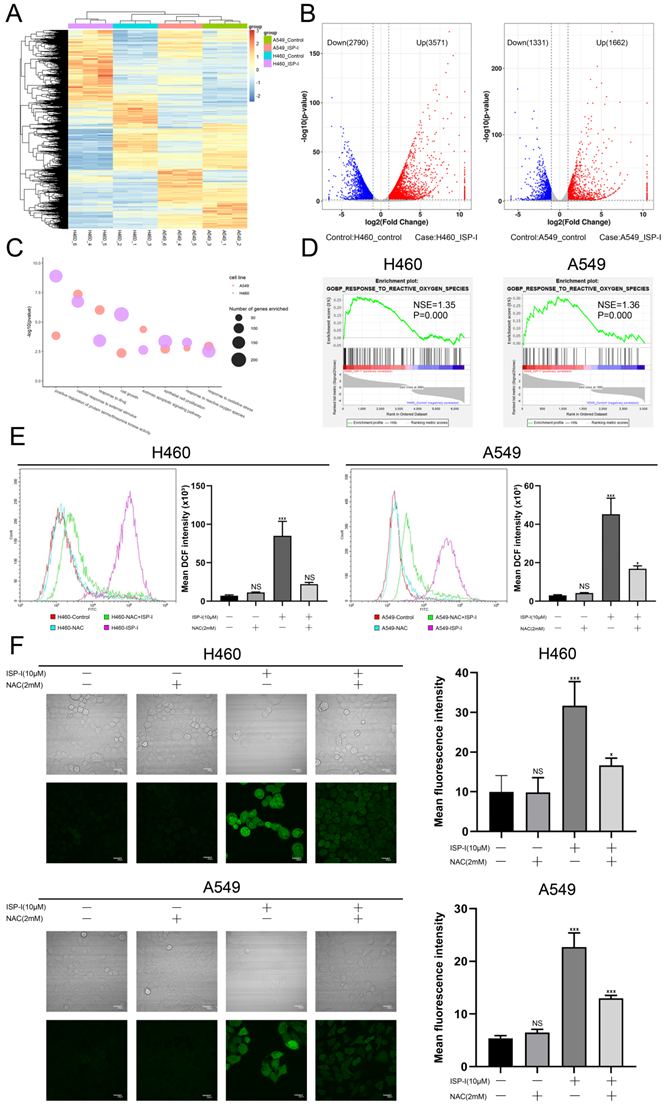
ROS accumulation has been implicated as an upstream regulator in inhibition of PI3K/AKT pathway [28, 29]. Circle plots from RNA-seq data depicted that the enrichment GO_BP terms, especially ROS related, were closely correlated with several essential genes involved in PI3K/AKT pathway (Fig. 6A). We further treated H460 and A549 with 10μM ISP-I alone or in combination with 2mM NAC for 48h. The results showed that compare to single ISP-I treatment group, the expression levels of PI3K, p-PI3K, AKT and p-AKT in H460 and A549 were restored after ISP-I + NAC treatment, which suggested that PI3K/AKT pathway inactivation was mediated by ROS accumulation (Fig. 6B). In conclusion, these above findings validated that the PI3K/AKT signaling pathway was involved in ROS-mediated tumor inhibition.
PI3K/AKT signaling pathway inactivation was ROS-dependent. (A) Circle plots from RNA-seq data revealed the relationship between enriched BP terms, including ROS related terms, and several essential genes involved in PI3K/AKT pathway. (B) The protein expression levels of PI3K, p-PI3K, AKT and p-AKT in H460 and A549 after ISP-I (10μM, 48h) treatment with or without NAC (2 mM) by western blotting.
ISP-I inhibited growth of tumor in vivo
In vivo effect of ISP-I on NSCLC was determined via intragastric administration in a tumor-transplanted nude mouse model established by subcutaneously injecting H460 and A549 cells. After treating with ISP-I for 18 days at doses of 30 and 60 mg/kg, the tumor volume and weight were both significantly inhibited compared to vehicle-treated group (Fig. 7A-7B), while the body weight of mice was stable with a slight upward trend (Fig. 7C). Furthermore, we also generated orthotopic lung tumors to directly detect the efficacy of ISP-I against NSCLC in situ. After 18 days of treatment, the number and size of tumors in the lung of each experimental group (30 and 60 mg/kg) were significantly less than those in control group (Supplementary Fig. S2).
