Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
RNA Modification
Delivery Systems for RNA...
RNA-based Tumor Therapeutics
Perspectives, Challenges, and...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(10):3159-3183. doi:10.7150/ijbs.83732 This issue Cite
Review
Emerging Progress of RNA-Based Antitumor Therapeutics
Jiayan Fu1,2,3§, Haiyang Dong1,2,3§, Jian Wu4, Yongfeng Jin1,2,3,5 ![]()
1. National Key Laboratory of Advanced Drug Delivery and Release Systems, Zhejiang University, 310058, Hangzhou, China.
2. MOE Laboratory of Biosystems Homeostasis & Protection, Innovation Center for Cell Signaling Network, College of Life Sciences, Zhejiang University, Hangzhou 310058, China.
3. Cancer Center, Zhejiang University, 310058, Hangzhou, Zhejiang, China.
4. Department of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital, Zhejiang University School of Medicine, 310006, Hangzhou, Zhejiang, China.
5. Department of Neurosurgery, The First Affiliated Hospital, School of Medicine, Zhejiang University, 310006, Hangzhou, China.
§These authors contributed equally
Received 2023-2-21; Accepted 2023-6-1; Published 2023-6-19
Abstract

RNA-based therapeutics (e.g., mRNAs, siRNAs, microRNAs, ASOs, and saRNAs) have considerable potential for tumor treatment. The development and optimization of RNA modifications and delivery systems enable the stable and efficient delivery of RNA cargos in vivo to elicit an antitumor response. Targeted RNA-based therapeutics with multiple specificities and high efficacies are now available. In this review, we discuss progress in RNA-based antitumor therapeutics, including mRNAs, siRNAs, miRNAs, ASOs, saRNAs, RNA aptamers, and CRISPR-based gene editing. We focus on the immunogenicity, stability, translation efficiency, and delivery of RNA drugs, and summarize their optimization and the development of delivery systems. In addition, we describe the mechanisms by which RNA-based therapeutics induce antitumor responses. Furthermore, we review the merits and limitations of RNA cargos and their therapeutic potential for cancers.
Keywords: RNA, therapeutics, modification, delivery, tumor-treatment
Introduction
RNA-based therapeutics, including messenger RNAs (mRNAs), small interfering RNAs (siRNAs), microRNAs (miRNAs), antisense oligomers (ASOs), small activating RNAs (saRNAs), RNA aptamers, and CRISPR-based gene editing—have considerable therapeutic potential for genetic diseases [1, 2, 3, 4, 5]. Nearly 200 drugs have entered clinical trials for cancer, infectious diseases, autoimmune diseases, and neurodegenerative diseases. Compared to small-molecule and DNA-based drugs, RNA-based therapeutics have several advantages. They can target almost any genetic component and upregulate, downregulate, or abolish the expression of genes encoding a variety of proteins, including those with functions in immunity. In addition, because they are not integrated into the host genome, they have little genotoxicity. Moreover, the high efficiency and controllability of their production facilitate their development.
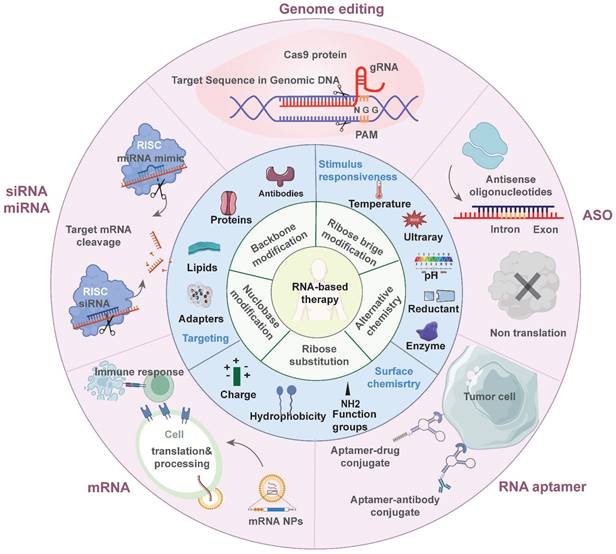
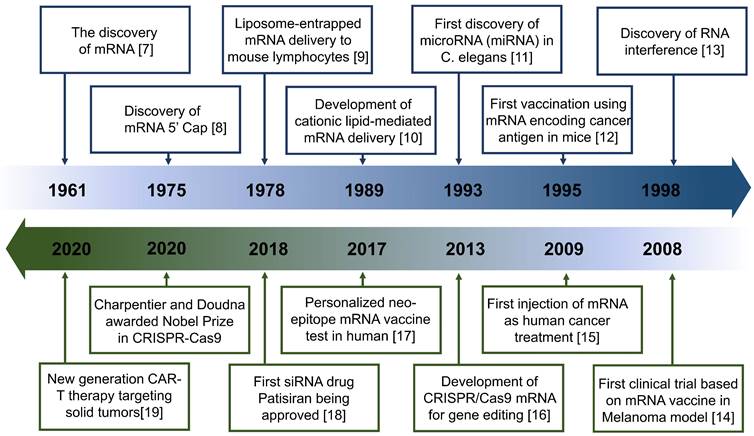
These advantages are particularly beneficial in tumor treatment. Such therapeutics can control the expression of target proteins, reshape the suppressive tumor microenvironment (TME) by regulating cytokine expression, and induce an innate or adaptive immune response [6] (Figure 1). These mechanisms provide a theoretical basis for the application of antitumor treatment. Indeed, the field of RNA-based therapeutics has expanded considerably in recent years. Several milestone events in RNA-based antitumor treatment are shown in Figure 2 [7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19].
Overview of RNA-based antitumor therapeutics. The inner ring discusses the chemical modification strategies of RNA molecules. The middle ring discusses the current optimization for delivery systems. The outer ring introduces various types of RNA-based therapeutics being applied in antitumor treatment.
Timeline of major RNA-based therapeutics and development milestones for antitumor treatment.
RNA-based therapeutics remains a significant challenge due to the inherent susceptibility of natural RNA to degradation by nucleases. As a result, there is a need to modify RNA structures and optimize delivery vehicles to efficiently deliver nucleic acid cargos to the target tissues and/or organs [20]. Chemical modifications can reduce the responses of cellular endogenous immunosensors to double-stranded RNA (dsRNA), greatly improving the safety of these drugs. Enhancing the stability of RNA drugs prevents their degradation by endogenous endonucleases and exonucleases, significantly improving efficacy [21]. Nanotechnology-based delivery systems have been a focus of development because viral vectors have genotoxicity and side effects. Nanoparticles (NPs) enable target-specific delivery of therapeutic agents due to their small size, physiological stability, structural tunability, high surface-to-volume ratio, and other favorable characteristics [22, 23]. NPs can be engineered to improve their therapeutic effectiveness by attaching cross-linkers and designing stimuli-responsive systems, thereby facilitating their accumulation at target sites and reducing off-target toxicity. Moreover, nanocarriers protect their cargo from degradation in the circulation, thus prolonging half-life.
Here we review recent progress in antitumor RNA-based therapeutics—mRNAs, siRNAs, miRNAs, ASOs, saRNAs, RNA aptamers, and CRISPR-based gene editing. We focus on the mechanisms by which RNA-based therapeutics induce antitumor immune responses. We also discuss RNA modification strategies and classify their carriers according to composition. Moreover, we review the relative merits and bottlenecks of various RNA-based therapeutics in tumor treatment (Figure 1).
RNA Modification
RNA molecules are inherently unstable. Exogenous RNA molecules trigger immune responses, leading to limited level of protein expression. RNA molecule modifications can be used to address these issues, which is the subject of the next section.
mRNA modification
mRNA drugs are synthetic versions of mature eukaryotic mRNAs and are typically produced by in vitro transcription (IVT). They consist of five main structures: the 5' cap, the 5' and 3' untranslated regions (UTRs), the open reading frames (ORFs) encoding the target proteins, and the 3' poly(A) tail. These structures affect the stability, translation efficiency, and immunogenicity of mRNA-based therapeutics [24].
The 5' and 3' UTRs are non-coding regions whose secondary structures, elements, and lengths affect ribosome recruitment and mRNA translation [25, 26, 27]. The 5' UTR directly affects the translation of its downstream ORFs [28]. Translated elements of the 5' UTR are referred to as upstream open reading frames (uORFs) [29]. For example, the so-called Kozak sequence improves the accuracy of translation initiation by surrounding the start codon with highly conserved nucleotides [30, 31]. A completely randomized 10-nucleotide-sequence preceding an uORF would drive translational output and determine mRNA stability, providing insight into the cis-regulatory code in the 5' UTR [32]. The complex secondary structure of the GC-rich 5' UTR, such as that of the ornithine decarboxylase mRNA [33], is associated with translation inhibition [27]. This explains in part the effect of the 5' UTR on oncogene expression. The presence of additional uAUG motifs in and the complex secondary structure of the 5' UTR prevent translation and suppress BRCA1 expression in breast cancer cells [34]. The 3' UTR also contains elements that regulate multiple aspects of mRNA metabolism, such as their nuclear export, cytoplasmic localization, translation efficiency, and stability [35]. There is an optimal length for the 3' UTRs: mRNAs with long 3' UTRs have short half-lives, whereas those with short 3' UTRs have low translation efficiency [36]. The most commonly used UTR sequences are from those of genes expressed at high levels, e.g., α-globin β-globin [37]. Repetitive concatenation of UTR sequences can enhance mRNA stability and translation efficiency [38].
The 5' cap structure contributes to mRNA stability and translation efficiency [1, 39]. The biological roles of cap-0 (m7GpppN-), cap-1 (m7GpppNm-), and cap-2 (m7GpppNmNm-) in mRNAs have been widely investigated [40, 41]. Uncapped or abnormally capped mRNAs can be recognized by the innate-immune receptor RIG-1, whereas cap-1 and cap-2 prevent recognition by innate-immune sensors [42]. Mainstream capping systems include enzymatic capping and the addition of cap analogs co-transcriptionally [43]. To date, the capping enzymes from the vaccinia virus are commercially available and widely used for post-transcriptional in vitro capping [44]. Under the catalysis of 2'-O-methyltransferase, cap-0 structure can be further modified to form cap-1 or cap-2 cap structures. Enzymatic capping exhibits high capping efficiency but cumbersome production. In contrast, co-transcriptional capping has limited efficiency due to competitive binding to GTP. To prevent reverse incorporation, anti-reverse cap analogs (ARCAs) have been developed to ensure capping only at non-methylated guanosines [45, 46]. Co-transcriptional capping using the novel CleanCap™ system does not affect indel formation and has a capping efficiency of 90-99%. The 3' poly(A) tail can also be optimized. Its deletion renders the mRNA molecule unstable [47]. Polyadenylation can be engineered by adding a fixed-length poly(A) sequence to the DNA template or enzymatically [1].
Nucleotide modification can also optimize mRNA stability, translation efficiency, and immunogenicity in vivo. Activation of Toll-like receptor (TLR) 3 can be inhibited by replacing the original nucleotide with 6-methyladenosine (m6A) or 2-thiouridine (s2U), whereas replacement with 5-methylcytidine (m5C), 5-methyluridine (m5U), s2U, m6A, or pseudouridine (Ψ) blocks the activation of TLR7 and TLR8, thus preventing an innate immune response [21, 48, 49, 50, 51, 52]. Indeed, Ψ and m5C enhance RNA stability and translational capability while diminishing its immunogenicity [48, 53]. N1-methyl Ψ-modified nucleotides are employed in IVT to improve the safety and stability of mRNA vaccines. Two mRNA vaccines against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) were modified using N1-methyl Ψ and each showed > 90% protection against coronavirus disease 2019 (COVID-19) [54]. By contrast, another mRNA vaccine candidate failed to reach the expected level of efficacy, potentially due to the lack of nucleotide modification [55].
siRNA modification
siRNA can be chemically modified to enhance the selectivity of their antisense strands for RNA-induced silencing complex (RISC) loading and to reduce off-target RNAi activity. However, unmodified siRNA may induce immune toxicity by activating TLR3. A variety of chemical modification strategies can be used to maximize the therapeutic efficacy and minimize the side effects of siRNAs.
Chemical modification typically targets the sugar ring, base, and phosphate skeleton of nucleotides. The goals are to improve the binding affinity of siRNA and protect it from nuclease degradation [56]. 2'-O-methyl (2'-O-Me) is the most commonly used modification of naturally occurring ribose [57]. Modification of 2'-O-methyl to 2'-methoxyethyl (MOE) increases binding affinity and nuclease resistance [58]. In addition, 2'-fluorine (2'-F) also improves siRNA binding affinity and is well tolerated due to its similar size and charge [59]. 2'-F modified siRNA shows excellent silencing of factor VII gene expression in mouse models.
The most common phosphate skeleton modification is phosphorothioate (PS), which prolongs the half-life in the circulation. Clinically approved siRNA therapeutics (e.g., Lumasiran and Inclisiran) have PS modifications. The phosphorodithioate (PS2) substitution in siRNAs involves the replacement of two non-bridged oxygen atoms. The potency and nuclease resistance of PS2-modified siRNAs are slightly higher than those of PS and unmodified siRNAs. Boron phosphate siRNA may be more effective than phosphorothioate [60]. Moreover, 5'-(E)-VP modification of siRNA stabilizes the 5'-end of the guide strand and promotes Ago2 loading, thereby improving tissue retention and gene silencing [61, 62].
Base replacement is another modification strategy for siRNA-based therapeutics. 5'-nitroindole modification of siRNAs reduces passenger strand-mediated off-target effects [63]. Due to concern about genome integration of metabolized non-natural residues, base structures present in natural nucleic acids, such as m5C and m6A, tend to be used [64]. Notably, 5-fluoro-2'-deoxyuridine modification of siRNA enhances cytotoxicity 10- to 100-fold and activates multiple apoptotic pathways, which shows therapeutic potential for cancer treatment [65].
N-acetylgalactosamine (GalNAc)-siRNA conjugates enable drug delivery to the liver. Tris-GalNAc binds to asialoglycoprotein receptor, which is highly expressed on the surface of hepatocytes, resulting in rapid endocytosis [66]. Several GalNAc-siRNA conjugates have exhibited therapeutic potential in clinical trials [67]. In one study, delivery of GalNAc-siRNA by a cholesterol-modified antimicrobial peptide silenced the expression of peptidyl-prolyl cis/trans isomerase (Pin) in a model of orthotopic liver cancer [68], and showed sustainable drug delivery.
ASO modification
ASOs are susceptible to nuclease degradation [69]. Various chemical modification strategies have been explored to increase their efficacy and enzymatic stability and reduce their immunogenicity and off-target toxicity. Similar to siRNAs, 2'-ribose modifications (2'-F, 2'-O-Me, and 2'-O-MOE) can improve binding affinity and resistance to enzymatic degradation. However, caution is needed with 2'-F modifications due to their potential toxicity [70]. A prior study showed that ASOs with 2'-F modifications exhibited hepatotoxicity. G-clamp is a cytosine analog that increases ASO binding affinity [71] by forming five hydrogen bonds with complementary guanine nucleobases in the target sequence.
An example of a modified ASO molecule is peptide nucleic acids (PNAs) [72], which have stronger binding affinity to RNA sequences than unmodified ASOs. Cationic engineering [73] and lysine modification [74] have been explored to resolve their low water solubility and poor cellular uptake. Locked nucleic acid (LNA) is a nucleotide derivative containing a disaccharide ring that locks the sugar ring into double ring molecular mode via a methylene bridge. This structure limits the flexibility of the sugar ring. LNA/DNA/RNA pairing products have higher unwinding temperatures and greater biological activities; they also activate RNase H [75]. Studies have found that the half-lives of nucleotides with three terminal LNAs are 10-fold longer than unmodified nucleotides [76].
RNA aptamer modification
The inherent susceptibility of RNA aptamers to nuclease degradation determines their stability [77]. Commonly used chemical modifications of the 3'- and 5'-ends, the sugar ring, the phosphodiester backbone, and bases can protect aptamers from degradation and prevent their renal clearance. The strategy of conjugating poly ethylene glycol (PEG)[78] at the 3'- or 5'- end can partially overcome obstacles such as short serum half-life, high renal clearance rate, and nuclease stability for oligonucleotides. However, long PEG chains reduce their binding affinity and increase their half-life in the circulation [79]. Thus, PEG-conjugation involves a trade-off between renal clearance and gene-silencing efficiency. Other terminal modifications can also enhance the stability of RNA aptamers. In vivo administration of 5'-cholesterol-modified aptamers significantly prolongs plasma half-life and exposure [80]. The most common 3'-end modification is inverted thymidine capping, which promotes RNA-aptamer stability in the circulation [81].
