Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Risk factors and prevention of EC
Established treatment options...
RCD subroutines and EC treatment
Targeting apoptotic pathways...
Targeting autophagy pathways...
Targeting ferroptosis pathways...
Targeting pyroptosis pathways...
Targeting necroptosis pathways...
Summary of EC clinical trials...
Conclusion and perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(12):3831-3868. doi:10.7150/ijbs.85753 This issue Cite
Review
Unraveling the Complexity of Regulated Cell Death in Esophageal Cancer: from Underlying Mechanisms to Targeted Therapeutics
Haowen Zhang1,2#, Jin Zhang2,3#, Siyuan Luan1,2#, Zhiying Liu3, Xiaokun Li1, Bo Liu2 ![]() , Yong Yuan1
, Yong Yuan1 ![]()
1. Department of Thoracic Surgery, West China Hospital, Sichuan University, Chengdu 610041, China.
2. State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu 610041, China.
3. School of Pharmaceutical Sciences of Medical School, Shenzhen University, Shenzhen, 518000, China.
#These authors contributed equally to this work.
Received 2023-4-30; Accepted 2023-7-13; Published 2023-7-31
Abstract

Esophageal cancer (EC) is the sixth most common and the seventh most deadly malignancy of the digestive tract, representing a major global health challenge. Despite the availability of multimodal therapeutic strategies, the existing EC treatments continue to yield unsatisfactory results due to their limited efficacy and severe side effects. Recently, knowledge of the subroutines and molecular mechanisms of regulated cell death (RCD) has progressed rapidly, enhancing the understanding of key pathways related to the occurrence, progression, and treatment of many types of tumors, including EC. In this context, the use of small-molecule compounds to target such RCD subroutines has emerged as a promising therapeutic strategy for patients with EC. Thus, in this review, we firstly discussed the risk factors and prevention of EC. We then outlined the established treatment regimens for patients with EC. Furthermore, we not only briefly summarized the mechanisms of five best studied subroutines of RCD related to EC, including apoptosis, ferroptosis, pyroptosis, necroptosis and autophagy, but also outlined the recent advances in the development of small-molecule compounds and long non-coding RNA (lncRNA) targeting the abovementioned RCD subroutines, which may serve as a new therapeutic strategy for patients with EC in the future.
Keywords: Esophageal cancer, Cancer therapy, Regulated cell death, Small-molecule compounds, Long non-coding RNA
Introduction
Globally, esophageal cancer (EC) is regarded as a fatal gastrointestinal malignancy. According to the most recent global cancer statistics released by the International Agency for Research on Cancer (IARC), Esophageal Cancer (EC) ranks seventh in terms of overall cancer prevalence and sixth in terms of cancer-related mortality worldwide. In 2020, an estimated 604,000 new cases of EC were reported, resulting in approximately 544,000 deaths [1]. EC can be classified into two broad histopathological subtypes: esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC). Globally, ESCC is the major subtype of EC, accounting for approximately 90% of all EC cases. Furthermore, while ESCC remains the main subtype of EC in eastern regions, South America and East Africa, the incidence of EAC has been on the rise in some developed western countries over the past decades, and EAC is now even the predominant subtype in Europe, the Americas and Australia [2,3]. Owing to the absence of distinctive symptoms during the initial stages and the constraints of diagnostic techniques, individuals with EC frequently receive a diagnosis at an advanced and untreatable phase. [4]. Depending on characteristics of the tumor (mainly the TNM stage) and patients (including fitness), different treatment options can be used to treat patients with EC, including endoscopic management, surgery, chemotherapy, radiotherapy and molecular targeted therapy. Despite significant advancements in medicine and cancer biology over the past decades, the existing EC treatments continue to yield unsatisfactory results due to their limited efficacy and severe side effects. The 5-year survival rate of EC remains less than 20%. Furthermore, decreased radiotherapy sensitivity and increased chemotherapy drug resistance of EC cells are becoming prominent, problems that urgently need to be solved. Therefore, the development of effective and rational therapies for patients with EC remains imperative [2,5,6].
Cell death, an inevitable destiny of all life, has physiological and pathological functions. According to functional characteristics, Cell death is commonly classified into two categories: regulated cell death (RCD) and accidental cell death (ACD). Unlike ACD, which is an uncontrolled process induced via unexpected attack and/or injury, RCD is closely related to precise signaling cascades involving a series of defined effector molecules [7]. Kerr et al. described the concept of apoptosis and its role in physiological and pathological processes in the 1970s [8]. Since then, RCD-related research has rapidly increased, and an increasing number of new nonapoptotic modes of RCD have been identified over the past few decades, including but not limited to autophagy, necroptosis, ferroptosis, pyroptosis, entosis, parthanatos, alkaliptosis, oxeiptosis, lysosome-dependent cell death, NETotic cell death and cuprotosis [9-11].
As one of the current research hotspots, dysregulation of single or mixed types of RCD subroutines has been linked to the development of various human diseases, especially cancer. The occurrence of malignant tumors significantly rises when there is a disruption in normal RCD or when abnormal cell proliferation is not properly controlled [12-15]. For EC, accumulating evidence has shown that dysregulation of RCD subroutines can not only affect the occurrence and development of EC but also reduce its treatment effectiveness, for example, causing increased resistance to chemotherapy and immunotherapy and decreased radiotherapy sensitivity. Recently, researchers have devoted much effort to developing EC treatments that function by targeting RCD to promote individualized treatment of EC, decrease the risk of disease progression, improve the prognosis of patients and reduce toxicity. Using small-molecule compounds to target RCD subroutines has emerged as a promising therapeutic option for patients with EC, and rapid progress has been made in the past few years [16-19].
In this review, we firstly discussed the risk factors and prevention of EC. Subsequently, we outline established treatment regimens for patients with EC. In addition, we briefly summarize the mechanisms of five main types of RCD subroutines related to EC: apoptosis, autophagy, ferroptosis, pyroptosis and necroptosis. Importantly, we provide an overview of the latest progress in the advancement of small-molecule compounds that specifically target these five regulated cell death (RCD) subroutines. These compounds hold potential as innovative therapeutic approaches for individuals diagnosed with EC. Furthermore, we discuss the opportunities and challenges in this research field. We will provide the latest advances in the abovementioned RCD subroutine research and developments in the area of EC therapy. These findings will enhance the understanding of EC and provide some promising treatment options for patients with EC.
Risk factors and prevention of EC
Risk factors differ between ESCC and EAC due to different mechanisms of carcinogenesis. Chronic irritation of carcinogens, mainly cigarette, alcohol and hot beverages, can increase the risk of the carcinogenesis of ESCC [20]. Additionally, nutritional deficiencies are also found to increase the risk of ESCC [21]. For EAC, the development of EAC is strongly linked to gastroesophageal reflux disease (GERD). Barrett's esophagus (BE) serves as a prominent risk factor for EAC [22]. In addition, obesity was reported to be associated with a decreased risk of ESCC and an increased risk of EAC [23] (Figure 1A).
Main risk factors, symptoms and current treatment algorithm of EC. (A) Main risk factors and symptoms of EC. (B) Current treatment algorithm for patients with locally advanced EC. (Created with BioRender.com)
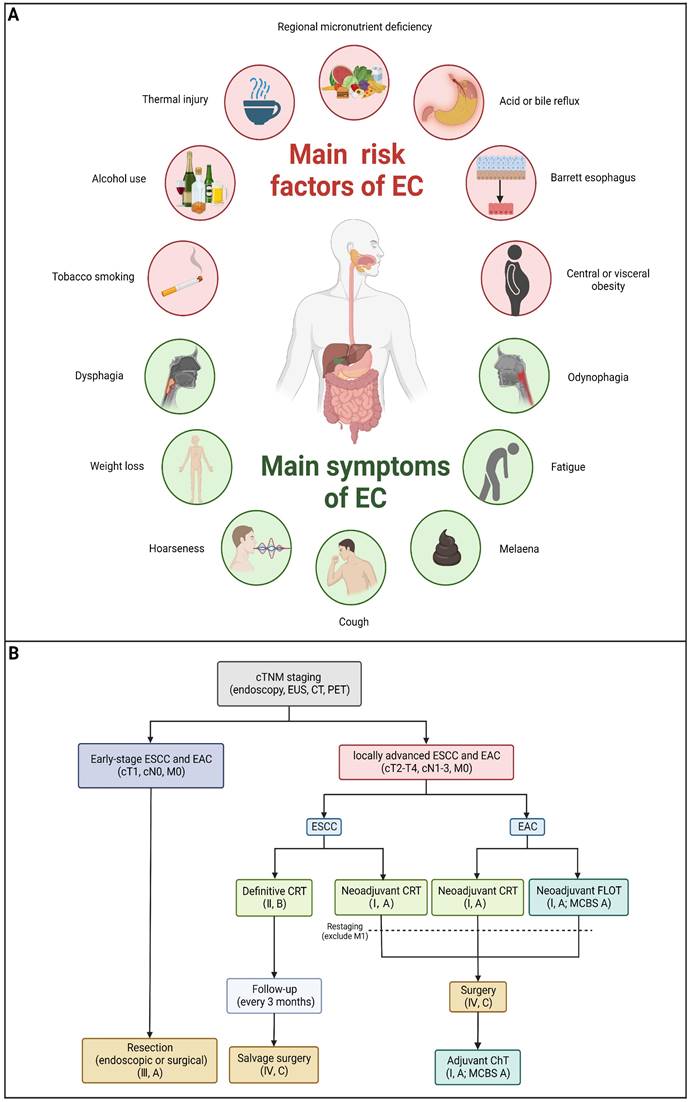
In order to prevent EC, it is important to identify and eliminate risk factors mentioned above as the primary means of prevention. Furthermore, screening precancerous and early cancerous lesions in high-risk population is crucial to prevent, which can make curative treatment possible.
Established treatment options for EC
To date, there are various methods to treat patients with EC, such as endoscopic resection, surgical resection, radiotherapy, chemotherapy, immunotherapy or some specific combination of these (Table 1). Clinically, clinicians can decide the treatment for patients with EC based on both personal characteristics (such as fitness) and characteristics of the tumor, primarily its TNM stage [2,24,25]. Patients with early-stage EC are treated primarily through surgery or endoscopic resection, and the therapeutic efficacy is astounding. In terminal cases, surgery is not sufficient and is often combined with preoperative or perioperative chemotherapy/chemoradiotherapy. Although the treatment options for patients with advanced EC are constantly being optimized, the poor quality of life and poor five-year survival rate are still disappointing. In addition, increased resistance to chemotherapy and immunotherapy and decreased radiotherapy sensitivity of EC cells are becoming prominent, problems that urgently need to be solved. Below, we summarize the current available treatment strategies for EC. As shown in Figure 1B, an algorithm has been formulated to address the treatment of individuals diagnosed with locally advanced EC.
Appropriate patient population, advantages and disadvantages of different types of EC treatment
| Type of EC treatment | Appropriate patient population | Advantages | Disadvantages |
|---|---|---|---|
| Endoscopic resection | Patients with Lesions of intraepithelial high-grade dysplasia and most T1 tumors | Minimally invasive treatment | May lead to complications (mainly perforation and bleeding). |
| Surgery | Patients with locally advanced EC (stages T1b-T4, N1-N3, M0) | Complete removal of the tumor to minimise recurrence | High surgical risk. Long postoperative recovery time. May lead to postoperative complications (such as anastomotic leakage, pulmonary complications etc.). |
| Radiotherapy | Patients with advanced EC | Can effectively control local tumors with minimal harm to healthy tissue nearby | The damage to healthy tissues may result in many side-effects (such as dysphagia, skin problems and hair loss etc.). |
| Chemotherapy | Patients with advanced EC | Can kill cancer cells anywhere in the body, even metastases | Chemoresistance. The damage to healthy tissues may result in many side-effects (such as organ damage, nausea and hair loss etc.). |
| Immunotherapy | Specific patients with advanced EC, who show favorable predictive value for immunotherapy | Has the potential for durable therapeutic effects by activating the immune system to attack EC cells | May be ineffective for certain patients. Could lead to immune-related adverse reactions. |
Endoscopic resection
Endoscopic resection has emerged as a treatment option for lesions with intraepithelial high-grade dysplasia as well as most T1 tumors. The indication for ER depends on lymph node metastasis risk. The development of lymph node metastases rarely occurs among lesions in which the depth of invasion does not extend beyond the mucosal layer (T1a); therefore, ER can be curative in these cases. ER is also an option if lesions extend into the muscularis mucosae or slightly infiltrate the submucosa (up to 200 mm), which are regarded as relative indications. At present, ER used for early EC patients can typically be categorized into cap-assisted endoscopic mucosal resection (EMRC), ligation-assisted endoscopic mucosal resection (EMRL), multiband mucosectomy (MBM) and endoscopic submucosal dissection (ESD) [25-27].
Surgery
Surgery is often required for nonmetastatic, locally advanced ESCC and EAC (stages T1b-T4, N1-N3, M0). Currently, various methods for EC resection based on different approaches and the extent of lymphadenectomy are used, such as McKeown esophagectomy, Ivor Lewis esophagectomy and Sweet esophagectomy [2,28,29]. Furthermore, in recent years, minimally invasive esophagectomy (MIO) techniques such as robotics techniques have become more common clinically and are reported to have quicker functional recovery, lower postoperative morbidity and improved quality of life (QoL) [25,30,31].
Preoperative and perioperative treatment
According to the latest ESMO Clinical Practice Guideline, all cases with locally advanced resectable EC should be carefully considered for preoperative or perioperative ChT/CRT, which are expected to reduce the primary tumor volume, increase rates of radical (R0) resection and survival and decrease the recurrence risk. Moreover, preoperative treatment using ChT or CRT can not only attenuate dysphagia and improve nutritional status among most patients but also reduce the requirement for feeding tube placement. Furthermore, it is worth mentioning that the need for preoperative treatment of cT2 N0 tumors is currently controversial, and further study is required [25,32,33].
