Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(7):2167-2197. doi:10.7150/ijbs.82191 This issue Cite
Review
Application of single-cell RNA sequencing on human testicular samples: a comprehensive review
Fan Dong1,2, Ping Ping1,2, Yi Ma1,2 ![]() , Xiang-Feng Chen1,2,3
, Xiang-Feng Chen1,2,3 ![]()
1. Center for Reproductive Medicine, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China.
2. Shanghai Key Laboratory for Assisted Reproduction and Reproductive Genetics, Shanghai, China.
3. Shanghai Human Sperm Bank, Shanghai, China.
Received 2022-12-27; Accepted 2023-3-25; Published 2023-4-9
Abstract

So far there has been no comprehensive review using systematic literature search strategies to show the application of single-cell RNA sequencing (scRNA-seq) in the human testis of the whole life cycle (from embryos to aging males). Here, we summarized the application of scRNA-seq analyses on various human testicular biological samples. A systematic search was conducted in PubMed and Gene Expression Omnibus (GEO), focusing on English researches published after 2009. Articles related to GEO data-series were also retrieved in PubMed or BioRxiv. 81 full-length studies were finally included in the review. ScRNA-seq has been widely used on different human testicular samples with various library strategies, and new cell subtypes such as State 0 spermatogonial stem cells (SSC) and stage_a/b/c Sertoli cells (SC) were identified. For the development of normal testes, scRNA-seq-based evidence showed dynamic transcriptional changes of both germ cells and somatic cells from embryos to adults. And dysregulated metabolic signaling or hedgehog signaling were revealed by scRNA-seq in aged SC or Leydig cells (LC), respectively. For infertile males, scRNA-seq studies revealed profound changes of testes, such as the increased proportion of immature SC/LC of Klinefelter syndrome, the somatic immaturity and altered germline autophagy of patients with non-obstructive azoospermia, and the repressed differentiation of SSC in trans-females receiving testosterone inhibition therapy. Besides, the re-analyzing of public scRNA-seq data made further discoveries such as the potential vulnerability of testicular SARS-CoV-2 infection, and both evolutionary conservatism and divergence among species. ScRNA-seq analyses would unveil mechanisms of testes' development and changes so as to help developing novel treatments for male infertility.
Keywords: single-cell RNA sequencing, human testis, male gonad, spermatogenesis, bioinformatic analysis
Background
Human testis is the core organ of the male reproductive system [1], and acts as the cornerstone of male fertility since it is the place of spermatogenesis [2]. On the one hand, the testis is lionized by developmental biologists because it's the start of sex determination [3] and the kindergarten of male germline cells [4]. Meanwhile, human testis is also the priority of andrologists because testicular failure is the leading cause of male infertility [4]. Therefore, scientists have delved into both normal [5, 6] and abnormal testes [7-9], and detected mechanisms behind them especially those related to male infertility [10, 11]. Although numerous approaches have been used to learn about human testis including cell experiments [12], animal experiments [13] or even new methods like testicular organoid [14], most of the above-mentioned methodologies are indirect. It is no doubt that the most ideal model to study human testes is the testis itself. Therefore, human testicular biological sample-based studies could be important sources of knowledge of changes in both normal and impaired testes.
As early as the 1910s, human sample-based studies had started to give direct evidence of testicular diseases [15]. Yet until the 1970s, most of those studies were restricted to anatomy and morphology/histology [16-19] perhaps because at that time nonspecific histological staining was the most popular way to study human testes. Then with the development of some more specific methods such as immunohistochemistry staining (IHC), researches on the human testis reached protein levels [20-22]. A huge breakthrough in this field was the application of microarray and high-throughput RNA-seq on human testicular samples [23-26], which gave scientists a transcriptional view of both the normal testis's development and the pathogenesis of spermatogenic dysfunctions [27, 28]. Nevertheless, both microarray and traditional RNA-seq are based on bulk tissue, for which the testicular sample is considered as a whole, so we could only obtain the mainstream of transcriptional changes in the sample but ignore the heterogeneity of different cells. To solve this problem, Tang et al. introduced single-cell RNA sequencing (scRNA-seq) [29], which could detect the transcriptome at a single-cell resolution. The technology was created on blastomere and oocytes and soon applied to human testicular cells [30] and then to unsorted testicular tissues [31]. So far, there have been plenty of studies using human testicular scRNA-seq data and several reviews partially or roughly touched upon this topic (human testicular scRNA-seq analysis) [32-39]. Nevertheless, some of these reviews didn't fully focus on “human testis” and/or “scRNA-seq” (for example, some included much more about female gonad or animal testis [32, 34, 40] and some focused much on techniques other than scRNA-seq [33, 37, 38]), which led to limited paragraphs and blurry outline on this topic. Other reviews were almost restricted to germ cells [35, 36] or after-birth gonads [39], which gave an incomplete depiction of this topic. Most importantly, none of these articles was with systematic literature searching strategies, which might result in a biased interpretation. Using systematic literature searching, this comprehensive review plans to fully summarize the new findings offered by scRNA-seq analyses of human testicular biological samples, concerning both testicular cells and unsorted testicular tissues, both prenatal and postnatal male gonads, both underage and adult testes, both young and aged testes, both normal spermatogenic and impaired spermatogenic patients, and both original sequencing data and reusage of published data. We hope this review will give scientists detailed and all-round information on testicular development, spermatogenesis as well as testicular pathogenesis in the single-cell resolution, and humbly suggest the future directions in this field.
Methods
Search methods
The searches were conducted on 29 Aug 2022. First, a systematic search in PubMed database was conducted. Based on the fact that this review mainly focused on two aspects, including “scRNA” and “human testicular samples”, we used a matched searching strategy to link these two aspects. In detail, for the former aspect, the terms “single-cell” (term A) or “scRNA” (term B) were selected. And each of these two terms was combined with one of the following testis-related (the latter aspect-related) search terms, including “testis” (term 1), “spermatogenesis” (term 2), “Klinefelter Syndrome” (term 3), “azoospermia” (term 4), “Y chromosome microdeletion” (term 5), “cryptozoospermia” (term 6), “oligozoospermia” (term 7), “asthenospermia” (term 8), “teratospermia” (term 9), “orchitis” (term 10), “cryptorchidism” (term 11) and “male and gonad” (term 12). In order to minimize the chance of omitting related studies especially preprint studies, we do an extra search on GEO dataset (https://www.ncbi.nlm.nih.gov/gds) using four combination of advanced search terms including “((testis) AND scRNA) AND Homo sapiens [Organism]”, “((testis) AND single-cell) AND Homo sapiens[Organism]”, “(((male) AND gonad) AND scRNA) AND Homo sapiens[Organism]” and “(((male) AND gonad) AND single-cell) AND Homo sapiens[Organism]”, which also referred to the data related to both focused aspects. For filtering, the “series” of Entry type and “Expression profiling by high throughput sequencing” of Study type were also chosen. For each identified GEO series, its related articles were obtained from GEO or bioRxiv (https://www.biorxiv.org/) if there was no published article at the searching time.
Selection criteria
The inclusion criteria were (1) studies with single-cell RNA sequencing applied on human testicular biological samples (including testicular tissues, cells and prenatal male gonads) and/or (2) studies with re-analyses of published/public scRNA-seq data of human testicular biological samples (not including pure literature review of published articles).The exclusion criteria were (1) articles not in English; (2) articles published before 2009 (the year when scRNA-seq technology was created [41]); (3) non-research articles (such as pure review/case report/guideline/protocol/technical note/correction/comment/chapter of book/letter) without scRNA-seq analysis on human testis; (4) full-length articles not available; (5) tumor studies without scRNA-seq data analyzing or not relevant to human testis; (6) studies based on sequencing methods other than scRNA-seq (e.g. single-cell small RNA-seq or spatial transcriptomics sequencing); (7) pure animal or botanic studies; (8) human-related studies but with no scRNA-seq analysis on human testicular samples.
Results
Application of scRNA-seq on human testicular samples: an overview
The workflow of literature searching and selections was in Figure 1. The searches returned about 1300 records (609 after de-duplication). By further filtering, 81 full-length articles were enrolled into the final analysis. There were 33 articles containing original scRNA-seq data (at least from one donor) on human testicular samples, while the rest 48 articles were based on the re-analysis of previously published data (no matter whether the data was previously published by the same team). Since the year of 2015, Guo et al. for the first time do scRNA-seq on human fetal testicular cells [30], followed by adult testicular cells sequenced by two studies in 2017[42, 43]. Interestingly, although those two adult studies were from different teams, both of them focused on spermatogonia of one accord. From 2018, numerous studies have done scRNA-seq on human testicular samples regarding both prenatal and postnatal, both normal and abnormal testes. Table 1 summarized articles that have originally published scRNA-seq data on human testicular samples. We could conclude that most of the studies were conducted by scientists from the USA and China. As for library techniques, most of the studies used the 10×Genomics platform (https://www.10xgenomics.com/) to carry out their works. While other studies chose techniques/plateforms such as BD Rhapsody [44], (modified) smart-seq2 [45-47], STRT-seq [48], Microwell-seq [49], Drop-seq [50], inDrop [51], Slingleron GEXSCOPE® [52, 53] and some earlier platform such as Fluidigm C1 [54] and Tang method [29, 41]. In some studies [55, 56], two or more methods simultaneously appeared for cell capturing. So far, no comparative study has focused on which technique is more suitable for scRNA-seq of human testicular samples. Nevertheless, some reviews and comparative studies based on scRNA-seq of other tissues have gradually revealed that the technological differences of scRNA-seq, including both the wet-lab parts (e.g., cell capturing/sequencing/barcoding) [44, 57-59] and the dry-lab parts (e.g., data-processing/batch-effect correction/dimension reduction/quality control) [60-64] might have profound impact on the final results. Especially, unavoidable technological batch effects will be generated using data generated from different sequencing platforms [65]. Although the data will go through batch effects correction before downstream analysis [60], we still recommend that researchers should choose one suitable techniques and consistent data-analyzing methods to do their testicular scRNA-seq works. If you need to do integrated analyses and have to combine your own scRNA-seq data with public scRNA-seq data, we suggest that you should choose the public data that used the same cell-capturing techniques as your own data, to minimize the non-biological batch effects.
Workflow of the literature searching and study selection
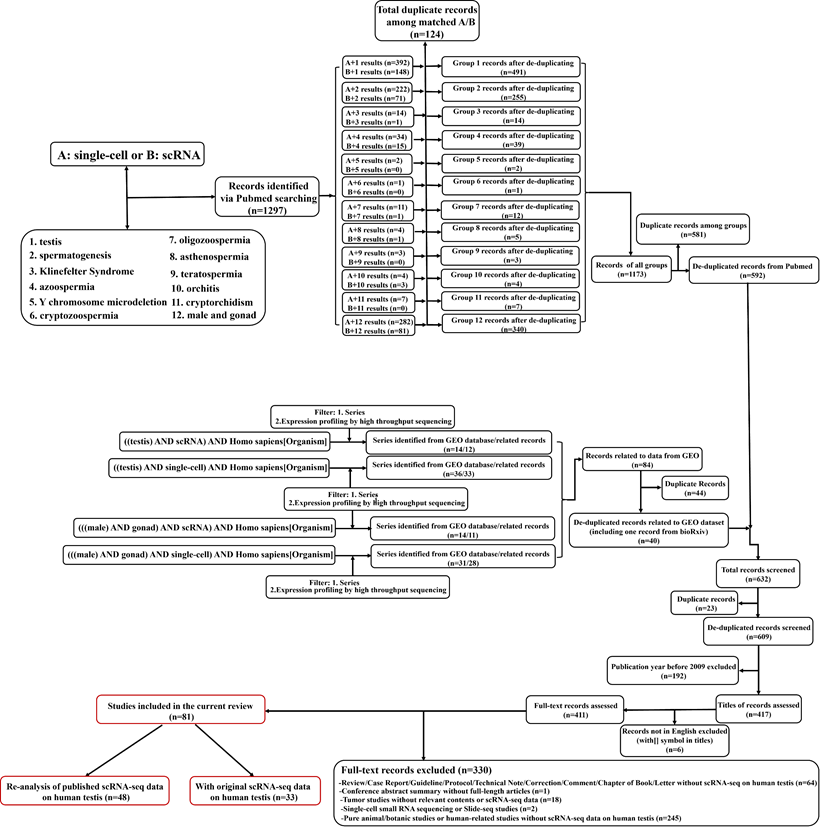
Studies containing originally reported scRNA-seq data of human testicular biological samples
| Author | Country* | Year | Techniques | Conditions | No. of donors | No. of cells | References |
|---|---|---|---|---|---|---|---|
| Guo et al. | China | 2015 | Tang method | Prenatal | 9 | 197 | [30] |
| Guo et al. | USA | 2017 | Fluidigm C1 | Normal | 5 | 175(92 filtered) | [42] |
| Guo et al. | China | 2017 | Tang method | Prenatal | 5 | 14 | [181] |
| Neuhaus et al. | Germany | 2017 | Tang method/Shallow RNA-seq | Normal spermatogenesis | 1 | 2 | [43] |
| Li et al. | China | 2017 | Modified smart-seq2 | Prenatal | 12 | 1204(1068 filtered) | [73] |
| Guo et al. | USA | 2018 | 10× Genomics | Normal (one from 17-year-old male)/Infant | 5 | 7830 filtered | [31] |
| Vértesy et al. | Netherlands | 2018 | Smart-seq2 | Prenatal | 1 | 9 | [182] |
| Wang et al. | China | 2018 | Modified Smart-seq2 | Normal/OB/NOA | 10 | 3243(3028 filtered) | [76] |
| Hermann et al. | USA | 2018 | 10× Genomics/Fluidigm C1 | OB/Normal | 16 | 635 for C1, 36451 for 10x (including 1 experiment with a 1:1 mixture of human and mouse cells) | [68] |
| Sohni et al. | USA | 2019 | 10× Genomics | OB/Neonatal | 4 | 33585 filtered | [67] |
| Laurentino et al. | Germany | 2019 | 10× Genomics | KS | 1 | 3289 filtered | [121] |
| Xia et al. | USA | 2020 | inDROP | OB | 2 | 2554 filtered | [70] |
| Zhao et al. | China | 2020 | 10× Genomics/ BD Rhapsody | OB/iNOA/KS/Juvenile/AZFa deletion | 17 | 88723 | [55] |
| Liu et al. | China | 2020 | Modified STRT-seq | NOA | 2 | 61 Sertoli cells | [159] |
| Han et al. | China | 2020 | Microwell-seq | Prenatal | 2 | 13211 filtered | [183] |
| Guo et al. | USA | 2020 | 10× Genomics | Juvenile/trans-females | 6 | 19223(12854 filtered) | [72] |
| Shami et al. | USA | 2020 | Drop-seq | Normal | 4 | 13837 filtered | [184] |
| Tan et al. | USA | 2020 | 10X Genomics | OB | 2 | 11159(8916 filtered) | [69] |
| Chitiashvili et al. | USA | 2020 | 10× Genomics | Prenatal | 5 | 24929 | [178] |
| Hwang et al. | USA | 2020 | 10× Genomics | Prenatal | 3 | 16429 filtered | [185] |
| Persio et al. | Germany | 2021 | 10× Genomics | OB/Cryptozoospermia | 6 | 28690 filtered | [66] |
| Wang et al. | China | 2021 | Modified Smart-seq2/STRT-seq | NOA | 1 | 480(432 filtered) | [110] |
| Mahyari et al. | USA | 2021 | 10× Genomics | iNOA/Ejaculatory dysfunction/KS | 4 | 10856 | [78] |
| Bhutani et al | USA | 2021 | Smart-seq2 | Normal | 2 | 466 | [186] |
| Alfano et al. | Italy | 2021 | 10× Genomics | OB/iNOA | 4 | 5200 filtered | [88] |
| Guo et al. | USA | 2021 | 10× Genomics | Prenatal/Infant | 2 | 6992 filtered (W12 and M5) | [79] |
| Chen et al. | China | 2022 | Slingleron GEXSCOPETM | OB/iNOA | 2 | 3844 | [56] |
| Nie at al. | USA | 2022 | 10× Genomics | Normal (young adult)/Aging | 12 | 44657 filtered | [75] |
| Cheng et al. | USA | 2022 | 10× Genomics | Prenatal | 6 | 89477(72257 filtered) including 4 fetal adrenal glands and 1 4wpf urogenital ridge | [80] |
| Wang et al. | China | 2022 | 10× Genomics/modified STRT-seq | Prenatal | 11 | 31006 for 10x 709 for modified STRT-seq | [82] |
| Voigt et al. | Canada | 2022 | 10× Genomics | Infant/Juvenile | 3 | 29125 | [87] |
| Wang et al. | China | 2022 | 10× Genomics | Prenatal | 4 | 53508 filtered for 4 male and 4 female | [84] |
| Garcia-Alonso et al. | UK | 2022 | 10× Genomics | Prenatal | 22 | 13381 | [74] |
*According to the first authors' information
Note: OB, obstruction; (i)NOA, (idiopathic) non-obstructive azoospermia; KS, Klinefelter Syndrome; AZFa, Azoospermia factor a; wpf, weeks postfertilization; W12, 12 weeks post-fertilization; M5, 5 months post birth.
Note2: Normal referred to normal adults; Prenatal referred to the fetus and/or embryo; Neonatal referred to several days after birth; Infant referred to several months (upto 1 year and 13 months) after birth; Juvenile referred to all underages (under the age of adult), and Aging referred to >60 years old.
Note3: The information of scRNA-seq data/samples in this table referred to the information of the original and first-time reported cases in each study. For example, Guo et al. integrated seven cases in [79], but five of them were first reported by Chitiashvili et al. [178] (these two studies were from the same research team). So, in the item of Guo's study, we only summarized the information of the rest two cases (w12 and M5) which were reported for the first time.
In terms of sample types, one-third of the studies choose prenatal male gonads (the youngest sample was from the gonad from a 4-week male embryo). For adult testes (the oldest sample was from a 76-year-old male for now), most of the studies included samples with complete/full/normal spermatogenesis. But the sources of these normal spermatogenic samples varied. Some collected deceased healthy male testicular samples [31] to serve as the normal groups, which were ideal controls. For many institutes, obstructive azoospermia (OA) samples with full spermatogenesis confirmed by histopathological diagnosis, including congenital bilateral absence of the vas deferens [66], post-vasectomy [67], and physical obstruction of the vas deferens [66] were considered as workarounds for normal/control groups. It is noteworthy that in the original manuscripts of Hermann et al. [68], Tan et al. [69] and Sohni et al [67], some of the adult testicular biopsies were obtained from patients receiving vasectomy reversal, and the samples in Xia et al.'s work [70] were from testicular sperm extraction (TESE) surgeries of patients with obstructive infertility (they were also post-vasectomy patients after confirming with the first author). Interestingly, none of these patients was clearly claimed as OA patients, although they all had obvious obstructive etiology. Frankly speaking, these post-vasectomy patients were extremely unlikely to have sperm in ejaculate (only if some rare situations happened such as duplication of vas deferens or natural recanalization [71]). But in order to be strict and consistent, in the current review, all patients with obstructive etiology (no matter they were diagnosed with obstructive azoospermia or not) were also unitedly abbreviated as obstruction (OB). Besides, testes removed from patients receiving transsexual operation might also be an option for sample collection [43, 72]. Additionally, normal testicular samples at age 17 were collected by both Zhao's and Guo's studies, but were assigned to different groups (adult for Guo's and underage for Zhao's studies) [31, 55], maybe due to the different definition of adult (biological adult versus legal adult).
