Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(6):1778-1790. doi:10.7150/ijbs.82217 This issue Cite
Research Paper
The mitochondrial protein YME1 Like 1 is important for non-small cell lung cancer cell growth
Yingchen Xia1*, Chunyan He2*, Zhi Hu3*, Zhichao Wu4*, Yin Hui5,6 ![]() , Yuan-yuan Liu7
, Yuan-yuan Liu7 ![]() , Chuanyong Mu8
, Chuanyong Mu8 ![]() , Jianhua Zha9
, Jianhua Zha9 ![]()
1. Department of Thoracic Surgery, The First Affiliated Hospital of University of Science and Technology of China (USTC), Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China.
2. Department of Clinical Laboratory, Kunshan Hospital of Chinese Medicine, Affiliated Hospital of Yangzhou University, Kunshan, China.
3. Department of Thoracic Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, China.
4. Department of Thoracic Surgery, Changshu Hospital Affiliated to Nanjing University of Chinese Medicine, Changshu, China.
5. Department of Thoracic Surgery, The First Affiliated Hospital of Shaoyang University, Shaoyang, China.
6. Department of Thoracic Surgery, The Central Hospital of Shaoyang, Shaoyang, China.
7. Department of Radiotherapy and Oncology, Affiliated Kunshan Hospital of Jiangsu University, Kunshan, China.
8. Department of Respiratory and Critical Care Medicine, First Affiliated Hospital of Soochow University, Suzhou, China.
9. Department of Thoracic Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, China.
*These authors contributed equally to this work.
Received 2022-12-28; Accepted 2023-3-3; Published 2023-3-21
Abstract

The expression and biological function of the mitochondrial inner membrane protease YME1L (YME1 Like 1 ATPase) in NSCLC are tested here. Bioinformatical analyses and results from local human tissues show that YME1L expression is elevated in NSCLC tissues. YME1L upregulation was observed in primary and immortalized NSCLC cells. In NSCLC cells, shRNA-mediated silence of YME1L or dCas9/sgRNA-induced knockout (KO) of YME1L robustly suppressed cell growth and migration, and provoking apoptosis. YME1L shRNA/KO resulted in mitochondrial dysfunctions in NSCLC cells, leading to mitochondrial depolarization, ROS accumulation and ATP depletion. Conversely, ectopic YME1L overexpression augmented NSCLC cell proliferation and motility. Akt-S6K1 phosphorylation was reduced after YME1L shRNA/KO in primary NSCLC cells, but augmented after YME1L overexpression. Importantly, YME1L KO-caused anti-NSCLC cell activity was attenuated by a constitutively-activate Akt1 (S473D) construct. In vivo, subcutaneous NSCLC xenograft growth was remarkably slowed following intratumoral YME1L shRNA AAV injection in nude mice. YME1L knockdown, Akt-mTOR inactivation and ATP reduction were detected in YME1L-silenced NSCLC xenografts. Taken together, overexpressed YME1L in NSCLC exerts pro-tumorigenic function.
Introduction
Over 13% of all new cancers are lung cancer [1, 2]. The most common non-small cell lung cancer (NSCLC) is one primary cause of global cancer mortality [3-5]. The advanced NSCLC has high metastatic potential and malignancy [3-5]. The present treatments, including surgical cancer resection, radiation, molecularly-targeted agents, immunotherapy (including PD1/PD-L1 blockers) and chemotherapies, have been failed significantly improve the prognosis of advanced NSCLC patients [3, 5].
The understanding of genetic alterations driving NSCLC is evolving in recent years [6-10]. For example, mutations of EGFR, BRAF and MET as well as translocations of ALK, RET and NTRK are currently incorporated in the diagnostic standards of NSCLC [4, 5]. Inhibitors and antibodies of these molecular targets have displayed promising efficiency in NSCLC patients, either alone or in combination of current chemotherapies [6-10]. Novel targeted therapies of NSCLC are being explored [8, 11-13].
Mitochondria are vital for oxidative phosphorylation (OXPHOS), ATP production and amino acid metabolism, as well as macromolecules biosynthesis, fatty acid oxidation and ion homeostasis [14-18]. Mitochondria act as the key hub for signaling transduction and apoptosis regulation [14-18]. Mitochondrial functions are altered in NSCLC and other human cancers. In particular, increased bioenergetics in the rapidly proliferating tumor cells can meet their energy demand by generating more ATP [14-18]. Increased mitochondrial respiration and ATP generation are vital for NSCLC tumorigenesis and progression [6, 19]. For example, enhanced synthesis and/or uptake of heme will fuel elevated OXPHOS in NSCLC. Whereas suppressing uptake/synthesis of heme inhibited mitochondrial OXPHOS and robustly reduced oxygen consumption, thereby suppressing NSCLC growth [6, 19]. Studies are focusing on understanding the mechanisms of mitochondrial alterations that are vital for tumorigenesis and development NSCLC [6, 14-19].
The mitochondrial protein YME1L (YME1 Like 1 ATPase) locates primarily at the inner mitochondrial (IM) membrane [20-24] and is important for maintaining mitochondrial morphology, functions and plasticity [22, 25, 26]. Studies revealed that YME1L can regulate the degradation of different mitochondrial proteins [20-22, 27]. Stiburek et al., showed that YME1L knockdown in HEK293 cells impaired cell proliferation and changed cristae morphology, while inducing oxidative injury and decreasing rotenone-sensitive respiration [23]. Silence of YME1L also induced accumulation of Ndufb6, ND1, Cox4 and other non-assembled respiratory chain subunits [23]. In the present study the expression and possible biological functions of YME1L in NSCLC were studied.
Methods
Reagents
All cell culture reagents were provided by Hyclone (Logan, UT, USA). All chemicals were obtained from Sigma-Aldrich Co. (St. Louis, Mo, USA). The YME1L antibody was provided by Dr. Cao [28, 29]. All other antibodies were reported previously [30, 31] or from Dr. Xu [32]. Fluorescence dyes, including JC-1, DAPI, EdU and TUNEL as well as CellROX, Hoechst 33342were all from Thermo-Fisher Invitrogen (Carlsbad, CA, USA). The Histone-bound DNA ELISA Kit was purchased from Roche Diagnostic (Indianapolis, IN, USA). The “Transwell” chambers were provided by Corning Co. (New York, NY, USA).
Cells
A549 cells were reported previously [30, 31] and were cultivated under RPMI medium plus serum. The primary NSCLC cells derived from three written-informed consent patients (pNSCLC-1, pNSCLC-2 and pNSCLC-3), the primary lung epithelial cells-derived from two written-informed consent donors were described early [30, 31], and cells cultured in medium described [30, 31]. The Ethics Committee of Nanchang University approved the the protocols of the present study.