ISP-I possessed a favorable tumor-suppressive effect as well as a good safety profile in vivo. (A) Xenograft tumors in vehicle-treated group and ISP-I-treated groups (30mg/kg, 60mg/kg) after 18 days' treatment. Tumor volumes were calculated every 3 days to plot tumor growth curve. (B) Tumors weight in vehicle-treated group and ISP-I-treated groups (30mg/kg, 60mg/kg) at the end of experiments. (C) Body weight curve in nude mice. (D) The HE staining images of major organs in mice after vehicle- and ISP-I-treatment (30mg/kg, 60mg/kg). (E) Liver and kidney function indicators between blank control group and ISP-I-treated groups. n=5, *** p < 0.001.
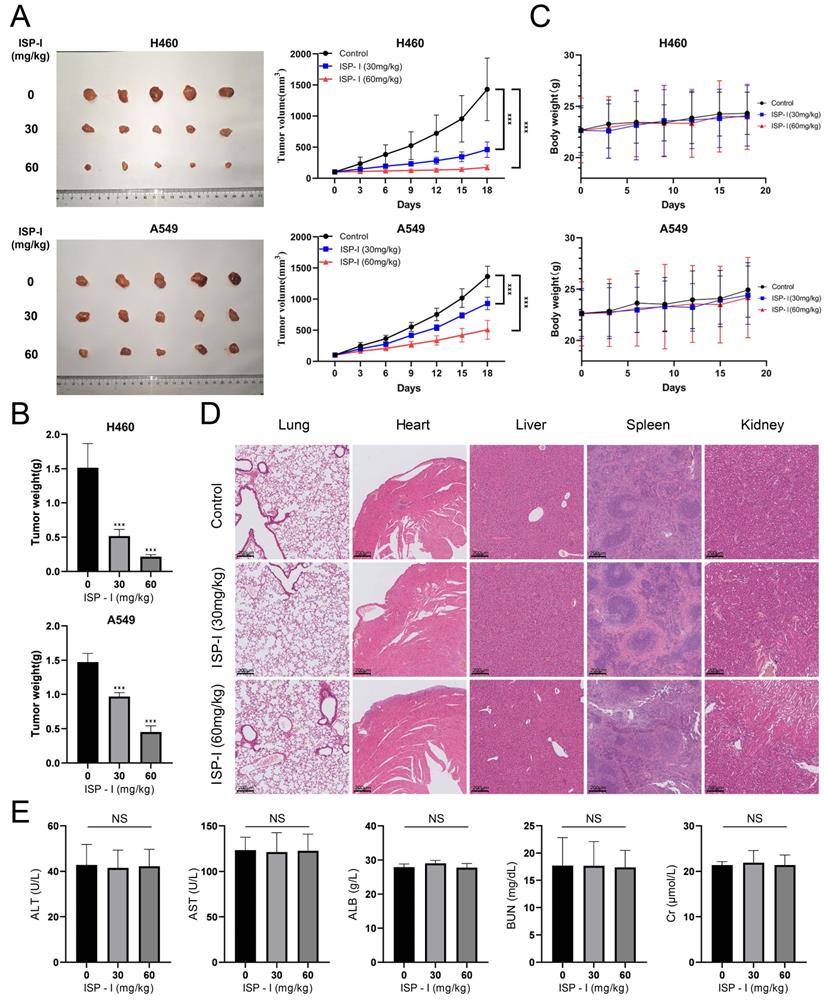
To further investigate whether ISP-I had potential cytotoxic effects on normal tissues, organs and blood samples of various treated groups (0mg/kg, 30mg/kg, 60mg/kg) were collected after oral administration for 18 consecutive days. HE staining of major organs exhibited no significant pathological changes, which revealed ISP-I was no noticeable major organ-related toxicities (Fig. 7D, Supplementary Fig. S3). Blood biochemical tests were carried out as well. The results showed that the liver and kidney function indicators between blank control group and ISP-I-treated groups were no statistical differences and remained at normal levels (Fig. 7E). In addition, to further evaluate ISP-I's effect on the more proliferating organs of the body, we added IHC staining analysis of proliferation markers Ki67 and apoptosis markers cleaved-caspase-3 of stomach, small intestine, skin, and eye. The results indicated that treatment with ISP-I did not exert an effect on the more proliferating organs in nude mice (Supplementary Fig. S3). To sum up, no significant tissue toxicity and inflammation were detected after treatment with ISP-I at the maximum daily dose of 60mg/kg. The above results indicated that ISP-I possesses a favorable tumor-suppressive effect as well as a good safety profile.