Definitive CRT
According to the latest ESMO Clinical Practice Guideline, definitive CRT (with salvage surgery and close surveillance) is a recommended treatment for patients with resectable EC. Furthermore, for patients with EC who are unwilling or unable to undergo surgery, definitive CRT should also be considered [25,34].
Palliative care
In recent years, palliative care has increasingly played a vital role in EC patients with numerous and complex symptoms caused by tumor location and required treatments, such as dysphagia, malnutrition, pain and psychological symptoms [35]. Palliative chemotherapy is initiated in patients with advanced EC who experience dysphagia and malnutrition caused by esophageal stenosis and aspiration induced by a fistula, with the goal of improving their symptom burden and QoL. Various approaches have been developed for the palliation of advanced EC, such as self-expanding metal stents (SEMSs), stents loaded with (125) iodine seeds, brachytherapy and external radiation therapy [36-38].
RCD subroutines and EC treatment
Despite the availability of multimodal therapeutic strategies at present, the five-year survival rate of patients with EC is consistently less than 20%. In addition, increased resistance to chemotherapy and immunotherapy as well as decreased radiotherapy sensitivity of EC cells is becoming prominent. Thus, there is a pressing need to optimize existing clinical treatments for patients with EC and to find novel and effective treatment options. In this context, accumulating evidence has shown that using small-molecule compounds to precisely target specific RCD patterns is a promising approach to treat patients with EC, which is expected to improve the clinical outcomes of existing treatment modalities, including chemotherapy, radiotherapy and immunotherapy. Here, we briefly summarize the mechanisms of five main types of RCD subroutines that are associated with EC and review recent advances in the development of RCD-related small-molecule compounds in the field of EC.
Targeting apoptotic pathways with small-molecule compounds in EC
Brief introduction to apoptosis
Apoptosis, a conventional type of regulated cell death (RCD), is distinguished by a sequence of morphological alterations and the generation of apoptotic bodies (ABs). It is predominantly triggered through the intrinsic pathway (also referred to as the mitochondrial pathway) and the extrinsic pathway (also known as the death receptor pathway) [10]. Additionally, apoptosis can also be induced by perforin and granzyme B released from immune cells, which is called the granzyme-perforin pathway [39]. In general, the intrinsic pathway can be activated via various intracellular stress signals, such as ionizing radiation, chemotherapy, targeted therapy, endoplasmic reticulum stress and growth factor deprivation [10]. The extrinsic pathway can be activated via interactions between members of the tumor necrosis factor receptor (TNFR) superfamily and their corresponding ligands (e.g., FAS can be activated by FAS ligand) [40] (Figure 2).
Core apoptotic pathways in EC and EC therapeutic approaches by targeting apoptosis pathways. (A) Core apoptotic pathways in EC. Apoptosis is a form of typical RCD characterized by a series of morphological changes and the formation of so-called apoptotic bodies and is mainly activated via the intrinsic pathway, the extrinsic pathway and the granzyme-perforin pathway. (B) Small-molecule compounds targeting apoptosis-related BCL-2 family proteins in EC. (C) Small-molecule compounds targeting apoptosis-related IAP proteins in EC. (D) Small-molecule compounds targeting apoptosis-related death receptor in EC. (E) Small-molecule compounds targeting apoptosis-related the JNK/p38 MAPK pathway in EC. (F) Small-molecule compounds targeting apoptosis-related the PI3K/AKT/mTOR pathway in EC. (G) Small-molecule compounds targeting apoptosis-related other regulators in EC. (H) Some lncRNAs targeting apoptosis-related pathways in EC. (Created with BioRender.com)
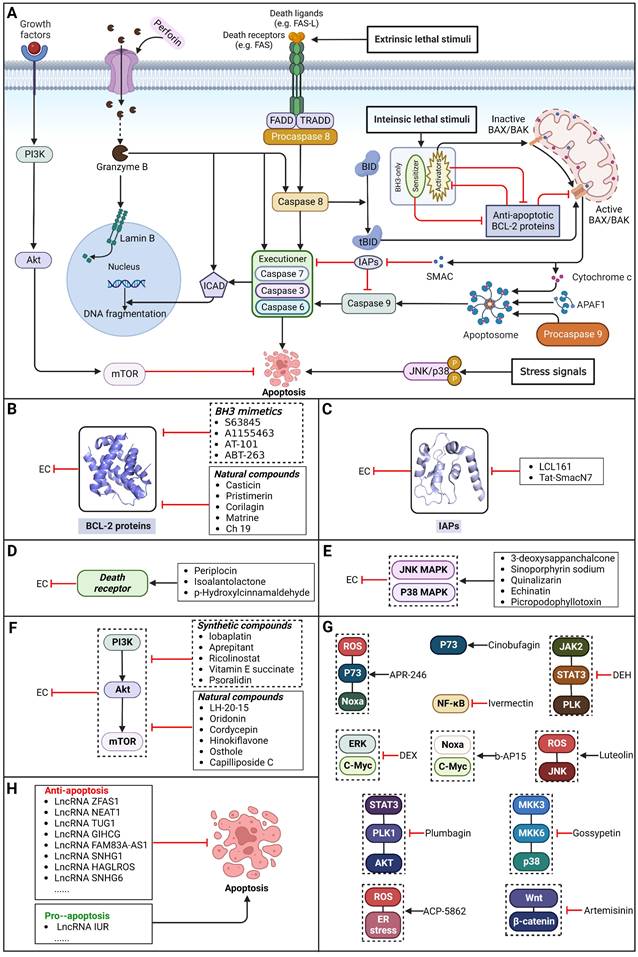
Targeting BCL-2 family proteins
During apoptosis, B-cell lymphoma-2 (BCL-2) family proteins act as central regulators. The intrinsic pathway of apoptosis is controlled tightly through balance between BCL-2 protein family members, including anti-apoptotic BCL-2 proteins (e.g., BCL-XL, BCL-2 and MCL-1), pro-apoptotic BH3-only proteins and pro-apoptotic effector proteins (e.g., BAX and BAK1). Normally, by binding and sequestering proapoptotic BH3-only 'activator' proteins and effector proteins, BCL-2 proteins can act against apoptosis [41]. BH3-only proteins can be upregulated to promote apoptosis in response to intracellular stress signals. Therefore, patients with EC may benefit from selectively targeting BCL-2 family proteins to induce apoptosis.
BH3 mimetics
Notably, BH3 mimetics are regarded as a novel class of antitumor agents that can directly target BCL-2 family proteins and induce MOMP in a BAK/BAX-dependent manner [42,43] and show certain potential in the treatment of EC. Researchers have tested BH3-mimetic drugs in preclinical cancer models, and some drugs that target MCL-1 and BCL-XL have been assessed in phase I clinical trials for certain cancers [44]. It is worth mentioning that the BCL-2-specific inhibitor venetoclax is approved by many regulatory authorities worldwide, including the US Food and Drug Administration, to treat acute myeloid leukemia and chronic lymphocytic leukemia, and there is potential for an expanding range of indications [45]. For EC, BCL-XL and MCL-1 were found to be highly expressed in EAC cell lines and tumor samples. The BH3 mimetics S63845 and A1155463 were further confirmed to be able to activate the BAX/BAK-dependent mitochondrial apoptotic pathway and sensitize EAC to chemotherapeutics by targeting MCL-1 and BCL-XL respectively [46]. In addition, a natural BH3-mimetic molecule named AT-101 has been reported to decrease the expression of MCL-1 and BCL-2, leading to apoptosis of EAC cells, and thus has a favorable anti-EC and clinical effect [47,48]. Moreover, a novel BH3 mimetic named ABT-263 with high affinity for BCL-XL, BCL-2 and BCL-w has been proven to exert both cytotoxic and cytostatic effects on EC cells in vitro and has entered clinical trials for the treatment of cancer. ABT-263 dose-dependently suppressed the viability of 3 EC cell lines with IC50 values of 8.2±1.6, 7.1±1.5 and 10.7±1.4 μmol/L in CaES-17, HKESC-2 and EC109 cells, respectively [49]. Hopefully, using BH3-mimetic drugs alone or in combination with other drugs has broad prospects in clinical application (Table 2).
Small-molecule compounds targeting apoptosis in EC
| Name | Structure | Regulatory mechanism | Function | EC subtype | Cancer cell line (activity) | References |
|---|---|---|---|---|---|---|
| S63845 |  | MCL-1↓, BCL-XL↓, BAX↑ | Induce apoptosis | EAC | FLO-1 (IC50=0.8μM) | [46] |
| A1155463 |  | MCL-1↓, BCL-XL↓, BAX↑ | Induce apoptosis | EAC | FLO-1 (IC50=1.13μM) | [46] |
| AT-101 |  | BCL-2↓, MCL-1↓ | Induce apoptosis | EAC | KATOⅢ (IC50=3011 nM) JHESO (IC50=1678 nM) | [47,48] |
| ABT-263 | 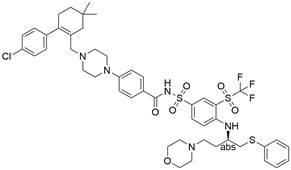 | BCL-xL↓, BCL-2↓, BCL-w↓, PARP↑, caspase-3/9↑ | Induce apoptosis | ESCC | EC109 (IC50=10.7±1.4 μM) HKESC-2 (IC50=7.1±1.5 μM) CaES-17 (IC50=8.2±1.6 μM) | [49] |
| Casticin | 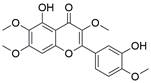 | BCL-2↓, BAX↑, PARP↑, caspase-3/9↑, JNK↑ | Induce apoptosis | ESCC | TE-1 ECA109 | [50] |
| Pristimerin |  | BCL-2↓, BAX↑, caspase-3/9↑ | Induce apoptosis | ESCC | ECA109 | [51] |
| Corilagin | 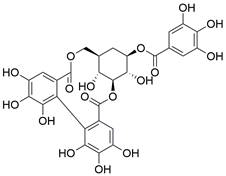 | BCL-2↓, BAX↑, caspase-3/8/9↑ | Induce apoptosis | ESCC | ECA109 (IC50=28.58 ± 2.08μM) KYSE-150 (IC50=35.05 ±2.86μM) | [52] |
| Matrine |  | BCL-2↓, BAX↑, caspase-3/8/9↑, ROS↑ | Induce apoptosis | ESCC | KYSE-150 (IC50=1.94 mg/ml) | [53] |
| Ch‑19 | 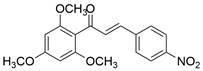 | BCL-2↓, Bad↑, Bim↑, PUMA↑, BAX↑ , PARP↑, caspase-3↑, ROS↑ | Induce apoptosis | ESCC | ECA109 (IC50=9.43μM) KYSE-450 (IC50=4.97μM) | [54] |
| LCL161 | 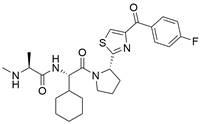 | cIAP1↓, XIAP↓ | Induce apoptosis | ESCC | KYSE-150 (IC50=34.5μM) | [62,63] |
| Tat-SmacN7 | Not known | cIAP1↓, XIAP↓ | Induce apoptosis | ESCC | EC109 | [64] |
| CPP (+TRAIL) | 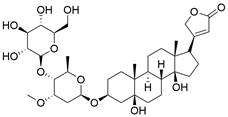 | FoxP3↓, DR4↑, DR5↑ | Induce apoptosis | ESCC | YES-2 KYSE-150 KYSE-510 | [80] |
| Isoalantolactone | 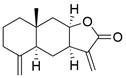 | DR5↑, ROS↑ | Induce apoptosis | ESCC | ECA109 EC9706 TE- 1 TE-13 | [81] |
| CMSP (+TRAIL) | 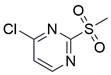 | DR4↑, DR5↑, p38 MAPK↑ | Induce apoptosis | ESCC | KYSE-30 | [82] |
| 3-DSC | 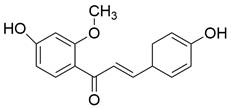 | JNK/p38 MAPKs↑ | Induce apoptosis | ESCC | KYSE-30 (IC50=19.8μM) KYSE-70 (IC50=12.2μM) KYSE-450 (IC50=24.7μM) KYSE-510 (IC50=24.8μM) | [86] |
| DVDMs |  | JNK/p38 MAPKs↑ | Induce apoptosis | ESCC | ECA109 | [87] |
| Quina | 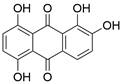 | JNK/p38 MAPKs↑, STAT3↓, NF-κB↓ | Induce apoptosis | ESCC | HCE-4 TE-2 | [88] |
| Echinatin |  | JNK/p38 MAPKs↑ | Induce apoptosis | ESCC | KYSE-30 (IC50=15μM) KYSE-70 (IC50=15μM) KYSE-410 (IC50=6μM) KYSE-450 (IC50=13μM) KYSE-510 (IC50=10μM) | [89] |
| PPT | 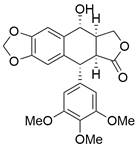 | JNK/p38 MAPKs↑ | Induce apoptosis | ESCC | KYSE-30 (IC50=0.15μM) KYSE-70 (IC50=0.32μM) KYSE-410 (IC50=0.15μM) KYSE-450, (IC50=0.26μM) KYSE-510 (IC50=0.24μM) | [90] |
| Lobaplatin | 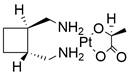 | PI3K/AKT↓ | Induce apoptosis | ESCC | ECA109 (IC50=3.99 ± 0.46μM) EC9706 (IC50=10.3 ± 1.9μM) KYSE-150 (IC50=7.91 ± 2.2μM) | [92] |
| Aprepitant | 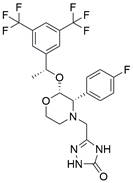 | PI3K/AKT/NF-κB↓ | Induce apoptosis | ESCC | KYSE-30 (IC50=48.21μM) | [93] |
| Ricolinostat | 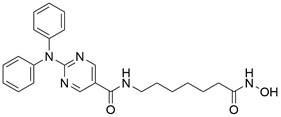 | PI3K/AKT/mTOR↓, ERK↓ | Induce apoptosis | ESCC | EC109 (IC50=46μM) KYSE-150 (IC50=57μM) TE-1 (IC50= 45 μM) TE-13 (IC50=37 μM) | [94] |
| VES | 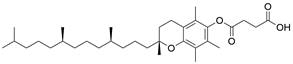 | PI3K/AKT/mTOR↓ | Induce apoptosis | ESCC | EC109 (IC50=25.1uM) | [95] |
| Psoralidin |  | PI3K/AKT↓, NF-κB↓ | Induce apoptosis | ESCC | EC9706 | [96] |
| LH-20-15 | Not known | PI3K/AKT/GLUT1↓ | Induce apoptosis | ESCC | EC9706 (IC50=54.3 mg/L) | [97] |
| Oridonin | 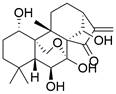 | PI3K/AKT/mTOR↓, Ras/Raf↓ | Induce apoptosis | ESCC | KYSE-150 (IC50=28.69 ± 1.45μM) EC9706 (IC50=34.43 ± 1.53μM) KYSE-30 (IC50=32.29 ± 1.51μM) | [98] |
| Cordycepin | 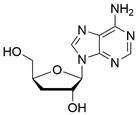 | PI3K/AKT/mTOR↓, AMPK↑ | Induce apoptosis | ESCC | HK (IC50=86.12μM) K180 (IC50=66.84μM) K70 (IC50=69.27μM) ECA109 (IC50=73.82μM) | [99] |
| Hinokiflavone | 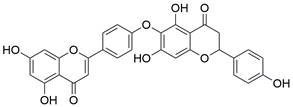 | PI3K/AKT/mTOR↓ | Induce apoptosis | ESCC | KYSE-150 (IC50=24.91μM) TE-14 (IC50=22.07μM) | [100] |
| Osthole |  | PI3K/AKT↓, | Induce apoptosis | ESCC | KYSE-150 (IC50=102.51μM) KYSE-410 (IC50=114.02μM) | [101] |
| Capilliposide C | 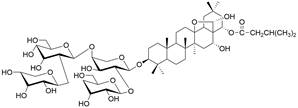 | PI3K/AKT/mTOR↓ | Induce apoptosis | ESCC | TE-1 (IC50=5.43±0.63 μM) TE-2 (IC50=6.64±0.91μM) | [102] |
| DEX | 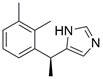 | ERK↓ | Induce apoptosis | ESCC | ECA109 | [103] |
