Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2022; 18(9):3908-3917. doi:10.7150/ijbs.71261 This issue Cite
Research Paper
Clinical and biochemical characteristics of 12 Chinese primary hypertrophic osteoarthropathy patients with HPGD mutations
Qi Lu1*, Yang Xu1*, Shanshan Li1, Zeng Zhang1, Jiagen Sheng2 ![]() , Zhenlin Zhang1
, Zhenlin Zhang1 ![]()
1. Shanghai Clinical Research Center of Bone Disease, Department of Osteoporosis and Bone Diseases, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, China.
2. Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, China.
*These authors contributed equally to this work.
Received 2022-1-20; Accepted 2022-5-24; Published 2022-6-6
Abstract

Primary hypertrophic osteoarthropathy (PHO) is a rare genetic disease mainly affecting the skeletal and skin. Two genes involved in prostaglandin degradation are known to be responsible for PHO: HPGD and SLCO2A1. HPGD gene mutation can cause PHO autosomal recessive 1 (PHOAR1). The purpose of the present study is to analyze the clinical and biochemical characteristics and HPGD gene mutations of 12 Chinese PHOAR1 patients. Twelve PHOAR1 patients from eleven families, including eleven males and one female, were enrolled in this study. Digital clubbing and periostosis came out to be the most common features, which always occur in the early childhood. We performed HPGD gene analysis and identified six novel (c.1A>G, c.34G>T, c.317T>A, c.475G>T, c.548C>T and c.421+1G>T) and one known (c.310_311delCT) HPGD mutations. The recurrent mutation c.310_311delCT were found in all eleven patients, suggesting it is a hotspot mutation. PHOAR1 patients are considered to have an autosomal recessive inheritance pattern. Here, in addition to nine compound heterozygous patients and two homozygous patients, we found one heterozygous patient and reviewed two heterozygous patients reported in other studies. In terms of biochemical characteristics, our PHOAR1 patients have elevated urinary prostaglandin E2 (PGE2) levels (P<0.001) and decreased urinary prostaglandin E metabolite (PGE-M) levels (P=0.04) compared with healthy controls. The patients' PGE2/PGE-M (E/M) ratio came out to be lower than normal subjects (P<0.001). This study provides a comprehensive description of the clinical phenotypes of Chinese PHOAR1 patients and expands the genotypic spectrum of the disease.
Keywords: Primary hypertrophic osteoarthropathy autosomal recessive 1, HPGD gene, Clinical manifestation, PGE2
Introduction
Primary hypertrophic osteoarthropathy (PHO, OMIM 167100) is a rare inherited disease. In 1868, Friedreich first reported this disease in two brothers [1]. Later, Touraine, Solente and Golé designated PHO as the primary form of hypertrophic osteoarthropathy [2]. PHO mainly involves the skin and skeletal, and the predominant characteristics include pachydermia, digital clubbing, periostosis and joint swelling, sometimes concomitant with arthralgia, affecting joints such as knees, ankles, and wrists. Additional features include hyperhidrosis, acne, acro-osteolysis, anemia, and gastrointestinal abnormalities. PHO should be distinguished from secondary hypertrophic osteoarthropathy (SHO), which has the same skeletal and skin patterns as the primary form. SHO is often associated with impaired lung function. For example, SHO can appear as a paraneoplastic syndrome in patients with lung cancer, which provides clues to the diagnosis. Besides, cardiovascular or other extrathoracic diseases such as inflammatory bowel disease can also cause SHO [3, 4].
Two genes were found to be associated with PHO: HPGD (OMIM 601688) and SLCO2A1 (OMIM 601460), which encode 15-hydroxyprostaglandin dehydrogenase (15-PGDH) and prostaglandin transporter (PGT, also known as solute carrier organic anion transporter family member 2A1), respectively. Uppal et al identified HPGD gene as the causal gene for PHO autosomal recessive type 1 (PHOAR1, OMIM 259100) in 2008 [5]. Subsequently, in 2012 and 2021, our team found that SLCO2A1 gene deficiency is responsible for both PHO autosomal recessive type 2 (PHOAR2, OMIM 614441) and PHO autosomal dominant (PHOAD, OMIM 167100) [6, 7]. These findings provide great breakthrough to clarify the pathogenesis of PHO. Both genes play roles in the prostaglandin metabolism pathway, suggesting the elevated level of prostaglandin E2 (PGE2) may be the pathogenesis. HPGD gene is ubiquitously present in mammalian tissues [8]. It localizes on the chromosome 4q34.1 and consists of seven exons. 15-PGDH, the enzyme encoded by HPGD gene, has been considered the key enzyme responsible for the inactivation of prostaglandins. It catalyzes the oxidation of the 15-hydroxyl group of prostaglandins to produce a 15-keto metabolite of greatly reduced biological activities [9]. Our previous study demonstrated that both HPGD deficient and SLCO2A1 deficient patients had elevated urinary levels of PGE2 than the normal controls [10]. PHO has a male predominance, the skewed male to female ratio is about 9:1. The onset age has a bimodal distribution, peaking during the first year of life in HPGD deficient patients and at puberty in SLCO2A1 deficient patients [11].
In this study, we summarized twelve PHOAR1 patients from eleven families and reviewed all reported patients with HPGD gene mutations. Among them, seven individuals were reported in our previous drug intervention study [12], but the detailed clinical phenotype has not been analyzed before. The other five were additional patients not previously reported. Our present study analyzes their clinical and biochemical characteristics and expands the mutational spectrum of PHOAR1.
Study subjects and Methods
Families
Twelve patients from eleven families diagnosed with PHOAR1 according to the standard criteria were involved in this study (Figure 1). Seven individuals were reported before in our previous drug intervention study [12]. Here, we especially focused on their clinical features, biochemical characteristics, and mutation analysis. All individuals came from nonconsanguineous families except patient 2. The medical history was collected by retrospective review of medical records and clinical inquiry, complete physical examination and radiological data were obtained and systematically analyzed. Bone mineral densities (BMD) of the lumbar spine and proximal femur were measured with a dual-energy X-ray absorptiometry (Prodigy Advance, GE Lunar Corporation, USA). As described in our previous study [12], the hyponychial angle was used as the index for grading digital clubbing as follows: grade 0 = absent (hyponychial angle <192 degrees), grade 1 = mild (192-202 degrees), grade 2 = moderate (203-207 degrees), and grade 3 = severe (>207 degrees) [13]. The scoring system for pachydermia mainly involved the depth of the winkles in the forehead: grade 0 = not furrowed, grade 1 = not furrowed but a ditch between slight swellings can be seen, grade 2 = furrowed but the bottom of the furrow is visible, and grade 3 = deeply furrowed so that the bottom of the furrow is invisible [14].