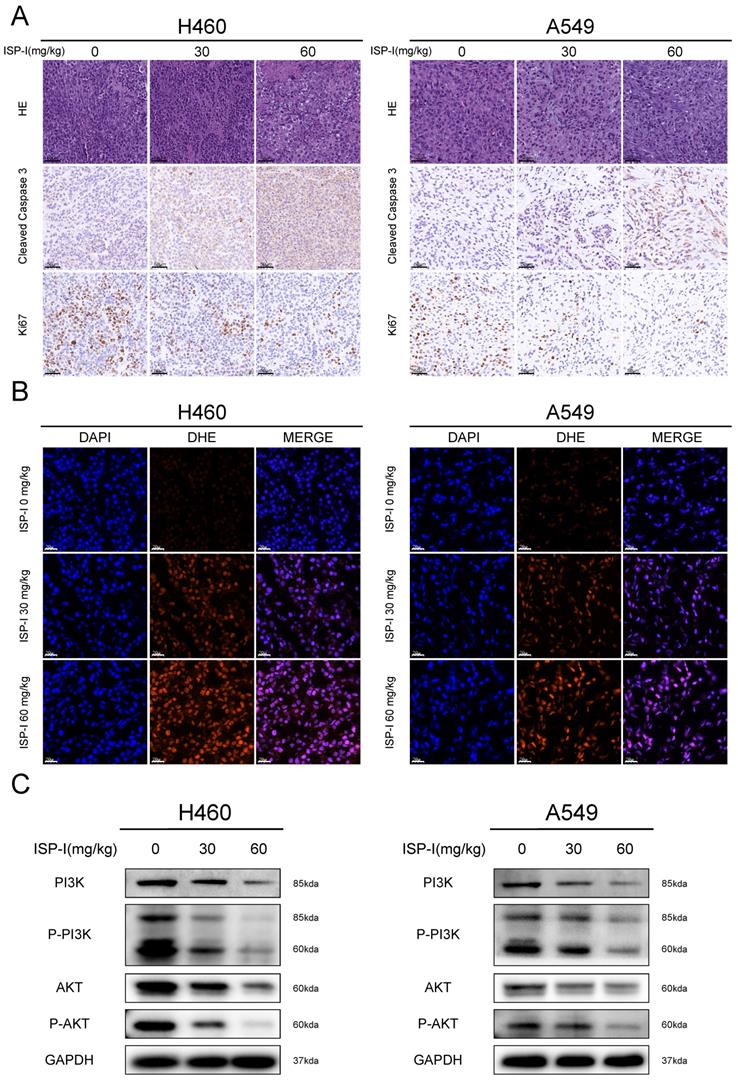
We next verified the mechanism of ISP-I's tumor-suppressing function in vivo. IHC was used to investigate the proliferation and apoptosis of xenograft tumor. ISP-I treated tumor tissues showed high cleaved Caspase-3 expression and low proliferation marker Ki-67 expression (Fig. 8A). DHE staining could be applied to detect the in situ ROS levels in frozen tissue sections [30]. In our research, we found that DHE was markedly increased in ISP-I treated tumors compared with the controls (Fig. 8B), indicating excessive ROS accumulation. In western blotting of xenograft samples, ISP-I treatment led to a decrease in p-PI3K, PI3K, AKT, p-AKT levels compared with vehicle-treated group (Fig.8C). In conclusion, ISP-I significantly inhibited tumor growth in vivo by generation of ROS resulting in inhibition of PI3K/AKT pathway. These results were consistent with that in vitro.
ISP-I inhibited tumor proliferation and promoted tumor apoptosis in vivo by generation of ROS resulting in PI3K/AKT pathway inhibition. (A) HE staining of tumor tissues and immunohistochemistry of Ki67, cleaved Caspase3 in vehicle-treated group and ISP-I-treated groups (30mg/kg, 60mg/kg). (B) Fluorescent microscopic observation of tumor tissues stained with DHE, which reflected ROS levels. (C) The protein expression of PI3K, p-PI3K, AKT and p-AKT in vehicle-treated group and ISP-I-treated groups (30mg/kg, 60mg/kg) by western blotting.
Discussion
The issues of toxic side effect and multidrug resistance (MDR) remain a challenge in the treatment and clinical management of NSCLC recently [31]. It is crucial to find some small molecules with good therapeutic potentials and low toxicity to act as a novel drug candidate for comprehensive treatment of NSCLC [24]. Repurposing old drugs in treating other diseases is a technique with many advantages including time and cost savings, since the safety and toxicity of candidate drugs have already been clearly studied [17]. Carrimycin is a Chinese Food and Drug Administration (CFDA)-approved macrolide antibiotic [32]. Here, we firstly isolated the active component of carrimycin, ISP-I, and verified its satisfying anti-NSCLC effect both in vitro and in vivo. Moreover, we identified that ISP-I possessed an excellent safety profile, based on its less toxic to human bronchial epithelial cell BEAS-2B in vitro and no significant weight loss or organ toxicities to oral administration mice in vivo. Therefore, we proposed that ISP-I represented a new therapeutic agent for NSCLC.