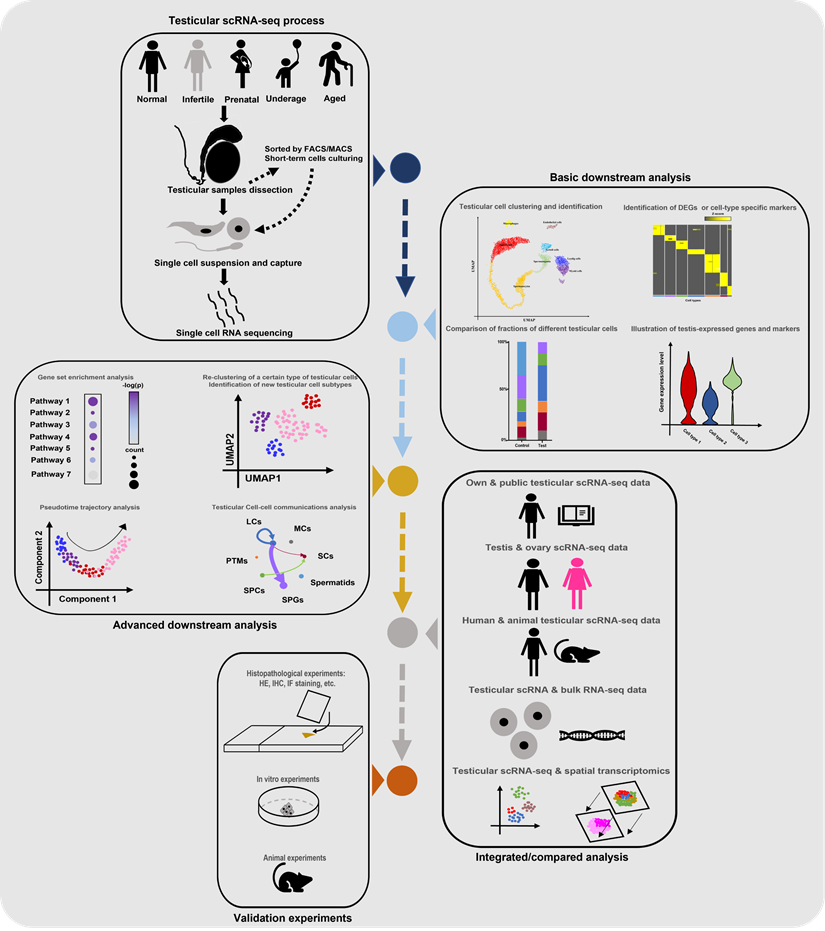
Till now, the number of scRNA-seq testicular samples in each study has been limited, maybe due to the high price of scRNA-seq commercial service and limited sample donors. To partially solve this dilemma, some studies chose to do combined analyses of their own scRNA-seq data and public scRNA data or bulk RNA-seq data. And most of the studies in Table 1 did extra experiments (such as histological experiments, animal or cell experiments) to validate the findings from scRNA-seq analyses. Additionally, some studies were conducted on both testes and ovaries [22, 73], or on testes from both human and animals [68], in order to do comparisons between genders or among species. Garcia-Alonso et al did integrated analyses by the combination of testicular scRNA-seq with spatial transcriptomics [74], which is a novel direction. Nevertheless, so far there has been no guideline for scRNA-seq studies on the human testis. By reviewing all related studies, we summarized a standard guideline(workflow) of testicular scRNA-seq-based studies (Figure 2). We believe that with the wider application of scRNA-seq techniques, there will be more scRNA-seq-based studies of testicular samples published, with more comprehensive analyzing methods used.
Standard guideline/workflow of single-cell RNA sequencing studies on human testicular biological samples. Some of the elements used in this figure were downloaded from Icons of Microsoft PowerPoint 2021(Microsoft, USA). Note: scRNA-seq, single-cell RNA sequencing; FACS, fluorescence activated cell sorting; MACs, magnetic activated cell sorting; DEGs, differentially expressed genes; HE, hematoxylin-eosin; IHC, immunohistochemistry; IF, immunofluorescence.
Cell identifications by scRNA-seq on human testicular samples
The fundament of scRNA-seq analysis is cell identification. Apart from a few studies based on isolated cells by fluorescence activated cell sorting (FACS) or magnetic activated cell sorting (MACS), the majority of studies in Table 1 were based on testicular tissues. For adult testes, the representatively identified cell types in scRNA-seq analyses were summarized in Table 2. ScRNA-seq-identified testicular cells could be roughly divided into three classifications, including spermatogenic cells (germ cells), non-immune somatic cells and immune cells [66]. The widely used and some newly identified markers for cell identification were also summarized (Table 2). Importantly, because of the different study purposes and different sample selections, the types and subtypes of testicular cells in different studies were slightly variant. For germ cells, spermatogonia (SPG), spermatocyte (SPC) and spermatid could be broadly identified in a full-spermatogenic sample if the focus of the study is not spermatogenic cells [55]. Nevertheless, a more detailed classification of these three germ cells could be done based on stage-specific markers. Here we recommended a moderate classification of germ cells used by Nie et al., in which SPG could be further divided into spermatogonial stem cell (SSC) (also called undifferentiated SPG) and differentiating SPG (diff_SPG), SPC into early primary SPC and late primary SPC, spermatid into round and elongating/elongated spermatid [75]. One can adjust the subdivision of each germ cell type according to your research focus. For example, if you were not interested in the development of SPC, you could easily keep SPC as an unclassified type but classifying SPG and spermatid into more detailed subtypes [56]. If the whole spermatogenesis is your attention, you might choose a more aggressive strategy for germ cell classification [66, 76].
Representative cell types/subtypes and candidate markers for adult testicular scRNA-seq data
| Cell classifications | Cell types | Sub-types | Candidate markers | References |
|---|---|---|---|---|
| Spermatogenic cells | DDX4, MAGEA4 | [66, 75, 76] | ||
| SPG | ||||
| SSC | FGFR3, ID4, UTF1, GFRA1, HMGA1, PIWIL4 | [31, 55, 56, 75, 76] | ||
| Diff_SPG | KIT, MKI67, DMRT1 | [55, 56, 76] | ||
| Diffed_SPG | STRA8 | [55, 76] | ||
| SPC | ||||
| L.SPC | DMC1, SPO11, SYCP3, SYCP2, SYCP1, TEX12, RAD51AP2, SCML1 | [55, 76] | ||
| Z.SPC | SYCP3, SYCP2, SYCP1, TEX12, SPO11, DMC1, RAD51AP2, TDRG1 | [55, 76] | ||
| P.SPC | OVOL1, OVOL2, CCNA1, CCDC112 | [55, 76] | ||
| D.SPC | OVOL1, OVOL2, CCNA1, AURKA | [55, 76] | ||
| Meiotic (1/2) and Secondary SPC | ACR, C9orf116, SLC26A3, SIRPG might express (Low specificity) | [55, 66, 76] | ||
| Spermatid | ||||
| Round (early) Spermatid | TEX29, SIRT1 | [55, 66, 76] | ||
| Elongated (late) Spermatid | PMR1, PMR2, PMR3, SPEM1 | [55, 66, 75, 76] | ||
| Non-immune somatic cells | VIM | [31, 75] | ||
| Sertoli cells | SOX9, AMH, WT1 | [55, 67, 73, 76] | ||
| Leydig cells | IGF1, DLK1, INHBA, INSL3, HSD17B3 | [56, 75, 76, 88] | ||
| Endothelial cells | VWF, PECAM1 | [31, 55] | ||
| Myoid cells | ACTA2, MYH11 | [31, 75] | ||
| Peritubular myoid cells (PTM) | ACTA2, MYH11 | [55, 56, 66, 76] | ||
| Perivascular smooth muscle cells | NOTCH3, FAM129A, MUSTN1 | [55, 66, 75] | ||
| Fibrotic PTM | CLEC3B, CFD | [66] | ||
| Immune cells | PTPRC | [55] | ||
| Macrophages | CD14, CD163, CD68 | [31, 55, 76] | ||
| Macrophage M1 | Not reported | [78] | ||
| Macrophage M2 | Not reported | [78] | ||
| Mast cells | TPSAB1, TPSB2 | [55, 56, 164] | ||
| T cells | CD3D, CD3E, CCL5 | [55, 66, 88, 164] | ||
| Tissue-resident cytotoxic T cells | CD8, CD69 | [88] | ||
| B cells | Not reported | [78] |
Note: SPG, spermatogonia; SSC, spermatogonial stem cell; diff_SPG, differentiating SPG; diffed_SPG, differentiated SPG; SPC, spermatocyte; L.SPC, Leptotene SPC; Z.SPC, Zygotene SPC; P.SPC, Pachytene SPC; D.SPC, Diplotene SPC
Apart from germ cells, it is well known that somatic cells also played important roles in spermatogenesis. Such somatic cells could be further divided into non-immune somatic cells and testicular immune cells. For non-immune somatic cells, Sertoli cells (SC) were one of the most important cell types in spermatogenesis, as they could not only form blood-testis barrier but also directly support germ cell survival [55]. And Leydig cells (LC) could affect spermatogenesis by producing testosterone [75]. Although the roles of testicular immune cells in spermatogenesis have not been fully revealed, more evidence indicated that they could interact with germ cells and take part in spermatogenesis [77].
The rules of cell identification in germ cells also worked when annotating somatic cells. For example, although the rest studies all keep testicular macrophages as a whole, Mahyari et al. chose to further divided them into M1 and M2 macrophages [78]. Nevertheless, the above-mentioned all classifications were “well-known” classification of adult testicular cells. One important function of scRNA-seq is to identify novel subtypes of cells. For example, traditionally SC were considered as one type of cells, but Zhao et al. and Guo et al. found three subtypes of SC in the human testis [55, 72]. Similar findings also occurred in LC [78]. Owing to the lack of unified understanding, sometimes similar novel subtypes of certain cells were independently reported by different teams with different nomenclatures. For instance, an important subtype of prenatal gonadal cells which could bifurcate into the developmental path of both SC and LC were reported by two different teams with different names [79, 80]. Meanwhile, samples with different pathological status might deeply influence the identified cell types and numbers. A case in point is testicular immune cells. In the microenvironment of testes with spermatogenic dysfunction, an increasing number of immune cells were observed [66]. Additionally, the main type of immune cells in full spermatogenic testes was macrophages, while a large number of mast cells were identified in testes from non-obstructive azoospermia (NOA) patients, especially in Sertoli cell-only syndrome (SCOS) [55, 56].
ScRNA-seq demonstrated the development and changes of testicular cells from all ages
Testicular development from prenatal to postnatal period
1. Overview
Before the year 2015 in which the first prenatal testicular sample was scRNA-seqed, the understanding of the development of different types of testicular cells in prenatal testes was limited to the changes of cell number, morphology and distribution [81]. So far, with over ten articles published their own scRNA-seq data on human prenatal testicular samples, we already have an outline of both germline and somatic development at single-cell transcriptional level.
2. Germline transition
For germline development pre- and postnatally, Guo et al pointed out that prenatal germs cells generally have two main forms of transcriptional profiling, including a form of primordial germ cells (PGC) (expressing PGC markers such as TFAP2C, KIT, NANOG, POUF51 and SOX17) and form of the most naïve SSC (expressing earliest SSC markers such as PIWIL4, EGR4, MSL3 and TSPAN33) [79]. PGC form mainly started at the embryotic time and then gradually stopped meiosis and inhibited pluripotency and at transcriptional level became cells with high similarity to the early status of adult SSC [79]. The latter form (named State f0) [79] strode across human birth and kept a quite similar transcriptional status until the adult time (named State 0 in adult) [31]. During the germline transition from PGC to (f)State 0, pathways related to gonad and stem cell development were downregulated while signaling related to transcription/homeobox were enhanced [79]. Similarly, Wang et al. found a transition from mitotic (mainly existing before 15 weeks) to meiotic arrest (mainly existing after 15 weeks) male fetal germ cells (FGC) [82], which was in line with Guo's [79] and Li's results [73], indicating that the downward status of meiosis (proliferative-quiescent-arrested) was the symbol of male fetal germ cell development. Garcia-Alonso et al. called the latter form of FGC prespermatogonia and also showed the activation of EGR4 in this stage [74]. Interestingly, Wang et al. also reported an extra subtype of FGC called SPARC+ POU5F1+ FGC, which might keep the ability to migrate locally within the fetal testis [82].
3. Somatic development
As for prenatal somatic cells, most of the researches focused on SC and LC due to the isogenesis of these two cells. The source of testicular somatic cells was embryotic coelomic epithelium (CE) [83]. By re-clustering of scRNA-seqed CE cells, Cheng et al. found two subclusters of CE, of which one was NR0B1+STAR+NR5A1+ but GATA4-LHX9- (annotated as adrenogenic CE), while the other was GATA4+LHX9+HOXA9+HOXD9+ but NR5A1- (known as posterior/gonadogenic CE) [74, 80]. The former subcluster will develop into the adrenal gland while the latter goes to the testis. Noteworthily, gonadogenic CE will step into a sort of bipotent somatic progenitors (symbolized by the expression of NR5A1), which had been recognized by several studies [79, 80]. In Wang et al.'s study, a subpopulation of KRT19+ somatic cells were identified mainly in 6- to 8-week testes, which also resembled these somatic progenitors [82]. Starting from 7-8 weeks, these somatic progenitors bifurcated into two different developmental paths, one of which is embryonic Sertoli progenitors (marked by SRY, similar to the “early supporting gonadal cells” in Garcia-Alonso et al.'s work [74]) and then became fetal SC [79]. The rest path, on the contrary, led to embryonic/fetal interstitial progenitors (marked by ARX and TCF21, also called Leydig precursor cells by Li et al. [73] and called somatic progenitor by Wang et al. [84]), and finally part of the fetal interstitial progenitors became fetal Leydig cells (marked by HSD3B2 and CYP17A1, also called differentiated LC by Li et al. [73]) [79, 80]. During the development of prenatal interstitial/Leydig lineage, the enriched molecular pathways altered from “extracellular matrix”/ “cell adhesion” to “steroid biosynthesis”. Interestingly, at the perinatal stage, fetal SC successfully passed the birth (with genes related to translation and respiratory chain enhanced, while genes related to the endoplasmic reticulum and steroid biosynthesis reduced) and became postnatal SC including stage_a and b (discussed in more detail below) [55, 79], while fetal Leydig cells disappeared in the postnatal time [79]. In fact, it was CYP17A1-negative fetal interstitial cells that might finally pass the birth and finally become the common progenitors of adult LC and PTM [79]. Regarding EC, although some studies proved the early appearance of EC in prenatal male gonad [73, 84], so far there have been few studies that focused on EC development. Like the previous murine studies [85, 86], scRNA-seq data on human the fetal testis also revealed two distinct types of macrophages in the fetal testis, of which one is named SIGLEC15+ fetal testicular macrophages (osteoclast-like) and the other is TREM2+ fetal testicular macrophages (microglia-like) [74]. Further study showed that SIGLEC15+ macrophages were usually located in the interstitium and could interact with EC/mesenchymal cells and play roles in mesonephric endothelial cell migration, while TREM2+ macrophages (located in the testis cord) interacted with fetal SC and FGC and worked as an immune regulator [74]. Unfortunately, the transition from fetal testicular macrophages to postnatal testicular macrophages remained unexplored. Further scRNA-seq-based studies might have the opportunity to reveal the relationship between fetal and infant testicular macrophages.
After-birth testicular maturation from underage to adult
1. Overview
Among all scRNA-seq studies on testicular samples, several studies did original scRNA-seq on underage testicular samples [31, 55, 67, 72, 79, 87]. And in some other studies [78, 88], previously published underage testicular scRNA-seq data were re-analyzed. Those studies pointed out that huge heterogeneity existed in both spermatogenic cells and somatic cells during the maturation of testes after birth.
2. Spermatogenic maturation
For analyzing the after-birth development of spermatogenic cells, pseudotime trajectory analysis was widely used. In fact, pseudotime trajectory analysis on spermatogenic cells could be divided into three distinct types. First, if we only focused on the normal testis of an adult man, spermatogenesis itself, starting from SSCs and ending at mature spermatids, was a continuous process running along a time axis of maturation [89]. Hence, the complete spermatogenesis process could be analyzed using pseudotime trajectory in scRNA-seq data. In 2018, Guo et al. did this task and showed there was only one no-branch pseudotime trajectory in spermatogenesis of healthy male [31], which started with SSCs, passed diff_SPGs, early and late SPCs, then round and elongated spermatids and ended up with testicular sperms (also known as mature spermatids [90]). The second type of pseudotime trajectory analysis on spermatogenic cells is to extract and re-cluster a certain type of germ cells and do pseudotime trajectory analysis on this type of germ cells. For instance, Sohni et al. did pseudotime trajectory analysis on SPGs and found a minor group of SPGs called “transitional cells” which was the linkage between SSCs and diff_SPGs during SPGs differentiation [67]. Besides, Guo et al. did pseudotime analysis on SPGs and reported a five-stage development of SPG, starting from state 0 (markered by PIWIL4, similar to the above-mentioned state f0 in the fetal testis), then state 1 (ID4, GFRA1 expressed), followed by state 2-3 (marked by KIT and MKI67) and finally reaching state 4 (marked by STRA8) [31, 42]. By comparing these 5 stages with the results from Wang et al. [76] and Sohni et al [67]., we could conclude that state 0 and 1 were two types of SSC (0 was more naïve and fetal-like, and resembled the “SSC-1” in Sohni's work, while 1 resembled “SSC-2” in Sohni's work), and state 2-3 were diff_SPG while state 4 were diffed_SPG. Third, pseudotime analysis could also be used to study the maturation of germ cells from underage to adult. In fact, the discovery of state 0 was based on pseudotime analysis on the combination of infant and adult spermatogenic cells, from which they found this special state of SPGs (which was close to the infant germ cell cluster) in adult [31]. Sohni et al. further re-clustered infant germ cell cluster into three subclusters, including primordial germ cells (PGCs)-like cells (PGCLs) (marked by POU5F1 and NANOG), Pre-SPG1 (marked by DOCK8 and SERINC2) and Pre-SPG2 (marked by COL1A2 and TIMP2) [67, 91]. And further pseutotime trajectory showed that the developmental order of fetal/infant/adult naïve germ cells was PGCL→Pre-SPG1→Pre-SPG2→adult SSC subcluster (SSC-1) [67]. During puberty, the spermatogenic status changed obviously with age, and pseutotime trajectory analysis of a series of testicular scRNA-seq data from 1- to 25-years old males showed the dynamic changes of germ cells at different ages [72]. In detail, during the infant and child period (1 and 7 years old), there were only SSC in testes (also known as undifferentiated SPG or SPG state 0 and 1, which all referred to the naïve status of SPG) [72], and this finding was further validated by scRNA-seq data on testes from 2- and 5- years old males [55]. At year 11, both diff_SPG and SPC appeared, while spermatid appeared at 13 years old samples [72]. Zhao et al. further showed that diff_SPG had already existed in the 8-year-old testis but SPC was still absent at that time [55]. The first maturity of spermatogenesis was observed at 14 years old, by which time the proportion of each type of spermatogenic cells was almost the same as adult testes [72]. In terms of molecular signaling, Activin pathway was downregulated in SSC but was obviously upregulated during spermatogonia differentiation [72]. In a word, scRNA-seq together with pseutotime trajectory analysis is very powerful in analyzing the after-birth maturation of spermatogenic cells, especially in the identification of new cell subclusters and finding new stages of cells.
3. Somatic maturation
For somatic cells, the development of Sertoli cells has been fully analyzed by scRNA-seq analysis. By integratedly analyzing testicular scRNA-seq data of five young adult with normal spermatogenesis (age from 23 to 31 years), three children (age 2, 5 and 8 years) as well as two adolescents (age 11 and 17 years), Zhao et al. revealed that the normal maturation of testicular Sertoli cells consisted of three stages (termed as stage_a, b and c) [55]. In detail, stage_a (marked by JUM) was the majority of Sertoli cells at infant time (including the neonatal testis) and gradually decreased with the age increasing. While stage_c (marked by DEFB119) appeared at the time of puberty (after 11 years old) and became the dominant type of Sertoli cells in adult people. Besides, they also found that the Wnt signaling pathway should be the key of regulating Sertoli cell maturation [55]. Such findings are outstanding because it focused on the normal development of postnatal somatic cells and revealed not only the phenotype development of Sertoli cells but their related molecular mechanisms as well. Similarly, Guo et al. also found two immature stages of SC, named immature 1# (marked by PDPN) and 2# (with higher expression of TOMM7 and ATP5E), which finally merged into mature SC after 11 years old [72]. By comparing the differentially expressed genes (DEGs) of stage_a/b SC (from Zhao's study) versus those of immature 1#/2# SC (from Guo's study), we concluded that stage_a was similar to immature 1# while stage_b was closer to immature 2#. A huge difference between these two studies is that Guo's study showed 1# and 2# as two independent paths of immature SC, which will merge into mature SC after 11 years old. Zhao's study, however, showed a potential sequence of stage_a, b and c, (from a to be to c) by both analyzing the proportion of these three cells at different ages and analyzing the functions of these three cells. In detail, they found that before the appearance of mature SC, the ratio of SC a to SC b decreased with age, suggesting there might be a transition from a to b [55]. But was this change of ratio caused by “transition from a to b” or by “proliferation of b/death of a”? Enrichment analysis and cycle-specific gene analysis further showed that stage a SC were the most proliferative and had more features as progenitors, which supported the hypothesis that stage a, b and c were three consecutive development stage of SC [55]. Since Zhao's study was more comprehensive in terms of SC, hereinafter we chose the nomenclature of SC from Zhao's study [55].
Moreover, Mahyari et al. found that in adult human testes, the maturation of Leydig cells also consisted of three types including progenitor LCs (PLCs), immature LCs (ILCs) and mature LCs (MLCs) [78]. And pseudotime analysis confirmed that the maturation of LCs started from PLCs, followed by ILCs and ended at MLCs [78]. However, in underage testes, the LCs seemed to be a unique type which was totally separated from the above-mentioned three adult LC types [78]. In line with that finding, using pseudotime analysis on both infant and adult LCs, another study also found that infant LCs and adult LCs appeared at two different ends of the trajectory, indicating that the progress of LCs from underage to adult might be a discontinuous alteration [67]. Further, Guo et al. for the first time showed that the so-called “infant or child LCs” might actually be the common progenitors of LC and PTM [72], which will finally develop into both LC and PTM during puberty [72]. As for PTMs, although the developmental trend was similar to LCs (because they were homologous), pseudotime analysis of scRNA-seq data identified a special subtype of adult PTMs which was clustered together with neonatal PTMs and showed a neonatal-like gene expression characteristic [67]. For the molecular signaling involved in the maturation of LC and PTM, Guo et al. found that the common progenitors expressed high level of genes associated to “transcription” signaling, and the enriched pathways went to cytoskeleton/ cell adhesion signaling (if the progenitor differentiated into PTM) or secretion signaling (when it differentiated into LC) [72]. So far, the development and maturation of other types of somatic cells, especially testicular ECs and immune cells, have not been fully revealed. Future scRNA-seq studies should pay special attention to the differences of these rare somatic cells between underage and adult human testes.