| APR-246 | 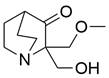 | ROS↑, p73/Noxa↑ | Induce apoptosis | ESCC | KYSE-410 (IC50=31.6μM) TE-1 (IC50=10.5μM) | [104] |
| Luteolin | 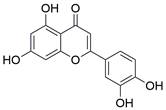 | ROS/JNK↑ | Induce apoptosis | ESCC | TE-1 EC109 | [105] |
| Plumbagin | 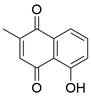 | STAT3/PLK1/AKT↓ | Induce apoptosis | ESCC | KYSE-150 (IC50=6.4μM) KYSE-450 (IC50=8.0μM) | [106] |
| Gossypetin |  | MKK3/MKK6/p38↓ | Induce apoptosis | ESCC | KYSE-30 KYSE-450 KYSE-510 | [107] |
| Artemisinin | 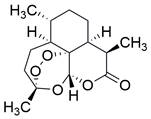 | Wnt/β-catenin↓ | Induce apoptosis | ESCC | EC109 (IC50=55.33μM) | [108] |
| Lvermectin | Not known | ROS↑, NF-κB↓ | Induce apoptosis | ESCC | KYSE-70 (IC50=10μM) KYSE-30 (IC50=6μM) | [109] |
| Dehydrocostus lactone |  | JAK2/STAT3/PLK ↓ | Induce apoptosis | ESCC | KYSE-150 (IC50=5-10μM) ECA109 (IC50=5-10μM) | [110] |
| Cinobufagin | 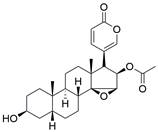 | p73↑ | Induce apoptosis | ESCC | EC109 (IC50=10.13 ± 0.02μM) KYSE-150 (IC50=0.09 ±0.04 μM) KYSE-520 (IC50=0.08 ± 0.03μM) | [111] |
| b-AP15 | 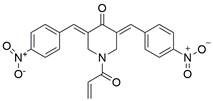 | c-Myc↑, Noxa↑ | Induce apoptosis | ESCC | EC1 (IC50=0.4-0.6μM) EC109 (IC50=0.1-0.2μM) KYSE-510 (IC50=0.1-0.2μM) KYSE-450 (IC50=0.4-0.6μM) | [112] |
| ACP-5862 | 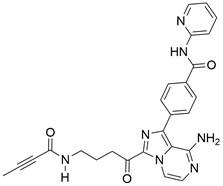 | ROS/ER stress↑ | Induce apoptosis | ESCC | EC109 (IC50=3.55μM) KYSE-270 (IC50=8.21μM) | [113] |
*↓, decrease/inhibition; ↑, increase/activation
Natural compounds
To regulate apoptosis, natural compounds and their derivatives have gradually become a new source of inspiration and have the advantages of easy preparation, low multidrug resistance and low cytotoxicity. Some of these natural compounds have been proven to exert anti-EC activity by targeting BCL-2 family proteins. For instance, a flavonoid named casticin has been reported to induce apoptosis of ESCC cells by repressing BCL-2 expression and upregulating BAX, cleaved caspase-3, caspase-9, and cleaved PARP, as well as activating the JNK signaling pathway [50]. Moreover, a natural quinonoid triterpene named pristimerin was found to downregulate BCL-2 expression and upregulate the expression of BAX and caspase-3 and caspase-9, resulting in apoptosis of ESCC cells. Further results of in vivo experiments demonstrated that pristimerin also inhibited tumor growth in an EC mouse model [51]. In addition, a tannin named corilagin was found to promote apoptosis of ESCC Eca109 and KYSE150 cells by increasing the BAX/BCL-2 ratio as well as the expression of cleaved caspase-3, caspase-8, and caspase-9[52]. Moreover, the Chinese traditional herbal medicine matrine induces apoptosis of ESCC cells by downregulating the expression of BCL-2 and upregulating the expression of BAX and caspase-3, caspase-8 and caspase-9, as well as increasing ROS [53]. In addition, 2,4,6‑trimethoxy‑4'‑nitrochalcone (Ch‑19), a new synthetic chalcone derivative, has been reported to exert an anti-EC effect by inducing ROS accumulation in and apoptosis of EC cells. In ESCC Eca109 and KYSE450 cells, Ch-19 promoted apoptosis by downregulating BCL-2 expression, upregulating Bad, Bim, PUMA and BAX expression, and activating PARP and caspase-3 [54] (Table 2).
Overexpression of BCL-2 family proteins renders cancer cells resistant to apoptotic signals, accelerating cancer progression and reducing treatment response. Thus, using synthetic chemical compounds or natural product-derived compounds to inhibit BCL-2 family proteins has critical importance in the treatment and prognosis of EC [55]. However, these compounds have ongoing crucial challenges for clinical application, which are in large part due to their on-target effects on normal tissues and organs. In addition, the poor stability and short half-life of some of the compounds mentioned above are also major issues that have seriously impaired their clinical application. These challenges can potentially be overcome by utilizing delivery systems based on stable, efficient and highly targeted vectors, such as exosomes derived from human tumor cells or normal cells. Consequently, targeting BCL-2 family proteins to induce apoptosis has great potential to treat patients with EC, and further study of delivery systems combined with other agents, such as exosome-based carriers combined with various BCL-2 inhibitory preparations, have much promise for EC treatment.
Targeting IAP proteins
As anti-apoptotic proteins, IAPs are reported to be overexpressed in various cancers, including EC, and to be linked to resistance to chemotherapy. Therefore, targeting IAPs, including XIAP, cIAP1, and cIAP2, is generally considered a promising therapeutic strategy [56-58]. The endogenous mitochondrial protein Smac can promote apoptosis by antagonizing IAP proteins. Researchers found that the expression of Smac in EC cell lines and in tumor specimens from patients with EC was downregulated, which was believed to be related to decreased sensitivity to EC chemotherapy [59]. Smac mimetics, synthetic small-molecule peptides, have been designed and developed as a new class of targeted drugs that can mimic the proapoptotic mitochondrial protein Smac for cancer treatment in recent years [60]. In general, Smac mimetics can be divided into two types: monovalent and bivalent, which contain one and two SMAC-mimicking units, respectively. To date, Smac mimetics, including but not limited to five monovalent compounds (CUDC-427/GDC-0917, Debio 1143/AT-406/SM-406, LCL161, ASTX660 and RG7419/GDC-0152) and three bivalent agents (AEG40826/HGS1029, APG-1387/SM-1387 and birinapant/TL32711), have been evaluated as therapeutic agents for solid tumors and hematological cancers in early clinical trials. In these clinical trials, Smac mimetics were used alone or in combination with other treatments and demonstrated clinical efficacy and acceptable safety [61]. The Smac mimetic LCL161 was reported to enhance apoptosis by downregulating the expression of cIAP1 and promoting the activation of caspase 8, which acts as a strong radiosensitizer to X-ray irradiation in ESCC cells [62]. Similarly, another study also reported that LCL161 could promote apoptosis of Eca109 cells in a dose-dependent manner by removing the repression of caspase by XIAP [63]. Furthermore, a Smac mimetic named Tat-SmacN7 was developed and shown to be a new and potent radiosensitizer of human EC cells because it induces apoptosis mediated by caspase activation by removing the negative blockers XIAP and cIAP-1 [64]. In summary, these findings indicate that using Smac mimetics alone or in combination with conventional anticancer drugs can produce better effects for the treatment of EC. Further studies of Smac mimetics may help develop novel therapeutic strategies for patients with EC (Table 2).
Targeting death receptor-induced apoptosis
Among TNFR superfamily members, DR4 and DR5 are significantly increased in cancer cells compared to normal cells and thus are regarded as the most promising candidates for targeted therapy for tumors. In humans, interaction between TNF-related apoptosis-inducing ligand (TRAIL) and DR4 or DR5 can lead to the assembly of DISC, which is followed by caspase-8-dependent apoptosis. Consequently, recombinant human TRAIL (RhTRAIL) protein or agonistic antibodies targeting DR4 and DR5, also known as proapoptotic receptor agonists (PARAs), have been developed and found to induce apoptosis of several different cancer cell lines [65]. Additionally, numerous studies have shown that PARAs and rhTRAIL can enhance the sensitivity of tumors to therapies, including radiotherapy, chemotherapy, and targeted therapy [66-69]. Nevertheless, clinical trials of TRAIL have not demonstrated satisfactory results, which is thought to be due to acquired resistance of tumor cells to TRAIL-induced apoptosis [70]. Researchers have thus shown a keen interest in compounds that are capable of increasing the expression of death receptors. Compounds that can sensitize cells to TRAIL-induced apoptosis can be divided into three groups: 1) clinically used anticancer drugs (e.g., etoposide, mitomycin c and mitoxantrone) [71-73], 2) natural compounds with anticancer properties (e.g., ginsenoside compound K, magnolol, polyphenol mixture and chikusetsusaponin IVa butyl ester) [74-76], and 3) anticancer agents that are currently being developed (e.g., Nutlin-3, capsazepin and GW280264X) [77-79]. For EC, a natural compound named periplocin (CPP) alone or in combination with TRAIL was found to dose-dependently increase the expression of DR4/DR5, FADD and cleaved caspase-3 by inhibiting FoxP3, inducing apoptosis of ESCC cells and making ESCC cells more susceptible to TRAIL-induced apoptosis [80]. Similarly, a major sesquiterpene lactone named isoalantolactone was reported to promote apoptosis by upregulating DR5 and ROS in human EC cells [81]. In addition, p-hydroxylcinnamaldehyde (CMSP) is an extract from traditional Chinese medicine that has been proven to enhance TRAIL-induced apoptosis of ESCC cells by increasing the expression of DR4 and DR5 and activating the p38 MAPK signaling pathway [82]. These sensitizers alone or in combination with a TRAIL-related agent might be novel clinical treatment options for patients with EC (Table 2).
Targeting the JNK/p38 MAPK pathway
Mitogen-activated protein kinase (MAPK) signaling pathways have crucial roles in various cellular networks related to the cell cycle, cell growth, cell survival and cell death [83]. Activation of MAPK pathways by ROS has been linked to induction of cell apoptosis. By sequential phosphorylation of MAPK, it is possible to activate members of the MAPK pathway, such as JNK and p38. Activating p38 in the MAPK signaling pathway can promote apoptosis of tumor cells and halt tumor formation [84]. JNKs can regulate the apoptotic signaling pathway by either regulating the activities of proapoptotic or antiapoptotic proteins or inducing the expression of apoptotic genes [85]. Therefore, patients with EC may benefit from selective targeting of the JNK/p38 MAPK pathway. A recent study reported that a chemical separated from Caesalpinia sappan L named 3-deoxysappanchalcone (3-DSC) can induce cell cycle arrest and ROS-mediated apoptosis of EC cells via the JNK/p38 MAPK signaling pathway [86]. Similarly, sinoporphyrin sodium (DVDMS), a novel sensitizer isolated from photofrin, can induce ROS-mediated apoptosis in combination with photodynamic therapy (PDT) via the JNK/p38 MAPK signaling pathway [87]. In addition, quinalizarin (Quina), a prominent constituent of several herbal medicines, is believed to exert antitumor effects in EC, which can induce apoptosis by modulating MAPK, STAT3, and NF-κB signaling pathways through the generation of ROS [88]. A retrochalone from licorice, echinatin (Ech), was found to promote apoptosis of ESCC cells by generating ROS/ER stress as well as activating the JNK/p38 MAPK signaling pathway [89]. Moreover, as an epimer of podophyllotoxin isolated from the roots of Podophyllum hexandrum, picropodophyllotoxin (PPT) has been reported to exert antitumor effects by inducing apoptosis via upregulation of ROS levels and activation of the JNK/p38 signaling pathways [90] (Table 2). Thus, it might be possible to treat EC by regulating JNK/p38 MAPK pathway-mediated apoptosis.
