Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Multi-omics aberrations of FL
Composition and function of TME...
POD24 and transformed FL:...
Novel approaches targeting...
Prospects
Conclusion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(6):1955-1967. doi:10.7150/ijbs.80401 This issue Cite
Review
Advances in the multi-omics landscape of follicular lymphoma
Tianyuan Xu, Zhong Zheng, Weili Zhao ![]()
Shanghai Institute of Hematology, State Key Laboratory of Medical Genomics, National Research Center for Translational Medicine at Shanghai, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China
Received 2022-11-1; Accepted 2023-3-11; Published 2023-3-27
Abstract

Follicular lymphoma (FL) is the most common indolent lymphoma originating from germinal center B cells. FL represents a clinically and biologically heterogeneous disease. Most patients have favorable outcomes, but a subset of patients experiences early progression or transformation and has a poor prognosis. Abnormalities in FL cells and tumor microenvironment have been revealed using multi-omics techniques, including genomic, epigenomic, transcriptomic and proteomic analysis. Recurrent somatic gene aberrations mainly involve epigenetic modifiers, transcription factors, oncogenic pathways and microenvironment modulators. Single-cell transcriptomic analysis show marked inter- and intra-patient FL subclone heterogeneity. In addition, a comprehensive profile of microenvironmental components is provided, unveiling the crosstalk between tumor and microenvironment that induce FL progression and facilitate immune escape. Together, these studies provide insights into the mechanisms and biomarkers of high-risk FL populations, as well as the potential targeted and immunotherapy options. Future research should focus on integrating multi-omics aberrations to optimize therapeutic strategies in FL.
Keywords: follicular lymphoma, genomics, transcriptomics, microenvironment, biomarkers, targeted therapy, immunotherapy
Introduction
Follicular lymphoma (FL) is the second most common type of non-Hodgkin's lymphoma, accounting for approximately 15-25% of adult lymphomas with an annual incidence of 3-5/100 000 [1, 2]. As an indolent lymphoma, about 80% of FL patients have an overall survival (OS) of more than 10 years, but most patients experience recurrent remission and relapse with increasing drug resistance [3]. Moreover, a subset of patients experiences early progression or transformation and has a poor prognosis [4, 5]. The underlying genetic, epigenetic, and microenvironmental mechanisms driving FL progression and resistance need to be further elucidated.
The pathogenesis of FL is a multistep process. The initial and hallmark genetic aberration in FL is the t (14;18)(q32;q21) translocation detected in >85% of patients. This occurs during V(D) J recombination of pro/pre B cells in the bone marrow, leading to sustained expression of the anti-apoptotic gene BCL2. Then naïve B cells enter the germinal center (GC) region of lymph nodes, where they undergo activation-induced cytidine deaminase (AID)-mediated somatic hypermutation (SHM) and class switch recombination (CSR). The overexpression of BCL2 provides a survival advantage for GC which is independent of BCR affinity [6]. These BCL2(+) FL-like cells traffic between secondary lymphoid organs and bone marrow, accumulating additional genetic alterations through repeated GC reentry, and eventually develop into overt FL [7].
FL exhibits a high dependence on the tumor microenvironment (TME) composed of different types of immune cells and stromal cells. In lymph nodes, the FL microenvironment mainly consists of CD4+ T follicular helper (Tfh) cells, CD4+ T regulatory cells (Tregs), CD8+ cytotoxic T cells (CTLs), macrophages, follicular dendritic cells (FDCs) and fibroblastic reticular cells (FRCs) [8, 9]. Through various soluble factors and ligand-receptor interactions, FL-TME crosstalk supports tumor cell survival and proliferation, promotes immune escape, and also serves as promising prognostic biomarker and therapeutic target [10].
The multi-omics approaches provide a comprehensive assessment of the biological features of disease by integrating information at different levels, including the genome, epigenome, transcriptome and proteome [11, 12]. These techniques have been applied to not only comprehensively elucidate the pathogenesis and progression of the disease, but also to identify the biomarkers for improving clinical management and explore novel treatment approaches. Here, this review discusses the multi-omics abnormalities in tumor cells and TME of FL, and their application in risk stratification and treatment approaches.
Multi-omics aberrations of FL
Through whole-genome sequencing, targeted deep sequencing and single-nucleotide polymorphism array, mutations and chromosomal structural variants have been identified (Table 1) [13-22].
Representative genetic aberrations in FL.
| Gene | Frequency (%) | Alterations | Function | Refs |
|---|---|---|---|---|
| Epigenetic modifiers | ||||
| KMT2D | 70-90 | LOF | Histone methyltransferase | 15,16,20,21,34,66 |
| CREBBP | 50-70 | LOF | Histone acetyltransferase | |
| EP300 | 20-30 | LOF | Histone acetyltransferase | |
| EZH2 | 20-30 | GOF | Histone methyltransferase | |
| Linker histones | 27-44 | LOF | linker histones; chromatin remodelling | |
| ARID1A | 11-15 | LOF | SWI/SNF complex; chromatin remodelling | |
| BCL7A | 10 | LOF | SWI/SNF complex; chromatin remodelling | |
| Transcription factors | ||||
| BCL6 | Mutations, 5 Translocations, 20 | GOF | Transcription factors | 15,16,17,18,20,21,34 |
| MEF2B | 7-15 | GOF | ||
| FOXO1 | 12 | GOF | ||
| EBF1 | 17 | unknown | ||
| IRF8 | 6-13 | unknown | ||
| IRF4 | 11 | unknown | ||
| BCR-NF-κB pathway | ||||
| CARD11 | 11 | GOF | Signals transducer downstream BCR activation | 16,20,21,44,45,48 |
| BTK | 7-10 | LOF | Tyrosine kinase | |
| TNFAIP3 | 10 | LOF | Inhibitor of NF-κB, immune response modulator | |
| CD79B | 10 | GOF | BCR complex | |
| IGV | 80 | GOF | N-glycosylation of IGV region of BCR | |
| JAK-STAT pathway | ||||
| STAT6 | 12 | GOF | Signal transduction transcription activation | 16,20,48,49 |
| SOCS1 | 8-10 | LOF | JAK-binding protein | |
| PI3K/AKT/mTOR pathway | ||||
| RRAGC | 10-20 | GOF | Guanine nucleotide-binding protein | 20,52,53,54,55 |
| ATP6V1B2 | 10-22 | LOF | V-ATPase complex | |
| ATP6AP1 | 10 | LOF | V-ATPase complex | |
| VMA21 | 12 | LOF | V-ATPase complex | |
| SESTRIN1 | Deletion, 20 | LOF | Negative regulator of mTOR activity | |
| NOTCH pathway | ||||
| NOTCH1, NOTCH2, NOTCH3, NOTCH4, DTX1, SPEN | 18 | LOF | NOTCH pathway | 21 |
| Migration | ||||
| GNA13 | 10 | LOF | Guanine nucleotide-binding proteins | 18,19,20,52 |
| Cell cycle | ||||
| RB1 | Deletion, 12 | LOF | Cell cycle regulator | 20,59 |
| CDK4 | Copy number gain, 29 | GOF | ||
| CCND3 | 15 | GOF | ||
| Survival | ||||
| BCL2 | Mutations, 50Translocations, 85 | GOF | anti-apoptosis | 25,26 |
| Immune evasion | ||||
| TNFRSF14 | 20-50 | LOF | Receptor | 16,20 |
| CTSS | 4-22 | GOF | Protease | 62,63 |
| EPHA7 | 70 | LOF | Receptor | 22 |
These are mutations unless specifically mentioned as translocations or copy number variations.
GOF: gain-of-function
LOF: loss-of function
Epigenetic modifiers
Mutations in epigenetic modifiers are key pathogenic factors of FL. These include histone modifiers (KMT2D, CREBBP/EP300, and EZH2), members of the linker histone H1 family (HIST1H1C and HIST1H1E), and SWI/SNF chromatin remodeling complex genes (ARID1A and BCL7A). These mutations inhibit critical transcriptional programs for B cell selection and differentiation, and are detected in almost all FL patients.
KMT2D (also called MLL2) is the most common mutation detected in 70-90% of patients. Inactivating mutations of KMT2D result in reduced lysine 4 of histone H3 (H3K4) methylation in FL cells, altering the expression of a wide range of genes including CD40, JAK-STAT, Toll-like receptor, B cell receptor signaling pathways and tumor suppressor genes [23]. Early loss of KMT2D during B cell development promotes proliferation and malignant outgrowth of B cells [24]. CREBBP and its paralogue EP300 are mutated in 50-70% and 10-20% of patients respectively. Loss-of-function mutations in CREBBP cause focal depletion of enhancer H3K27 acetylation, resulting in transcriptional repression of genes regulating cell differentiation, BCR/CD40 signaling and MHC presentation [25, 26]. CREBBP-deficient hematopoietic stem and progenitor cells are unable to acetylate P53 and exhibit a defective DNA damage response, thereby facilitating the accumulation of subsequent mutations [27]. EP300 regulates transcriptional programs that partially overlap with CREBBP, and deletion of both produces synthetic lethal effects [28]. Histone-lysine N-methyltransferase EZH2 is the catalytic subunit of the PRC2/EED-EZH2 complex and increases H3K27 trimethylation to silence gene transcription [29]. Mutations in EZH2 are observed in 20-30% of patients. Gain-of-function mutations in EZH2 repress the expression of proliferation checkpoint CDKN1A and differentiation-related gene PRDM1 and IRF4 [30, 31].
Inactivating mutations in the linker-histone H1 family members and SWI/SNF chromatin remodeling complex genes are observed in 28-44% and 11-15% of patients respectively. These tumor suppressors organize chromatin structure by regulate DNA-ribosome topology and histone modifications [32, 33]. The combined deletion of H1c/H1e in mice can enhance the self-renewal properties of GC B cells through focal chromatin relaxation and stem cell gene upregulation [32].