Pedigrees of the eleven families with PHOAR1. Arrows indicate the probands. Filled black circles refer to patients with PHO. Half-black circles refer to healthy relatives with a heterozygous mutation.
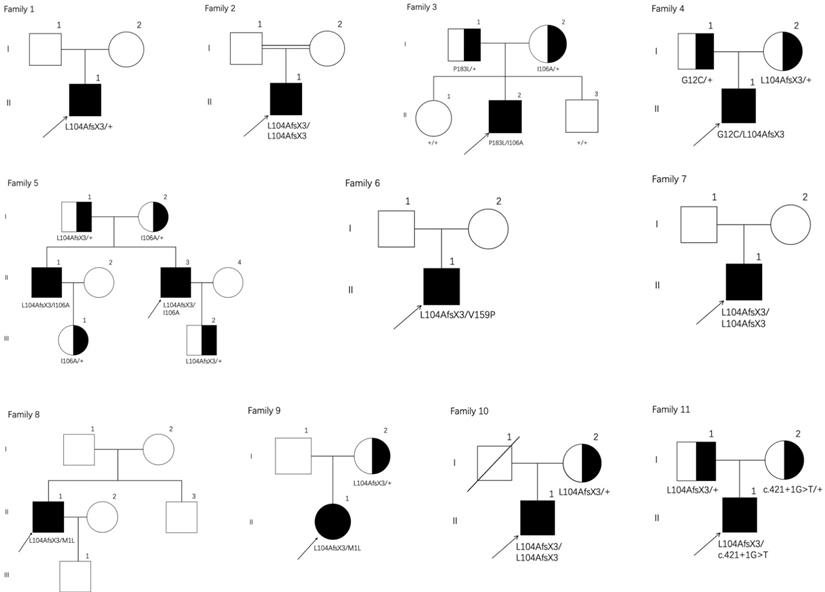
The study was approved by the Ethics Committee of the Shanghai Jiao Tong University Affiliated Sixth People's Hospital (Ethic Number: 2015-KY-001), and written informed consent was obtained from all the subjects who contributed clinical information and blood and urine samples to the study.
HPGD gene analysis
For pathogenic gene analysis of affected individuals, genomic DNA was extracted and purified from peripheral blood leukocytes with QuickGene DNA whole blood kit L by Nucleic Acid Isolation System (QuickGene-610L; FUJI FILM, Tokyo, Japan), the exons and exon-intron boundaries of HPGD (NM_000860.5) were amplified through PCR. Direct sequencing was performed using the BigDye Terminator cycle sequencing ready reaction kit, version 3.1 (Applied Biosystems; Foster City, CA, USA), and the reaction was performed using an ABI Prism 3130 automated sequencer (Applied Biosystems). UniProt (http://beta.uniprot.org/), Poly Phen-2 (Polymorphism Phenotyping V2; http://genetics.bwh.harvard.edu/pph2/) and SIFT (Sorting Intolerant from Tolerant; http://sift.jcvi.org/) were used to predict the conservation and pathogenicity of missense mutations found in our patients.
Biochemical Measurements
Serum bone metabolism markers and sex hormones, including total 25-hydroxyvitamin D (25OHD), intact parathyroid hormone (PTH), Beta-CrossLaps of type 1 collagen containing cross-linked C-telopeptide (β-CTX), osteocalcin (OC), total testosterone, estradiol, luteinizing hormone (LH), follicle stimulating hormone (FSH), and sex hormone-binding globulin (SHBG) were measured using an automated Roche electrochemiluminescence system (Roche Diagnostic GmbH), following both the manufacturer's protocol and specialized assay laboratory quality control procedures. Early morning urine samples were collected at the patients' first visit. Urinary levels of PGE2 and PGE-M were detected using competitive enzyme-linked immunosorbent assays (ELISA; Cayman Chemical, Ann Arbor, MI, USA), which was described in our previous study [12]. The values were normalized to creatinine according to the manufacturer's instructions (Item 500141 for PGE2, Item 514531 for PGE-M, Item 500701 for creatinine; Cayman Chemicals, Ann Arbor, MI, USA).
Statistical analysis
Normally distributed data were expressed as the mean ± standard deviation (SD) while non-normally distributed data were expressed as the median (25th and 75th percentiles). Independent-samples t test and Mann-Whitney U test were used to compare two groups. Pearson correlation test was used to explore the relationships between continuous variables. Statistical significance was set at p <0.05, and SPSS version 23 for Mac (IBM Corp, Armonk, NY, USA) was used for analysis.
Results
Clinical manifestations
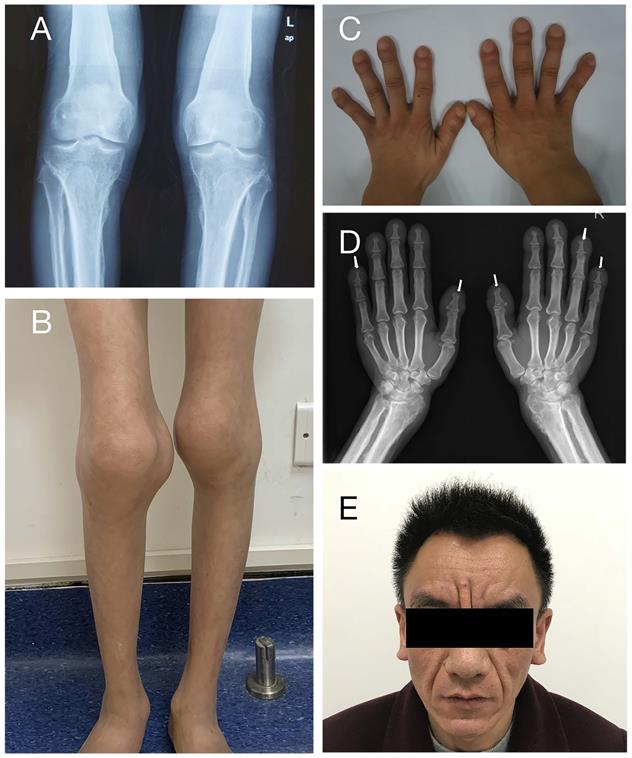
A total of 12 patients from 11 families were enrolled in this study, including eleven males and one female, with a median onset age of 2.0 years (range: 1.0-18.0 years). Table 1 summarizes the patients' clinical characteristics. The initial symptom of all patients is digital clubbing, which always occurs in infancy or early childhood. Digital clubbing and periostosis are the most frequent characteristics as they were presented in all patients. The degrees of digital clubbing are listed in Table 1, and more than half patients presented with severe digital clubbing. As for skin manifestations, eight patients have pachydermia and most of them were with a mild degree. Only patient 9 characterizes with severe pachydermia (Figure 2E). Ten patients (83.3%) had joint swelling and four of them (33.3%) were accompanied by arthralgia. The knee joint is most often involved, as all ten individuals complained about knee joint swelling, followed by ankles (seven patients) and wrist (one patient). Patient 10 and patient 12 were found to have patent ductus arteriosus (PDA) at birth. It closed spontaneously in patient 10 at the age of four without any treatment. Patient 12 took surgery and recovered. The incidences of other clinical features are listed in Table 1. None of our patients presented with hypoalbuminemia or gastrointestinal hemorrhage.