Targeting the PI3K/AKT/mTOR pathway
The phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway regulates some common cellular functions that are also crucial for tumorigenesis, such as cell metabolism, angiogenesis, cell proliferation, cell cycle progression and apoptosis [91].
Synthetic compounds
As a third-generation platinum antineoplastic drug, lobaplatin (LBP) can effectively induce apoptosis, repress proliferation and enhance radiosensitivity by inhibiting the PI3K/AKT pathway in ESCC [92]. Furthermore, a nonpeptide NK1R antagonist named aprepitant has been reported to induce apoptosis and G2/M arrest via the PI3K/Akt/NF-κB axis in ESCC spheres [93]. Furthermore, ricolinostat (ACY-1215), a selective HDAC6 inhibitor, has been shown to induce apoptosis and G2/M arrest in ESCC cells through the PI3K/AKT/mTOR and ERK pathways, which were then confirmed in vivo [94]. As a potential chemical agent for cancer prevention and therapy, vitamin E succinate (VES) was found to induce apoptosis by blocking the PI3K/AKT/mTOR axis in ESCC cells [95]. Moreover, psoralidin could effectively inhibit proliferation and enhance apoptosis by inhibiting the PI3K/Akt and NF-κB signaling pathways in ESCC cells [96] (Table 2).
Natural compounds
The use of natural compounds for the treatment of human diseases has a long history, and these compounds are thus an important resource for developing drugs. For EC, LH-20-15, a cytotoxic extract obtained from Gekko japonicus, demonstrates the ability to inhibit the proliferation and induce apoptosis of ESCC cells, which are achieved through the inhibition of the PI3K/Akt/GLUT1 signaling pathway [97]. In addition, as an active diterpenoid isolated from Rabdosia rubescens, oridonin was also found to cause mitochondria-dependent apoptosis of ESCC cells by inhibiting the Ras/Raf and PI3K/AKT/mTOR pathways. An in vivo experiment demonstrated that treatment with oridonin inhibited tumor growth in an EC mouse model [98]. Cordycepin, a nucleoside analog, has been shown to augment the chemosensitivity of ESCC cells to cisplatin. This effect is achieved by inhibiting the PI3K/AKT/mTOR signaling pathway and activating AMPK, representing a novel potential treatment for EC [99]. In addition, hinokiflavone, a natural biflavonoid compound, has been reported to induce the apoptosis and inhibit the proliferation of ESCC cells by blocking the PI3K/AKT/mTOR signaling pathway [100]. In addition, an active component extracted from the fruit of Fructus cnidii named osthole promoted cell cycle arrest in and the apoptosis of ESCC cells by inhibiting the PI3K/AKT signaling pathway [101]. Capilliposide C (CPS-C) is an extract of a traditional Chinese medicine that can sensitize ESCC cells to chemotherapy by promoting apoptosis by inhibiting the PI3K/Akt/mTOR pathway [102]. In summary, regulating apoptosis by targeting the PI3K/AKT/mTOR axis is a novel and promising therapeutic strategy for patients with EC (Table 2).
Targeting other apoptosis regulators
New targets for the treatment of EC have also been studied in recent years along with the above common targets. For instance, dexmedetomidine (DEX) has been shown to suppress the proliferation and induce apoptosis of EC cells by inhibiting the expression of the C-Myc gene through the blockade of the ERK signaling pathway [103]. In addition, in a recent library screening using cells harboring mutant p53, the small-molecule compound APR-246 was identified. Further study demonstrated that APR-246 induced pronounced antitumor effects by promoting ROS-p73-Noxa-mediated apoptosis of ESCC cell lines bearing p53 missense mutations. These observations were confirmed using xenografts and patient-derived xenograft (PDX) models of p53-mutant ESCC [104]. Moreover, in vitro and in vivo experiments were performed to confirm that combining luteolin with low-dose paclitaxel promotes ROS/JNK pathway-mediated apoptosis of ESCC cells [105]. Additionally, a natural quinoid constituent named plumbagin was found to inhibit the proliferation and promote the apoptosis of ESCC cells in vivo and in vitro by inhibiting STAT3-PLK1-AKT signaling [106]. In addition, as a hexahydroxylated flavonoid found in many flowers and hibiscus, gossypetin was found to induce apoptosis of ESCC cells in vivo by inhibiting the MKK3/MKK6/p38 signaling pathway, which might prevent the development of EC among healthy individuals, especially those who are at high risk of developing this cancer [107]. Artemisinin (ART), an antimalarial compound, has been reported to exhibit inhibitory effects on cell proliferation, migration, and invasion in addition to inducing apoptosis of ESCC cells. These effects are attributed to the repression of the Wnt/β-catenin signaling pathway [108]. Additionally, a polycyclic lactone pesticide produced by Streptomyces avermitilis named ivermectin was found to promote apoptosis of ESCC cells by increasing ROS accumulation and repressing the NF-κB signaling pathway [109]. A recent study demonstrated that one of the sesquiterpene lactones, dehydrocostus lactone (DEH), could induce cell cycle arrest in and promote the apoptosis of ESCC cells by repressing the JAK2/STAT3/PLK signaling pathway, inducing a potent antitumor effect [110]. Furthermore, the traditional Chinese herbal extract cinobufagin has been reported to promote cell cycle arrest and apoptosis to block the growth of ESCC cells by activating the p73 signaling pathway [111]. The deubiquitinase inhibitor b-AP15 has been identified as capable of inducing apoptosis in ESCC cells by upregulating c-Myc and Noxa. These findings suggest its potential utility as a therapeutic agent for ESCC [112]. Moreover, as a major metabolite of acalabrutinib, ACP-5862 has been reported to exert antitumor effects in ESCC by promoting ER stress-mediated apoptosis by upregulating the production of ROS [113] (Table 2). These findings provide additional potential novel therapeutic apoptosis-related targets and pathways for treating EC that may have far-reaching ramifications in the future.
LncRNAs regulating apoptosis in EC
Notably, lncRNAs, which are endogenous cellular RNAs longer than 200 nucleotides and do not encode proteins, have been shown to play a significant role in the initiation and progression of several cancers, including EC. For instance, lncRNA SNHG7 was significantly upregulated in EC cells and tissues, and SNHG7 was found to enhance ESCC cell proliferation and inhibit ESCC cell apoptosis through the modulation of p15 and p16 expression [114]. In addition, downregulation of lncRNA IUR has been reported in ESCC, and it is associated with an unfavorable prognosis in ESCC patients. Further study proved that IUR could regulate cancer cell proliferation and the apoptosis of ESCC cells via miR‑21 and PTEN [115]. Moreover, lncRNA ZFAS1 was found to be upregulated in ESCC tissues, and further study proved that ZFAS1 could activate the proliferation, migration and invasion of ESCC cells and suppress their apoptosis by regulating miR-124 and STAT3 [116]. In addition to the lncRNAs mentioned above, other lncRNAs that can regulate the apoptosis of EC cells are also summarized in Table 6 [117-138]. LncRNAs may be useful biomarkers for diagnosing EC and predicting prognosis and relapse, and the ongoing development of drugs targeting these lncRNAs might produce new therapeutic targets.
LncRNAs targeting RCD subroutines in EC
| Name | Regulatory mechanism | Function | EC subtype | References |
|---|---|---|---|---|
| LncRNA SNHG7 | P15/16 | Inhibit apoptosis | ESCC | [114] |
| LncRNA IUR | miR‑21/PTEN | Induce apoptosis | ESCC | [115] |
| LncRNA ZFAS1 | miR-124/STAT3 | Inhibit apoptosis | ESCC | [116] |
| LncRNA NEAT1 | miR-377/E2F3 | Inhibit apoptosis | ESCC | [117] |
| LncRNA FER1L4 | Unspecified | Induce apoptosis | ESCC | [118] |
| LncRNA LINC01535 | JAK/STAT3 | Inhibit apoptosis | ESCC | [119] |
| LncRNA TUG1 | miR-1294/PLK1 | Inhibit apoptosis | ESCC | [120] |
| LncRNA GIHCG | miR-29b-3p/ANO1 | Inhibit apoptosis | ESCC | [121] |
| LncRNA FAM83A-AS1 | miR-214/CDC25B | Inhibit apoptosis | ESCC | [122] |
| LncRNA MIR22HG | STAT3/c-Myc/p-FAK | Inhibit apoptosis | EAC | [123] |
| LncRNA LINC01234 | Unspecified | Inhibit apoptosis | ESCC | [124] |
| LncRNA SNHG1 | miR-204/HOXC8 | Inhibit apoptosis | ESCC | [125] |
| LncRNA HAGLROS | miR-206/NOTCH3 | Inhibit apoptosis | ESCC | [126] |
| LncRNA TUG1 | miR-498/XBP1 | Inhibit apoptosis | ESCC | [127] |
| LncRNA SNHG6 | miR-101-3p/EZH2 | Inhibit apoptosis | ESCC | [128] |
| LncRNA CDKN2B-AS1 | TFAP2A /SCN1 | Inhibit apoptosis | ESCC | [129] |
| LncRNA LINC00467 | miR-485-5p/DPAGT1 | Inhibit apoptosis | ESCC | [130] |
| LncRNA LINC00491 | Unspecified | Inhibit apoptosis | ESCC | [131] |
| LncRNA PVT1 | miR-145/FSCN1 | Inhibit apoptosis | ESCC | [132] |
| LncRNA GAS5 | miR-301a/CXCR4 | Inhibit apoptosis | ESCC | [133] |
| LncRNA LINC00511 | microRNA-150-5p | Inhibit apoptosis | ESCC | [134] |
| LncRNA LINC00857 | STAT3, MET | Inhibit apoptosis | EAC | [135] |
| LncRNA C9orF139 | miR-661/HDAC11 | Inhibit apoptosis | ESCC | [136] |
| LncRNA MIR205HG | miR-214/SOX4 | Inhibit apoptosis | ESCC | [137] |
| LncRNA DLEU2 | miR-30e-5p/E2F7 | Inhibit apoptosis | ESCC | [138] |
| LncRNA DRAIC | miR-149-5p/NFIB | Inhibit autophagy | ESCC | [189] |
| LncRNA LINC00337 | E2F4/TPX2 | Induce autophagy | ESCC | [190] |
| LncRNA OIP5-AS1 | GPX4 | Inhibit ferroptosis | ESCC | [236] |
| LncRNA BBOX1-AS1 | miR-513a-3p/SLC7A11 | Inhibit ferroptosis | ESCC | [237] |
Developing drugs that target apoptotic pathways directly has great potential to cause tumor regression in difficult-to-treat EC, and thus, these drugs are expected to be used in combination with other treatments, such as immunotherapy, drugs targeting oncogenic survival pathways, radiotherapy, and chemotherapy. Furthermore, lncRNAs may be useful in the diagnosis and prognostication of EC and even be drug targets. Undoubtedly, targeting apoptotic pathways with small-molecule compounds is a promising method for EC therapy, warranting further in-depth investigations.
Targeting autophagy pathways with small-molecule compounds in EC
Brief introduction to autophagy
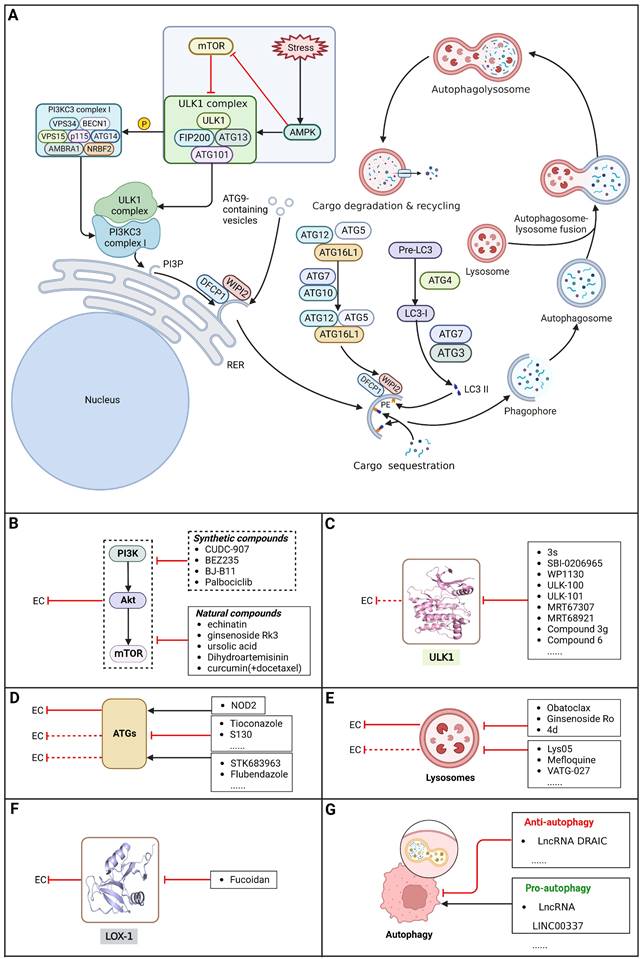
Autophagy, first coined by Christian de Duve in 1963, refers to an evolutionarily conserved intracellular catabolic process involving the formation of double-membraned vesicles named autophagosomes that causes cellular components (e.g., organelles, proteins and lipids) to be delivered to lysosomes for degradation and recycling, contributing to the maintenance of cellular homeostasis. The autophagy process can be conceptualized as consisting of five distinct stages: 1) initiation, 2) nucleation of the phagophore, 3) formation of the autophagosome, 4) fusion of the autophagosome and lysosome, and 5) cargo degradation and recycling (Figure 3) [139-141]. As a multistep lysosomal degradation process, autophagy has a dynamic role in the progression of cancer based on the context. In the earliest stages of tumorigenesis, autophagy can serve as a suppressor of tumor growth by eliminating mutated proteins and oncogenes that possess the ability to induce cellular mutations and/or cancer. Nevertheless, as the tumor advances to an advanced stage, autophagy can transition into a defensive and survival mechanism for cancer cells. It contributes to the survival, growth, and metastasis of established tumors by providing essential nutrients to cope with environmental stresses, including hypoxia, DNA damage, metabolic stress, nutrient shortage and cancer therapy [142,143]. In this context, determining how to take advantage of this "double-edged sword" in the treatment of EC via external interventions such as small-molecule compounds have been a hot research topic among cancer researchers.