Based on the published studies, we can conclude that during the maturation of after-birth testes, huge differences existed in both somatic cells and germ cells between underage and adult testis in terms of cell subtypes, expressed markers and regulating mechanisms.
Testicular senescence from adult to aged males
1. Overview
Unlike the deep studies on differences between underage and adult spermatogenesis, few scRNA-seq researches focused on the changes in aging testes. Until last May, Nie et al. published the world's first testicular scRNA-seq study based on the comparison between young and old adult men [75]. In this study, by integrated analysis of 4 young adults (age 17-22 years) and 8 older adults (age 62-76 years), this team found huge age-related changes in both spermatogenic cells and somatic cells.
2. Germ cell senescence
In terms of germ cells, they found that spermatogenic cells were affected by age in an inconsistent way (some old people still have complete spermatogenesis, while others showed impaired spermatogenesis), and in old males with obviously impaired spermatogenesis, the decreased number of SSC might be one of the causes of age-related spermatogenic dysfunction [75]. Mechanistically, using Cellchat analysis, a powerful function of scRNA-seq data to detect cell-cell communications [92], Nie et al found that Activin and KIT signaling pathways weakened between SCs and SSC/diff_SPGs, which might lead to damaging effects on germ cells [75].
3. Somatic cell senescence
For somatic cells, scRNA-seq analyses revealed pan-somatic cell changes, including inflammation changes of SCs, loss and testosterone decline of LCs, decreased contraction and integrity of PTMs-based tubular walls and the senility of both ECs and Macrophages [75]. Mechanistically, Nie found that for SCs, metabolic signaling was significantly altered in aged males, while in senescent LCs, key components of the Hedgehog pathway were decreased [75].
4. Young infertile testes showing onsets of senescent process
For some infertile males who were still young, Alfano et al. found that their testes showed early onsets of senescent process [88]. Such abnormal senescence was observed in almost all somatic cells of idiopathic germ cell aplasia, and was characterized by the upregulation of proteins of innate immunity, the overexpression of UBA52/NACA and decreased pathways of amino acids metabolism [88]. This phenomenon suggested that the senescence of human testes was not only associated to the exact age of male, but affected by their pathological status as well.
Transitions of SC/LC/PTM in the context of the whole life cycle
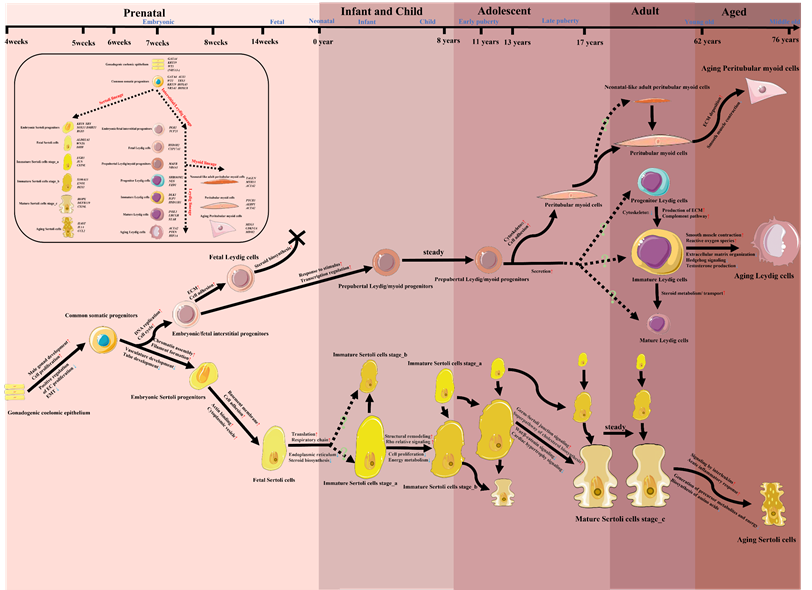
So far, there has been limited scRNA-seq study that focused on the all-age process of the origin, development, maturation and senescence of a certain type of testicular cells by analyzing prenatal, infant, children, adolescent, adult and aged testicular samples. Due to the profound studies on the transitions of SC/LC/PTM and the low inconsistency among these studies, we tried to summarize the development and changes of these three main somatic cells in the context of the whole life cycle (from embryotic to aged) in Figure 3, based on the current evidence retrieved from human testicular scRNA-seq studies [55, 67, 72-75, 78-80]. Future studies should try to integrate the transcriptional phenotypes of testicular cells from all periods of the life cycle, and do a coherent study on the other types of testicular cells, especially testicular immune cells, to which less attention was paid.
A model map of the potential developing paths of Sertoli cells/Leydig cells/Peritubular myoid cells in the whole life cycle, together with changing cell functions/pathways and highly expressed markers. The information of this diagram was retrieved from studies listed in Table 1, especially from [55, 67, 72-75, 78-80]. Icons of various cells in this figure were downloaded from Servier Medical Art (https://smart.servier.com/) or redrawn based on the elements of Servier Medical Art. EMT, epithelial-mesenchymal transition; ECM, extracellular matrix.
ScRNA-seq mapped the testicular microenvironment in infertile males but with complete spermatogenesis
Obstructive azoospermia
Obstructive azoospermia was clinically diagnosed by semen tests (no sperm in ejaculates) and the existence of excurrent duct obstruction [93]. According to traditional opinion, OA patients were with rather conserved spermatogenesis [94]. Therefore, in traditional studies, due to the difficulty in obtaining completely healthy and fertile male testicular samples [95, 96], researchers would like to choose OA samples with confirmed normal histopathologic features as the controls [97-100]. In testicular scRNA-seq studies, this workaround was also widely used [55, 66, 76]. Although we could conclude that these OA control samples were with complete or full spermatogenesis because of the histopathologic confirmation, could these OA testes strictly be called as “normal” testes? In the past, due to the limitation of research methods, this question was not fully answered. By the combination of healthy and OA testicular scRNA-seq data, Chen et al. revealed an obvious loss of late spermatids and an increase of early SPCs in OA testes compared with testes from the healthy man [56]. Mechanistically, the changes were found in both germ cells and somatic cells. First, SPCs of OA testes might suffer from the weakening meiotic process and enhanced apoptosis, which eventually leading to the loss of spermatids [56]. As for somatic cells, PTMs might be the matchmaker between obstruction-caused pressure and the damage of spermatogenesis [56]. This study partially solved the unsettled issue and suggested that although the spermatogenesis of OA testes was functionally and pathologically normal, it was not technically normal. Meanwhile, this study only used scRNA-seq data of post-epididymitis and post-vasectomy OA testes. Is there a difference in testicular function among OA patients with various causes (e.g., congenital bilateral absence of the vas deferens and post-vasectomy)? Whether the changes of OA testes found by scRNA-seq are obstructive time-dependent? These questions remain to be answered.
Ejaculation dysfunction
Ejaculation dysfunction contains a wide range of situations in which the normal ejaculation was disordered or even failed [101]. Infertility caused by ejaculatory dysfunction or retrograde ejaculation could also be accompanied by testes with complete/full spermatogenesis. Using testicular scRNA-seq data of a patient with retrograde ejaculation (post-colectomy), Mahyari et al. pointed out that the characteristics of cell score residuals (a statistic reflecting testicular cells' pattern) of the retrograde ejaculation sample and adult control samples were alike, which indicating that the patient with retrograde ejaculation had undisturbed spermatogenic status and could be considered as another type of control samples [78]. Nevertheless, the above-mentioned scRNA-seq study did not especially focus on ejaculation dysfunction, so the evaluation of testicular changes of ejaculation dysfunction was not that comprehensive. Further scRNA-seq studies should take several situations of ejaculation dysfunction, including “total anejaculation”, “situational anejaculation” [102] and “retrograde ejaculation”, into consideration, in order to profoundly reveal the potential alterations caused by ejaculation dysfunction.
ScRNA-seq revealed cellular and molecular disorders in patients with spermatogenic dysfunctions
Non-obstructive azoospermia
Spermatogenic dysfunction was characterized by the impairment of intratesticular spermatogenesis, and NOA is the most severe condition of it [103]. NOA was clinically diagnosed with no sperm in ejaculates along with evidence reflecting/causing the impairment of testicular spermatogenesis (e.g., elevated FSH or history of chemotherapy) [104]. Maybe due to the relatively easy availability of NOA testicular samples (a large number of NOA patients need testicular sperm retrieval surgeries [105, 106]), so far there have been seven studies that did their own scRNA-seq on at least one NOA testicular sample (Table 3). It should be noted that in this part (Table 3) we only discussed NOA samples without evidence of chromosome abnormality (NOA patients with chromosome abnormality will be discussed separately). Among these studies, over 70% (five) were conducted by Chinese research teams. Interestingly, due to the heterogeneity of NOA, the pathological status of scRNA-seq NOA samples used in each study varied across studies. Besides, maybe due to the decrease or lack of germ cells in the NOA testis, most of these studies focused on changes in somatic cells, especially SCs and LCs. Notably, most of the scRNA-seq studies did analyses on NOA samples in the following two ways: 1) identifying DEGs (in a certain type of cells) between control and NOA testes, and 2) finding new/major subtypes of a certain cell in NOA. For instance, Zhao et al. found that in iNOA patients, Sertoli cells were basically in stage a and stage b (mentioned above), and they revealed iNOA SCs showed different gene expressions pattern compared with normal testes [55], which were independently confirmed by Wang et al. [76] and Chen et al. [56]. In terms of somatic cells, two main somatic cells of the human testis, SCs and LCs, showed similar status in NOA patients, both were immature but with enhanced proliferative/divisive ability [55, 78], which were in line with the fact that immature somatic cells have proliferative ability [107, 108]. Alfano et al. further found that in SCOS (the severe pathology of NOA), the transcriptional and phenotypic features of LC were more like pre-pubertal LC, indicating the repressed maturation of LC in NOA [88]. As for germ cells in the NOA sample (if it had germ cells), Cst3 mediated autophagy might play an important role in the development of NOA by harming SSC maintenance (the ability of SSC to keep its population by self-renewal [109]), and knockdown of this gene might cause the impairment of SSC which might further lead to spermatogenic dysfunctions [110].
Studies with original scRNA-seq data on human testes from NOA patients without evidence of chromosomal abnormality
| Year | No. of NOA donors | Age | Technique on NOA samples | Cell numbers of NOA samples | Pathology of NOA samples | NOA etiology | FSH | Testicular volumes | Main findings on NOA spermatogenic cells | Main findings on NOA somatic cells | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2018 | 1 | 24 | Modified Smart-seq2 | 174 total | SCOS | Idiopathic* | Unknown | Unknown | Not focused | 1.Identifying DEGs of somatic cells (SC, PTM and LC) between NOA and control samples 2.The enrichment of γH2AX signal in NOA somatic cells, indicating their DNA damage response | [76] |
| 2020 | 3 | 26/31/32 | BD Rhapsody | 20507 total | SCOS | Idiopathic | Increased (based on average data) | Unknown | Not focused | 1.Sertoli cells in iNOA patients showed an immature phenotype and suffered from maturation arrest 2.Inhibition of the Wnt signal pathway could reverse the immature status of iNOA Sertoli cells and enhance their assistance to spermatogenic cells | [55] |
| 2020 | 2 | 29/30 | Modified STRT-seq | 61 Sertoli cells | Spermatogenic Arrest | Unknown | Unknown | Unknown | Not focused | 1.SARS-CoV-2-related gene ACE2 was significantly downregulated in NOA Sertoli cells | [159] |
| 2021 | 1 | 36 | Modified Smart-seq2/STRT-Seq | 432 total | Hypospermatogenesis* | Idiopathic* | Unknown | Unknown | 1.Identifying changes of autophagy-related genes in NOA germ cells, such as the upregulation of SQSTM1 and the downregulation of LC3A in spermatids 2.Cst3-mediated autophagy could help with SSC maintenance by regulating SSC maintenance-related genes (Oct4, Id1 and Nanos3) 3.Inhibition of Cst3 disturbed meiosis and spermatid formation | Not focused | [110] |
| 2021 | 1 | 26 | 10× Genomics | 3301 total | Hypospermatogenesis?** | Idiopathic/primary hypogonadism | Increased | Small | Not focused | 1.Lower mature LCs in NOA patients but more LCs experiencing cell division/differentiation 2.Identifying DEGs of immature LCs | [78] |
| 2021 | 3 | 37/37/41 | 10× Genomics | 3880 total | SCOS | Idiopathic | Increased | Small* | Not focused | 1.Finding the special transcriptional characteristics of somatic cells in SCOS, including immaturity of LC (p16/CDKN2A+), senescence of somatic cells, pro-inflammatory status of the microenvironment 2.The circulatory inflammatory markers such as CCL4 and CCL5 were enhanced in SCOS patients 3.Reporting a high level of DNA damage in SCOS LC and a genetic mutation in LMNA of SCOS testes | [88] |
| 2022 | 1 | 31 | Slingleron GEXSCOPETM | 1212 total | SCOS | Idiopathic | Increased | Small | Not focused | 1.Identifying the special transcriptomic pattern of NOA Sertoli cells, including the decrease of cell junction genes (SYNE2, ATP2B1, MTDH) and increase of FATE1 | [56] |
*Confirmed by checking with the first author of original articles.
**Successful testicular sperm retrieval but only few sperm were found, which inferring the pathology of hypospermatogenesis.
Note: FSH, follicle-stimulating hormone; SCOS, Sertoli cell-only syndrome; DEGs, differential expressed genes.
Note2: NOA phenotype patients with clear evidence of chromosomal abnormality (such as KS or Y chromosome microdeletion) were excluded from this table and will be specially discussed in a separate part.
Unfortunately, maybe due to the reduction or absence of germ cells in the NOA testis, most of these studies merely focused on somatic cells, which made the changes of residual spermatogenic cells in NOA patients largely unknown. Future studies could especially select samples with spermatogenic arrest or hypospermatogenesis to do scRNA-seq, which will give a chance to solve this problem.
Cryptozoospermia
In the area of Oligo-astheno-teratozoospermia (OAT), so far there have been few scRNA-seq data reported. This phenomenon is easy to understand because the scRNA-seq data was based on testicular tissue which is usually dissected in sperm retrieval surgeries such as TESE or microdissection TESE (mTESE) [111], and for OAT patients, many of them could use ejaculated sperm to do in vitro fertilization (IVF). Only severe OAT patients might need to undergo testicular sperm retrieval surgeries [112]. Cryptozoospermia, which was characterized by the appearance of isolated sperms in semen under the microscopic examination after centrifuging [106], could be considered as the transitional situation between severe oligozoospermia and azoospermia. The previous study pointed out that for cryptozoospermia patients, surgically retrieved testicular sperm was better than ejaculated sperm when doing intracytoplasmic sperm injection (ICSI) [113], which could also provide more convenience for obtaining testicular samples to do scRNA-seq. Using three cryptozoospermia testicular samples versus three controls (OA samples), Persio et al. profoundly revealed the extensive changes of testicular microenvironment in cryptozoospermia patients [66]. Deeper changes occurred in spermatogenic cells, including the reduction of germ cells such as pachytene spermatocytes and beyond, the changes of transcriptional profiles of all-type spermatogenic cells, an increase of PIWIL4 positive spermatogonia (representing the most naïve germ cells [114], just like the above-mentioned state 0 SPG or SSC-1), and a decrease of Adark SPGs (reserved SSCs [115]) and UTF1 positive SPGs (a SSC/undifferentiated SPG marker [116]) [66]. More importantly, an enhanced and prolonged expression of EGR4 was noticed in SPGs (also resembled the SSC-1 cells in Sohni's study [67]), which could act as a transcriptional factor and inhibit the expression of UTF1 in part of SPGs from cryptozoospermia patients [66]. Besides, scRNA-seq data also revealed perturbed somatic cells in cryptozoospermia, including an obvious increase of PTMs (including fibrotic PTMs) and macrophages, a deep transcriptional change of PTMs, an elevated ratio of MUSTN1 postitive (a pan-musculoskeletal cell marker [117]) blood vessels and the gathering of CD3 T cells around blood vessels [66]. Besides, two crosstalk networks between SPGs and adjacent microenvironments including “SPGs-FGFR1/3-FGF2-pachytene/diplotene SPCs” and “SPGs-ACKR2-CCL2/3/4/5/3L1/14-EC/immune cells/perivascular cells” were detected in cryptozoospermia rather than normal patients [66].
Trans-females after gender-affirming hormone therapy
The testis removed from patients receiving sexual reassignment surgery was another important tissue source for scRNA-seq studies [43, 72]. Of course, if a physically healthy patient did not receive any medical treatment before transsexual surgery, the removed testis was definitely an ideal sample of the “normal” group. Unfortunately, many transgender individuals received gender-affirming hormone therapy [118], which might profoundly impair the spermatogenesis of their testes [119]. Guo et al. drew the single-cell transcriptional profiles of two trans-females who received long-term (over a year) testosterone antagonist and estradiol treatment before transsexual surgeries [72], and found huge disorders in both germ cells and somatic cells. In terms of germ cells, although they still existed in both donors, the proportions of advanced spermatogenic cells including spermatids, SPC and diff_SPG were extremely low or even absent, and such changes stopped at the level of undifferentiated SPG (or SSC, or state 0 & 1 SPG as mentioned above) [72], indicating that testosterone inhibition might have less impact on SSC itself at the transcriptional level but could stop its differentiation. We found that such status was similar to the testis of prepuberty boys, for whom diff_SPG and more developed germ cells did not appear before 8 years old [55]. As for somatic cells, SC was found to be immature with higher expression of AMH and HES1 in these two patients, and the transcriptional profiles of SC were more like pubertal males (stage_b SC was predominant during this period [55]) rather than adults [72].
ScRNA-seq detected impairment of testicular somatic cells in patients with chromosome abnormalities
Klinefelter syndrome
Klinefelter syndrome (KS) makes up the most common part of male chromosome disorders [120], which is also widely studied by scRNA-seq studies [55, 78, 121]. Importantly, usually the studies on KS patients were considered as a part of works on NOA patients because of the popular azoospermic phenotype [122]. Nevertheless, due to the special karyotype of KS (one or more extra X [123]), the changes and relative mechanisms in KS testes were distinguishable from the above-mentioned iNOA patients [124]. Interestingly, for scRNA-seq data of KS testicular samples with the presence of germ cells, there seemed to be no huge changes at the transcriptional level of KS germ cells compared with normal germ cells [121]. But such a conclusion was drawn indirectly based on the co-clustering of KS germ cells with normal germ cells, rather than differential expression analysis (because there was only one KS sample sequenced with only 39 germ cells identified in Laurentino's study [121]). Unlike the other type of male infertility (in which the scientist could deeply analyze germ cell changes), Laurentino's study was almost the “best” work of KS germ cells because most of the scRNA-seq samples on KS from other teams lacked spermatogenic cells and showed a SCOS or even tubular atrophy pattern [55, 78]. Although previous study pointed out that spermatogenic cells were not entirely lost among all KS patients [125], we found it was hard to accurately get one sample with germ cells to do scRNA-seq. According to our experience, for KS patients, if the sample was coming from a random biopsy, we can't guarantee there were germ cells in it. And if the sample was coming from TESE or mTESE surgery (which we could judge the spermatogenic status by the form of tubules during surgeries), although it was possible for us to find a sample that contains germ cells or even spermatozoa, most of such “good” samples should be used for finding sperms and ICSI (since such good samples were quite rare and precious), so it was unlikely to have an extra “good” sample for scientists to do further studies including scRNA-seq. By combining scRNA-seq with bulk RNA-seq data and IHC staining, Winge et al. found that such germ cell loss in KS patients might start as early as in the fetal testis [126]. And another study found that KIF2C might be a germ-cell-related gene to regulate spermatogenic development in KS testes [127]. Frankly, based on the current evidence from scRNA-seq data, it's too early to answer the question that what was the transcriptional change in the residual germ cells of KS patients.
As for KS somatic cells, scRNA-seq data has offered us abundant findings which we didn't know before. SCs were found to be the most affected somatic cells in KS testes [126]. In detail, KS SCs were almost stage_b SCs and showed altered energy metabolism characteristics such as enhanced expression level of glycolysis and oxidative phosphorylation-related genes but rather low triglyceride metabolism level [55]. Huge transcriptional changes were observed in KS SCs including the upregulation of immune-related genes such as B2M [128] and MIF [129], the decreasing of sex hormone -regulating gene GNRH1 and the increasing expression of X-linked genes [55, 78]. Notably, Mahyari et al. found a special subpopulation of KS SCs which showed lacking expression of XIST, which could partially explain the X-linked genes' enhancement in SCs [78]. The involvement of immune activation in KS testes found by Zhao et al. [55] were also validated in peripheral blood of KS patients [130], indicating the profound impact of immune-related pathogenic factors in the pathogenesis of KS. Similarly, the proportion of immature LCs was also increased in KS LC population and more LCs were found to undergo division/development (with higher expression level of RNA processing/splicing-regulating genes such as CCNL1, DDX17, PNISR and FUS) [78]. Interestingly, when comparing KS-NOA somatic cells with iNOA somatic cells, both shared and heterogenous features were found. For example, the expression of β-catenin protein level was both significantly enhanced in KS-NOA and iNOA SCs, indicating a shared pattern of activated Wnt/β-catenin pathways in both type of NOA compared with normal adults [55]. While SERPINE1, a tissue plasminogen blocker, was highly expressed in KS immature LCs but not in iNOA LCs, indicating the heterogenous pathogenesis of KS-NOA compared to NOA with other etiology [78].