Apoptosis and cell cycle arrest are two main reasons in suppression of tumor growth [33, 34]. In our study, we found that ISP-I could cause apoptosis and G2/M arrest. Apoptosis can be triggered via two canonical pathways: the intrinsic mitochondrial pathway, and the extrinsic apoptotic pathway [35]. PUMA, BAX and BCL-2 are prominent members of BCL-2 family, which can trigger mitochondria-induced endogenous apoptosis through BCL-2/Caspase-9/Caspase-3 cascade reaction [23]. Sequential activation of caspase-8 is the central event in the execution-phase of extrinsic apoptosis, and followed by the direct activation of effector caspases3 [36, 37]. Our results indicated that ISP-I led to both intrinsic and extrinsic apoptosis through caspase-dependent cell signaling cascades.
Cell cycle deregulation is a common mechanism of tumorigenesis and can help tumor cells escape from tumor-suppressive pathways [38, 39]. Three major cell cycle checkpoints (G1/S, S phase and G2/M checkpoints) are pivotal steps in the process of cell cycle [40], which can be validated as targets for anti-tumor drugs [41]. Our results showed that ISP-I suppressed cell proliferation associated with G2/M arrest. Activation of the cyclin B1/cdc2 complex is central to facilitate G2/M cell cycle transition, mainly mediated by dephosphorylation of cdc2 [42, 43]. P21, as a vital member of CDK inhibitor, suppresses cyclin B1/cdc2 complex activity by interacting with many important proteins, including cdc2 and GADD45A [44], thereby inhibiting the G2/M transition in various cancer types both p53-dependent and p53-independent [45, 46]. In our study, we demonstrated that ISP-I markedly up-regulated the expression of p-cdc2 and p21, followed by decreasing the expression of cyclin B1 and cdc2. Overall, our results revealed that ISP-I could act as a cell cycle blocker regardless whether p53 whether present or not.
As is well known, induction of apoptosis and G2/M arrest is tightly controlled by various biological processes and regulatory molecules [47]. Among them, excessively accumulated ROS is involved in tumor growth inhibition and acts as an important mediator which can cause apoptosis and cell cycle arrest via multiple routes [48, 49]. According to our results, we found that ISP-I could significantly enhance ROS accumulation, whilst the ROS scavenger NAC almost entirely reversed the antitumor activity of ISP-I in NSCLC cells. Hence, ISP-I suppressed NSCLC cell growth through ROS over-accumulation, which may serve as a critical upstream regulator to the anti-cancer potentials of ISP-I. The baseline ROS levels of tumor cells are generally much higher than normal cells and increase along with the tumor progression due to its vigorous cell metabolism and proliferation [50, 51]. These findings indicate that cancer cells may be more sensitive to agents that induce further ROS accumulation compared to normal cells [52], which provide a specific therapeutic window for tumor targeting. Taken together, ISP-I may act as a novel ROS inducer for anti-NSCLC treatment.
The PI3K/AKT pathway inhibition has been identified to strongly associate with ROS accumulation and involved in ROS-induced tumor suppression [53, 54]. In our research, we confirmed that the PI3K/AKT pathway was significantly repressed and could be rescued efficiently by NAC. Our results therefore revealed the detailed mechanism underlying ISP-I-mediated cell growth inhibition caused by ROS over-accumulation. Aberrant activation of the PI3K/AKT pathway is prevalent across various cancer types, and often associates with the tumor progression and poor prognosis [55, 56]. Activated AKT, as a key downstream effector of the P13K signaling pathway, can modulate numerous substrates' function involved in a variety of biological activities including cell growth, proliferation, death and metabolism (bad, procaspase-9, p21Cip/Waf1, mTOR, FOXOs etc.) [57]. FOXOs are discovered as a tumor suppressor through activating G2/M arrest related genes CDKN1A and GADD45A, apoptosis related genes FAS, DR4 and DR5, respectively [58-62]. FAS, DR4 and DR5 are important cell surface death receptors, which recruit FADD to induce death-inducing signaling complex (DISC) formation and subsequent caspase-8 activation [63]. Activation of caspase 8 is an essential marker for extrinsic apoptosis. mTOR, which is another important downstream protein of PI3K/AKT signaling pathway, modulates cell growth and metabolism [64]. According to the RNA-seq data and qRTPCR, we confirmed that PI3K/AKT pathway is inactivated through ROS accumulation caused by ISP-I, which in turn induced corresponding changes in numerous downstream genes related to apoptosis and G2/M arrest. The diversity of alterations in PI3K/AKT pathway (PIK3CB, AKT1, MTOR, FOXOs, CCKN1A etc.) provides several molecular therapeutic targets [65] and ISP-I exhibits an excellent potential of multiple-target inhibitions. These results indicated that ISP-I was a promising anti-tumor agent due to this unique multi-target ability, which may compensate for the deficiency of traditional single-target agents [66] and decrease the probability of chemotherapy resistance.