Human tissues
Fifteen (15) fresh NSCLC tissues and the matched adjacent normal epithelial tissues were from primary written-informed consent NSCLC patients (stage-III-IV) in authors' institutions. Tissue slides were tested via immunohistochemistry (IHC) staining using the described protocols [32].
YME1L shRNA or overexpression
The lentivirus encoding YME1L shRNA ((5'-GATCCCCGTGGCAGAGGAATTCATATTTCAAGAGAATATGAGTTCCTCTGCCACTTTTTGGAAA-3') or YME1L cDNA were provided by Dr. Cao [28] and were added to the described cells. After 48h, cells were maintained under puromycin-containing medium for another four passages. YME1L silencing or overexpression was verified at mRNA and protein levels. For xenograft studies, YME1L shRNA sequence/shC sequence was inserted into an AAV (adeno-associated virus) vector [28, 29]. The construct and the AAV envelope plasmids were co-transfected to HEK-293 cells to generate shRNA-expressing AAV.
YME1L knockout (KO) by Cas9-sgRNA (single guide RNA) method
Cells were first transduced with a dCas9-expressing construct [33] to generate dCas9-expressing stable cells [34]. Next, the lentivirus with the sgRNA-CRISPR/dCas-9-YME1L-KO construct (Target DNA Sequence: ATGATGTCGATACAAGCAAG, PAM Sequence: AGG, provided by Dr. Cao [28]) was added to dCas9-expressing NSCLC cells, and stable cells established following selection and YME1L KO screening. The lenti-CRISPR/dCas-9 vector encoding the non-sense sgRNA (“Cas9C”) was transduced to the control cells.
Thiobarbituric acid reactive substance (TBAR) assaying of lipid peroxidation
Tissue lysates, at 20 proteins per sample, were measured using a commercial TBAR kit (Cayman Chemical, MI) specifically quantifying lipid peroxidation and malondialdehyde (MDA) contents colorimetrically. TBAR intensity was examined at 555 nm with the reference of 590 nm.
Other cell functional assays and gene/protein expression/interaction detection
NSCLC cells/epithelial cells with the designated YEM1L genetic treatment were seeded at optimized confluence and cultivated. CCK-8 cell viability, colony formation, the nuclear EdU/DAPI staining assay of cell proliferation, the caspase-3 activity assay, cell apoptosis detection by nuclear TUNEL/Hoechst 33342 staining, in vitro cell migration “Transwell” assays, JC-1 staining of mitochondrial depolarization were described in detail in our previous studies [30, 31]. Trypan blue staining of cell death and ssDNA (single strand DNA) ELISA (Merck, Shanghai, China) were described in a previous study [35], with the Histone-bound DNA ELISA assays described in another study [36]. Quantitative real time-PCR (qPCR), Western blotting and co-immunoprecipitation (Co-IP) was described early as well [30, 31]. CellROX staining of ROS content, tissue/cellular ATP contents and the mitochondrial complex I activity were measured using the described protocols [29, 37]. Figure S1 listed the uncropped blotting images.
Constitutively-active mutant Akt1 (caAkt1)
The caAkt1 (S473D)-expressing adenovirus (from Dr. Li [38, 39]) was added to cultured NSCLC cells for 48h. With selection the single stable cells were formed, and caAkt1 expression always verified.
Animal studies
The nude mice were purchased from the animal center described [30, 31]. The pNSCLC-1 cells (3 × 10 6 cells of each mouse) were subcutaneously (s.c.) injected to mice's right flanks. After 21 days the pNSCLC-1 xenograft tumors were formed, with tumors close to 100 mm3. The pNSCLC-1 xenograft mice were intratumorally injected with the described AAV (2.5 μL virus per xenograft, 0.85×10 9 PFU). Tumor xenografts were measured formulas described [30, 31]. All animal experiments were approved by Nanchang University's IACUC and Animal Ethics Board.
Statistical analysis
The detailed procedures of statistical analyses were reported in our previous studies [30, 31]. All in vitro experiments were repeated five times. Error bars were mean ± standard deviation (SD).
Results
YME1L expression is elevated in NSCLC
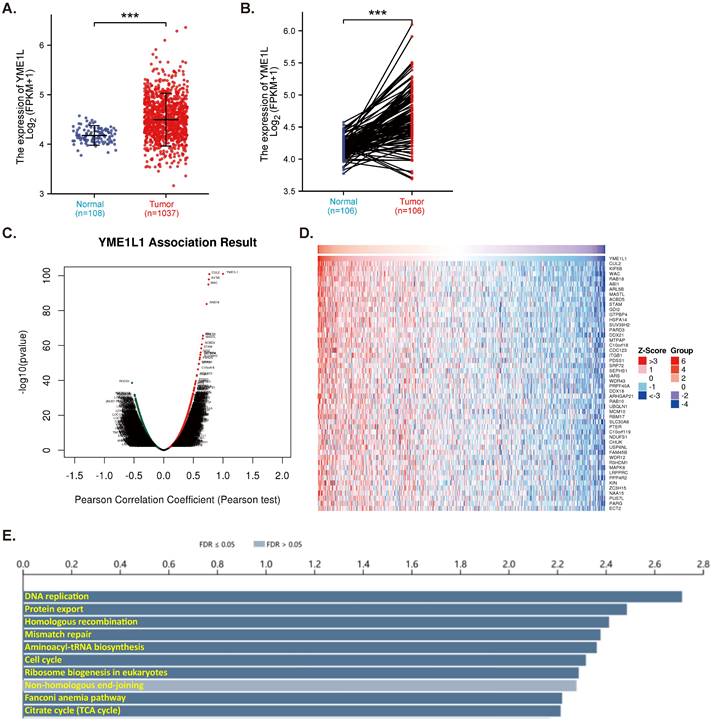
TCGA-LUAD database reveals that expression of YME1L is significantly elevated in NSCLC tissues (“Tumor”, Figure 1A). The relatively low YME1L expression was detected in lung tissues (“Normal”, Figure 1A). In NSCLC tumor tissues YME1L transcripts' number is robustly higher than its number in the adjacent normal tissues (Figure 1B). The mRNA sequencing data from a total of 515 NSCLC-LUAD patients in TCGA were analyzed using the LinkedOmics functional module. Figure 1C, the volcano plot, shows red dot genes that were positively correlated with YME1L, whereas green dot genes were negatively correlated with YME1L (false discovery rate/FDR < 0.01). The top fifty genes positively correlating with YME1L expression were presented in a heat map (Figure 1D). Significant KEGG term annotation by overrepresentation enrichment analysis (ORA) showed the top ten pathways enriched by YME1L-co-expressing genes (Figure 1E). Many of these pathways are vital for cancer progression, including DNA replication, mismatch repair, cell cycle progression and citrate cycle (TCA cycle) (Figure 1E). These bioinformatics studies show that overexpressed YME1L might exert a tumorigenic role in NSCLC.