Modifications of the sugar ring can improve nuclease stability and prolong serum half-life. Similarly, the 2'-OH positions of RNA aptamers can be substituted with 2'-F [82, 83, 84], 2'-OMe [85], 2'-NH2, 2'-LNA [86, 87], and 2'-d-/l-isonucleoside [88, 89]. Substitution of non-natural nucleotides into oligonucleotides is achieved by mutating T7 RNA polymerase. However, such substitutions can lead to nonspecific immune reactions or toxicity. Phosphate-backbone modifications can be introduced to stabilize the phosphodiester bonds, including PS [90] and PS2 [91] bonds. The substitution in PS bonds is chiral (Sp or Rp configuration), unlike the natural conformation, and may therefore have adverse effects on biological function.
Others
Chemical modification strategies are under development for other RNA-based therapeutics. The nucleotide sugar ring, base, and phosphate skeleton are modified in anti-miRNA oligonucleotides (AMOs). Most widely used chemical modifications on AMOs are LNA, 2'-F-RNA, 2'-OMe, PNAs. Modified AMOs with higher binding affinity and superior stability have greater regulatory potency [92, 93]. Oligonucleotides with 2'-F modifications show increased thermal stability (Tm +1.6°C, higher than the 2'-OMe modification) [94]. The 2'-F modification can be combined with the 2'-MOE substitution to enhance AMO stability in vivo [95]. Substituting ribonucleotides with LNAs can endow antimir drugs with increased resistance to nuclease degradation and enhanced target affinity [96]. This modification strategy has been used for the clinical trial of Miravirsen, an antagomir of miR-122 (NCT01200420). Moreover, adding a non-base modifier to the end of AMO have been reported to increase the Tm by mediating hydrophobic stacking interactions [97].
Delivery Systems for RNA Therapeutics
RNA therapeutics must be delivered to the correct tissues without triggering an immune reaction. However, the high molecular weight and negative charge of RNA hampers their delivery to target sites. RNA delivery systems can be divided to viral or nonviral. Below we summarize several nonviral RNA delivery systems being extensively studied (Figure 3).
Schematic representation of different types of nanocarriers used in RNA delivery. (A) Lipid-based delivery system mainly include liposome and lipid nanoparticle, etc. (B) Polymer-based delivery system mainly include polymeric nanoparticle, polymer micelles, etc. (C) Inorganic nanodelivery system mainly include carbon, metal NPs, core-shell, MSN, etc. (D) Others includes extracellular vesicles, DNA origami, hydrogel, etc.
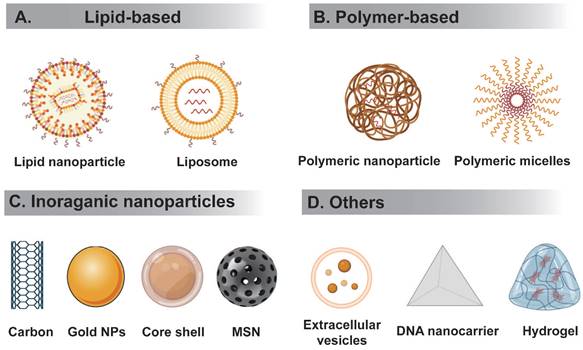
Polymer-based NPs
Polymeric NPs are typically prepared from biocompatible and biodegradable polymers, in which the drug is dissolved, entrapped, encapsulated, or attached to a nanoparticle matrix. Polymers bind nucleic acids to form polymeric complexes at physiological pH, facilitating gene delivery. Polymeric NPs promote electrostatic binding to nucleic acid cargo by interacting with positively charged units. Nucleic acids and polymers can be covalently linked using degradable linkers. The addition of cationic groups such as chitosan to polylactic-co-glycolic acid (PLGA) enables its use for siRNA delivery [98]. In addition, polyethyleneimine (PEI) and poly(l-lysine) (PLL) form complexes with RNA via electrostatic interactions. Because unmodified PEI and PLL are not well-tolerated [99], they have been chemically modified to enhance their in vivo transfection capability and reduce their toxicity [100]. The synthetic bio-reducible polymer poly(beta-amino ester) is synthesized by conjugating amine monomers to diacrylates [101]. Polymer-based NPs have low nonspecific toxicity due to degradation by hydrolysis as well as by bio-reduction in the reducing environment of the cytoplasm [102].
Lipid-based NPs
Lipid-based NPs have high biocompatibility and biosafety, and their production is simple. They include liposomes, micelles, emulsions, and solid lipid NPs (SLNs). LNPs have been evaluated as mainstream delivery systems in multiple preclinical trials.
Because the size, shape, surface charge, and materials of NPs affect their cellular uptake [103], LNPs with different molecular structures have been developed for RNA delivery. Helper lipids, such as DOPE and cholesterol, are critical components of LNPs. The structural characteristics of cholesterol are determinants of their intracellular delivery and the efficiency of gene transfection [104]. Study revealed that incorporation of C-24 alkyl phytosterols into LNPs enhances gene transfection. The length of the alkyl tail, flexibility of the sterol ring, and polarity are required to maintain high transfection efficiency [104]. There are also some concerns about the use of cationic lipids, for those cationic lipids bearing quaternary ammonium groups have potential cytotoxicity and relatively short blood circulation time stemming from the positive charge, which hindered their clinical translation [105]. In contrast, the neutrality of ionizable lipids at physiological pH help reduces the toxicity and, to some extent, increases the circulation half-life of ionizable LNPs [106]. The loss of mRNA activity in LNP delivery systems is caused by electrophilic impurities derived from the oxidation of ionizable cationic lipid components and subsequent hydrolysis of the tertiary amine [107]. Another report speculated that the highly pro-inflammatory effect of LNP-based systems is likely caused by their ionizable lipid components, the removal of which abolishes visible skin inflammation [108]. However, mitigating this toxicity by reducing the charge of cationic lipids seem to be unwise because of the descending level of nucleic acid encapsulation and transfection efficiency of LNP-based systems. Therefore, toxicity, immunity, and therapeutic effectiveness must be balanced.
Inorganic NPs
Inorganic NPs (gold NPs [AuNPs], silver NPs [AgNPs], carbon nanotubes, mesoporous silica NPs [MSNs] and so on) have a narrow size distribution and a surface chemistry suitable for ligand conjugation [109]. The physicochemical properties of inorganic NPs are not susceptible to the environment, which makes them suitable for photothermal or photodynamic therapy of solid tumors [110].
AuNPs typically have diameters of 1 to 100 nm, a large surface-to-volume ratio, good optoelectronic properties, excellent biocompatibility, and low toxicity [111]. Modifications of the shape, diameter, PEGylation, and surface charge of AuNPs affect their drug-loading capacities [110]. Due to their unique properties, AuNPs have been applied in conjunction with chemotherapy or photothermal therapy for cancer [112]. Currently, various optimization strategies have been developed in vivo for extending the plasma half-lives of AuNPs and enhance their targeted accumulation and controllable release [113]. Similarly, AgNPs are NPs 1-100 nm in diameter composed of silver atoms [114]. In addition to their antibacterial properties, L-cysteine AgNPs have potential for drug delivery and excellent biocompatibility [115].
MSNs are biodegradable and chemically stable nanostructured materials composed of silica particles with pore channels [116]. The tunable pore size and mesoporous structure of MSNs facilitate drug dissolution and encapsulation. Furthermore, they exhibit high chemical, thermal, and mechanical stability under physiological conditions, across broad ranges of pH and temperature. Exterior and interior surface modifications of MSNs can improve their therapeutic efficacy and pharmacokinetics. For example, structure-optimized silica nanocarriers coloaded with a TLR9 agonist and antigen exhibit increased accumulation in draining lymph nodes, thereby enhancing antigen-specific B- and T-cell immunity in a murine tumor model [117].
Others
Versatile multifunctional nanomaterials whose synthesis is simple and inexpensive, as well as hybrids that integrate the advantages of different materials, have been developed [118]. An example is exosomes, a type of extracellular vesicle (EV) secreted by most types of cells [119]. Exosomes mediate intercellular communication and have functional and structural similarities with synthetic drug carriers such as liposomes [120], thus can serve as potent candidates for drug delivery [121, 122, 123, 124]. Exosomes could be used as biomimetic nano-vehicles for gene therapy. The synthesis of DNA nanostructure-based carriers is simple, and they have excellent biocompatibility. DNA nanostructure-based carriers are fabricated [125] as tetrahedrons [126], prisms [127], nanotubes [128], and planar origami [129, 130].
Hydrogels consist of a three-dimensional network swollen with water [131]. Compared to directly delivering naked RNA using hydrogels, loading RNA into nanocarriers encapsulated in the hydrogel network improves RNA stability with no need for chemical modifications. In one study, local delivery of mRNA using a chitosan-alginate gel scaffolded lipoplex promoted T-cell proliferation and antibody secretion [132]. Similar results were obtained for a COVID-19 subunit vaccine containing CpG/Alum as adjuvants. Injectable polymeric NP-based hydrogels provide broad protection against SARS-CoV-2 variants [133]. Hydrogels can deliver RNA cargos packaged in polymers [134, 135] or inorganic NPs [136, 137]. Controlled continuous RNA release can be achieved by adjusting the cross-linking density, hydrophilicity, pore size, and other parameters of hydrogels. Modified hydrogels can prolong the retention of antitumor drugs in the tumor tissue [138], thereby enhancing their uptake by cancer cells and reducing their toxic effects on nontarget cells [131, 139]. Optimization of their degradability, clearance rate, and controlled release will make hydrogel-based systems more suitable for in vivo delivery of RNA-based therapeutics in clinical applications.
RNA-based Tumor Therapeutics
mRNA
mRNA vaccines
mRNA-based therapeutics are promising alternatives to DNA for cancer immunotherapy due to their lower mutagenicity and easier transient expression. Furthermore, the in vitro production and purification of mRNAs prevents host protein and virus contamination [3, 140]. When delivered to antigen-presenting cells (APCs), mRNAs encoding tumor antigens escape to the cytoplasm, where they are translated and processed into peptide epitopes. Subsequently, those peptides bind major histocompatibility complex (MHC) class I and are transferred to the APC surface, activating CD8 T cells and inducing antitumor immune responses [140] (Figure 4). However, the large size, structural instability, and negative charge of naked mRNAs hinder their ability to reach target sites. LNPs are self-assembled nanocarriers that prevent in vivo degradation and promote the intracellular delivery and endosomal escape of mRNAs [141]. Nucleoside-modified mRNA-LNPs were used by Pfizer/BioNTech and Moderna in their COVID-19 mRNA vaccines [142, 143], and lipid-based systems have subsequently been a focus of interest. Other mRNA LNP formulations have been widely evaluated in preclinical and clinical trials for cancer [144, 145].
Principles of mRNA-based antitumor therapeutics. (A) Antigen-encoding mRNA-based nanoparticles enter the cytoplasm through endocytosis and then translated to protein with the help of ribosome. Those antigen proteins are degraded to peptides by the proteasome and further presented to the APCs via MHC processing. (B) mRNA-based nanoparticles being delivered could be translated to proinflammatory cytokines and chemokines to activate the immune signal pathway downstream, they could also reshape the TME by restoring tumor suppressor expression. (C) Nanocarriers help effectively deliver CAR-encoding mRNA to the T cell, induce T cell activation and subsequently lead to antigen-specific recognition and tumor tissue elimination.
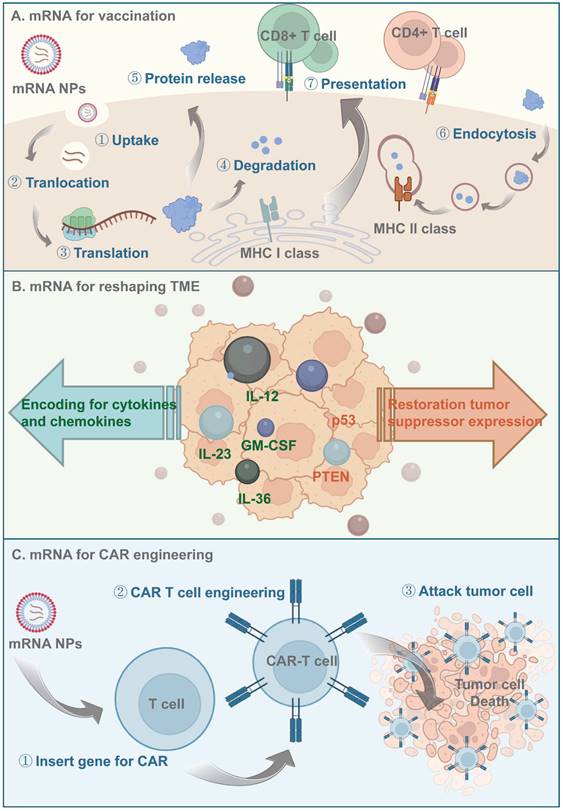
LNPs consist of phospholipids, cholesterol, PEGylated lipids, and cationic or ionizable lipids. Phospholipids and cholesterol mediate LNP endocytosis and accelerate mRNA release during endocytosis. PEGylated lipids prolong the half-life in the circulation [146] and act as a steric barrier, preventing aggregation during storage. Thus, the particles could be controlled to an appropriate size. Cationic/ionizable lipids, which serve as the core component of LNPs [147], facilitate binding to negatively charged mRNA molecules and promote their transfer from the endosome to the cytosol for translation via pH-triggered electrostatic interactions with the anionic endosomal membrane [147].
Acute inflammatory responses, such as pain, swelling, and fever, can be caused by mRNA-LNP vaccines [148, 149, 150, 151]. These effects are associated with the pro-inflammatory properties of LNPs, and may provide a basis for their adjuvant properties. Previous preclinical data suggested mRNA/LNP complexes show adjuvant activity [152], whereas mRNAs undergo nucleoside modification to attenuate the activation of innate inflammatory pathways. Several cationic/ionizable lipids can induce inflammation by activating TLR pathways [153, 154]. In preclinical research on nucleoside-modified mRNA vaccines, LNPs induced considerable neutrophil infiltration and production of inflammatory cytokines and chemokines in mice, independent of the administration route [108]. Though ionizable lipids may overcome the pro-inflammatory and cytotoxic effects caused by permanently charged cationic lipids [155]. LNP formulations containing ionizable lipids still elicit an innate immune response by releasing IL-l, triggering the secretion of the pro-inflammatory cytokine IL-6 [156]. The ionizable lipid SM-102 induces inflammasome activation. In addition, PEG, a component of LNPs, is reported to be immunogenic. Repeated administration led those pre-existing PEG antibodies induce complement activation-related pseudo-allergic (CARPA) reactions [157]. Therefore, booster shots induce severe adverse reactions, possibly due to further strengthening of the immune memory against LNPs [158]. In this sense, LNPs may function not only as carriers of RNAs but also as adjuvants that trigger innate and adaptive immune responses.
Depending on their charge and composition, LNPs can be broadly classified as cationic LNPs, ionizable LNPs, and lipid calcium phosphate (LCP) NPs. Classical mRNA cancer vaccines target tumor-associated antigens (TAAs) preferentially expressed in malignant cells. For instance, Oberli et al. [159] reported a lipid NP formulation loaded with the tumor-associated antigens gp100 and TRP2 for the delivery of mRNA vaccines to induce a cytotoxic CD8 T-cell response. The optimized mRNA formulation overcame self-tolerance and significantly prolong survival in transgenic and aggressive mouse melanoma models. More excitingly, replacement of 1% of the molar composition of PEG in the optimized LNP formulation with lipopolysaccharide enhanced the immune response by activating TLRs. Therefore, further investigation of LNPs combined with adjuvants as mRNA vaccine vectors is warranted. However, adding LPS renders manufacture difficult and there is a risk of toxicity. To possibly avoid systemic toxicity, a minimalist vaccine uses heterocyclic lipids as mRNA carriers and self-adjuvants, thereby triggering a stimulator of interferon genes (STING)-mediated type I interferon innate immune response [160]. The 12-C-tailed C1 LNP stimulates the expression of inflammatory cytokines such as IL-12 via the TLR4 signaling pathway [161]. Cationic lipid-assisted NPs (CLAN) fabricated with biocompatible and biodegradable block copolymer poly (ethylene glycol)-block-poly (lactic-co-glycolic acid) (PEG-b-PLGA) and cationic lipid have been developed for mRNA delivery. They have been used to package OVA-encoded mRNA in dendritic cells in vitro, enhancing CD11c-cell maturation and CD8 T-cell proliferation in aggressive E·G7-OVA lymphoma models [162]. To further improve intracellular mRNA delivery and the immune response, liposomes modified with a novel cationic and hydrophilic antimicrobial peptide, DP7-C, have been developed. As an immune adjuvant, DP7-C promotes DC maturation and enhances the immune response by stimulating the TLR2-MyD88-IKK-IκB-NF-κB signaling pathway. The carrier and immunoadjuvant functions of the system increase the antitumor effect of a neoantigen-based mRNA vaccine [163].