Clinical and genetic features of PHOAR1 patients
| Family | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | F11 | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 | P12 | |
| Sex | male | male | male | male | male | male | male | male | male | female | male | male | 11/1* |
| Age | 42 | 36 | 33 | 35 | 53 | 58 | 32 | 39 | 48 | 31 | 29 | 12 | 35.5 (12.0-58.0)** |
| Onset age | 15 | 1 | 16 | 2 | 18 | 12 | 1 | 2 | 4 | 1 | 1 | 1 | 2.0 (1.0-18.0)** |
| Consanguineous | - | + | - | - | - | - | - | - | - | - | - | - | 1/12 (8.3%) |
| Clubbing Fingers | mild | severe | moderate | severe | severe | mild | severe | + | severe | moderate | severe | severe | 12/12 (100%) |
| Pachydermia | mild | mild | - | mild | mild | - | mild | + | severe | - | mild | - | 8/12 (66.7%) |
| Periostosis | + | + | + | + | + | + | + | + | + | + | + | + | 12/12 (100%) |
| Joint swelling | - | + | + | + | + | + | + | - | + | + | + | + | 10/12 (83.3%) |
| Arthralgia | - | - | - | + | + | - | - | - | - | + | + | - | 4/12 (33.3%) |
| Acro-osteolysis | - | - | - | + | + | - | + | - | + | - | - | - | 4/12 (33.3%) |
| Hyperhidrosis | - | - | - | + | + | - | + | + | + | + | - | + | 7/12 (58.3%) |
| Acne | - | - | - | + | + | - | + | - | - | - | + | - | 4/12 (33.3%) |
| Seborrhea | + | - | + | + | + | - | + | + | + | + | + | - | 9/12 (75%) |
| Anemia | - | - | + | - | - | - | - | - | - | - | - | + | 2/12 (16.7%) |
| Patent ductus arteriosus | - | - | - | - | - | - | - | - | - | + | - | + | 2/12 (16.7%) |
| Watery diarrhea | - | - | + | + | + | + | - | - | - | - | - | - | 4/12 (33.3%) |
| DNA change | c.310_311delCT(heterozygous) | c.310_311delCT/c.310_311delCT | c.317T>A/c.548C>T | c.310_311delCT/c.34G>T | c.310_311delCT/c.317T>A | c.310_311delCT/c.317T>A | c.310_311delCT/c.475G>T | c.310_311delCT/c.310_311delCT | c.310_311delCT/c.1A>G | c.310_311delCT/c.1A>G | c.310_311delCT/c.310_311delCT | c.310_311delCT/c.421+1G>T | / |
| Parents | NA | NA | F: c.548C>T M: c.317T>A | F: c.34G>T M: c.310_311delCT | F: c.310_311delC M: c.317T>A | NA | NA | NA | F:NA M: c.310_311delCT | F:NA M: c.310_311delCT | F: c.310_311delC M: c.421+1G>T | / | |
NA, not available; F, father; M, mother.
*male/female; **median (range).
Clinical features of PHOAR1 patients. (A) Periostosis in knee joints (Patient 3). (B) Joint swelling (Patient 11). (C) Digital clubbing (Patient 3). (D) Acro-osteolysis (Patient 9). (E) Severe facial pachydermia (Patient 9).
Serum biochemical characteristics and urinary PGE2 and PGE-M levels
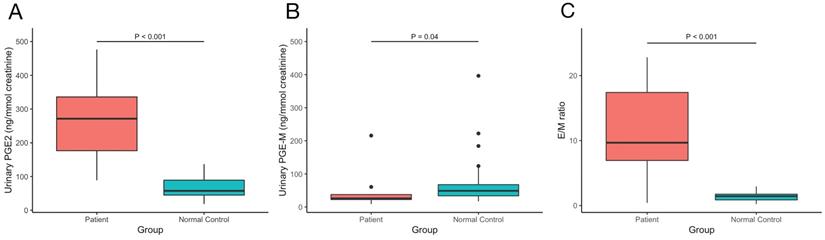
The values of serum bone metabolism markers including β-CTX, OC, PTH, ALP, 25OHD and serum sex hormones including testosterone, estradiol, LH, FSH, SHBG were measured, BMDs at lumber spine, femoral neck and total hip were detected. The results are shown in Table 2. We also measured our patients' urinary levels of PGE2 and PGE-M and calculated the PGE2/PGE-M (E/M) ratio. The results are shown in Figure 3. Compared with normal controls, our PHOAR1 patients had elevated PGE2 levels (271.28 ng/mmol creatinine vs. 57.41 ng/mmol creatinine, p<0.001) and decreased PGE-M levels (26.05 ng/mmol creatinine vs. 48.83 ng/mmol creatinine, p=0.04). And the patients' E/M ratios were higher than healthy controls (9.68 vs 1.44, p<0.001). We did not find any relationships between urinary PGE2 and serum estradiol, FSH, or LH, but the urinary PGE2 level is related to serum testosterone using Pearson correlation analysis (p=0.024).