YME1L expression is elevated in local NSCLC
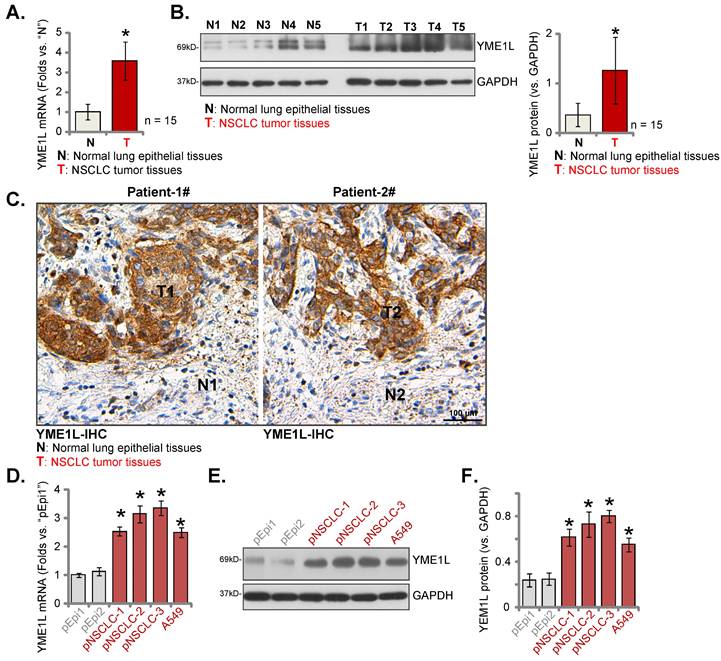
YME1L expression was measured in local surgery-resection NSCLC tissues. NSCLC tissues (“T”) and matched adjacent normal tissues (“N”) were obtained from fifteen (n = 15) primary NSCLC patients (LUAD, stage III-IV). Analyzing tissue lysates confirmed that YME1L mRNA levels in NSCLC tumor tissues were dramatically higher than those in normal tissues (Figure 2A). Moreover, increased protein expression of YME1L was observed in NSCLC tissues of five patients (“T1” to “T5”) (Figure 2B). Quantified results combining YME1L protein blotting data of the 15 sets of tissues demonstrated that the protein expression of YME1L was significantly elevated in NSCLC tissues (Figure 2B, the right panel). The tissue IHC images further supported the protein upregulation of YME1L in NSCLC tissues (“T1” and “T2”) of “Patient-1# and Patient-2#” (Figure 2C). The protein expression of YME1L in normal lung epithelial tissues was low (Figure 2C).
In the primary pNSCLC-1, pNSCLC-2 and pNSCLC-3 cells [30, 31] and the immortalized A549 cells, YME1L mRNA expression was higher than its expression in “pEpi1” and “pEpi2” primary lung epithelial cells (Figure 2D). The protein expression of YME1L was upregulated in the NSCLC cells (Figure 2E and F), and low expression detected in the lung epithelial cells (Figure 2E and F). Thus, elevated YME1L expression is detected in local NSCLC tissues and NSCLC cells.
YME1L expression is elevated in NSCLC. TCGA-LUAD cohort showed YME1L expression (RNA-Seq) in 1037 NSCLC tissues (“Tumor”) and 108 normal lung tissues (“Normal” (A). TCGA-LUAD cohort showed YME1L expression in 106 NSCLC tissues (“Tumor”) and matched 106 adjacent normal tissues (“Normal”) (B). LinkedOmics functional assays demonstrated the YME1L-co-expressed genes (C). Top 50 co-expressed genes positively correlated with YME1L (D) and top the enriched pathways (through KEGG, E) were shown. *** P < 0.001 (A and B).
YME1L depletion induces robust anti-NSCLC cell activity
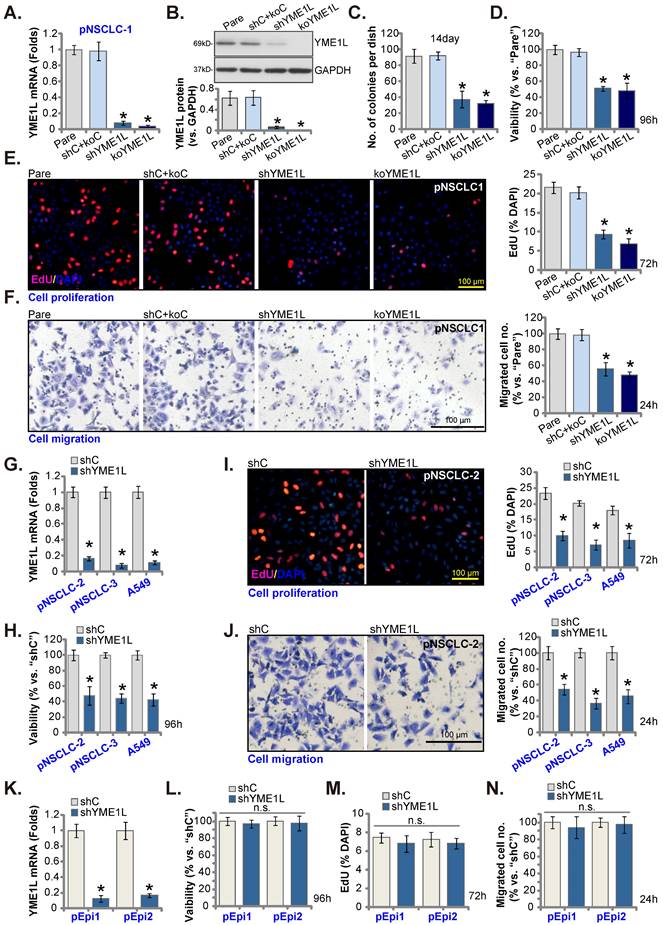
Aiming to knockdown YME1L, the YME1L shRNA-encoding lentivirus was added to pNSCLC-1 primary cells [30, 31], and stable cells, “shYME1L” cells, formed following selection. Alternatively, the CRISPR/dCas9-YME1L-KO construct-expressing lentivirus was added to dCas9-expressing pNSCLC-1 cells, and single stable “koYME1L” cells formed following selection and YME1L KO verification. When compared to pNSCLC-1 cells with the non-sense scramble control shRNA and the CRISPR/dCas9-KO control construct (“shC+koC”), YME1L mRNA (Figure 3A) and protein (Figure 3B) were substantially decreased in shYME1L and koYME1L pNSCLC-1 cells. As a result, cell colony formation ability was robustly inhibited (Figure 3C). YME1L shRNA/KO also potently decreased viability in pNSCLC-1 cells (Figure 3D). Further studies showed that depletion of YME1L significantly inhibited pNSCLC-1 cell proliferation by decreasing the percentage of EdU nuclei (Figure 3E). Furthermore, pNSCLC-1 cell motility was suppressed after YME1L depletion. The in vitro cell migration (Figure 3F) was largely inhibited in shYME1L and koYME1L cells. Therefore, depletion of YME1L inhibited pNSCLC-1 cell survival, proliferation and motility.