Several ionizable lipoplex-type mRNA carriers are available. Tateshita et al. [164] combined the ssPalmE and KALA peptides to modify NPs for DC-based cancer immunotherapy. Their amphiphilic material consists of a series of ionizable lipids and an SS-cleavable and pH-activated lipid-like material (ssPalm). ssPalm enables cytoplasmic delivery of loaded nucleic acids, and the α-helical cationic KALA-peptide synergistically increases mRNA adjuvant activity by triggering the cytoplasmic nucleic acid sensor. LCP NPs have been used for mRNA delivery in cancer immunology, e.g., LCP for the codelivery of an mRNA encoding a melanoma-associated antigen (TRP2) and an immune checkpoint-targeting siRNA to DCs in vivo [165]. The calcium phosphate core promotes acid-mediated dissolution in the endolysosomal compartment, triggering rapid cargo release after cellular internalization. The codelivery of a PD-L1 siRNA and an mRNA vaccine elicits a robust and durable antigen-specific immune response in the melanoma model. More stringent requirements have been proposed for delivery-system modification. To overcome the need for repeated administration of NP-based vaccines, in one study, graphene oxide (GO) and low-molecular-weight polyethyleneimine (LPEI) were mixed and used to fabricate an injectable GO-LPEI hydrogel. Use of this injectable hydrogel nanocarrier to deliver OVA-encoding mRNA generated ovalbumin and adjuvant-laden nanovaccines and markedly increased the number of antigen-specific CD8 T cells, thereby inhibiting tumor growth [166].
The clinical applications of the above delivery system are hampered by the lack of specific targeting, which make them useful only for local inoculation and liver-targeted therapy. Efficient lymphatic drainage and accumulation can be promoted by PEGylation and modifying NP size and surface charge [167] of LNP-based mRNA therapeutics. Xu et al. [168] found that the delivery and targeting of LNPs are modulated by the head chemical structure. They screened an endogenously LN-targeting lipid NP, 113-O12B. Compared to Pfizer/BioNTech's mRNA vaccine, ALC-0315, 113-O12B targeted lymph nodes and the liver at a 3:1 ratio, showing increased lymph-node and significantly decreased liver mRNA expression. Encapsulation of a TRP-2 peptide-encoding mRNA markedly inhibited tumor growth. Moreover, mice with complete remission did not show new tumor formation after injection of metastatic tumor cells, indicating induction of long-term immune memory [168]. Kranz et al. reported that a decreased cationic lipid-to-DOPE ratio of mRNA-loaded lipoplexes affected organ specificity. Based on this rationale, they developed a lipoplex-based system for cargo delivery to splenic DCs [169]. These RNA-LPX complexes showed synchronized induction of highly potent adaptive and type-I-IFN-mediated innate immune responses for cancer immunotherapy.
mRNA for reshaping the TME
Use of mRNAs to restore tumor-suppressor expression has therapeutic potential for cancer. An example is a polymeric NP platform for delivering mRNA encoding phosphatase and tensin homolog deleted on chromosome ten (PTEN), a cancer-inhibiting factor. Reactivating PTEN in PTEN-mutated melanoma cells and PTEN-null prostate cancer cells by mRNA delivery reversed the immunosuppressive TME by promoting CD8 T-cell infiltration and lifting the expression level of proinflammatory cytokines [170]. The results suggested that this PTEN-NP platform elicited a robust and safe antitumor immune response by inducing tumor-cell autophagy and releasing damage-associated molecular patterns (DAMPs), thereby triggering tumor immunogenic cell death (ICD) and sensitizing cancers to immune checkpoint blockade (ICB) therapy.
p53 is a tumor suppressor involved in cell cycle arrest, apoptosis, senescence, and other cellular pathways [171]. Beyond its autonomous tumor-suppressive effect, it regulates the TME by modulating the interactions between tumor cells and immune cells. In one study, a modified lipid-polymer hybrid NP platform for mRNA delivery enhanced the selectivity of CXCR4 targeting. A series of ionizable lipid-like compounds and varying densities of CXCR4-targeting ligands were screened for mRNA translation efficiency and HCC-targeting specificity in vivo. The CXCR4-targeting NP system transported p53 mRNA to HCC cells, restoring p53 activity and decreasing HCC cell viability. This mRNA nanotherapy-based p53 restoration strategy in combination with anti-PD-1 therapy induced a potent antitumor effect in intrahepatic and ectopic models of HCC with p53 loss. Therefore, combining p53 mRNA therapeutics with ICB could reverse immunosuppression in HCC [171]. The introduction of LCOR mRNA into tumor cells can restore the expression of LCOR, a tumor suppressor, by modulating IFN sensitivity [172]. In that study, mice serially administered EV-based LCOR mRNA and anti-PD-L1 therapies in combination showed significantly longer survival and complete elimination of lung metastasis. Therefore, LCOR mRNA delivery in conjunction with ICB has potential for specifically modulating antigen presentation in tumor cells.
mRNAs encoding cytokines or chemokines can also induce APC maturation and activation, activate T-cell-mediated immunity, and adjust the dysfunctional immune TME. The mRNA-based adjuvant TriMix consists of mRNAs encoding the costimulatory molecule CD70, the activation stimulator CD40 ligand (CD40L), and constitutively active TLR4 (caTLR4). Upon codelivery of tumor-associated antigen (TAA), the TriMix mRNA reprograms CD8+ TiDCs in vivo into stimulatory cells that efficiently process spontaneously engulfed TAAs, upregulate costimulatory molecules, and migrate to TDLNs to activate cytotoxic T lymphocytes (CTLs), ultimately delaying the growth of established tumors [173].
Moderna has collaborated with AstraZeneca to develop a local intratumoral mRNA therapy for IL-12 delivery [174]. As a crucial mediator of the Th1 immune response, IL-12 facilitates the activation and cytotoxicity of natural killer (NK) cells and CTLs via the IFN-γ signaling pathway. Intratumoral injection of LNP-formulated mIL12 (MEDI1191) induced tumor regression in superficial and deep-seated lesion models, with upregulation of CD8+ T-cell infiltration and IFN-γ expression. The systematic administration of such cytokines leads to high exposure, possibly resulting in toxicity. For this reason, MEDI1191, an optimized IL-12 mRNA, was developed for tumor-targeted local delivery. To minimize off-target liver toxicity, the miRNA-mediated binding site was incorporated into the 3'-UTR of the IL-12 mRNA to promote its elimination [175].
Preclinical data suggest limited antitumor activity for IL-12 mRNA monotherapy. For this reason, other mRNA therapeutics encoding mixtures of cytokines and chemokines have been developed. Intratumoral injection of DAL4-LNP-IL-12 and IL-27 mRNAs showed synergistic suppression of tumor growth with robust infiltration of immune effector cells [176]. Another research investigated the optimal combination of different cytokines, and demonstrated that a mixture of mRNAs encoding IL-12 single chain, interferon-α (IFN-α), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-15 increased systemic antigen-specific T-cell expansion and granzyme B T-cell infiltration, thereby promoting tumor regression [177]. Moderna has announced two other local-injection mRNA therapeutics (mRNA-2416 and mRNA-2572), which encode multiple immunoregulatory factors. mRNA-2416 encodes OX40L, dosed alone or in combination with the intravenously administered PD-L1 inhibitor durvalumab for the treatment of lymphoma and metastatic ovarian cancer (NCT03323398). mRNA-2572 includes OX40L, IL-23, and IL-36γ mRNAs and is intended for the treatment of lymphoma (NCT03739931). OX40L enhances the expansion and survival of CD4 and CD8 T cells, and IL-23 and IL-36γ are pro-inflammatory cytokines of the IL-12 and IL-1 families, respectively, which activate and mature DCs and other immune cells. Compared to mono-cytokine mRNA therapy, addition of an mRNA encoding the T-cell costimulator OX40L increased the complete response rates of treated and untreated distal tumors. Mice treated with the mixture exhibited complete immune responses and effective protection [178]. In summary, the above multi-cytokine or chemokine strategies elicit durable and robust antitumor protection (Figure 4, Table 1).
Overview of clinical trials of RNA-based therapeutics discussed in this review
| RNA Types | Name | NCT Number | Phase | Description | Treating diseases | Delivery system |
|---|---|---|---|---|---|---|
| mRNA | CV9201 | NCT00923312 | I/II | Encoding TAA MAGE-C1, MAGE-C2, NY-SEO-1, survivin, 5 T4 | NSCLC | Protamine |
| CV9202 | NCT03164772 | I/II | Encoding TAA: MAGE-C1、NY-ESO-1、MAGE-C2、survivin、5T4, MUC-1 | NSCLC | Protamine | |
| CV9103 | NCT00831467 | I/II | Encoding TAA: PSA, PSCA, PSMA, STEAP1 | Prostate cancer | Protamine | |
| CV9104 | NCT01817738 | I/II | Encoding TAA: PSA, PSCA, PSMA, STEAP1, PAP, MUC1 | Prostate cancer | Protamine | |
| BNT111 | NCT02410733 | I | Encoding TAA: NY-ESO-1, Tyrosinase, MAGE-A3, TPTE | Advanced melanoma | Lipoplex | |
| NCT04526899 | II | Encoding TAA: NY-ESO-1, Tyrosinase, MAGE-A3, TPTE | Advanced melanoma | Lipoplex | ||
| BNT112 | NCT04382898 | I/II | Encoding TAA: PAP, PSA, three undisclosed antigens | Prostate | Lipoplex | |
| BNT113 | NCT04534205 | II | Encoding HPV16 E6 and E7 oncoproteins | Head and neck squamous cell carcinoma | Lipoplex | |
| BNT116 | NCT05142189 | I | Encoding NSCLC tumor-associated antigens | NSCLC | Lipoplex | |
| BNT122 | NCT04161755 | I | Encoding personalized tumor mutation antigens | Pancreatic | Lipoplex | |
| NCT03815058 | I | Encoding personalized tumor mutation antigens | Advanced melanoma | Lipoplex | ||
| NCT04486378 | II | Encoding personalized tumor mutation antigens | Colorectal | Lipoplex | ||
| NCT03289962 | I | Encoding personalized tumor mutation antigens | Solid tumors | Lipoplex | ||
| mRNA-4157 | NCT03313778 | I | Encoding several neoantigens | Solid tumors | LNP | |
| NCT03897881 | II | Encoding 20 different mutated neoepitopes | Melanoma | LNP | ||
| mRNA-5671 | NCT03948763 | I | Encoding KRAS gene driver mutations (G12C, G12D, G12V, G13C) | NSCLC, pancreatic, colorectal neoplasms | LNP | |
| CARVac | NCT04503278 | I/II | Encoding CLDN6 | Solid tumors | Lipoplex | |
| IVAC | NCT02316457 | I | Encoding gp100 3 TAAs selected | TNBC | Lipoplex | |
| BNT141 | NCT04683939 | I/II | Encoding IgG antibody | CLDN18.2-positive solid tumors/ solid tumor | Lipoplex | |
| BNT152+153 | NCT04710043 | I | Encoding IL-7 and IL12 | Solid tumors | Lipoplex | |
| MEDI1191 | NCT03946800 | I | Encoding IL-12 | Solid tumors | LNP | |
| mRNA-2416 | NCT03323398 | I | Encoding OX40L, IL-23, IL-36γ | Solid Tumor Malignancies or Lymphoma; Ovarian Cancer | LNP | |
| BNT151 | NCT04455620 | I/II | Encoding IL-2 | Solid tumor | Lipoplex | |
| siRNA | ALN-VSP02 | NCT01158079; NCT00882180 | I | Target ACSL4 and PLK1 | Solid Tumors | LNP |
| CALAA-01 | NCT00689065 | I | Target Fibronectin | Solid Tumor | cyclodextrin polymer | |
| TKM-080301 | NCT01437007; NCT02191878; NCT01262235 | I/II | Target TGFBR2 | Colorectal Cancer with Hepatic Metastases Pancreas Cancer with Hepatic Metastases Gastric Cancer with Hepatic Metastases | LNP | |
| siG12D LODER | NCT01676259; NCT01188785 | II | Target K-ras | Pancreatic Ductal Adenocarcinoma Pancreatic Cancer | Polymers | |
| iExosomes | NCT03608631 | I | Target K-ras | Metastatic Pancreatic Adenocarcinoma Pancreatic Ductal Adenocarcinoma | Exosomes | |
| Atu027 | NCT01808638; NCT00938574 | I/II | Target 4E-BP1 | Carcinoma, Pancreatic Ductal Advanced Solid Tumors | LNP | |
| DCR-MYC | NCT02314052; NCT02110563 | I/II | Target MYC | Hepatocellular Carcinoma/ Solid Tumors | LNP | |
| saRNA | MTL-CEBPA | NCT05097911; NCT02716012; NCT04105335 | I | Target CCAAT/enhancer binding protein alpha (C/EBP-α). | Hepatocellular Carcinoma Liver Cancer | |
| miRNA | MRX34 | NCT01829971; NCT02862145; | I/II | Target miR-34 | Primary Liver Cancer SCLC Lymphoma Melanoma Multiple Myeloma Renal Cell Carcinoma NSCLC | |
| MesomiR-1 | NCT02369198 | I | Target miR-16 | Malignant Pleural Mesothelioma Non-Small Cell Lung Cancer | ||
| Aptamer | AS1411 | NCT00512083; NCT00881244; NCT01034410 | I/II | Target nucleolin | Leukemia, Myeloid/solid tumors | |
| NOX-A12 | NCT03168139; NCT00976378; NCT01194934 | I/II | Target CXCL-12 | Metastatic Colorectal Cancer/ Metastatic Pancreatic Cancer/ Hematopoietic Stem Cell Transplantation |
mRNAs for CAR engineering
Engineered T cells that express chimeric antigen receptors (CARs) for adoptive cell therapy (ACT) have considerable benefits for the treatment of certain blood malignancies [179, 180]. CARs are recombinant receptor constructs containing replaceable intracellular T-cell signaling domains, targeting domains, and transmembrane domains, enabling the substitution of antigen-binding domains encoded by single-chain variable fragments (scFv). Thus, different signal transduction pathways activate different T-cell functions and properties. The activation mediated by intra-cytoplasmic signaling domains could promote tumor targeting by inducing the release of granzyme and perforin, as well as facilitating tumor killing via activation of other immune components. Although CAR-T therapy has much therapeutic potential for cancer, challenges remain in terms of “on-target, off-tumor” cytotoxicity and feasibility for individuals with severe immunodeficiency [181, 182].
NPs as vehicles for CAR delivery
Clinical-scale manufacturing of engineered T cells requires their isolation, transfection, modification, amplification, and re-injection, which is difficult and costly. Although virus-mediated transfer can prolong transgene expression by T cells [183] and has been used for the transduction of CARs into T cells [184], it is time-consuming and its clinical application is hampered by safety concerns, such as mutagenicity and genome-insertion toxicity. NPs have potential for CAR mRNA delivery and have higher transfection efficiency, lower cost, and fewer off-target effects than viral vectors (Figure 4).
Nanocarrier-mediated targeted delivery of an mRNA encoding a rare-cleaving megaTAL nuclease disrupts T-cell receptor expression [185]. Surface-anchored targeting ligands of anti-CD3 and anti-CD8 antibodies mediate selective binding of the NPs to T cells and initiate rapid receptor-induced endocytosis. Polyglutamic acid (PGA)-coated surfaces were designed to minimize off-target binding by shielding surface charges. This lymphocyte-targeted NP system improves the therapeutic activity of CAR-T cells by reprogramming them towards a TCM-like phenotype. mRNA NPs transiently expressing the transcription factor Foxo1, which mediates effector-cell differentiation into functionally competent memory cells, induce persistent changes in surface markers and improved antitumor efficacy.
Ionizable lipid NPs have been used to deliver mRNAs to primary human T cells ex vivo [145] and they show equivalent CAR expression but less cytotoxicity than electroporation. Ionizable lipids in ethanol were combined with cholesterol (NP stability and membrane fusion), DOPE (endosomal escape), and C14-PEG (suppresses aggregation and nonspecific endocytosis. In another orthogonal experiment for optimizing lipid nanoparticles, the results also showed the impact of excipient on LNP performance and CAR-T reprogramming efficiency [186].