KEAP1 loss-of-function mutations occur in various cancer types and can decrease NRF2 degradation to activate the KEAP1/NRF2 antioxidant response pathway, which confers a poor prognosis [67]. 17% of lung adenocarcinomas possess KEAP1/NRF2 alterations against oxidative stress [68]. Some studies have demonstrated that combined aberrant activation of PI3K and KEAP1/NRF2 pathways can substantially promote NSCLC development and is often linked with chemotherapy drug resistance [69]. In our study, we used NSCLC cell lines A549 and H460, which carried KEAP1 inactive homozygous mutations [70]. The excellent anti-tumor effect shows that ISP-I provide a new therapeutic option targeting to NSCLC patients carrying KEAP1 mutation through excessive ROS accumulation and play dual roles as a PI3K/AKT pathway inhibitor simultaneously. In addition, numerous studies have shown that compounds leading to even higher oxidative stress can sensitize MDR cancer cells to certain chemotherapeutic drugs or even induce MDR cancer cells death, regardless of the underlying mechanisms of MDR [71]. As a result, ISP-I is considered a promising alternative to overcome the drug resistance of MDR cancer cells. Moreover, ROS inducing agents are reported to be used in combination with traditional therapies, immunotherapy or other pro-oxidant agents to exert synergistic effects [9, 10, 54, 72]. Therein, combined ISP-I with traditional chemoradiotherapy, immunotherapy or other pro-oxidant agents such as doxorubicin, β-phenylethyl isothiocyanates can probably exhibit a higher tumor killing activity and further increase their clinical efficacy along with a reduction of side effects to improve survival. These need to be further studied.
To sum up, our study shows that ISP-I, the active component of CFDA-approved macrolide antibiotic carrimycin, induces apoptosis and G2/M arrest through ROS-mediated PI3K/AKT signaling pathway inhibition, ultimately leading to NSCLC cell death. ISP-I is a novel ROS inducer and may be a promising candidate with low biological toxicity and multi-target ability for anti- NSCLC treatment.
Abbreviations
ISP-I: isovalerylspiramycin I; NSCLC: non-small cell lung cancer; RTKs: multiple receptor tyrosine kinases; PEG: polyethylene glycol; DHE: dihydroethidium; NAC: N-acetyl-l-cysteine; FBS: fetal bovine serum; DEGs: differentially expressed genes; GO: Gene Ontology; BP: biological processes; KEGG: Kyoto Encyclopedia of Genes and Genomes; MFI: mean fluorescence intensity; GSEA: Gene Set Enrichment Analysis; qRTPCR: quantitative real-time PCR analysis; HE: hematoxylin and eosin; IHC: immunohistochemistry; ALT: alanine transaminase; AST: aspartate transaminase; ALB: albumin; BUN: blood urea nitrogen; Cr: creatinine; MDR: multidrug resistance; CFDA: Chinese Food and Drug Administration.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Shanghai Tonglian Pharmaceutical Co., Ltd. (China) for providing Isovalerylspiramycin I for research. We appreciate Shanghai Personal Biotechnology Cp. Ltd. (Shanghai, China), for technical assistance on sequencing assays. This work was funded by National Natural Science Foundation of China (81930001, 81870055, 81673014,81900059), Three-year Action Plan of Shanghai Shenkang Hospital Development Center (SHDC2020CR3063B), Medicine and Engineering Interdisciplinaty Research Fund of Shanghai Jiao Tong University (YG2020YQ18, ZH2018ZDA21), Shanghai Leading Talent Plan (2020, 064).