Next, to pNSCLC-2/3 cells and immortalized A549 cells, the lentivirus with YME1L shRNA was added and stable “shYME1L” cells were formed following selection. Expression of YME1L mRNA was indeed robustly decreased in the shYME1L NSCLC cells (Figure 3G). Silence of YME1L in the NSCLC cells decreased viability (Figure 3H), arrested cell proliferation (Figure 3I) and hindered in vitro cell migration (Figure 3J).
The pEpi1 and pEpi2 primary epithelial cells [30, 31] were also transduced with the lentiviral YME1L shRNA (“shYME1L”). The latter led to dramatic YME1L silencing (Figure 3K). Yet YME1L shRNA failed to significantly inhibit viability (Figure 3L), EdU incorporation (Figure 3M) and in vitro cell migration (Figure 3N) in pEpi1/2 epithelial cells.
YME1L depletion provokes NSCLC cell apoptosis
We tested whether YME1L depletion could induce apoptosis activation. As shown, in shYME1L and koYME1L pNSCLC-1 cells (see Figure 3), Caspase-3 activity was significantly increased when compared to that in shC+koC control cells (Figure 4A). Moreover, in pNSCLC-1 cells YME1L shRNA/KO caused Caspase-3, PARP1 and Caspase-9 cleavages (Figure 4B). The contents of Histone-bound DNA were increased in YME1L-silenced/-KO pNSCLC-1 cells (Figure 4C). Importantly, following YME1L shRNA/KO, the ratio of nuclei with TUNEL staining was significantly increased, supporting apoptosis induction (Figure 4D and E). Increased cell death was detected in shYME1L and koYME1L pNSCLC-1 cells and Trypan blue-positive staining was significantly increased (Figure 4F).
YME1L expression is elevated in local NSCLC. YME1L expression in the descried NSCLC tumor tissues (“T”) and matched adjacent normal lung epithelial tissues (“N”) of fifteen (n = 15) primary local NSCLC patients was examined (A and B); YME1L IHC images in the described tissue slides were shown (C). YME1L expression in the descried NSCLC cells/epithelial cells was measured (D-F). * P < 0.05 versus “N” tissues or “pEpi1” cells. Scale bar = 100 μm.
YME1L depletion induces robust anti-NSCLC cell activity. YME1Lexpression in pNSCLC-1cells with the described genetic modification of YME1L or with control treatment was shown (A and B), and GAPDH tested as the internal control. Cells were cultivated, clonogenicity (C), CCK-8 viability (D), EdU incorporation (E) and cell migration (F) were measured. pNSCLC-2/3 cells, A549 cells, or pEpi1/2 epithelial cells, stably expressing YME1L shRNA (“shYME1L”) or control shRNA (“shC”) were formed, and YME1L mRNA expression examined (G and K). Cells were cultivated, CCK-8 viability (H and L), nuclear EdU incorporation (I and M) and migration (J and N) were measured, with results quantified. “Pare” stands for the parental control cells (same for all Figures). * P < 0.05 versus “Pare”/ “shC”. “n.s.” stands for P > 0.05 (same for all Figures). Scale bar=100 μm.
In pNSCLC-2/3 cells and A549 cells, YME1L shRNA-induced silence of YME1L augmented Caspase-3 activity (Figure 4G) and increased TUNEL nuclei ratio (Figure 4H and I). Moreover, YME1L silencing provoked cell death and increased Trypan blue staining in the NSCLC cells (Figure 4J). Whereas in pEpi1 and pEpi2 lung epithelial cells, YME1L knockdown by YME1L shRNA (see Figure 3) failed to provoke apoptosis (Figure 4K and L).
YME1L depletion provokes NSCLC cell apoptosis. pNSCLC-1cells with the described genetic modification of YME1L or with control treatment were cultivated, the caspase-3 activity was tested (A), and listed proteins measured (B); The histone-DNA contents were tested (C). Cell apoptosis was tested via the nuclear TUNEL staining assay (D and E), and cell death measured by the Trypan blue staining assay (F). pNSCLC-2/3 cells, A549 cells, or pEpi1/2 epithelial cells, stably expressing YME1L shRNA (“shYME1L”) or control shRNA (“shC”) were cultured, the caspase-3 activity was measured (G and K), with cell apoptosis measured via the TUNEL staining assays (H, I and L). Cell death was measured as well (J). * P < 0.05 versus “Pare”/ “shC” cells. Scale bar=100 μm.

NSCLC cell mitochondrial functions are impaired after YME1L depletion
YME1L locates in the inner mitochondrial membrane [20-24]. It maintains mitochondrial morphology and is vital for mitochondrial function and plasticity [22, 25, 26]. We therefore analyzed whether mitochondrial functions were impaired in YME1L-depleted NSCLC cells. As shown, the CellROX red fluorescence intensity was substantially increased in shYME1L and koYME1L pNSCLC-1 cells, supporting ROS accumulation (Figure 5A). In addition, YME1L shRNA/KO resulted in depolarization of mitochondria in pNSCLC-1 cells, causing JC-1 red fluorescence conversion to green JC-1 monomers (Figure 5B). Furthermore, ssDNA accumulation and increased DNA breaks were detected in YME1L-depleted pNSCLC-1 cells (Figure 5C). Increased ATM and ATR phosphorylation further supported DNA damage in YME1L-depleted cells (Figure 5D). The activity of mitochondrial complex I was substantially decreased in shYME1L and koYME1L pNSCLC-1 cells (Figure 5E). Consequently, the ATP contents were decreased (Figure 5F).
In pNSCLC-2/3 cells and A549 cells, YME1L shRNA also induced ROS production and increased the CellROX red fluorescence intensity (Figure 5G). Moreover, depolarization of mitochondria, reflected by green monomer JC-1 accumulation, was detected in YME1L-silenced primary and A549 NSCLC cells (Figure 5H). In addition, ATP depletion was detected in the NSCLC cells with YME1L silencing (Figure 5I). Thus, YME1L depletion impaired mitochondrial functions in NSCLC cells.