NPs combined with CAR
Most solid malignancies failed to effectively respond to CAR-T cell infusion because of tumor resistance, tumor-antigen-escape relapse, and the suppressive TME [187]. On the one hand, the dense tumor tissue and compact extracellular matrix are tightly crosslinked, and the resulting pressure hampers the infiltration of CAR-T cells into tumors [188]. On the other hand, the TME is not conducive to CAR-T cell survival because of hypoxia, low levels of nutrients, acidic pH, and high permeability. In addition, a variety of immunosuppressive cells and immune checkpoints (PD1, CTLA4) inhibit the killing activity of CAR-T cells [189]. Therefore, NPs, with their intrinsic properties, could improve the anticancer efficacy of CAR-T by enhancing cargo activity and stability, stimulating CAR‐T cell proliferation and survival, and increasing in vivo delivery efficiency.
In one study, a liposomal antigen-encoding RNA was intravenously administered to stimulate tumor-associated T cells in patients with cancer [19]. This CAR-T cell-amplifying RNA vaccine, referred to as CARVac, induced the expression of CLDN6 on DCs, thereby stimulating cytokine secretion and the proliferation of co-cultured CLDN6 CAR-T cells in a dose-dependent manner. The RNA vaccine completely induced tumor regression in an ovarian cancer model compared to CLDN6 CAR-T therapy alone. This pioneering method led to the development of the next-generation drug BNT211, which targets solid tumors. Preliminary results from a dose-exploration clinical trial showed that RNA vaccine (CARVac) comprising CLDN6 CAR-T cells combined with CAR-T showed good safety and efficacy. After 6 weeks of treatment, 4 of 14 patients with testicular cancer and 2 with ovarian cancer showed partial remission. In addition, the target lesions were reduced in size. One patient had no change compared to pretreatment, and two patients showed progression. The objective remission rate was 43%, and the disease control rate was 86% (NCT04503278).
CAR targeting macrophages rather than T cells has emerged as another meaningful strategy for the treatment of solid tumors. MT-101 is a new class of non-T cell CARs produced by transforming monocytes with mRNAs. Most monocytes in blood differentiate into macrophages after migrating to tissues. MT-101 targets tumor cells in peripheral tissues by expressing CARs targeting CD5 on the surface of monocytes. A phase 1/2 clinical study demonstrated the safety and tolerability of MT-101 in patients with refractory or relapsed peripheral T-cell lymphoma (PTCL) at day 28, with no dose-limiting toxicity, cytokine release syndrome (CRS), or immune effector cell-associated neurotoxicity syndrome (ICANS) (NCT05138458). In 2020, Moderna and Carisma Therapies established a cooperative relationship to combine Carisma's engineered macrophage technology with Moderna's mRNA and LNP technology and launched the first clinical study of their so-called CAR-M therapy (CT-0508). The combination of CT-0508 with an anti-PD-1 antibody (pembrolizumab) was used in the phase 1 clinical development stage to treat solid tumors with HER-2 overexpression.
siRNA
siRNAs are double-stranded RNA molecules of 21 to 23 nucleotides, typically with two free bases at the 3'-end, that silence target genes by RNA interference. The precursor is recognized by Dicer RNase and subsequently binds to the target mRNA via the RISC and cleaves it at bases 10 to 11 from the 5'-end, resulting in post-transcriptional gene silencing [190] (Figure 5). Unlike other RNAi technologies, each siRNA can bind to only one mRNA target. Owing to its well-tolerated nature and few side effects, siRNAs have been used to treat various tumors in rodent models [191, 192]. However, the development of siRNA-based tumor therapies is hampered by the selection of a suitable targeted delivery method with few systemic side effects.
Schematic of the mechanisms of siRNA, miRNA and ASO. (A) siRNA inhibits the expression of target genes through Dicer and the RISC, leading to mRNA breakdown and avoiding the expression of the corresponding proteins. (B) The pri-miRNA is processed twice to form a mature miRNA. miRNA complementarily binds the 3' UTR of the target gene during transcription or translation and then directly cut the mRNA or directly inhibit the translation process. (C) ASO specifically binds to the target mRNA, forming a DNA-RNA hybrid that then triggers mRNA cleavage via RNase H recruitment, ultimately leading to the mRNA level reduction.
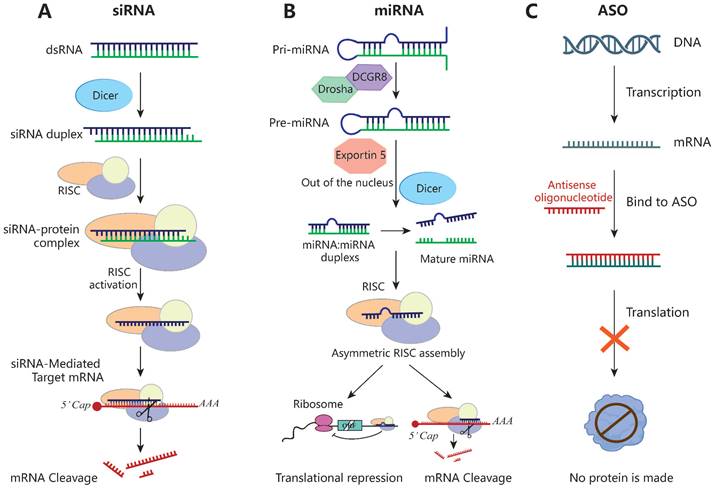
siRNAs can be used in combination with nanomaterials. Currently, various lipid-based delivery systems have been used for co-delivering siRNA and drugs (Table 2). Cationic liposomes protect the siRNA cargo from enzymatic digestion and prevent its renal clearance. Guo et al. [193] used 50 nm cationic lipid-polymer hybrid NPs (LPHs) packed with siRNA in combination with microbubble-enhanced focused ultrasound (MB-FUS) to enhance siRNA delivery in the preclinical brain TME in children and adults by more than 10-fold. In a smoothened (SMO)-activated medulloblastoma model, MB-FUS delivery of SMO-targeted siRNA significantly reduced the production of SMO protein and promoted tumor-cell death [193]. In addition, the combination of As2O3 and HER2-siRNA shows an excellent antitumor effect in an orthotopic gastric tumor model. As2O3 induces apoptosis and suppresses tumor metastasis, and HER2-siRNA blocks the expression of the oncogene HER2, inhibiting tumor invasion and metastasis [194]. The cRGD peptide-modified nanocarrier enabled pH-triggered drug release—the pH-sensitive shell rapidly dissolved in the acidic lysosome, enabling efficient lysosomal escape and release of siRNA to achieve efficient gene silencing [195, 196]. A cationic amphiphile containing an endosomal pH-sensitive imidazole ring can be used to deliver both paclitaxel and a Bcl-2 siRNA, significantly inhibiting cellular proliferation and reducing tumor growth [197].
Nanoplatform for delivery of siRNA in anti-tumor therapy
| Types | Nanocarriers | Function of the nanocarriers | Targeted gene | Treating diseases | Injection methods | Ref |
|---|---|---|---|---|---|---|
| Targeting | Cationic lipid nanoparticles | Enhance tumor-targeted delivery | CD47 | Triple negative breast cancer; Melanoma | Intravenous injection | [210] |
| Apolipoprotein E3-reconstituted high-density lipoprotein with a CaP | Enhance the Ras-activated cancer cells to swallow drugs | ATF5 | Glioblastoma | Intravenous injection | [281] | |
| Hybrid nanocomplex | Target CD44 on TNBC cells; higher cellular uptake and better tumor penetration of the encapsulated cargos | IKBKE | Triple-negative breast cancer | Peritumoral injection | [198] | |
| Tumor-targeted lipid-dendrimer-calcium-phosphate nanoparticles | Enhanced gene delivery capacity and immune adjuvant properties by activating the STING-cGAS pathway | PD-1 | Hepatocellular carcinoma | Intravenous injection | [200] | |
| Nanoliposomes | Delivery to target cells and affect tumor cells and infiltrating lymphocytes | PD-1 | Melanoma | Intravenous injection | [203] | |
| Stimulus responsiveness | CaP-phospholipid complex nano delivery system | PH sensitivity; Protects siRNA by endogenous RNases | As2O3; HER2 | Gastric cancer | Intravenous injection | [194] |
| Liposomal nanocarrier | PH-sensitive | Bcl-2 | Melanoma | Intravenous injection | [197] | |
| Nucleic acid nanogel | Kill tumor cells photodynamically and induce remarkable immunogenic cell death | PD-1 | Melanoma | Intravenous injection | [208] | |
| Photolabile spherical nucleic acid | NIR-sensitive, designable and biocompatible merits | HIF-1α; Bcl-2 | Cervical cancer | Intravenous injection | [282] | |
| Surface chemistry | Lipidoid nanoparticles | Facilitate interaction with the cell membrane; Endosomal escape in other tissues | HoxA1 | Premalignant breast lesion | Direct nipple injection | [192] |
| Cationic nanoparticles | Prolong RNA circulation and augment cell uptake | SMO | Glioma; medulloblastoma | Intravenous injection | [193] | |
| Solid lipid nanoparticles | Engineered with lecithin and cholesterol and were surface-modified with acid-sensitive sheddable PEG | PD-1 | Melanoma | Intratumoral injection | [206] | |
| Stable nucleic acid-lipid nanoparticle | Enable high encapsulation Efficiency of nucleic acids with improved cellular uptake and subsequent release | CD47 | Colon cancer | Intravenous injection | [212] | |
| Neutral nano-liposomal carrier | Process a high rate of in vivo tumor reducing capability | mTOR | Mammary carcinogenesis | Intravenous injection | [207] |
Suitable modification of nanocarriers enables siRNA delivery to target tissues for cancer treatment. One study introduced a versatile codelivery platform for the treatment of triple-negative breast cancer [198]. The nanocomplex was modified with hyaluronic acid to specifically target CD44 on TNBC cells. The codelivery of cabazitaxel (a microtubule stabilizer) and IKBKE siRNA (a TNBC oncogene) showed high tumor accumulation and antitumor activity in an orthotopic TNBC mouse model. In addition, integrative hybrid nanoplexes (EhCv/siRNA NPs) prepared from endoplasmic reticulum membranes isolated from cancer cells transported an siRNA via the endosome-Golgi-ER pathway. This method avoids lysosomal degradation and enhances siRNA silencing and antitumor activity against MCF-7 human breast cancer cells in nude mice [199]. An NP for dual-targeted immune therapy with a tumor-targeting peptide (SP94) enhances the tumor accumulation of NPs and the intracellular delivery of the therapeutic pDNA/siRNA to HCC cells [200].
Silencing key elements of tumor progression or downregulating immunosuppressive genes can induce an antitumor immune response [201]. siRNA-based nanotherapeutics targeting tumor cells downregulate immune-checkpoint proteins (e.g., PD-L1), so-called “don't-eat-me” signals (e.g., CD47), and anti-inflammatory cytokines to induce an antitumor immune response [202]. Programmed cell death protein-1 (PD-1) is an immune checkpoint molecule that impairs T-cell activity and induces T-cell depletion. Injection of liposomal nanoparticles loaded with PD-1 siRNA into B16F10 tumor-bearing mice enhances the T cell-mediated antitumor immune response and improves survival [203]. PD-1 is also expressed by B cells, macrophages, and NK cells [204]. Tumor-associated macrophages (TAMs) overexpressing PD-1 inhibit the phagocytosis by TAMs of PD-L1-expressing tumor cells [205]. Hanafy et al. [206] reported that PD-1 siRNA encapsulated in SLNs downregulated PD-1 expression in TAMs and mouse tumor tissues and inhibited tumor growth in a mouse model. In another study, delivery by neutral nanoliposomes of mTOR-siRNA to rats with breast cancer enhanced antitumor efficacy by silencing the oncogenic gene mTOR and promoting apoptosis [207].
Research is now focusing on combination therapy or codelivery via nanomaterials with siRNA. Guo et al. [208] grafted the photosensitizer pheophorbide A (PPA) onto the DNA backbone at the phosphorothioate modification site and to a PD-1 siRNA linker via supramolecular self-assembly to form a siRNA and PPA co-packaged nanogel. The nanogel photodynamically killed tumor cells, and inhibited PD-L1 expression in tumor cells, thereby synergistically increasing the antitumor immune response. Tumor-targeted lipid-dendrimer-calcium-phosphate NPs with thymosin-functional dendritic polymers have been used to deliver PD-L1 siRNA and immunostimulatory IL-2 encoding plasmid DNA to HCC. They increase tumor infiltration and CD8+ T-cell activation, enhancing the effectiveness of cancer immunotherapy and inhibiting HCC progression [200]. Selective targeting and reshaping of the immunosuppressive TME by dual delivery of siRNA and plasmid DNA has been attempted to improve cancer immunotherapy [200]. In another study, an in situ-injectable chitosan hydrogel containing C-C chemokine ligand 5 (CCL5) siRNA-loaded NPs, together with mRNA encoding lipid-immune regulatory factor 5 (IRF5), reshaped the TME in a model of pancreatic cancer by promoting M1 macrophage polarization [209]. Delivery of JQ1 (indirect inhibitor of MYC) and a CD47 siRNA in cationic lipid NPs significantly inhibits the expression of PD-L1 and CD47, significantly improving therapeutic efficacy in a mouse model of triple-negative breast cancer [210].
Combination therapy with siRNA and chemotherapeutic drugs has shown promise for some types of cancer. Chen et al. [211] reported that chemotherapeutic drugs induce cancer cells to overexpress Xkr8 (a scrambling enzyme activated during apoptosis) at the transcriptional level in vitro and in vivo. Coadministration of nanocarriers loaded with Xkr8 siRNA and the FuOXP prodrug into tumors via intravenous injection significantly inhibited tumor growth in colon and pancreatic cancer models and enhanced antitumor immune responses. In addition, doxorubicin (Dox) induces ICD in tumors, a type of apoptosis that enhances the protective immune response. Stable nucleic acid-lipid particles (SNALPs) have been used for the simultaneous delivery of Dox and an siRNA that knocks down CD47 (siCD47) [212], the combination therapy synergistically enhances ICD and shows potent antitumor activity [212]. Codelivery of Dox and an siRNA using a hydrogel containing an enzyme-cleavable peptide motif overcomes drug resistance by enabling controlled spatiotemporal release [213].
ASOs
ASOs are synthetic single-strand nucleic acids of 12 to 30 nucleotides. Upon entering a cell, they specifically bind to the target mRNA, forming a DNA-RNA hybrid that triggers mRNA cleavage via recruitment of RNase H, reducing the mRNA level [214]. The single strand of ASOs enables targeting and specificity via binding interactions (Figure 5). Because ASOs are based on nucleotide sequences, they can be used to develop protein-associated inhibitors suitable for use in conjunction with traditional therapeutic approaches. Furthermore, they can be chemically modified to improve their stability and resistance to DNases [215]. In addition to the 2'-MOE modification, next-generation ASOs have a phosphorothioate (PS) backbone and 2'-4' constrained ethyl (cEt) chemistry at both ends [216], which improve their effectiveness.
Because conventional methods cannot downregulate the protein level of STAT3, Hong et al. used a 2.5-generation cEt-ASO (AZD9150) to suppress STAT3 expression, resulting in antitumor activity in lymphoma and lung cancer models [217]. In a preclinical study, AZD4785 [218], a high-affinity cEt-ASO targeting KRAS mRNA, selectively depleted KRAS mRNA and protein, selectively inhibited the downstream pathways, and suppressed the proliferation of KRAS-mutant cells. AZD4785 targeted KRAS in tumors and showed antitumor activity in mouse and primate models [218].
The limited cell membrane permeability and lack of nuclear targeting of ASOs hinder their clinical application. To address these issues, Cheng et al. [219] implemented a gapmer-based design for ASOs by adding 2'-O-methyl modifications with PS linkages, which protected ASOs from nuclease degradation and enhanced their RNase H-mediated cleavage. ASOs targeting both Bcl-2 and Akt-1 were loaded onto lipid NPs to increase their stability. Several studies have evaluated ASO-conjugated nanocarriers for cancer treatment. For instance, the T7 peptide, which has high affinity for the transferrin receptor, was conjugated to a nanocarrier for specific tumor targeting. An ASO-based gene therapy drug for homozygous familial hypercholesterolemia [220] received O-methyl moieties on the terminal ribose groups. The ASO together with a nucleus-targeting TAT peptide packaged in Au NPs shows promise for controlling cancer metastasis [221]. Codelivery of ASOs with siRNAs or other drugs may have a synergistic effect. Codelivery siRNA and ASO responds to NIR light [222]. Upon NIR light irradiation, the oxygen-cleavable linker between the siRNA and pASO promotes the lysosomal escape of the siRNA and pASO. A multifunctional DNA origami-based nanocarrier for codelivery of doxorubicin and dual-targeted ASOs significantly silences Bcl2 and P-gp and induces apoptosis, enhancing therapeutic effectiveness [130].
miRNAs
miRNAs are single-stranded, non-coding RNAs typically of 20 to 24 nucleotides. They complementarily bind the 3' UTR of the target mRNA, and then cleave the mRNA or inhibit its translation [223, 224] (Figure 5). miRNAs regulate gene expression via silencing, upregulation, translation activation, or post-transcriptionally. They modulate tumor progression by affecting the interactions between tumor cells and immune cells. miRNA-based therapeutics can restore tumor-suppressor miRNA levels (miRNA mimics and other small-molecule drugs) or block oncomiR function (locked nucleic acids or miRNA sponges) [225]. However, naked miRNA mimics are unstable and easily degraded by nucleases, thus requiring a suitable delivery system.