Author Contributions
Tao Ren, Zhengping Zhuang and Shan Shan designed the project. Zeyu Liu performed the experiments, interpreted the data, and wrote the manuscript. Moli Huang performed bioinformatics analysis. Tao Ren and Yue Hong read the manuscript critically and gave helpful comments. Shaoyang Wang, Yongle Xu, Cheng Zhong, Jingyuan Zhang participated in part of the experiments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. WHO. Cancer. [EB/OL]. (2021.3.3) [2021.9.11]. https://www.who.int/zh/news-room/fact-sheets/detail/cancer.
2. Miller KD, Nogueira L, Mariotto AB, Rowland JH, Yabroff KR, Alfano CM. et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69:363-85
3. Kim JW, Marquez CP, Kostyrko K, Koehne AL, Marini K, Simpson DR. et al. Antitumor activity of an engineered decoy receptor targeting CLCF1-CNTFR signaling in lung adenocarcinoma. Nat Med. 2019;25:1783-95
4. Tucker ZC, Laguna BA, Moon E, Singhal S. Adjuvant immunotherapy for non-small cell lung cancer. Cancer Treat Rev. 2012;38:650-61
5. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
6. Yan Y, Chen B, Wang Z, Yin Q, Wang Y, Wan F. et al. Sequential Modulations of Tumor Vasculature and Stromal Barriers Augment the Active Targeting Efficacy of Antibody-Modified Nanophotosensitizer in Desmoplastic Ovarian Carcinoma. Adv Sci (Weinh). 2021;8:2002253
7. Schirrmacher V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int J Oncol. 2019;54:407-19
8. Remon J, Lopes G, Camps C. How sustainable are new treatment strategies for NSCLC? Lancet Respir Med. 2019;7:733-5
9. Masui K, Gini B, Wykosky J, Zanca C, Mischel PS, Furnari FB. et al. A tale of two approaches: complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis. 2013;34:725-38
10. Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ Jr, Wu YL. et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389:299-311
11. Doroshow DB, Sanmamed MF, Hastings K, Politi K, Rimm DL, Chen L. et al. Immunotherapy in Non-Small Cell Lung Cancer: Facts and Hopes. Clin Cancer Res. 2019;25:4592-602
12. Teng JF, Qin DL, Mei QB, Qiu WQ, Pan R, Xiong R. et al. Polyphyllin VI, a saponin from Trillium tschonoskii Maxim. induces apoptotic and autophagic cell death via the ROS triggered mTOR signaling pathway in non-small cell lung cancer. Pharmacol Res. 2019;147:104396
13. Song X, Wang Z, Liang H, Zhang W, Ye Y, Li H. et al. Dioscin Induces Gallbladder Cancer Apoptosis by Inhibiting ROS-Mediated PI3K/AKT Signalling. Int J Biol Sci. 2017;13:782-93
14. Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E. et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011;1:248-59
15. Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S. et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4:452-65
16. Voss L, Guttek K, Reddig A, Reinhold A, Voss M, Schraven B. et al. Screening of FDA-Approved Drug Library Identifies Adefovir Dipivoxil as Highly Potent Inhibitor of T Cell Proliferation. Front Immunol. 2020;11:616570
17. Chong CR, Sullivan DJ Jr. New uses for old drugs. Nature. 2007;448:645-6
18. Liang SY, Zhao TC, Zhou ZH, Ju WT, Liu Y, Tan YR. et al. Anti-tumor effect of carrimycin on oral squamous cell carcinoma cells in vitro and in vivo. Transl Oncol. 2021;14:101074
19. Wang R, Shi M, Xu F, Qiu Y, Zhang P, Shen K. et al. Graphdiyne-modified TiO(2) nanofibers with osteoinductive and enhanced photocatalytic antibacterial activities to prevent implant infection. Nat Commun. 2020;11:4465
20. Li C, Zhao Y, Cheng J, Guo J, Zhang Q, Zhang X. et al. A Proresolving Peptide Nanotherapy for Site-Specific Treatment of Inflammatory Bowel Disease by Regulating Proinflammatory Microenvironment and Gut Microbiota. Adv Sci (Weinh). 2019;6:1900610
21. Lu M, Xia L, Liu YC, Hochman T, Bizzari L, Aruch D. et al. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood. 2015;126:972-82
22. Dong EL, Wang C, Wu S, Lu YQ, Lin XH, Su HZ. et al. Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol Neurodegener. 2018;13:36
23. Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ. et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348-58