Biochemical features of PHOAR1 patients
| Patient | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 | P12 | median (25th and 75th percentiles) or mean ± SD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Height (cm) | NA | NA | 167.00 | 173.90 | 181.10 | 172.90 | 174.50 | 158.00 | 173.20 | 153.40 | 172.50 | 137.00 | 172.70 (156.85, 174.05)a |
| Weigh (kg) | NA | NA | 52.00 | 69.00 | 83.00 | 70.50 | 57.00 | 49.00 | 60.00 | 53.00 | 80.00 | 23.50 | 59.70 ± 17.30b |
| BMI (kg/m2) | NA | NA | 18.65 | 22.82 | 25.31 | 23.58 | 18.72 | 19.63 | 20.00 | 22.52 | 26.89 | 12.52 | 21.06 ± 4.10 |
| PGE2 (ng/mmol creatinine) | 88.77 | NA | 185.64 | 261.50 | 370.65 | 281.06 | 476.64 | NA | 324.38 | NA | 149.53 | NA | 271.28 (158.56, 359.08) |
| PGE-M (ng/mmol creatinine) | 215.78 | NA | 25.68 | 30.02 | 60.67 | 26.41 | 25.18 | NA | 14.22 | NA | 8.85 | NA | 26.05 (16.96, 53.00) |
| E/M ratio | 0.41 | NA | 7.22 | 8.71 | 6.11 | 10.64 | 18.93 | NA | 22.81 | NA | 16.90 | NA | 9.68 (6.39, 18.42) |
| ALP (U/L) | NA | NA | 68.00 | 86.00 | 113.00 | 88.00 | 78.00 | 82.00 | 84.00 | 61.00 | 102.00 | 189.00 | 85.00 (75.50, 104.75) |
| β-CTX (ng/L) | NA | NA | NA | NA | 132.00 | 310.00 | 718.00 | 726.20 | 412.10 | NA | 861.20 | 2033.00 | 741.79 ± 625.74 |
| OC (ng/mL) | NA | NA | NA | NA | 11.95 | 27.58 | 23.78 | 35.26 | 24.16 | NA | 37.24 | 146.90 | 27.58 (23.78, 37.24) |
| PTH (pg/mL) | NA | NA | 41.47 | NA | 42.43 | 50.67 | 34.31 | 39.14 | 49.41 | 35.66 | 57.05 | 21.15 | 41.25 ± 10.57 |
| 25OHD (ng/mL) | NA | NA | 16.74 | NA | 19.60 | 14.78 | 26.42 | 28.99 | 18.30 | 22.29 | 14.29 | 22.89 | 20.48 ± 5.09 |
| Estradiol (pmol/L) | NA | NA | 95.00 | NA | 146.72 | 144.06 | 276.06 | 155.75 | 220.5 | 474.16 | 140.78 | NA | 151.24 (143.24, 234.39) |
| FSH (IU/L) | NA | NA | 4.68 | NA | 8.47 | 5.25 | 1.12 | 9.09 | 4.42 | 4.08 | 3.78 | 1.02 | 4.65 ± 2.77 |
| LH (IU/L) | NA | NA | 2.86 | NA | 6.40 | 3.60 | 3.21 | 3.71 | 4.75 | 4.74 | 6.26 | 0.28 | 3.98 ± 1.87 |
| Testosterone (nmol/L) | NA | NA | 4.64 | NA | 15.94 | 7.63 | 18.22 | 12.23 | 8.59 | 1.48 | 8.09 | 0.12 | 8.55 ± 6.11 |
| SHBG (nmol/L) | NA | NA | NA | NA | 45.50 | 34.50 | 35.00 | 58.40 | 67.00 | 42.60 | 20.10 | 119.80 | 52.86 ± 30.71 |
| L1-L4 BMD (g/cm2) (Z score) | NA | NA | 1.133 (0.8) | 1.273 (0.5) | 1.140 (0.4) | NA | 1.199 (0.5) | 1.173 (0.9) | 1.340 (2.4) | 1.374 (2.2) | 1.089 (-0.4) | 0.601 (0.1c) | 1.147 ± 0.226 (0.8 ± 0.9) |
| Femoral neck BMD (g/cm2) (Z score) | NA | NA | 0.906 (-0.3) | 0.964 (0.0) | 0.920 (-0.2) | NA | 1.014 (0.5) | 1.047 (0.7) | 0.967 (0.3) | 1.250 (2.7) | 0.745 (-2.1) | 0.615 (-0.1c) | 0.936 ± 0.180 (0.2 ± 1.2) |
| Total hip BMD (g/cm2) (Z score) | NA | NA | 0.997 (0.2) | 1.219 (2.0) | 1.054 (0.5) | NA | 1.040 (0.8) | 1.031 (0.3) | 1.058 (0.6) | 1.333 (2.8) | 0.758 (-1.9) | 0.649 (-0.1c) | 1.015 ± 0.208 (0.6 ± 1.3) |
NA, not available.
a. Non-normally distributed data are shown as the median (25th and 75th percentiles). b. Normally distributed data are shown as the mean ± SD. c. The Z score at L1-L4, femoral neck and total hip of young patients were calculated by comparison with the age-specific BMD reference value of Chinese children.'
The comparison of urinary (A) PGE2, (B) PGE-M levels and (C) E/M ratio between our PHOAR1 patients and normal controls.
HPGD mutations
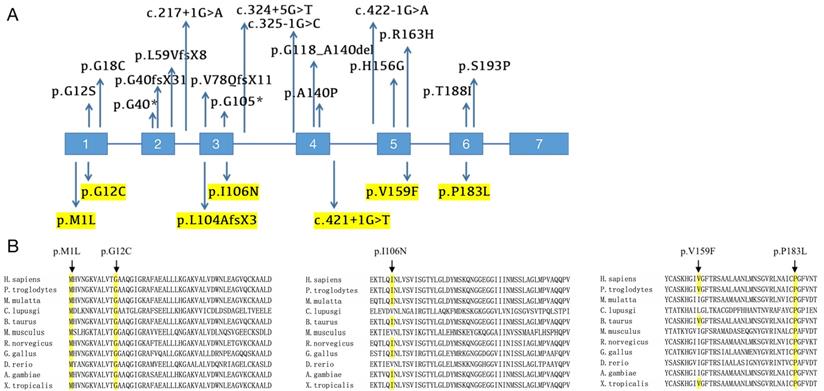
We screened for HPGD mutations in affected individuals and identified one known and six novel mutations, including five missense mutations, one frameshift mutation and one splicing site mutation. Five mutations were mentioned before in our previous study [12]. The mutation sites of each patient are listed in Table 1, and the locations of these mutations on the HPGD gene are shown in Figure 4A (highlight). Eleven patients have the c.310_311delCT mutation in HPGD gene, suggesting it is a hotspot mutation. Among them, one was heterozygous (patient 1), two were homozygous and the remaining eight were compound heterozygous. The only patient without c.310_311delCT mutation had compound heterozygous mutations (c.317T>A/c.548C>T). We also screened for SLCO2A1 mutation and did not identify any pathogenic variants in all the individuals. We verified the mutations in the patients' family members if they agreed (Family 3, 4, 5, 9, 10 and 11). The parents of our probands available for blood samples were carriers carrying monoallelic mutation site of the HPGD gene, and the results are presented in Figure 1. They don't have any PHO symptoms according to the available data. Evolutionary conservation analyses of missense mutations in HPGD gene by comparing the corresponding sequences of different species is shown in Figure 4B. Three amino changes (M1L, G12C and P183L) in HPGD gene were highly conserved according to UniProt. All the point mutations were predicted to be pathogenic by Polyphen-2 or SIFT.