The first clinical miRNA-based therapeutic was the liposomal formulation of miR-34a, named MRX34 [226]. miR-34a is a tumor-suppressor miRNA that is dysregulated by p53. In phase 1 studies, delivery of miR-34a using liposome restored a normal tumor-suppressor pathway, inducing apoptosis in tumor cells in vitro and in mouse models of cancer. In addition, mice treated with MRX34 exhibited significant tumor growth inhibition, and a significantly increased survival rate. However, liposomal delivery systems have issues with liver and kidney accumulation and acute hypersensitivity reactions.
Inorganic NPs are superior to liposomes in terms of their adjustable size and superior pharmacokinetics. For example, a lipid-coated calcium phosphonate NP has been designed for macrophage-targeted miRNA delivery in tumor immunotherapy [227]. Mannose conjugation and a pH-responsive steric shielding enable this nanocarrier to reach macrophages and release miRNA-155 in the acidic TME, reactivating protumor TAMs to antitumor macrophages, thereby inhibiting tumor growth without marked off-target effects. Similar TAM-targeting strategies have been used in a non-small cell lung cancer mouse model [228] and epithelial ovarian cancer model [229]. HA-based polymeric NPs have been modified to deliver miRNA-125b to CD44 macrophages. The augmented expression of miR-125b markedly inhibits primary tumor growth and repolarizes TAMs [228], suggestive of therapeutic potential.
Vesicular exosomes could be used for miRNA delivery. Exosomes, the smallest extracellular vesicles (EVs) (40 to 160 nm), can transport miRNAs in a paracrine and endocrine manner [230]. miRNA-containing exosomes are taken up via receptor-ligand interaction and subsequently regulate gene expression in recipient cells [231]. Exosomes can overcome the limitations of liposomes, such as their membrane toxicity and low biocompatibility with target ligands. Their small size promotes the penetration of, and retention in, solid tumors, suggesting potential for tumor immunotherapy. EVs from patients with melanoma can prevent tumor relapse by downregulating β-catenin and blocking tumor-cell proliferation in an miR-34a-dependent manner [232]. In hepatoma cells, insulin-like growth factor 1 (IGF1) secretion prevents the intercellular exosomal transfer of miR-122, thus promoting the proliferation of neighboring cells by suppressing the expression of miR-122 [233]. These findings suggest a link between the loss of tumor-suppressor miRNAs in cancer and exosome secretion.
saRNAs
Small activating RNAs (saRNAs) are small dsRNAs of approximately 21 nucleotides. After generating active Ago-RNA complexes, they trigger gene expression activation by targeting promoter regions. In addition, they have an effect in any genomic region with antisense transcripts [234, 235]. SaRNAs have received much attention in the field of cancer therapy, as they can enhance the transcriptional activation of tumor-suppressor genes such as p21 [236, 237], Wt-1 [238], E-cadherin [239], NKX3-1 [240], and PTEN [241] through various mechanisms [236, 242, 243], by inducing cell-cycle arrest, inhibiting proliferation, inducing apoptosis, inhibiting metastasis, and reversing multidrug resistance [236, 242, 243]. Chemical modification could overcome the endonuclease resistance, serum stability, and off-target effects of saRNAs and nano-delivery systems.
Lipid-based delivery systems are commonly used for systemic or local saRNA delivery. Take target gene P21 as example, results illustrated that using targeting saRNA transcriptionally activated p21 expression and promoted cell-cycle arrest in the G1/G0 phase, inhibiting cell proliferation and enhancing chemosensitivity [236]. Local administration of saRNAs has minimal off-target potential. Regarding the therapeutic potential of saRNAs targeting p21, intravesicular delivery of LNP/dsP21-322 results in approximately 40% tumor regression and prolongs survival in a mouse orthotopic model of bladder cancer [244].
Surface modification, for example, with biodegradable polymers such as PEG, can overcome the toxicity and off-target effects of LNP delivery systems [244]. Indeed, 2'-fluoro modification of the saRNA backbone improves duplex stability in urine [244]. To further increase tumor tissue specificity, a selective rectal delivery system has been designed for colorectal cancer. In this system, saRNA-322 is loaded onto an HA covalently anchored anionic lipid shell to accumulate at the lesion site and effectively target CD44-rich tumor cells [245]. In another study, to achieve targeted delivery, pancreatic ductal adenocarcinoma (PDAC)-specific adapters have been used to deliver 2'‑fluoropyrimidine-modified saRNA to tumor cells to activate C/EBPα expression [246]. Due to their ability to maintain their structural conformation even under physiologically harsh reducing conditions, aptamers exhibit better structural stability, lower toxicity, and lower immunogenicity. Compared to lipid-mediated delivery, high-affinity aptamers better deliver saRNA to tumor nodules, markedly inhibiting tumor growth and significantly reducing the tumor burden in a xenograft model [246].
RNA aptamers
RNA aptamers are sequences 20-100 nucleotides in length with complex three-dimensional structures that bind to target molecules with high affinity and specificity via non-covalent pocket interactions [247]. Aptamers have high structural flexibility, stability, and specificity. In addition, targeted ligands can confer cancer tissue specificity to antitumor drugs [248, 249]. RNA aptamers are classified into three functional categories: antagonists that block the interactions of disease-related targets, agonists that activate target receptor function, and those that target moieties that deliver drugs to cancer tissue [250].
Several clinical trials of aptamers for tumor therapy are ongoing. The guanosine-rich AS1411 is the first oligodeoxynucleotide aptamer to be evaluated in phase I and II clinical trials. It suppresses tumor-cell proliferation by interfering with the stability of Bcl-2 mRNA [251, 252]. NOX-A12 is an L-type RNA antagonistic aptamer known as Spiegelmer® that targets chemokine (C-X-C motif) ligand 12 (CXCL-12) [253], mobilizes cells from the protective TME, and induces apoptosis and chemosensitization to have an antitumor effect [254]. NOX-A12, a mirror-image oligonucleotide with 40 kDa PEG branches, resists nuclease degradation and is immunologically passive [254, 255].
RNA aptamers are used in combination with other therapeutic agents (e.g., siRNAs, microRNAs, and peptides) for targeted delivery. For example, the combination of a PSMA-specific aptamer for prostate cancer with therapeutic oligonucleotides inhibited oncogene activity in PSMA-expressing cells, thereby having an antitumor effect [256]. Combining this aptamer with other functional units can achieve multiple biological effects [250, 257, 258]. Similarly, in one study, linking a FOXP3-blocking peptide to an aptamer targeting CD28 functionally inactivated Tregs and enhanced the efficacy of cancer immunotherapy [259].
CRISPR-Cas9 system
The clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system has unprecedented therapeutic potential for genetic diseases [16]. This adaptive and heritable immune defense system was discovered in bacteria and archaea, and uses short RNAs to guide the degradation of invading viruses, plasmids, or foreign mobile genetic elements [260]. CRISPR/Cas9 has progressed since the first report in 2013 of its use for simultaneous precise editing of several sites in the mammalian genome [16, 261]. The 2020 Nobel Prize in chemistry was awarded for the discovery of CRISPR RNA (crRNA) and transactivating-crRNA (tracrRNA) [262]. The interaction between these two elements forms a two-RNA guide RNA (gRNA), which directs the Cas9 nuclease to the target site (Figure 6).
Schematic diagram of CRISPR-Cas9 technology for gene editing. Cas9 can specifically target any genomic locus and induce double-strand breaks under the guidance of gRNA. Cells then initiate the repair mechanism by non-homologous end joining (NHEJ) or homologous directed repair (HDR).
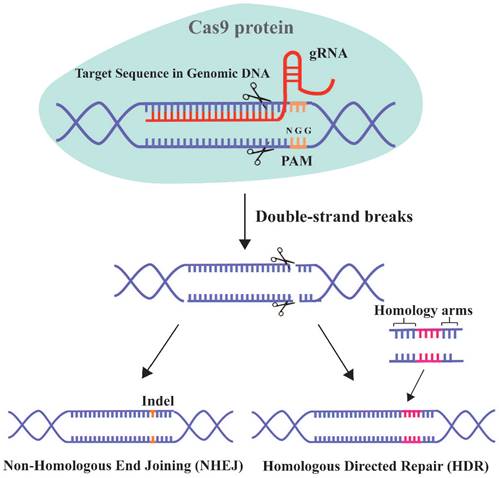
The two critical components of the CRISPR/Cas9 system are the Cas9 protein and gRNA. The principle of this system involves the insertion of a specific DNA sequence (spacer) from the invading virus and plasmid into the CRISPR locus. Upon re-infection, the CRISPR sites containing the spacer acquired previously are transcribed and the products are processed into mature gRNAs. The gRNA targets Cas9 to a particular genomic locus, where it induces double-strand breaks (DSBs). For binding to and cleavage of DNA by Cas9, the 3'-side of the target sequence must have an NGG protospacer adjacent motif (PAM) [263]. The DSB triggers DNA repair by non-homologous end joining (NHEJ) or homologous directed repair (HDR) [264].
Cancer arises from the accumulation of genetic/epigenetic abnormalities. CRISPR-Cas9-mediated genome editing enables precise manipulation of a genomic sequence, enabling the identification of genes involved in carcinogenesis and correction of oncogenic mutations [265, 266]. In addition, CRISPR-Cas9 gene editing allows permanent disruption of genes that are essential for tumor survival, potentially circumventing the need for repeated dosing of chemotherapeutics and thereby improving treatment outcomes [267, 268]. To selectively kill cancer cells without affecting surrounding normal cells, Kwon et al. [269] developed a cancer-specific insertions-deletions (InDels) attacker (CINDELA), which targets cancer-specific CRISPR-mediated DSBs to promote cell death. Notably, CINDELA with CRISPR/Cas9 targets multiple InDels, generating many DNA DSBs. The CINDELA method has been used to kill cancer cells, xenograft cancer cells in mice, patient-derived glioblastomas, and patient-driven xenograft (PDX) lung cancer models without affecting normal cells [269].
However, the CRISPR/Cas9 system has limitations related to cell injury, limited packaging capacity, and immune activation. In addition, the large sizes of Cas9 (160 kDa, 4300 bases) and sgRNA (~31 kDa, 130 bases) preclude the use of viral and nonviral delivery systems [267]. The CRISPR-Cas9 gene editing system can be delivered intracellularly using arginine NPs (ArgNPs) to generate SIRP-α knockout macrophages [270]. The technique enables tumor penetration and codelivery of the single gRNA and Cas9 to knockdown the “don't-eat-me” signal in macrophages, which prevents engulfment of cancer cells. The technique enhances the phagocytic capacity of macrophages fourfold [270]. To address the low editing efficiency and high toxicity of CRISPR-Cas9, Rosenblum et al. [267] used lipid NPs to deliver Cas9 mRNAs and sgRNAs. A single intracerebral injection of CRISPR-LNPs for PLK1 (sgPLK1-cLNPs) into an aggressive orthotopic glioblastoma resulted in ~70% gene editing in vivo, promoting tumor-cell apoptosis. In addition, the technique allows antibody-targeted delivery for the treatment of diffuse tumors. Intraperitoneal injection of EGFR-targeted sgPLK1-cLNPs resulted in selective uptake into diffuse ovarian tumors, leading to up to ~80% gene editing in vivo, inhibiting tumor growth and improving survival [267]. A CRISPR/Cas9 codelivery strategy has been used for the treatment of immunological diseases and cancers. In one study, MSNs were encapsulated in lipid layers to form virus-like nanoparticles (VLNs), which protected the RNP complex from enzymatic degradation and prolonged their half-life in the circulation [271]. Simultaneous delivery of an RNP complex targeting the PD-L1 gene and the antitumor drug axitinib achieved PD-L1 knockout in cancer cells, significantly reduced immunosuppressive Tregs, and enhanced tumor growth inhibition [271]. Zhang et al. [272] developed a gene-drug combination that targeted EGFR by specifically inhibiting CRISPR/Cas9 and Sora. Similarly, gene-drug coloaded NPs have been used to stimulate the antitumorigenic pathway in hepatoma carcinoma by inhibiting pro-inflammatory cytokines (IL-6 and IL-8) by regulating the downstream tumorigenic pathway (NF-κB p65) [273]. Stimulus-responsive nano systems using NIR-responsive and reducing agent-responsive NPs for codelivery of the Cas9/sgRNA RNP and the antitumor photosensitizer chlorin e6 (ce6) result in the generation of reactive oxygen species (ROS) upon NIR irradiation, facilitating the release of Cas9/sgRNA targeting Nrf2 and enhancing tumor-cell sensitivity to ROS [274]. The therapeutic potential of NIR light-triggered systems has been verified by others [275, 276]. Although they target different oncogenes, these platforms specifically inhibit the proliferation of cancer cells.
Perspectives, Challenges, and Conclusion
Although cell and antibody-based therapies currently dominate tumor immunotherapy, more work is needed to overcome the risk for side effects, induction of an inappropriate and potentially harmful immune response, the complexity of treatment, and the high cost of production. RNA-based therapeutics (mRNAs, siRNAs, miRNAs, ASOs, saRNAs, RNA aptamers, and CRISPR gene editing) can modulate the expression of target genes to varying degrees. In addition, they can stimulate an immune response or reshape the suppressive TME by producing antigens or restoring the levels of beneficial proteins. The ability to target multiple genetic components is an advantage of RNA therapeutics over other small-molecule or protein-based drugs. Furthermore, once the chemical structure of the RNA molecule and the in vivo delivery system have been developed, RNA-based drugs can be rapidly designed and synthesized. However, several technical bottlenecks affect RNA-based antitumor therapies, including their targeting specificity, safety, and efficacy. To address these issues, a variety of optimization and modification strategies for RNA molecules and delivery systems have been explored.
Therapeutic effectiveness is influenced by off-target effects induced by nonspecific accumulation. To prolong accumulation at tumor sites, promote target uptake, and control drug release, surface modifications with targeting ligands have been developed [277]. Target ligand length, density, hydrophobicity, and avidity are determinants of the efficacy of such surface modifications [278]. Furthermore, optimization of the particle size, surface charge, and other properties of delivery vehicles would promote selective accumulation of cargo in target organs or tissues. Strategies based on microenvironment-specific targeting release are feasible, including on-demand delivery of CRISPR-Cas9 for precise genome editing. Tumor heterogeneity results in significant variability in the responses—including nonspecific responses—to internal stimuli. The design of next-generation RNA delivery systems should focus not only on reducing the potential toxicity of byproducts but also on overcoming the problems of irreversibility and weak selectivity. Delivery systems will gradually shift from passively responding to actively modulating via several mechanisms, such as regulating tumor-associated immune cells, enhancing tumor cell immunogenicity, and blocking tumor immune-escape mechanisms.
The safety of RNA cargos is a key consideration for their clinical application. The innate immunostimulatory properties of RNA molecules and their function as immune adjuvants enhance the immune response to antigens during vaccine development. However, excessive immunogenicity can lead to severe adverse reactions. Clinical translation requires the striking of a balance between safety and efficacy. The development of more biocompatible delivery vehicles with better biodegradability would accelerate the clinical translation of nanomedicines [279]. Novel carriers such as exosomes, bacteriophages, and macrophages could be used to deliver therapeutics such as siRNAs, ASOs, antibodies, and small-molecule drugs. These have considerable potential for gene therapy based on their transport characteristics, prolonged half-life in the circulation, and excellent biocompatibility. Because the components of RNA-based therapeutics are from different cell sources and have different biological functions [280], their safety needs to be systematically evaluated.
Another crucial issue is therapeutic effectiveness. Tumorigenesis involves complex regulatory networks and immune-escape mechanisms. Treating tumors with a single modality is often unsatisfactory. Treatment of tumors requires multiple modalities, potentially including codelivery of cytokines and chemokines, alleviating immunosuppressive signaling in the TME, and in vivo targeting of immune cells. Such approaches could enhance the effectiveness of immunotherapy and so reduce the RNA-based drug dose required. Because drugs have different pharmacokinetic and pharmacological characteristics, as well as interindividual differences in drug distribution after systemic administration, multiple-drug treatment strategies must consider the optimal dosage ratio and sequential release to reduce toxicity and enhance efficacy. P-gp inhibitors should be released by delivery systems prior to any co-delivered drugs. Otherwise, the co-delivered drug(s) might be exported extracellularly by P-gp, thus reducing their therapeutic efficacy.
