Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Aging liver
Aging myeloid cells
Myeloid cells and aging-related...
Promising therapeutics targeting...
Conclusion and outlooks
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(5):1564-1578. doi:10.7150/ijbs.82352 This issue Cite
Review
The Roles of Myeloid Cells in Aging-related Liver Diseases
Jinghao Yao1,2, Yang Li3, Hua Wang1,2, ![]()
1. Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei 230022, China
2. Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Anhui Medical University, Hefei 230032, China
3. Department of Genetics, School of Life Science, Anhui Medical University, Hefei 230032, China
Received 2023-1-4; Accepted 2023-2-20; Published 2023-3-5
Abstract

Aging is a necessary process of life associated with various mechanisms, such as genomic instability, loss of proteostasis, deregulated nutrient sensing, and cellular senescence, causing progressive dysregulation of the microenvironment, organ homeostasis and biological functions. The hepatic microenvironment is essential for maintaining liver homeostasis, in which hepatocytes, sinusoidal endothelial cells, stellate cells and immune cells are closely associated with the development of aging-related liver diseases. There is increasing evidence that immunocytes, especially myeloid cells, are involved in aging-related liver diseases such as alcoholic liver disease, nonalcoholic liver disease, liver fibrosis or cirrhosis and liver cancer, becoming promising treatment targets of these diseases. This review summarizes the phenotypic and functional alterations associated with aging liver and myeloid cells, as well as the roles of myeloid cells in the progression of aging-related liver diseases.
Keywords: Aging, Myeloid Cells, Microenvironment, Inflammation, Liver Disease
Introduction
Aging is commonly characterized as an organism's functional decline over time, with characteristics such as loss of physiological integrity, disruption of organ homeostasis, and progressive degradation of many biological systems [1]. These changes reshape the immune landscape in both physiological and pathological states, increasing the susceptibility of the organism to pathogenic factors [2]. With the improvement in living standards and advances in medicine, human life expectancy is significantly increased, leading to the prominent aging of the population accompanied by various chronic diseases, which demonstrate a natural relationship between aging and the pathogenic processes of many chronic diseases [3, 4].
Liver diseases cause approximately 1.2 million deaths each year, accounting for 3.5% of deaths and affecting the lives of 1.5 billion people worldwide [5]. The liver is a complex metabolic organ that maintains the homeostasis of the body by regulating energy metabolism, biosynthesis, and xenobiotic removal [6-8]. The liver is also an important immune organ, where a variety of immune cells are temporarily or permanently distributed to perform immune surveillance of pathogens of intestinal origin as well as senescent cells [7]. The aged liver typically exhibits a higher infiltration of immune cells such as macrophages, neutrophils, lymphocytes and natural killer (NK) cells, resulting in a higher inflammatory state [9] which aggravates liver aging and increases the susceptibility to aging-related liver diseases, including nonalcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), liver fibrosis or cirrhosis, and liver cancer [10]. Among the immune cells, myeloid cells such as macrophages and neutrophils play major roles in the development of liver diseases independently or through crosstalk with nonimmune mesenchymal cells [11]. However, the specific mechanisms by which myeloid cells induce aging-related liver diseases remain largely unclear.
The incidence of aging-related liver diseases will continue to rise as the population ages, which poses new requirements for further in-depth studies on the mechanisms between myeloid cells and liver aging. This review summarizes the phenotypic and functional alterations of aging liver as well as myeloid cells and presents the latest research progress of myeloid cells in affecting aging-related liver diseases, which will provide new insights for further exploration of the clinical strategies of aging-related liver diseases.
Aging liver
Aging liver encompasses most of the hallmarks of aging (Figure 1) and is associated with progressive changes in hepatic structure and function, as well as various hepatocyte alterations, including reduced proliferative capacity, metabolic and nutritional imbalances and altered liver microenvironment, which ultimately leads to increased disease susceptibility.
The hallmarks of aging hepatocytes. Aging hepatocytes encompass most of the hallmarks of aging. With age, DNA damage such as DSBs can occur spontaneously or be induced by ionizing radiation or ROS, causing pathological features of aging. Epigenetic alterations such as DNA methylation and histone acetylation were significantly reduced in aged livers, which possibly contributed to aging-related metabolic dysfunction. Deregulation of the trophic and metabolic pathways is another hallmark of aging hepatocytes, and decreased expression of MANF may promote lipid accumulation in the aged liver. Mitochondria in hepatocytes are also altered with age and are associated with a decrease in mitochondrial membrane potential and an increase in volume and peroxide production, supporting the key role of mitochondrial damage during aging. Senescent hepatocytes significantly accumulate in aged liver due to the accumulation of DNA damage and activation of the p53-p21 and p16ink4a-pRb pathways. In addition, abnormal intercellular communication between hepatocytes and immune cells such as macrophages and neutrophils can cause persistent sterile inflammation, known as inflammaging. DSBs: DNA double-strand breaks; MANF: mesencephalic-astrocyte-derived neurotrophic factor; ROS: reactive oxygen species.
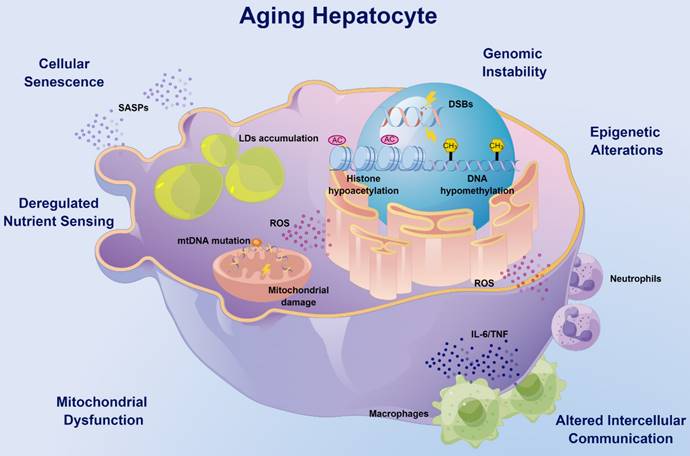
The liver is the most resilient organ in adults and is characterized by its unique regenerative capacity. From their maturity, the volume of hepatocytes starts to decrease gradually with age [4]. On the one hand, senescent hepatocytes accumulate in the liver as a result of DNA damage accumulation and the activation of the p53-p21 and p16ink4a-pRb pathways [12, 13]. On the other hand, DNA double-strand breaks (DSBs) [14] and mutations in mitochondrial DNA (mtDNA) [15] caused by ionizing radiation or reactive oxygen species (ROS) may cause aging phenotypes of the liver, including expression of aging markers, mitochondrial fusion and elongation, and altered gene expression profiles. Moreover, the process of liver diseases and natural aging is accompanied by increased hepatocyte polyploidy [13, 16], which limits the ability of hepatocytes to proliferate and may affect the recovery from inflammatory liver injury and partial hepatectomy [17].
Deregulation of the trophic and metabolic pathways is another hallmark of the aging liver, which significantly affects liver morphology, physiology, and oxidative capacity. Adenosine 5'-monophosphate activated protein kinase (AMPK), a serine-threonine kinase, is essential for regulating the homeostasis of energy metabolism in cells, and phosphorylated AMPK (activation) stimulates the physiological process of energy production and inhibits energy utilization [18]. The AMPK signaling declines with age in a variety of tissues, thus affecting mitochondrial function and energy homeostasis [19]. Interestingly, although increased levels of phosphorylated AMPK can be observed in the aging liver, the signaling pathway is impaired because of the inactivated downstream signals [20]. Multiple approaches including exercise, dietary interventions and probiotic supplementation can improve liver aging and steatosis by activating the AMPK pathway [21, 22]. Moreover, decreased expression of mesencephalic astrocyte-derived neurotrophic factor (MANF) with age induces alterations in the transcriptome of lipid metabolism genes in mouse hepatocytes, promotes lipid accumulation in the liver through the induction of G0/G1 switch gene 2 (G0S2), and causes hepatic steatosis in response to metabolic stresses such as a high-fat diet (HFD) [23]. As the metabolic center, aging-related mitochondrial damage may decrease the metabolic capacity of hepatocytes. Recent studies have shown that the Hedgehog pathway is downregulated with age, leading to a decrease in the quantity and function of mitochondria, which results in reduced hepatocyte elasticity, impaired liver regeneration, and greater susceptibility to inflammation [24]. Epigenetic alterations, including DNA methylation and histone acetylation, may also affect liver metabolism [25-27]. Histone hypoacetylation is upregulated with age, leading to impaired hepatocyte plasticity, and selective inhibition of histone deacetylase (HDAC) can significantly restore plasticity and repopulation of aged hepatocytes confronting liver injury [28, 29]. Our previous studies have revealed that sirtuin-1 (SIRT-1), another class of histone deacetylases with antiaging effects [30, 31], is downregulated with age and increases the susceptibility of hepatocytes to alcohol damage [32]. The different regulation between SIRT-1 and HDAC may reflect the functional diversity of acetylation modification.
The hepatic microenvironment comprises mainly liver sinusoidal endothelial cells (LSECs) [33], hepatic stellate cells (HSCs) [34] and Kupffer cells (KCs) [35], which synergistically influence the aging process of the liver. Pseudocapillarization, a typical morphological change in aging LSECs [36], results in drug metabolism impairment [33] and lipoproteins [37] and insulin clearance [38], causing hyperlipidemia and hepatic insulin resistance, which may lead to cardiovascular and metabolic diseases. Hepatocyte growth factor (HGF) released by HSCs is significantly reduced with age, resulting in decreased regenerative capacity of the liver [34]. KCs are liver resident macrophages [39], the number of which gradually increases with age [40]. Autophagy and autophagy-associated protein 5 (ATG5) expression is reduced in KCs with age, promoting their polarization toward the pro-inflammatory M1 phenotype [35]. In aged rats, the numbers of both pro-inflammatory M1 and anti-inflammatory M2 KCs have been reported to be increased [41], which may reflect the continued activation of the immune response in advanced age.