24. Lu H, Zhu G, Tang T, Ma Z, Chen Q, Chen Z. Anticancer Molecule Discovery via C2-Substituent Promoted Oxidative Coupling of Indole and Enolate. iScience. 2019;22:214-28
25. Pise-Masison CA, Radonovich M, Dohoney K, Morris JC, O'Mahony D, Lee MJ. et al. Gene expression profiling of ATL patients: compilation of disease-related genes and evidence for TCF4 involvement in BIRC5 gene expression and cell viability. Blood. 2009;113:4016-26
26. Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R. et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A. 2001;98:10833-8
27. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627-44
28. Wen C, Wang H, Wu X, He L, Zhou Q, Wang F. et al. ROS-mediated inactivation of the PI3K/AKT pathway is involved in the antigastric cancer effects of thioredoxin reductase-1 inhibitor chaetocin. Cell Death Dis. 2019;10:809
29. Guo C, He J, Song X, Tan L, Wang M, Jiang P. et al. Pharmacological properties and derivatives of shikonin-A review in recent years. Pharmacol Res. 2019;149:104463
30. Cai Z, Wang Z, Yuan R, Cui M, Lao Y, Wang Y. et al. Redox-sensitive enzyme SENP3 mediates vascular remodeling via de-SUMOylation of β-catenin and regulation of its stability. EBioMedicine. 2021;67:103386
31. He Q, Shi J. MSN anti-cancer nanomedicines: chemotherapy enhancement, overcoming of drug resistance, and metastasis inhibition. Adv Mater. 2014;26:391-411
32. Yan H, Sun J, Wang K, Wang H, Wu S, Bao L. et al. Repurposing carrimycin as an antiviral agent against human coronaviruses, including the currently pandemic SARS-CoV-2. Acta Pharm Sin B. 2021;11:2850-8
33. Khan M, Ding C, Rasul A, Yi F, Li T, Gao H. et al. Isoalantolactone induces reactive oxygen species mediated apoptosis in pancreatic carcinoma PANC-1 cells. Int J Biol Sci. 2012;8:533-47
34. Xia X, Huang C, Liao Y, Liu Y, He J, Shao Z. et al. The deubiquitinating enzyme USP15 stabilizes ERα and promotes breast cancer progression. Cell Death Dis. 2021;12:329
35. Zhang B, Zhang T, Zhang TY, Wang N, He S, Wu B. et al. A Novel Methoxybenzyl 5-Nitroacridone Derivative Effectively Triggers G1 Cell Cycle Arrest in Chronic Myelogenous Leukemia K562 Cells by Inhibiting CDK4/6-Mediated Phosphorylation of Rb. Int J Mol Sci. 2020 21
36. Pozzesi N, Fierabracci A, Liberati AM, Martelli MP, Ayroldi E, Riccardi C. et al. Role of caspase-8 in thymus function. Cell Death Differ. 2014;21:226-33
37. Fritsch M, Günther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP. et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575:683-7
38. Castelli M, Piobbico D, Chiacchiaretta M, Brunacci C, Pieroni S, Bartoli D. et al. HOPS/TMUB1 retains p53 in the cytoplasm and sustains p53-dependent mitochondrial apoptosis. EMBO Rep. 2020;21:e48073
39. Lee Y, Lee CE, Oh S, Kim H, Lee J, Kim SB. et al. Pharmacogenomic Analysis Reveals CCNA2 as a Predictive Biomarker of Sensitivity to Polo-Like Kinase I Inhibitor in Gastric Cancer. Cancers (Basel). 2020 12
40. Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256:42-9
41. Jiang L, Wang Y, Liu G, Liu H, Zhu F, Ji H. et al. C-Phycocyanin exerts anti-cancer effects via the MAPK signaling pathway in MDA-MB-231 cells. Cancer Cell Int. 2018;18:12
42. Wang HX, Qin XH, Shen J, Liu QH, Shi YB, Xue L. Proteomic Analysis Reveals That Placenta-Specific Protein 9 Inhibits Proliferation and Stimulates Motility of Human Bronchial Epithelial Cells. Front Oncol. 2021;11:628480
43. Cheng YM, Tsai CC, Hsu YC. Sulforaphane, a Dietary Isothiocyanate, Induces G₂/M Arrest in Cervical Cancer Cells through CyclinB1 Downregulation and GADD45β/CDC2 Association. Int J Mol Sci. 2016 17
44. Kovalsky O, Lung FD, Roller PP, Fornace AJ Jr. Oligomerization of human Gadd45a protein. J Biol Chem. 2001;276:39330-9
45. Dash BC, El-Deiry WS. Phosphorylation of p21 in G2/M promotes cyclin B-Cdc2 kinase activity. Mol Cell Biol. 2005;25:3364-87
46. Dong C, Li Q, Lyu SC, Krensky AM, Clayberger C. A novel apoptosis pathway activated by the carboxyl terminus of p21. Blood. 2005;105:1187-94