In conclusion, we reviewed the modifications of, delivery systems for, and potential applications of RNA-based therapeutics for cancers. Although RNA delivery platforms have limitations, advances in RNA-based bioengineering mean considerable therapeutic potential for cancer treatment. Indeed, these modalities offer hope for types of cancers with few or no treatment options.
Acknowledgements
We thank Y. Yang for comments and suggestions on the manuscript. This work was supported by research grants from the National Key Research and Development Program of China (2021YFE0114900), the National Natural Science Foundation of China (91940303), the Natural Science Foundation of Zhejiang Province (LD21C050002), the Starry Night Science Fund at Shanghai Institute for Advanced Study of Zhejiang University (SN-ZJU-SIAS-009), the Science Technology Department of Zhejiang Province (2023C03063). Some images in this paper are created with BioRender.com.
Authors Contributions
J.F., H.D., J.W., Y.J. conceived and co-wrote the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fang E, Liu X, Li M. et al. Advances in COVID-19 mRNA vaccine development. Signal transduction and targeted therapy. 2022;7:94
2. Hill SF, Meisler MH. Antisense Oligonucleotide Therapy for Neurodevelopmental Disorders. Dev Neurosci. 2021;43:247-252
3. Xu S, Yang K, Li R. et al. mRNA Vaccine Era-Mechanisms, Drug Platform and Clinical Prospection. International journal of molecular sciences. 2020 21
4. He B, Zhao Z, Cai Q. et al. miRNA-based biomarkers, therapies, and resistance in Cancer. International journal of biological sciences. 2020;16:2628-2647
5. Gavrilov K, Saltzman WM. Therapeutic siRNA: principles, challenges, and strategies. The Yale journal of biology and medicine. 2012;85:187-200
6. Yang R, Yu S, Xu T. et al. Emerging role of RNA sensors in tumor microenvironment and immunotherapy. Journal of hematology & oncology. 2022;15:43
7. Brenner S, Jacob F, Meselson M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature. 1961;190:576-581
8. Muthukrishnan S, Both GW, Furuichi Y. et al. 5'-Terminal 7-methylguanosine in eukaryotic mRNA is required for translation. Nature. 1975;255:33-37
9. Dimitriadis GJ. Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature. 1978;274:923-924
10. Malone RW, Felgner PL, Verma IM. Cationic liposome-mediated RNA transfection. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:6077-6081
11. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843-854
12. Conry RM, LoBuglio AF, Wright M. et al. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995;55:1397-1400
13. Fire A, Xu S, Montgomery MK. et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806-811
14. Weide B, Pascolo S, Scheel B. et al. Direct Injection of Protamine-protected mRNA: Results of a Phase 1/2 Vaccination Trial in Metastatic Melanoma Patients. 2009; 32: 498-507.
15. Weide B, Pascolo S, Scheel B. et al. Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. J Immunother. 2009;32:498-507
16. Cong L, Ran FA, Cox D. et al. Multiplex genome engineering using CRISPR/Cas systems. Science (New York, NY). 2013;339:819-823
17. Guo Y, Lei K, Tang L. Neoantigen Vaccine Delivery for Personalized Anticancer Immunotherapy. Frontiers in immunology. 2018;9:1499
18. Adams D, Gonzalez-Duarte A, O'Riordan WD. et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. The New England journal of medicine. 2018;379:11-21
19. Reinhard K, Rengstl B, Oehm P. et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science (New York, NY). 2020;367:446-453
20. Rosenblum D, Joshi N, Tao W. et al. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9:1410
21. Roundtree IA, Evans ME, Pan T. et al. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187-1200
22. Jung HN, Lee SY, Lee S. et al. Lipid nanoparticles for delivery of RNA therapeutics: Current status and the role of in vivo imaging. Theranostics. 2022;12:7509-7531
23. Alfagih IM, Aldosari B, AlQuadeib B. et al. Nanoparticles as Adjuvants and Nanodelivery Systems for mRNA-Based Vaccines. Pharmaceutics. 2020 13
24. Qin S, Tang X, Chen Y. et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal transduction and targeted therapy. 2022;7:166
25. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406-3415
26. Lorenz R, Bernhart SH, Höner Zu Siederdissen C. et al. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26
27. Pelletier J, Sonenberg N. Insertion mutagenesis to increase secondary structure within the 5' noncoding region of a eukaryotic mRNA reduces translational efficiency. Cell. 1985;40:515-526
28. Pickering BM, Willis AE. The implications of structured 5' untranslated regions on translation and disease. Semin Cell Dev Biol. 2005;16:39-47
29. Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15:423-437
30. Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283-292
31. Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol. 1987;196:947-950
32. Jia L, Mao Y, Ji Q. et al. Decoding mRNA translatability and stability from the 5' UTR. Nat Struct Mol Biol. 2020;27:814-821
33. Manzella JM, Blackshear PJ. Regulation of rat ornithine decarboxylase mRNA translation by its 5'-untranslated region. J Biol Chem. 1990;265:11817-11822
34. Fernandes LR, Costa EC, Vargas FR. et al. Influence of estrogen and variations at the BRCA1 promoter region on transcription and translation. Mol Biol Rep. 2014;41:489-495
35. Moore MJ. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514-1518
36. Schwanhäusser B, Busse D, Li N. et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337-342
37. Weissman D. mRNA transcript therapy. Expert Rev Vaccines. 2015;14:265-281
38. Holtkamp S, Kreiter S, Selmi A. et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108:4009-4017
39. Ramanathan A, Robb GB, Chan SH. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016;44:7511-7526
40. Ghosh A, Lima CD. Enzymology of RNA cap synthesis. Wiley Interdiscip Rev RNA. 2010;1:152-172
41. Decroly E, Ferron F, Lescar J. et al. Conventional and unconventional mechanisms for capping viral mRNA. Nat Rev Microbiol. 2011;10:51-65
42. Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 2012;86:2900-2910
43. Grudzien-Nogalska E, Kowalska J, Su W. et al. Synthetic mRNAs with superior translation and stability properties. Methods in molecular biology (Clifton, NJ). 2013;969:55-72
44. Muttach F, Muthmann N, Rentmeister A. Synthetic mRNA capping. Beilstein journal of organic chemistry. 2017;13:2819-2832
45. Shanmugasundaram M, Senthilvelan A, Kore AR. Recent Advances in Modified Cap Analogs: Synthesis, Biochemical Properties, and mRNA Based Vaccines. Chemical record (New York, NY). 2022;22:e202200005
46. Henderson JM, Ujita A, Hill E. et al. Cap 1 Messenger RNA Synthesis with Co-transcriptional CleanCap(®) Analog by In Vitro Transcription. Current protocols. 2021;1:e39
47. Whitelaw E, Coates A, Proudfoot NJ. Globin gene transcripts can utilize histone gene 3' end processing signals. Nucleic Acids Res. 1986;14:7059-7070
48. Karikó K, Muramatsu H, Welsh FA. et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008;16:1833-1840
49. Karikó K, Buckstein M, Ni H. et al. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165-175
50. Mauer J, Luo X, Blanjoie A. et al. Reversible methylation of m(6)A(m) in the 5' cap controls mRNA stability. Nature. 2017;541:371-375
51. Carlile TM, Rojas-Duran MF, Zinshteyn B. et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143-146
52. Gilbert WV, Bell TA, Schaening C. Messenger RNA modifications: Form, distribution, and function. Science (New York, NY). 2016;352:1408-1412
53. Arango D, Sturgill D, Alhusaini N. et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell. 2018;175:1872-1886.e1824
54. Nance KD, Meier JL. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Cent Sci. 2021;7:748-756
55. Baker N, Dolgin E. Coronapod: CureVac disappoints in COVID vaccine trial. Nature. 2021
56. Dong Y, Siegwart DJ, Anderson DG. Strategies, design, and chemistry in siRNA delivery systems. Adv Drug Deliv Rev. 2019;144:133-147
57. Kraynack BA, Baker BF. Small interfering RNAs containing full 2'-O-methylribonucleotide-modified sense strands display Argonaute2/eIF2C2-dependent activity. RNA. 2006;12:163-176
58. Lavergne T, Baraguey C, Dupouy C. et al. Synthesis and preliminary evaluation of pro-RNA 2'-O-masked with biolabile pivaloyloxymethyl groups in an RNA interference assay. The Journal of organic chemistry. 2011;76:5719-5731
59. Cummins LL, Owens SR, Risen LM. et al. Characterization of fully 2'-modified oligoribonucleotide hetero- and homoduplex hybridization and nuclease sensitivity. Nucleic Acids Res. 1995;23:2019-2024
60. Yang X, Sierant M, Janicka M. et al. Gene silencing activity of siRNA molecules containing phosphorodithioate substitutions. ACS Chem Biol. 2012;7:1214-1220
61. Haraszti RA, Roux L, Coles AH. et al. 5΄-Vinylphosphonate improves tissue accumulation and efficacy of conjugated siRNAs in vivo. Nucleic Acids Res. 2017;45:7581-7592
62. Parmar R, Willoughby JL, Liu J. et al. 5'-(E)-Vinylphosphonate: A Stable Phosphate Mimic Can Improve the RNAi Activity of siRNA-GalNAc Conjugates. ChemBioChem. 2016;17:985-989
63. Zhang J, Zheng J, Lu C. et al. Modification of the siRNA passenger strand by 5-nitroindole dramatically reduces its off-target effects. ChemBioChem. 2012;13:1940-1945
64. Hu B, Zhong L, Weng Y. et al. Therapeutic siRNA: state of the art. Signal transduction and targeted therapy. 2020;5:101
65. Wu SY, Chen TM, Gmeiner WH. et al. Development of modified siRNA molecules incorporating 5-fluoro-2'-deoxyuridine residues to enhance cytotoxicity. Nucleic Acids Res. 2013;41:4650-4659
66. Thangamani L, Balasubramanian B, Easwaran M. et al. GalNAc-siRNA conjugates: Prospective tools on the frontier of anti-viral therapeutics. Pharmacol Res. 2021;173:105864
67. Springer AD, Dowdy SF. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic acid therapeutics. 2018;28:109-118
68. Zhao B, Zhou B, Shi K. et al. Sustained and targeted delivery of siRNA/DP7-C nanoparticles from injectable thermosensitive hydrogel for hepatocellular carcinoma therapy. Cancer Sci. 2021;112:2481-2492
69. Bennett CF, Baker BF, Pham N. et al. Pharmacology of Antisense Drugs. Annu Rev Pharmacol Toxicol. 2017;57:81-105
70. Shen W, De Hoyos CL, Sun H. et al. Acute hepatotoxicity of 2' fluoro-modified 5-10-5 gapmer phosphorothioate oligonucleotides in mice correlates with intracellular protein binding and the loss of DBHS proteins. Nucleic Acids Res. 2018;46:2204-2217
71. Rapireddy S, Bahal R, Ly DH. Strand invasion of mixed-sequence, double-helical B-DNA by γ-peptide nucleic acids containing G-clamp nucleobases under physiological conditions. Biochemistry. 2011;50:3913-3918
72. Nielsen PE, Egholm M, Berg RH. et al. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science (New York, NY). 1991;254:1497-1500
73. Lundin P, Johansson H, Guterstam P. et al. Distinct uptake routes of cell-penetrating peptide conjugates. Bioconjug Chem. 2008;19:2535-2542
74. Swenson CS, Heemstra JM. Peptide nucleic acids harness dual information codes in a single molecule. Chemical communications (Cambridge, England). 2020;56:1926-1935
75. Braasch DA, Corey DR. Locked nucleic acid (LNA): fine-tuning the recognition of DNA and RNA. Chem Biol. 2001;8:1-7
76. Kurreck J, Wyszko E, Gillen C. et al. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. 2002;30:1911-1918
77. Ulrich H. RNA aptamers: from basic science towards therapy. Handbook of experimental pharmacology. 2006:305-326
78. Cosmi B. ARC-1779, a PEGylated aptamer antagonist of von Willebrand factor for potential use as an anticoagulant or antithrombotic agent. Current opinion in molecular therapeutics. 2009;11:322-328
79. Winkler J. Therapeutic oligonucleotides with polyethylene glycol modifications. Future medicinal chemistry. 2015;7:1721-1731
80. Lee CH, Lee SH, Kim JH. et al. Pharmacokinetics of a Cholesterol-conjugated Aptamer Against the Hepatitis C Virus (HCV) NS5B Protein. Molecular therapy Nucleic acids. 2015;4:e254
81. Ortigão JF, Rösch H, Selter H. et al. Antisense effect of oligodeoxynucleotides with inverted terminal internucleotidic linkages: a minimal modification protecting against nucleolytic degradation. Antisense research and development. 1992;2:129-146
82. London GM, Mayosi BM, Khati M. Isolation and characterization of 2'-F-RNA aptamers against whole HIV-1 subtype C envelope pseudovirus. Biochem Biophys Res Commun. 2015;456:428-433
83. Gruenke PR, Alam KK, Singh K. et al. 2'-fluoro-modified pyrimidines enhance affinity of RNA oligonucleotides to HIV-1 reverse transcriptase. RNA. 2020;26:1667-1679
84. Thirunavukarasu D, Chen T, Liu Z. et al. Selection of 2'-Fluoro-Modified Aptamers with Optimized Properties. J Am Chem Soc. 2017;139:2892-2895
85. Awachat R, Wagh AA, Aher M. et al. Favorable 2'-substitution in the loop region of a thrombin-binding DNA aptamer. Bioorg Med Chem Lett. 2018;28:1765-1768
86. Jørgensen AS, Hansen LH, Vester B. et al. Improvement of a streptavidin-binding aptamer by LNA- and α-l-LNA-substitutions. Bioorg Med Chem Lett. 2014;24:2273-2277
87. Pal R, Deb I, Sarzynska J. et al. LNA-induced dynamic stability in a therapeutic aptamer: insights from molecular dynamics simulations. J Biomol Struct Dyn. 2023;41:2221-2230