For healthy aging, alterations in hepatocytes themselves reduce the proliferation and function, while the roles of pathological factors such as inflammation in liver aging have been understudied. As an important immune organ, in addition to KCs, a variety of immune cells also colonize the liver. Aging not only leads to a decrease in the defensive capacity of the liver but is also accompanied by greater inflammatory injury, known as inflammaging. Myeloid cells, including macrophages and neutrophils, have been reported to play significant roles in inflammatory liver injuries [42], which may be exacerbated by aging. Therefore, this review will further focus on the impact of aging immune cells, especially myeloid cells, on liver aging and disease susceptibility.
Aging myeloid cells
Aging of the immune system, which refers to an age-related decline in immune surveillance and clearance, dramatically increases the susceptibility to diseases [43]. Inflammaging, one of the hallmarks of immune aging, is the chronic progressive elevation of a low inflammatory response in naturally aging organisms in the absence of pathogens [44]. A major feature of inflammaging is the chronic activation of the innate immune system [45] with an imbalance in the secretion of pro- and anti-inflammatory factors [46], which may drive the aging of solid organs [47].
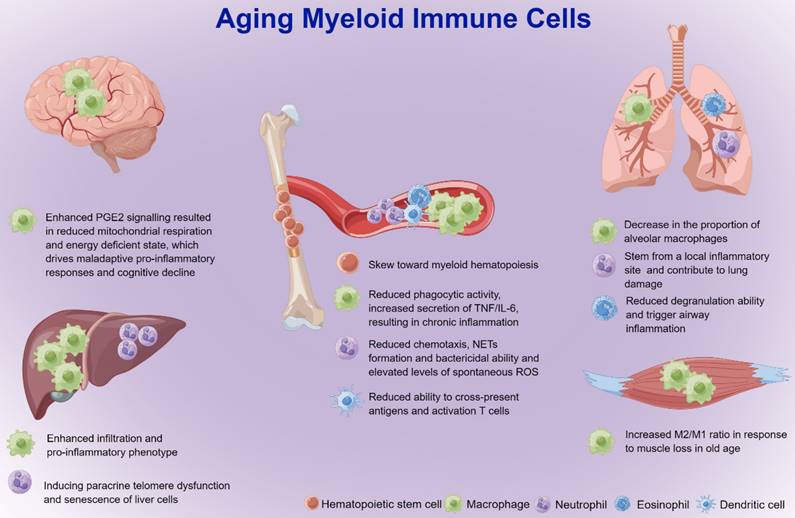
The functional integrity of the immune system is shaped by hematopoietic stem cells, the source of immune cells [48]. Aging hematopoietic stem cells exhibit reduced output of lymphoid and erythroid lineages, whereas output of myeloid lineage is maintained or even increased compared with young hematopoietic stem cells [49]. This skew toward myeloid hematopoiesis appears to be related to early differentiation of hematopoietic stem cells, as old mice have increased numbers of common myeloid progenitor cells (CMPs) and decreased numbers of common lymphoid progenitor cells (CLP) compared to young mice [50, 51]. Myeloid cells include mainly monocytes/macrophages, neutrophils, eosinophils, basophils, myeloid-derived suppressor cells (MDSCs) and dendritic cells (DCs). Aging increases the accumulation of myeloid cells and causes a significant reduction in the capacity of movement and surveillance, which further leads to organ injuries and diseases (Figure 2) [52-54]. Therefore, further studies focusing on aging myeloid cells in specific inflammatory settings are needed to better understand how such cells contribute to organ damage and diseases in the elderly population.
Effect of myeloid cells on aging-related organ injuries and diseases. Hematopoietic stem cells in the aged body exhibit reduced output of lymphoid and erythroid lineages but relatively more output of myeloid lineage cells than those in the young body. With age, macrophages with reduced phagocytic activity and increased secretion of TNF/IL-6 result in polarization to the pro-inflammatory M1 phenotype as well as chronic inflammation in the liver and central nervous system, and the anti-inflammatory M2 phenotype in muscles. Aging neutrophils may induce paracrine telomere dysfunction and senescence of liver cells, and loss of neutrophil-directed motility at epithelial junctions may lead to re-entry of neutrophils into the circulation and cause distant lung tissue injury. The degranulation capacity of eosinophils is significantly reduced in elderly individuals, which may trigger airway inflammation. Aging DCs are significantly less efficient in cross-presenting antigens and activating CD8+ T cells, which may lead to a decrease in adaptive immune responses in aged hosts. DCs: dendritic cells; IL-6: interleukin-6; M1: M1 macrophages; M2: M2 macrophages; NETs: neutrophil extracellular traps; PGE2: prostaglandin E2; ROS: reactive oxygen species; TNF: tumor necrosis factor.
Macrophages
Macrophages are versatile immunocytes that perform critical roles in innate immunity, inflammation and tissue homeostasis [55, 56]. Macrophages can be classified as either tissue-resident or monocyte-derived populations. Tissue-resident macrophages, such as KCs, are differentiated from erythro-myeloid progenitors (EMPs) in the yolk sac and fetal liver, while monocyte-derived macrophages are differentiated from bone marrow [57]. During inflammation or tissue injury, depending on the tissue type and stimuli, both resident and monocyte-derived macrophages engage in migration, expansion, and signaling. During extravasation into inflammatory sites under chemokine guidance [58], monocytes differentiate into macrophages to perform immune functions, largely based on various signaling molecules presented in the local microenvironment [59]. Depending on the stimulus, macrophages can be classified as the pro-inflammatory M1 phenotype, which is polarized by either lipopolysaccharide (LPS) alone or in combination with T helper 1 (Th1) cell cytokines, or the anti-inflammatory M2 phenotype, which is polarized by T helper 2 (Th2) cell cytokines such as interleukin-4 (IL-4) [60, 61]. However, the scenario in vivo is often more complex, and they are generally accepted as transient, reversible, and occurring along a spectrum [62].
The number of monocytes in the bone marrow and peripheral circulation rises with age, creating chronic inflammation by secreting more cytokines such as tumor necrosis factor (TNF) and IL-6 [63]. Macrophages are found to infiltrate more in liver and white adipose tissue with age [64] but less in bone and skeletal muscle [65, 66]. Aged macrophages have a significantly altered transcriptional profile and often exhibit a stronger pro-inflammatory phenotype (M1) than younger macrophages [64, 65]. Therefore, the M1/M2 ratio could reflect the age-related functional status of macrophages.
The phagocytic capacity of macrophages decreases with age, which reduces bacterial killing and inflammatory clearance. 1A/1B-light chain 3 (LC3)-associated phagocytosis (LAP) is an important innate immune response of macrophages [67]. Bone marrow-derived macrophages (BMDMs) in aged mice not only lack LAP and bacterial killing but also produce higher levels of pro-inflammatory cytokines, resulting in susceptibility to pathogens [67]. Increased macrophage p38 mitogen-activated protein kinase (MAPK) activity in elderly individuals leads to a decrease in T-cell immunoglobulin mucin receptor-4 (TIM-4) on macrophages, causing reduced efferocytosis of apoptotic vesicles [68]. Therefore, although the onset of inflammation is similar between young and elderly individuals, the ability to eliminate inflammation is greatly diminished in elderly individuals, resulting in persistent chronic inflammation.
Recent studies have highlighted the critical role of metabolism in the programming of the immune response [69]. In aging macrophages and microglia, prostaglandin E2 (PGE2), a typical senescence-associated secretory phenotype (SASP) [70], signals through the EP2 receptor to promote glycogen synthesis and reduce glucose flux and mitochondrial respiration [71]. This energy-deficient state triggers an undesirable pro-inflammatory response. The NAD+ of macrophages is derived mainly from the kynurenine pathway metabolism of tryptophan. Although the immune response triggers upstream kynurenine pathway activation, aging inhibits NAD+ de novo synthesis in macrophages and suppresses mitochondrial NAD+-dependent signaling and respiration, which shift macrophages toward a pro-inflammatory phenotype and impair phagocytosis and resolution of inflammation [72].
There is great heterogeneity of macrophages from different organs during aging [73-75]. Single-cell RNA sequencing (scRNA-seq) analysis revealed a significant increase in the proportion of interstitial lung macrophages but a decrease in the proportion of alveolar macrophages. There was an increase in the proportion of hepatic macrophages expressing chemokine (C-X3-C motif) receptor-1 (CX3CR-1), but the number of peritoneal macrophages expressing intercellular adhesion molecule 2 (ICAM2) was significantly reduced [74]. In addition, scRNA-seq analysis of aged mice showed enhanced inflammatory function of KCs, which may exacerbate inflammatory injury in the liver [75]. Recent research revealed that the number and transcriptional characteristics of alveolar macrophages that change with age are not cell autonomous, but rather are formed by the alveolar milieu in which they live, irrespective of signaling molecules or cells in the circulation [76]. This finding emphasized that the local microenvironment of tissues may play a dominant role in macrophage aging.
Although aging is irreversible, the inflammatory state of an aging organism can be improved to some extent by reducing food intake and avoiding malnutrition [77]. Recent studies revealed that caloric restriction (CR) [64] and fibroblast growth factor-21 [78] could regulate macrophage polarization from the M1 to M2 phenotype in several organs of an aged mouse model, thereby suppressing the persistent inflammation associated with aging. Therefore, strategies targeting macrophages may extend human lifespan and improve susceptibility to aging-related diseases.
Neutrophils
Neutrophils, the most prevalent leukocytes in the body, serve as the first line of defense against pathogens and respond to a variety of inflammatory stimuli [79]. Neutrophils originate from CMPs in the bone marrow and extramedullary tissues such as spleen [79]. After differentiation into granulocyte macrophage progenitor cells (GMPs) [80], neutrophil-committed differentiation starts with neutrophilic promyelocytes and myelocytes, which both have the propensity to divide and are part of the so-called “mitotic neutrophil pool”. The presence of mature neutrophils in the bone marrow represents the end of the “postmitotic pool”, where neutrophils undergo terminal differentiation before being liberated into the peripheral blood. Then, mature neutrophils in the “mature neutrophil pool” enter a state of exchange between the bone marrow, blood, and other tissues [81]. When stimulated by pathogens, neutrophils in blood can rapidly migrate to the site of infection through cytokine-mediated chemotaxis [82], β2 integrin-mediated vascular endothelial adhesion, ICAM-1- and ICAM-2-mediated transendothelial migration [83], and defend against invading pathogens [84] through the release of cytotoxic proteins, peptides and enzymes in the phagolysosome [85], ROS [86] and neutrophil extracellular traps (NETs) formation (NETosis) [87]. Eventually, neutrophils that have engulfed bacteria accumulate in the liver and are taken up by KCs, which reduces the generation of inflammatory cytokines by macrophages and promotes inflammation resolution once infections have been removed [88]. However, if pathogens are not cleared promptly, neutrophils may induce or aggravate many inflammatory diseases, including both acute and chronic diseases pertaining to the liver, lung and other organs [89, 90].