Mutational analysis of PHOAR1 patients. (A) Distribution of all mutations in HPGD gene identified so far (the mutation sites identified in our study are highlighted). (B) Evolutionary conservation analyses of missense mutations in HPGD gene, as shown by comparing the corresponding sequences of 11 vertebrates.
Discussion
In the present study, the clinical features and laboratory findings of twelve genetically diagnosed Chinese PHOAR1 patients from eleven families were carefully collected and analyzed. We identified six novel (c.1A>G, c.34G>T, c.317T>A, c.475G>T, c.548C>T and c.421+1G>T) and one known (c.310_311delCT) HPGD mutations and described their clinical phenotypes. Although seven individuals were mentioned in our previous drug intervention study [12], our present study provides more detailed information and summarized the features of all twelve PHO patients with HPGD mutations we have up to now. We reviewed all PHOAR1 cases reported so far. A total of 77 cases have been reported to date since HPGD mutations in PHO patients were first identified by Uppal et al in 2008 [5, 15-29]. We summarized the clinical manifestations of all PHOAR1 patients reported in Table 3. The most common clinical features are digital clubbing and periostosis, which is consistent with our present study. According to our previous study and clinical observation, both PHOAR1 and PHOAR2 patients were affected with periostosis, and the severity did not appear to be strongly related to subtypes. Multilayer or irregular new bone formation occurs mainly at the diaphysis in both subtypes [12]. Arthralgia and arthritis are constantly found in PHOAR1 patients, resulting in limitation of motion and decreases of life quality. Knees are most often involved, followed by ankles and wrists. Joint manifestations always occur after digital clubbing and get worse progressively. Pachydermia, one of the triad of PHO, were observed in 52% of the reported PHOAR1 cases, which is much lower than those in PHO patients with SLCO2A1 mutations. More than half (58%) of the patients available for an X-ray examination have acro-osteolysis. Anemia in PHO patients is commonly considered to be due to myelofibrosis or gastrointestinal hemorrhage. However, our two patients presented with anemia don't have gastrointestinal hemorrhage or myelofibrosis. It is well known that PGE2 has the proliferative capacity, enhanced PGE2 synthesis results in increased number of hematopoietic stem cell (HSC), and blocking PGE2 synthesis decreases stem cell numbers, suggesting that PGE2 plays an important role in HSC formation [30]. A previous study demonstrated that inhibition of 15-PGDH increases bone marrow PGE2 levels, expands hematopoietic stem cell and progenitor cell numbers, and accelerates hematologic reconstitution after bone marrow transplantation in mice [31].
Summary of phenotypes and genotypes in reported patients with HPGD mutations
| Reference | Uppal et al. | Seifert et al | Tariq et al | Yuksel-Konuk et al | Diggle et al | Sinibaldi et al | Bergmann et al | Tuysuz et al | Erken et al | Nakazawa et al | Yuan et al | Chen et al | Khan et al | Stephan et al | Pang et al | Radhakrishnan et al | Total | The present study |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex ratio (F/M) | 8/5 | 3/1 | 7/4 | 5/1 | 3/5 | 0/1 | 0/3 | 0/2 | 1/1 | 0/1 | 1/8 | 1/6 | 2/2 | 0/1 | 0/1 | 0/4 | 31/46 | 1/11 |
| heterozygous | 0/13 | 0/4 | 0/11 | 0/6 | 0/8 | 0/1 | 0/3 | 0/2 | 0/2 | 0/1 | 2/9 | 0/7 | 0/4 | 0/1 | 0/1 | 0/4 | 2/77 (2.6%) | 1/12 (8.3%) |
| Clubbing finger | 13/13 | 4/4 | 11/11 | 6/6 | 8/8 | 1/1 | 3/3 | 2/2 | 2/2 | 1/1 | 9/9 | 7/7 | 4/4 | 1/1 | 1/1 | 4/4 | 77/77 (100%) | 12/12 (100%) |
| Periostosis | 8/8 | 0/2 | 0/2 | 4/4 | 4/4 | 1/1 | 3/3 | 2/2 | 2/2 | 1/1 | 8/9 | 0/7 | 1/1 | NE | 1/1 | 3/4 | 38/51 (74.5%) | 12/12 (100%) |
| Pachydermia | 9/13 | 0/4 | 0/11 | 4/6 | 1/8 | 0/1 | 2/3 | 0/2 | 2/2 | 1/1 | 8/9 | 7/7 | 2/4 | 1/1 | 1/1 | 1/2 | 39/75 (52%) | 8/12 (66.7%) |
| Joint swelling | 5/13 | 0/4 | 0/11 | 3/6 | 5/8 | 0/1 | 1/3 | 2/2 | 2/2 | 0/1 | 7/9 | 1/7 | 4/4 | 0/1 | 1/1 | 2/4 | 33/77 (42.9%) | 10/12 (83.3%) |
| Arthralgia | 10/13 | 1/4 | 0/11 | NM | 3/8 | 1/1 | 1/3 | 2/2 | 2/2 | 1/1 | 0/9 | 0/7 | 2/4 | 0/1 | 1/1 | 4/4 | 28/71 (39.4%) | 4/12 (33.3%) |
| Acro-osteolysis | 8/8 | 2/2 | 0/2 | 4/4 | 2/4 | 1/1 | 2/3 | 0/2 | 2/2 | NE | 1/9 | 1/7 | 1/1 | NE | 1/1 | 4/4 | 29/50 (58%) | 4/12 (33.3%) |
| Hyperhidrosis | 12/13 | 4/4 | 0/11 | 5/6 | 6/8 | 1/1 | 2/3 | 2/2 | 2/2 | 1/1 | 7/9 | NM | 4/4 | 1/1 | 1/1 | 0/2 | 48/68 (70.6%) | 7/12 (58.3%) |
| Patent ductus arteriosus | 4/13 | 1/4 | 0/11 | 1/6 | 2/8 | 0/1 | 0/3 | 1/2 | 0/2 | 0/1 | 0/9 | 2/7 | 0/4 | 0/1 | 0/1 | 1/4 | 12/77 (15.6%) | 2/12 (16.7%) |
| Anemia | NM | NM | 0/11 | 0/6 | NM | 1/1 | NM | 0/2 | 1/2 | 0/1 | 0/9 | 0/7 | 0/4 | 0/1 | 0/1 | NM | 2/45 (4.4%) | 2/12 (16.7%) |
| Acne | 4/13 | 0/4 | 0/11 | NM | NM | 1/1 | 2/3 | 0/2 | 2/2 | NM | NM | NM | 2/4 | 0/1 | 0/1 | NM | 11/42 (26.2%) | 4/12 (33.3%) |
| Seborrhea | 10/13 | 0/4 | 0/11 | NM | NM | 1/1 | 2/3 | 0/2 | NM | 1/1 | 1/9 | NM | 4/4 | 0/1 | 1/1 | NM | 20/50 (40%) | 9/12 (75%) |
| DNA change | c.418G>C* c.232_241delinsCA* c.175_176delCT* | c.52G>T* c.120delA* | c.577T>C* | c.418G>C c.1A>T* | c.120delA c.175_176delCT c.325-1G>C* | c.217+1G>A* | c.118G>T* c.563C>T* c.175_176delCT | c.310_311delCT* | c.310_311delCT | c.422-1G>A* | c.310_311delCT c.488G>A* | c.310_311del CT c.324+5G>A* a Deletion of exon 4* | c.577T>C | c.468T>A* | c.310_311delCT | c.34G>A* c.418G>C c.313C>T* | / | c.310_311delCT c.317T>A* c.548C>T* c.475G>T* c.34G>T* c.1A>G* c.421+1G>T* |
NM, not mentioned; NE, not evaluated.