Y chromosome microdeletion
The testicular evidence got from single-cell studies on patients with Y chromosome microdeletion was limited and less than the evidence from bulk studies [131, 132], especially for AZFc microdeletion (the most common form of Y chromosome microdeletion in male infertility [133]). So far, there has been only one scRNA-seq testicular sample with azoospermia factor a (AZFa) deletion reported [55]. Unfortunately, this sample was with SCOS pathology, so we can't gather enough data on germ cells of patients with Y chromosome microdeletion. Pseudotime trajectory analysis in the AZFa testis found its SCs were in the early part of stage_c (a form of mature SCs), together with upregulation of MIF and DEFB119 (two important factors in immunoregulation), suggesting the involvement of immune regulating pathways in the etiology of AZFa SCs [55]. Considering the limited research on this field, we suggest future scRNA-seq studies should focus on the pathogenesis of azoospermia factor b (AZFb) and azoospermia factor c (AZFc) testes (especially those AZFc testes with germ cells), so as to profoundly reveal the special changes in patients with Y chromosome microdeletions.
Re-analysis of published scRNA-seq data on human testicular samples met diversified scientific requests
Overview
With more and more scRNA-seq data on human testicular samples published, so far there have been plenty of studies that chose to re-analyze those published data in various aspects. Table 4 summarized the studies that re-analyzed the previously reported scRNA-seq data on human testicular samples. A special case is Chen et al.'s spatial transcriptomic work on testes [134]. In that Slide-seq-based study, the authors only employed published scRNA-seq data as a reference for cell-type assignment, and as an input for pseudotime value assignment, which (strictly speaking) were not the real “re-analyses” of scRNA-seq data itself. Therefore, we chose to exclude that study from Table 4. Among the reviewed 48 studies in Table 4, the majority of them combined the human testicular scRNA-seq data with other types of scRNA-seq data or bulk RNA-seq/microarray data in order to get integrated results, and most of them chose to experiments such as IHC/immunofluorescence (IF) staining on testicular sections to validate their findings. Hence, in Table 4 we also offered the important public data (other than human testicular scRNA-seq data) that the study used and listed the experiments/clinical studies or sequencing works conducted on their own. Although those studies had various designs and different findings, we still found universalities among these studies and categorized them into several categories. The detailed classifications as well as representative studies were discussed hereinafter, and many of the studies were with multi-categories.
Studies only with reanalysis of previously published scRNA-seq data of human testicular samples
| Authors | Year | Country* | Human testicular scRNA-seq data type | Other key public/published datasets/databases used** | Main findings | Important experiments/clinical studies and original sequencing data | Reference |
|---|---|---|---|---|---|---|---|
| Gomes Fernandes et al. | 2018 | Netherlands | Prenatal | 1.ScRNA-seq data of gonadal cells from fetal ovaries (GEO) | 1.PIWIL1 was barely expressed in fetal testes but located paranuclearly as a single large dense satellite-like body in oocytes 2.PIWIL2 was detected in the cytoplasm of both female and male germ cells but shrank in intermitochondrial cement in oocytes in primordial follicles 3.PIWIL3 was absent in germ cells of both genders 4.PIWIL4 was found in the cytoplasm of germ cells and localized in intermitochondrial cement of both oocytes in primordial follicles and spermatogonia | 1.Immunofluorescence (IF) staining of fetal testes/ovary, mouse testes and adult ovary 2.Fluorescence-activated cell sorting (FACS) analysis of fetal testicular or ovarian cells | [187] |
| Pont et al. | 2019 | France | Normal | 1.ScRNA-seq data of other human tissue/cells (GEO/ArrayExpress/ 10× Genomics website [188]) | 1.Developing a method (Single Cell Signature Explorer) for gene signature scoring and its visualization at a single-cell resolution (https://sites.google.com/site/fredsoftwares/products/single-cell-signature-explorer) | No | [174] |
| Reznik et al. | 2019 | USA | Prenatal | 1.Human transposable element consensus sequences from GIRI Repbase dataset [189] | 1.Drawing the dynamic maps of PIWI proteins' expression and identifying transposon-derived piRNAs of fetal testes across development, and detecting an active piRNA pathway and transposable elements repression 2.Finding pre-pachytene piRNAs amplified in human fetal testes, which were mostly derived from transposable elements especially long interspersed element type 1 family (L1) 3.Finding L1-ORF1p (coded by L1 gene open reading frame 1) were highly expressed at mid-gestation and decreased in germ cells along with the enhancement of piRNAs, H3K9me3 and HIWI2 nuclear localization 4.Finding the evidence that L1 expression is heterogeneous, in other words, a subset of L1-expressing fetal germ cells utilized the PIWI-piRNA pathway to cause epigenetic silencing of L1 via H3K9me3, but the rest cells were L1-resistant | 1.Human fetal testes-based experiments including IF staining and small RNA-seq, etc. 2.Xenograft model (mice)-based experiments including model construction (implanting human fetal testes into immunocompromised mice) and xenograft samples IF staining, etc. | [190] |
| Wang et al. | 2020 | USA | OB/Normal | No | 1.Revealing the expression of ACE2 (SARS-CoV-2 target) in SPGs, LCs and SCs | No | [145] |
| Pan et al. | 2020 | China | Normal | No | 1.SARS-CoV-2 was not detected in the semen of patients recovering from COVID-19 2.There was barely any co-expression of ACE2 and TMPRSS2 based on scRNA-seq data | 1.Collecting semen samples from COVID-19-recovered male Chinse patients (along with clinical information) and do RT-qPCR of SARS-CoV-2 in semen samples | [146] |
| Xia et al. | 2020 | China | OB | No | 1.Finding endosialin as a specific marker of human stem Leydig cells 2.Finding that endosialin+ cells were with obvious proliferation (self-renewal) and differentiation (to testosterone-producing LCs) ability in vitro 3. Finding that transplanted human endosialin+ cells could localize in the interstitial region of murine testes and gain proliferation and differentiation ability in vivo | 1.Cell experiments of human testicular endosialin+ cells including cell isolation, cell culture, cell proliferation assay, in vitro cell differentiation, RT-qPCR, lentiviral vector infection, enzyme-linked immunosorbent assay (ELISA), flow cytometry, IF staining, etc. 2.IF staining of human and mouse testicular tissue 3.In vivo experiments by transplanting human endosialin+ cells in mouse testes | [191] |
| Ceyhan et al. | 2020 | USA | Normal | 1.Gene microarray data of testes from cryptorchid/infertile or normal persons (ArrayExpress) | 1.INPP4B is predominantly expressed in post-meiotic spermatogenic cells of both human and mice testes 2.Inpp4b-/- male mice were with smaller testes and fewer sperm, which could be worsened by aging and high-fat diet, and decreased steroidogenic enzymes and LH receptor gene were also found 3.Inpp4b-/- male mice had decreased elongated spermatids with increased diploid cells in testes 4.Finding enhanced apoptosis rate in spermatogenic cells and reduced meiotic marker γH2A.X expression in Inpp4b-/- mice testes | 1.Animal experiments including the establishment of gene knockout mice models, different feeding, sperm count and hormone measurement 2.RT-qPCR, immunohistochemical (IHC) staining, Western blotting (WB), propidium iodide staining and flow cytometry of testicular tissue of mice | [170] |
| Zhou et al. | 2020 | China | OB | 1.ScRNA-seq data of multi organs/cells (GEO/HPA) 2. IHC staining of ACE2/ TMPRSS2/Furin proteins from HPA [192], and bulk RNA expression level of these three genes from HPA [192] and GTEx [193] | 1.Ranking the organs and cells which were easier to be harmed by SARS-CoV-2 and finding the candidates with high risk including lung AT2 cells and macrophages, cardiomyocytes, stromal cells of the adrenal gland, the testis, the ovary and the thyroid 2.Finding that an acid condition could inhibit the activity of SARS-CoV-2 pseudovirus, which indicated the defensive role of the stomach against SARS-CoV-2 infection | 1.Infecting 293T-ACE2 and Hela-ACE2 cell lines with pseudovirus of SARS-CoV-2 pretreated with pH of 1.0, 2.0, 4.0, or 7.0 and luciferase assay of infected cells (24h or 48h) | [147] |
| Jiang et al. | 2020 | USA/China | OB | 1.ScRNA-seq data of mouse testes (GEO) | 1.Finding the total opposite survival status of Trf2Fl/Fl;SFTPC-CreYo (100%viability) and Trf2Fl/Fl;SFTPC-CreBlh (100%lethality), which was driven by heterogeneity in embryonic expression of those two transgenes 2.Finding the activity of both SFTPC-cre lines in the male germline, indicating premeioticly expressed Cre recombinase 3. Finding that SFTPC only expressed in late spermatogenesis of human and were absent in murine spermatogenesis | 1.Animal experiments based on Trf2 floxed, SFTPC-creYo, SFTPC-creBlh and Rosa26mT/mG mice and their crosses 2.Hematoxylin-Eosin (HE), IHC and IF staining of mouse testes | [171] |
| Winge et al. | 2020 | Denmark | Normal/OB/KS | 1.Bulk RNA-seq or gene/methylation microarray data of testis/blood/cultured lymphocytes of KS patients (GEO/EGA [194], etc.) | 1.Reviewing the histopathological changes of KS patients (fetal, underage and adult), such as the loss of germ cells starting from fetal time, hyalinized tubules, loss of SCs and LCs hyperplasia 2.Identifying a series of genes (by comparing analyses on DEGs/DMRs among KS studies) which is related to escape X-inactivation and might lead to the pathogenesis of KS 3.Revealing that SCs might be the most altered cells in terms of transcriptional changes | 1.HE staining of testes from normal karyotype and KS patients | [126] |
| Lau et al. | 2020 | Singapore | Normal/OB | 1.ScRNA-seq data of mouse testes (ArrayExpress) | 1.Drawing the map of spermatogenesis of cynomolgus macaque 2.Identifying different subtypes of SPG, revealing their marker genes and detecting the self-renewal versus differentiation dynamics of SPG in cynomolgus macaque 3.Showing both the similar and different genes expressed among human, mouse and cynomolgus macaque during the spermatogenic process and finding the conservatism of meiotic sex chromosome inactivation among three species | 1.Original scRNA-seq data of the cynomolgus macaque testis 2.HE, IHC and IF staining on testes of cynomolgus macaque | [167] |
| Shen et al. | 2020 | China | Normal/OB/NOA | 1.Interaction network data of ACE2 from GeneCards [195] | 1.Detecting the expression of ACE2 (SARS-CoV-2 target) in germ cells and somatic cells 2. Detecting the higher positive rate of ACE2 in infertile men than in normal persons | No | [148] |
| Chen et al. | 2020 | China | Normal | 1.In-frame indels in tandem repeat regions from the UCSC genome browser database [196] 2.Gene information from Consensus Coding Sequence (CCDS) database [197], and gene variants from databases including 1000 Genomes Project [198], ESP6500siv2 [199] and gnomAD [200] 3.Gene expression data from GTEx and HPA 4. Mouse phenotype information from the MGI database | 1. Identifying six pathogenic/likely pathogenic variants and four unknown-significance variants (VUS) in genes that could cause NOA or severe oligospermia (SO) 2. Reporting 20 new genes which might be related to NOA/SO, and five of them (BRDT, CHD5, MCM9, MLH3 and ZFX) were considered as strong candidates based on testicular single-cell data and previously reported murine models | 1. Whole-exome sequencing of 314 NOA or severe oligospermic Chinese patients and sanger sequencing | [139] |
| Hikmet et al. | 2020 | Sweden | Normal | 1.ScRNA-seq data of other tissues (GEO/ COVID-19 Cell Atlas [201]) 2.RNA expression data of testes and other organs from FANTOM5, GTEx and HPA 3.Protein expression data of testes and other organs from PaxDB [202] and ProteomicsDB [203] | 1.Comparing the expression level of ACE2 (SARS-CoV-2 target) among different tissues and finding the high expression of ACE2 mRNA and protein in the testis, especially LCs and SCs | 1.IHC staining of ACE2 in the human testis and other organs 2.WB of ACE2 in human lung/tonsil/kidney/colon | [149] |
| Ren et al. | 2020 | China | OB | 1.Clinical and epidemiological data of COVID-19 patients from public databases [204, 205] 2.Transcriptional data and microarray of testes and other organs from FANTOM5 and GTEx and GEO database 3.Transcriptional data of normal and testicular cancer and other cancers from TCGA 4.IHC staining data of testes and other organs from HPA 5.ScRNA-seq data of kidney from KIT [206, 207]and HCL [183] databases | 1.Identifying the high expression of ACE2 and TMPRSS2 in kidney with chronic renal diseases or diabetic nephropathy 2.Identifying enrichment of ACE2 in testicular germ cells and renal proximal tubules 3.Identifying the concentration of pro-inflammatory cytokines (like IL6ST) in testicular EC, macrophages, SSC as well as renal endothelial cells | No | [150] |
| Stanley et al. | 2020 | UK | Normal | 1.ScRNA-seq data of cynomolgus monkeys' ovary (GEO) 2.Transcriptional data and protein expression level of testes and other organs from HPA and HPM database [208] 3.Transcriptional data of EFO-21, AN3-Ca and BEWO cell lines from human cell atlas [209] | 1.Reporting the lack of co-expression of ACE2 and TMPRSS2 among testicular cells and ovarian somatic cells and the expression of both in a subpopulation of oocytes 2.Showing the wide expression of ACE2 with a lack of expression of TMPRSS2 in human cumulus cells 3.Suggesting the unlikeliness of long-term impact on human reproductive glands caused by SARS-CoV-2 | 1.Original RNA-seq of human cumulus cells from nine individuals | [151] |
| Qi et al. | 2021 | China | Normal | 1.ScRNA-seq data of other human organs (GEO/Tissue Stability Cell Atlas [210]) | 1.Investigating ACE2 and TMPRSS2 expression in different cell types of 31 organs and finding gall bladder and fallopian tube might be harmed by SARS-CoV-2 infection 2.Recognizing human nose, heart, small intestine, large intestine, esophagus, brain, testis, and kidney as “high-risk organs” due to high expression levels of ACE2 and TMPRSS2 | No | [152] |
| Soraggi et al. | 2020 | Denmark | Normal/OB | No | 1.Reviewing the genetic causes of spermatogenic dysfunction 2.Reviewing common data-processing procedures and visualization methods of scRNA-seq data 3.Doing integrated analysis of three independent testicular scRNA-seq data and analysis of some NOA-related genes 4.Building up an interactive website (https://testis.cells.ucsc.edu/) of integrated testicular scRNA-seq data | 1.HE staining of the human testis (OA and NOA) | [177] |
| Zheng et al. | 2021 | China | Normal | 1.Microarray of NOA testes and related controls (GEO) 2.IHC staining of testes from HPA | 1.Showing the largest group of immune cells in the normal testis was macrophages which kept an anti-inflammatory status 2.Reporting and validating a negative correlation of both M1/M2 macrophages with testicular Johnsen scores and an enhanced number of both M1/M2 macrophages in NOA | 1.IHC staining (CD163/CD68/CD86/INOS) of the human testis (NOA and normal spermatogenesis) | [142] |
| Olivieri et al. | 2021 | USA | OB | 1.ScRNA-seq data of various tissues from human (Tabula Sapiens [211]), mouse lemur (Tabula Microcebus [212]) and mouse (Tabula Muris [213]/GEO) | 1.Drawing the RNA splicing profiles of 12 human tissues using the SpliZ method and showing that splicing is regulated cell-type-specifically 2.Finding a number of cell-type-specifically spliced genes based on 10×Chromium scRNA-seq data, including MYL6 and RPS24, which were validated by FISH, Smart-seq2 and single-cell RT-PCR 3.Revealing conserved regulated splicing in spermatogenesis among human, mouse and mouse lemur (such as CEP112 and SPTY2D1OS) 4.Identifying subpopulations of monocytes (using SpliZ) with subpopulation-specific splicing of an ultraconserved exon of SAT1 | 1.Fluorescence in situ hybridization (FISH) tests on human lung/muscle tissue and isolated muscle cells 2.Sanger sequencing and single-cell RT-PCR | [165] |
| He et al. | 2021 | China | OB | 1.Gene microarray data of testes from NOA/OA patients or pooled controls (GEO) | 1.Identifying three hub genes including C22orf23, TSACC, and TTC25 which were related to spermatogenesis 2.Finding that TTC25 kept a low level through spermatogenesis, and C22orf23 had two peaks (one in diff_SPGs and one in late primary spermatocytes), while TSACC had an increasing trend during spermatogenesis | No | [143] |
| Hadziselimovic et al. | 2021 | Switzerland | Normal/OB/NOA | 1.Bulk RNA expression data of human testes and other tissues from GTEx 2. Genome-wide annotations of regulatory sites information in the SwissRegulon dataset [214] | 1.Finding NHLH2 to be decreased in pre-pubertal high infertility risk patients and reversed by GnRHa treatment | No | [215] |
| Zhang et al. | 2021 | China | OB | 1.Bulk RNA expression of ACE2 from testes and other organs based on GEPIA2 and Expression Atlas [216] 2. IHC staining of ACE2 on human testes from HPA | 1.Finding the high expression level of ACE2 mRNA in the testis compared with other organs 2.Finding that Leydig cells might be the target of SARS-CoV-2 and could be infected by pseudovirus SARS-CoV-2 in mice 3.Recovered COVID-19 patients had significantly lower testosterone levels than healthy controls | 1.Animal experiments including intratesticular injection with pseudovirus SARS-CoV-2 in mice (including infection detecting by Immunofluorescence), and IHC staining of ACE2 on mice testes 2.Testing sexual hormone levels in patients recovered from COVID-19 and controls | [153] |
| Yang et al. | 2021 | China | OB/NOA | No | 1.ELAVL2 was highly expressed in the testis of human and mice but decreased in NOA testes 2.ELAVL2 was predominantly located in SSCs of human and mice 3.ELAVL2 could facilitate the proliferation and reduce apoptosis of C18-4 and TCam-2 cell lines by sensitizing ERK and AKT pathways 4.ELAVL2 as an RNA-binding protein could bind mRNAs that were important in regulating SSC survival and proliferation and enhance their protein level post-transcriptionally 5.ELAVL2 could interact with DAZL in human and mouse testes | 1.Cell experiments based on primary testicular cells or testicular cell lines including cell culturing, IF staining, RT-qPCR, WB, Cell Counting Kit-8 (CCK-8) and EdU incorporation assay, lentivirus infection, Annexin-V/PI staining with flow cytometry, TdT-mediated dUTP Nick-End Labeling (TUNEL) Assay and co-IP, etc. 2.Human/mouse tissue-based experiments including IF staining, RT-qPCR, WB, Co-Immunoprecipitation (Co-IP), RNA Binding Protein Immunoprecipitation (RIP) and mass spectrometry, etc. | [160] |
| Han et al. | 2021 | China | Normal/NOA | 1.Gene microarray of NOA/control testes (GEO) | 1.Revealing the DEGs and associated signal pathways in NOA patients compared to control 2.Identifying CHD5 and SPTBN2 as potential biomarkers of NOA pathogenesis and showing its obvious downregulation in NOA testes among various testicular cell types | No | [144] |
| Salehi et al. | 2021 | Iran | Normal/OB | No | 1.Identifying ten bridge genes (highest betweenness centrality) among different stages of spermatogenesis, including DNAJC5B, C1orf194, HSP90AB1, BST2, EEF1A1, CRISP2, PTMS, NFKBIA, CDKN3, and HLA-DRA | No | [217] |
| Fan et al. | 2021 | Netherlands | Normal/OB | 1.ScRNA-seq data of fetal female gonads (GEO) 2.Gene lists related to male and female infertility from the DisGeNET v6.0 database [218] | 1.Drawing the maps of molecular signatures (marker genes, DEGs among subclusters, dynamic expression changes of functional genes such as cytoskeleton-associated genes, etc.) during female meiotic prophase I stages 2.Revealing both conserved and sex-distinctive transcriptional features as well as DNA methylational regulation during meiotic prophase I stages between female and male 3.Revealing a momentary increase of X-linked expression during female pachytene (which is opposite to meiotic sex chromosome inactivation of males) and revealing that it was due to a lower turnover or higher stability of X-linked genes rather than enhanced X-linked transcription | 1.IF staining on fetal ovaries/testes and adult ovaries/testes 2.RNA/DNA FISH on fetal ovaries and RNA FISH on adult testes | [172] |