Ectopic YME1L overexpression further promotes NSCLC cell growth
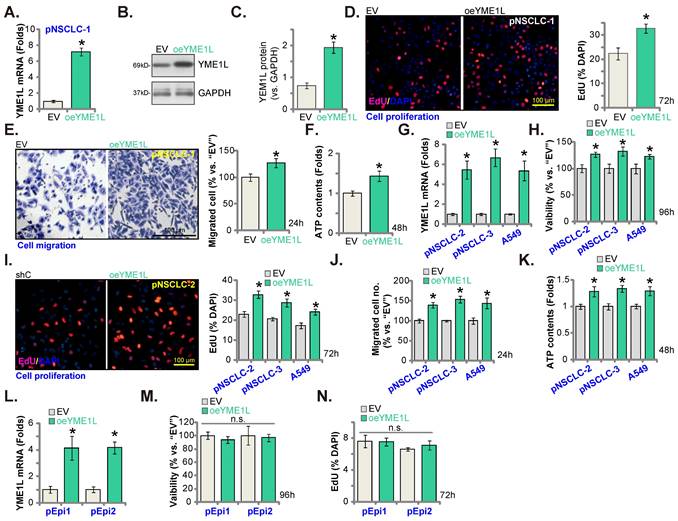
The lentivirus-packed YME1L-expressing construct was stably transduced to pNSCLC-1 cells, establishing YME1L-ovexpressed cells (“oeYME1L”). Comparing to the empty vector (“EV”)-expressing pNSCLC-1, expression of YME1L mRNA (Figure 6A) and protein (Figure 6B and C) was substantially elevated in the oeYME1L pNSCLC-1 cells. YME1L overexpression potentiated pNSCLC-1 cell proliferation and augmented nuclear EdU incorporation (Figure 6D). Moreover, the in vitro cell migration (Figure 6E) was augmented and ATP content (Figure 6F) was increased in oeYME1L cells.
The lentivirus-packed YME1L-expressing construct was also transduced to pNSCLC-2/pNSCLC-3 and A549 cells. Thereafter YME1L-ovexpressed NSCLC cells (“oeYME1L”) were formed, showing significantly-elevated YME1L mRNA expression (Figure 6G). With overexpression of YME1L, cell viability (Figure 6H) and proliferation (Figure 6I) were augmented in the NSCLC cells. Moreover, the in vitro migration was accelerated (Figure 6J), and the cellular ATP content increased in oeYME1L NSCLC cells (Figure 6K). To lung epithelial cells (pEpi1 and pEpi2), the lentivirus-packed YME1L-expressing construct was transduced and YME1L-ovexpressed stable epithelial cells (“oeYME1L”) were formed. YME1L mRNA expression was elevated in oeYME1L lung epithelial cells (Figure 6L). YME1L overexpression however failed to increase viability (Figure 6M) and nuclear EdU incorporation (Figure 6N) in the lung epithelial cells.
YME1L depletion in NSCLC cells disrupts mTOR complex assembling and Akt-mTOR activation
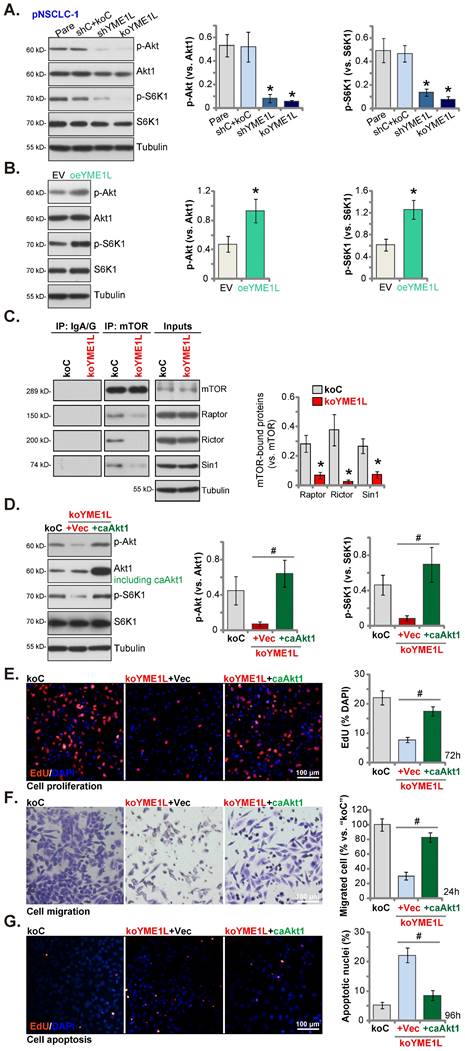
The mitochondrial function is required for the activation of Akt-mTOR and other pro-cancerous oncogenic cascades in NSCLC [40, 41]. In pNSCLC-1 cells, shRNA-induced silence of YME1L (“shYME1L”) or dCas9/sgRNA-mediated KO of YME1L (“koYME1L”) remarkably decreased phosphorylation of Akt (Ser-473) and S6K1 (Figure 7A). Total Akt1 and S6K1 were unchanged in YME1L-silenced/-KO pNSCLC-1 cells (Figure 7A). Contrarily, in YME1L-overexpressing pNSCLC-1 cells (“oeYME1L”) Akt (Ser-473) and S6K1 phosphorylation were augmented (Figure 7B).
NSCLC cell mitochondrial functions are impaired after YME1L depletion. pNSCLC-1cells with the described genetic modification of YME1L or with control treatment were cultivated for 48h, CellROX intensity (A), depolarization of mitochondria (JC-1 monomer intensity, (B), DNA breaks (ssDNA contents, (C) and ATM/ATR phosphorylation and expression (D) were tested. The activity of mitochondrial complex I (E) and intracellular ATP contents (F) were examined as well. pNSCLC-2/3 cells, A549 cells, or pEpi1/2 epithelial cells, stably expressing YME1L shRNA (“shYME1L”) or control shRNA (“shC”) were cultured for 48h, CellROX intensity (G), JC-1 monomer intensity (H) and ATP levels (I) were measured. * P < 0.05 versus “Pare”/ “shC” cells. Scale bar=100 μm.

Ectopic YME1L overexpression further promotes NSCLC cell growth. pNSCLC-1/2/3 cells (A-K), A549 cells (G-K) or pEpi1/2 epithelial cells (L-N), stably expressing the lentivirus-packed YME1L-expressing construct (“oeYME1L”) or the vector (“EV”) were formed, mRNA and protein expression of YME1L was measured (A, B, C, G and L); After further cell culturing, CCK-8viability (H and M), nuclear EdU incorporation (D, I and N) and in vitro migration (E and J) were examined, with the cellular ATP contents measured (F and K). * P < 0.05 versus “EV” cells. Scale bar=100 μm.
There are two mTOR complexes, mTORC1 and mTORC2 [42, 43]. mTORC1, a complex including mTOR, mLST8, Raptor, and several others, is responsible for phosphorylating S6K1 and 4E-BP1 [42, 43]. mTORC2 is composed of mTOR, mSIN1, Rictor and mLST8 [44-46], and phosphorylates Akt (at Ser-473) [47, 48]. Both mTOR complexes are important for NSCLC tumorigenesis and progression [30, 39, 49, 50]. Here the co-immunoprecipitation assay results showed that the assembles mTORC1 (association of mTOR-Raptor) and mTORC2 (association of mTOR-Rictor-mSin1) were disrupted in YME1L-KO pNSCLC-1 cells (Figure 7C). mTOR, Raptor, mSin1 and Rictor protein expression however unchanged with YME1L KO (Figure 7C, “Inputs”). Thus, disruption of the assembling of mTORC1/2 could be the primary mechanism of Akt-mTOR inactivation by YME1L depletion in NSCLC cells.