Core autophagy pathways in EC and EC therapeutic approaches by targeting autophagy pathways. (A) Core autophagy pathways in EC. The autophagy process can be conceptualized as consisting of five distinct stages: 1) initiation, 2) nucleation of the phagophore, 3) formation of the autophagosome, 4) fusion of the autophagosome and lysosome, and 5) cargo degradation and recycling. (B) Small-molecule compounds targeting autophagy-related the PI3K/AKT/mTOR pathway in EC. (C) Small-molecule compounds targeting autophagy-related ULK1 in other cancers. (D) Small-molecule compounds targeting autophagy-related ATG proteins in EC and other cancers. (E) Small-molecule compounds targeting autophagy-related lysosomes in EC and other cancers. (F) Small-molecule compounds targeting autophagy-related LOX-1 in EC. (G) Some lncRNAs targeting autophagy-related pathways in EC. (Created with BioRender.com)
Targeting the PI3K/AKT/mTOR pathway
Besides its role in regulating apoptosis, the PI3K/Akt/mTOR pathway is reported as the central pathway regulating autophagy and is regarded as a crucial survival pathway in tumor cells related to aggressive growth and malignant progression. Mounting evidence supports the ability of small-molecule compounds to induce autophagy in EC cells through modulation of the PI3K/AKT/mTOR pathway [144].
Synthetic compounds
A recent study demonstrated that HDACs and the PI3K/Akt/mTOR signaling pathway were highly activated in EC cell lines and ESCC patients. As an innovative artificial small-molecule inhibitor that can suppress both the PI3K/Akt/mTOR signaling pathway and HDACs, CUDC-907 has been reported to activate autophagy in ESCC cells by suppressing the PI3K/Akt/mTOR signaling pathway or LCN2 to induce accumulation of ROS, suggesting a potential targeted therapy option for patients with EC [145]. Moreover, it has been reported that the dual PI3K/mTOR inhibitor BEZ235 enhances antitumor activities by promoting autophagy in ESCC cells through the inhibition of the PI3K/Akt/mTOR signaling pathway [146]. In addition, BJ-B11, a selective Hsp90 inhibitor, exhibited effective and potent antitumor activity in ESCC cells by inducing autophagy by suppressing the Akt/mTOR/p70S6K signaling pathway in a time- and concentration-dependent manner [147]. Additionally, a recent study showed that the CDK4/6 inhibitor palbociclib can effectively improve the radiosensitivity of ESCC cells in vivo and in vitro by suppressing mTOR and thus has great potential to act as a radiosensitization agent for ESCC treatment [148] (Table 3).
Small-molecule compounds targeting autophagy in EC
| Name | Structure | Regulatory mechanism | Function | EC subtype | Cancer cell line (activity) | References |
|---|---|---|---|---|---|---|
| CUDC-907 | 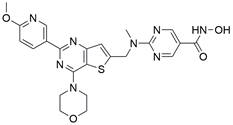 | PI3K/Akt/mTOR↓, LCN2↓ | Induce autophagy | ESCC | KYSE-150 (IC50=14.27 nM) KYSE-450 (IC50=6.893 nM) KYSE-510 (IC50=8.703 nM) KYSE-30 (IC50=16.95 nM) | [145] |
| BEZ235 | 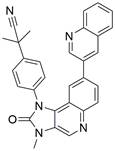 | PI3K/Akt/mTOR↓ | Induce autophagy | ESCC | ECA109 (IC50=160.7 nM) TE-1 (IC50=109.4 nM) | [146] |
| BJ-B11 | Not known | Akt/mTOR/p70S6K↓ | Induce autophagy | ESCC | ECA109 (IC50=0.31±0.01 μM) | [147] |
| Palbociclib |  | mTOR↓ | Induce autophagy | ESCC | EC109 KYSE-150 KYSE-30 KYSE-70 | [148] |
| Echinatin |  | Akt/mTOR↓ | Induce autophagy | ESCC | KYSE-30 KYSE-270 | [149] |
| Rk3 | 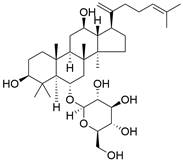 | PI3K/Akt/mTOR↓ | Induce autophagy | ESCC | ECA109 KYSE-150 | [150] |
| Ursolic acid | 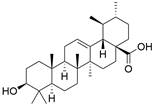 | PI3K/Akt/mTOR↓ | Induce autophagy | ESCC | TE-8 (IC50=39.01 nM) TE-12 (IC50=29.65 nM) | [151] |
| DHA | 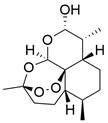 | Akt/mTOR↓ | Induce autophagy | ESCC | ECA109 TE-1 | [152] |
| CUR(+DTX) | 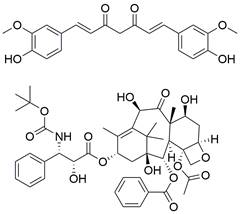 | PI3K/Akt/mTOR↓ | Induce autophagy | ESCC | KYSE-150 (IC50=5.80 ± 0.98 μg/ml) KYSE-510 (IC50=8.91 ± 0.88 μg/ml) | [153] |
| NOD2 | Not known | ATG16L1↑ | Induce autophagy | EAC | OE33 SEG-1 BIC-1 | [168,191] |
| Obatoclax | 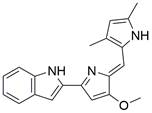 | Obatoclax inhibits the fusion of autophagosomes and lysosomes | Inhibit autophagy | ESCC | EC109 (IC50=0.3μM) | [173,183] |
| Ginsenoside Ro | 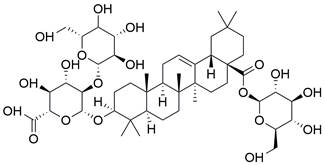 | Ginsenoside Ro inhibits the fusion of autophagosomes and lysosomes | Inhibit autophagy | ESCC | EC9706 ECA109 TE-1 | [184] |
| 4d | 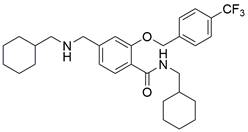 | 4d inhibits the fusion of autophagosomes and lysosomes | Inhibit autophagy | ESCC | ECA109 | [185] |
| Fucoidan | 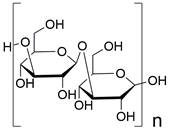 | LOX-1↓ | Inhibit autophagy | ESCC | TE-1 KYSE-150 | [186] |
| 3-MA | 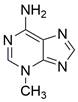 | Unspecified | Inhibit autophagy | ESCC | TE-1 EC9706 | [187,188] |
*↓, decrease/inhibition; ↑, increase/activation
Natural compounds
An active component of licorice named echinatin was found to promote autophagy in ESCC cells through suppressing the Akt/mTOR signaling pathway, resulting in sensitization of ESCC cells to 5-FU treatment [149]. In addition, as a bioactive component derived from Panax notoginseng and ginseng, it was reported that ginsenoside Rk3 could exhibit a significant inhibitory effect on the proliferation and colony formation of ESCC cells by activating autophagy through the inhibition of the PI3K/Akt/mTOR signaling pathway. Experiments with the KYSE150 xenograft model demonstrated that Rk3 significantly suppressed tumor growth and caused little organ toxicity, suggesting that Rk3 may be a promising antitumor agent for EC [150]. Furthermore, ursolic acid (UA) has demonstrated effectiveness in inhibiting the growth and metastasis of ESCC cells by inducing autophagy mediated by reactive oxygen species (ROS) through the inhibition of the PI3K/Akt/mTOR signaling pathway [151]. Dihydroartemisinin (DHA), the main active derivative of artemisinin, has been documented to induce autophagy in a dose-dependent manner by reducing Akt phosphorylation and suppressing the Akt/mTOR signaling pathway, which could lead to the inhibition of ESCC cell migration [152]. Furthermore, the combination of curcumin (CUR) and docetaxel (DTX) was found to induce autophagy in ESCC cells through the PI3K/AKT/mTOR signaling pathway, enhancing the antitumor effect of DTX in vivo [153] (Table 3).
As the PI3K/AKT/mTOR pathway is hyperactive in EC, there are opportunities for the discovery and research of anti-EC drugs. Targeting the PI3K/AKT/mTOR pathway selectively with small-molecule compounds, such as mTOR inhibitors, PI3K inhibitors, AKT inhibitors, and dual PI3K/mTOR inhibitors, to induce autophagy in EC cells represents a promising therapeutic approach for individuals diagnosed with EC.
Targeting ULK1
ULKs are serine/threonine protein kinases that have the ability to form complexes with multiple regulators. Among ULKs, ULK1 is a component of the ULK1 complex (ULK1-ATG13-FIP200-ATG101), which has important functions in autophagy induction. Interestingly, upregulation of ULK1 has been observed in various cancers, including non-small cell lung cancer, colorectal cancer, nasopharyngeal carcinoma, and clear cell renal carcinoma. This upregulation has been associated with treatment resistance and unfavorable prognosis [154-159]. As such, inhibiting ULK1 to regulate autophagy may be an effective therapeutic strategy. A growing number of ULK1 inhibitors have been researched and reported to suppress tumor growth and metastasis and promote autophagy in various cancer types, such as 3s [158], SBI-0206965 [160], WP1130 [161], ULK-100 [162], ULK-101 [162], MRT67307[163], MRT68921 [163], compound 3g [164], and compound 6 [165]. For EC, a study found that ESCC samples had higher levels of ULK1 protein than normal EC cells and tissues, as well as higher ULK1 protein stabilization. Further study showed that the overall survival time was shorter among patients with higher ULK1 expression. Using specific small interfering RNA to inhibit ULK1 expression in ESCC cell lines can block cell proliferation, which supports the idea that blocking ULK1 may be a beneficial therapy for patients with EC [166]. However, there are few relevant studies assessing the effect of ULK1 inhibitors as EC therapy. Therefore, trying to regulate autophagy using existing ULK1 inhibitors that have been applied to other kinds of cancers or developing new ULK1 inhibitors is a promising direction for EC therapeutic research and may have far-reaching ramifications in the future.
Targeting ATG proteins
Membrane PI3P produced by VPS34 recruits PI3P-binding ATG proteins and other factors contributing to the elongation of the phagophore. ATGs such as ATG16L1 play important roles in ubiquitin-like conjugation system 1 and ubiquitin-like conjugation system 2, contributing to the formation of autophagosomes. Thus, targeting ATG proteins may be useful for efficient EC treatment because it would regulate autophagy [167]. As an innate immune receptor for bacteriogenic components activated by muramyl dipeptide (MDP), nucleotide binding oligomerization domain containing 2 (NOD2) was found to be decreased in EC cells, and further study demonstrated that NOD2 overexpression promoted autophagy in and inhibited the proliferation of EAC cells by acting on the ATG16L1 pathway [168]. In addition, there has been very little research on the usage of ATG modulators in EC. Notably, a growing number of ATG modulators have been discovered and tested in other diseases, including various cancers, such as tioconazole [169], S130 [170], STK683963 [171] and flubendazole [172]. Collectively, trying to regulate autophagy using existing ATG modulators or developing new ATG modulators is a novel and promising direction for EC therapeutic research and may have far-reaching ramifications in the future (Table 3).