*Novel mutations first identified by the referenced study.
These studies suggest that prostaglandins play a positive role in hematopoiesis, which is opposite to anemia. Further investigations will be required to answer why PHO patients with elevated PGE2 levels suffer from anemia. Delayed cranial suture closure can presented in affected infants in some of the previously reported cases, this phenotype cannot be seen in our patients since most of them were adults.
The pathogenic HPGD mutations reported so far are listed in Table 3 and presented in Figure 4A. Twenty-five distinct HPGD pathogenic alterations have been identified up to now. The most common mutation appears to be c.310_311delCT, the 2-bp deletion contributes to a frameshift after codon 104 and truncates the protein (p.L104AfsX2), and the lost part includes the proton acceptor site and the putative substrate binding sites [26]. This mutation was first reported by Tuysuz et al in a Turkish family [17]. Subsequently, 19 additional PHOAR1 patients with c.310_311delCT mutation were reported by four independent studies, including two Turkish patients and seventeen Chinese patients [16, 22, 26, 28]. Eleven patients in our present study also have this recurrent mutation, suggesting it is a hotspot mutation, especially in Chinese population. The variant c.175_176del is also a common recurrent mutation. It was reported in eight families and most of them have European origins [5, 27, 29]. All the mutation sites reported up to now were summarized in Figure 4A, including eight missense mutations (c.1A>T, c.34G>A, c.52G>T, c.418G>C, c.468T>A, c.488G>A, c.563C>T and c.577T>C), two nonsense mutations (c.118G>T and c.313C>T), four frameshift mutations (c.120delA, c.175_176delCT, c.232_241delinsCA and c.310_311delCT), four splicing site mutation (c.217+1G>A, c.324+5G>A, c.325-1G>C and c.422-1G>A), and one large fragment deletion (deletion of exon 4). Here we identified six novel mutations, including five missense mutations (c.1A>G, c.34G>T, c.317T>A, c.475G>T and c.548C>T), which cause protein changes M1L (identical to the mutation site c.1A>T), G12C, I106N, V159F and P183L, and one splicing site mutation (c.421+1G>T). Most patients are compound heterozygous or homozygous, which fit an autosomal recessive heritance pattern. But in some cases, individuals with monoallelic mutations of HPGD gene can also presented with PHO symptoms, which will be discuss later in this article.
The identification of the causative genes brought us a novel classification of PHO. It can be classified into three subtypes so far: PHOAR1 [5], PHOAR2 [6] and PHOAD [7]. It is important to distinguish between HPGD deficient patients and SLCO2A1 deficient patients in clinical practice as they differ in onset age, sex ratio, clinical features, and biochemical manifestations. The onset age is peaking during the early childhood in HPGD deficient patients and at puberty in SLCO2A1 deficient patients. Although the urinary PGE2 levels were elevated in all three subtypes, it was notably higher in PHOAR2 patients than those in PHOAR1 and PHOAD patients. The urinary levels of PGE-M can also help us distinct PHO subtypes as it usually decreases in HPGD deficient patients and increases in SLCO2A1 deficient patients. And the E/M ratio in HPGD deficient patients is higher than normal people, but in SLCO2A1 deficient patients, it is similar to normal people. Moreover, the clinical manifestation profile differs between subtypes. Pachydermia and clubbing fingers are usually milder in PHOAR1 and PHOAD patients than in PHOAR2 patients. Besides, patients with SLCO2A1 deficiency are more likely to develop gastrointestinal symptoms than those with HPGD deficiency [7, 12].
The pathogenesis of PHO has been well studied since the pathogenic genes were identified. PGE2 degradation requires two steps: first, the prostaglandin transporter mediates cellular PGE2 uptake, and then cytoplasmic oxidation is performed by 15-PGDH [32]. Defects of either gene contribute to the elevation of circulation or local levels of prostaglandins (mainly PGE2). We tested urinary PGE2 and PGE-M levels of our patients and found that the defected HPGD gene resulted in elevated urinary PGE2 and decreased urinary PGE-M levels. Skeletal features such as periostosis or acro-osteolysis might be the consequence of elevated PGE2 levels in the circulation or microenvironment since prostaglandins can stimulate both bone resorption and formation [33].
There are gender differences among different subtypes of PHO. It is commonly considered that there is an equal sex ratio in PHOAR1 patients while almost all the PHOAR2 and PHOAD patients are males. The total sex ratio (Female:Male) in reported PHOAR1 cases up to now is 31:46. However, in our cohort, there is only one female patient, which is accordance to two other studies with a Chinese population [16, 28]. This makes the sex ratio 3:26 in Chinese population. One explanation might be an incomplete penetrance in females. The skewed sex ratio in PHO patients has long been discovered and discussed. It is proposed that sex hormone may play a potential role, but in PHOAR2 patients, no significant associations of urinary PGE2 or PGE-M with sex hormones, including estradiol, FSH, LH, testosterone, and free testosterone were detected [12]. In the present study, we found there is a correlation between levels of urinary PGE2 and serum testosterone, but not other sex hormones. However, this result does not make much sense since the sample size is too small. The relationship between sex hormones and PGE2 metabolism remains to be investigated and data from more patients will be needed. Patient 10 in this study is the only female. She came to our department complaining about arthralgia of her ankles. The patient has special features of cerebral palsy. She was born prematurely, and asphyxia occurred after birth. At one year old, the patient presented with digital clubbing and patent ductus arteriosus was found. The ductus arteriosus closed spontaneously at the age of four. The pain in ankle joints occurred two years before her first visit, and it was obvious several days before menarche and relieved after menarche, which may be explained by the change of PGE2 level related to menstrual cycle. She had obvious periostosis and clubbing fingers, but her skin symptoms were mild. We cannot compare female patients to male patients because of the limited female PHOAR1 case. More PHOAR1 families with both male and female patients are required to help us reveal the effect of gender on PHOAR1, which will be our future direction.