YME1L depletion in NSCLC cells disrupts mTOR complex assembling and Akt-mTOR activation. Listed proteins in pNSCLC-1cells with the described genetic modification of YME1L or with control treatment were tested (A and B). The mTOR-immunoprecipitated proteins (Raptor, Rictor and mSin1) were measured by Co-IP assays, with expression of the described proteins measured in “Inputs” (C). The koYME1L pNSCLC-1 cells were further stably transduced with caAkt1 (S473D) or the vector (“Vec”), with the listed proteins tested (D); After further culturing nuclear EdU incorporation (E), in vitro migration (F) and apoptosis (TUNEL-nuclei staining, (G) were tested. * P < 0.05 versus “shC+Cas9-C”/“EV” cells. # P < 0.05. Scale bar=100 μm.
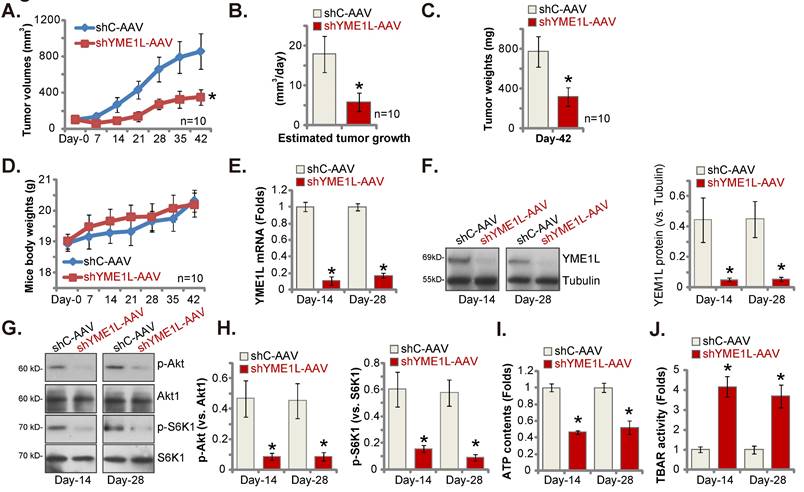
Silence of YME1L inhibits NSCLC xenograft growth. ThepNSCLC-1 xenograft nude mice were injected with the YME1L shRNA AAV (“shYME1L-AAV”) or control shRNA AAV (“shC-AAV”). The virus was intratumorally injected every 48h for a total of five rounds. The weekly tumor volumes (A) and mice body weights (D) were weekly recorded. The estimated daily pNSCLC-1 xenograft growth was shown (B). At Day-42, pNSCLC-1 xenografts were carefully isolated and individually weighted (C). Listed mRNAs and proteins in the described pNSCLC-1 xenografts were measured (E-H), with ATP contents (I) and TBAR (J) activity tested as well. * P < 0.05 “shC-AAV” group. Scale bar=100 μm.
Next a caAkt1 (S473D) construct [30, 31] was stably transduced to koYME1L pNSCLC-1 cells (Figure 7D) and completely restored Akt (Ser-473) and S6K1 phosphorylation in the YME1L KO cells (Figure 7D). Significantly YME1L KO-induced proliferation (EdU incorporation) suppression (Figure 7E), migration inhibition (Figure 7F) and apoptosis (TUNEL nuclei increasing, Figure 7G) in pNSCLC-1 cells were ameliorated by caAkt1.
Silence of YME1L inhibits NSCLC xenograft growth
Lastly, pNSCLC-1 cells were subcutaneously (s.c.) injected to nude mice. The pNSCLC-1 xenograft tumors were formed after 21 days (“Day-0”). The YME1L shRNA-expressing AAV (“shYME1L-AAV”) were then intratumorally injected to the nude mice. Whereas in control mice the control shRNA AAV (“shC-AAV”) was injected. Virus injection was carried every 48h for five rounds. shYME1L-AAV injection robustly hindered pNSCLC-1 xenograft growth (Figure 8A). The estimated daily pNSCLC-1 xenograft growth, expressed as mm3 per day, was calculated [30, 31]. The results again showed that shYME1L-AAV treatment suppressed pNSCLC-1 xenograft growth (Figure 8B). All the pNSCLC-1 xenografts were isolated carefully at Day-42 and individually weighted. The shYME1L-AAV-injected pNSCLC-1 xenografts injection were substantially lighter than the shC-AAV xenografts (Figure 8C). When comparing the animal body weights, we failed to detect any significant difference between the two groups (Figure 8D).
Signaling changes in the pNSCLC-1 xenografts were examined. Specifically at Day-14 and Day-28, one pNSCLC-1 xenograft from the shYME1L-AAV group and the shC-AAV group was carefully isolated. Part of the fresh tumor xenografts were cut into five pieces and signalings were tested. YME1L expression was remarkably decreased in shYME1L-AAV pNSCLC-1 xenograft tissues (Figure 8E and F), where Akt (Ser-473) and S6K1 phosphorylation was robustly decreased (Figure 8G and H). Total Akt1 and S6K1 was again unaffected by shYME1L-AAV (Figure 8G and H). Moreover, ATP reduction was detected in shYME1L-AAV xenografts (Figure 8I). Lipid peroxidation, or increased TBAR activity, was detected in YME1L-silenced pNSCLC-1 xenograft tissues (Figure 8J).
Discussion
Srinivasainagendra et al., reported that in human colorectal cancer YME1L could be frequently mutated, and its mutation also occurring in other human cancers to a less degree [51]. YME1L inhibition led to significant death of cancer cells [23, 52]. Silence of YME1L caused accumulation of Ndufb6, ND1, and Cox4, thereby suppressing cell proliferation [23].
Recent studies have proposed a possible tumorigenic role of YME1L. Liu et al., have shown that overexpressed YME1L is important for orthotopic glioma xenograft growth in mice [28]. The same group further reported that TIMM44, another mitochondrial protein, promoted glioma cell growth possibly by increasing YME1L transcription and expression [29]. Liao et al., have implied that YME1L could be a promising biomarker for diagnosis and prognosis prediction in ovarian cancer [53]. YME1L is upregulated in ovarian cancer and is associated with worse overall survival [53]. Moreover, YME1L and it co-expressing genes are enriched in immune-related signaling pathways, supporting a possible inhibitory role of YEM1L in cancer immunotherapy [53]. Kakehashi et al., reported that expression of YME1L, together with other cytoskeletal proteins involved in endoplasmic reticulum stresses and mitochondrial dysfunctions, are overexpressed in HCV-associated hepatocellular carcinomas (HCC) [54].