During the aging process, neutrophil infiltration is increased in a variety of organs [64], but neutrophil chemotaxis [91], phagocytosis and bacterial killing activity are significantly reduced [92]. This decrease in chemotactic directionality causes neutrophil elastase-mediated tissue degradation and neutrophil-mediated local inflammation, leading to additional tissue injury [93, 94]. Neutrophils produce a burst of ROS that can directly kill pathogens. Compared to neutrophils in younger individuals, acute ROS generation is substantially lower in older individuals [95], whereas spontaneous ROS is significantly higher [96, 97], which may indirectly lead to persistent inflammation and a reduced immune response to pathogens. Aging can also cause excessive neutrophil activation, which is involved in liver injury and mortality associated with viral infection by inducing IL-17 production from natural killer T (NKT) cells [98]. In addition, NETosis is the ability of neutrophils to expel genomic DNA to capture pathogens. With age, NETosis decreases and leads to delayed pathogen clearance and increased susceptibility to infection [99].
The aging inflammatory microenvironment significantly reprograms neutrophils and in turn affects multiple organs throughout the body. Aging neutrophils cannot efficiently clear inflammation but instead induce aggravated telomere damage and cellular senescence in organ parenchymal cells through the release of matrix metalloproteinases (MMPs) and ROS, resulting in chronic tissue and organ damage [100]. Enhanced reverse trans-epithelial migration of neutrophils can be observed in aged mice. Due to the accumulation of mast cells expressing C-X-C chemokine ligand-1 (CXCL-1), neutrophil C-X-C chemokine receptor-2 (CXCR-2) was desensitized and lost neutrophil-directed motility at epithelial junctions, resulting in neutrophil re-entry into the circulation and causing distant lung tissue damage [101].
In summary, aging-related neutrophil hypofunction delays pathogen clearance and injury repair, increases local inflammation, and exacerbates aging-related inflammatory diseases [102]. Further exploration of the interactions between neutrophils and multiple inflammatory microenvironments may help explore the underlying mechanism of aging-related multiorgan dysfunction syndrome.
Other myeloid cells
In contrast to macrophages and neutrophils, the functional alterations and mechanisms of aging in other myeloid cells still lack in-depth studies. For example, aging DCs are significantly less efficient in cross-presenting antigens and activating CD8+ T cells [103], leading to a decrease in adaptive immune responses in aged hosts [104]. However, it is still controversial whether different DC subtypes exhibit altered antigen-presenting ability [105]. Studies in human patients have found that eosinophil degranulation capacity is significantly reduced in elderly patients, which may explain the differences in disease manifestations and drug sensitivity of airway inflammation in older and younger patients [106], but its role in diverse aging-related inflammatory diseases remains poorly understood. Activation of NF-κB, a key transcription factor in the cellular response to injury and stress, partially causes a significant increase in the proportion of MDSCs in the bone marrow and spleen of aged animals, thereby suppressing the immune response and leading to an impaired response to stress [107]. Currently, the role of different subpopulations of myeloid cells in aging-related diseases remains unclear due to low cell abundance, and future omics tools such as single-cell sequencing may better explore the potential roles and mechanisms of myeloid cell subpopulations in aging.
Interaction among myeloid cells and nonimmune systems
As one of the main features of cellular senescence, the production and secretion of SASPs not only triggers local inflammation but also induces infiltration of immune cells such as macrophages, neutrophils, NK cells and T cells [100, 108, 109]. The surveillance and clearance of senescent cells by immune cells is crucial for preserving tissue homeostasis and preventing hazardous inflammation [108, 110]. Senescent cells accumulate as a result of age-related immune system degradation, which may cause age-related functional decline and diseases [111]. Elimination of senescent cells by gene editing prevents or delays tissue dysfunction and extends healthy lifespan, suggesting that immunosurveillance and clearance of senescent cells can help ameliorate aging-related diseases [112, 113].
The hypothesis that an aging immune system can drive solid organ aging is directly evidenced by the study from Yousefzadeh et al [47]. The investigators established a mouse model of premature immune failure from deletion of Ercc1 in hematopoietic stem cells and observed that along with aging of the immune system, nonlymphoid organs also showed increased aging and injury, whereas transplantation of immune cells from young mice blocked organ aging [47]. In liver studies, infiltration of macrophages and NK cells in aged liver can inhibit hepatocyte DNA synthesis and liver regeneration by producing excess interferon-γ (IFN-γ), while depletion of such cells can significantly improve liver regeneration [114]. In a mouse model of acute liver injury (ALI), the telomeres of hepatocytes are highly susceptible to oxidative damage caused by neutrophil infiltration, and depletion of neutrophils reduces telomere dysfunction and cellular senescence in hepatocytes [100]. Therefore, the aging immune system can drive systemic aging and may serve as a therapeutic target to extend healthy lifespan.
Senescent nonimmune cells can also regulate the functional state of immune cells. scRNA-seq analysis revealed that the age-related changes in the number and transcriptional characteristics of alveolar macrophages are regulated mainly by the alveolar microenvironment but not by circulating growth factors and cells [76]. During aging, senescent cells gradually accumulated in the liver and visceral white adipose tissue may induce macrophage proliferation with SASPs secretion [115]. The interaction between immune and nonimmune cells in the aging process is still not sufficiently studied. A better understanding of the interaction during aging will help to develop new therapeutic interventions to eliminate harmful senescent cells and maintain the youthfulness of the organism.
Myeloid cells and aging-related liver diseases
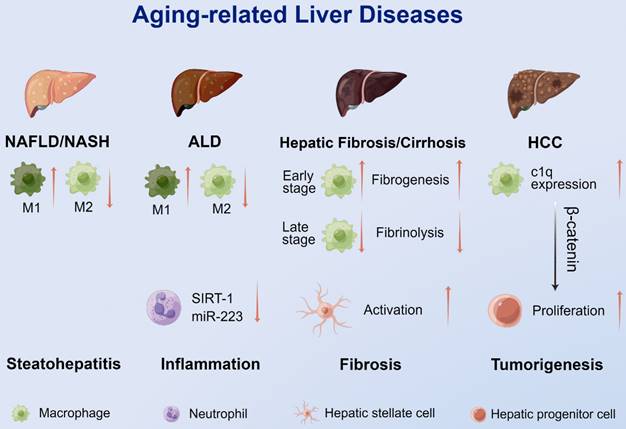
The immune system plays an integral role in developing liver diseases [116]. There is growing evidence supporting that aging immune cells, especially myeloid cells, directly or indirectly contribute to liver aging and increased susceptibility to aging-related diseases, including NAFLD/nonalcoholic steatohepatitis (NASH), ALD, liver fibrosis/cirrhosis, hepatocellular carcinoma (HCC) and comorbidities involving the liver (Figure 3).
The roles of myeloid cells in aging-related liver disease. An increase in M1 macrophages and a decrease in M2 macrophages were observed in NAFLD and ALD. Reduced SIRT-1 expression in aged neutrophils suppressed miR-223 expression, causing more severe inflammatory injury in ALD. Increased macrophage infiltration and fibrogenesis can be observed in the early stage of chronic injury, while decreased macrophage infiltration leads to decreased fibrinolysis and promotes liver fibrosis through activation of HSCs in the late stage. During aging, macrophages express increased complement C1q which promotes the proliferation and dedifferentiation of HPCs and tumorigenesis through the β-catenin pathway. ALD: alcoholic liver disease; HCC: hepatocellular carcinoma; M1: M1 macrophages; M2: M2 macrophages; miR-223: microRNA-223; NAFLD: nonalcoholic fatty liver disease; NASH: nonalcoholic steatohepatitis; SIRT-1: sirtuin-1.
Nonalcoholic fatty liver disease
NAFLD is the leading cause of chronic liver disease worldwide [117]. Compared to young people, elderly people are more likely to progress to NASH [118]. The mechanisms underlying the development of aging-related steatosis are not fully understood and may be attributed to the accumulation of toxic free fatty acids caused by mitochondrial dysfunction [119], reduced autophagy [35, 120] and endoplasmic reticulum stress [121] in aged hepatocytes.
During the disease process of NAFLD, macrophages accumulate in the liver through C-C motif ligand-2 (CCL-2)/CCR-2 chemotaxis [122]. M1 macrophages promote the development of aging-related NAFLD/NASH, while M2 KCs promote the apoptosis of M1 KCs and reduce hepatocyte apoptosis and steatosis, thereby alleviating the disease progression of NAFLD [123]. Fontana et al. [124] used an HFD to trigger NAFLD in mice of different ages. Liver injury occurred mostly in older mice with more pronounced M1 macrophages in the liver and adipose tissue than in younger mice. Similarly, a significant positive correlation between CD163, a marker of M1 macrophages, and the severity of NAFLD was observed in human patients [125]. Therefore, the infiltration and polarization of macrophages influences the course of aging-related NAFLD/NASH, and therapies targeting macrophages may improve the incidence and severity of this disease in the elderly population.
Alcoholic liver disease
ALD is one of the leading causes of death from liver diseases, and its pathological changes include a series of processes, including steatosis, steatohepatitis, cirrhosis, and HCC [126, 127]. In recent years, the incidence of ALD has tended to be stable, but the incidence of alcohol-related cirrhosis and HCC and the need for liver transplantation are continuing to rise [128]. Elderly people are prone to excessive alcohol consumption, and the risk of alcoholism is further increased due to altered metabolism or tobacco and illicit drug use [129]. Old individuals tend to have a chronic low-grade inflammatory state, as evidenced by elevated circulating levels of pro-inflammatory cytokines and local infiltration of inflammatory cells, which further exacerbates ALD [130, 131].