Both dominant and recessive inheritance patterns have been suggested in PHO patients. Recently, our team identified a novel dominant pattern of PHO with SLCO2A1 mutation [7]. Up to now, all patients with HPGD mutations showed an autosome recessive inheritance pattern except three Chinese individuals with c.310_311delCT mutation reported in Yuan's study and ours [16], indicating an autosomal dominant inheritance pattern may also exists in PHO patients with HPGD mutations. The heterozygous patient in our study had mild symptoms which is listed in Table 1 (patient 1). We detected his urinary levels of PGE2 and PGE-M, which were 88.77 ng/mmol creatinine and 215.78 ng/mmol creatinine, respectively. His urinary PGE2 level was not elevated. And the PGE-M level was higher than normal controls, which confused us. Maybe there exist other enzymes that can degrade prostaglandins. Unfortunately, we do not have any information about his parents and more families or cases with monoallelic HPGD mutation will be needed to clarify this phenomenon.
Since the elevated PGE2 level is considered to be the pathogenesis, the selective cyclooxygenase-2 (COX-2) inhibitor which can suppress PGE2 biosynthesis was used for treatment. Two clinical trials confirmed the efficacy and safety of etoricoxib [12, 34]. Seven PHOAR1 patients were recruited in our previous trial [12]. The results showed that the urinary PGE2 levels in our patients were decreased after 6-month etoricoxib treatment and the biochemical bone markers have also been improved. The PHO symptoms including pachydermia and digital clubbing were relieved at the evaluation at 6 months. However, periostosis did not improve at 6 months according to the X-ray examination.
Recently, Palla et al pointed out that 15-PGDH may influence the muscle strength and identified elevated 15-PGDH as a hallmark of aged muscles [35]. The inhibition of 15-PGDH increases aged muscle mass, strength, and exercise performance in mice, indicating that it may be a potential treatment of sarcopenia. We wonder if this phenotype can also be presented in our patients with 15-PGDH deficiency. We contacted our patients and asked if they think their muscle strength is superior to that of their peers, but they all denied that. On the contrary, some of these patients have slender limbs and look weak. We measured their BMIs, with a median of 21.26 kg/m2, which is in the normal range. However, the exact muscle strength data were unavailable because we did not pay much attention to their muscle strength before. We plan to collect more detailed data and use precise methods to measure their muscle strength for further investigation.
In conclusion, here we described eleven PHOAR1 families. The early onset digital clubbing and periostosis are shown to be the most frequent symptoms. Mutational analysis was performed and seven mutations in HPGD gene were identified. Among them, six were novel mutations. One recurrent mutation c.310_311delCT were found in eleven patients, indicating it is a hotspot mutation in Chinese population. Elevated urinary PGE2 and decreased urinary PGE-M levels were found in our patients. Etoricoxib, a selected COX-2 inhibitor, was used for treatment and was beneficial to our patients. This study analyzed clinical and biochemical characteristics of PHOAR1 patients and expands the mutational spectrum.
Abbreviations
PHO: primary hypertrophic osteoarthropathy; SHO: secondary hypertrophic osteoarthropathy; PHOAR1: primary hypertrophic osteoarthropathy, autosomal recessive 1; PHOAR2: primary hypertrophic osteoarthropathy, autosomal recessive 2; PHOAD: primary hypertrophic osteoarthropathy, autosomal dominant; PGE2: prostaglandin E2; PGE-M: prostaglandin E metabolite; E/M ratio: PGE2/ PGE-M ratio; SLCO2A1: solute carrier organic anion transporter family member 2A1; PGT: prostaglandin transporter; HPGD or 15-PGDH: 15-hydroxyprostaglandin dehydrogenase; BMD: Bone mineral density; 25OHD: 25-hydroxyvitamin D; PTH: parathyroid hormone; β-CTX: Beta-CrossLaps of type 1 collagen containing cross-linked C-telopeptide; OC: osteocalcin; LH: luteinizing hormone; FSH: follicle stimulating hormone; SHBG: sex hormone-binding globulin; COX-2: cyclooxygenase-2; HSC: hematopoietic stem cell.
Acknowledgements
This study was supported by the National Key Research and Development Program of China (2018YFA0800801), National Natural Science Foundation of China (NSFC) (81974123, 81570794, 81900807), Clinical Science and Technology Innovation Project of Shanghai Shenkang Hospital Development Center (SHDC12018120), Shanghai Key Clinical Center for Metabolic Disease, Shanghai Health Commission Grant (2017ZZ01013), and Shanghai Municipal Key Clinical Specialty.