Increased mitochondrial respiration and ATP generation are extremely important for NSCLC tumorigenesis and progression. Here we found that the mitochondrial protein YME1L exerted tumorigenic activity in NSCLC. TCGA database and local human tissues/cells results demonstrated that YME1L expression is elevated in NSCLC tissues and cells. YME1L shRNA or KO potently suppressed NSCLC cell viability, proliferation and in vitro migration, and provoking apoptosis. In addition, YME1L depletion caused mitochondrial dysfunctions, leading to depolarization of mitochondria, oxidative injury, DNA breaks and ATP depletion in different NSCLC cells. In vivo, the growth of subcutaneous primary NSCLC xenografts was hindered following YME1L shRNA AAV injection in nude mice. ATP reduction and oxidative injury were observed in YME1L-depleted xenograft tissues.
Due to mutation and other genetic alterations, increased activation of PI3K-Akt-mTOR cascade is often detected in NSCLC [7, 10, 55-57]. Conversely, small molecular inhibitors or genetic modifications that can inactivate this cascade have shown promising anti-NSCLC efficiency [7, 10, 55-57]. PQR620, a mTOR kinase inhibitor, prevented mTORC1/2 activation and arrested NSCLC cell growth [30]. ASP4132, the highly effective AMPK activator, suppressed NSCLC cell growth possibly via inhibiting Akt-mTOR signaling [31].
YME1L was recently shown to promote Akt activation [28]. Here activation of Akt-mTOR was reduced after YME1L shRNA/KO in primary NSCLC cells. It was augmented after ectopic YME1L overexpression. Our results supported that YME1L should be important for the integrity of mTORC1/2 and YME1L depletion disrupted the assembling of mTORC1/2. Importantly, YME1L KO-mediated anti-NSCLC cell activities, including proliferation arrest, migration inhibition, and apoptosis, were largely ameliorated by caAkt1. Akt-mTOR inhibition was observed in YME1L-silenced NSCLC xenograft tissues. Thus, Akt-mTOR activation is important for YME1L-promoted NSCLC cell growth.
Conclusion
The mitochondrial protein YME1L protein is overexpressed in NSCLC and exerts significant pro-tumorigenic activity possibly by supporting mitochondrial function and promoting Akt-mTOR activation.
Supplementary Material
Supplementary figures.
Acknowledgements
Funding Statement
This study was supported by NSCF.
Author Contributions
All authors performed experiments, organized data, drafted the article and revised it critically for important intellectual content, and with final approval of the version submitted to the journal.
Ethics Statement
This study was approved by the Ethics Committee of Nanchang University.
Data Availability Statement
All data are available upon request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7-34
3. Arbyn M, Weiderpass E, Bruni L, de Sanjose S, Saraiya M, Ferlay J. et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Health. 2020;8:e191-e203
4. Hiley CT, Le Quesne J, Santis G, Sharpe R, de Castro DG, Middleton G. et al. Challenges in molecular testing in non-small-cell lung cancer patients with advanced disease. Lancet. 2016;388:1002-11
5. Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet. 2021;398:535-54
6. Ghosh P, Vidal C, Dey S, Zhang L. Mitochondria Targeting as an Effective Strategy for Cancer Therapy. Int J Mol Sci. 2020 21
7. Heavey S, O'Byrne KJ, Gately K. Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC. Cancer Treat Rev. 2014;40:445-56
8. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446-54
9. Jonna S, Subramaniam DS. Molecular diagnostics and targeted therapies in non-small cell lung cancer (NSCLC): an update. Discov Med. 2019;27:167-70
10. Vestergaard HH, Christensen MR, Lassen UN. A systematic review of targeted agents for non-small cell lung cancer. Acta Oncol. 2018;57:176-86
11. Metzger-Filho O, Moulin C, Awada A. Molecular targeted therapy in prevalent tumors: learning from the past and future perspectives. Curr Clin Pharmacol. 2010;5:166-77
12. Forde PM, Brahmer JR, Kelly RJ. New strategies in lung cancer: epigenetic therapy for non-small cell lung cancer. Clin Cancer Res. 2014;20:2244-8
13. Horn L, Reck M, Spigel DR. The Future of Immunotherapy in the Treatment of Small Cell Lung Cancer. Oncologist. 2016;21:910-21
14. Porporato PE, Filigheddu N, Bravo-San Pedro JM, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Research. 2018;28:265-80
15. Burke PJ. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer. 2017;3:857-70
16. Vyas S, Zaganjor E, Haigis MC. Mitochondria and Cancer. Cell. 2016;166:555-66
17. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9-15
18. Viale A, Corti D, Draetta GF. Tumors and mitochondrial respiration: a neglected connection. Cancer Res. 2015;75:3685-6
19. Sohoni S, Ghosh P, Wang T, Kalainayakan SP, Vidal C, Dey S. et al. Elevated Heme Synthesis and Uptake Underpin Intensified Oxidative Metabolism and Tumorigenic Functions in Non-Small Cell Lung Cancer Cells. Cancer Res. 2019;79:2511-25
20. Hartmann B, Wai T, Hu H, MacVicar T, Musante L, Fischer-Zirnsak B. et al. Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. Elife. 2016 5
21. Rainbolt TK, Saunders JM, Wiseman RL. YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Rep. 2015;16:97-106
22. Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E. et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. 2014;204:919-29
23. Stiburek L, Cesnekova J, Kostkova O, Fornuskova D, Vinsova K, Wenchich L. et al. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol Biol Cell. 2012;23:1010-23
24. Coppola M, Pizzigoni A, Banfi S, Bassi MT, Casari G, Incerti B. Identification and characterization of YME1L1, a novel paraplegin-related gene. Genomics. 2000;66:48-54
25. Ohba Y, MacVicar T, Langer T. Regulation of mitochondrial plasticity by the i-AAA protease YME1L. Biol Chem. 2020;401:877-90
26. MacVicar T, Ohba Y, Nolte H, Mayer FC, Tatsuta T, Sprenger HG. et al. Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature. 2019;575:361-5
27. Van Dyck L, Langer T. ATP-dependent proteases controlling mitochondrial function in the yeast Saccharomyces cerevisiae. Cell Mol Life Sci. 1999;56:825-42
28. Liu F, Chen G, Zhou L-N, Wang Y, Zhang Z-q, Qin X. et al. YME1L overexpression exerts pro-tumorigenic activity in glioma by promoting Gαi1 expression and Akt activation. Protein & Cell. 2022 doi.org/10.1093/procel/pwac011
29. Guo YZ, Chen G, Huang M, Wang Y, Liu YY, Jiang Q. et al. TIMM44 is a potential therapeutic target of human glioma. Theranostics. 2022;12:7586-602
30. Zha JH, Xia YC, Ye CL, Hu Z, Zhang Q, Xiao H. et al. The Anti-Non-Small Cell Lung Cancer Cell Activity by a mTOR Kinase Inhibitor PQR620. Front Oncol. 2021;11:669518
31. Xia YC, Zha JH, Sang YH, Yin H, Xu GQ, Zhen J. et al. AMPK activation by ASP4132 inhibits non-small cell lung cancer cell growth. Cell Death Dis. 2021;12:365
32. Zhang JZ, Liu J, Xu YX, Pu WY, Shen MJ, Jiang KQ. et al. Identification of the mitochondrial protein ADCK2 as a therapeutic oncotarget of NSCLC. Int J Biol Sci. 2022;18:6163-75
33. Wang Y, Liu F, Wu J, Zhang MQ, Chai JL, Cao C. G protein inhibitory alpha subunit 2 is a molecular oncotarget of human glioma. Int J Biol Sci. 2023;19:865-79
34. Yang J, Xia A, Zhang H, Liu Q, You H, Ding D. et al. Up-Regulating ERIC by CRISPR-dCas9-VPR Inhibits Cell Proliferation and Invasion and Promotes Apoptosis in Human Bladder Cancer. Frontiers In Molecular Biosciences. 2021;8:654718
35. Gao YY, Ling ZY, Zhu YR, Shi C, Wang Y, Zhang XY. et al. The histone acetyltransferase HBO1 functions as a novel oncogenic gene in osteosarcoma. Theranostics. 2021;11:4599-615
36. Xing ZY, Wang Y, Cheng L, Chen J, He XZ, Xing W. Bromodomain-Containing Protein 4 (BRD4) Inhibition Sensitizes Palomid 529-Induced Anti-Renal Cell Carcinoma Cell Activity in vitro and in vivo. Cell Physiol Biochem. 2018;50:640-53
37. Yin DP, Zheng YF, Sun P, Yao MY, Xie LX, Dou XW. et al. The pro-tumorigenic activity of p38γ overexpression in nasopharyngeal carcinoma. 2022; Cell Death Dis.