Transcription factors
Approximately half of FL patients harbor non-silent mutations in lymphoid transcription factors such as BCL6, MEF2B and FOXO1[34]. BCL6 is mutated or translocated in 20% of patients. BCL6 is a transcriptional repressor of DNA damage checkpoints and B cell differentiation genes, and can cooperate with histone modifiers through recruiting histone demethylases and deacetylases [35, 36]. In FL, highly expressed BCL6 drives the survival and growth of FL cells in part through repressing NOTCH pathway genes [37, 38]. MEF2B, a transcriptional activator of BCL6 and MYC, is mutated in 7-15% of patients. Mutations in MEF2B disrupt its interaction with the corepressor CABIN1 and reduce the sensitivity to inhibitory signals, resulting in increased transcriptional activity [39, 40]. Gain-of-function mutations in FOXO1 are detected in 12% of patients, leading to hyperactivation of PI3K and SAPK/JNK signaling [41].
Oncogenic pathways
Aberrant activating or inhibitory mutations in oncogenic pathways in FL include BCR-NF-κB (BTK, CARD11, TNFAIP3, IGV region), JAK-STAT (SOCS1, STAT6), PI3K/AKT/mTOR (RRAGC, ATP6V1B2, VMA21, SESTRIN1) [42] (Figure 1). Besides, mutations in the NOTCH pathway, S1PR2-Gα13 axis, and cell cycle regulators are also detected. All of these contribute to the prolonged survival and sustained proliferation of FL cells.
Epigenetic modifiers and signaling pathways in follicular lymphoma cells. Several epigenetic modifiers and signaling pathways are deregulated in FL cells, such as histone modifiers, BCR-NF-κB, JAK-STAT and PI3K/AKT/mTOR pathway, which serve as targets for novel precision therapies.
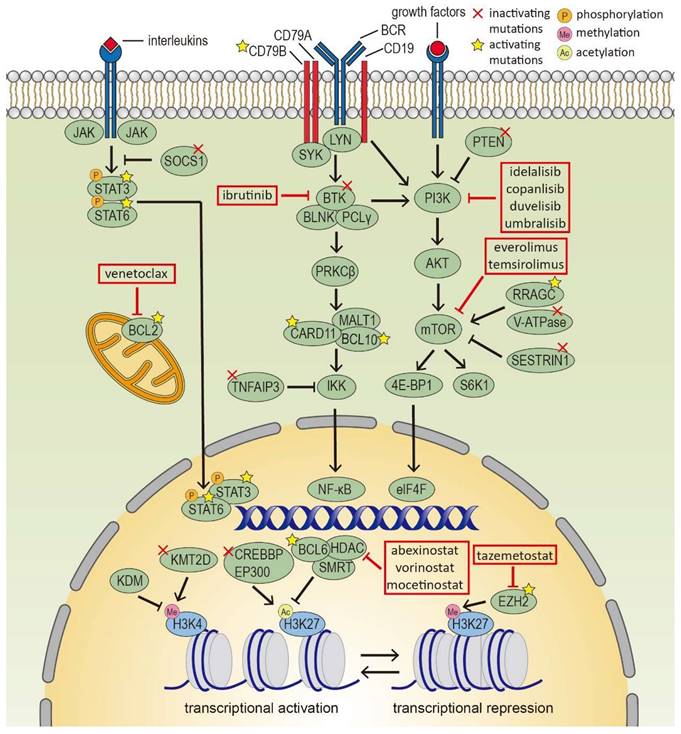
About half of FL patients harbor mutations in genes associated with the BCR-NF-κB pathway. Most FL cells express IgM and mediate antigen-independent BCR signal that promotes the survival and proliferation of B cells. Activating mutations in CARD11 can initiate a spontaneous, receptor-independent activation of downstream NF-κB signaling [43]. Inactivating mutations in BTK upregulate AKT phosphorylation [44]. Loss-of-function mutations in TNFAIP3 activate NF-κB function [45]. Besides, a BCR N-glycosylation sites asparagine-X-serine/threonine (N-gly sites) motif is present in over 80% of patients, which can interact with lectins in the TME and activate downstream BCR pathways [46]. Interestingly, N-gly sites are early events in FL which are conserved during diagnosis, progression and transformation. N-gly sites provide a clear survival advantage for FL cells and are rarely found on normal B cells, suggesting therapeutic potential [46].
The JAK-STAT pathway genes are mutated in about 20% of patients. It can be activated by IL-4, IL-10 and IL-21 in the microenvironment, leading to downstream phosphorylation of STAT6 and STAT3 [47]. Activating mutations in STAT6 increase nuclear residency, and subsequently induce the expression of responsive genes CISH, FCER2, NFIL3 and CCL17 [48]. Inactivating mutations in SOCS1, an inhibitor of STAT, leads to the dysregulation of STAT activity [49].
The PI3K/AKT/mTOR pathway genes are mutated in 25% of patients, regulating cell growth and nutrient sensing. Mutated RRAGC proteins make mTORC1 insensitive to amino acid deprivation, enhance B-cell response and accelerate lymphomagenesis [50-52]. ATP6V1B2 mutations activate mTOR, allowing cells to survive in low albumin concentrations [53]. VMA21 mutations impair lysosomal function and lead to compensatory autophagy activation, a process that can be blocked by autophagy-inhibiting compounds [54]. SESTRIN1 can be genetically inactivated or epigenetically silenced by EZH2, which affects p53-mediated regulation of mTORC1 and induces mRNA translation under genotoxic stress. Interestingly, mutations in EZH2 and RRAGC and loss of SESTRIN1 display mutual exclusivity, indicating that mTORC1 insufficiency can be generated by various mechanisms, and justifies the use of mTOR inhibitors [55].
Mutations in other oncogenic pathways are also observed in FL. NOTCH pathway is mutated in 18% of patients [56]. Loss-of-function mutations in GNA13 of the S1PR2-Gα13 axis can be found in 10% of patients, promoting the dissemination, survival and SHM frequency of FL cells [57, 58]. Nearly half of FLs have retinoblastoma (RB) pathway disruptions, including deletions of p16/CDKN2A/N and RB1, and gains of CDK4 [19, 59, 60].
Microenvironment modulators
A number of somatic mutations not only affect tumor cells, but also modulate the microenvironment development. These microenvironment modulators include TNFRSF14, CTSS, TNFAIP3, CREBBP, EZH2 and RRAGC. Depending on the effect on tumor-supporting Tfh cells, these genes exhibit two strategies of TME reprogramming. Mutations in TNFRSF14, CTSS, and TNFAIP3 recruit more Tfh cells to support FL proliferation. Loss-of-function mutations in TNFRSF14 (HVEM) disrupt normal interaction with inhibitory receptor BTLA expressed on Tfh cells, leading to higher Tfh infiltration and IL-4 activity [61]. Activating mutations in CTSS increase MHC-II presentation and CD4+ T cell recruitment [62, 63]. Inactivating mutations in TNFAIP3 exhibit a higher frequency in Th1 cells and CD8+ T cells [64]. On the other hand, mutations in CREBBP, EZH2, and RRAGC reduce the frequency of infiltrating Tfh cells. Inactivation of CREBBP reduced MHC-II expression and antigen presentation, leading to decreased Tfh cells and cytotoxic T cell infiltration [65]. Activating mutations in EZH2 downregulate MHC expression and limit the interaction of FL cells with Tfh cells [66, 67]. Activating mutations in RRAGC are associated with decreased abundance of Tfh cells [68]. Both RRAGC and CTSS mutations were mutually exclusive with TNFRSF14 mutations, suggesting the divergent evolution regarding dependence on TME [50, 63].
Transcriptional heterogeneity
Single-cell RNA sequencing provided insights into the transcriptional heterogeneity both inter- and intra-patient. Genes differentially expressed among patient samples are mainly related to BCR and NF-κB signaling [69]. In single patient lymph node biopsies of different locations, cluster analysis identifies marked site-to-site diversity in the expression profile of NF-κB signaling, antigen presentation, MAPK pathway, and cell adhesion, which indicate spatial heterogeneity within individual patients [46, 69-73]. Therapeutic targets HDAC9 and CD79B are also differentially expressed, which could affect response to HDAC inhibitors or CD79B-directed therapies [69]. BCR phylogenetic trees suggest that FL cells can migrate freely in vivo, undergo divergent evolution and independently develop between sites [46, 69]. Overall, the degree of site-to-site variations in transcriptome profiles is consistent with BCR sequence variations.
Single-cell RNA sequencing also elucidates the diverse functional status of FL cells in the GC region within a single lymph node. FL cells present a continuous gene expression profile across the dark zone (DZ)/light zone (LZ) phenotype. In addition to conventional LZ and DZ phenotypes, approximately 30% of FL B cells exhibit a transitional state [74-76]. Besides, FL cells exhibit GC desynchronization, highlighted by different co-expression patterns at the single-cell level [74]. Furthermore, FL cells share a heterogeneous differentiation state across PC-, GC- and Mem-like B cells, with a small proportion exhibiting plasma cell-like features, while the majority span a continuous state between proliferating GC-like and quiescent Mem-like states [75]. In single biopsies, transcriptional heterogeneity is independent of evolutionary subclones marked by BCR sequence [74].
Together, the simultaneous presence of multiple phenotypes suggests that FL cells may dynamically alternate between states. Epigenetics and TME can act as potential regulators in this process, paralleling normal GC reactions. Whether the functional status by transcriptome can reflect evolution pattern by the BCR sequence is still unclear. Further efforts are required to precisely define the high-risk populations in heterogeneous FL subclones for targeted therapies.
Composition and function of TME in FL
The TME consists of T cells, macrophages, stromal cells, and a smaller proportion of neutrophils and natural killer (NK) cells [8, 77-79]. Recently, single-cell RNA sequencing, mass cytometry by time of flight (CyTOF) and reverse phase protein array (RPPA) studies provide a comprehensive picture of microenvironmental cell components at the transcriptomics and proteomics levels [69, 71, 72].