Targeting lysosomes
Lysosomes have key functions in the growth and metabolism of cells. During degradation of the lysosomal content, the catalytic functions of lysosomal hydrolytic enzymes can only work when the pH is in the range of 4.5 to 5. At present, there are only two autophagy inhibitors approved for clinical use: chloroquine (CQ) and hydroxyquinoline (HCQ), which can inhibit autophagy by deacidifying the lysosome and blocking autophagosome‐lysosome fusion. Nevertheless, the clinical utility of these drugs is restricted due to high doses of CQ and HCQ are needed for effective autophagy inhibition in vitro, which is difficult to accomplish in humans; in addition, there is a lack of selectivity, and there are side effects [173]. Notably, some new drugs targeting lysosomes have been discovered, such as Lys05 [174,175], mefloquine [176,177], VATG-027 [178], DQ661 [179], bafilomycin A1 [180,181], and Ganoderma lucidum polysaccharide (GLP) [182]. For EC, Obatoclax, a newly developed drug with potential as an innovative anticancer agent, was observed to become sequestered within lysosomes. This resulted in the accumulation of LC3 II and p62 proteins, indicating disruption of the autophagosome-lysosome fusion process. The resulting IC50 values were 0.13 and 0.24 µM in HKESC-1 and EC109 cells (ESCC cancer cell lines), respectively [173,183]. Moreover, a new autophagy inhibitor extracted from Panax ginseng named ginsenoside Ro (Ro) can effectively inhibit the fusion of autophagosomes and lysosomes by upregulating the pH in lysosomes and decreasing lysosomal cathepsin activity. This mechanism suppresses autophagy in a variety of EC cell lines, indicating that Ro might serve as an anticancer agent to overcome chemoresistance because of its role as an autophagy inhibitor [184]. In addition, N-(cyclohexylmethyl)-5-(((cyclohexylethyl)amino)methyl)-2-((4-(trifluoromethyl)benzyl)oxy)benzamide (4 d), a synthesized compound, was found to inhibit autophagy by suppressing the autophagosome-lysosome fusion process, which enhanced the chemosensitivity of vincristine in vincristine-resistant ESCC cells, thus serving as a novel autophagy inhibitor[185]. Thus, using existing lysosome inhibitors that have been applied in other kinds of cancers or developing new lysosome inhibitors to improve the outcomes of patients with EC may be promising options (Table 3).
Targeting other autophagy regulators
EC treatments directed at new targets related to autophagy have also been a focus of research in recent years in addition to the above targets. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) has been reported to be overexpressed in EC tissues. A sulfated polysaccharide named fucoidan has been reported to activate autophagy in ESCC cells by inhibiting LOX-1[186]. Both in vitro and in vivo studies have demonstrated that radiation-induced autophagy plays a protective role in preventing cell death. When ESCC EC9706 cells were treated with 10 mM 3-MA and ionizing radiation, the sensitization enhancement ratio was 1.76, and the effect was proven in a xenograft ESCC mouse model in vivo, in which there was an obvious decrease in tumor volume relative to that achieved by single treatment. Unfortunately, the poor solubility profile of 3-MA necessitates administration of high doses to obtain sufficient autophagy inhibition [187,188] Targeting these autophagy-related targets and pathways to regulate autophagy in EC cells may help develop novel therapeutic strategies for patients with EC (Table 3).
LncRNAs regulating autophagy in EC
A study identified downregulation of miR-149-5p and upregulation of lncRNA NFIB and DRAIC in EC cells. Subsequent investigations have revealed that targeting the miR-149-5p/NFIB axis by inhibiting DRAIC can induce autophagy in EC cells, leading to suppressed proliferation and invasion of ESCC cells. Consequently, DRAIC emerges as a potential key gene for diagnosis and treatment of EC [189]. Furthermore, lncRNAs are involved in the regulation of drug resistance in EC. A report has shown that lncRNA LINC00337 is overexpressed in ESCC cells and tissues. Downregulating LINC00337 inhibited autophagy in ESCC cells and enhanced sensitivity to cisplatin via the upregulation of TPX2 by recruiting E2F4[190]. In the context of EC treatment, regulating autophagy in EC cells by targeting lncRNAs has been shown to be a novel and promising approach (Table 6).
Targeting ferroptosis pathways with small-molecule compounds in EC
Brief introduction to ferroptosis
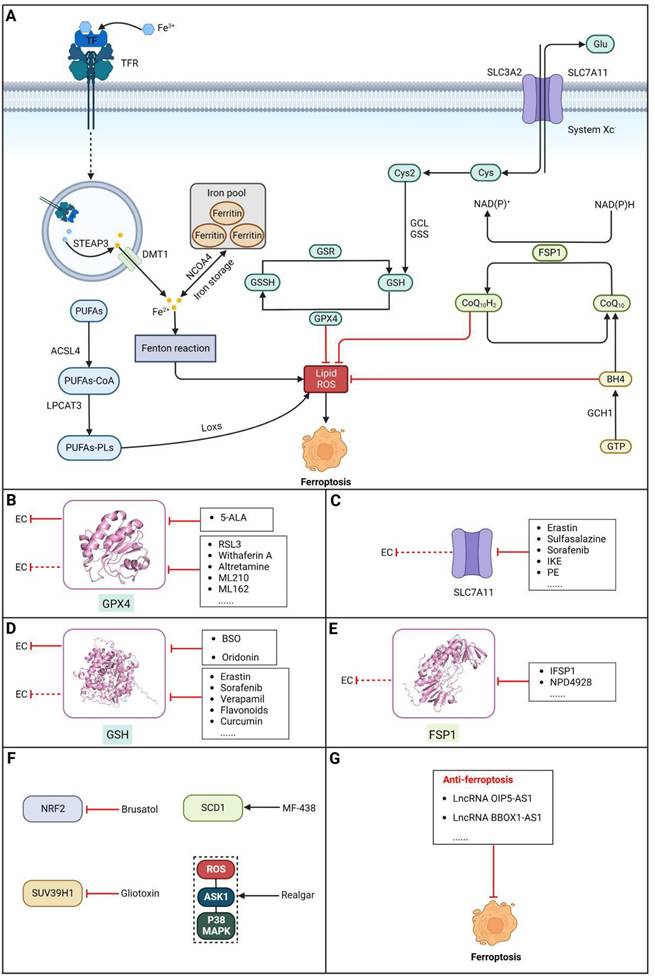
Ferroptosis, proposed by Dixon in 2012, is an iron- and lipid ROS-dependent form of RCD with ruptured outer mitochondrial membrane, condensed mitochondrial membrane, reduced mitochondrial volume and diminished or vanished mitochondrial cristae as the main morphological features [192]. Ferroptosis has been extensively associated with numerous diseases, particularly cancer. Based on accumulating evidence, several pathways are related to the regulation of ferroptosis, including pathways related to iron metabolism, the system Xc-GSH-GPX4 pathway, pathways related to lipid peroxidation, the FSP1-NADPH-CoQ10 pathway and the GCH1-BH4 pathway (Figure 4). Ferroptosis is an iron- and lipid ROS-dependent type of RCD. Nevertheless, some types of cancer cells can resist ferroptosis via at least three mechanisms, including restricting the availability of labile iron, limiting the synthesis and peroxidation of PUFA-PL and upregulating cellular defense systems against ferroptosis [193-195]. Thus, how to breakdown antioxidant defenses to induce ferroptosis in tumor cells has garnered great interest in cancer research communities. Recently, there have been numerous studies on designing and developing anticancer drugs that induce ferroptosis in cancer cells. In this context, it is becoming increasingly clear that inducing ferroptosis in EC cells may be a feasible and practical strategy to improve clinical therapy and give hope to patients with EC.
Core ferroptosis pathways in EC and EC therapeutic approaches by targeting ferroptosis pathways. (A) Core ferroptosis pathways in EC. Ferroptosis is an iron- and lipid ROS-dependent form of RCD, which is determined by several pathways, including pathways related to iron metabolism, the system Xc-GSH-GPX4 pathway, pathways related to lipid peroxidation, the FSP1-NADPH-CoQ10 pathway and the GCH1-BH4 pathway. (B) Small-molecule compounds targeting ferroptosis-related GPX4 in EC and other cancers. (C) Small-molecule compounds targeting ferroptosis-related SLC7A11 in other cancers. (D) Small-molecule compounds targeting ferroptosis-related GSH in EC and other cancers. (E) Small-molecule compounds targeting ferroptosis-related FSP1 in other cancers. (F) Small-molecule compounds targeting ferroptosis-related other regulators in EC. (G) Some lncRNAs targeting autophagy-related pathways in EC. (Created with BioRender.com)
Targeting GPX4
Glutathione peroxidase 4 (GPX4), an antioxidative enzyme, acts as a critical negative regulator of ferroptosis due to its ability to prevent membrane lipid peroxidation [196]. According to a study by Hangauer et al., persistent drug-resistant cancer cells acquire dependency on GPX4, indicating that preventing acquired drug resistance may be achievable by targeting GPX4 [197]. Thus, inducing ferroptosis in cancer cells via inactivation or degradation of GPX4 is being intensely pursued as a novel cancer treatment strategy. A growing number of experimental compounds that can inhibit GPX4 to induce ferroptosis in cancer cells and/or certain normal cells have been identified, such as RSL3, withaferin A, altretamine, ML210, ML162 and some diverse pharmacological inhibitor (DPI) compounds, and these ferroptosis inducers (FINs) inhibit GPX4 activity by covalently and irreversibly binding the selenocysteine (Sec) in its active site, resulting in the buildup of lipid peroxides, ultimately culminating in ferroptosis[198]. Among these compounds, it is noteworthy that the FDA has granted approval for altretamine to treat ovarian cancer, which could induce ferroptosis via GPX4 inhibition [199]. For EC, a recent study demonstrated that ESCC cells escape ferroptosis caused by elevated lipid peroxidation by upregulating GPX4 and SLC7A11, indicating that targeting GPX4 to block this intrinsic protective mechanism against ferroptosis has great potential for the treatment of EC [200]. In a recent study, an unfavorable prognosis was correlated with increased GPX4 expression and decreased HMOX1 expression. The induction of ferroptosis in ESCC cells by 5-Aminolevulinic acid (5-ALA) was demonstrated through the regulation of GPX4 and HMOX1, which was further validated in vivo [201]. However, although the initial in vitro results are promising, the poor pharmacokinetic properties of most GPX4 inhibitors remain a major barrier to their use in vivo [202]. Moreover, some research has confirmed that GPX4 is important for the development of mice, and inactivation of GPX4 can induce acute renal failure or even death in mice [203,204]. Hence, it is imperative to develop and optimize GPX4 inhibitors with improved pharmacokinetics and selectivity for clinical use (Table 4).
Small-molecule compounds targeting ferroptosis in EC
| Name | Structure | Regulatory mechanism | Function | EC subtype | Cancer cell line (activity) | References |
|---|---|---|---|---|---|---|
| 5-ALA | 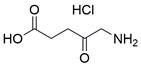 | GPX4↓, HMOX1↑ | Induce ferroptosis | ESCC | KYSE-30 KYSE-510 | [201] |
| BSO |  | GSH↓ | Induce ferroptosis | ESCC | NA | [223] |
| Oridonin | 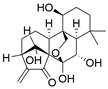 | GSH↓ | Induce ferroptosis | ESCC | TE-1 (IC50=18.95μM) | [225-227] |
| Gliotoxin | 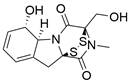 | SUV39H1↓ | Induce ferroptosis | ESCC | KYSE-150 TE-14 | [232] |
| Realgar | Not known | ROS/ASK1/p38 MAPK↑ | Induce ferroptosis | ESCC | KYSE-150 (IC50=48.012μM) ECA109 (IC50=61.336μM) | [233] |
| Brusatol | 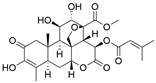 | NRF2↓ | Induce ferroptosis | EAC | FLO-1 (IC50=0.075μM) OE33 (IC50=0.058μM) | [234] |
| MF-438 |  | SCD1↓ | Induce ferroptosis | ESCC | KYSE-30 KYSE-70 KYSE-140 KYSE-150 KYSE-410 KYSE-450 KYSE-510 (IC50=1-2.4μM) | [235] |
*↓, decrease/inhibition; ↑, increase/activation
Targeting SLC7A11
The cystine/glutamate transporter (also known as system Xc-) imports extracellular cystine into cells in exchange for intracellular glutamate (Glu) at a ratio of 1:1. Once in cells, cystine (Cys2) can be converted into cysteine (Cys), which is then used to generate glutathione (GSH) in a reaction catalyzed by glutathione synthetase (GSS) as well as glutamate-cysteine ligase (GCL) [196,205]. As a core component of system Xc-, solute carrier family 7 member 11 (SLC7A11, also known as xCT) promotes the uptake of cystine and the biosynthesis of glutathione, thereby terminating the lipid peroxidation reaction and inhibiting the occurrence of ferroptosis. Interestingly, because tumor cells are often exposed to high levels of oxidative stress, SLC7A11 is more important for enhancing their antioxidant defense and inhibiting ferroptosis, which is beneficial for tumor growth [206]. Extensive research has consistently demonstrated the overexpression of SLC7A11 in several cancer types, including colorectal cancer, ovarian cancer, lung cancer, and ESCC. This upregulation of SLC7A11 is associated with the occurrence, progression and drug resistance of tumors [207,208]. Similarly, a recent study demonstrated that overexpression of SLC7A11 was linked to poorer survival and therapy outcomes among patients with ESCC [207]. Therefore, SLC7A11 is regarded as a promising target in therapy for different cancers, including EC. Some compounds have been identified and validated as SLC7A11 inhibitors, including but not limited to erastin, sulfasalazine, sorafenib, imidazole ketone erastin (IKE) and piperazine erastin (PE) [192,196,209]. For EC, as mentioned above, a recent study found that damage from increased lipid peroxidation can be avoided by upregulating GPX4 and SLC7A11, which rescues ESCC cells from ferroptosis, indicating that targeting SLC7A11 to block this intrinsic protective mechanism against ferroptosis has great potential in the treatment of EC [200]. Undoubtedly, SLC7A11 is a promising target in EC therapy. However, all currently available SLC7A11 inhibitors have either bioavailability issues or off-target effects, which limit the clinical application of these inhibitors [210]. To overcome these limitations, packaging and delivering these drugs with biocompatible nanoparticles might be a good strategy, as it not only increases their bioavailability and efficacy but also may reduce or eliminate associated side effects [211].