38. Zhang D, Xia H, Zhang W, Fang B. The anti-ovarian cancer activity by WYE-132, a mTORC1/2 dual inhibitor. Tumour Biol. 2016;37:1327-36
39. Yang H, Zhao J, Zhao M, Zhao L, Zhou LN, Duan Y. et al. GDC-0349 inhibits non-small cell lung cancer cell growth. Cell Death Dis. 2020;11:951
40. Liu X, Jiang Q, Liu H, Luo S. Vitexin induces apoptosis through mitochondrial pathway and PI3K/Akt/mTOR signaling in human non-small cell lung cancer A549 cells. Biol Res. 2019;52:7
41. Han SY, Jeong YJ, Choi Y, Hwang SK, Bae YS, Chang YC. Mitochondrial dysfunction induces the invasive phenotype, and cell migration and invasion, through the induction of AKT and AMPK pathways in lung cancer cells. Int J Mol Med. 2018;42:1644-52
42. Zaytseva YY, Valentino JD, Gulhati P, Evers BM. mTOR inhibitors in cancer therapy. Cancer Lett. 2012;319:1-7
43. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274-93
44. Huang TT, Sarkaria SM, Cloughesy TF, Mischel PS. Targeted therapy for malignant glioma patients: lessons learned and the road ahead. Neurotherapeutics. 2009;6:500-12
45. Lefranc F, Rynkowski M, DeWitte O, Kiss R. Present and potential future adjuvant issues in high-grade astrocytic glioma treatment. Adv Tech Stand Neurosurg. 2009;34:3-35
46. Li XM, Wu CJ, Chen NC, Gu HD, Yen A, Cao L. et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016;7:33440-50
47. Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010;7:209-19
48. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729-34
49. Li C, Dong Y, Wang L, Xu G, Yang Q, Tang X. et al. Ginsenoside metabolite compound K induces apoptosis and autophagy in non-small cell lung cancer cells via AMPK-mTOR and JNK pathways. Biochem Cell Biol. 2019;97:406-14
50. Zhang G, Wang C, Sun M, Li J, Wang B, Jin C. et al. Cinobufagin inhibits tumor growth by inducing intrinsic apoptosis through AKT signaling pathway in human nonsmall cell lung cancer cells. Oncotarget. 2016;7:28935-46
51. Srinivasainagendra V, Sandel MW, Singh B, Sundaresan A, Mooga VP, Bajpai P. et al. Migration of mitochondrial DNA in the nuclear genome of colorectal adenocarcinoma. Genome Med. 2017;9:31
52. Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P. et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. 2015;350:aad0116
53. Liao WT, Chu PY, Su CC, Wu CC, Li CJ. Mitochondrial AAA protease gene associated with immune infiltration is a prognostic biomarker in human ovarian cancer. Pathol Res Pract. 2022;240:154215
54. Kakehashi A, Suzuki S, Shiota M, Raymo N, Gi M, Tachibana T. et al. Canopy Homolog 2 as a Novel Molecular Target in Hepatocarcinogenesis. Cancers (Basel). 2021 13
55. Tan AC. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac Cancer. 2020;11:511-8
56. Perez-Ramirez C, Canadas-Garre M, Molina MA, Faus-Dader MJ, Calleja-Hernandez MA. PTEN and PI3K/AKT in non-small-cell lung cancer. Pharmacogenomics. 2015;16:1843-62
57. Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem Pharmacol. 2014;90:197-207
Author contact
![]() Corresponding authors: Dr. Jianhua Zha, Department of Thoracic Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, China. Email: zhajianhuancu.edu.cn; Dr. Chuan-yong Mu, Department of Respiratory and Critical Care Medicine, the First Affiliated Hospital of Soochow University, Suzhou, Jiangsu, China. Email: mu_chuan_yongcom; Dr. Yin Hui, Department of Thoracic Surgery, The First Affiliated Hospital of Shaoyang University, Shaoyang, China. Yin Hui Email: 191618538com; Dr. Yuan-yuan Liu, Department of Radiotherapy and Oncology, Affiliated Kunshan Hospital of Jiangsu University, Kunshan, China. E-mail: liuyuanyuanszcom.
Corresponding authors: Dr. Jianhua Zha, Department of Thoracic Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, China. Email: zhajianhuancu.edu.cn; Dr. Chuan-yong Mu, Department of Respiratory and Critical Care Medicine, the First Affiliated Hospital of Soochow University, Suzhou, Jiangsu, China. Email: mu_chuan_yongcom; Dr. Yin Hui, Department of Thoracic Surgery, The First Affiliated Hospital of Shaoyang University, Shaoyang, China. Yin Hui Email: 191618538com; Dr. Yuan-yuan Liu, Department of Radiotherapy and Oncology, Affiliated Kunshan Hospital of Jiangsu University, Kunshan, China. E-mail: liuyuanyuanszcom.