T cells
Tumor-infiltrating T cells (TILs) can be classified into three main populations, Tfh, Treg and CD8+ T cells. A prominent feature of FL TME is the higher concentration of CD4+ Tfh and Treg cells. TILs can also be divided into distinct subgroups based on functional distribution, including naive, effector, exhausted and memory cells [69, 71, 72, 80, 81]. CYTOF studies revealed that TILs are skewed towards an exhausted phenotype in FL, marked by high expression of inhibitory checkpoints such as PD-1, LAG-3 and TIGIT, as well as low expression of co-stimulatory receptors CD27 and CD28, leading to reduced interaction with antigen-presenting cells (APCs) and impaired TCR signaling responses [73, 80, 82-87]. However, the expression of immune checkpoints does not fully represent a defective state, since PD-1 is also highly expressed in active Tfh cells [47, 84]. CYTOF studies also identified that specific subpopulations of PD-1(+) T cells and deficient CD4(+) memory T cells are related to poor prognosis [80]. Besides, a recent study has defined four FL TME subtypes based on T-cell subsets population, in which a T cell-depleted subset exhibited an inferior outcome [88].
Tfh cells are necessary for the selection and differentiation of B cells in normal GC, and are marked by a CD4+CXCR5+ICOS+PD1+BCL6+ immunophenotype. FL Tfh maintain a high production of cytokines and chemokines such as IL-4, IL21, TNFα, IFNγ, and CXCL13. IL4 and IL21 activate STAT6 in FL B cells, supporting FL growth and survival. IL-4 also drives macrophages toward the M2 phenotype and triggers upregulation of CXCL12 in lymphoid stromal cells (LSCs) [89, 90]. RPPA studies verified high concentrations of IL-4 and the elevated downstream phosphorylation of STAT6 and ERK in FL at the protein level [91]. Besides, Tfh highly expresses CD40L, IL6R and CTLA. CD40L binds to CD40 on FL B cells, activates the NF-κB signaling pathway, and induces FL B cells to respond to macrophage-derived IL-15 [92-94]. Interestingly, single-cell analysis revealed that Tfh abundance is the only microenvironmental cell subpopulation that correlated with FL spatial heterogeneity, and is associated with CD40 expression, reinforcing the role of Tfh in FL development [69].
Treg cells are characterized by a CD4+CD25+FOXP3+ immunophenotype with a frequent expression of CD25, GITR, CTLA-4 and CD45RO [85, 95]. FL B cells convert conventional T cells into Treg, recruit and promote the proliferation of Treg through a variety of chemokines (CXCL13, CCL17, CCL19 and CCL22) and membrane molecules (CD70, CD80, CD86 and ICOSL) [85]. Treg cells inhibit CD4+ T cell proliferation and CD8+ T cell cytotoxic function [82, 85]. Notably, in addition to classical Treg, a population of CXCR5+PD-1+ICOS+ T follicular regulatory (Tfr) cells was found. Tfr cells inhibit Tfh and B cell activation, suggesting their potential to exert antitumor effects [96-98].
CD8+ T cells are the major population that exerts specific cytotoxic antitumor immunity [99] CYTOF and CITE-seq analysis demonstrated that CD8+ T cells in FL are skewed towards effector cells which exhibit an enhanced capacity to produce granules (granzyme B and perforin) and cytokines (IFNγ and TNFα). However, these cells are more immunologically depleted due to sustained antigenic stimulation, marked by high expression of PD-1, TIGIT, TIM-3, ICOS, BTLA, and LAG3 [82, 100, 101]. Besides, FL CD8+ T cells show downregulated TCR-induced ERK phosphorylation and IFNγ production, while TCR proximal signaling is not affected [86]. Furthermore, PI3K inhibitors downregulate transcription factors responsible for effector cell differentiation, indicating a therapeutic mechanism of PI3K inhibitors on the TME [100].
Macrophages
Depending on the stimulatory signals received, macrophages can be divided into M1 and M2 subtypes, and are generally polarized to M2 subtypes in tumors [102]. M2 macrophages express DC-SIGN, which binds to the N-glycosylation site on the BCR of malignant B cells to promote cell survival [103]. M2 macrophages also produce various cytokines to enhance angiogenesis (VEGF and angiopoietin), recruit monocytes and neutrophils (CCL2, CLL3 and IL-8), and promote dissemination (CXCL12, CCL18 and MMP9) [104]. Moreover, phagocytosis by macrophages is hampered by the interaction of SIRPα with the "do not eat me" receptor CD47, which is abundantly expressed on FL B cells. Using CYTOF, FL macrophages can be further divided into three subpopulations based on the expression levels of SIRPα and CD14 ((CD14(+)SIRPα(hi), CD14(-)SIRPα(low), and CD14(-)SIRPα(neg)), which exhibit distinctive differentiation, migration, immune function and prognostic values [105].
Stromal cells
Stromal cells support the survival and dissemination of tumor cells, and are involved in the reprogramming of the immune compartment. In FL, malignant B cells reprogram LSC precursors into cancer-associated fibroblasts (CAFs) through expressing CCR7, CXCR4, TNF, LT and TGF-β. FL stromal cells overexpress a variety of chemokines such as CXCL12, CCL19 and CCL2 [79]. The two most prominent stromal cell subpopulations are FRCs and FDCs. FRCs are present in the T-cell region around the follicle and secret extracellular matrix [106]. FRCs contribute to the polarization of FL-Tfh by enhancing IL-4 secretion in a Notch- and ICAM1/LFA1-dependent manner [106]. FRCs also promote B cell activation and adhesion through the expression of CXCL12, CXCL13, IL-7 and BAFF, which can be antagonized by BTK and PI3K inhibitors [79, 90]. FDCs are specialized APCs enriched in the B-cell zone. The FDC network is often disrupted compared to the FRC network in FL, and malignant B cells reduced expression of the GC-confinement receptors S1PR2 and P2RY8, suggesting a less role for FDCs to support FL B cells [79, 107]. FDCs activate B cells through presenting lectins or antigens to BCR. Besides, FDCs express TIGIT ligand, reinforcing their immunosuppressive function [82, 86].
A recent study classified LSCs into four major clusters by expression of CD49a (ITGA1), podoplanin (PDPN) and CD21, including cell populations of FRCs (PDPN+CD21-CD49a+), FDCs (PDPN+CD21+CD49a-), double-negative (DN) perivascular cells (PDPN-CD21-CD49a+), and a less defined fourth subset (PDPN+CD49a-). DNs are similar to cells with precursor capabilities, expressing nestin and markers of adipocytes and osteoblasts, and can differentiate toward FRC-like cells in response to TNF and LT [79]. By single-cell clustering analysis, lymph node stromal cells can also be divided into blood endothelial cells (BECs), lymphatic endothelial cells (LECs), and non-endothelial SCs (NESCs). Upregulation of a number of previously unidentified genes and interaction pairs such as CD70-CD27 was observed [78]. Using histology examination, the location and quantification of stromal cells was assessed in situ and showed higher abundance than common sequencing methods [12].
Together, these results indicate that the heterogeneity of microenvironment cells lies far beyond our understanding and remains to be further functionally explored. The cellular components of TME in the bone marrow are very different from those in lymph nodes (reviewed in separate articles [108, 109]). FL subclones with different gene expression profiles are detected between bone marrow and lymph nodes [109, 110]. Currently no systematic study has combined the relationship between TME components and genomic and transcriptomic abnormalities of FL B cells to study how TME contributes to the spatial heterogeneity between lymph node sites and extranodal involvement. In addition, it remains to be elucidated how FL-TME interactions alter throughout the course of the disease. A better understanding of how specific TME components regulate the onset, progression, therapeutic response and relapse of FL would provide novel opportunities for harnessing TME as treatment options and potential biomarkers.
POD24 and transformed FL: underlying mechanism and biomarkers
The clinical course and outcomes of FL are heterogeneous. A subset of patients who develop early disease progression within 24 months of diagnosis (POD24) or histological transformation has a poor prognosis. Approximately one-third of POD24 patients also undergo transformation [111, 112]. The mechanisms underlying these high-risk FL populations are not yet fully understood. Prognostic models remain to be explored to identify these patients in order to provide personalized therapeutic interventions.
As to genomic aberrations, comparative analysis of paired samples revealed an increase in genomic complexity in patients with transformed FL (t-FL) and POD24, including mutations and copy number variations (CNVs). t-FL is especially enriched in genetic aberrations involving the NF-κB pathway (MYD88, TNFAIP3), DNA damage responses (TP53), transcription factors (EBF1, MYC, IRF4), immune surveillance (CD58, B2M), cell-cycle (CDKN2A/B, CCND3) and dissemination (S1PR2, GNA13). t-FL also shows a dysregulation of miRNA (miR-142, miR-150, miR-7e-5p) and a higher level of SHM [14, 16-20, 113-117]. About 80% of t-FL is of the GC B-cell-like (GCB) subtype and about 16% is of activated B-cell-like (ABC) subtype. ABC subtype has a lower incidence of t(14;18) and increased mutations in BCR-NF-κB signaling [18, 19]. For POD24 patients, single-cell RNA sequencing identified the upregulation of BCR signaling pathway [12]. The overall SHM burden in samples from early versus late/never progressors is not significantly different. A ten-gene list was found to be mutated more commonly in POD24 patients, including KMT2C, TP53, BTG1, MKI67, XBP1, SOCS1, B2M, FAS, IKZF3 and MYD88. Gains in VRK2 and FANCL by small insertions were also observed. About 80% of POD24 patients had mutations in any of the ten genes despite low mutation frequencies, suggesting a role of rare genetic events in FL progression [19, 115]. Notably, a clonal evolution study shows distinct patterns underlying transformation and early progression, in which t-FL is generated from clones that are rare or absent in diagnostic samples, while POD24 arisen from detective treatment-resistant clones that already exist at diagnosis [19].
The prognostic value of FL TME is also identified by immunohistochemistry, flow cytometry and RNA sequencing. TME factors associated with t-FL include a low lymphocyte-to-monocyte ratio (LMR), diffuse pattern of PD1+ T cells and CD14+ FDCs, follicular distribution of FOXP3+ cells, and increased vessel density [118-122]. POD24 events are significantly enriched in patients with low immune infiltration level marked by low expression of the immune checkpoint PD-L2 [123]. Fibroblasts, DC-SIGN+ cells and extracellular matrix (ECM) were found enriched in early progression FL patients, as verified by quantitative imaging.[12] Other TME predictors for POD24 include lack of intrafollicular CD4 expression as well as low expression of CD8+ T-cell markers and genes regulating Th1 and Th2 responses [81, 124].