KCs are the major defense of hepatic innate immunity. Increased intestinal permeability and high levels of endotoxin in portal blood were found to lead to the activation of KCs [132]. KCs play key roles in early alcohol-induced liver injury by recognizing endotoxins in the portal circulation and inducing an innate immune response through polarization to the pro-inflammatory M1 phenotype [132]. Wan et al. [133] found that promoting the polarization of KCs to the anti-inflammatory M2 type prevented alcohol-induced hepatocyte steatosis and apoptosis. A high M2/M1 ratio may be a protective feature of ALD [123].
Emerging evidence supports liver infiltration of neutrophils as a key mechanism in promoting ALD, possibly by producing ROS and inflammatory mediators [134, 135]. SIRT-1 is a recognized antiaging protein, and knockdown of SIRT-1 may exacerbate ALD [136]. Our recent study found that neutrophilic SIRT-1 expression was significantly downregulated in aged and ethanol-fed mice, and myeloid cell-specific sirt-1 knockout mice had more severe ALD [89]. In addition, downregulation of SIRT-1 decreased the expression of anti-inflammatory/antifibrotic microRNA-223 in neutrophils [137, 138], leading to an increase in the production of ROS and inflammatory mediators, which ultimately resulted in acute alcoholic liver injury in alcohol-fed mice and patients with chronic alcohol consumption.
Recent study from Ma and Gao et al. [139] have expanded existing knowledge on the roles of hepatic neutrophils in mediating liver injury. Based on the extent of inflammatory cell infiltration, severe alcoholic hepatitis was divided into high intrahepatic neutrophils with low CD8+ T cells (NeuhiCD8lo) and NeuloCD8hi subtypes, and the effects of the neutrophil cytosolic factor-1 (NCF-1)/SIRT-1/AMPK axis on lipid metabolism and the NCF-1/p-38 MAPK/miR-223 pathway on alcohol-induced inflammation and fibrosis were further revealed [139]. Consistent with the notion that neutrophils in young adults have a greater capacity to release ROS [95], NeuhiCD8lo patients were relatively younger and had more severe liver injury compared with those with NeuloCD8hi. However, considering the inclusion of patients who are mostly middle-aged in the two groups, it remains unknown whether hepatic neutrophils exert pro-inflammatory effects through similar pathways in the truly aged population. But one thing is clear that treatments targeting neutrophil must be a promising direction for future translational research, which is expected to improve the prevention and treatment of ALD as well as its complications.
Liver fibrosis and cirrhosis
Liver fibrosis, which can progress to cirrhosis, HCC, and eventually liver failure, is one of the main outcomes of various chronic liver diseases. Liver fibrosis is a dynamic and reversible process of wound repair, including progression and regression stages [140]. Various cytokines and chemokines secreted by macrophages, including TGF-β, TNF-α, IL-1β and CCL-2, activate HSCs and enhance myofibroblast proliferation through NF-κB-dependent signaling pathways, eventually resulting in liver fibrosis [141].
The incidence of liver fibrosis increases with age and is typically characterized by age-related changes in macrophage infiltration. In the early stages of chronic injury, macrophages infiltrate more in the aged liver than in the young liver, which promotes inflammation and fiber formation. In the later stages, macrophage infiltration of the aged liver is reduced and insufficient to adequately lyse fibers, thereby exacerbating liver fibrosis [142]. The direction of macrophage polarization may also influence the liver fibrosis process. Mohammed et al. [143] found that markers of M1 macrophages, pro-inflammatory cytokines (TNF-α, IL-6 and IL-1β), and markers of fibrosis were significantly upregulated in aged mice with necroptosis-induced liver injury. To inhibit macrophage recruitment, a recent clinical trial using CCR-2/CCR-5 antagonists found that, as compared to placebo, twice as many participants achieved fibrosis improvement and no exacerbation of steatohepatitis [144]. Subgroup analysis showed that the therapy was equally effective in patients under and over 56 years of age, indicating its potential therapeutic value in aging-related liver fibrosis [144].
Hepatocellular carcinoma
HCC occurs mostly on the basis of viral infection or noninfectious chronic hepatitis [145]. These chronic inflammatory conditions create a pro-tumorigenic microenvironment that is an important factor in inducing the transformation of hepatocytes into cancer cells [146]. Advanced age is a high-risk factor for HCC due to the constant low-grade inflammation that comes with aging [44]. Research from Ho et al. [147] revealed that aged mice with knockout of hepatocyte β-catenin had an inflammatory microenvironment contribute to the development of HCC. C1q, a complement released from macrophages in the inflammatory microenvironment, is thought to activate the nonclassical pathway of β-catenin in periportal hepatic progenitor cells (HPCs), promoting the proliferation and dedifferentiation of HPCs and thus inducing hepatocarcinogenesis. C1q inhibitors blocked the β-catenin pathway in HPCs and HCC but not the classical pathway in normal hepatocytes. The above mechanism was also verified in liver specimens from patients with chronic hepatitis. Therefore, C1q in macrophages may be a new target for blocking carcinogenesis in elderly patients.
Other liver diseases
In addition to the abovementioned diseases, aging may also affect liver diseases pertaining to the dysregulation of proliferation, healing, and tolerance. During severe acute injury, the liver regenerative capacity may be impaired, leading to ALI and even acute liver failure (ALF). Compared with young people, ALI in elderly individuals is more likely to develop into ALF with a worse prognosis. Our recent study, using the thioacetamide (TAA)-induced ALI mouse model (TAA-ALI), demonstrated that aging can polarize macrophages to the M1 phenotype through the secretion of inflammatory factors and SASPs and thus exacerbate liver injury [35]. Macrophages are also involved in postoperative ischemia and reperfusion (IR) liver injury. In aged IR mice, NLRP3 activation can be observed in the liver and macrophages, while knockdown of the STING-NLRP3 axis in macrophages eliminates the deleterious role of aging in exacerbating intrahepatic inflammation and IR injury [148]. Other aging-related liver comorbidities, such as multiorgan dysfunction, may also be regulated by KCs [149], and more studies are needed to reveal the impact of the immune system on aging-related liver diseases.
Promising therapeutics targeting aging
As the population ages, the rise of aging-related liver diseases is not just a medical issue but also a significant social issue without new preventive and curative therapeutics specifically for not only young and middle-aged populations but also elderly people. In this scenario, several strategies focused on the aging immune system, a novel therapeutic target, to treat aging-related liver diseases have shown promising prospects, and some of them may have potential in clinical application (Table 1).
Clinical trials with anti-aging strategies for liver diseases.
| Strategy | Study ID | Indication | Regimen | Status |
|---|---|---|---|---|
| Senolytic Therapy | NCT00382668 | Liver diseases | Dasatinib | Completed |
| NCT00459108 | HCC | Dasatinib | Terminated | |
| NCT02143401 | HCC | Navitoclax | Active, not recruiting | |
| Caloric Restriction | NCT04230655 | NAFLD | Low energy diet | Recruiting |
| NCT05041673 | Fatty liver | Metformin | Active, not recruiting | |
| NCT04972396 | NASH | Metformin | Recruiting | |
| NCT04033107 | HCC | Metformin | Recruiting | |
| Microbiological Therapy | NCT03796598 | Liver cirrhosis | FMT | Recruiting |
| NCT04594954 | NAFLD | FMT | Recruiting | |
| NCT04758806 | Alcoholic hepatitis | FMT | Recruiting | |
| NCT05007470 | ALD | VSL #3 Capsule | Recruiting |
Aberrations: ALD: alcoholic liver disease; FMT: fecal microbiota transplantation; HCC: hepatocellular carcinoma; NAFLD: Nonalcoholic fatty liver disease: NASH: nonalcoholic steatohepatitis.
Senolytic therapy
As mentioned above, aging-related immune senescence, which leads to a reduction in the phagocytosis capacity of macrophages and neutrophils, may result in the accumulation of senescent cells and increased susceptibility to diseases. Senolytic therapy was designed to compensate for immune clearance and maintain organ rejuvenation. Using a hypothesis-driven strategy that targeted senescent cell antiapoptotic pathways which were more highly expressed by senescent than nonsenescent cells, the first generation senolytic medicines dasatinib, quercetin, fisetin, and navitoclax were discovered [150]. In preclinical models, senolytics prevent or alleviate cardiovascular [151], liver [119], lung [152], kidney disorders [153] as well as complications of organ transplantation [154], age-dependent fracture healing [155] and disc degeneration [156].
Preclinical studies of senolytic therapies for liver diseases have shown promising results. The classic senolytic cocktail of dasatinib plus quercetin (D + Q) reduced overall hepatic steatosis, thus alleviating aging-related NAFLD [119] and CCl4-induced liver fibrosis [157] in mice. Fisetin has also been shown to improve APAP-induced hepatotoxicity by promoting autophagy and inhibiting inflammasome activation [158]. In addition to chemical drugs, chimeric antigen receptor (CAR) T cells that target senescent cells have also shown promising results in ameliorating liver fibrosis induced chemically or by diet [159]. However, senolytic therapy is not a panacea. Research from Cheng et al. [160] showed that ABT263 (navitoclax) may impair liver regeneration following partial hepatectomy through elimination of senescent HSCs, which induces multiple signaling pathways to stimulate liver regeneration by secretion of IL-6 and CXCR-2 ligands. Moreover, D + Q was ineffective against age-associated NAFLD-induced HCC [161]. Therefore, until the pharmacological mechanism, efficacy, and safety of senolytics are well clarified, there is still a long way to go before senolytic therapies can be clinically applied in aging-related liver disease.
Caloric restriction
CR is typically a daily decreased nutrient uptake intervention without malnutrition [162]. CR extends the lifespan of various model organisms ranging from yeast, worms and flies to mice and primates [163] by regulating hallmarks of aging, including deregulated nutrient sensing, cellular senescence, stem cell exhaustion and altered intercellular communication [162]. Recently, scRNA-seq analysis revealed that aging may be delayed by CR by reversing the aging-disturbed immune ecosystem, which has excessive pro-inflammatory ligand-receptor interplay [64] and extends health lifespan [164]. In humans, a two-year study of CR identified a reduction in the rate of living and systemic oxidative stress and improved biomarkers of aging [165]. CR also leads to a marked improvement in glucose metabolism [166] and reverse fatty liver caused by saturated fat overeating [167].