| Persio et al. | 2021 | Germany | OB/Cryptozoospermia | 1.Whole-genome bisulfite sequencing (WGBS) of sperm/ embryonal stem cells/SSC/ primordial germ cells (GEO/ENA [219]) | 1.Finding no obvious difference between control and cryptozoospermia patients in terms of whole genome levels' or imprinted regions' DNA methylation 2.Identifying a series of differentially methylated regions (DMRs) (in cryptozoospermia) and their related genes, most of which were hypermethylated 3.Further recognizing 13 DMR-associated (most were hypermethylated DMRs) genes (BBS5, C2orf92, C10orf120, CFAP299, DHX16, MTCP1, PACRG, PPP1R36, PLAC8L1, PPP3CC, SPAG16, TCP10L2, ZFAND4), which were differentially expressed (most were downregulated) in meiosis and/or spermiogenesis of cryptozoospermia patients 4.Finding that four genes (C2orf92, C10orf120, PPP3CC, and PPP1R36) had hypermethylated promoters in cryptozoospermia while the rest might have methylated germ cell-specific enhancers | 1.Periodic Acid-Schiff (PAS) staining of human testes (control/ cryptozoospermia) 2.Isolation and culturing of primary testicular cells and ploidy analysis 3.Targeted deep bisulfite sequencing and WGBS of testicular cells (control/cryptozoospermia) | [220] |
| Martin-Inaraja et al. | 2021 | Spain | Prenatal | No | 1.Evaluating the effects of two different culture media (Shinohara-medium and Zhou-medium) and four substrates (laminin, gelatin, vitronectin and matrigel) on culturing human fetal germ cells and finding the huge differences of hFGC numbers in different culture environment 2.Finding that the Shinohara-medium with gelatin-coated substrate could lead to the highest number of hFGCs (10% in day 6) while vitronectin-coated substrate could lead to similar hFGCs number (day 6) in two media | 1.Isolation, FACS analysis and culturing of male human fetal germ cells from fetal testes 2.IF staining of human fetal testes and cultured human fetal germ cells | [141] |
| Hardy et al. | 2021 | USA | Normal | 1.Mouse model phenotypes in MGI database 2. Gene expression data of testes using GTEx, Ace-View [221] and BioGPS [222-224] 3. SprT-like domain 3D modeling and intrici disordered region motif search of GCNA via several online database/tools such as UniProt [225], HHpred[226], MODELLER[227] , MetaDisorder[228], etc. | 1.Identifying seven potential pathogenic variants (NOA or cryptozoospermia) of GCNA, such as p.Ala115ProfsTer7(which caused early frameshift), p.Ser659Trp/p.Arg664Cys(located in SprT-like domain), and variants such as p.Ser295Pro which were located in predicted consensus IDR motif 2.Finding the expression of GCNA throughout spermatogenesis, especially in differentiating SPGs | 1.Orignial whole genome sequencing (WGS) of 2225 NOA patients (with clinical information analyses) from multi-centers 2.PAS and IF staining of human testicular tissue (normal/GCNA variants) | [229] |
| Zhou et al. | 2021 | China | NOA | 1.Gene microarray of NOA/OA testes (GEO) | 1.Building up a five-gene based random forest diagnosis model (CCT8/CDC6/PSMD1/RPS4X/RPL36A) for NOA and OA 2.Validated those five genes' expression and the efficacy of the random forest model in local cohort | 1.IHC staining of RPS4X on human testes (20 OA and 20 NOA samples) 2.RT-qPCR on seminal plasma (20 OA and 20 NOA) of five genes (CCT8, CDC6, PSMD1, RPL36A, RPS4X) 3.Analysis of clinical (age, sex hormones) and pathological (Johnsen scores) parameters of the above-mentioned 40 patients | [161] |
| Cai et al. | 2021 | China | Normal/Infant | 1.Transcriptional data of normal testes from GTEx database | 1.Analyzing the expression of of ACE2 (SARS-CoV-2 target) in both adult testes and infant testes 2.Validating the high expression of ACE2 protein in testes | IHC staining of ACE2 on human normal testes | [154] |
| Wu et al. | 2022 | China | Normal | No | 1.Drawing the original single-cell Assay for transposase-accessible chromatin sequencing (scATAC-seq) based map of human spermatogenesis (including accessibility of chromosomes, TF-binding sites and motifs with high frequencies during spermatogenesis, etc.) and showing scATAC-seq's advantages over scRNA-seq 2.Identifying two spermatogenically functional genes including TLE3 (exclusively expressed in diff_SPGs) and PFN4 (relevant to actin cytoskeletal organization during meiosis) and finding the motifs (such as CTCF, CTCT, etc) with high accessibility which were enriched in the upstream of these two genes | 1.Original scATAC-seq of human normal testes | [230] |
| Tang et al. | 2022 | China | OB/NOA | No | 1.Showing a significant increase of LC and macrophages in iNOA patients' testes 2.Identifying of LC-specific transcription factors (TFs) (including LHX9, KLF8, KLF4, ARID5B and RXRG) in NOA, and macrophages-specific TFs (such as POU2F2, SPIB, IRF5, CEBPA, ELK4 and KLF6) in NOA, which might be related to the function of LC and macrophages and finally lead to impaired spermatogenesis | No | [164] |
| Zhang et al. | 2022 | China | Juvenile/NOA | 1.Gene microarray of mouse testicular Sertoli cells exposed to Bisphenol A | 1.Finding the proliferation and maturation of spermatogenic cells caused by Bisphenol A, which might be triggered by secretory proteins from SC 2.Finding that Bisphenol A might be able to harm SC by dysreguating its secretory proteins, and might be a potential cytotoxic factor related to NOA | No | [163] |
| Rengaraj et al. | 2022 | South Korea | Prenatal | No | 1.Establishing a transgenic chicken model (DAZL::GFP) with GFP expressing but not changing the expression of endogenous DAZL 2.Identifying four male-specific and five female-specific stages during the development of chicken germ cells, and revealing the paths as well as their related transcriptional patterns of chicken germ cells' stages transition (one path in male and two paths including meiosis and apoptosis in female) 3.Revealing both conserved and species-specific transcriptional patterns between human and chicken germ cell development | 1.Isolation (using MACS) and culturing of chicken primordial germ cells from male gonads 2.Building up DAZL::GFP PGCs and DAZL::GFP transgenic chickens 3.Validation of the transgenic PGCs and chickens by WB, RT-qPCR and IF staining, etc. 4.Original scRNA-seq of the DAZL::GFP germ cells collected from embryos or embryonic gonads (both male and female) at different ages | [166] |
| Wu et al. | 2022 | China/USA | Normal/OB/NOA | No | 1.Reviewing the role of laminin and collagen chains (bioactive peptides) in regulating rodents' spermatogenesis 2.Showing the different expression level of important genes encoding laminin and collagen chains or basement membrane proteins among normal, OA and NOA human testes, suggesting their supporting roles in human spermatogenesis | 1.HE staining of human testes with complete spermatogenesis 2.IF staining of α-tubulin and F-actin on human SC | [137] |
| He et al. | 2022 | China | Normal | 1.ScRNA-seq data of human head and neck carcinoma (GEO) 2. ScRNA-seq data (PLIN2 expression level) of various organs (lung/pancreas/prostate) from HPA 3.Data of tumor infiltrating immune cell markers of oral squamous cell carcinoma (OSCC) from TIMER [231] 4.Gene expression data of head and neck squamous cell cancer in cBioPortal [232, 233] | 1.PLIN2 is highly expressed in macrophages in human testes and other organs (lung, pancreas and prostate) and CD68+ tumor-associated macrophages of OSCC stroma 2.OSCC patients with higher CD68+ TAM-derived PLIN2 expression were with advanced stages, more malignant phenotypes and poorer prognosis 3.An immune suppressed features (low CD8+ T cells/high CD68+ TAMs and FOXP3+ Tregs with enhanced immune checkpoints) were observed in OSCC patients with high PLIN2 expression | 1.IHC, IF and Oil Red O staining of human oral squamous cell carcinoma tissues 2.Clinicopathological data analysis and survival analysis of OSCC patients | [138] |
| Zhang et al. | 2022 | China | Juvenile/Normal | No | 1.Identifying five SC subtypes, five LC subtypes and four PTM subtypes in pigs during somatic development 2.Identifying PRND as a new maker of SCs 3.Revealing the high conservatism of somatic cell development between human and pigs (except for LCs) and screening some transcription factors involved in the development of somatic cells of pigs and human | 1.Origial scRNA-seq of pig testes at different ages 2.HE and IF staining (SOX9/PCNA/AMH/ STAR/ACTA2/PRND) of pig testes | [169] |
| Zhou et al. | 2022 | China | NOA | 1.Gene microarray data of OA/NOA and pooled control testes (GEO) 2. Gene symbols of transcription factors (TFs) from Human Transcription Factor database [234] | 1.Building up a three-gene based random forest diagnosis model (ETV2/TBX2/ZNF689) for NOA 2.Validated those three genes' expression and the efficacy of the random forest model in local cohort | 1.RT-qPCR (ETV2, TBX2 and ZNF689) of seminal plasma from 20 NOA and 20 OA (with normal spermatogenesis) 2.IHC staining of ETV2/TBX2/ZNF689 on testicular samples of the above-mentioned 40 patients 3.Analysis of clinical (age, FSH, LH, T) and pathological (Johnsen scores) parameters of the above-mentioned 40 patients | [140] |
| Chitiashvili et al. | 2022 | USA | Prenatal | 1.ScRNA-seq data of fetal ovaries, fetal germ cells and day 4 aggregate cells (GEO) | 1.Showing that FGFR3 were enriched in primordial germ cells (PGCs) of both ovaries and testes, but decreased with the appearance of primordial oocytes 2.Based on the evidence from fluorescence-activated cell sorting and scRNA, finding that FGFR3 could work as an in vitro biomarker to enrich FGFR3-positive PGCs from the human embryonic and fetal ovaries | 1.IF staining of fetal ovaries and day 4 aggregate cells differentiated from human embryonic stem cells (hESC) 2.Fluorescence activated cell sorting of fetal ovarian cells 3.Culturing of hESC and differentiating into primordial germ cell-like cells (PGCLC) 4.Original scRNA-seq of FGFR3-sorted fetal ovarian cells and PGCLC | [235] |
| Guo et al. | 2022 | China | Normal | 1.Clinicopathological, transcriptional, DNA-methylation data and copy number of TCGA testicular germ cell tumor (TGCT) from UCSC XENA database [236] and microarray data of TGCT (GEO) 2. Expression levels of RFPL3S in different tumors from GEPIA2 database, and immune data of TGCT (immune scores, tumor infiltrating immune cells, immune, and reactions to immune therapy, etc.) from Sangerbox [237], GSCA [238] and TIDE databases [239]. | 1.Revealing that long non-coding RNA RFPL3S might be a tumor suppressor gene in TGCT (with low expression, hypermethylation and low copy number in TGCT, and was negatively associated with the stage of TGCT) and validating this phenomenon by cell experiments 2.Showing that RFPL3S was mainly expressed in testicular germ cells and were positively associated with the infiltration of immune-activating cells and immunotherapy benefits, while negatively related to immunosuppressive cells | 1.Cell experiments including transfection of si-RFPL3S, RT-qPCR of RFPL3S, transwell assay and CCK-8 assay based on NCCIT and Tcam-2 cell lines | [136] |
| Tian et al. | 2022 | China | Normal | 1.ScRNA-seq data of testes of mice and cynomolgus macaque | 1.Constructing the profile of spermatogenesis of Mongolia sheep using scRNA-seq data 2.Comparing the spermatogenic process of human, mice, sheep and cynomolgus macaque and finding the conserved meiotic sex chromosome inactivation and genetic dynamics of spermatogenesis among four species 3.Revealing the high conservatism of spermatogenic cell development, gene expression and testicular cell-cell communication signaling between human and sheep | 1.Original scRNA-seq of the Mongolia sheep testis 2.IF staining (DMRT1/ TEX101/KLF5/WWTR1) on the Mongolia sheep testis | [168] |
| Yang et al. | 2022 | China | OB | 1.Exome Aggregation Consortium (ExAC) data [240] and 1000 Genomes Project [198] 2. Known pathogenic genes of murine azoospermia from MGI database 3. Human testes enriched genes from HPA database | 1.Identifying a new copy number variation (CNV) (seq [GRCh37] del(19) (19q13.33) chr19: g.49894043-49903011del) and a heterozygous loss of function variant (NM_144688: c.979_980del: p.R327Sfs*21) in KASH5 gene of a NOA patient (with meiotic arrest pathology) and his sister 2.Reporting the expression of KASH5 in human testes (mostly in SPCs) and finding the similar expression pattern of it between human and murine testes 3.Validating the similar phenotype (NOA) using Kash5-null mouse model | 1.Original scRNA-seq data of mouse testes 2.HE and IF staining on human (OA and meiotic arrest) testes 3.Whole-exome sequencing, PCR, RT-qPCR, sanger sequencing, CNV array together with clinical data analysis of an NOA (meiotic arrest) patient with bi-allelic variants in KASH5 and his family members 4.Establishing Kash5-heterozygotes and Kash5-null mice model (together with wide type mice) and HE/IF/TUNEL staining & WB of mice testes 5.Meiotic chromosomal spread experiments on human and murine testes | [135] |
| He et al. | 2022 | China | OB/Neonatal/KS | 1.Bulk RNA-seq data of adult/fetal testes from Klinefelter syndrome (KS) patients with adult/fetal normal controls (GEO) | 1.Identifying four un-reported hub genes (KIF2C, MRPS2, RPS15 and TSFM) which might be functional in KS 2.Finding that KIF2C showed an increasing trend in the development of spermatogenetic cells and might be strongly relevant to germ cell development of KS patients | No | [127] |
| Calonga-Solís et al. | 2022 | Germany | Fetal/Neonatal/Infant/Juvenile/OB | 1.ScRNA-seq data of prenatal ovaries 2.Binding site information in GeneHancer database [241] | 1.Detailed case report of a patient with de novo stop-gain variant in MYRF (p.Q838*), which was associated with the patient's Scimitar syndrome, 46,XY partial gonadal dysgenesis and severe hyperopia 2.Reporting the expression status of MYRF using scRNA-seq data of human gonads at different ages, and finding a high expression of MYRF in subsets of coelomic epithelium cells for both male and female 3.Suggesting that MYRF might involve in early development of gonad and heart by upregulating CITED2 | 1.Detailed case report of a patient with Scimitar syndrome, 46,XY partial gonadal dysgenesis and severe hyperopia 2.Trio-WGS of the patient's family (mother, father and affected child) | [242] |
| Stow et al. | 2022 | USA | Normal | 1.ScRNA-seq data of mouse testes and MCF7 cell line (NCBI Sequence Read Archive [243]) | 1.Developing and testing a new method named SCIFER (including technical parameters needed), to quantify expression of long interspersed element-1 at the single-locus resolution in scRNA-Seq datasets 2.Pointing out that unsupervised analysis of L1 expression in single cells exponentially inflates the levels of L1 expression and the number of expressed L1 loci 3.Finding that mouse round spermatids and human SPGs, SPCs, and round Spermatids had the highest levels of L1 mRNA | 1.Original scRNA-seq of MCF7 and HEK293-FRT-LacZeo combined cells 2.Original bulk RNA-seq of human testicular bulk RNA samples (commercial) | [175] |
| Luo et al. | 2022 | China | Normal/OB | No | 1.Recognizing the upstream trend of FOXP4 with the development of SSC, and showing it as a marker of a subset of SPG that had stem cell features 2.Proving that the inhibition of FOXP4 could significantly repress SSC's proliferation and enhance its apoptosis 3.Showing that FOXP4 were significantly downregulated in human testes with spermatogenic dysfunction, especially in those with severe pathological pattern | 1.Tissue based experiments including HE and IHC (FOXP4)/ IF staining (FOXP4/GRFA1/UCHL/PCNA/KIT) and WB (FOXP4) of testicular samples from OA and NOA patients 2.Cell experiments including cuturing of human SCC cell line, si-FOXP4 transfection, RT-qPCR (FOXP4), CCK-8 assay, EdU assay, Flow cytometry with Annexin V-APC/PI staining, TUNEL assay and WB (FOXP4/PCNA) | [162] |
*Country information was based on the institutional information of the first author
**To be more brief, the references of some databases employed by multi-studies were listed here, including GEO (Gene Expression Omnibus) [244], ArrayExpress[245], HPA(Human Protein Atlas [192, 246], GTEx (Genotype-Tissue Expression) [193], FANTOM5 (Function Annotation of The Mammalian Genome) [247], TCGA (The Cancer Genome Atlas) [248], GEPIA2 (Gene Expression Profiling Interactive Analysis) [249] and MGI (Mouse Genome Informatics) [250] databases.
Detecting the transcriptional pattern of interested genes in human testes
As a transcriptome-level technology, the fundamental usage of scRNA-seq was to detect genes' transcriptional patterns such as the mRNA expression level of a certain gene in different types of cells. A typical study was carried out by Yang et al. [135], in which they first found a new variant of KASH5 by Whole Genome Sequencing (WGS) in an NOA patient, but did not know the detailed KASH5 expression pattern in the human testis. So, they chose scRNA-seq data (the data used here were previously reported by the same team) to see the expression of KASH5 and found it to be enriched in SPCs, especially leptotene to pachytene SPCs, and finally validated it by IF staining. This process of “finding target gene” and then “detecting it in human testicular scRNAs-seq data” (sometimes with a third step as “validating genes using experiments”) was quite common for studies with interested genes [136-141]. Meanwhile, the interested genes were not restricted to one gene or several genes, the researchers could detect a gene family (such as Interleukins) or a series of function-related genes (such as inflammatory cytokine receptors) in scRNA-seq data [142], which could give researchers a view on macroscale. Moreover, an updated usage of this process was to not only show the type of cells that a certain gene was expressed in, but also show its transcriptional changes during spermatogenesis [143] or among different pathological groups [127, 144]. A good example came from He et al.'s work [127], in which they identified four interested genes (KIF2C, MRPS2, RPS15 and TSFM), and then based on human scRNA-seq data of both KS testes and normal testes, they showed that KIF2C were downregulated while RPS15 were upregulated in KS testes. Additionally, pseudotime trajectories showed a growing tendency of KIF2C and a downtrend of RPS15 with the maturation of spermatogenic cells [127]. We need to point out that no matter what method the study used and no matter how complicated the study procedure was, the nature and essence of this type of re-analyzing were to show the expression of given genes in human testes, no matter how these interested genes were found out (by other technologies/experiments, by bulk RNA-seq/microarray data, by scRNA-seq data of other organs/species, by literature reviews or even by the authors' own idea).
Indirectly revealing the vulnerability of testes to SARS-CoV-2 infection
Due to the Corona Virus Disease 2019 (COVID-19) pandemic and the high proportion of SARS-CoV-2-related articles in Table 4 (10 out of 48), we especially discussed the usage of human testicular scRNA-seq data here. As for the usage of testicular scRNA-seq data, most of these studies were actually in the form of “detecting the transcriptional pattern of interested genes in human testes” [145-154], since several important proteins, such as ACE2 [155], TMPRSS2 [156], Furin [157] and CD147 [158], were deeply involved in the entry of SARS-CoV-2. So far, the opinions on this topic have been controversial. In some studies, ACE2 was reported to be expressed in both germ cells (especially SSCs&SPGs) and somatic cells, including SCs, LCs as well as myoid cells [145, 147, 149, 154]. And one study showed a higher positive rate of ACE2 in testes from infertile patients (OA and NOA) [148]. Those results indicated the potential vulnerability of human testes to SARS-CoV-2 infection, especially in infertile men. And some studies even called the human testis a “high-risk organ” for SARS-CoV-2 infection [152]. But based on the scRNA-seq-derived evidence that there was no co-expression of ACE2 and TMPRSS2 in the human testis, other two studies gave the opposite view that human testes might not be able to be long-term affected by SARS-CoV-2 infection [146, 151]. From our point of view, those ten scRNA-seq-based studies only gave indirect evidence based on previously published data. By combing original and published scRNA-seq data on human testes, Liu et al. partially overcame the shortcomings of those studies and for the first time showed an expression of ACE2 in primordial germ cells of prenatal testes and a downward trend of ACE2 with the age increasing [159]. More importantly, although ACE2 had the highest expression in SCs, it significantly shrank in SCs of NOA patients [159]. But the results of Liu's study were still based on scRNA-seq of patients without histories of COVID-19, which were also indirect. So far, there has been no report of scRNA-seq data on human testes from COVID-19 recovered patients, maybe due to the huge difficulty of sample obtaining. But with the pandemic continuing, there must be patients who simultaneously suffered from COVID-19 and male infertility and needed testicular biopsy or TESE surgery. Therefore, in the future, researchers can use scRNA-seq to compare testicular samples from COVID-19-recovered patients with those from non-COVID-19 patients, which will offer direct evidence of the impact of SARS-CoV-2 on human testes.