Prognostic models of FL regarding clinical factors include FLIPI1, FLIPI2, PRIMA-PI and FLEX, which are designated based on different indexes and treatment regimens [125-128]. Integrated models that combine clinical and molecular factors have also been proposed, including m7-FLIPI, POD24-PI and a 23-gene model [34, 129, 130]. The sensitivity and specificity of these models in predicting POD24 range from 43% to 86%, with the highest sensitivity for POD24-PI (78%) and specificity for m7-FLIPI (86%) [129, 131]. t-FL is also found relative to a range of clinical factors, including high histological grade and high FLIPI score, while the response to therapy had no impact on the risk of transformation [112].
To date, no genetic, microenvironmental, clinical or combined models have shown adequate efficiency as biomarkers for t-FL or POD24 to direct clinical practice. The integration of multi-omics data is hopeful to incorporate multiple levels of information for detailed patient stratification and accurate prediction.
Novel approaches targeting multi-omics alterations
With a better understanding of FL and its microenvironmental, significant progress has been made in FL treatment approaches. New targeted agents and immunotherapies offer alternatives for relapsed/refractory (R/R) patients following traditional rituximab-based chemoimmunotherapy (Figure 1).
Since epigenetic modifiers deregulations are key events in FL, epigenetic therapies play an important role in FL treatment. EZH2 inhibitor tazemetostat demonstrated favorable clinical response with an ORR of 69% in EZH2 mutant patients and 35% in EZH2 wild-type patients, with a median PFS of 13.8 and 11.1 months. EZH2 inhibitor is well tolerated, with only 4% of patients showed serious adverse effects [132]. Tazemetostat has received accelerated approval from the FDA, and its combination therapies with CD20 monoclonal antibodies or immunomodulatory agents are also in clinical trials [133]. Inactivation of CREBBP and KMT2D are difficult to target directly, but can be rescued by inhibiting their antagonists. Histone deacetylase 3 (HDAC3) inhibitors (abexinostat, vorinostat, mocetinostat) compensate for CREBBP/EP300 deficiency by targeting the BCL6 /SMRT/HDAC3 complex [25, 134-137]. KDM5 inhibition can restore histone methylation and the expression of genes repressed by KMT2D loss [138]. Other epigenetic agents include DNMT inhibitors, PRMT inhibitors and BET inhibitors, which have shown efficacy in other hematological malignancies such as acute myeloid leukemia and diffuse large B-cell lymphoma, but have not yet been implemented in FL [133, 139].
BCL2 overexpression is considered a major driver of FL, but the BCL2 inhibitor venetoclax failed to show a significant benefit in monotherapy or combination with rituximab/bendamustine-rituximab [140]. The reasons could be BCL2 mutations, heterogeneity of BCL2 expression among subclones, expression of other anti-apoptotic genes, and a decrease in BCL2 dependence at advanced stages of the disease [141]. Similarly, although BCR signaling-related genes are highly activated in FL, the effect of the BTK inhibitor ibrutinib was unsatisfactory with ibrutinib monotherapy showing ORR and CR rates of 21-38% and 11-13% [142, 143]. Possible explanation is an alternative BCR pathway independent of BTK [144]. Other BTK inhibitors (acalabrutinib and zanubrutinib) and SYK/JAK inhibitor cerdulatinib are under testing in single agent or combination trials [145-147].
The PI3K-AKT-mTOR pathway is highly activated in FL. Four PI3K inhibitors (Idelalisib, Copanlisib, Duvelisib, Umbralisib) have been approved with varying efficacy and toxicity profiles, depending on the specific subunit targeted [148-152]. The four agents showed moderate activity with ORR ranging from 42% to 59% and the median PFS about 10 months. Clinical trials for other PI3K inhibitors and combination regimens are ongoing [133, 153, 154]. Besides, mTORC1 inhibitors everolimus and temsirolimus exhibited an ORR of 61% and 54% in R/R indolent lymphoma [155, 156].
New immunotherapy approaches that harness TME to activate anti-tumor immunity are being explored and have shown promising potential, including immune modulators, immune-checkpoint inhibitors, bispecific antibodies (BSAbs) and chimeric antigen receptor (CAR) T cell therapy.
Lenalidomide is a non-specific immunomodulatory agent that binds cereblon, induces degradation of transcriptional factors, and increases the cytotoxicity effect of CTLs, NKs and antibodies [157]. The combination of lenalidomide plus rituximab (R2) significantly improved PFS for R/R FL than rituximab alone, and R2 has been approved as the standard of care for the relapsed setting [158-160]. In untreated FL patients, R2 shows comparable efficacy to conventional immunochemotherapy as the first-line treatment of FL [159]. A similar high response rate can also be observed in lenalidomide and obinutuzumab [161], suggesting the chemo-free regimen can be a less-toxic first-line option for FL patients to avoid the side effects of chemotherapy.
Despite the enrichment of PD-1+ TILs in FL TME, the effect of PD-1 inhibitors (nivolumab and pembrolizumab) in FL has been disappointing. In a phase II study of R/R FL, nivolumab only had an ORR of 4% and median PFS of 2.2 months, and the response had no correlation with PD-1 or PD-L1 expression [162, 163]. The efficacy of nivolumab was improved when combined with ibrutinib, with an ORR of 33% [162]. Combining Urelumab, an agonist of co-stimulatory receptor 4-1BB (CD137), fails to enhance clinical activity compared to rituximab alone [164]. Other immune checkpoints such as LAG-3, TIM-3, and TIGIT are also highly expressed in FL TILs, and relative combination therapies are under investigation [165-167].
The phagocytosis of macrophages can be suppressed by CD47 on tumor cells that bind with SIRPα on macrophages [168]. In a phase Ib/II study, CD47 monoclonal antibody Magrolimab (Hu5F9-G4) in combination with rituximab induced an ORR and CR rate of 71% and 43% in 7 patients with R/R FL [169]. Other agents of interest to target macrophages include ALX148, TTI-622, and CD19-CD47 bispecific antibodies [154].
Bispecific antibodies (BSAbs) and autologous CAR-T cells generate immune responses by targeting B cell markers on the surface of lymphoma cells. BSAbs consists of two binding domains, usually CD3 for T cells and CD20 or CD19 for lymphoma cells. A variety of BSAbs has shown remarkable efficacy in clinical trials, including mosunetuzumab, odronextamab, epcoritamab, and glofitamab, with ORR of 64%-93% [170-175]. CAR-T cells are engineered T cells that can specifically recognize and eradicate cells expressing particular antigens. Based on the ZUMA-5 trial, axicabtagene ciloleucel, a CD19 CAR-T therapy, has been approved for R/R FL with ORR and CR rates of 94% and 60% [176, 177]. The ELARA study of tisagenlecleuce has a similar high ORR of 86% and a CRR of 66% [178, 179]. In addition, a series of clinical trials combining CAR-T with other targeted agents and BSAbs are under evaluation. Serious side effects for CAR-T and BSAbs therapies are cytokine release syndrome and neurological toxicity, which will require further product optimization and adjuvant drug application. Long-term data are needed to support the durability of these therapies. Due to the impressive efficacy demonstrated in clinical trials, the future of immunotherapies is highly promising.
Despite the wide range of available treatments for FL patients, from first-line rituximab-based regimens (including R-CVP, R-CHOP, RB and R2) to a multitude of targeted agents and immunotherapies, there is currently no uniform standard for optimal sequencing and combination of therapies for the individual. There are very limited biomarkers to predict patient response except for EZH2 inhibitors. In addition, the mechanism of action of many agents is not limited to their known targets, particularly with effects on TME. Tazemetostat can enhance T-cell recruitment through modulating MHC II and chemokine expression [180]. HDAC inhibitors induce expression of IFN pathway and antigen-presenting genes, and work synergistically with checkpoint inhibitors to restore the cytotoxic ability of T cells [181]. Idelalisib can reduce the recruitment of Tfh and Treg cells, inactivate the suppressive activity of Treg cells, impair FDCs proliferation, and restore FL response to venetoclax [182, 183]. Activity of CD47 inhibitors also involves DCs and CTLs [184]. Response to PD-1 blockade is associated with NK cells and CD4+ T cells [185]. Therefore, further studies on the mechanisms of action can help to better understand the drivers of response and resistance for patients. In addition, the potential alternative mechanism is thought to possibly interfere with the efficacy of other therapies, and further clinical trials are needed to confirm the efficacy of combination and sequential therapy in practice.
Prospects
Multiple approaches can be applied to unravel key molecular mechanisms that contribute to FL. Correlation analysis between transcriptome and genome or epigenome can help identify differentially regulated pathways that are subject to gene mutations or epigenetic modifications. Integrating proteomic data with other omics can reveal functional consequences of upstream alterations, as well as the relationship between intracellular abnormalities and FL-TME crosstalk. In addition, network analysis forms a web of interactions between genes, proteins and other molecules. Machine learning can analyze large-scale datasets to generate potential biomarkers and personalized therapy through new algorithms. With these knowledge, subsequent experimental validation can be performed to elucidate primary alterations and secondary functional effects. Therefore, the integration of multiple omics provides a more comprehensive view of FL malignant cells and the interaction with TME, leading to the development of novel diagnostic and therapeutic strategies.