Given that a healthy lifestyle requires sustained efforts and discipline, it is easier said than done for routine application of CR. Therefore, several pharmaceutical compounds or dietary supplements that mimic the effects of CR are being explored. Metformin, previously used as an antidiabetic drug, is viewed as a CR mimetic via activation of AMPK [168]. Studies have shown evidence of metformin attenuating aging hallmarks [169] and ameliorating age-related changes in LSECs via AMPK and endothelial nitric oxide pathways, which may improve liver insulin sensitivity, especially in old age [170]. In addition to metformin, resveratrol, another classic CR mimetic, has shown promising results in preclinical studies pertaining to ALI [171] and NAFLD [172]. However, current evidence, including results from clinical trials, does not support supplementation with resveratrol for the management of NAFLD [173-175]. We believe that healthy lifestyle and dietary habits may contribute more to delaying aging and improving susceptibility to aging-related diseases than the use of synthetic drugs [176]. Individualized strategies that combine genetic alteration, disease pathology and lifestyles will bring the greatest benefit to patients.
Microbiological therapy
Gut microbes occupy the interface between the external environment and the host [177], which significantly influences human aging and diseases [178]. Dysbiosis is characterized by an imbalance in the microbiota including local distribution, functional composition and metabolic activities [179]. Aging-related dysbiosis of the gut microbiome was proven to contribute to a global inflammatory state and diseases in the elderly [180] through chronic upregulation of pro-inflammatory mediators (TNF-α and IL-6), thus serving as a catalyst for fueling inflammaging [44]. These mediators activate many signaling pathways influencing immune function, leading to a gradual deterioration of the immune system, known as immunosenescence [181]. Accumulating evidence suggests that both inflammaging and immunosenescence are responsible for most aging-related diseases, including but not limited to disorders pertaining to the cardiovascular [182], neurological [183], respiratory [184] and digestive systems [143].
Changes in the composition and function of the gut microbiota have profound effects on the development and management of liver disorders, including steatosis [185], cirrhosis [186], liver failure [187] and even HCC [188]. Recently, new promising probiotics, such as Saccharomyces boulardii, have been discovered and have potential therapeutic effects in ALI, ALF and liver fibrosis [189]. Living materials of a hierarchy-assembled dual probiotic system containing bulgaricus and Lactobacillus rhamnosus GG, fabricated by Chen et al. [190], effectively prevented cholestatic drug-induced liver injury through inhibition of hepatic bile acid synthesis and facilitation of bile acid excretion. Fecal microbiota transplantation (FMT), which involves the transfer of donor stool into recipients, is increasingly being explored as a potential treatment in gastric intestinal and liver diseases [191]. Clinical trials supported the safety of FMT and have shown improvement of survival and clinical severity in patients with severe alcoholic hepatitis [192, 193]. However, FMT can be lethal for elderly people because of serious complications, such as infections from multidrug-resistant organisms [194]. Therefore, FMT should be used with appropriate monitoring and careful donor selection in treating aging-related liver diseases in elderly population. With a further understanding of the crosstalk among the microbiome, aging and disease, individualized treatment focusing on different disease conditions and populations will better balance the efficacy and safety when being administered in aging-related liver diseases.
Conclusion and outlooks
Aging is the process of gradual decline of organisms over time. Liver, the largest digestive and immune organ in the body, also deteriorates with age, increasing the risk of chronic liver diseases. Immune surveillance and clearance maintain the homeostasis and normal functioning of the liver, whereas the aging immune microenvironment inappropriately releases excessive inflammatory factors, causing and exacerbating various aging-related liver diseases.
There are still many unknowns in the field of liver aging, such as whether immune cells that form different liver structures affect the course of aging-related diseases differently, the interaction between immune cells and nonimmune mesenchymal cells and the functional or metabolic changes they cause, and the unique roles of low-abundance myeloid cells such as DCs and eosinophils in liver aging. In addition, the lack of clinical exploration has limited the translation of results from animal research. Distinguishing physiological aging from age-related conditions can be challenging, and more studies are needed to assess the effects of aging on the liver prior to the onset of age-related conditions to separate the mechanisms of aging from the manifestations of the pathological conditions themselves. In the future, as new technologies become more available, multiomics analysis at the single-cell level will further reveal the roles immune cells play in regulating aging at the organ and even systemic level, providing an updated understanding and individualized treatment strategies for aging-related diseases.
Acknowledgements
All figures were created by Figdraw (www.figdraw.com).
Abbreviations
ALD: alcoholic liver disease; ALI: acute liver injury; ALF: acute liver failure; AMPK: Adenosine 5'-monophosphate activated protein kinase; ATG5: autophagy-associated protein 5; BMDMs: bone marrow-derived macrophages; CLP: common lymphoid progenitor; CMPs: common myeloid progenitor cells; CR: caloric restriction; DCs: dendritic cells; DSBs: DNA double-strand breaks; EMPs: erythro-myeloid progenitors; FMT: fecal microbiota transplantation; GMPs: granulocyte macrophage progenitor cells; HCC: hepatocellular carcinoma; HDAC: histone deacetylase; HFD: high-fat diet; HGF: hepatocyte growth factor; HPCs: hepatic progenitor cells; HSCs: hepatic stellate cells; ICAM-2: intercellular adhesion molecule-2; IL-6: interleukin-6; IR: ischemia and reperfusion; KCs: kupffer cells; LPS: lipopolysaccharide; LSECs: liver sinusoidal endothelial cells; MANF: mesencephalic-astrocyte-derived neurotrophic factor; MAPK: mitogen-activated protein kinase; MDSCs: myeloid-derived suppressor cells; MMPs: matrix metalloproteinases; mtDNA: mitochondrial DNA; NAFLD: nonalcoholic fatty liver disease; NASH: nonalcoholic steatohepatitis; NCF-1: neutrophil cytosolic factor-1; NETs: neutrophil extracellular traps; NK: natural killer; PGE2: prostaglandin E2; ROS: reactive oxygen species; SASP: senescence-associated secretory phenotype; SIRT-1: sirtuin-1; TIM-4: T-cell immunoglobulin mucin receptor-4; TNF: tumor necrosis factor.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194-217
2. Partridge L, Deelen J, Slagboom PE. Facing up to the global challenges of ageing. Nature. 2018;561:45-56
3. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular Senescence: Aging, Cancer, and Injury. Physiol Rev. 2019;99:1047-78
4. Maeso-Diaz R, Gracia-Sancho J. Aging and Chronic Liver Disease. Semin Liver Dis. 2020;40:373-84
5. Moon AM, Singal AG, Tapper EB. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin Gastroenterol Hepatol. 2020;18:2650-66
6. Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4:177-97
7. Kubes P, Jenne C. Immune Responses in the Liver. Annu Rev Immunol. 2018;36:247-77
8. Ficht X, Iannacone M. Immune surveillance of the liver by T cells. Sci Immunol. 2020 5
9. Singh P, Coskun ZZ, Goode C, Dean A, Thompson-Snipes L, Darlington G. Lymphoid neogenesis and immune infiltration in aged liver. Hepatology. 2008;47:1680-90
10. Stahl EC, Haschak MJ, Popovic B, Brown BN. Macrophages in the Aging Liver and Age-Related Liver Disease. Front Immunol. 2018;9:2795
11. Wan J, Weiss E, Ben Mkaddem S, Mabire M, Choinier PM, Thibault-Sogorb T. et al. LC3-associated phagocytosis in myeloid cells, a fireman that restrains inflammation and liver fibrosis, via immunoreceptor inhibitory signaling. Autophagy. 2020;16:1526-8
12. Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311-23
13. Wang MJ, Chen F, Li JX, Liu CC, Zhang HB, Xia Y. et al. Reversal of hepatocyte senescence after continuous in vivo cell proliferation. Hepatology. 2014;60:349-61
14. White RR, Milholland B, de Bruin A, Curran S, Laberge RM, van Steeg H. et al. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat Commun. 2015;6:6790
15. Niemann J, Johne C, Schroder S, Koch F, Ibrahim SM, Schultz J. et al. An mtDNA mutation accelerates liver aging by interfering with the ROS response and mitochondrial life cycle. Free Radic Biol Med. 2017;102:174-87
16. Donne R, Saroul-Ainama M, Cordier P, Celton-Morizur S, Desdouets C. Polyploidy in liver development, homeostasis and disease. Nat Rev Gastroenterol Hepatol. 2020;17:391-405
17. Wilkinson PD, Delgado ER, Alencastro F, Leek MP, Roy N, Weirich MP. et al. The Polyploid State Restricts Hepatocyte Proliferation and Liver Regeneration in Mice. Hepatology. 2019;69:1242-58
18. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC. et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056-60
19. Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ. et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151-6
20. Mulligan JD, Gonzalez AA, Kumar R, Davis AJ, Saupe KW. Aging elevates basal adenosine monophosphate-activated protein kinase (AMPK) activity and eliminates hypoxic activation of AMPK in mouse liver. J Gerontol A Biol Sci Med Sci. 2005;60:21-7