Finding novel genes involved in normal testicular function and male infertility
Testicular scRNA-seq data could not only be used to draw expression patterns of certain genes in the testis, but could directly be used as the sources of such novel genes. The difference between this type of usage and the type “Detecting the transcriptional pattern of interested genes in human testes” is that this type of study directly used testicular scRNA-seq data as a tool to find novel genes, while the other one only used scRNA-seq data as a tool of validation or display. For example, by comparing the DEGs in SPG between OA and NOA scRNA-seq data, Yang et al. reported a novel SPG-specific gene ELAVL2 and found it to be significantly downregulated in SPGs of NOA patients [160]. Further experiments proved this gene as an important factor in regulating SSC maintenance and apoptosis [160]. Like that study, scientists could use testicular scRNA-seq data as a tool to reveal functional genes that may be related to the development of testicular diseases [161-164]. We recommend that this type of usage should be “cell-oriented”, which means the researchers should focus on a certain type of testicular cells and try to dig up novel genes or mechanisms of this type of cells, rather than just comparing the differences between disease and control groups without considering cell heterogeneity (in other words don't use scRNA-seq data to do tasks that bulk RNA-seq can do).
Comparing conserved and species- or sex-specific transcriptional features
Another important type of re-analyses of human testicular scRNA-seq data is to do species or sex comparisons. For species comparison, so far, the spermatogenesis of mouse [165], mouse lemur [165], chicken [166], cynomolgus macaque [167], sheep [168] and the somatic development of pigs [169] has been fully compared with that of human. The similarities of these studies were that they reported both conserved (especially the conservatism of X chromosome inactivation) and species-specific features among different animals and human. A good example would be Tian et al.'s work, in which they compared the spermatogenesis of four different species (human, mouse, sheep and cynomolgus macaque) [168]. In that study, they found the spermatogenesis between sheep and human were partially conserved at the transcriptional level (e.g., similar regulators were expressed in the same stages of spermatogenesis in human and sheep) and further showed the high conservatism of X chromosome inactivation during meiosis among four species [168], which was in line with Lau et al.'s report [167]. On the other hand, Rengaraj et al. reported a large proportion of species-specific genes between human and chickens (24 out of 56 orthologs) during the development of male fetal germ cells [166]. Additionally, the species comparison (usage type 4) could be combined with detecting certain genes' expression patterns (usage type 1). For example, INPP4B was found to be highly expressed in postmeiotic spermatogenic cells in the testis of both human and mice [170], which remained evolutionary conservatism, while SFTPC was expressed in late spermatogenesis of human but were absent in murine spermatogenesis [171], which indicated evolutionary divergence. And the tasks of those two studies were using testicular scRNA-seq data to show the expression of certain genes, but such tasks were done cross-species by combining two types of usage. In this area, the spermatogenesis among mammalian species or some other common animals (such as chicken) has been largely compared, but two subjects remain to be discovered. First, are there any highly conserved genes in spermatogenesis between humans and rare species or other non-mammalian animals (which are evolutionarily far)? Second, what's the conserved mechanism of testicular somatic cells' development among species? We believe scRNA-seq analysis will give the answers in the future. Besides, it's well known that ovary is the female counterpart of the testis, thus scientists tried to compare gonads of two genders. The conservatism of germ cells for both genders was observed during meiotic prophase I, such as the high expression of the same marker genes at each stage of primary SPC/oocytes (TEX19 in leptotene; SPO11 in zygotene; BRDT in pachytene and H1FOO in diplotene) [172]. Wang et al. even identified shared subtypes of germ cells (SPARC+ fetal germ cells) in prenatal testes and ovaries [82]. But huge heterogeneity of germ cells was also reported in the expression of X-linked genes, including the high expression of SMS in diplotene oocytes but not SPC and the lack of XIST in adult testes [73, 172]. Both homogeneity and diversity between genders were also reported in somatic cells during scRNA-seq analyses. For example, shared transcriptional expressions of NR5A1/SOX9 in both fetal SC and granulosa cells [73] and shared type of somatic cells (KRT19+cells) [82] reflected the homogeneity between genders, while the divergent somatic expression pattern of DMRT1 (high in SC but not granulosa cells) suggested the diversity [73].
Bioinformatic usages including algorithm developing and database/portal establishing
Since developmental biology is one of the most important areas in which scRNA-seq analysis is applied [173], so far there has been a huge accumulation of related raw data deposited online. Therefore, the raw data could then be used to develop or validate scRNA data-related bioinformatic tools, such as bioinformatic algorithms, packages, softwares and databases. Pont et al. developed a method for single-cell based scoring system and visualization of gene set signature, and a scRNA-seq set of normal human spermatogenesis was employed to show the function of this method [174]. Recently, Stow et al. developed another scRNA-seq-based bioinformatic tool called SCIFER to analyze long interspersed element-1 (L1) mRNA level from individual L1 loci in single cells, and further employed human testicular scRNA-seq data to show its function [175]. Theoretically, the role of scRNA-seq data could either be a developer (to build up the tool) or a validator (to confirm the tool works well or to show how to use it), while the current studies seemed to prefer to use testicular scRNA-seq data as a validator. Moreover, for many life scientists (especially clinicians) who would like to use scRNA-seq analysis to validate their findings or interested genes in human testes, it's too hard for them to start from the raw data due to the high demand for coding skills [176]. Therefore, it's more convenient for researchers to use some interact-friendly or “zero-code” online websites/portals. So far there have been several popular interactive online portals containing human testicular scRNA-seq data (listed in Table 5), some of which were testis-specific [31, 72, 78, 177], two were human gonad-based (with female's samples and in vitro differentiated gonadal cells) in terms of scRNA-seq data [79, 178-180], while the rest were based on various organs with testicular scRNA-seq data included. The function of these online portals was basically Uniform Manifold Approximation and Projection (UMAP)/t-distributed stochastic neighbor embedding (t-SNE) visualization of cells and illustration of the expression patterns of certain genes. Some advanced functions such as pseudotime trajectory of testicular cells or enrichment analysis are only supported by few portal [78]. As a result, bioinformatic engineers should pay more attention to the realization of integrated analysis and comprehensive analysis through interact-friendly (non-code) website tools.
Popular interaction-friendly online portals/websites containing scRNA-seq data of human testicular samples
| Author | Year | Country | Portal name | Website | Testicular data type | gonad-specific portal? | Reference |
|---|---|---|---|---|---|---|---|
| Guo et al. | 2018 | USA | UCSC cell browser-(ID)adult-testis | https://cells.ucsc.edu/?ds=adult-testis | Normal | Yes | [31] |
| Franzén et al. | 2019 | Sweden | PanglaoDB | https://panglaodb.se/ | Normal/Underage | No | [251] |
| Darde et al. | 2019 | France | ReproGenomics Viewer | https://rgv.genouest.org/ | Normal/OB/NOA/Prenatal | Yes* | [180] |
| Soraggi et al. | 2020 | Denmark | UCSC cell browser-(ID)testis | https://cells.ucsc.edu/?ds=testis | Normal/OB | Yes | [177] |
| Singh et al. | 2020 | USA | UCSC cell browser-(ID) scarface | https://cells.ucsc.edu/?ds=scarface | OB | No | [252] |
| Han et al. | 2020 | China | Human cell landscape (HCL) | http://bis.zju.edu.cn/HCL/ | Normal/Prenatal | No | [183] |
| Chen/Chitiashvili/Guo et al. | 2020 | USA | Human germline developmental atlas | https://germline.mcdb.ucla.edu/ | Prenatal | Yes | [79, 178, 179] |
| Cairns Lab/Guo et al. | 2020 | USA | Human Testis Atlas | https://humantestisatlas.shinyapps.io/humantestisatlas1/ | Normal/Underage | Yes | [31, 72] |
| Karlsson et al. | 2021 | Sweden | The human protein atlas (HPA) | https://www.proteinatlas.org/humanproteome/single+cell+type | Normal | No | [246] |
| Zheng et al. | 2021 | China | ColorCells | https://rna.sysu.edu.cn/colorcells/ | Normal/OB/NOA | No | [253] |
| Mahyari et al. | 2021 | USA | Human infertility single-cell transcription atlas (HISTA) | https://conradlab.shinyapps.io/HISTA/ | Normal/OB/KS/NOA/Ejaculatory dysfunction/Underage | Yes | [78] |
| Li et al. | 2022 | Singapore | Deeply integrated human single-Cell omics data (DISCO) | https://www.immunesinglecell.org/ | Normal/Prenatal/Underage/OB/NOA/KS | No | [254] |
| Chan Zuckerberg Initiative | - | USA | CZ CELLxGENE Discover | https://cellxgene.cziscience.com/ | Prenatal | No | [255] |
* In terms of scRNA-seq data
Conclusions and future perspectives
The main purpose of the current review is to gather the updated evidence on human testicular samples revealed by scRNA-seq data, in order to gain a “single-cell” view of both normal and pathological development of the testis. So far, there have been a large number of novel findings in terms of normal spermatogenesis, age-related testicular microenvironment's development and changes, testis-related male infertility, as well as new bioinformatic methods suitable for testicular scRNA-seq data analysis. In terms of normal status, the whole development process of spermatogenic cells (from the most naïve primordial germ cells to testicular spermatozoa), together with their transcriptional patterns at each stage were largely revealed (including their similarity and differences among species). The researches on normal somatic cells were basically restricted to SCs, LCs and PTMs, while fewer scRNA-seq-based findings about human testicular macrophages, ECs or other rare cells (e.g., T cells) were reported. Additionally, numerous transcriptional changes and potential mechanisms have been revealed in disease data including spermatogenic dysfunction, OA, ejaculatory disorder and chromosomal abnormalities, giving us insight into the heterogenous pathogenesis of male infertility and providing us with innovative targets of treatments. Last but not least, more and more publicly available testicular scRNA-seq datasets give researchers more opportunities to do their individualized data analysis for different research needs. We believe this review will not only pave the way to the transcriptionally understanding of both normal and abnormal spermatogenesis, but also be a reference for the future usage of scRNA-seq analysis in the human testis.
One of the most prospective research highlights in this area is the combination of testicular scRNA-seq analysis with spatial transcriptomic analysis of the testis. Due to the dissociation of testicular cells in scRNA-seq, it could neither spatially illustrate spermatogenic cells in seminiferous tubules, nor analyze the spatial interaction between somatic and spermatogenic cells [134]. To solve this problem, Chen et al employed the novel technology “spatial transcriptomic analysis” to create spatial atlas that spatially illustrate testicular gene expression at near-single-cell resolution in the human testis [134]. More importantly, Garcia-Alonso et al did integrated analyses of the development of male gonads by combining scRNA-seq and spatial transcriptomics [74], which was more comprehensive and in-depth. We believe that the integration of these two approaches will improve future researches of the human testis and male infertility, allowing for a more complete and nuanced understanding of the functional/dysregulated spermatogenesis in the human testis. So far, there has been a limited number of such integrated studies, leading to a novel “Blue Ocean” of testicular researches.
Although scRNA-seq analysis has proved the existence of immune cells, such as macrophages (both M1 and M2), T cells, mast cells, or even B cells, in testicular microenvironment, the role of these minor cells and their transcriptional patterns in the development of male infertility remain unknown.
Meanwhile, more types of testicular diseases, including cryptorchidism, AZFc deletions, azoospermia after chemo/radiotherapy, as well as teratospermia, should be focused on and will benefit from the further usage of scRNA-seq technologies. Due to the high cost of scRNA-seq, few of the above-reviewed articles were directly out of clinical purpose. With the development and cost reduction of this technology, clinical usages such as diagnosis, classification, prediction of sperm retrieval or the effect of hormone treatment might come true.
Abbreviations
ScRNA-seq: single-cell RNA sequencing; GEO: Gene Expression Omnibus; OA: obstructive azoospermia; TESE: testicular sperm extraction; OB: obstruction; FACS: fluorescence-activated cell sorting; MACS: magnetic activated cell sorting; SPG: spermatogonia; SPC: spermatocyte; SSC: spermatogonial stem cell; diff_SPG: differentiating SPG; diffed_SPG: differentiated SPG; L.SPC: leptotene SPC; Z.SPC: zygotene SPC; P.SPC: pachytene SPC; D.SPC: diplotene SPC; SC: Sertoli cell; LC: Leydig cell; NOA: non-obstructive azoospermia; SCOS: Sertoli cell-only syndrome; PGC: primordial germ cell; FGC: fetal germ cell; CE: coelomic epithelium; PLCs: progenitor LCs; ILCs: immature LCs; MLCs: mature LCs; PGCLs: primordial germ cells-like cells; iNOA: idiopathic NOA; OAT: oligo-astheno-teratozoospermia; mTESE: microdissection TESE; ICSI: intracytoplasmic sperm injection; KS: Klinefelter Syndrome; AZFa: azoospermia factor a; AZFb: azoospermia factor b; AZFc: azoospermia factor c; IHC: immunohistochemistry; IF: immunofluorescence; WGS: whole genome sequencing; COVID-19: corona virus disease 2019; L1: long interspersed element-1; UMAP: Uniform manifold approximation and projection; t-SNE: t-distributed stochastic neighbor embedding; FSH: follicle-stimulating hormone; DEGs: differential expressed genes; HPA: Human Protein Atlas; GTEx: Genotype-Tissue Expression; FANTOM5: Function Annotation of The Mammalian Genome; TCGA: The Cancer Genome Atlas; GEPIA2: Gene Expression Profiling Interactive Analysis; MGI: Mouse Genome Informatics; HE: hematoxylin-eosin; PTM: peritubular myoid cell; wpf: weeks postfertilization; ELISA: enzyme-linked immunosorbent assay; RT-qPCR: reverse transcription real-time polymerase chain reaction; WB: western blotting; VUS: unknown-significance variants; SO: severe oligospermia; FISH: fluorescence in situ hybridization; CCK-8: cell counting kit-8; TUNEL: TdT-mediated dUTP Nick-End Labeling; Co-IP: co-immunoprecipitation; RIP: RNA binding protein immunoprecipitation; DMR: differentially methylated region; PAS: periodic acid-schiff; WGBS: whole-genome bisulfite sequencing; ENA: European Nucleotide Archive; scATAC-seq: single-cell assay for transposase-accessible chromatin sequencing; OSCC: oral squamous cell carcinoma; TFs: transcription factors; hESC: human embryonic stem cells; PGCLC: primordial germ cell-like cells; TGCT: testicular germ cell tumor; ExAC: Exome Aggregation Consortium; CNV: copy number variation; EMT: epithelial-mesenchymal transition; ECM: extracellular matrix.
Acknowledgements
Author contributions
FD, YM and XC conceived and designed the study. FD did the literature researches and wrote the first draft. YM, PP and XC reviewed, rewrote and revised the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mawhinney M, Mariotti A. Physiology, pathology and pharmacology of the male reproductive system. Periodontol 2000. 2013;61:232-51
2. Zheng W, Zhang Y, Sun C, Ge S, Tan Y, Shen H. et al. A Multi-Omics Study of Human Testis and Epididymis. Molecules. 2021 26
3. Koopman P. The Curious World of Gonadal Development in Mammals. Curr Top Dev Biol. 2016;116:537-45
4. Bittman EL. Timing in the Testis. J Biol Rhythms. 2016;31:12-36
5. Dores C, Alpaugh W, Dobrinski I. From in vitro culture to in vivo models to study testis development and spermatogenesis. Cell Tissue Res. 2012;349:691-702
6. Shalet SM. Normal testicular function and spermatogenesis. Pediatr Blood Cancer. 2009;53:285-8
7. Das A, Halpern JA, Darves-Bornoz AL, Patel M, Wren J, Keeter MK. et al. Sperm retrieval success and testicular histopathology in idiopathic nonobstructive azoospermia. Asian J Androl. 2020;22:555-9
8. Ma Y, Zhou Y, Xiao Q, Zou SS, Zhu YC, Ping P. et al. Seminal exosomal miR-210-3p as a potential marker of Sertoli cell damage in Varicocele. Andrology. 2021;9:451-9
9. Tian En L, Brougham MFH, Wallace WHB, Mitchell RT. Impacts of platinum-based chemotherapy on subsequent testicular function and fertility in boys with cancer. Hum Reprod Update. 2020;26:874-85
10. Xie C, Wang W, Tu C, Meng L, Lu G, Lin G. et al. Meiotic recombination: insights into its mechanisms and its role in human reproduction with a special focus on non-obstructive azoospermia. Hum Reprod Update. 2022
11. Aydin S, Billur D, Kizil S, Ozkavukcu S, Topal Celikkan F, Aydos K. et al. Evaluation of blood-testis barrier integrity in terms of adhesion molecules in nonobstructive azoospermia. Andrologia. 2020;52:e13636
12. Ma Y, Zhou Y, Zou SS, Sun Y, Chen XF. Exosomes released from Sertoli cells contribute to the survival of Leydig cells through CCL20 in rats. Mol Hum Reprod. 2022 28
13. Rahmani F, Movahedin M, Mazaheri Z, Soleimani M. Transplantation of mouse iPSCs into testis of azoospermic mouse model: in vivo and in vitro study. Artif Cells Nanomed Biotechnol. 2019;47:1585-94
14. Alves-Lopes JP, Stukenborg JB. Testicular organoids: a new model to study the testicular microenvironment in vitro? Hum Reprod Update. 2018;24:176-91
15. Mills RG. The Pathological Changes in the Testes in Epidemic Pneumonia. J Exp Med. 1919;30:505-29
16. Johnsen SG. Testicular biopsy score count-a method for registration of spermatogenesis in human testes: normal values and results in 335 hypogonadal males. Hormones. 1970;1:2-25
17. Meinhard E, McRae CU, Chisholm GD. Testicular biopsy in evaluation of male infertility. Br Med J. 1973;3:577-81
18. Friberg J, Kjessler B. Sperm-agglutinating antibodies and testicular morphology in fifty-nine men with azoospermia or cryptozoospermia. American journal of obstetrics and gynecology. 1975;121:987-90
19. Ansbacher R, Gangai MP. Testicular biopsy: sperm antibodies. Fertil Steril. 1975;26:1239-42
20. Bouhdiba M, Leroy-Martin B, Peyrat JP, Saint Pol P, Djiane J, Leonardelli J. Immunohistochemical detection of prolactin and its receptors in human testis. Andrologia. 1989;21:223-8
21. Daar AS, Fuggle SV, Fabre JW, Ting A, Morris PJ. The detailed distribution of HLA-A, B, C antigens in normal human organs. Transplantation. 1984;38:287-92
22. Forti G, Vannelli GB, Barni T, Balboni GC, Orlando C, Serio M. Sertoli-germ cells interactions in the human testis. The Journal of steroid biochemistry and molecular biology. 1992;43:419-22
23. Malcher A, Rozwadowska N, Stokowy T, Kolanowski T, Jedrzejczak P, Zietkowiak W. et al. Potential biomarkers of nonobstructive azoospermia identified in microarray gene expression analysis. Fertil Steril. 2013;100:1686-94 e1-7
24. Feig C, Kirchhoff C, Ivell R, Naether O, Schulze W, Spiess AN. A new paradigm for profiling testicular gene expression during normal and disturbed human spermatogenesis. Mol Hum Reprod. 2007;13:33-43
25. Winge SB, Dalgaard MD, Belling KG, Jensen JM, Nielsen JE, Aksglaede L. et al. Transcriptome analysis of the adult human Klinefelter testis and cellularity-matched controls reveals disturbed differentiation of Sertoli- and Leydig cells. Cell Death Dis. 2018;9:586
26. Tang D, Li K, Lv M, Xu C, Geng H, Wang C. et al. Altered mRNAs Profiles in the Testis of Patients With "Secondary Idiopathic Non-Obstructive Azoospermia". Front Cell Dev Biol. 2022;10:824596
27. He Z, Chan WY, Dym M. Microarray technology offers a novel tool for the diagnosis and identification of therapeutic targets for male infertility. Reproduction. 2006;132:11-9
28. Lecluze E, Jegou B, Rolland AD, Chalmel F. New transcriptomic tools to understand testis development and functions. Mol Cell Endocrinol. 2018;468:47-59
29. Tang F, Lao K, Surani MA. Development and applications of single-cell transcriptome analysis. Nat Methods. 2011;8:S6-11
30. Guo F, Yan L, Guo H, Li L, Hu B, Zhao Y. et al. The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells. Cell. 2015;161:1437-52
31. Guo J, Grow EJ, Mlcochova H, Maher GJ, Lindskog C, Nie X. et al. The adult human testis transcriptional cell atlas. Cell Res. 2018;28:1141-57
32. La H, Yoo H, Lee EJ, Thang NX, Choi HJ, Oh J. et al. Insights from the Applications of Single-Cell Transcriptomic Analysis in Germ Cell Development and Reproductive Medicine. Int J Mol Sci. 2021 22
33. Shiraishi K. Genome medicine in male infertility: From karyotyping to single-cell analysis. J Obstet Gynaecol Res. 2021;47:2586-96
34. Tan K, Wilkinson MF. A single-cell view of spermatogonial stem cells. Curr Opin Cell Biol. 2020;67:71-8
35. Gille AS, Lapoujade C, Wolf JP, Fouchet P, Barraud-Lange V. Contribution of Single-Cell Transcriptomics to the Characterization of Human Spermatogonial Stem Cells: Toward an Application in Male Fertility Regenerative Medicine? Int J Mol Sci. 2019 20
36. Wen L, Tang F. Human Germline Cell Development: from the Perspective of Single-Cell Sequencing. Molecular cell. 2019;76:320-8
37. Li L, Yang R, Yin C, Kee K. Studying human reproductive biology through single-cell analysis and in vitro differentiation of stem cells into germ cell-like cells. Hum Reprod Update. 2020;26:670-88
38. Wu X, Zhou L, Shi J, Cheng CY, Sun F. Multiomics analysis of male infertilitydagger. Biology of reproduction. 2022;107:118-34
39. Di Persio S, Neuhaus N. Human spermatogonial stem cells and their niche in male (in)fertility: novel concepts from single-cell RNA-sequencing. Hum Reprod. 2023;38:1-13
40. Rabbani M, Zheng X, Manske GL, Vargo A, Shami AN, Li JZ. et al. Decoding the Spermatogenesis Program: New Insights from Transcriptomic Analyses. Annu Rev Genet. 2022
41. Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377-82
42. Guo J, Grow EJ, Yi C, Mlcochova H, Maher GJ, Lindskog C. et al. Chromatin and Single-Cell RNA-Seq Profiling Reveal Dynamic Signaling and Metabolic Transitions during Human Spermatogonial Stem Cell Development. Cell Stem Cell. 2017;21:533-46 e6
43. Neuhaus N, Yoon J, Terwort N, Kliesch S, Seggewiss J, Huge A. et al. Single-cell gene expression analysis reveals diversity among human spermatogonia. Mol Hum Reprod. 2017;23:79-90
44. Gao C, Zhang M, Chen L. The Comparison of Two Single-cell Sequencing Platforms: BD Rhapsody and 10x Genomics Chromium. Curr Genomics. 2020;21:602-9
45. Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171-81
46. Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096-8
47. Spaethling JM, Sanchez-Alavez M, Lee J, Xia FC, Dueck H, Wang W. et al. Single-cell transcriptomics and functional target validation of brown adipocytes show their complex roles in metabolic homeostasis. FASEB J. 2016;30:81-92
48. Islam S, Kjallquist U, Moliner A, Zajac P, Fan JB, Lonnerberg P. et al. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011;21:1160-7
49. Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S. et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell. 2018;172:1091-107 e17
50. Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M. et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202-14
51. Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187-201
52. Singleron Biotechnologies. GEXSCOPE® Single Cell RNA Library Kit. https://singleron.bio/product/common/assets/upload/2022/0803/16422395.pdf. Acess Date: 2022/9/3.