47. Schultz DR, Harrington WJ Jr. Apoptosis: programmed cell death at a molecular level. Semin Arthritis Rheum. 2003;32:345-69
48. You L, Yang C, Du Y, Liu Y, Chen G, Sai N. et al. Matrine Exerts Hepatotoxic Effects via the ROS-Dependent Mitochondrial Apoptosis Pathway and Inhibition of Nrf2-Mediated Antioxidant Response. Oxid Med Cell Longev. 2019;2019:1045345
49. Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH. et al. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol Cancer. 2017;16:10
50. Xia Y, Liu S, Li C, Ai Z, Shen W, Ren W. et al. Discovery of a novel ferroptosis inducer-talaroconvolutin A-killing colorectal cancer cells in vitro and in vivo. Cell Death Dis. 2020;11:988
51. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931-47
52. Zhao Y, Hu X, Liu Y, Dong S, Wen Z, He W. et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol Cancer. 2017;16:79
53. Kim KY, Park KI, Kim SH, Yu SN, Park SG, Kim YW. et al. Inhibition of Autophagy Promotes Salinomycin-Induced Apoptosis via Reactive Oxygen Species-Mediated PI3K/AKT/mTOR and ERK/p38 MAPK-Dependent Signaling in Human Prostate Cancer Cells. Int J Mol Sci. 2017 18
54. Liu Y, Shi C, He Z, Zhu F, Wang M, He R. et al. Inhibition of PI3K/AKT signaling via ROS regulation is involved in Rhein-induced apoptosis and enhancement of oxaliplatin sensitivity in pancreatic cancer cells. Int J Biol Sci. 2021;17:589-602
55. Haddadi N, Lin Y, Travis G, Simpson AM, Nassif NT, McGowan EM. PTEN/PTENP1: 'Regulating the regulator of RTK-dependent PI3K/Akt signalling', new targets for cancer therapy. Mol Cancer. 2018;17:37
56. Salvesen HB, Carter SL, Mannelqvist M, Dutt A, Getz G, Stefansson IM. et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci U S A. 2009;106:4834-9
57. Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193-204
58. Zhao F, Lam EW. Role of the forkhead transcription factor FOXO-FOXM1 axis in cancer and drug resistance. Front Med. 2012;6:376-80
59. Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27:352-60
60. Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ Jr, DiStefano PS. et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530-4
61. Wang F, Reece EA, Yang P. Advances in revealing the molecular targets downstream of oxidative stress-induced proapoptotic kinase signaling in diabetic embryopathy. Am J Obstet Gynecol. 2015;213:125-34
62. Gupta M, Hendrickson AE, Yun SS, Han JJ, Schneider PA, Koh BD. et al. Dual mTORC1/mTORC2 inhibition diminishes Akt activation and induces Puma-dependent apoptosis in lymphoid malignancies. Blood. 2012;119:476-87
63. Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev. 2017;277:76-89
64. Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018;18:744-57
65. Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15:273-91
66. Caino MC, Altieri DC. Molecular Pathways: Mitochondrial Reprogramming in Tumor Progression and Therapy. Clin Cancer Res. 2016;22:540-5
67. Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M. et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303-9
68. Swanton C, Govindan R. Clinical Implications of Genomic Discoveries in Lung Cancer. N Engl J Med. 2016;374:1864-73
69. Best SA, De Souza DP, Kersbergen A, Policheni AN, Dayalan S, Tull D. et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018;27:935-43.e4
70. Wang J, Lu Q, Cai J, Wang Y, Lai X, Qiu Y. et al. Nestin regulates cellular redox homeostasis in lung cancer through the Keap1-Nrf2 feedback loop. Nat Commun. 2019;10:5043
71. Cui Q, Wang JQ, Assaraf YG, Ren L, Gupta P, Wei L. et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat. 2018;41:1-25
72. Mármol I, Quero J, Rodríguez-Yoldi MJ, Cerrada E. Gold as a Possible Alternative to Platinum-Based Chemotherapy for Colon Cancer Treatment. Cancers (Basel). 2019 11
Author contact
![]() Corresponding authors: liuyuanrentaoedu.cn (T.R.), shanshan_shcncom (S.S.), zpzhuacom (Z.-P. Z.)
Corresponding authors: liuyuanrentaoedu.cn (T.R.), shanshan_shcncom (S.S.), zpzhuacom (Z.-P. Z.)