21. Lew LC, Hor YY, Jaafar MH, Lau AS, Lee BK, Chuah LO. et al. Lactobacillus Strains Alleviated Hyperlipidemia and Liver Steatosis in Aging Rats via Activation of AMPK. Int J Mol Sci. 2020 21
22. Gao Y, Zhang W, Zeng LQ, Bai H, Li J, Zhou J. et al. Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol. 2020;36:101635
23. Sousa-Victor P, Neves J, Cedron-Craft W, Ventura PB, Liao CY, Riley RR. et al. MANF regulates metabolic and immune homeostasis in ageing and protects against liver damage. Nat Metab. 2019;1:276-90
24. Maeso-Diaz R, Dalton GD, Oh S, Du K, Tang L, Chen T. et al. Aging reduces liver resiliency by dysregulating Hedgehog signaling. Aging Cell. 2022;21:e13530
25. Bacalini MG, Franceschi C, Gentilini D, Ravaioli F, Zhou X, Remondini D. et al. Molecular Aging of Human Liver: An Epigenetic/Transcriptomic Signature. J Gerontol A Biol Sci Med Sci. 2019;74:1-8
26. Kronfol MM, Jahr FM, Dozmorov MG, Phansalkar PS, Xie LY, Aberg KA. et al. DNA methylation and histone acetylation changes to cytochrome P450 2E1 regulation in normal aging and impact on rates of drug metabolism in the liver. Geroscience. 2020;42:819-32
27. Price AJ, Manjegowda MC, Kain J, Anandh S, Bochkis IM. Hdac3, Setdb1, and Kap1 mark H3K9me3/H3K14ac bivalent regions in young and aged liver. Aging Cell. 2020;19:e13092
28. Sato S, Solanas G, Peixoto FO, Bee L, Symeonidi A, Schmidt MS. et al. Circadian Reprogramming in the Liver Identifies Metabolic Pathways of Aging. Cell. 2017;170:664-77 e11
29. Nie YZ, Zheng YW, Taniguchi H. Improving the repopulation capacity of elderly human hepatocytes by decoding aging-associated hepatocyte plasticity. Hepatology. 2022
30. Nakagawa T, Guarente L. SnapShot: sirtuins, NAD, and aging. Cell Metab. 2014;20:192-e1
31. You M, Jogasuria A, Taylor C, Wu J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg Nutr. 2015;4:88-100
32. Ramirez T, Li YM, Yin S, Xu MJ, Feng D, Zhou Z. et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J Hepatol. 2017;66:601-9
33. Mitchell SJ, Huizer-Pajkos A, Cogger VC, McLachlan AJ, Le Couteur DG, Jones B. et al. Age-related pseudocapillarization of the liver sinusoidal endothelium impairs the hepatic clearance of acetaminophen in rats. J Gerontol A Biol Sci Med Sci. 2011;66:400-8
34. Rohn F, Kordes C, Buschmann T, Reichert D, Wammers M, Poschmann G. et al. Impaired integrin alpha5 /beta1 -mediated hepatocyte growth factor release by stellate cells of the aged liver. Aging Cell. 2020;19:e13131
35. Liu R, Cui J, Sun Y, Xu W, Wang Z, Wu M. et al. Autophagy deficiency promotes M1 macrophage polarization to exacerbate acute liver injury via ATG5 repression during aging. Cell Death Discov. 2021;7:397
36. McLean AJ, Cogger VC, Chong GC, Warren A, Markus AM, Dahlstrom JE. et al. Age-related pseudocapillarization of the human liver. J Pathol. 2003;200:112-7
37. Hilmer SN, Cogger VC, Fraser R, McLean AJ, Sullivan D, Le Couteur DG. Age-related changes in the hepatic sinusoidal endothelium impede lipoprotein transfer in the rat. Hepatology. 2005;42:1349-54
38. Mohamad M, Mitchell SJ, Wu LE, White MY, Cordwell SJ, Mach J. et al. Ultrastructure of the liver microcirculation influences hepatic and systemic insulin activity and provides a mechanism for age-related insulin resistance. Aging Cell. 2016;15:706-15
39. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol. 2013;3:785-97
40. Hilmer SN, Cogger VC, Le Couteur DG. Basal activity of Kupffer cells increases with old age. J Gerontol A Biol Sci Med Sci. 2007;62:973-8
41. Bloomer SA, Moyer ED, Brown KE, Kregel KC. Aging results in accumulation of M1 and M2 hepatic macrophages and a differential response to gadolinium chloride. Histochem Cell Biol. 2020;153:37-48
42. Lian M, Selmi C, Gershwin ME, Ma X. Myeloid Cells and Chronic Liver Disease: a Comprehensive Review. Clin Rev Allergy Immunol. 2018;54:307-17
43. Nikolich-Zugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. 2018;19:10-9
44. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14:576-90
45. Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E. et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244-54
46. Morrisette-Thomas V, Cohen AA, Fulop T, Riesco E, Legault V, Li Q. et al. Inflamm-aging does not simply reflect increases in pro-inflammatory markers. Mech Ageing Dev. 2014;139:49-57
47. Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE. et al. An aged immune system drives senescence and ageing of solid organs. Nature. 2021;594:100-5
48. Leins H, Mulaw M, Eiwen K, Sakk V, Liang Y, Denkinger M. et al. Aged murine hematopoietic stem cells drive aging-associated immune remodeling. Blood. 2018;132:565-76
49. Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. 2013;13:376-89
50. Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ. et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102:9194-9
51. Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D. et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A. 2010;107:5465-70
52. Rawji KS, Kappen J, Tang W, Teo W, Plemel JR, Stys PK. et al. Deficient Surveillance and Phagocytic Activity of Myeloid Cells Within Demyelinated Lesions in Aging Mice Visualized by Ex vivo Live Multiphoton Imaging. J Neurosci. 2018;38:1973-88
53. Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P. et al. Aging and cancer: The role of macrophages and neutrophils. Ageing Res Rev. 2017;36:105-16
54. Esfahani NS, Wu Q, Kumar N, Ganesan LP, Lafuse WP, Rajaram MVS. Aging influences the cardiac macrophage phenotype and function during steady state and during inflammation. Aging Cell. 2021;20:e13438
55. Mowat AM, Scott CL, Bain CC. Barrier-tissue macrophages: functional adaptation to environmental challenges. Nat Med. 2017;23:1258-70
56. Uderhardt S, Martins AJ, Tsang JS, Lammermann T, Germain RN. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell. 2019;177:541-55 e17
57. Kielbassa K, Vegna S, Ramirez C, Akkari L. Understanding the Origin and Diversity of Macrophages to Tailor Their Targeting in Solid Cancers. Front Immunol. 2019;10:2215
58. Dey A, Allen J, Hankey-Giblin PA. Ontogeny and polarization of macrophages in inflammation: blood monocytes versus tissue macrophages. Front Immunol. 2014;5:683
59. Brown BN, Haschak MJ, Lopresti ST, Stahl EC. Effects of age-related shifts in cellular function and local microenvironment upon the innate immune response to implants. Semin Immunol. 2017;29:24-32
60. De Santis M, Locati M, Selmi C. The elegance of a macrophage. Cell Mol Immunol. 2018;15:196-8
61. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F. et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233:6425-40
62. Li C, Xu MM, Wang K, Adler AJ, Vella AT, Zhou B. Macrophage polarization and meta-inflammation. Transl Res. 2018;191:29-44
63. Puchta A, Naidoo A, Verschoor CP, Loukov D, Thevaranjan N, Mandur TS. et al. TNF Drives Monocyte Dysfunction with Age and Results in Impaired Anti-pneumococcal Immunity. PLoS Pathog. 2016;12:e1005368
64. Ma S, Sun S, Geng L, Song M, Wang W, Ye Y. et al. Caloric Restriction Reprograms the Single-Cell Transcriptional Landscape of Rattus Norvegicus Aging. Cell. 2020;180:984-1001 e22
65. Clark D, Brazina S, Yang F, Hu D, Hsieh CL, Niemi EC. et al. Age-related changes to macrophages are detrimental to fracture healing in mice. Aging Cell. 2020;19:e13112
66. Cui CY, Driscoll RK, Piao Y, Chia CW, Gorospe M, Ferrucci L. Skewed macrophage polarization in aging skeletal muscle. Aging Cell. 2019;18:e13032
67. Inomata M, Xu S, Chandra P, Meydani SN, Takemura G, Philips JA. et al. Macrophage LC3-associated phagocytosis is an immune defense against Streptococcus pneumoniae that diminishes with host aging. Proc Natl Acad Sci U S A. 2020;117:33561-9
68. De Maeyer RPH, van de Merwe RC, Louie R, Bracken OV, Devine OP, Goldstein DR. et al. Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat Immunol. 2020;21:615-25
69. van Beek AA, Van den Bossche J, Mastroberardino PG, de Winther MPJ, Leenen PJM. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. 2019;40:113-27
70. Wiley CD, Brumwell AN, Davis SS, Jackson JR, Valdovinos A, Calhoun C. et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight. 2019 4
71. Minhas PS, Latif-Hernandez A, McReynolds MR, Durairaj AS, Wang Q, Rubin A. et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature. 2021;590:122-8
72. Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S. et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat Immunol. 2019;20:50-63
73. Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC, Schultze JL. The Myeloid Cell Compartment-Cell by Cell. Annu Rev Immunol. 2019;37:269-93
74. Mogilenko DA, Shpynov O, Andhey PS, Arthur L, Swain A, Esaulova E. et al. Comprehensive Profiling of an Aging Immune System Reveals Clonal GZMK(+) CD8(+) T Cells as Conserved Hallmark of Inflammaging. Immunity. 2021;54:99-115 e12
75. Tabula Muris C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature. 2020;583:590-5
76. McQuattie-Pimentel AC, Ren Z, Joshi N, Watanabe S, Stoeger T, Chi M. et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J Clin Invest. 2021 131
77. Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell. 2015;161:106-18
78. Kang K, Xia A, Meng F, Chunyu J, Sun X, Ren G. et al. FGF21 alleviates chronic inflammatory injury in the aging process through modulating polarization of macrophages. Int Immunopharmacol. 2021;96:107634
79. Hidalgo A, Chilvers ER, Summers C, Koenderman L. The Neutrophil Life Cycle. Trends in immunology. 2019;40:584-97
80. Görgens A, Radtke S, Möllmann M, Cross M, Dürig J, Horn PA. et al. Revision of the human hematopoietic tree: granulocyte subtypes derive from distinct hematopoietic lineages. Cell reports. 2013;3:1539-52