Targeting GSH
Glutathione (GSH), a natural tripeptide that includes cysteine, glycine and glutamic acid, is a pivotal intracellular antioxidant that can scavenge reactive oxygen species (ROS), causing the maintenance of cellular homeostasis. In contrast to normal cells, most cancer cells tend to have an elevated level of ROS to support their survival, metastasis, proliferation and growth in different conditions or microenvironments, and this can be exploited in cancer therapy [212,213]. Correspondingly, the expression of GSH is elevated in some cancers, such as lung cancer, head and neck cancers, breast cancer and ovarian cancer [214]. Further studies have confirmed that GSH is not only a pivotal regulator of cancer cell metastasis, progression and development but also is involved in resistance to chemotherapeutic drugs [215,216]. Hence, many researchers believe that depleting intracellular GSH might be a useful strategy to increase oxidative stress in tumor cells and improve cancer therapy outcomes. Various strategies have been proposed to reduce intracellular levels of glutathione (GSH), considering the therapeutic needs of cancer treatment and the processes involved in GSH metabolism, including 1) blocking the supply of raw materials for GSH synthesis (e.g., erastin, imidazole ketone erastin, sorafenib and sulfasalazine) [192,196,209]; 2) delivering certain substances to consume GSH; 3) promoting GSH efflux (e.g., verapamil, flavonoids and staurosporine) [217-219]; and 4) inhibiting GSH synthesis or regeneration (e.g., BSO, sulfinosine and curcumin) [220-222]. For EC, ESCC cells were found to have high enrichment of the GSH metabolism pathway compared to normal cells, and the ESCC tumor burden in mice was greatly relieved by depleting GSH using a γ-glutamyl cysteine synthetase inhibitor (BSO) [223]. Researchers have considered terpenoids due to their excellent pharmacological effects, including antitumor, antioxidant, and anti-inflammatory effects [224]. One such compound is called oridonin (ORI). The combined use of BSO and ORI was found to cause irreversible ROS accumulation and cell death by depleting GSH in ESCC cells [225]. Similarly, the combined use of ORI and cisplatin (CIS) was proven to synergistically inhibit the proliferation of ESCC cells and induce cell death mediated by GSH/ROS systems [226]. In addition, a recent study also suggested that ORI could indeed cause dysfunction of GSH synthesis, which further contributed to ferroptosis in TE1 cells [227]. Based on the reports and cases discussed above, using drugs that function via different strategies to exhaust intracellular GSH has great potential in therapy for different cancers, including EC. However, long-term administration can cause increased synthesis or regeneration of GSH in cells, which may induce drug resistance [228]. The problem may be solved by using strategies such as simultaneously inhibiting GSH synthesis from the upstream pathway and consuming existing GSH within cancer cells. In addition, most drugs that deplete GSH, for example, BSO, have short half-lives (less than 2 hours) and are usually abundantly enriched in normal tissues. Therefore, there is an urgent need for strategies to enhance the targeted delivery efficiency and bioavailability of free drugs. Nanomedicine may be a means to solve these problems and is gaining increasing attention for its use in synergistic cancer treatment and GSH depletion; for example, manganese dioxide-based nanocarriers, gold nanoclusters, iron-linked nanoscale metal-organic frameworks and polyoxometalate-based nanoplatforms have been tested and have shown a good ability to simultaneously deliver therapeutic components and GSH-depleting agents [229]. In summary, improved strategies targeting GSH with small-molecule compounds may be promising for patients with EC (Table 4).
Targeting FSP1
Ferroptosis suppressor protein 1 (FSP1) acts as an oxidoreductase by reducing CoQ10 to CoQ10H2, which can function as a lipophilic radical trap within membranes, inhibiting ferroptosis. Thus, FSP1 is a major ferroptosis defense factor. A study has demonstrated the widespread expression of FSP1 in a majority of cancer cell lines. Moreover, the FSP1 inhibitor iFSP1 exhibited significant efficacy in sensitizing these cells to ferroptosis induced by RSL3 [230]. Furthermore, Yoshioka et al. screened a chemical library and identified a compound called NPD4928 that could induce ferroptosis by directly inhibiting FSP1 and enhancing the sensitivity of a variety of cancer cells to GPX4 inhibitors [231]. At present, few modulators of FSP1 have been studied and identified. In addition, few studies of how FSP1 inhibitors affect EC therapy have been conducted. However, the previously mentioned studies targeting FSP1 with small-molecule compounds have shown great potential for efficient EC treatment.
Targeting other factors related to ferroptosis
New targets based on ferroptosis have also been a focus of EC treatment in recent years along with the abovementioned targets. A natural compound called gliotoxin has shown antitumor properties in a variety of cancers. In a recent study, it was revealed that gliotoxin induced ferroptosis in ESCC cells through the downregulation of SUV39H1 expression. This finding suggests the potential of gliotoxin as a novel natural therapy for ESCC [232]. In addition, realgar (REA), a Chinese herbal decoction, was found to not only decrease the proliferation, migration and invasion of KYSE150 and Eca109 cells but also promote ferroptosis by activating the ROS-ASK1-p38 MAPK signaling pathway [233]. Moreover, high expression of NRF2 protein in both primary tissues and EAC cell lines was detected by Ballout et al., and further studies showed that treatment with the NRF2 inhibitor brusatol alone or in combination with cisplatin induced ferroptosis of EAC cells by targeting NRF2; these results were evidenced by reduced expression of the ferroptosis markers GPX4 and xCT, which was confirmed in vivo [234]. Furthermore, analyses revealed that stearoyl-CoA desaturase (SCD1) conferred radiation resistance by inhibiting ferroptosis in ESCC cells and was related to unfavorable survival in patients with ESCC. The utilization of MF-438, an inhibitor of SCD1, for targeting SCD1 has been demonstrated to substantially enhance the radiosensitivity of ESCC cells through the induction of ferroptosis [235]. These findings provide additional potential novel therapeutic ferroptosis-related targets and pathways for the treatment of EC and may have far-reaching ramifications in the future (Table 4).
LncRNAs regulating ferroptosis in EC
One recent study found that the expression of both lncRNA OIP5-AS1 and its direct target GPX4 was upregulated in ESCC cells, and subsequent experiments confirmed that knockdown of lncRNA OIP5-AS1 promoted ferroptosis in ESCC cells by regulating GPX4[236]. Additionally, there have been reports indicating a significant upregulation of long non-coding RNA BBOX1 antisense 1 (BBOX1-AS1) in ESCC tissues, with this upregulation being associated with an unfavorable prognosis. Further study proved that downregulation of BBOX1-AS1 suppressed cell proliferation and metastasis and promoted ferroptosis in ESCC cells by upregulating miR-513a-3p to inhibit the expression of SLC7A11 [237]. The lncRNAs involved in ferroptosis in EC cells deserve further study, which may uncover potential therapeutic targets for the treatment of patients with EC (Table 6).
Targeting pyroptosis pathways with small-molecule compounds in EC
Brief introduction to pyroptosis
Pyroptosis is a lytic and inflammatory type of RCD usually caused by intracellular or extracellular factors including chemotherapy drugs, toxins, viruses and bacteria, leading to cell swelling, chromatin fragmentation, plasma membrane lysis and secretion of proinflammatory cytokines such as IL-18 and IL-1β. First observed in 1992 in infected macrophages by Zychlinsky et al., pyroptosis was subsequently identified as a gasdermin (GSDM)-mediated type of PCD in 2015 [238,239]. To date, four different pathways have been identified to be involved in pyroptosis: the caspase-3/8-mediated pathway, the granzyme-mediated pathway, the canonical pathway and the noncanonical pathway [240] (Figure 5). Pyroptosis is considered to play roles in both tumor progression and tumor suppression. On the one hand, intense, acute activation of pyroptosis causes massive infiltration of immune cells, which represses tumor development by directly inducing substantial cancer cell death and activating antitumor immunity. On the other hand, long-term chronic pyroptosis of cancer cells induced by an unfavorable TME is more conducive to tumor progression [241]. Therefore, pyroptosis-based therapies may be promising cancer therapies because they induce intense, acute pyroptosis in cancer cells. To date, several methods for improving therapeutic efficacy by targeting pyroptosis or combining pyroptosis-targeted agents with other cancer treatment methods have been explored. In this context, it is becoming increasingly clear that targeting pyroptosis with small-molecule compounds has great potential to improve therapeutic outcomes for patients with EC.
Core pyroptosis pathways in EC and EC therapeutic approaches by targeting pyroptosis pathways. (A) Core pyroptosis pathways in EC. Pyroptosis is a lytic and inflammatory type of RCD caused by intracellular or extracellular factors, such as toxins, viruses, bacteria and chemotherapy drugs, which is determined by several pathways, including the canonical pathway, the noncanonical pathway, the granzyme-mediated pathway and the caspase-3/8-mediated pathway. (B) Small-molecule compounds targeting pyroptosis-related gasdermins in EC. (C) Small-molecule compounds targeting pyroptosis-related inflammasomes in EC and other cancers. (Created with BioRender.com)
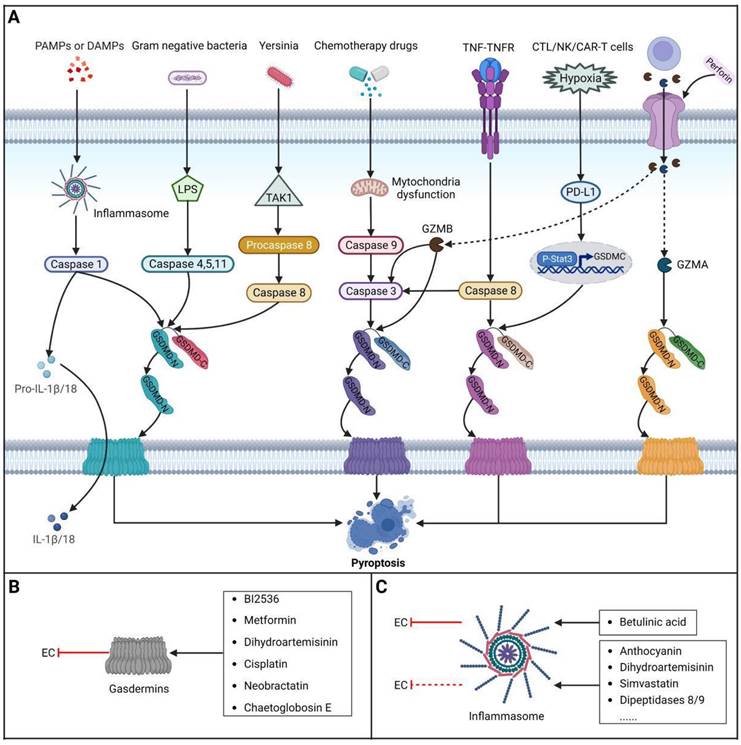
Targeting gasdermins
The GSDM family is composed of six members in humans: gasdermin A (GSDMA), gasdermin B (GSDMB), gasdermin C (GSDMC), gasdermin D (GSDMD), gasdermin E (GSDME) and DFNB59. Except for DFNB59, all members are made up of two conserved domains: the GSDM-CT (C-terminal) and GSDM-NT (N-terminal) domains. The activity of GSDM-NT can be inhibited by GSDM-CT via FLTD peptides (linkers). Once the connection is cleaved by an activation signal, plasma membrane pores are formed by GSDM-NT, which causes pyroptosis. As a family of pore-forming proteins, GSDMs are regarded as effectors of pyroptosis, and activation of GSDMs may improve the outcomes of cancer therapy. Among all GSDMs, GSDMD and GSDME are two effectors that have been extensively studied in pyroptosis. Previous studies have shown that some drugs or molecules can trigger GSDM-mediated pyroptosis in various cancer types, including but not limited to ovarian cancer [242], breast cancer [243], colon cancer [244], lung cancer [245,246] and melanoma [247]. In line with these studies, pyroptosis can also be induced by certain drugs or molecules in ESCC cells by targeting certain GSDMs. A recent study indicated that the PLK1 inhibitor BI2536 has the potential to trigger pyroptosis in ESCC cells by enhancing the expression of GSDME in the cytoplasm and activating the caspase-3/GSDME pathway. This leads to a noteworthy enhancement in the effectiveness of cisplatin. Interestingly, ESCC tissues exhibited elevated levels of GSDME expression compared to the surrounding normal tissues. Additionally, the group with higher GSDME expression demonstrated a more favorable five-year survival rate than the group with lower GSDME expression. These findings suggest that GSDME could serve as a potential prognostic factor [248]. Furthermore, a report demonstrated that metformin could induce pyroptosis in ESCC cells by regulating the miR-497/PELP1 axis, ultimately resulting in GSDMD-mediated pyroptosis, indicating that metformin may be a potential treatment for patients with cancers resistant to chemotherapy and radiotherapy but sensitive to pyroptosis [249]. In addition, GSDME was reported to be overexpressed in EC tissue. As an effective and approved treatment option for a variety of cancers, photodynamic therapy was found to promote pyroptosis in human ESCC cells by targeting the PKM2/caspase-8/caspase-3/GSDME axis [250]. Moreover, a recent study showed that dihydroartemisinin (DHA) could promote pyroptosis in ESCC cells through the PKM2/caspase-8/caspase-3/GSDME axis, providing new insights into the role of DHA in ESCC [251]. Moreover, treatment with cisplatin was proven to cause the activation of CAPN1 and CAPN2 upstream of BAX/BAK cleavage, leading to activation of caspase-9/3, which in turn resulted in GSDME-dependent pyroptosis in ESCC cells [252]. In addition, a caged prenylxanthone named neobractatin (NBT) has been reported to activate GSDME-mediated pyroptosis in ESCC cells [253]. Furthermore, a recent study showed that chaetoglobosin E, which is derived from fungal secondary metabolites, could induce GSDME-mediated pyroptosis in ESCC cells by inhibiting PLK1 and thus has the potential to improve the outcomes of EC treatment [254]. In summary, these findings suggest that drugs or molecules that target GSDM-related pathways can be utilized as therapeutic options for patients with EC. Furthermore, nanomaterials are being increasingly studies as a means to better target GSDMs, as they have great potential in delivering GSDM cytotoxic peptides/expression constructs into tumors and inducing targeted activation of intrinsic GSDM pro-cell death functions in tumor cells. This may have far-reaching ramifications for treating patients with EC in the future (Table 5).