Conclusion
FL is an indolent but still incurable disease. Over the last decade, the understanding of the abnormalities in FL and its microenvironment has been dramatically improved. The multi-omics analysis provides a panorama view of the genome, epigenome, transcriptome, and microenvironment abnormalities of FL. Future research is required to elucidate how these complex factors work together to promote disease development and predict therapeutic responses. The combination of molecular subtype with clinical heterogeneity will lead to precise risk stratification and personalized therapy in FL.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cerhan JR. Epidemiology of Follicular Lymphoma. Hematol Oncol Clin North Am. 2020;34:631-46
2. Dreyling M, Ghielmini M, Rule S, Salles G, Ladetto M, Tonino SH. et al. Newly diagnosed and relapsed follicular lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32:298-308
3. Follicular lymphoma. Nat Rev Dis Primers. 2019; 5: 84.
4. Casulo C, Dixon JG, Le-Rademacher J, Hoster E, Hochster H, Hiddemann W. et al. Validation of POD24 As a Robust Early Clinical Endpoint of Poor Survival in FL from 5,225 Patients on 13 Clinical Trials. Blood. 2021
5. Lossos IS, Gascoyne RD. Transformation of follicular lymphoma. Best Pract Res Clin Haematol. 2011;24:147-63
6. Sungalee S, Mamessier E, Morgado E, Grégoire E, Brohawn PZ, Morehouse CA. et al. Germinal center reentries of BCL2-overexpressing B cells drive follicular lymphoma progression. J Clin Invest. 2014;124:5337-51
7. Kumar E, Pickard L, Okosun J. Pathogenesis of follicular lymphoma: genetics to the microenvironment to clinical translation. Br J Haematol. 2021;194:810-21
8. Dobaño-López C, Araujo-Ayala F, Serrat N, Valero JG, Pérez-Galán P. Follicular Lymphoma Microenvironment: An Intricate Network Ready for Therapeutic Intervention. Cancers (Basel). 2021 13
9. Lackraj T, Goswami R, Kridel R. Pathogenesis of follicular lymphoma. Best Pract Res Clin Haematol. 2018;31:2-14
10. Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC. et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351:2159-69
11. Nam AS, Chaligne R, Landau DA. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nature Reviews Genetics. 2021;22:3-18
12. Radtke AJ, Postovalova E, Varlamova A, Bagaev A, Sorokina M, Kudryashova O. et al. A Multi-scale, Multiomic Atlas of Human Normal and Follicular Lymphoma Lymph Nodes. bioRxiv. 2022. 2022 06.03.494716
13. Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL. et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121:1604-11
14. Bouska A, McKeithan TW, Deffenbacher KE, Lachel C, Wright GW, Iqbal J. et al. Genome-wide copy-number analyses reveal genomic abnormalities involved in transformation of follicular lymphoma. Blood. 2014;123:1681-90
15. Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S. et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014;123:1487-98
16. Okosun J, Bödör C, Wang J, Araf S, Yang CY, Pan C. et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176-81
17. Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB. et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6:130-40
18. Kridel R, Mottok A, Farinha P, Ben-Neriah S, Ennishi D, Zheng Y. et al. Cell of origin of transformed follicular lymphoma. Blood. 2015;126:2118-27
19. Kridel R, Chan FC, Mottok A, Boyle M, Farinha P, Tan K. et al. Histological Transformation and Progression in Follicular Lymphoma: A Clonal Evolution Study. PLoS Med. 2016;13:e1002197
20. Bouska A, Zhang W, Gong Q, Iqbal J, Scuto A, Vose J. et al. Combined copy number and mutation analysis identifies oncogenic pathways associated with transformation of follicular lymphoma. Leukemia. 2017;31:83-91
21. Krysiak K, Gomez F, White BS, Matlock M, Miller CA, Trani L. et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood. 2017;129:473-83
22. Oricchio E, Nanjangud G, Wolfe AL, Schatz JH, Mavrakis KJ, Jiang M. et al. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell. 2011;147:554-64
23. Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang YW, Zhao CY. et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nature Medicine. 2015;21:1199 -+
24. Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M. et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21:1190-8
25. Jiang Y, Ortega-Molina A, Geng H, Ying HY, Hatzi K, Parsa S. et al. CREBBP Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discov. 2017;7:38-53
26. Zhang J, Vlasevska S, Wells VA, Nataraj S, Holmes AB, Duval R. et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-cell Lymphoma. Cancer Discov. 2017;7:322-37
27. Horton SJ, Giotopoulos G, Yun H, Vohra S, Sheppard O, Bashford-Rogers R. et al. Early loss of Crebbp confers malignant stem cell properties on lymphoid progenitors. Nat Cell Biol. 2017;19:1093-104
28. Meyer SN, Scuoppo C, Vlasevska S, Bal E, Holmes AB, Holloman M. et al. Unique and Shared Epigenetic Programs of the CREBBP and EP300 Acetyltransferases in Germinal Center B Cells Reveal Targetable Dependencies in Lymphoma. Immunity. 2019;51:535-47.e9
29. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R. et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nature Genetics. 2010;42:181-U24
30. Beguelin W, Rivas MA, Fernandez MTC, Teater M, Purwada A, Redmond D. et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nature Communications. 2017 8
31. Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M. et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677-92
32. Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, Chin CR. et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature. 2021;589:299 -+
33. Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35-43
34. Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M. et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16:1111-22
35. Cardenas MG, Oswald E, Yu WB, Xue FT, MacKerell AD, Melnick AM. The Expanding Role of the BCL6 Oncoprotein as a Cancer Therapeutic Target. Clinical Cancer Research. 2017;23:885-93
36. Hatzi K, Geng H, Doane AS, Meydan C, LaRiviere R, Cardenas M. et al. Histone demethylase LSD1 is required for germinal center formation and BCL6-driven lymphomagenesis. Nat Immunol. 2019;20:86-96
37. Horn H, Kohler C, Witzig R, Kreuz M, Leich E, Klapper W. et al. Gene expression profiling reveals a close relationship between follicular lymphoma grade 3A and 3B, but distinct profiles of follicular lymphoma grade 1 and 2. Haematologica. 2018;103:1182-90
38. Valls E, Lobry C, Geng H, Wang L, Cardenas M, Rivas M. et al. BCL6 Antagonizes NOTCH2 to Maintain Survival of Human Follicular Lymphoma Cells. Cancer Discov. 2017;7:506-21
39. Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M. et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol. 2013;14:1084-92
40. Pon JR, Wong J, Saberi S, Alder O, Moksa M, Grace Cheng SW. et al. MEF2B mutations in non-Hodgkin lymphoma dysregulate cell migration by decreasing MEF2B target gene activation. Nat Commun. 2015;6:7953
41. Roberto MP, Varano G, Vinas-Castells R, Holmes AB, Kumar R, Pasqualucci L. et al. Mutations in the transcription factor FOXO1 mimic positive selection signals to promote germinal center B cell expansion and lymphomagenesis. Immunity. 2021;54:1807-24.e14
42. Huet S, Sujobert P, Salles G. From genetics to the clinic: a translational perspective on follicular lymphoma. Nat Rev Cancer. 2018;18:224-39
43. Lamason RL, McCully RR, Lew SM, Pomerantz JL. Oncogenic CARD11 Mutations Induce Hyperactive Signaling by Disrupting Autoinhibition by the PKC-Responsive Inhibitory Domain. Biochemistry. 2010;49:8240-50
44. Hu N, Wang F, Sun T, Xu Z, Zhang J, Bernard D. et al. Follicular Lymphoma-associated BTK Mutations are Inactivating Resulting in Augmented AKT Activation. Clin Cancer Res. 2021;27:2301-13
45. Schmitz R, Hansmann ML, Bohle V, Martin-Subero JI, Hartmann S, Mechtersheimer G. et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med. 2009;206:981-9
46. Odabashian M, Carlotti E, Araf S, Okosun J, Spada F, Gribben JG. et al. IGHV sequencing reveals acquired N-glycosylation sites as a clonal and stable event during follicular lymphoma evolution. Blood. 2020;135:834-44
47. Myklebust JH, Irish JM, Brody J, Czerwinski DK, Houot R, Kohrt HE. et al. High PD-1 expression and suppressed cytokine signaling distinguish T cells infiltrating follicular lymphoma tumors from peripheral T cells. Blood. 2013;121:1367-76
48. Yildiz M, Li H, Bernard D, Amin NA, Ouillette P, Jones S. et al. Activating STAT6 mutations in follicular lymphoma. Blood. 2015;125:668-79
49. Mottok A, Renné C, Seifert M, Oppermann E, Bechstein W, Hansmann ML. et al. Inactivating SOCS1 mutations are caused by aberrant somatic hypermutation and restricted to a subset of B-cell lymphoma entities. Blood. 2009;114:4503-6
50. Ortega-Molina A, Deleyto-Seldas N, Carreras J, Sanz A, Lebrero-Fernández C, Menéndez C. et al. Oncogenic Rag GTPase signaling enhances B cell activation and drives follicular lymphoma sensitive to pharmacological inhibition of mTOR. Nat Metab. 2019;1:775-89
51. Ying ZX, Jin MY, Peterson LF, Bernard D, Saiya-Cork K, Yildiz M. et al. Recurrent Mutations in the MTOR Regulator RRAGC in Follicular Lymphoma. Clinical Cancer Research. 2016;22:5383-93
52. Okosun J, Wolfson RL, Wang J, Araf S, Wilkins L, Castellano BM. et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat Genet. 2016;48:183-8
53. Wang F, Gatica D, Ying ZX, Peterson LF, Kim P, Bernard D. et al. Follicular lymphoma-associated mutations in vacuolar ATPase ATP6V1B2 activate autophagic flux and mTOR. J Clin Invest. 2019;129:1626-40
54. Wang F, Yang Y, Boudagh G, Eskelinen EL, Klionsky DJ, Malek SN. Follicular lymphoma-associated mutations in the V-ATPase chaperone VMA21 activate autophagy creating a targetable dependency. Autophagy. 2022
55. Oricchio E, Katanayeva N, Donaldson MC, Sungalee S, Pasion JP, Béguelin W. et al. Genetic and epigenetic inactivation of SESTRIN1 controls mTORC1 and response to EZH2 inhibition in follicular lymphoma. Sci Transl Med. 2017 9
56. Karube K, Martinez D, Royo C, Navarro A, Pinyol M, Cazorla M. et al. Recurrent mutations of NOTCH genes in follicular lymphoma identify a distinctive subset of tumours. Journal of Pathology. 2014;234:423-30
57. Healy JA, Nugent A, Rempel RE, Moffitt AB, Davis NS, Jiang XY. et al. GNA13 loss in germinal center B cells leads to impaired apoptosis and promotes lymphoma in vivo. Blood. 2016;127:2723-31
58. Muppidi JR, Schmitz R, Green JA, Xiao WM, Larsens AB, Braun SE. et al. Loss of signalling via G alpha 13 in germinal centre B-cell-derived lymphoma. Nature. 2014;516:254 -+
59. Oricchio E, Ciriello G, Jiang M, Boice MH, Schatz JH, Heguy A. et al. Frequent disruption of the RB pathway in indolent follicular lymphoma suggests a new combination therapy. Journal of Experimental Medicine. 2014;211:1375-87
60. Pae J, Ersching J, Castro TBR, Schips M, Mesin L, Allon SJ. et al. Cyclin D3 drives inertial cell cycling in dark zone germinal center B cells. Journal of Experimental Medicine. 2021 218
61. Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E. et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell. 2016;167:405-18.e13
62. Bararia D, Hildebrand JA, Stolz S, Haebe S, Alig S, Trevisani CP. et al. Cathepsin S Alterations Induce a Tumor-Promoting Immune Microenvironment in Follicular Lymphoma. Cell Reports. 2020 31
63. Dheilly E, Battistello E, Katanayeva N, Sungalee S, Michaux J, Duns G. et al. Cathepsin S Regulates Antigen Processing and T Cell Activity in Non-Hodgkin Lymphoma. Cancer Cell. 2020;37:674-89.e12
64. Wang N, Qin W, Zheng Z, Zhao M, Xiong J, Fang H. et al. Hepatitis B virus-associated follicular lymphoma presents T-cell inflamed phenotype and response to lenalidomide. Cancer Commun (Lond). 2022;42:170-4
65. Green MR, Kihira S, Liu CL, Nair RV, Salari R, Gentles AJ. et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A. 2015;112:E1116-25
66. Béguelin W, Teater M, Meydan C, Hoehn KB, Phillip JM, Soshnev AA. et al. Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell. 2020;37:655-73.e11