53. Dura B, Choi JY, Zhang K, Damsky W, Thakral D, Bosenberg M. et al. scFTD-seq: freeze-thaw lysis based, portable approach toward highly distributed single-cell 3' mRNA profiling. Nucleic Acids Res. 2019;47:e16
54. Kim J, Marignani PA. Single-Cell RNA Sequencing Analysis Using Fluidigm C1 Platform for Characterization of Heterogeneous Transcriptomes. Methods Mol Biol. 2022;2508:261-78
55. Zhao L, Yao C, Xing X, Jing T, Li P, Zhu Z. et al. Single-cell analysis of developing and azoospermia human testicles reveals central role of Sertoli cells. Nat Commun. 2020;11:5683
56. Chen S, An G, Wang H, Wu X, Ping P, Hu L. et al. Human obstructive (postvasectomy) and nonobstructive azoospermia - Insights from scRNA-Seq and transcriptome analysis. Genes & Diseases. 2022;9:766-76
57. Lee J, Hyeon DY, Hwang D. Single-cell multiomics: technologies and data analysis methods. Exp Mol Med. 2020;52:1428-42
58. Mylka V, Matetovici I, Poovathingal S, Aerts J, Vandamme N, Seurinck R. et al. Comparative analysis of antibody- and lipid-based multiplexing methods for single-cell RNA-seq. Genome Biol. 2022;23:55
59. Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet-Adkins A, Smets M. et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular cell. 2017;65:631-43 e4
60. Chen G, Ning B, Shi T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front Genet. 2019;10:317
61. Tran HTN, Ang KS, Chevrier M, Zhang X, Lee NYS, Goh M. et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020;21:12
62. Feng C, Liu S, Zhang H, Guan R, Li D, Zhou F. et al. Dimension Reduction and Clustering Models for Single-Cell RNA Sequencing Data: A Comparative Study. Int J Mol Sci. 2020 21
63. Jiang P. Quality Control of Single-Cell RNA-seq. Methods Mol Biol. 2019;1935:1-9
64. Galow AM, Kussauer S, Wolfien M, Brunner RM, Goldammer T, David R. et al. Quality control in scRNA-Seq can discriminate pacemaker cells: the mtRNA bias. Cell Mol Life Sci. 2021;78:6585-92
65. Jovic D, Liang X, Zeng H, Lin L, Xu F, Luo Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin Transl Med. 2022;12:e694
66. Di Persio S, Tekath T, Siebert-Kuss LM, Cremers JF, Wistuba J, Li X. et al. Single-cell RNA-seq unravels alterations of the human spermatogonial stem cell compartment in patients with impaired spermatogenesis. Cell Rep Med. 2021;2:100395
67. Sohni A, Tan K, Song HW, Burow D, de Rooij DG, Laurent L. et al. The Neonatal and Adult Human Testis Defined at the Single-Cell Level. Cell Rep. 2019;26:1501-17 e4
68. Hermann BP, Cheng K, Singh A, Roa-De La Cruz L, Mutoji KN, Chen IC. et al. The Mammalian Spermatogenesis Single-Cell Transcriptome, from Spermatogonial Stem Cells to Spermatids. Cell Rep. 2018;25:1650-67 e8
69. Tan K, Song HW, Thompson M, Munyoki S, Sukhwani M, Hsieh TC. et al. Transcriptome profiling reveals signaling conditions dictating human spermatogonia fate in vitro. Proc Natl Acad Sci U S A. 2020;117:17832-41
70. Xia B, Yan Y, Baron M, Wagner F, Barkley D, Chiodin M. et al. Widespread Transcriptional Scanning in the Testis Modulates Gene Evolution Rates. Cell. 2020;180:248-62 e21
71. Labrecque M, Hays M, Chen-Mok M, Barone MA, Sokal D. Frequency and patterns of early recanalization after vasectomy. BMC Urol. 2006;6:25
72. Guo J, Nie X, Giebler M, Mlcochova H, Wang Y, Grow EJ. et al. The Dynamic Transcriptional Cell Atlas of Testis Development during Human Puberty. Cell Stem Cell. 2020;26:262-76 e4
73. Li L, Dong J, Yan L, Yong J, Liu X, Hu Y. et al. Single-Cell RNA-Seq Analysis Maps Development of Human Germline Cells and Gonadal Niche Interactions. Cell Stem Cell. 2017;20:858-73 e4
74. Garcia-Alonso L, Lorenzi V, Mazzeo CI, Alves-Lopes JP, Roberts K, Sancho-Serra C. et al. Single-cell roadmap of human gonadal development. Nature. 2022;607:540-7
75. Nie X, Munyoki SK, Sukhwani M, Schmid N, Missel A, Emery BR. et al. Single-cell analysis of human testis aging and correlation with elevated body mass index. Dev Cell. 2022;57:1160-76 e5
76. Wang M, Liu X, Chang G, Chen Y, An G, Yan L. et al. Single-Cell RNA Sequencing Analysis Reveals Sequential Cell Fate Transition during Human Spermatogenesis. Cell Stem Cell. 2018;23:599-614 e4
77. Wang M, Yang Y, Cansever D, Wang Y, Kantores C, Messiaen S. et al. Two populations of self-maintaining monocyte-independent macrophages exist in adult epididymis and testis. Proc Natl Acad Sci U S A. 2021 118
78. Mahyari E, Guo J, Lima AC, Lewinsohn DP, Stendahl AM, Vigh-Conrad KA. et al. Comparative single-cell analysis of biopsies clarifies pathogenic mechanisms in Klinefelter syndrome. Am J Hum Genet. 2021;108:1924-45
79. Guo J, Sosa E, Chitiashvili T, Nie X, Rojas EJ, Oliver E. et al. Single-cell analysis of the developing human testis reveals somatic niche cell specification and fetal germline stem cell establishment. Cell Stem Cell. 2021;28:764-78 e4
80. Cheng K, Seita Y, Moriwaki T, Noshiro K, Sakata Y, Hwang YS. et al. The developmental origin and the specification of the adrenal cortex in humans and cynomolgus monkeys. Sci Adv. 2022;8:eabn8485
81. O'Shaughnessy PJ, Fowler PA. Development of the human fetal testis. Ann Endocrinol (Paris). 2014;75:48-53
82. Wang R, Liu X, Li L, Yang M, Yong J, Zhai F. et al. Dissecting human gonadal cell lineage specification and sex determination using a single-cell RNA-seq approach. Genomics Proteomics Bioinformatics. 2022
83. Karl J, Capel B. Sertoli cells of the mouse testis originate from the coelomic epithelium. Dev Biol. 1998;203:323-33
84. Wang Y, Guo B, Guo Y, Qi N, Lv Y, Ye Y. et al. A spatiotemporal steroidogenic regulatory network in human fetal adrenal glands and gonads. bioRxiv. 2021. 2021 12.22.473776
85. DeFalco T, Potter SJ, Williams AV, Waller B, Kan MJ, Capel B. Macrophages Contribute to the Spermatogonial Niche in the Adult Testis. Cell Rep. 2015;12:1107-19
86. Mossadegh-Keller N, Gentek R, Gimenez G, Bigot S, Mailfert S, Sieweke MH. Developmental origin and maintenance of distinct testicular macrophage populations. J Exp Med. 2017;214:2829-41
87. Voigt AL, Dardari R, Su L, Lara NLM, Sinha S, Jaffer A. et al. Metabolic transitions define spermatogonial stem cell maturation. Hum Reprod. 2022
88. Alfano M, Tascini AS, Pederzoli F, Locatelli I, Nebuloni M, Giannese F. et al. Aging, inflammation and DNA damage in the somatic testicular niche with idiopathic germ cell aplasia. Nat Commun. 2021;12:5205
89. Neto FT, Bach PV, Najari BB, Li PS, Goldstein M. Spermatogenesis in humans and its affecting factors. Seminars in cell & developmental biology. 2016;59:10-26
90. Schulze W, Thoms F, Knuth UA. Testicular sperm extraction: comprehensive analysis with simultaneously performed histology in 1418 biopsies from 766 subfertile men. Hum Reprod. 1999;14(Suppl 1):82-96
91. Saitou M, Yamaji M. Primordial germ cells in mice. Cold Spring Harb Perspect Biol. 2012 4
92. Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH. et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021;12:1088
93. Wosnitzer MS, Goldstein M. Obstructive azoospermia. Urol Clin North Am. 2014;41:83-95
94. Wang H, Peng Y, Fu W, Hu X, Li C, Guan J. MRI findings of obstructive azoospermia: lesions in and out of pelvic cavity. Abdom Radiol (NY). 2020;45:851-64
95. Lv MQ, Zhou L, Ge P, Li YX, Zhang J, Zhou DX. Over-expression of hsa_circ_0000116 in patients with non-obstructive azoospermia and its predictive value in testicular sperm retrieval. Andrology. 2020;8:1834-43
96. Li Z, Chen S, Yang Y, Zhuang X, Tzeng CM. Novel biomarker ZCCHC13 revealed by integrating DNA methylation and mRNA expression data in non-obstructive azoospermia. Cell Death Discov. 2018;4:36
97. Ge P, Zhang J, Zhou L, Lv MQ, Li YX, Wang J. et al. CircRNA expression profile and functional analysis in testicular tissue of patients with non-obstructive azoospermia. Reprod Biol Endocrinol. 2019;17:100
98. Zhu F, Luo Y, Bo H, Gong G, Tang R, Fan J. et al. Trace the profile and function of circular RNAs in Sertoli cell only syndrome. Genomics. 2021;113:1845-54
99. Yao C, Yuan Q, Niu M, Fu H, Zhou F, Zhang W. et al. Distinct Expression Profiles and Novel Targets of MicroRNAs in Human Spermatogonia, Pachytene Spermatocytes, and Round Spermatids between OA Patients and NOA Patients. Mol Ther Nucleic Acids. 2017;9:182-94
100. Zhang Z, Wu H, Zheng L, Zhang HT, Yang YZ, Mao JM. et al. Identification and characterization of circular RNAs in the testicular tissue of patients with non-obstructive azoospermia. Asian J Androl. 2022
101. Vale J. Ejaculatory dysfunction. BJU Int. 1999;83:557-63
102. Orum MH, Egilmez OB. Successfully treatment of intercourse anejaculation with psychosexual counseling: A very rare case of situational anejaculation specific to penetrative sex with the wife. Rev Int Androl. 2020;18:79-83
103. Caroppo E, Campagna C, Colpi EM, D'Amato G, Colpi GM. Sperm source does not affect the ICSI outcome of patients with severely compromised spermatogenesis. Andrologia. 2020;52:e13884
104. Pena VN, Kohn TP, Herati AS. Genetic mutations contributing to non-obstructive azoospermia. Best Pract Res Clin Endocrinol Metab. 2020;34:101479
105. Chen X, Ma Y, Zou S, Wang S, Qiu J, Xiao Q. et al. Comparison and outcomes of nonobstructive azoospermia patients with different etiology undergoing MicroTESE and ICSI treatments. Transl Androl Urol. 2019;8:366-73
106. Corona G, Minhas S, Giwercman A, Bettocchi C, Dinkelman-Smit M, Dohle G. et al. Sperm recovery and ICSI outcomes in men with non-obstructive azoospermia: a systematic review and meta-analysis. Hum Reprod Update. 2019;25:733-57
107. Tian L, Li X, Wang Y, Chen Q, Li X, Ge RS. et al. Oncostatin M stimulates immature Leydig cell proliferation but inhibits its maturation and function in rats through JAK1/STAT3 signaling and induction of oxidative stress in vitro. Andrology. 2022;10:354-66
108. Meroni SB, Galardo MN, Rindone G, Gorga A, Riera MF, Cigorraga SB. Molecular Mechanisms and Signaling Pathways Involved in Sertoli Cell Proliferation. Front Endocrinol (Lausanne). 2019;10:224
109. Cafe SL, Skerrett-Byrne DA, De Oliveira CS, Nixon B, Oatley MJ, Oatley JM. et al. A regulatory role for CHD4 in maintenance of the spermatogonial stem cell pool. Stem Cell Reports. 2021;16:1555-67
110. Wang M, Xu Y, Zhang Y, Chen Y, Chang G, An G. et al. Deciphering the autophagy regulatory network via single-cell transcriptome analysis reveals a requirement for autophagy homeostasis in spermatogenesis. Theranostics. 2021;11:5010-27
111. Achermann APP, Pereira TA, Esteves SC. Microdissection testicular sperm extraction (micro-TESE) in men with infertility due to nonobstructive azoospermia: summary of current literature. Int Urol Nephrol. 2021;53:2193-210
112. Nordhoff V, Fricke RK, Schüring AN, Zitzmann M, Kliesch S. Treatment strategies for severe oligoasthenoteratozoospermia (OAT) (<0.1 million/mL) patients. Andrology. 2015;3:856-63
113. Kang YN, Hsiao YW, Chen CY, Wu CC. Testicular sperm is superior to ejaculated sperm for ICSI in cryptozoospermia: An update systematic review and meta-analysis. Sci Rep. 2018;8:7874
114. Bao J, Zhang Y, Schuster AS, Ortogero N, Nilsson EE, Skinner MK. et al. Conditional inactivation of Miwi2 reveals that MIWI2 is only essential for prospermatogonial development in mice. Cell Death Differ. 2014;21:783-96
115. Waheeb R, Hofmann MC. Human spermatogonial stem cells: a possible origin for spermatocytic seminoma. Int J Androl. 2011;34:e296-305 discussion e
116. von Kopylow K, Staege H, Schulze W, Will H, Kirchhoff C. Fibroblast growth factor receptor 3 is highly expressed in rarely dividing human type A spermatogonia. Histochem Cell Biol. 2012;138:759-72
117. Hadjiargyrou M. Mustn1: A Developmentally Regulated Pan-Musculoskeletal Cell Marker and Regulatory Gene. Int J Mol Sci. 2018 19
118. Nguyen HB, Chavez AM, Lipner E, Hantsoo L, Kornfield SL, Davies RD. et al. Gender-Affirming Hormone Use in Transgender Individuals: Impact on Behavioral Health and Cognition. Curr Psychiatry Rep. 2018;20:110
119. Schneider F, Kliesch S, Schlatt S, Neuhaus N. Andrology of male-to-female transsexuals: influence of cross-sex hormone therapy on testicular function. Andrology. 2017;5:873-80
120. Giltay JC, Maiburg MC. Klinefelter syndrome: clinical and molecular aspects. Expert Rev Mol Diagn. 2010;10:765-76
121. Laurentino S, Heckmann L, Di Persio S, Li X, Meyer Zu Horste G, Wistuba J. et al. High-resolution analysis of germ cells from men with sex chromosomal aneuploidies reveals normal transcriptome but impaired imprinting. Clin Epigenetics. 2019;11:127
122. Kanakis GA, Nieschlag E. Klinefelter syndrome: more than hypogonadism. Metabolism. 2018;86:135-44
123. Visootsak J, Graham JM Jr. Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet J Rare Dis. 2006;1:42
124. Kim SY, Lee BY, Oh AR, Park SY, Lee HS, Seo JT. Clinical, Hormonal, and Genetic Evaluation of Idiopathic Nonobstructive Azoospermia and Klinefelter Syndrome Patients. Cytogenetic and genome research. 2017;153:190-7
125. Willems M, Gies I, Van Saen D. Germ cell loss in Klinefelter syndrome: When and why? Am J Med Genet C Semin Med Genet. 2020;184:356-70
126. Winge SB, Soraggi S, Schierup MH, Rajpert-De Meyts E, Almstrup K. Integration and reanalysis of transcriptomics and methylomics data derived from blood and testis tissue of men with 47,XXY Klinefelter syndrome indicates the primary involvement of Sertoli cells in the testicular pathogenesis. Am J Med Genet C Semin Med Genet. 2020;184:239-55
127. He H, Huang T, Yu F, Chen K, Guo S, Zhang L. et al. KIF2C affects sperm cell differentiation in patients with Klinefelter syndrome, as revealed by RNA-Seq and scRNA-Seq data. FEBS Open Bio. 2022;12:1465-74
128. del Campo AB, Kyte JA, Carretero J, Zinchencko S, Mendez R, Gonzalez-Aseguinolaza G. et al. Immune escape of cancer cells with beta2-microglobulin loss over the course of metastatic melanoma. Int J Cancer. 2014;134:102-13
129. Sumaiya K, Langford D, Natarajaseenivasan K, Shanmughapriya S. Macrophage migration inhibitory factor (MIF): A multifaceted cytokine regulated by genetic and physiological strategies. Pharmacol Ther. 2022;233:108024
130. Liu X, Tang D, Zheng F, Xu Y, Guo H, Zhou J. et al. Single-Cell Sequencing Reveals the Relationship between Phenotypes and Genotypes of Klinefelter Syndrome. Cytogenetic and genome research. 2019;159:55-65
131. Cappallo-Obermann H, von Kopylow K, Schulze W, Spiess AN. A biopsy sample reduction approach to identify significant alterations of the testicular transcriptome in the presence of Y-chromosomal microdeletions that are independent of germ cell composition. Hum Genet. 2010;128:421-31
132. Gatta V, Raicu F, Ferlin A, Antonucci I, Scioletti AP, Garolla A. et al. Testis transcriptome analysis in male infertility: new insight on the pathogenesis of oligo-azoospermia in cases with and without AZFc microdeletion. BMC Genomics. 2010;11:401
133. Colaco S, Modi D. Genetics of the human Y chromosome and its association with male infertility. Reprod Biol Endocrinol. 2018;16:14
134. Chen H, Murray E, Sinha A, Laumas A, Li J, Lesman D. et al. Dissecting mammalian spermatogenesis using spatial transcriptomics. Cell Rep. 2021;37:109915
135. Yang C, Lin X, Ji Z, Huang Y, Zhang L, Luo J. et al. Novel bi-allelic variants in KASH5 are associated with meiotic arrest and non-obstructive azoospermia. Mol Hum Reprod. 2022
136. Guo J, Wang S, Jiang Z, Tang L, Liu Z, Cao J. et al. Long Non-Coding RNA RFPL3S Functions as a Biomarker of Prognostic and Immunotherapeutic Prediction in Testicular Germ Cell Tumor. Front Immunol. 2022;13:859730
137. Wu X, Gao S, Wang L, Bu T, Wu S, Zhou L. et al. Role of laminin and collagen chains in human spermatogenesis - Insights from studies in rodents and scRNA-Seq transcriptome profiling. Seminars in cell & developmental biology. 2022;121:125-32
138. He Y, Dong Y, Zhang X, Ding Z, Song Y, Huang X. et al. Lipid Droplet-Related PLIN2 in CD68(+) Tumor-Associated Macrophage of Oral Squamous Cell Carcinoma: Implications for Cancer Prognosis and Immunotherapy. Front Oncol. 2022;12:824235
139. Chen S, Wang G, Zheng X, Ge S, Dai Y, Ping P. et al. Whole-exome sequencing of a large Chinese azoospermia and severe oligospermia cohort identifies novel infertility causative variants and genes. Hum Mol Genet. 2020;29:2451-9
140. Zhou R, Liang J, Chen Q, Tian H, Yang C, Liu C. A 3-Gene Random Forest Model to Diagnose Non-obstructive Azoospermia Based on Transcription Factor-Related Henes. Reprod Sci. 2022
141. Martin-Inaraja M, Ferreira M, Taelman J, Eguizabal C, Chuva De Sousa Lopes SM. Improving In vitro Culture of Human Male Fetal Germ Cells. Cells. 2021 10
142. Zheng W, Zhang S, Jiang S, Huang Z, Chen X, Guo H. et al. Evaluation of immune status in testis and macrophage polarization associated with testicular damage in patients with nonobstructive azoospermia. Am J Reprod Immunol. 2021;86:e13481
143. He H, Yu F, Shen W, Chen K, Zhang L, Lou S. et al. The Novel Key Genes of Non-obstructive Azoospermia Affect Spermatogenesis: Transcriptomic Analysis Based on RNA-Seq and scRNA-Seq Data. Front Genet. 2021;12:608629
144. Han B, Yan Z, Yu S, Ge W, Li Y, Wang Y. et al. Infertility network and hub genes for nonobstructive azoospermia utilizing integrative analysis. Aging (Albany NY). 2021;13:7052-66
145. Wang Z, Xu X. scRNA-seq Profiling of Human Testes Reveals the Presence of the ACE2 Receptor, A Target for SARS-CoV-2 Infection in Spermatogonia, Leydig and Sertoli Cells. Cells. 2020 9
146. Pan F, Xiao X, Guo J, Song Y, Li H, Patel DP. et al. No evidence of severe acute respiratory syndrome-coronavirus 2 in semen of males recovering from coronavirus disease 2019. Fertil Steril. 2020;113:1135-9
147. Zhou L, Niu Z, Jiang X, Zhang Z, Zheng Y, Wang Z. et al. SARS-CoV-2 Targets by the pscRNA Profiling of ACE2, TMPRSS2 and Furin Proteases. iScience. 2020;23:101744