81. Dancey JT, Deubelbeiss KA, Harker LA, Finch CA. Neutrophil kinetics in man. The Journal of clinical investigation. 1976;58:705-15
82. McKinstry WJ, Li CL, Rasko JE, Nicola NA, Johnson GR, Metcalf D. Cytokine receptor expression on hematopoietic stem and progenitor cells. Blood. 1997;89:65-71
83. Filippi MD. Neutrophil transendothelial migration: updates and new perspectives. Blood. 2019;133:2149-58
84. Scapini P, Marini O, Tecchio C, Cassatella MA. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol Rev. 2016;273:48-60
85. Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. 2016;273:11-28
86. Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest. 1973;52:741-4
87. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134-47
88. Holub M, Cheng CW, Mott S, Wintermeyer P, van Rooijen N, Gregory SH. Neutrophils sequestered in the liver suppress the proinflammatory response of Kupffer cells to systemic bacterial infection. J Immunol. 2009;183:3309-16
89. Ren R, He Y, Ding D, Cui A, Bao H, Ma J. et al. Aging exaggerates acute-on-chronic alcohol-induced liver injury in mice and humans by inhibiting neutrophilic sirtuin 1-C/EBPalpha-miRNA-223 axis. Hepatology. 2022;75:646-60
90. Winslow S, Odqvist L, Diver S, Riise R, Abdillahi S, Wingren C. et al. Multi-omics links IL-6 trans-signalling with neutrophil extracellular trap formation and Haemophilus infection in COPD. Eur Respir J. 2021 58
91. Simell B, Vuorela A, Ekstrom N, Palmu A, Reunanen A, Meri S. et al. Aging reduces the functionality of anti-pneumococcal antibodies and the killing of Streptococcus pneumoniae by neutrophil phagocytosis. Vaccine. 2011;29:1929-34
92. Butcher S, Chahel H, Lord JM. Review article: ageing and the neutrophil: no appetite for killing? Immunology. 2000;100:411-6
93. Nomellini V, Brubaker AL, Mahbub S, Palmer JL, Gomez CR, Kovacs EJ. Dysregulation of neutrophil CXCR2 and pulmonary endothelial icam-1 promotes age-related pulmonary inflammation. Aging Dis. 2012;3:234-47
94. Naccache PH, Lefebvre JS. A straight neutrophil path to healthy aging? Blood. 2014;123:154-6
95. Sauce D, Dong Y, Campillo-Gimenez L, Casulli S, Bayard C, Autran B. et al. Reduced Oxidative Burst by Primed Neutrophils in the Elderly Individuals Is Associated With Increased Levels of the CD16bright/CD62Ldim Immunosuppressive Subset. J Gerontol A Biol Sci Med Sci. 2017;72:163-72
96. Ogawa K, Suzuki K, Okutsu M, Yamazaki K, Shinkai S. The association of elevated reactive oxygen species levels from neutrophils with low-grade inflammation in the elderly. Immun Ageing. 2008;5:13
97. Kovalenko EI, Boyko AA, Semenkov VF, Lutsenko GV, Grechikhina MV, Kanevskiy LM. et al. ROS production, intracellular HSP70 levels and their relationship in human neutrophils: effects of age. Oncotarget. 2014;5:11800-12
98. Stout-Delgado HW, Du W, Shirali AC, Booth CJ, Goldstein DR. Aging promotes neutrophil-induced mortality by augmenting IL-17 production during viral infection. Cell Host Microbe. 2009;6:446-56
99. Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A. et al. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell. 2014;13:690-8
100. Lagnado A, Leslie J, Ruchaud-Sparagano MH, Victorelli S, Hirsova P, Ogrodnik M. et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 2021;40:e106048
101. Barkaway A, Rolas L, Joulia R, Bodkin J, Lenn T, Owen-Woods C. et al. Age-related changes in the local milieu of inflamed tissues cause aberrant neutrophil trafficking and subsequent remote organ damage. Immunity. 2021;54:1494-510 e7
102. Brubaker AL, Rendon JL, Ramirez L, Choudhry MA, Kovacs EJ. Reduced neutrophil chemotaxis and infiltration contributes to delayed resolution of cutaneous wound infection with advanced age. J Immunol. 2013;190:1746-57
103. Agrawal A, Gupta S. Impact of aging on dendritic cell functions in humans. Ageing Res Rev. 2011;10:336-45
104. Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS. et al. Loss of Phagocytic and Antigen Cross-Presenting Capacity in Aging Dendritic Cells Is Associated with Mitochondrial Dysfunction. J Immunol. 2015;195:2624-32
105. Wong C, Goldstein DR. Impact of aging on antigen presentation cell function of dendritic cells. Curr Opin Immunol. 2013;25:535-41
106. Mathur SK, Schwantes EA, Jarjour NN, Busse WW. Age-related changes in eosinophil function in human subjects. Chest. 2008;133:412-9
107. Flores RR, Clauson CL, Cho J, Lee BC, McGowan SJ, Baker DJ. et al. Expansion of myeloid-derived suppressor cells with aging in the bone marrow of mice through a NF-kappaB-dependent mechanism. Aging Cell. 2017;16:480-7
108. Kale A, Sharma A, Stolzing A, Desprez PY, Campisi J. Role of immune cells in the removal of deleterious senescent cells. Immun Ageing. 2020;17:16
109. Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020;18:e3000599
110. Burton DGA, Stolzing A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res Rev. 2018;43:17-25
111. Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A. et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun. 2018;9:5435
112. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232-6
113. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J. et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184-9
114. Singh P, Goode T, Dean A, Awad SS, Darlington GJ. Elevated interferon gamma signaling contributes to impaired regeneration in the aged liver. J Gerontol A Biol Sci Med Sci. 2011;66:944-56
115. Covarrubias AJ, Kale A, Perrone R, Lopez-Dominguez JA, Pisco AO, Kasler HG. et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat Metab. 2020;2:1265-83
116. Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwalder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits. J Hepatol. 2022;77:1136-60
117. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73-84
118. Noureddin M, Yates KP, Vaughn IA, Neuschwander-Tetri BA, Sanyal AJ, McCullough A. et al. Clinical and histological determinants of nonalcoholic steatohepatitis and advanced fibrosis in elderly patients. Hepatology. 2013;58:1644-54
119. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A. et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017;8:15691
120. Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME. et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5:e1179
121. Xiong X, Wang X, Lu Y, Wang E, Zhang Z, Yang J. et al. Hepatic steatosis exacerbated by endoplasmic reticulum stress-mediated downregulation of FXR in aging mice. J Hepatol. 2014;60:847-54
122. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N. et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61:416-26
123. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F. et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59:130-42
124. Fontana L, Zhao E, Amir M, Dong H, Tanaka K, Czaja MJ. Aging promotes the development of diet-induced murine steatohepatitis but not steatosis. Hepatology. 2013;57:995-1004
125. Kazankov K, Moller HJ, Lange A, Birkebaek NH, Holland-Fischer P, Solvig J. et al. The macrophage activation marker sCD163 is associated with changes in NAFLD and metabolic profile during lifestyle intervention in obese children. Pediatr Obes. 2015;10:226-33
126. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol. 2019;70:151-71
127. Avila MA, Dufour JF, Gerbes AL, Zoulim F, Bataller R, Burra P. et al. Recent advances in alcohol-related liver disease (ALD): summary of a Gut round table meeting. Gut. 2020;69:764-80
128. Dang K, Hirode G, Singal AK, Sundaram V, Wong RJ. Alcoholic Liver Disease Epidemiology in the United States: A Retrospective Analysis of 3 US Databases. Am J Gastroenterol. 2020;115:96-104
129. Blazer DG, Wu LT. The epidemiology of at-risk and binge drinking among middle-aged and elderly community adults: National Survey on Drug Use and Health. Am J Psychiatry. 2009;166:1162-9
130. Pawelec G, Goldeck D, Derhovanessian E. Inflammation, ageing and chronic disease. Curr Opin Immunol. 2014;29:23-8
131. Adrover JM, Nicolas-Avila JA, Hidalgo A. Aging: A Temporal Dimension for Neutrophils. Trends Immunol. 2016;37:334-45
132. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66:1300-12
133. Wan J, Benkdane M, Alons E, Lotersztajn S, Pavoine C. M2 kupffer cells promote hepatocyte senescence: an IL-6-dependent protective mechanism against alcoholic liver disease. Am J Pathol. 2014;184:1763-72
134. Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol. 2019;70:249-59
135. Cho Y, Szabo G. Two Faces of Neutrophils in Liver Disease Development and Progression. Hepatology. 2021;74:503-12
136. Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R. et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology. 2014;146:801-11
137. He Y, Rodrigues RM, Wang X, Seo W, Ma J, Hwang S. et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. J Clin Invest. 2021 131
138. Calvente CJ, Tameda M, Johnson CD, Del Pilar H, Lin YC, Adronikou N. et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J Clin Invest. 2019;129:4091-109
139. Ma J, Guillot A, Yang Z, Mackowiak B, Hwang S, Park O. et al. Distinct histopathological phenotypes of severe alcoholic hepatitis suggest different mechanisms driving liver injury and failure. J Clin Invest. 2022 132
140. Cheng D, Chai J, Wang H, Fu L, Peng S, Ni X. Hepatic macrophages: Key players in the development and progression of liver fibrosis. Liver Int. 2021;41:2279-94
141. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH. et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461-73
142. Collins BH, Holzknecht ZE, Lynn KA, Sempowski GD, Smith CC, Liu S. et al. Association of age-dependent liver injury and fibrosis with immune cell populations. Liver Int. 2013;33:1175-86