67. Mutant EZH2 Drives Follicular Lymphoma by Altering B-cell Dependencies. Cancer Discov. 2020; 10: 900.
68. Ortega-Molina A, Deleyto-Seldas N, Carreras J, Sanz A, Lebrero-Fernandez C, Menendez C. et al. Oncogenic Rag GTPase signalling enhances B cell activation and drives follicular lymphoma sensitive to pharmacological inhibition of mTOR. Nature Metabolism. 2019;1:775-89
69. Haebe S, Shree T, Sathe A, Day G, Czerwinski DK, Grimes SM. et al. Single-cell analysis can define distinct evolution of tumor sites in follicular lymphoma. Blood. 2021;137:2869-80
70. Araf S, Wang J, Korfi K, Pangault C, Kotsiou E, Rio-Machin A. et al. Genomic profiling reveals spatial intra-tumor heterogeneity in follicular lymphoma. Leukemia. 2018;32:1261-5
71. Andor N, Simonds EF, Czerwinski DK, Chen J, Grimes SM, Wood-Bouwens C. et al. Single-cell RNA-Seq of follicular lymphoma reveals malignant B-cell types and coexpression of T-cell immune checkpoints. Blood. 2019;133:1119-29
72. Roider T, Seufert J, Uvarovskii A, Frauhammer F, Bordas M, Abedpour N. et al. Dissecting intratumour heterogeneity of nodal B-cell lymphomas at the transcriptional, genetic and drug-response levels. Nat Cell Biol. 2020;22:896-906
73. Milpied P, Gandhi AK, Cartron G, Pasqualucci L, Tarte K, Nadel B. et al. Follicular lymphoma dynamics. Adv Immunol. 2021;150:43-103
74. Milpied P, Cervera-Marzal I, Mollichella ML, Tesson B, Brisou G, Traverse-Glehen A. et al. Human germinal center transcriptional programs are de-synchronized in B cell lymphoma. Nat Immunol. 2018;19:1013-24
75. Milpied P, Nadel B, Spinelli L, Navarro J-M, Dong C, Gil L. et al. Single-Cell RNA Sequencing Identifies a Pseudo-Immune Differentiation Axis As the Main Source of Functional Heterogeneity in Follicular Lymphoma B-Cells. Blood. 2019;134:548 -
76. Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z. et al. Single-cell analysis of germinal-center B cells informs on lymphoma cell of origin and outcome. J Exp Med. 2020 217
77. Lamaison C, Tarte K. B cell/stromal cell crosstalk in health, disease, and treatment: Follicular lymphoma as a paradigm. Immunol Rev. 2021;302:273-85
78. Abe Y, Sakata-Yanagimoto M, Fujisawa M, Miyoshi H, Suehara Y, Hattori K. et al. A single-cell atlas of non-haematopoietic cells in human lymph nodes and lymphoma reveals a landscape of stromal remodelling. Nat Cell Biol. 2022
79. Mourcin F, Verdière L, Roulois D, Amin R, Lamaison C, Sibut V. et al. Follicular lymphoma triggers phenotypic and functional remodeling of the human lymphoid stromal cell landscape. Immunity. 2021;54:1788-806.e7
80. Yang ZZ, Kim HJ, Villasboas JC, Price-Troska T, Jalali S, Wu H. et al. Mass Cytometry Analysis Reveals that Specific Intratumoral CD4(+) T Cell Subsets Correlate with Patient Survival in Follicular Lymphoma. Cell Rep. 2019;26:2178-93.e3
81. Mondello P, Fama A, Larson MC, Feldman AL, Villasboas JC, Yang ZZ. et al. Lack of intrafollicular memory CD4 + T cells is predictive of early clinical failure in newly diagnosed follicular lymphoma. Blood Cancer J. 2021;11:130
82. Yang ZZ, Kim HJ, Wu H, Jalali S, Tang X, Krull JE. et al. TIGIT Expression Is Associated with T-cell Suppression and Exhaustion and Predicts Clinical Outcome and Anti-PD-1 Response in Follicular Lymphoma. Clin Cancer Res. 2020;26:5217-31
83. Yang ZZ, Kim HJ, Villasboas JC, Chen YP, Price-Troska T, Jalali S. et al. Expression of LAG-3 defines exhaustion of intratumoral PD-1(+) T cells and correlates with poor outcome in follicular lymphoma. Oncotarget. 2017;8:61425-39
84. Yang ZZ, Grote DM, Ziesmer SC, Xiu B, Novak AJ, Ansell SM. PD-1 expression defines two distinct T-cell sub-populations in follicular lymphoma that differentially impact patient survival. Blood Cancer J. 2015;5:e281
85. Le KS, Thibult ML, Just-Landi S, Pastor S, Gondois-Rey F, Granjeaud S. et al. Follicular B Lymphomas Generate Regulatory T Cells via the ICOS/ICOSL Pathway and Are Susceptible to Treatment by Anti-ICOS/ICOSL Therapy. Cancer Res. 2016;76:4648-60
86. Josefsson SE, Huse K, Kolstad A, Beiske K, Pende D, Steen CB. et al. T Cells Expressing Checkpoint Receptor TIGIT Are Enriched in Follicular Lymphoma Tumors and Characterized by Reversible Suppression of T-cell Receptor Signaling. Clin Cancer Res. 2018;24:870-81
87. Gravelle P, Do C, Franchet C, Mueller S, Oberic L, Ysebaert L. et al. Impaired functional responses in follicular lymphoma CD8(+)TIM-3(+) T lymphocytes following TCR engagement. Oncoimmunology. 2016 5
88. Han G, Deng Q, Marques-Piubelli ML, Dai E, Dang M, Ma MCJ. et al. Follicular Lymphoma Microenvironment Characteristics Associated with Tumor Cell Mutations and MHC Class II Expression. Blood Cancer Discov. 2022;3:428-43
89. Pangault C, Amé-Thomas P, Ruminy P, Rossille D, Caron G, Baia M. et al. Follicular lymphoma cell niche: identification of a preeminent IL-4-dependent T(FH)-B cell axis. Leukemia. 2010;24:2080-9
90. Pandey S, Mourcin F, Marchand T, Nayar S, Guirriec M, Pangault C. et al. IL-4/CXCL12 loop is a key regulator of lymphoid stroma function in follicular lymphoma. Blood. 2017;129:2507-18
91. Calvo KR, Dabir B, Kovach A, Devor C, Bandle R, Bond A. et al. IL-4 protein expression and basal activation of Erk in vivo in follicular lymphoma. Blood. 2008;112:3818-26
92. Epron G, Ame-Thomas P, Le Priol J, Pangault C, Dulong J, Lamy T. et al. Monocytes and T cells cooperate to favor normal and follicular lymphoma B-cell growth: role of IL-15 and CD40L signaling. Leukemia. 2012;26:139-48
93. Amé-Thomas P, Hoeller S, Artchounin C, Misiak J, Braza MS, Jean R. et al. CD10 delineates a subset of human IL-4 producing follicular helper T cells involved in the survival of follicular lymphoma B cells. Blood. 2015;125:2381-5
94. Pangault C, Amé-Thomas P, Rossille D, Dulong J, Caron G, Nonn C. et al. Integrative Analysis of Cell Crosstalk within Follicular Lymphoma Cell Niche: Towards a Definition of the FL Supportive Synapse. Cancers (Basel). 2020 12
95. Nedelkovska H, Rosenberg AF, Hilchey SP, Hyrien O, Burack WR, Quataert SA. et al. Follicular Lymphoma Tregs Have a Distinct Transcription Profile Impacting Their Migration and Retention in the Malignant Lymph Node. Plos One. 2016 11
96. Ame-Thomas P, Le Priol J, Yssel H, Caron G, Pangault C, Jean R. et al. Characterization of intratumoral follicular helper T cells in follicular lymphoma: role in the survival of malignant B cells. Leukemia. 2012;26:1053-63
97. Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T. et al. A distinct subpopulation of CD25(-) T-follicular regulatory cells localizes in the germinal centers. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E6400-E9
98. Sage PT, Ron-Harel N, Juneja VR, Sen DR, Maleri S, Sungnak W. et al. Suppression by T-FR\ cells leads to durable and selective inhibition of B cell effector function. Nature Immunology. 2016;17:1436-46
99. Chu F, Li HS, Liu X, Cao J, Ma W, Ma Y. et al. CXCR5(+)CD8(+) T cells are a distinct functional subset with an antitumor activity. Leukemia. 2019;33:2640-53
100. Wu H, Tang X, Kim HJ, Jalali S, Pritchett JC, Villasboas JC. et al. Expression of KLRG1 and CD127 defines distinct CD8(+) subsets that differentially impact patient outcome in follicular lymphoma. J Immunother Cancer. 2021 9
101. Josefsson SE, Beiske K, Blaker YN, Forsund MS, Holte H, Ostenstad B. et al. TIGIT and PD-1 Mark Intratumoral T Cells with Reduced Effector Function in B-cell Non-Hodgkin Lymphoma. Cancer Immunology Research. 2019;7:355-62
102. Jayasingam SD, Citartan M, Thang TH, Zin AAM, Ang KC, Ch'ng ES. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Frontiers in Oncology. 2020 9
103. Amin R, Mourcin F, Uhel F, Pangault C, Ruminy P, Dupré L. et al. DC-SIGN-expressing macrophages trigger activation of mannosylated IgM B-cell receptor in follicular lymphoma. Blood. 2015;126:1911-20
104. Valero JG, Matas-Céspedes A, Arenas F, Rodriguez V, Carreras J, Serrat N. et al. The receptor of the colony-stimulating factor-1 (CSF-1R) is a novel prognostic factor and therapeutic target in follicular lymphoma. Leukemia. 2021;35:2635-49
105. Chen YP, Kim HJ, Wu H, Price-Troska T, Villasboas JC, Jalali S. et al. SIRPα expression delineates subsets of intratumoral monocyte/macrophages with different functional and prognostic impact in follicular lymphoma. Blood Cancer J. 2019;9:84