We are grateful to the affected subjects and their families and the volunteers for participating in this study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Friedreich N. Hyperostose des gesammten Skelettes. Archiv für pathologische Anatomie und Physiologie und für klinische Medicin. 1868;43:83-7
2. Touraine A, Solente G, Gole L. Un syndrome osteodermopathique: La pachydermie plicaturee avec pachyperiostose des extremites. Presse Med. 1935;43:1820-1824
3. Martínez-Lavín M. Hypertrophic osteoarthropathy. Best Pract Res Clin Rheumatol. 2020;34:101507
4. Yap FY, Skalski MR, Patel DB, Schein AJ, White EA, Tomasian A. et al. Hypertrophic Osteoarthropathy: Clinical and Imaging Features. Radiographics. 2017;37:157-95
5. Uppal S, Diggle CP, Carr IM, Fishwick CW, Ahmed M, Ibrahim GH. et al. Mutations in 15-hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nat Genet. 2008;40:789-93
6. Zhang Z, Xia W, He J, Zhang Z, Ke Y, Yue H. et al. Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. Am J Hum Genet. 2012;90:125-32
7. Xu Y, Zhang Z, Yue H, Li S, Zhang Z. Monoallelic mutations in SLCO2A1 cause autosomal dominant primary hypertrophic osteoarthropathy. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2021;36:1459-68
8. Tai H-H, Ensor CM, Tong M, Zhou H, Yan F. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 2002;68-69:483-93
9. Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr Pharm Des. 2006;12:955-62
10. Zhang Z, He JW, Fu WZ, Zhang CQ, Zhang ZL. Mutations in the SLCO2A1 gene and primary hypertrophic osteoarthropathy: a clinical and biochemical characterization. J Clin Endocrinol Metab. 2013;98:E923-33
11. Zhang Z, Zhang C, Zhang Z. Primary hypertrophic osteoarthropathy: an update. Front Med. 2013;7:60-4
12. Li S-S, He J-W, Fu W-Z, Liu Y-J, Hu Y-Q, Zhang Z-L. Clinical, Biochemical, and Genetic Features of 41 Han Chinese Families With Primary Hypertrophic Osteoarthropathy, and Their Therapeutic Response to Etoricoxib: Results From a Six-Month Prospective Clinical Intervention. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2017;32:1659-66
13. Regan GM, Tagg B, Thomson ML. Subjective assessment and objective measurement of finger clubbing. Lancet. 1967;1:530-2
14. Pineda CJ, Martinez-Lavin M, Goobar JE, Sartoris DJ, Clopton P, Resnick D. Periostitis in hypertrophic osteoarthropathy: relationship to disease duration. AJR Am J Roentgenol. 1987;148:773-8
15. Yuksel-Konuk B, Sirmaci A, Ayten GE, Ozdemir M, Aslan I, Yilmaz-Turay U. et al. Homozygous mutations in the 15-hydroxyprostaglandin dehydrogenase gene in patients with primary hypertrophic osteoarthropathy. Rheumatol Int. 2009;30:39-43
16. Yuan L, Chen L, Liao RX, Lin YY, Jiang Y, Wang O. et al. A Common Mutation and a Novel Mutation in the HPGD Gene in Nine Patients with Primary Hypertrophic Osteoarthropathy. Calcif Tissue Int. 2015;97:336-42
17. Tuysuz B, Yilmaz S, Kasapcopur O, Erener-Ercan T, Ceyhun E, Bilguvar K. et al. Primary hypertrophic osteoarthropathy caused by homozygous deletion in HPGD gene in a family: changing clinical and radiological findings with long-term follow-up. Rheumatol Int. 2014;34:1539-44
18. Tariq M, Azeem Z, Ali G, Chishti MS, Ahmad W. Mutation in the HPGD gene encoding NAD+ dependent 15-hydroxyprostaglandin dehydrogenase underlies isolated congenital nail clubbing (ICNC). J Med Genet. 2009;46:14-20
19. Stephan C, Hanna E, Nemer G, Abbas O, Kurban M. A novel mutation in the HPGD gene results in the unusual phenotype of palmoplantar keratoderma with digital clubbing and hyperhidrosis. JAAD Case Rep. 2018;4:950-2
20. Seifert W, Beninde J, Hoffmann K, Lindner TH, Bassir C, Aksu F. et al. HPGD mutations cause cranioosteoarthropathy but not autosomal dominant digital clubbing. Eur J Hum Genet. 2009;17:1570-6
21. Radhakrishnan P, Jacob P, Nayak SS, Gowrishankar K, Prakash Soni J, Shukla A. et al. Digital clubbing as the predominant manifestation of hypertrophic osteoarthropathy caused by pathogenic variants in HPGD in three Indian families. Clin Dysmorphol. 2020;29:123-6
22. Pang Q, Xu Y, Qi X, Jiang Y, Wang O, Li M. et al. The first case of primary hypertrophic osteoarthropathy with soft tissue giant tumors caused by HPGD loss-of-function mutation. Endocr Connect. 2019;8:736-44
23. Nakazawa S, Niizeki H, Matsuda M, Nakabayashi K, Seki A, Mori T. et al. Involvement of prostaglandin E2 in the first Japanese case of pachydermoperiostosis with HPGD mutation and recalcitrant leg ulcer. J Dermatol Sci. 2015;78:153-5
24. Sinibaldi L, Harifi G, Bottillo I, Iannicelli M, El Hassani S, Brancati F. et al. A novel homozygous splice site mutation in the HPGD gene causes mild primary hypertrophic osteoarthropathy. Clin Exp Rheumatol. 2010;28:153-7
25. Khan AK, Muhammad N, Khan SA, Ullah W, Nasir A, Afzal S. et al. A novel mutation in the HPGD gene causing primary hypertrophic osteoarthropathy with digital clubbing in a Pakistani family. Ann Hum Genet. 2018;82:171-6
26. Erken E, Koroglu C, Yildiz F, Ozer HT, Gulek B, Tolun A. A novel recessive 15-hydroxyprostaglandin dehydrogenase mutation in a family with primary hypertrophic osteoarthropathy. Mod Rheumatol. 2015;25:315-21
27. Diggle CP, Carr IM, Zitt E, Wusik K, Hopkin RJ, Prada CE. et al. Common and recurrent HPGD mutations in Caucasian individuals with primary hypertrophic osteoarthropathy. Rheumatology (Oxford). 2010;49:1056-62
28. Chen Y, Li G, Xu Y, Yu T, Zhang Y, Li N. et al. Targeted exome sequencing identified a novel mutation hotspot and a deletion in Chinese primary hypertrophic osteoarthropathy patients. Clin Chim Acta. 2018;487:264-9
29. Bergmann C, Wobser M, Morbach H, Falkenbach A, Wittenhagen D, Lassay L. et al. Primary hypertrophic osteoarthropathy with digital clubbing and palmoplantar hyperhidrosis caused by 15-PGHD/HPGD loss-of-function mutations. Exp Dermatol. 2011;20:531-3
30. North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM. et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007-11
31. Smith JNP, Otegbeye F, Jogasuria AP, Christo KF, Antczak MI, Ready JM. et al. Inhibition of 15-PGDH Protects Mice from Immune-Mediated Bone Marrow Failure. Biol Blood Marrow Transplant. 2020;26:1552-6
32. Nomura T, Lu R, Pucci ML, Schuster VL. The two-step model of prostaglandin signal termination: in vitro reconstitution with the prostaglandin transporter and prostaglandin 15 dehydrogenase. Mol Pharmacol. 2004;65:973-8
33. Pilbeam C. Prostaglandins and Bone. Handb Exp Pharmacol. 2020;262:157-75
34. Yuan L, Liao R-X, Lin Y-Y, Jiang Y, Wang O, Li M. et al. Safety and efficacy of cyclooxygenase-2 inhibition for treatment of primary hypertrophic osteoarthropathy: A single-arm intervention trial. J Orthop Translat. 2019;18:109-18
35. Palla AR, Ravichandran M, Wang YX, Alexandrova L, Yang AV, Kraft P. et al. Inhibition of prostaglandin-degrading enzyme 15-PGDH rejuvenates aged muscle mass and strength. Science. 2021 371
Author contact
![]() Corresponding authors: Zhenlin Zhang, E-mail: zhangzledu.cn; Jiagen Sheng, E-mail: shengjiagencom.
Corresponding authors: Zhenlin Zhang, E-mail: zhangzledu.cn; Jiagen Sheng, E-mail: shengjiagencom.