88. Cai B, Yang X, Sun L. et al. Stability and bioactivity of thrombin binding aptamers modified with D-/L-isothymidine in the loop regions. Organic & biomolecular chemistry. 2014;12:8866-8876
89. Li L, Yang X, Li K. et al. d-/l-Isothymidine incorporation in the core sequence of aptamer BC15 enhanced its binding affinity to the hnRNP A1 protein. Organic & biomolecular chemistry. 2018;16:7488-7497
90. Volk DE, Lokesh GLR. Development of Phosphorothioate DNA and DNA Thioaptamers. Biomedicines. 2017 5
91. Wang C, Sun Y, Zhao Q. A sensitive thrombin-linked sandwich immunoassay for protein targets using high affinity phosphorodithioate modified aptamer for thrombin labeling. Talanta. 2020;207:120280
92. Lennox KA, Behlke MA. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011;18:1111-1120
93. Lennox KA, Owczarzy R, Thomas DM. et al. Improved Performance of Anti-miRNA Oligonucleotides Using a Novel Non-Nucleotide Modifier. Molecular therapy Nucleic acids. 2013;2:e117
94. Freier SM, Altmann KH. The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res. 1997;25:4429-4443
95. Davis S, Propp S, Freier SM. et al. Potent inhibition of microRNA in vivo without degradation. Nucleic Acids Res. 2009;37:70-77
96. Gebert LF, Rebhan MA, Crivelli SE. et al. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2014;42:609-621
97. Lennox KA, Vakulskas CA, Behlke MA. Non-nucleotide Modification of Anti-miRNA Oligonucleotides. Methods in molecular biology (Clifton, NJ). 2017;1517:51-69
98. Xiao B, Zhang Z, Viennois E. et al. Combination Therapy for Ulcerative Colitis: Orally Targeted Nanoparticles Prevent Mucosal Damage and Relieve Inflammation. Theranostics. 2016;6:2250-2266
99. Gao X, Yao L, Song Q. et al. The association of autophagy with polyethylenimine-induced cytotoxicity in nephritic and hepatic cell lines. Biomaterials. 2011;32:8613-8625
100. Tan L, Zheng T, Li M. et al. Optimization of an mRNA vaccine assisted with cyclodextrin-polyethyleneimine conjugates. Drug delivery and translational research. 2020;10:678-689
101. Paunovska K, Loughrey D, Dahlman JE. Drug delivery systems for RNA therapeutics. Nat Rev Genet. 2022;23:265-280
102. Kozielski KL, Ruiz-Valls A, Tzeng SY. et al. Cancer-selective nanoparticles for combinatorial siRNA delivery to primary human GBM in vitro and in vivo. Biomaterials. 2019;209:79-87
103. Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. Journal of controlled release: official journal of the Controlled Release Society. 2010;145:182-195
104. Patel S, Ashwanikumar N, Robinson E. et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat Commun. 2020;11:983
105. Fischer D, Li Y, Ahlemeyer B. et al. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials. 2003;24:1121-1131
106. Cullis PR, Hope MJ. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol Ther. 2017;25:1467-1475
107. Packer M, Gyawali D, Yerabolu R. et al. A novel mechanism for the loss of mRNA activity in lipid nanoparticle delivery systems. Nat Commun. 2021;12:6777
108. Ndeupen S, Qin Z, Jacobsen S. et al. The mRNA-LNP platform's lipid nanoparticle component used in preclinical vaccine studies is highly inflammatory. iScience. 2021;24:103479
109. Ho W, Gao M, Li F. et al. Next-Generation Vaccines: Nanoparticle-Mediated DNA and mRNA Delivery. Advanced healthcare materials. 2021;10:e2001812
110. Li W, Cao Z, Liu R. et al. AuNPs as an important inorganic nanoparticle applied in drug carrier systems. Artificial cells, nanomedicine, and biotechnology. 2019;47:4222-4233
111. Hong GL, Zhao HL, Deng HH. et al. Fabrication of ultra-small monolayer graphene quantum dots by pyrolysis of trisodium citrate for fluorescent cell imaging. International journal of nanomedicine. 2018;13:4807-4815
112. Khutale GV, Casey A. Synthesis and characterization of a multifunctional gold-doxorubicin nanoparticle system for pH triggered intracellular anticancer drug release. European journal of pharmaceutics and biopharmaceutics: official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV. 2017;119:372-380
113. Chuang CC, Cheng CC, Chen PY. et al. Gold nanorod-encapsulated biodegradable polymeric matrix for combined photothermal and chemo-cancer therapy. International journal of nanomedicine. 2019;14:181-193
114. Liu L, Cai R, Wang Y. et al. Polydopamine-Assisted Silver Nanoparticle Self-Assembly on Sericin/Agar Film for Potential Wound Dressing Application. International journal of molecular sciences. 2018 19
115. Wojnicki M, Luty-Błocho M, Kotańska M. et al. Novel and effective synthesis protocol of AgNPs functionalized using L-cysteine as a potential drug carrier. Naunyn-Schmiedeberg's archives of pharmacology. 2018;391:123-130
116. Yan Y, Fu J, Wang T. et al. Controlled release of silyl ether camptothecin from thiol-ene click chemistry-functionalized mesoporous silica nanoparticles. Acta Biomater. 2017;51:471-478
117. An M, Li M, Xi J. et al. Silica Nanoparticle as a Lymph Node Targeting Platform for Vaccine Delivery. ACS Appl Mater Interfaces. 2017;9:23466-23475
118. Guo K, Zhao X, Dai X. et al. Organic/inorganic nanohybrids as multifunctional gene delivery systems. J Gene Med. 2019;21:e3084
119. Nam GH, Choi Y, Kim GB. et al. Emerging Prospects of Exosomes for Cancer Treatment: From Conventional Therapy to Immunotherapy. Advanced materials (Deerfield Beach, Fla). 2020;32:e2002440
120. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373-383
121. Alhasan AH, Patel PC, Choi CH. et al. Exosome encased spherical nucleic acid gold nanoparticle conjugates as potent microRNA regulation agents. Small (Weinheim an der Bergstrasse, Germany). 2014;10:186-192
122. Alvarez-Erviti L, Seow Y, Yin H. et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341-345
123. Fu W, Lei C, Liu S. et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat Commun. 2019;10:4355
124. Wang S, Li F, Ye T. et al. Macrophage-tumor chimeric exosomes accumulate in lymph node and tumor to activate the immune response and the tumor microenvironment. Science translational medicine. 2021;13:eabb6981
125. Hu Q, Li H, Wang L. et al. DNA Nanotechnology-Enabled Drug Delivery Systems. Chemical reviews. 2019;119:6459-6506
126. Li Q, Zhao D, Shao X. et al. Aptamer-Modified Tetrahedral DNA Nanostructure for Tumor-Targeted Drug Delivery. ACS Appl Mater Interfaces. 2017;9:36695-36701
127. Bujold KE, Hsu JCC, Sleiman HF. Optimized DNA "Nanosuitcases" for Encapsulation and Conditional Release of siRNA. J Am Chem Soc. 2016;138:14030-14038
128. Sellner S, Kocabey S, Nekolla K. et al. DNA nanotubes as intracellular delivery vehicles in vivo. Biomaterials. 2015;53:453-463
129. Zhuang X, Ma X, Xue X. et al. A Photosensitizer-Loaded DNA Origami Nanosystem for Photodynamic Therapy. ACS nano. 2016;10:3486-3495
130. Pan Q, Nie C, Hu Y. et al. Aptamer-Functionalized DNA Origami for Targeted Codelivery of Antisense Oligonucleotides and Doxorubicin to Enhance Therapy in Drug-Resistant Cancer Cells. ACS Applied Materials & Interfaces. 2020;12:400-409
131. Zhong R, Talebian S, Mendes BB. et al. Hydrogels for RNA delivery. Nat Mater. 2023
132. Yan J, Chen R, Zhang H. et al. Injectable Biodegradable Chitosan-Alginate 3D Porous Gel Scaffold for mRNA Vaccine Delivery. 2019; 19: 1800242.
133. Gale EC, Powell AE, Roth GA. et al. Hydrogel-Based Slow Release of a Receptor-Binding Domain Subunit Vaccine Elicits Neutralizing Antibody Responses Against SARS-CoV-2. Advanced materials (Deerfield Beach, Fla). 2021;33:e2104362
134. Lei Y, Huang S, Sharif-Kashani P. et al. Incorporation of active DNA/cationic polymer polyplexes into hydrogel scaffolds. Biomaterials. 2010;31:9106-9116
135. Zhou YL, Yang QQ, Yan YY. et al. Localized delivery of miRNAs targets cyclooxygenases and reduces flexor tendon adhesions. Acta Biomater. 2018;70:237-248
136. Zhang ZQ, Kim YM, Song SC. Injectable and Quadruple-Functional Hydrogel as an Alternative to Intravenous Delivery for Enhanced Tumor Targeting. ACS Appl Mater Interfaces. 2019;11:34634-34644
137. Zhang L, Jean SR, Ahmed S. et al. Multifunctional quantum dot DNA hydrogels. Nat Commun. 2017;8:381
138. McMillan A, Nguyen MK, Huynh CT. et al. Hydrogel microspheres for spatiotemporally controlled delivery of RNA and silencing gene expression within scaffold-free tissue engineered constructs. Acta Biomater. 2021;124:315-326
139. Yu T, Wang H, Zhang Y. et al. The Delivery of RNA-Interference Therapies Based on Engineered Hydrogels for Bone Tissue Regeneration. Frontiers in bioengineering and biotechnology. 2020;8:445
140. Pardi N, Hogan MJ, Porter FW. et al. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. 2018;17:261-279
141. Kiaie SH, Majidi Zolbanin N, Ahmadi A. et al. Recent advances in mRNA-LNP therapeutics: immunological and pharmacological aspects. Journal of nanobiotechnology. 2022;20:276
142. Mulligan MJ, Lyke KE, Kitchin N. et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature. 2020;586:589-593
143. Jackson LA, Anderson EJ, Rouphael NG. et al. An mRNA Vaccine against SARS-CoV-2 - Preliminary Report. The New England journal of medicine. 2020;383:1920-1931
144. Cafri G, Gartner JJ, Zaks T. et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J Clin Invest. 2020;130:5976-5988
145. Billingsley MM, Singh N, Ravikumar P. et al. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020;20:1578-1589
146. Schoenmaker L, Witzigmann D, Kulkarni JA. et al. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int J Pharm. 2021;601:120586
147. Samaridou E, Heyes J, Lutwyche P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv Drug Deliv Rev. 2020;154-155:37-63
148. Ripabelli G, Tamburro M, Buccieri N. et al. Active Surveillance of Adverse Events in Healthcare Workers Recipients After Vaccination with COVID-19 BNT162b2 Vaccine (Pfizer-BioNTech, Comirnaty): A Cross-Sectional Study. J Community Health. 2022;47:211-225
149. Riad A, Pokorná A, Attia S. et al. Prevalence of COVID-19 Vaccine Side Effects among Healthcare Workers in the Czech Republic. Journal of clinical medicine. 2021 10
150. García-Grimshaw M, Ceballos-Liceaga SE, Hernández-Vanegas LE. et al. Neurologic adverse events among 704,003 first-dose recipients of the BNT162b2 mRNA COVID-19 vaccine in Mexico: A nationwide descriptive study. Clin Immunol. 2021;229:108786
151. Klein NP, Lewis N, Goddard K. et al. Surveillance for Adverse Events After COVID-19 mRNA Vaccination. Jama. 2021;326:1390-1399
152. Pardi N, Hogan MJ, Naradikian MS. et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J Exp Med. 2018;215:1571-1588
153. Tanaka T, Legat A, Adam E. et al. DiC14-amidine cationic liposomes stimulate myeloid dendritic cells through Toll-like receptor 4. Eur J Immunol. 2008;38:1351-1357
154. Lonez C, Vandenbranden M, Ruysschaert JM. Cationic lipids activate intracellular signaling pathways. Adv Drug Deliv Rev. 2012;64:1749-1758
155. Kulkarni JA, Cullis PR, van der Meel R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic acid therapeutics. 2018;28:146-157
156. Tahtinen S, Tong AJ, Himmels P. et al. IL-1 and IL-1ra are key regulators of the inflammatory response to RNA vaccines. Nat Immunol. 2022;23:532-542
157. Kozma GT, Shimizu T, Ishida T. et al. Anti-PEG antibodies: Properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv Drug Deliv Rev. 2020;154-155:163-175
158. Igyártó BZ, Jacobsen S, Ndeupen S. Future considerations for the mRNA-lipid nanoparticle vaccine platform. Current opinion in virology. 2021;48:65-72
159. Oberli MA, Reichmuth AM, Dorkin JR. et al. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017;17:1326-1335
160. Miao L, Li L, Huang Y. et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat Biotechnol. 2019;37:1174-1185
161. Zhang H, You X, Wang X. et al. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through Toll-like receptor 4 signaling. Proceedings of the National Academy of Sciences of the United States of America. 2021 118
162. Fan YN, Li M, Luo YL. et al. Cationic lipid-assisted nanoparticles for delivery of mRNA cancer vaccine. Biomaterials science. 2018;6:3009-3018
163. Zhang R, Tang L, Tian Y. et al. DP7-C-modified liposomes enhance immune responses and the antitumor effect of a neoantigen-based mRNA vaccine. Journal of controlled release: official journal of the Controlled Release Society. 2020;328:210-221
164. Tateshita N, Miura N, Tanaka H. et al. Development of a lipoplex-type mRNA carrier composed of an ionizable lipid with a vitamin E scaffold and the KALA peptide for use as an ex vivo dendritic cell-based cancer vaccine. Journal of controlled release: official journal of the Controlled Release Society. 2019;310:36-46
165. Carlsson L, Clarke JC, Yen C. et al. Biocompatible, Purified VEGF-A mRNA Improves Cardiac Function after Intracardiac Injection 1 Week Post-myocardial Infarction in Swine. Molecular therapy Methods & clinical development. 2018;9:330-346
166. Chen Z, Pan H, Luo Y. et al. Nanoengineered CAR-T Biohybrids for Solid Tumor Immunotherapy with Microenvironment Photothermal-Remodeling Strategy. Small (Weinheim an der Bergstrasse, Germany). 2021;17:e2007494
167. Son S, Nam J, Zenkov I. et al. Sugar-Nanocapsules Imprinted with Microbial Molecular Patterns for mRNA Vaccination. Nano Lett. 2020;20:1499-1509