106. Misiak J, Jean R, Rodriguez S, Deleurme L, Lamy T, Tarte K. et al. Human Lymphoid Stromal Cells Contribute to Polarization of Follicular T Cells Into IL-4 Secreting Cells. Front Immunol. 2020;11:559866
107. Pepe G, Di Napoli A, Cippitelli C, Scarpino S, Pilozzi E, Ruco L. Reduced lymphotoxin-beta production by tumour cells is associated with loss of follicular dendritic cell phenotype and diffuse growth in follicular lymphoma. J Pathol Clin Res. 2018;4:124-34
108. Jalali S, Ansell SM. Role of the Bone Marrow Niche in Supporting the Pathogenesis of Lymphoid Malignancies. Front Cell Dev Biol. 2021;9:692320
109. Dumontet E, Mancini SJC, Tarte K. Bone Marrow Lymphoid Niche Adaptation to Mature B Cell Neoplasms. Front Immunol. 2021;12:784691
110. Dumontet E, Pangault C, Roulois D, Desoteux M, Léonard S, Marchand T. et al. Extracellular vesicles shed by follicular lymphoma B cells promote polarization of the bone marrow stromal cell niche. Blood. 2021;138:57-70
111. Mozessohn L, Cheung MC, Crump M, Buckstein R, Berinstein N, Imrie K. et al. Chemoimmunotherapy resistant follicular lymphoma: predictors of resistance, association with transformation and prognosis. Leukemia & Lymphoma. 2014;55:2502-7
112. Sarkozy C, Trneny M, Xerri L, Wickham N, Feugier P, Leppa S. et al. Risk Factors and Outcomes for Patients With Follicular Lymphoma Who Had Histologic Transformation After Response to First-Line Immunochemotherapy in the PRIMA Trial. Journal of Clinical Oncology. 2016;34:2575-82
113. Brodtkorb M, Lingjærde OC, Huse K, Trøen G, Hystad M, Hilden VI. et al. Whole-genome integrative analysis reveals expression signatures predicting transformation in follicular lymphoma. Blood. 2014;123:1051-4
114. Loeffler M, Kreuz M, Haake A, Hasenclever D, Trautmann H, Arnold C. et al. Genomic and epigenomic co-evolution in follicular lymphomas. Leukemia. 2015;29:456-63
115. Qu X, Li H, Braziel RM, Passerini V, Rimsza LM, Hsi ED. et al. Genomic alterations important for the prognosis in patients with follicular lymphoma treated in SWOG study S0016. Blood. 2019;133:81-93
116. Musilova K, Devan J, Cerna K, Seda V, Pavlasova G, Sharma S. et al. miR-150 downregulation contributes to the high-grade transformation of follicular lymphoma by upregulating FOXP1 levels. Blood. 2018;132:2389-400
117. Lou X, Fu J, Zhao X, Zhuansun X, Rong C, Sun M. et al. MiR-7e-5p downregulation promotes transformation of low-grade follicular lymphoma to aggressive lymphoma by modulating an immunosuppressive stroma through the upregulation of FasL in M1 macrophages. J Exp Clin Cancer Res. 2020;39:237
118. Smeltzer JP, Jones JM, Ziesmer SC, Grote DM, Xiu B, Ristow KM. et al. Pattern of CD14+ follicular dendritic cells and PD1+ T cells independently predicts time to transformation in follicular lymphoma. Clin Cancer Res. 2014;20:2862-72
119. Mozas P, Rivero A, Rivas-Delgado A, Nadeu F, Clot G, Corre JG. et al. A low lymphocyte-to-monocyte ratio is an independent predictor of poorer survival and higher risk of histological transformation in follicular lymphoma. Leukemia & Lymphoma. 2021;62:104-11
120. Farinha P, Al-Tourah A, Gill K, Klasa R, Connors JM, Gascoyne RD. The architectural pattern of FOXP3-positive T cells in follicular lymphoma is an independent predictor of survival and histologic transformation. Blood. 2010;115:289-95
121. Farinha P, Kyle AH, Minchinton AI, Connors JM, Karsan A, Gascoyne RD. Vascularization predicts overall survival and risk of transformation in follicular lymphoma. Haematologica. 2010;95:2157-60
122. Kridel R, Sehn LH, Gascoyne RD. Can histologic transformation of follicular lymphoma be predicted and prevented? Blood. 2017;130:258-66
123. Tobin JWD, Keane C, Gunawardana J, Mollee P, Birch S, Hoang T. et al. Progression of Disease Within 24 Months in Follicular Lymphoma Is Associated With Reduced Intratumoral Immune Infiltration. J Clin Oncol. 2019;37:3300-9
124. Rai S, Inoue H, Sakai K, Hanamoto H, Matsuda M, Maeda Y. et al. Decreased expression of T-cell-associated immune markers predicts poor prognosis in patients with follicular lymphoma. Cancer Sci. 2021
125. Federico M, Bellei M, Marcheselli L, Luminari S, Lopez-Guillermo A, Vitolo U. et al. Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol. 2009;27:4555-62
126. Solal-Céligny P, Roy P, Colombat P, White J, Armitage JO, Arranz-Saez R. et al. Follicular lymphoma international prognostic index. Blood. 2004;104:1258-65
127. Bachy E, Maurer MJ, Habermann TM, Gelas-Dore B, Maucort-Boulch D, Estell JA. et al. A simplified scoring system in de novo follicular lymphoma treated initially with immunochemotherapy. Blood. 2018;132:49-58
128. Mir F, Mattiello F, Grigg A, Herold M, Hiddemann W, Marcus R. et al. Follicular Lymphoma Evaluation Index (FLEX): A new clinical prognostic model that is superior to existing risk scores for predicting progression-free survival and early treatment failure after frontline immunochemotherapy. Am J Hematol. 2020;95:1503-10
129. Jurinovic V, Kridel R, Staiger AM, Szczepanowski M, Horn H, Dreyling MH. et al. Clinicogenetic risk models predict early progression of follicular lymphoma after first-line immunochemotherapy. Blood. 2016;128:1112-20
130. Huet S, Tesson B, Jais JP, Feldman AL, Magnano L, Thomas E. et al. A gene-expression profiling score for prediction of outcome in patients with follicular lymphoma: a retrospective training and validation analysis in three international cohorts. Lancet Oncol. 2018;19:549-61
131. Wallace D, Casulo C. Early Progressing Follicular Lymphoma. Curr Oncol Rep. 2021;23:149
132. Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S. et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020;21:1433-42
133. Hanel W, Epperla N. Evolving therapeutic landscape in follicular lymphoma: a look at emerging and investigational therapies. J Hematol Oncol. 2021;14:104
134. Ogura M, Ando K, Suzuki T, Ishizawa K, Oh SY, Itoh K. et al. A multicentre phase II study of vorinostat in patients with relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. Br J Haematol. 2014;165:768-76
135. Evens AM, Balasubramanian S, Vose JM, Harb W, Gordon LI, Langdon R. et al. A Phase I/II Multicenter, Open-Label Study of the Oral Histone Deacetylase Inhibitor Abexinostat in Relapsed/Refractory Lymphoma. Clin Cancer Res. 2016;22:1059-66
136. Ribrag V, Kim WS, Bouabdallah R, Lim ST, Coiffier B, Illes A. et al. Safety and efficacy of abexinostat, a pan-histone deacetylase inhibitor, in non-Hodgkin lymphoma and chronic lymphocytic leukemia: results of a phase II study. Haematologica. 2017;102:903-9
137. Kirschbaum M, Frankel P, Popplewell L, Zain J, Delioukina M, Pullarkat V. et al. Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin's lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29:1198-203
138. Heward J, Konali L, D'Avola A, Close K, Yeomans A, Philpott M. et al. KDM5 inhibition offers a novel therapeutic strategy for the treatment of KMT2D mutant lymphomas. Blood. 2021;138:370-81
139. Wang L, Qin W, Huo YJ, Li X, Shi Q, Rasko JEJ. et al. Advances in targeted therapy for malignant lymphoma. Signal Transduct Target Ther. 2020;5:15
140. Zinzani PL, Flinn IW, Yuen SLS, Topp MS, Rusconi C, Fleury I. et al. Venetoclax-rituximab with or without bendamustine vs bendamustine-rituximab in relapsed/refractory follicular lymphoma. Blood. 2020;136:2628-37
141. Blombery P, Birkinshaw RW, Nguyen T, Gong JN, Thompson ER, Xu Z. et al. Characterization of a novel venetoclax resistance mutation (BCL2 Phe104Ile) observed in follicular lymphoma. Br J Haematol. 2019;186:e188-e91
142. Gopal AK, Schuster SJ, Fowler NH, Trotman J, Hess G, Hou JZ. et al. Ibrutinib as Treatment for Patients With Relapsed/Refractory Follicular Lymphoma: Results From the Open-Label, Multicenter, Phase II DAWN Study. J Clin Oncol. 2018;36:2405-12
143. Bartlett NL, Costello BA, LaPlant BR, Ansell SM, Kuruvilla JG, Reeder CB. et al. Single-agent ibrutinib in relapsed or refractory follicular lymphoma: a phase 2 consortium trial. Blood. 2018;131:182-90
144. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M. et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. 2018;560:387-91