143. Mohammed S, Thadathil N, Selvarani R, Nicklas EH, Wang D, Miller BF. et al. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell. 2021;20:e13512
144. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J. et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67:1754-67
145. Miyanishi K, Tanaka S, Sakamoto H, Kato J. The role of iron in hepatic inflammation and hepatocellular carcinoma. Free Radic Biol Med. 2019;133:200-5
146. Heinrich B, Gertz EM, Schaffer AA, Craig A, Ruf B, Subramanyam V. et al. The tumour microenvironment shapes innate lymphoid cells in patients with hepatocellular carcinoma. Gut. 2021
147. Ho TC, Wang EY, Yeh KH, Jeng YM, Horng JH, Wu LL. et al. Complement C1q mediates the expansion of periportal hepatic progenitor cells in senescence-associated inflammatory liver. Proc Natl Acad Sci U S A. 2020;117:6717-25
148. Zhong W, Rao Z, Rao J, Han G, Wang P, Jiang T. et al. Aging aggravated liver ischemia and reperfusion injury by promoting STING-mediated NLRP3 activation in macrophages. Aging Cell. 2020;19:e13186
149. Kawabata T, Kinoshita M, Inatsu A, Habu Y, Nakashima H, Shinomiya N. et al. Functional alterations of liver innate immunity of mice with aging in response to CpG-oligodeoxynucleotide. Hepatology. 2008;48:1586-97
150. Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med. 2020;288:518-36
151. Lewis-McDougall FC, Ruchaya PJ, Domenjo-Vila E, Shin Teoh T, Prata L, Cottle BJ. et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell. 2019;18:e12931
152. Barnes PJ, Baker J, Donnelly LE. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am J Respir Crit Care Med. 2019;200:556-64
153. Kim SR, Jiang K, Ogrodnik M, Chen X, Zhu XY, Lohmeier H. et al. Increased renal cellular senescence in murine high-fat diet: effect of the senolytic drug quercetin. Transl Res. 2019;213:112-23
154. Iske J, Seyda M, Heinbokel T, Maenosono R, Minami K, Nian Y. et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat Commun. 2020;11:4289
155. Beerman I, Basisty N, de Cabo R. Short-term senolytic treatment: a paradigm to promote fracture repair during aging. J Clin Invest. 2022 132
156. Novais EJ, Tran VA, Johnston SN, Darris KR, Roupas AJ, Sessions GA. et al. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat Commun. 2021;12:5213
157. Luo J, Li L, Chang B, Zhu Z, Deng F, Hu M. et al. Mannan-Binding Lectin via Interaction With Cell Surface Calreticulin Promotes Senescence of Activated Hepatic Stellate Cells to Limit Liver Fibrosis Progression. Cell Mol Gastroenterol Hepatol. 2022;14:75-99
158. Zhang J, Zhao L, Hu C, Wang T, Lu J, Wu C. et al. Fisetin Prevents Acetaminophen-Induced Liver Injury by Promoting Autophagy. Front Pharmacol. 2020;11:162
159. Li JH, Chen YY. A Fresh Approach to Targeting Aging Cells: CAR-T Cells Enhance Senolytic Specificity. Cell Stem Cell. 2020;27:192-4
160. Cheng N, Kim KH, Lau LF. Senescent hepatic stellate cells promote liver regeneration through IL-6 and ligands of CXCR2. JCI Insight. 2022
161. Raffaele M, Kovacovicova K, Frohlich J, Lo Re O, Giallongo S, Oben JA. et al. Mild exacerbation of obesity- and age-dependent liver disease progression by senolytic cocktail dasatinib + quercetin. Cell Commun Signal. 2021;19:44
162. Erbaba B, Arslan-Ergul A, Adams MM. Effects of caloric restriction on the antagonistic and integrative hallmarks of aging. Ageing Res Rev. 2021;66:101228
163. Martel J, Chang SH, Wu CY, Peng HH, Hwang TL, Ko YF. et al. Recent advances in the field of caloric restriction mimetics and anti-aging molecules. Ageing Res Rev. 2021;66:101240
164. Spadaro O, Youm Y, Shchukina I, Ryu S, Sidorov S, Ravussin A. et al. Caloric restriction in humans reveals immunometabolic regulators of health span. Science. 2022;375:671-7
165. Redman LM, Smith SR, Burton JH, Martin CK, Il'yasova D, Ravussin E. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018;27:805-15 e4
166. Willmann C, Heni M, Linder K, Wagner R, Stefan N, Machann J. et al. Potential effects of reduced red meat compared with increased fiber intake on glucose metabolism and liver fat content: a randomized and controlled dietary intervention study. Am J Clin Nutr. 2019;109:288-96
167. Rosqvist F, Kullberg J, Stahlman M, Cedernaes J, Heurling K, Johansson HE. et al. Overeating Saturated Fat Promotes Fatty Liver and Ceramides Compared With Polyunsaturated Fat: A Randomized Trial. J Clin Endocrinol Metab. 2019;104:6207-19
168. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577-85
169. Kulkarni AS, Gubbi S, Barzilai N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020;32:15-30
170. Hunt NJ, Lockwood GP, Kang SWS, Pulpitel T, Clark X, Mao H. et al. The Effects of Metformin on Age-Related Changes in the Liver Sinusoidal Endothelial Cell. J Gerontol A Biol Sci Med Sci. 2020;75:278-85
171. de Moraes ACN, de Andrade CBV, Ramos IPR, Dias ML, Batista CMP, Pimentel CF. et al. Resveratrol promotes liver regeneration in drug-induced liver disease in mice. Food Res Int. 2021;142:110185
172. Teng W, Zhao L, Yang S, Zhang C, Liu M, Luo J. et al. The hepatic-targeted, resveratrol loaded nanoparticles for relief of high fat diet-induced nonalcoholic fatty liver disease. J Control Release. 2019;307:139-49
173. Zeraattalab-Motlagh S, Jayedi A, Shab-Bidar S. The effects of resveratrol supplementation in patients with type 2 diabetes, metabolic syndrome, and nonalcoholic fatty liver disease: an umbrella review of meta-analyses of randomized controlled trials. Am J Clin Nutr. 2021;114:1675-85
174. Chachay VS, Macdonald GA, Martin JH, Whitehead JP, O'Moore-Sullivan TM, Lee P. et al. Resveratrol does not benefit patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2014;12:2092-103 e1-6
175. Poulsen MK, Nellemann B, Bibby BM, Stodkilde-Jorgensen H, Pedersen SB, Gronbaek H. et al. No effect of resveratrol on VLDL-TG kinetics and insulin sensitivity in obese men with nonalcoholic fatty liver disease. Diabetes Obes Metab. 2018;20:2504-9
176. Le Couteur DG, Solon-Biet SM, Parker BL, Pulpitel T, Brandon AE, Hunt NJ. et al. Nutritional reprogramming of mouse liver proteome is dampened by metformin, resveratrol, and rapamycin. Cell Metab. 2021;33:2367-79 e4
177. Bana B, Cabreiro F. The Microbiome and Aging. Annu Rev Genet. 2019;53:239-61
178. Haran JP, McCormick BA. Aging, Frailty, and the Microbiome-How Dysbiosis Influences Human Aging and Disease. Gastroenterology. 2021;160:507-23
179. Khan A, Ding Z, Ishaq M, Bacha AS, Khan I, Hanif A. et al. Understanding the Effects of Gut Microbiota Dysbiosis on Nonalcoholic Fatty Liver Disease and the Possible Probiotics Role: Recent Updates. Int J Biol Sci. 2021;17:818-33
180. Keebaugh ES, Ja WW. Breaking Down Walls: Microbiota and the Aging Gut. Cell Host Microbe. 2017;21:417-8
181. Santoro A, Zhao J, Wu L, Carru C, Biagi E, Franceschi C. Microbiomes other than the gut: inflammaging and age-related diseases. Semin Immunopathol. 2020;42:589-605
182. Bonora BM, Palano MT, Testa G, Fadini GP, Sangalli E, Madotto F. et al. Hematopoietic progenitor cell liabilities and alarmins S100A8/A9-related inflammaging associate with frailty and predict poor cardiovascular outcomes in older adults. Aging Cell. 2022;21:e13545
183. Passamonti L, Tsvetanov KA, Jones PS, Bevan-Jones WR, Arnold R, Borchert RJ. et al. Neuroinflammation and Functional Connectivity in Alzheimer's Disease: Interactive Influences on Cognitive Performance. J Neurosci. 2019;39:7218-26
184. D'Souza SS, Zhang Y, Bailey JT, Fung ITH, Kuentzel ML, Chittur SV. et al. Type I Interferon signaling controls the accumulation and transcriptomes of monocytes in the aged lung. Aging Cell. 2021;20:e13470
185. Delzenne NM, Bindels LB. Microbiome metabolomics reveals new drivers of human liver steatosis. Nat Med. 2018;24:906-7
186. Trebicka J, Bork P, Krag A, Arumugam M. Utilizing the gut microbiome in decompensated cirrhosis and acute-on-chronic liver failure. Nat Rev Gastroenterol Hepatol. 2021;18:167-80
187. Kolodziejczyk AA, Federici S, Zmora N, Mohapatra G, Dori-Bachash M, Hornstein S. et al. Acute liver failure is regulated by MYC- and microbiome-dependent programs. Nat Med. 2020;26:1899-911
188. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018 360
189. Cui B, Lin L, Wang B, Liu W, Sun C. Therapeutic potential of Saccharomyces boulardii in liver diseases: from passive bystander to protective performer? Pharmacol Res. 2022;175:106022
190. Chen QW, Li QR, Cao MW, Yan JH, Zhang XZ. Hierarchy-Assembled Dual Probiotics System Ameliorates Cholestatic Drug-Induced Liver Injury via Gut-Liver Axis Modulation. Adv Sci (Weinh). 2022;9:e2200986
191. Vaughn BP, Rank KM, Khoruts A. Fecal Microbiota Transplantation: Current Status in Treatment of GI and Liver Disease. Clin Gastroenterol Hepatol. 2019;17:353-61
192. Bajaj JS, Gavis EA, Fagan A, Wade JB, Thacker LR, Fuchs M. et al. A Randomized Clinical Trial of Fecal Microbiota Transplant for Alcohol Use Disorder. Hepatology. 2021;73:1688-700
193. Sharma A, Roy A, Premkumar M, Verma N, Duseja A, Taneja S. et al. Fecal microbiota transplantation in alcohol-associated acute-on-chronic liver failure: an open-label clinical trial. Hepatol Int. 2022;16:433-46
194. DeFilipp Z, Bloom PP, Torres Soto M, Mansour MK, Sater MRA, Huntley MH. et al. Drug-Resistant E. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N Engl J Med. 2019;381:2043-50
Author contact
![]() Corresponding author: Hua Wang, Ph.D., M.D. Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei, 230022, China. Tel/fax: +86-13505690896. Email address: wanghuaedu.cn
Corresponding author: Hua Wang, Ph.D., M.D. Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei, 230022, China. Tel/fax: +86-13505690896. Email address: wanghuaedu.cn