148. Shen Q, Xiao X, Aierken A, Yue W, Wu X, Liao M. et al. The ACE2 expression in Sertoli cells and germ cells may cause male reproductive disorder after SARS-CoV-2 infection. J Cell Mol Med. 2020;24:9472-7
149. Hikmet F, Mear L, Edvinsson A, Micke P, Uhlen M, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol Syst Biol. 2020;16:e9610
150. Ren X, Wang S, Chen X, Wei X, Li G, Ren S. et al. Multiple Expression Assessments of ACE2 and TMPRSS2 SARS-CoV-2 Entry Molecules in the Urinary Tract and Their Associations with Clinical Manifestations of COVID-19. Infect Drug Resist. 2020;13:3977-90
151. Stanley KE, Thomas E, Leaver M, Wells D. Coronavirus disease-19 and fertility: viral host entry protein expression in male and female reproductive tissues. Fertil Steril. 2020;114:33-43
152. Qi J, Zhou Y, Hua J, Zhang L, Bian J, Liu B. et al. The scRNA-seq Expression Profiling of the Receptor ACE2 and the Cellular Protease TMPRSS2 Reveals Human Organs Susceptible to SARS-CoV-2 Infection. Int J Environ Res Public Health. 2021 18
153. Zhang J, Wu Y, Li S, Wang X, Wang R, Wang X. Bioinformatic and mouse model reveal the potential high vulnerability of Leydig cells on SARS-CoV-2. Ann Transl Med. 2021;9:678
154. Cai T, Mao G, Zheng R, Fang M, Yang X, Wang L. et al. Testicular injury during SARS-CoV-2 infection may be neglected: An assessment from scRNA-seq profiling and protein detection of angiotensin-converting enzyme II. Exp Ther Med. 2021;22:1485
155. Scialo F, Daniele A, Amato F, Pastore L, Matera MG, Cazzola M. et al. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung. 2020;198:867-77
156. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271-80 e8
157. Essalmani R, Jain J, Susan-Resiga D, Andreo U, Evagelidis A, Derbali RM. et al. Distinctive Roles of Furin and TMPRSS2 in SARS-CoV-2 Infectivity. J Virol. 2022;96:e0012822
158. Behl T, Kaur I, Aleya L, Sehgal A, Singh S, Sharma N. et al. CD147-spike protein interaction in COVID-19: Get the ball rolling with a novel receptor and therapeutic target. Sci Total Environ. 2022;808:152072
159. Liu X, Chen Y, Tang W, Zhang L, Chen W, Yan Z. et al. Single-cell transcriptome analysis of the novel coronavirus (SARS-CoV-2) associated gene ACE2 expression in normal and non-obstructive azoospermia (NOA) human male testes. Sci China Life Sci. 2020;63:1006-15
160. Yang C, Yao C, Ji Z, Zhao L, Chen H, Li P. et al. RNA-binding protein ELAVL2 plays post-transcriptional roles in the regulation of spermatogonia proliferation and apoptosis. Cell Prolif. 2021;54:e13098
161. Zhou R, Lv X, Chen T, Chen Q, Tian H, Yang C. et al. Construction and external validation of a 5-gene random forest model to diagnose non-obstructive azoospermia based on the single-cell RNA sequencing of testicular tissue. Aging (Albany NY). 2021;13:24219-35
162. Luo SW, Tang L, Zhou D, Bo H, Fan LQ. FOXP4 promotes proliferation of human spermatogonial stem cells. Asian J Androl. 2022
163. Zhang N, Wang Y, Chen Z, Ren J, Rehman A, Ahmad DW. et al. Single-cell transcriptome analysis of Bisphenol A exposure reveals the key roles of the testicular microenvironment in male reproduction. Biomed Pharmacother. 2022;145:112449
164. Tang XJ, Xiao QH, Wang XL, He Y, Tian YN, Xia BT. et al. Single-Cell Transcriptomics-Based Study of Transcriptional Regulatory Features in the Non-Obstructive Azoospermia Testis. Front Genet. 2022;13:875762
165. Olivieri JE, Dehghannasiri R, Wang PL, Jang S, de Morree A, Tan SY. et al. RNA splicing programs define tissue compartments and cell types at single-cell resolution. Elife. 2021 10
166. Rengaraj D, Cha DG, Lee HJ, Lee KY, Choi YH, Jung KM. et al. Dissecting chicken germ cell dynamics by combining a germ cell tracing transgenic chicken model with single-cell RNA sequencing. Comput Struct Biotechnol J. 2022;20:1654-69
167. Lau X, Munusamy P, Ng MJ, Sangrithi M. Single-Cell RNA Sequencing of the Cynomolgus Macaque Testis Reveals Conserved Transcriptional Profiles during Mammalian Spermatogenesis. Dev Cell. 2020;54:548-66 e7
168. Tian Y, Sun P, Liu WX, Shan LY, Hu YT, Fan HT. et al. Single-cell RNA sequencing of the Mongolia sheep testis reveals a conserved and divergent transcriptome landscape of mammalian spermatogenesis. FASEB J. 2022;36:e22348
169. Zhang L, Guo M, Liu Z, Liu R, Zheng Y, Yu T. et al. Single-cell RNA-seq analysis of testicular somatic cell development in pigs. J Genet Genomics. 2022
170. Ceyhan Y, Zhang M, Guo J, Sandoval CG, Vacher J, Kaftanovskaya EM. et al. Deletion of inositol polyphosphate 4-phosphatase type-II B affects spermatogenesis in mice. PLoS One. 2020;15:e0233163
171. Jiang M, Roth MG, Chun-On P, Sullivan DI, Alder JK. Phenotypic Diversity Caused by Differential Expression of SFTPC-Cre-Transgenic Alleles. Am J Respir Cell Mol Biol. 2020;62:692-8
172. Fan X, Moustakas I, Torrens-Juaneda V, Lei Q, Hamer G, Louwe LA. et al. Transcriptional progression during meiotic prophase I reveals sex-specific features and X chromosome dynamics in human fetal female germline. PLoS Genet. 2021;17:e1009773
173. Williams CG, Lee HJ, Asatsuma T, Vento-Tormo R, Haque A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022;14:68
174. Pont F, Tosolini M, Fournie JJ. Single-Cell Signature Explorer for comprehensive visualization of single cell signatures across scRNA-seq datasets. Nucleic Acids Res. 2019;47:e133
175. Stow EC, Baddoo M, LaRosa AJ, LaCoste D, Deininger P, Belancio V. SCIFER: approach for analysis of LINE-1 mRNA expression in single cells at a single locus resolution. Mob DNA. 2022;13:21
176. Via A, Blicher T, Bongcam-Rudloff E, Brazas MD, Brooksbank C, Budd A. et al. Best practices in bioinformatics training for life scientists. Brief Bioinform. 2013;14:528-37
177. Soraggi S, Riera M, Rajpert-De Meyts E, Schierup MH, Almstrup K. Evaluating genetic causes of azoospermia: What can we learn from a complex cellular structure and single-cell transcriptomics of the human testis? Hum Genet. 2021;140:183-201
178. Chitiashvili T, Dror I, Kim R, Hsu FM, Chaudhari R, Pandolfi E. et al. Female human primordial germ cells display X-chromosome dosage compensation despite the absence of X-inactivation. Nat Cell Biol. 2020;22:1436-46
179. Chen D, Sun N, Hou L, Kim R, Faith J, Aslanyan M. et al. Human Primordial Germ Cells Are Specified from Lineage-Primed Progenitors. Cell Rep. 2019;29:4568-82 e5
180. Darde TA, Lecluze E, Lardenois A, Stevant I, Alary N, Tuttelmann F. et al. The ReproGenomics Viewer: a multi-omics and cross-species resource compatible with single-cell studies for the reproductive science community. Bioinformatics. 2019;35:3133-9
181. Guo H, Hu B, Yan L, Yong J, Wu Y, Gao Y. et al. DNA methylation and chromatin accessibility profiling of mouse and human fetal germ cells. Cell Res. 2017;27:165-83
182. Vertesy A, Arindrarto W, Roost MS, Reinius B, Torrens-Juaneda V, Bialecka M. et al. Parental haplotype-specific single-cell transcriptomics reveal incomplete epigenetic reprogramming in human female germ cells. Nat Commun. 2018;9:1873
183. Han X, Zhou Z, Fei L, Sun H, Wang R, Chen Y. et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581:303-9
184. Shami AN, Zheng X, Munyoki SK, Ma Q, Manske GL, Green CD. et al. Single-Cell RNA Sequencing of Human, Macaque, and Mouse Testes Uncovers Conserved and Divergent Features of Mammalian Spermatogenesis. Dev Cell. 2020;54:529-47 e12
185. Hwang YS, Suzuki S, Seita Y, Ito J, Sakata Y, Aso H. et al. Reconstitution of prospermatogonial specification in vitro from human induced pluripotent stem cells. Nat Commun. 2020;11:5656
186. Bhutani K, Stansifer K, Ticau S, Bojic L, Villani AC, Slisz J. et al. Widespread haploid-biased gene expression enables sperm-level natural selection. Science. 2021 371
187. Gomes Fernandes M, He N, Wang F, Van Iperen L, Eguizabal C, Matorras R. et al. Human-specific subcellular compartmentalization of P-element induced wimpy testis-like (PIWIL) granules during germ cell development and spermatogenesis. Hum Reprod. 2018;33:258-69
188. 10xGenomics. https://www.10xgenomics.com/. Acess Date: 2022/9/2
189. Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015;6:11
190. Reznik B, Cincotta SA, Jaszczak RG, Mateo LJ, Shen J, Cao M. et al. Heterogeneity of transposon expression and activation of the repressive network in human fetal germ cells. Development. 2019 146
191. Xia K, Ma Y, Feng X, Deng R, Ke Q, Xiang AP. et al. Endosialin defines human stem Leydig cells with regenerative potential. Hum Reprod. 2020;35:2197-212
192. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A. et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419
193. Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580-5
194. Freeberg MA, Fromont LA, D'Altri T, Romero AF, Ciges JI, Jene A. et al. The European Genome-phenome Archive in 2021. Nucleic Acids Res. 2022;50:D980-D7
195. Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: integrating information about genes, proteins and diseases. Trends in genetics: TIG. 1997;13:163
196. Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ. et al. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 2019;47:D853-D8
197. Pujar S, O'Leary NA, Farrell CM, Loveland JE, Mudge JM, Wallin C. et al. Consensus coding sequence (CCDS) database: a standardized set of human and mouse protein-coding regions supported by expert curation. Nucleic Acids Res. 2018;46:D221-D8
198. Fairley S, Lowy-Gallego E, Perry E, Flicek P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020;48:D941-D7
199. Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP). http://evs.gs.washington.edu/EVS/ Acess Date: 2022/9/7
200. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434-43
201. COVID-19 Cell Atlas. https://www.covid19cellatlas.org/.Acess Date: 2022/9/7
202. Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO. et al. PaxDb, a database of protein abundance averages across all three domains of life. Mol Cell Proteomics. 2012;11:492-500
203. Samaras P, Schmidt T, Frejno M, Gessulat S, Reinecke M, Jarzab A. et al. ProteomicsDB: a multi-omics and multi-organism resource for life science research. Nucleic Acids Res. 2020;48:D1153-D63
204. Xu B, Kraemer MUG, Open C-DCG. Open access epidemiological data from the COVID-19 outbreak. Lancet Infect Dis. 2020;20:534
205. Johns Hopkins Coronavirus Resource Center (CRC). https://coronavirus.jhu.edu/. Acess Date: 2022/9/6
206. Wu H, Uchimura K, Donnelly EL, Kirita Y, Morris SA, Humphreys BD. Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem Cell. 2018;23:869-81 e8
207. Wu H, Malone AF, Donnelly EL, Kirita Y, Uchimura K, Ramakrishnan SM. et al. Single-Cell Transcriptomics of a Human Kidney Allograft Biopsy Specimen Defines a Diverse Inflammatory Response. J Am Soc Nephrol. 2018;29:2069-80
208. Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R. et al. A draft map of the human proteome. Nature. 2014;509:575-81
209. Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E. et al. The Human Cell Atlas. Elife. 2017 6
210. Madissoon E, Wilbrey-Clark A, Miragaia RJ, Saeb-Parsy K, Mahbubani KT, Georgakopoulos N. et al. scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 2019;21:1
211. Tabula Sapiens C, Jones RC, Karkanias J, Krasnow MA, Pisco AO, Quake SR. et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376:eabl4896
212. Ezran C, Liu S, Chang S, Ming J, Botvinnik O, Penland L. et al. Tabula Microcebus: A transcriptomic cell atlas of mouse lemur, an emerging primate model organism. bioRxiv. 2022. 2021 12.12.469460
213. Tabula Muris C, Overall c, Logistical c, Organ c, processing, Library p. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562:367-72
214. Pachkov M, Balwierz PJ, Arnold P, Ozonov E, van Nimwegen E. SwissRegulon, a database of genome-wide annotations of regulatory sites: recent updates. Nucleic Acids Res. 2013;41:D214-20
215. Hadziselimovic F, Verkauskas G, Stadler MB. Molecular clues in the regulation of mini-puberty involve neuronal DNA binding transcription factor NHLH2. Basic Clin Androl. 2021;31:6
216. Papatheodorou I, Moreno P, Manning J, Fuentes AM, George N, Fexova S. et al. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 2020;48:D77-D83
217. Salehi N, Karimi-Jafari MH, Totonchi M, Amiri-Yekta A. Integration and gene co-expression network analysis of scRNA-seq transcriptomes reveal heterogeneity and key functional genes in human spermatogenesis. Sci Rep. 2021;11:19089
218. Pinero J, Ramirez-Anguita JM, Sauch-Pitarch J, Ronzano F, Centeno E, Sanz F. et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020;48:D845-D55
219. European Nucleotide Archive (ENA). https://www.ebi.ac.uk/ena/. Acess Date: 2022/9/7
220. Di Persio S, Leitao E, Woste M, Tekath T, Cremers JF, Dugas M. et al. Whole-genome methylation analysis of testicular germ cells from cryptozoospermic men points to recurrent and functionally relevant DNA methylation changes. Clin Epigenetics. 2021;13:160
221. Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 2006;7(Suppl 1):S12 1-4
222. Wu C, Jin X, Tsueng G, Afrasiabi C, Su AI. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 2016;44:D313-6
223. Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S. et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130
224. Wu C, Macleod I, Su AI. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 2013;41:D561-5
225. UniProt C. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480-D9
226. Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244-8
227. Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23:318-26
228. Kozlowski LP, Bujnicki JM. MetaDisorder: a meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinformatics. 2012;13:111
229. Hardy JJ, Wyrwoll MJ, McFadden W, Malcher A, Rotte N, Pollock NC. et al. Variants in GCNA, X-linked germ-cell genome integrity gene, identified in men with primary spermatogenic failure. Hum Genet. 2021;140:1169-82
230. Wu X, Lu M, Yun D, Gao S, Chen S, Hu L. et al. Single-cell ATAC-Seq reveals cell type-specific transcriptional regulation and unique chromatin accessibility in human spermatogenesis. Hum Mol Genet. 2022;31:321-33
231. Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS. et al. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017;77:e108-e10
232. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
233. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
234. Hu H, Miao YR, Jia LH, Yu QY, Zhang Q, Guo AY. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019;47:D33-D8
235. Chitiashvili T, Hsu FM, Dror I, Plath K, Clark A. FGFR3 is expressed by human primordial germ cells and is repressed after meiotic initiation to form primordial oocytes. Stem Cell Reports. 2022;17:1268-78
236. Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38:675-8
237. Shen W, Song Z, Zhong X, Huang M, Shen D, Gao P. et al. Sangerbox: A comprehensive, interaction-friendly clinical bioinformatics analysis platform. iMeta. 2022;1:e36
238. Liu CJ, Hu FF, Xia MX, Han L, Zhang Q, Guo AY. GSCALite: a web server for gene set cancer analysis. Bioinformatics. 2018;34:3771-2
239. Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550-8
240. Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D. et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017;45:D840-D5
241. Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T. et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford). 2017. 2017
242. Calonga-Solis V, Fabbri-Scallet H, Ott F, Al-Sharkawi M, Kunstner A, Wunsch L. et al. MYRF: A New Regulator of Cardiac and Early Gonadal Development-Insights from Single Cell RNA Sequencing Analysis. J Clin Med. 2022 11
243. Leinonen R, Sugawara H, Shumway M, International Nucleotide Sequence Database C. The sequence read archive. Nucleic Acids Res. 2011;39:D19-21
244. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207-10
245. Athar A, Fullgrabe A, George N, Iqbal H, Huerta L, Ali A. et al. ArrayExpress update - from bulk to single-cell expression data. Nucleic Acids Res. 2019;47:D711-D5
246. Karlsson M, Zhang C, Mear L, Zhong W, Digre A, Katona B. et al. A single-cell type transcriptomics map of human tissues. Sci Adv. 2021 7
247. Lizio M, Harshbarger J, Shimoji H, Severin J, Kasukawa T, Sahin S. et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015;16:22
248. The Cancer Genome Atlas. https://www.cancer.gov/tcga
249. Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47:W556-W60
250. Blake JA, Baldarelli R, Kadin JA, Richardson JE, Smith CL, Bult CJ. et al. Mouse Genome Database (MGD): Knowledgebase for mouse-human comparative biology. Nucleic Acids Res. 2021;49:D981-D7
251. Franzen O, Gan LM, Bjorkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford). 2019. 2019
252. Singh M, Bansal V, Feschotte C. A Single-Cell RNA Expression Map of Human Coronavirus Entry Factors. Cell Rep. 2020;32:108175
253. Zheng LL, Xiong JH, Zheng WJ, Wang JH, Huang ZL, Chen ZR. et al. ColorCells: a database of expression, classification and functions of lncRNAs in single cells. Brief Bioinform. 2021 22
254. Li M, Zhang X, Ang KS, Ling J, Sethi R, Lee NYS. et al. DISCO: a database of Deeply Integrated human Single-Cell Omics data. Nucleic Acids Res. 2022;50:D596-D602
255. Chan Zuckerberg Initiative. CZ CELLxGENE Discover. https://cellxgene.cziscience.com/. Access Date: 2023/02/15.
Author contact
![]() Corresponding author: Dr. Xiang-Feng Chen. Address: 845 Lingshan Road, Shanghai, P. R. China, 200135. Telephone: +86-21-20284500; Fax: +86-21-58394262; Email address: allanbaconcom. Dr. Yi Ma. Address: 845 Lingshan Road, Shanghai, P. R. China, 200135. Telephone: +86-21-20284500; Fax: +86-21-58394262; Email address: my_julycom
Corresponding author: Dr. Xiang-Feng Chen. Address: 845 Lingshan Road, Shanghai, P. R. China, 200135. Telephone: +86-21-20284500; Fax: +86-21-58394262; Email address: allanbaconcom. Dr. Yi Ma. Address: 845 Lingshan Road, Shanghai, P. R. China, 200135. Telephone: +86-21-20284500; Fax: +86-21-58394262; Email address: my_julycom