145. Tam CS, Quach H, Nicol A, Badoux X, Rose H, Prince HM. et al. Zanubrutinib (BGB-3111) plus obinutuzumab in patients with chronic lymphocytic leukemia and follicular lymphoma. Blood Adv. 2020;4:4802-11
146. Fowler NH, Coleman M, Stevens DA, Smith SM, Venugopal P, Martin P. et al. Acalabrutinib alone or in combination with rituximab (R) in follicular lymphoma (FL). Journal of Clinical Oncology. 2018;36:7549 -
147. Hamlin PA, Flinn IW, Wagner-Johnston N, Burger JA, Coffey GP, Conley PB. et al. Efficacy and safety of the dual SYK/JAK inhibitor cerdulatinib in patients with relapsed or refractory B-cell malignancies: Results of a phase I study. American Journal of Hematology. 2019;94:E90-E3
148. Flinn IW, Miller CB, Ardeshna KM, Tetreault S, Assouline SE, Mayer J. et al. DYNAMO: A Phase II Study of Duvelisib (IPI-145) in Patients With Refractory Indolent Non-Hodgkin Lymphoma. J Clin Oncol. 2019;37:912-22
149. Salles G, Schuster SJ, de Vos S, Wagner-Johnston ND, Viardot A, Blum KA. et al. Efficacy and safety of idelalisib in patients with relapsed, rituximab- and alkylating agent-refractory follicular lymphoma: a subgroup analysis of a phase 2 study. Haematologica. 2017;102:e156-e9
150. Dreyling M, Santoro A, Mollica L, Leppä S, Follows G, Lenz G. et al. Long-term safety and efficacy of the PI3K inhibitor copanlisib in patients with relapsed or refractory indolent lymphoma: 2-year follow-up of the CHRONOS-1 study. Am J Hematol. 2020;95:362-71
151. Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ. et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008-18
152. Fowler NH, Samaniego F, Jurczak W, Ghosh N, Derenzini E, Reeves JA. et al. Umbralisib, a Dual PI3Kδ/CK1ε Inhibitor in Patients With Relapsed or Refractory Indolent Lymphoma. J Clin Oncol. 2021;39:1609-18
153. Phillips TJ, Forero-Torres A, Sher T, Diefenbach CS, Johnston P, Talpaz M. et al. Phase 1 study of the PI3Kδ inhibitor INCB040093 ± JAK1 inhibitor itacitinib in relapsed/refractory B-cell lymphoma. Blood. 2018;132:293-306
154. Qualls D, Salles G. Prospects in the management of patients with follicular lymphoma beyond first-line therapy. Haematologica. 2022;107:19-34
155. Bennani NN, LaPlant BR, Ansell SM, Habermann TM, Inwards DJ, Micallef IN. et al. Efficacy of the oral mTORC1 inhibitor everolimus in relapsed or refractory indolent lymphoma. Am J Hematol. 2017;92:448-53
156. Smith SM, van Besien K, Karrison T, Dancey J, McLaughlin P, Younes A. et al. Temsirolimus has activity in non-mantle cell non-Hodgkin's lymphoma subtypes: The University of Chicago phase II consortium. J Clin Oncol. 2010;28:4740-6
157. Gribben JG, Fowler N, Morschhauser F. Mechanisms of Action of Lenalidomide in B-Cell Non-Hodgkin Lymphoma. J Clin Oncol. 2015;33:2803-11
158. Leonard JP, Trneny M, Izutsu K, Fowler NH, Hong XN, Zhu J. et al. AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. Journal of Clinical Oncology. 2019;37:1188 -+
159. Morschhauser F, Fowler NH, Feugier P, Bouabdallah R, Tilly H, Palomba ML. et al. Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N Engl J Med. 2018;379:934-47
160. Morschhauser F, Le Gouill S, Feugier P, Bailly S, Nicolas-Virelizier E, Bijou F. et al. Obinutuzumab combined with lenalidomide for relapsed or refractory follicular B-cell lymphoma (GALEN): a multicentre, single-arm, phase 2 study. Lancet Haematol. 2019;6:e429-e37
161. Bachy E, Houot R, Feugier P, Bouabdallah K, Bouabdallah R, Nicolas-Virelizier E. et al. Obinutuzumab plus lenalidomide (GALEN) in advanced, previously untreated follicular lymphoma in need of systemic therapy. Blood. 2021
162. Armand P, Janssens A, Gritti G, Radford J, Timmerman J, Pinto A. et al. Efficacy and safety results from CheckMate 140, a phase 2 study of nivolumab for relapsed/refractory follicular lymphoma. Blood. 2021;137:637-45
163. Younes A, Brody J, Carpio C, Lopez-Guillermo A, Ben-Yehuda D, Ferhanoglu B. et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: a phase 1/2a study. Lancet Haematol. 2019;6:e67-e78
164. Timmerman J, Herbaux C, Ribrag V, Zelenetz AD, Houot R, Neelapu SS. et al. Urelumab alone or in combination with rituximab in patients with relapsed or refractory B-cell lymphoma. Am J Hematol. 2020;95:510-20
165. Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O'Gorman WE. et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8(+) T cell responses. Immunity. 2022;55:512-26.e9
166. Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. Journal for Immunotherapy of Cancer. 2020 8
167. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989-1004
168. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S. et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699-713
169. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N. et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin's Lymphoma. N Engl J Med. 2018;379:1711-21
170. The Bispecific Antibody Epcoritamab Produces Responses in Lymphoma. Cancer Discov. 2021; 11: 2671.
171. Hutchings M, Mous R, Clausen MR, Johnson P, Linton KM, Chamuleau MED. et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label, phase 1/2 study. Lancet. 2021;398:1157-69
172. Hutchings M, Morschhauser F, Iacoboni G, Carlo-Stella C, Offner FC, Sureda A. et al. Glofitamab, a Novel, Bivalent CD20-Targeting T-Cell-Engaging Bispecific Antibody, Induces Durable Complete Remissions in Relapsed or Refractory B-Cell Lymphoma: A Phase I Trial. J Clin Oncol. 2021;39:1959-70
173. Bannerji R, Allan JN, Arnason JE, Brown JR, Advani R, Ansell SM. et al. Odronextamab (REGN1979), a Human CD20 x CD3 Bispecific Antibody, Induces Durable, Complete Responses in Patients with Highly Refractory B-Cell Non-Hodgkin Lymphoma, Including Patients Refractory to CAR T Therapy. Blood. 2020;136:42-3
174. Assouline SE, Kim WS, Sehn LH, Schuster SJ, Cheah CY, Nastoupil LJ. et al. Mosunetuzumab Shows Promising Efficacy in Patients with Multiply Relapsed Follicular Lymphoma: Updated Clinical Experience from a Phase I Dose-Escalation Trial. Blood. 2020;136:42-4
175. van der Horst HJ, de Jonge AV, Hiemstra IH, Gelderloos AT, Berry D, Hijmering NJ. et al. Epcoritamab induces potent anti-tumor activity against malignant B-cells from patients with DLBCL, FL and MCL, irrespective of prior CD20 monoclonal antibody treatment. Blood Cancer J. 2021;11:38
176. Jacobson C, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G. et al. Primary Analysis of Zuma-5: A Phase 2 Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients with Relapsed/Refractory (R/R) Indolent Non-Hodgkin Lymphoma (iNHL). Blood. 2020 136
177. Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G. et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022;23:91-103
178. Tisagenlecleucel Is Safe and Effective in Relapsed/Refractory Follicular Lymphoma. Cancer Discov. 2022.
179. Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J. et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med. 2021
180. Yuan H, Nishikori M, Otsuka Y, Arima H, Kitawaki T, Takaori-Kondo A. The EZH2 inhibitor tazemetostat upregulates the expression of CCL17/TARC in B-cell lymphoma and enhances T-cell recruitment. Cancer Sci. 2021;112:4604-16
181. Mondello P, Tadros S, Teater M, Fontan L, Chang AY, Jain N. et al. Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discovery. 2020;10:440-59
182. Chellappa S, Kushekhar K, Munthe LA, Tjonnfjord GE, Aandahl EM, Okkenhaug K. et al. The PI3K p110 delta Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. Journal of Immunology. 2019;202:1397-405
183. Serrat N, Guerrero-Hernandez M, Matas-Cespedes A, Yahiaoui A, Valero JG, Nadeu F. et al. PI3K delta inhibition reshapes follicular lymphoma-immune microenvironment cross talk and unleashes the activity of venetoclax. Blood Advances. 2020;4:4217-31
184. Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA. et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21:1209-15
185. Cader FZ, Hu XH, Goh WL, Wienand K, Ouyang J, Mandato E. et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nature Medicine. 2020 26
Author contact
![]() Corresponding author: Weili Zhao. Email: zwl10909com.cn
Corresponding author: Weili Zhao. Email: zwl10909com.cn