168. Pan-cancer analysis of whole genomes. Nature. 2020; 578: 82-93.
169. Kranz LM, Diken M, Haas H. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396-401
170. Lin YX, Wang Y, Ding J. et al. Reactivation of the tumor suppressor PTEN by mRNA nanoparticles enhances antitumor immunity in preclinical models. Science translational medicine. 2021 13
171. Xiao Y, Chen J, Zhou H. et al. Combining p53 mRNA nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat Commun. 2022;13:758
172. Pérez-Núñez I, Rozalén C, Palomeque J. et al. LCOR mediates interferon-independent tumor immunogenicity and responsiveness to immune-checkpoint blockade in triple-negative breast cancer. Nature cancer. 2022;3:355-370
173. Van Lint S, Renmans D, Broos K. et al. Intratumoral Delivery of TriMix mRNA Results in T-cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol Res. 2016;4:146-156
174. Hewitt SL, Bailey D, Zielinski J. et al. Intratumoral IL12 mRNA Therapy Promotes TH1 Transformation of the Tumor Microenvironment. Clin Cancer Res. 2020;26:6284-6298
175. Jain R, Frederick JP, Huang EY. et al. MicroRNAs Enable mRNA Therapeutics to Selectively Program Cancer Cells to Self-Destruct. Nucleic acid therapeutics. 2018;28:285-296
176. Liu JQ, Zhang C, Zhang X. et al. Intratumoral delivery of IL-12 and IL-27 mRNA using lipid nanoparticles for cancer immunotherapy. Journal of controlled release: official journal of the Controlled Release Society. 2022;345:306-313
177. Hotz C, Wagenaar TR, Gieseke F. et al. Local delivery of mRNA-encoded cytokines promotes antitumor immunity and tumor eradication across multiple preclinical tumor models. Science translational medicine. 2021;13:eabc7804
178. Hewitt SL, Bai A, Bailey D. et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Science translational medicine. 2019 11
179. June CH, O'Connor RS, Kawalekar OU. et al. CAR T cell immunotherapy for human cancer. Science (New York, NY). 2018;359:1361-1365
180. Khalil DN, Smith EL, Brentjens RJ. et al. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016;13:273-290
181. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321-3330
182. Mamonkin M, Rouce RH, Tashiro H. et al. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126:983-992
183. Sadelain M, Rivière I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35-45
184. Kalos M, Levine BL, Porter DL. et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science translational medicine. 2011;3:95ra73
185. Moffett HF, Coon ME, Radtke S. et al. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat Commun. 2017;8:389
186. Billingsley MM, Hamilton AG, Mai D. et al. Orthogonal Design of Experiments for Optimization of Lipid Nanoparticles for mRNA Engineering of CAR T Cells. Nano Lett. 2022;22:533-542
187. Abdalla AME, Xiao L, Miao Y. et al. Nanotechnology Promotes Genetic and Functional Modifications of Therapeutic T Cells Against Cancer. Advanced science (Weinheim, Baden-Wurttemberg, Germany). 2020;7:1903164
188. Nia HT, Liu H, Seano G. et al. Solid stress and elastic energy as measures of tumour mechanopathology. Nat Biomed Eng. 2016 1
189. Mardiana S, Solomon BJ, Darcy PK. et al. Supercharging adoptive T cell therapy to overcome solid tumor-induced immunosuppression. Science translational medicine. 2019 11
190. Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188-200
191. Tekedereli I, Alpay SN, Akar U. et al. Therapeutic Silencing of Bcl-2 by Systemically Administered siRNA Nanotherapeutics Inhibits Tumor Growth by Autophagy and Apoptosis and Enhances the Efficacy of Chemotherapy in Orthotopic Xenograft Models of ER (-) and ER (+) Breast Cancer. Mol Ther Nucleic Acids. 2013;2:e121
192. Brock A, Krause S, Li H. et al. Silencing HoxA1 by intraductal injection of siRNA lipidoid nanoparticles prevents mammary tumor progression in mice. Science translational medicine. 2014;6:217ra212
193. Guo Y, Lee H, Fang Z. et al. Single-cell analysis reveals effective siRNA delivery in brain tumors with microbubble-enhanced ultrasound and cationic nanoparticles. Science advances. 2021 7
194. Wang Q, Tian Y, Liu L. et al. Precise Targeting Therapy of Orthotopic Gastric Carcinoma by siRNA and Chemotherapeutic Drug Codelivered in pH-Sensitive Nano Platform. Advanced healthcare materials. 2021;10:e2100966
195. Tang X, Sheng Q, Xu C. et al. pH/ATP cascade-responsive nano-courier with efficient tumor targeting and siRNA unloading for photothermal-immunotherapy. Nano Today. 2021;37:101083
196. Xu J, Liu Y, Li Y. et al. Precise targeting of POLR2A as a therapeutic strategy for human triple negative breast cancer. Nature nanotechnology. 2019;14:388-397
197. Reddy TL, Garikapati KR, Reddy SG. et al. Simultaneous delivery of Paclitaxel and Bcl-2 siRNA via pH-Sensitive liposomal nanocarrier for the synergistic treatment of melanoma. Scientific reports. 2016;6:35223
198. Zhao Z, Li Y, Liu H. et al. Co-delivery of IKBKE siRNA and cabazitaxel by hybrid nanocomplex inhibits invasiveness and growth of triple-negative breast cancer. Science advances. 2020;6:eabb0616
199. Qiu C, Han HH, Sun J. et al. Regulating intracellular fate of siRNA by endoplasmic reticulum membrane-decorated hybrid nanoplexes. Nat Commun. 2019;10:2702
200. Huang KW, Hsu FF, Qiu JT. et al. Highly efficient and tumor-selective nanoparticles for dual-targeted immunogene therapy against cancer. Science advances. 2020;6:eaax5032
201. Ghafouri-Fard S, Ghafouri-Fard S. siRNA and cancer immunotherapy. Immunotherapy. 2012;4:907-917
202. Lin YX, Wang Y, Blake S. et al. RNA Nanotechnology-Mediated Cancer Immunotherapy. Theranostics. 2020;10:281-299
203. Barati M, Mirzavi F, Nikpoor AR. et al. Enhanced antitumor immune response in melanoma tumor model by anti-PD-1 small interference RNA encapsulated in nanoliposomes. Cancer Gene Ther. 2022;29:814-824
204. Jiang X, Wang J, Deng X. et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18:10
205. Gordon SR, Maute RL, Dulken BW. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-499
206. Hanafy MS, Hufnagel S, Trementozzi AN. et al. PD-1 siRNA-Encapsulated Solid Lipid Nanoparticles Downregulate PD-1 Expression by Macrophages and Inhibit Tumor Growth: PD-1 siRNA-Encapsulated Solid Lipid Nanoparticles. AAPS PharmSciTech. 2021;22:60
207. Sahu R, Jha S, Pattanayak SP. Therapeutic silencing of mTOR by systemically administered siRNA-loaded neutral liposomal nanoparticles inhibits DMBA-induced mammary carcinogenesis. Br J Cancer. 2022;127:2207-2219
208. Guo Y, Zhang Q, Zhu Q. et al. Copackaging photosensitizer and PD-L1 siRNA in a nucleic acid nanogel for synergistic cancer photoimmunotherapy. Sci Adv. 2022;8:eabn2941
209. Gao C, Cheng K, Li Y. et al. Injectable Immunotherapeutic Hydrogel Containing RNA-Loaded Lipid Nanoparticles Reshapes Tumor Microenvironment for Pancreatic Cancer Therapy. Nano Lett. 2022;22:8801-8809
210. Li Y, Meng X, Chen G. et al. Lipid-mediated delivery of CD47 siRNA aids JQ1 in ensuring simultaneous downregulation of PD-L1 and CD47 and improves antitumor immunotherapy efficacy. Biomater Sci. 2022;10:6755-6767
211. Chen Y, Huang Y, Li Q. et al. Targeting Xkr8 via nanoparticle-mediated in situ co-delivery of siRNA and chemotherapy drugs for cancer immunochemotherapy. Nat Nanotechnol. 2022
212. Abdel-Bar HM, Walters AA, Lim Y. et al. An "eat me" combinatory nano-formulation for systemic immunotherapy of solid tumors. Theranostics. 2021;11:8738-8754
213. Chen LH, Liang NW, Huang WY. et al. Supramolecular hydrogel for programmable delivery of therapeutics to cancer multidrug resistance. Biomaterials advances. 2023;146:213282
214. Mendonça MCP, Kont A, Aburto MR. et al. Advances in the Design of (Nano)Formulations for Delivery of Antisense Oligonucleotides and Small Interfering RNA: Focus on the Central Nervous System. Mol Pharm. 2021;18:1491-1506
215. Crooke ST, Baker BF, Crooke RM. et al. Antisense technology: an overview and prospectus. Nat Rev Drug Discov. 2021;20:427-453
216. Linnane E, Davey P, Zhang P. et al. Differential uptake, kinetics and mechanisms of intracellular trafficking of next-generation antisense oligonucleotides across human cancer cell lines. Nucleic Acids Res. 2019;47:4375-4392
217. Hong D, Kurzrock R, Kim Y. et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7:314ra185
218. Ross SJ, Revenko AS, Hanson LL. et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci Transl Med. 2017 9
219. Cheng X, Yu D, Cheng G. et al. T7 Peptide-Conjugated Lipid Nanoparticles for Dual Modulation of Bcl-2 and Akt-1 in Lung and Cervical Carcinomas. Mol Pharm. 2018;15:4722-4732
220. Lee SJ, Lim JH, Choi YH. et al. Interleukin-28A triggers wound healing migration of bladder cancer cells via NF-κB-mediated MMP-9 expression inducing the MAPK pathway. Cell Signal. 2012;24:1734-1742
221. Gong N, Teng X, Li J. et al. Antisense Oligonucleotide-Conjugated Nanostructure-Targeting lncRNA MALAT1 Inhibits Cancer Metastasis. ACS Applied Materials & Interfaces. 2019;11:37-42
222. Chen L, Li G, Wang X. et al. Spherical Nucleic Acids for Near-Infrared Light-Responsive Self-Delivery of Small-Interfering RNA and Antisense Oligonucleotide. ACS nano. 2021
223. Parayath NN, Gandham SK, Amiji MM. Tumor-targeted miRNA nanomedicine for overcoming challenges in immunity and therapeutic resistance. Nanomedicine (London, England). 2022;17:1355-1373
224. Zhang Z, Huang Q, Yu L. et al. The Role of miRNA in Tumor Immune Escape and miRNA-Based Therapeutic Strategies. Frontiers in immunology. 2021;12:807895
225. Lai X, Eberhardt M, Schmitz U. et al. Systems biology-based investigation of cooperating microRNAs as monotherapy or adjuvant therapy in cancer. Nucleic Acids Res. 2019;47:7753-7766
226. Bouchie A. First microRNA mimic enters clinic. Nat Biotechnol. 2013;31:577
227. Zang X, Zhang X, Zhao X. et al. Targeted Delivery of miRNA 155 to Tumor Associated Macrophages for Tumor Immunotherapy. Mol Pharm. 2019;16:1714-1722
228. Parayath NN, Parikh A, Amiji MM. Repolarization of Tumor-Associated Macrophages in a Genetically Engineered Nonsmall Cell Lung Cancer Model by Intraperitoneal Administration of Hyaluronic Acid-Based Nanoparticles Encapsulating MicroRNA-125b. Nano Lett. 2018;18:3571-3579
229. Parayath NN, Gandham SK, Leslie F. et al. Improved anti-tumor efficacy of paclitaxel in combination with MicroRNA-125b-based tumor-associated macrophage repolarization in epithelial ovarian cancer. Cancer Lett. 2019;461:1-9
230. Yu X, Odenthal M, Fries JW. Exosomes as miRNA Carriers: Formation-Function-Future. International journal of molecular sciences. 2016 17
231. Zhang J, Li S, Li L. et al. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics, proteomics & bioinformatics. 2015;13:17-24
232. Lee JH, Dindorf J, Eberhardt M. et al. Innate extracellular vesicles from melanoma patients suppress β-catenin in tumor cells by miRNA-34a. Life science alliance. 2019 2
233. Basu S, Bhattacharyya SN. Insulin-like growth factor-1 prevents miR-122 production in neighbouring cells to curtail its intercellular transfer to ensure proliferation of human hepatoma cells. Nucleic Acids Res. 2014;42:7170-7185
234. Meng X, Jiang Q, Chang N. et al. Small activating RNA binds to the genomic target site in a seed-region-dependent manner. Nucleic Acids Res. 2016;44:2274-2282
235. Wang J, Place RF, Portnoy V. et al. Inducing gene expression by targeting promoter sequences using small activating RNAs. Journal of biological methods. 2015 2
236. Wei J, Zhao J, Long M. et al. p21WAF1/CIP1 gene transcriptional activation exerts cell growth inhibition and enhances chemosensitivity to cisplatin in lung carcinoma cell. BMC Cancer. 2010;10:632
237. Dong Z, Dang Y, Chen Y. Small double-stranded RNA mediates the anti-cancer effects of p21WAF1/ClP1 transcriptional activation in a human glioma cell line. Yonsei medical journal. 2014;55:324-330
238. Qin Q, Lin YW, Zheng XY. et al. RNAa-mediated overexpression of WT1 induces apoptosis in HepG2 cells. World journal of surgical oncology. 2012;10:11
239. Junxia W, Ping G, Yuan H. et al. Double strand RNA-guided endogeneous E-cadherin up-regulation induces the apoptosis and inhibits proliferation of breast carcinoma cells in vitro and in vivo. Cancer Sci. 2010;101:1790-1796
240. Ren S, Kang MR, Wang J. et al. Targeted induction of endogenous NKX3-1 by small activating RNA inhibits prostate tumor growth. The Prostate. 2013;73:1591-1601
241. Li M, Peng Z, Ren W. et al. Small activating ribonucleic acid reverses tyrosine kinase inhibitor resistance in epidermal growth factor receptor-mutant lung cancer by increasing the expression of phosphatase and tensin homolog. Thoracic cancer. 2016;7:481-485
242. Kosaka M, Kang MR, Yang G. et al. Targeted p21WAF1/CIP1 activation by RNAa inhibits hepatocellular carcinoma cells. Nucleic acid therapeutics. 2012;22:335-343
243. Chen Z, Place RF, Jia ZJ. et al. Antitumor effect of dsRNA-induced p21(WAF1/CIP1) gene activation in human bladder cancer cells. Mol Cancer Ther. 2008;7:698-703
244. Kang MR, Yang G, Place RF. et al. Intravesical delivery of small activating RNA formulated into lipid nanoparticles inhibits orthotopic bladder tumor growth. Cancer Res. 2012;72:5069-5079
245. Wang LL, Feng CL, Zheng WS. et al. Corrigendum to "Tumor-selective lipopolyplex encapsulated small active RNA hampers colorectal cancer growth in vitro and in orthotopic murine" [Biomaterials 141 (2017) 13-28]. Biomaterials. 2020;245:119994
246. Yoon S, Huang KW, Reebye V. et al. Targeted Delivery of C/EBPα -saRNA by Pancreatic Ductal Adenocarcinoma-specific RNA Aptamers Inhibits Tumor Growth In Vivo. Mol Ther. 2016;24:1106-1116
247. Song KM, Lee S, Ban C. Aptamers and their biological applications. Sensors (Basel, Switzerland). 2012;12:612-631
248. Li Z, Fu X, Huang J. et al. Advances in Screening and Development of Therapeutic Aptamers Against Cancer Cells. Front Cell Dev Biol. 2021;9:662791
249. Yang C, Jiang Y, Hao SH. et al. Aptamers: an emerging navigation tool of therapeutic agents for targeted cancer therapy. Journal of materials chemistry B. 2021;10:20-33
250. Zhou J, Rossi JJ. Cell-type-specific, Aptamer-functionalized Agents for Targeted Disease Therapy. Molecular therapy Nucleic acids. 2014;3:e169
251. Mongelard F, Bouvet P. AS-1411, a guanosine-rich oligonucleotide aptamer targeting nucleolin for the potential treatment of cancer, including acute myeloid leukemia. Current opinion in molecular therapeutics. 2010;12:107-114
252. Soundararajan S, Chen W, Spicer EK. et al. The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res. 2008;68:2358-2365
253. Hoellenriegel J, Zboralski D, Maasch C. et al. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood. 2014;123:1032-1039
254. Marasca R, Maffei R. NOX-A12: mobilizing CLL away from home. Blood. 2014;123:952-953
255. Vater A, Klussmann S. Toward third-generation aptamers: Spiegelmers and their therapeutic prospects. Current opinion in drug discovery & development. 2003;6:253-261
256. Dassie JP, Liu XY, Thomas GS. et al. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27:839-849
257. Pastor F, Kolonias D, McNamara JO 2nd. et al. Targeting 4-1BB costimulation to disseminated tumor lesions with bi-specific oligonucleotide aptamers. Mol Ther. 2011;19:1878-1886
258. Lao YH, Phua KK, Leong KW. Aptamer nanomedicine for cancer therapeutics: barriers and potential for translation. ACS nano. 2015;9:2235-2254
259. Lozano T, Soldevilla MM, Casares N. et al. Targeting inhibition of Foxp3 by a CD28 2'-Fluro oligonucleotide aptamer conjugated to P60-peptide enhances active cancer immunotherapy. Biomaterials. 2016;91:73-80
260. Terns MP, Terns RM. CRISPR-based adaptive immune systems. Curr Opin Microbiol. 2011;14:321-327
261. Mali P, Yang L, Esvelt KM. et al. RNA-guided human genome engineering via Cas9. Science (New York, NY). 2013;339:823-826
262. Jinek M, Chylinski K, Fonfara I. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (New York, NY). 2012;337:816-821
263. Katti A, Diaz BJ, Caragine CM. et al. CRISPR in cancer biology and therapy. Nat Rev Cancer. 2022;22:259-279
264. Ran FA, Hsu PD, Wright J. et al. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281-2308
265. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096
266. Charpentier E, Marraffini LA. Harnessing CRISPR-Cas9 immunity for genetic engineering. Curr Opin Microbiol. 2014;19:114-119
267. Rosenblum D, Gutkin A, Kedmi R. et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci Adv. 2020 6
268. Hamis S, Nithiarasu P, Powathil GG. What does not kill a tumour may make it stronger: In silico insights into chemotherapeutic drug resistance. J Theor Biol. 2018;454:253-267
269. Kwon T, Ra JS, Lee S. et al. Precision targeting tumor cells using cancer-specific InDel mutations with CRISPR-Cas9. Proc Natl Acad Sci U S A. 2022 119
270. Ray M, Lee YW, Hardie J. et al. CRISPRed Macrophages for Cell-Based Cancer Immunotherapy. Bioconjug Chem. 2018;29:445-450
271. Liu Q, Wang C, Zheng Y. et al. Virus-like nanoparticle as a co-delivery system to enhance efficacy of CRISPR/Cas9-based cancer immunotherapy. Biomaterials. 2020;258:120275
272. Zhang B-C, Luo B-Y, Zou J-J. et al. Co-delivery of Sorafenib and CRISPR/Cas9 Based on Targeted Core-Shell Hollow Mesoporous Organosilica Nanoparticles for Synergistic HCC Therapy. ACS Applied Materials & Interfaces. 2020;12:57362-57372
273. Zhang B-C, Wu P-Y, Zou J-J. et al. Efficient CRISPR/Cas9 gene-chemo synergistic cancer therapy via a stimuli-responsive chitosan-based nanocomplex elicits anti-tumorigenic pathway effect. Chem Eng J. 2020;393:124688
274. Deng S, Li X, Liu S. et al. Codelivery of CRISPR-Cas9 and chlorin e6 for spatially controlled tumor-specific gene editing with synergistic drug effects. Science advances. 2020;6:eabb4005
275. Pan Y, Yang J, Luan X. et al. Near-infrared upconversion-activated CRISPR-Cas9 system: A remote-controlled gene editing platform. Science advances. 2019;5:eaav7199
276. Chen X, Chen Y, Xin H. et al. Near-infrared optogenetic engineering of photothermal nanoCRISPR for programmable genome editing. Proceedings of the National Academy of Sciences of the United States of America. 2020;117:2395-2405
277. Donahue ND, Acar H, Wilhelm S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv Drug Deliv Rev. 2019;143:68-96
278. Akinc A, Querbes W, De S. et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther. 2010;18:1357-1364
279. Patil V, Patel A. Biodegradable Nanoparticles: A Recent Approach and Applications. Curr Drug Targets. 2020;21:1722-1732
280. Zhang Y, Liu Q, Zhang X. et al. Recent advances in exosome-mediated nucleic acid delivery for cancer therapy. Journal of nanobiotechnology. 2022;20:279
281. Huang JL, Jiang G, Song QX. et al. Lipoprotein-biomimetic nanostructure enables efficient targeting delivery of siRNA to Ras-activated glioblastoma cells via macropinocytosis. Nat Commun. 2017;8:15144
282. Chen L, Li G, Wang X. et al. Spherical Nucleic Acids for Near-Infrared Light-Responsive Self-Delivery of Small-Interfering RNA and Antisense Oligonucleotide. ACS Nano. 2021;15:11929-11939
Author contact
![]() Corresponding author: Yongfeng Jin, College of Life Sciences, Zhejiang University, Hangzhou 310058, China. Tel.: +86-571-88206478; Email: jinyfedu.cn
Corresponding author: Yongfeng Jin, College of Life Sciences, Zhejiang University, Hangzhou 310058, China. Tel.: +86-571-88206478; Email: jinyfedu.cn