Small-molecule compounds targeting pyroptosis in EC
| Name | Structure | Regulatory mechanism | Function | EC subtype | Cancer cell line (activity) | References |
|---|---|---|---|---|---|---|
| BI2536 |  | Caspase-3↑, GSDME↑ | Induce pyroptosis | ESCC | YES2 KYSE-30 KYSE-70 KYSE-140 KYSE-150 KYSE-180 KYSE-410 KYSE-450 KYSE-510 | [248] |
| Metformin | 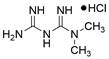 | miR-497↑, PELP1↓, GSDMD↑ | Induce pyroptosis | ESCC | KYSE-140 KYSE-510 | [249] |
| Dihydroartemisinin | 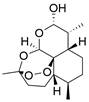 | PKM2↓, Caspase-8/3↑, GSDME↑ | Induce pyroptosis | ESCC | ECA109 EC9706 | [251] |
| Cisplatin | 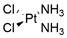 | CAPN1/2↑, BAK/BAX↑, Caspase-9/3↑, GSDME↑ | Induce pyroptosis | ESCC | KYSE-30 KYSE-510 | [252] |
| Neobractatin |  | Caspase-3↑, GSDME↑ | Induce pyroptosis | ESCC | KYSE-150 KYSE-450 ECA109 | [253] |
| Chaetoglobosin E | 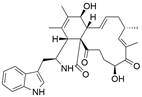 | PLK1↓, GSDME↑ | Induce pyroptosis | ESCC | KYSE-30 (IC50=2.57μM) | [254] |
| Betulinic acid |  | ASC↑, Caspase-1↑ | Induce pyroptosis | ESCC | TE-11 (IC50=6-10μM) | [262] |
*↓, decrease/inhibition; ↑, increase/activation
Targeting inflammasomes
Inflammasomes are multimeric cytosolic protein complexes composed of the adaptor protein apoptosis-associated speck-like protein (ASC), cytosolic pattern recognition receptors (PRRs) and pro-caspase-1 that assemble in response to danger signals, triggering caspase-1 activation and pyroptosis. In recent years, it has been proposed that improving inflammasome activity may prevent the development of related tumors. A protective role of inflammasomes has mainly been found in colitis-related cancers. NLRP3-/- mice were found to be more susceptible to acute and recurrent colitis-associated cancer [255]. Furthermore, the proliferation and invasion of renal carcinoma can be inhibited by increasing AIM2 expression [256]. Excitingly, some PRR activators have been reported thus far, including some that either activate PRRs directly or activate inflammasome components or related signaling events indirectly. For instance, anthocyanin was reported to reduce the viability of oral squamous cell carcinoma cells and inhibit migration and invasion abilities by activating NLRP3 inflammasome-based pyroptosis [257]. A recent study indicated that dihydroartemisinin could promote pyroptosis in breast cancer cells by targeting the AIM2/caspase-3/DFNA5 pathway [258]. Furthermore, simvastatin was proven to inhibit non-small cell lung cancer cell proliferation and migration through pyroptosis by activating the NLRP3-caspase-1 axis [259]. In addition, dipeptidases 8/9 (DPP8/9) were reported to activate NLRP1, leading to pyroptosis in human acute myeloid leukemia (AML) [260]. Conversely, some other reports have indicated that inflammasomes can also promote development of tumor. A report has shown that inflammasome/IL-1 pathways promote tumor cell proliferation and migration in both human and animal breast cancer models, and tumor growth and lung metastasis are significantly inhibited in mice lacking inflammasome components [261]. As a result, some potential inhibitors of inflammasomes have been developed to treat cancer. Considering the aforementioned findings, how inflammasomes function in human cancer remains controversial. Inflammasome's dual effects in cancer seem to be dependent on several factors, such as expression, the stage of tumorigenesis, cancer type, and the presence of mutations affecting PRR expression and downstream effector molecules (e.g., IL-1β/18). For EC, a lupine pentacyclic triterpene compound named betulinic acid (BA) was found to induce pyroptosis in ESCC cells by upregulating the protein expression of ASC and caspase-1, resulting in improved sensitivity of ESCC cells to cisplatin, which was further confirmed in vivo [262]. Because this area of study is fairly new in EC research, further studies are needed. However, the aforementioned studies utilizing small-molecule compounds to target inflammasomes have exhibited significant promise for effective treatment of EC (Table 5).
Targeting necroptosis pathways with small-molecule compounds in EC
Brief introduction to necroptosis
Necroptosis is a highly regulated caspase-independent form of RCD that bears a morphological resemblance to necrosis and a mechanistic resemblance to apoptosis. At present, it is widely believed that receptor-interacting protein kinase 3 (RIPK3), receptor-interacting protein kinase 1 (RIPK1) and mixed lineage kinase domain-like protein (MLKL) are critical regulators of necroptosis [263,264] (Figure 6).
Core necroptosis pathways in EC and EC therapeutic approaches by targeting necroptosis pathways. (A) Core necroptosis pathways in EC. Necroptosis is a highly regulated caspase-independent form of RCD. And it is widely believed that mixed lineage kinase domain-like protein (MLKL), receptor-interacting protein kinase 1 (RIPK1) and receptor-interacting protein kinase 3 (RIPK3) are critical regulators of necroptosis. (B) Small-molecule compounds targeting necroptosis-related the RIPK1/RIPK3/MLKL axis in other cancers. (Created with BioRender.com)
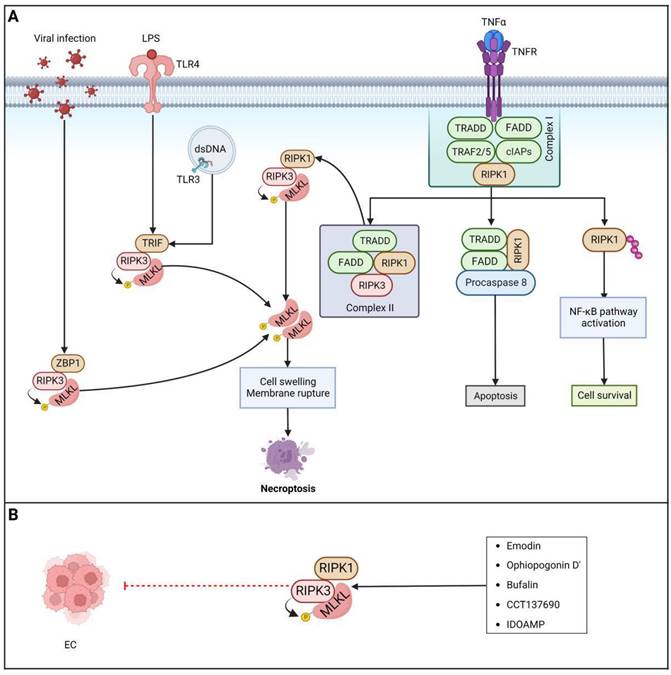
Currently, necroptosis is thought to exert dual effects in different types of cancer. On the one hand, necroptosis can serve as a "fail-safe" mechanism that protects against tumor development in the absence of apoptosis [264,265]. On the other hand, there is some evidence that necroptosis can cause inflammation and promote immunosuppression and cancer metastasis [266-268]. Thus, understanding how necroptosis functions in EC will enable exploration of the potential of targeting necroptosis with small-molecule compounds in patients with EC as a new EC therapy method.
Targeting the RIPK1/RIPK3/MLKL axis
There is widespread recognition that RIPK1, RIPK3 and MLKL are critical therapeutic targets for necroptosis. It is worth noting that a growing body of research suggests that necroptosis serves to suppress tumors in most cases. Interestingly, a study of over 60 cancer cell lines found that RIPK3 expression was downregulated or silenced in two-thirds of the samples, and decreased RIPK3 expression resulted in significant suppression of RIP3-dependent activation of MLKL and downstream programmed necrosis during chemotherapy-induced cell death, indicating that cancer cells can avoid necroptosis and survive [269]. Likewise, RIPK3 expression was proven to be significantly downregulated in primary colorectal cancer cells compared with paired normal colorectal mucosa cells [270]. In the majority of AML samples, there was a notable decrease in RIP3 expression when compared to healthy samples [271]. Moreover, RIPK3 was observed to be largely downregulated in EC, and decreased RIPK3 expression was related to a better response to chemotherapy and prolonged survival [272]. Furthermore, both RIPK1 mRNA and protein were downregulated in HNSCC cell lines, which was thought to enhance protumorigenic properties such as cell migration [267]. In addition, in resectable primary ovarian cancer [273], pancreatic adenocarcinoma [274], gastric cancer [275] and colon cancer [276], there was an association between decreased overall survival and disease-free survival and downregulated expression of MLKL. Therefore, inducing necroptosis by activating key regulators of necroptosis, including RIPK1, RIPK3 and MLKL, has emerged as a promising option for therapy for different cancers, including EC. Furthermore, an increasing number of compounds and some therapeutic agents have been reported to exhibit potential antitumor efficacy by manipulating necroptosis. The anthraquinone compound emodin has been proven to trigger necroptosis in glioma U251 cells and inhibit the proliferation of U251 cells by activating the TNF-α/RIP1/RIP3 axis [277]. A natural compound extracted from Ophiopogon japonicus named ophiopogonin D' (OPD') was proven to induce significant necroptosis by upregulating RIPK1 in prostate cancer cells [278]. Moreover, bufalin has been reported to induce necroptosis of human breast cancer cells by upregulating both RIPK1 and RIPK3 and to inhibit tumorigenesis in a mouse xenograft model engrafted with MDA-MB-231 cells [279]. A previous study also showed that the Aurora kinase inhibitor CCT137690 can trigger necrosis-like death of PDAC cells by activating MLKL, RIPK1 and RIPK3[280]. Furthermore, a rosin derivative known as IDOAMP was found to inhibit prostate cancer growth by activating the RIPK1/RIPK3/MLKL signaling pathway [281]. To date, a series of necroptosis inducers have been identified, as mentioned above. Nevertheless, it is noteworthy that the majority of studies were conducted in vitro, and the clinical feasibility of necroptosis inducers and their selectivity in killing tumor cells remain to be further explored. In summary, activating the necroptosis pathway with various agents, compounds, and drugs seems to be a promising approach for bypassing apoptosis resistance in cancer treatment. However, there are very few relevant experimental studies in the context of EC, and further exploration is needed to specifically design corresponding drugs for patients with EC.
Summary of EC clinical trials targeting RCD modalities
Although there have been recent numerous publications on novel activators and inhibitors of RCD subroutines, clinical trials assessing the impact of these modulators on RCD in the context of EC are still lacking and ongoing. Currently, clinical trials for patients with EC have primarily focused on modulators of apoptosis. However, as new and high-quality research articles on various RCD modalities including apoptosis ferroptosis, pyroptosis, necroptosis and autophagy continue to emerge, it is expected that more clinical trials will be conducted with the aim of understanding and exploring these modalities. Consequently, we anticipate that in the near future, there will be a greater utilization of these five RCD modalities to optimize anti-EC treatments (Table 7).
EC clinical trials targeting RCD modalities
| Trial number | Drug/treatment | Number of included patients | Function | Phase |
|---|---|---|---|---|
| NCT00319735 | Cetuximab, radiotherapy | 41 | Induce apoptosis | Phase 2 |
| NCT00130689 | Cetuximab | 43 | Induce apoptosis | Phase 2 |
| NCT00735826 | Vorinostat | 10 | Induce apoptosis | NA |
| NCT00037817 | Decitabine/Depsipeptide | 34 | Induce apoptosis | Phase 1 |
Conclusion and perspectives
Currently, the current mainstream treatments for patients with EC include endoscopic resection, surgical resection, radiotherapy, chemotherapy and immuno. Patients with early-stage EC are treated primarily through surgery or endoscopic resection, and the therapeutic efficacy is astounding. In terminal cases, surgery is not sufficient and is often combined with preoperative or perioperative ChT/CRT. Although the treatment options for patients with advanced EC are constantly being optimized, the poor quality of life and poor five-year survival rate are still disappointing. In addition, increased resistance to chemotherapy and immunotherapy and decreased radiotherapy sensitivity of EC cells are becoming prominent, problems that urgently need to be solved. Targeted therapy has recently gained prominence in various cancer treatment modalities for its ability to identify and attack cancer cells precisely, which can be applied in combination with other treatment modalities including surgical resection, radiotherapy, chemotherapy. However, in comparison to other solid tumors, EC has lagged significantly in the field of the targeted therapy. Over the previous decade, the options and availability of targeted therapies for EC is limited, including those that target VEGF, HER2, EGFR and PD-1. In this context, developing new small-molecule drugs that target such RCD subroutines has emerged as a promising therapeutic strategy for patients with EC.
Since apoptosis was discovered in 1972, knowledge of the subroutines and molecular mechanisms of cell death has increased rapidly, enhancing our understanding of pathways associated with the occurrence, progression and treatment of tumors. These advances have further prompted researchers to study RCD in the field of cancer therapy. In this context, accumulating evidence has shown that precisely targeting specific cell death patterns is a promising strategy for treating EC. During the past few decades, many new nonapoptotic forms of RCDs have been discovered, including but not limited to necroptosis, NETotic cell death, ferroptosis, autophagy, pyroptosis, oxeiptosis, entosis, parthanatos, alkaliptosis, lysosome-dependent cell death and cuprotosis, each of which has a different mechanism. In this review, present a summary of the mechanisms underlying five well-studied RCD subroutines in relation to EC, including apoptosis, ferroptosis, pyroptosis, necroptosis, and autophagy. Additionally, we outline recent advancements in the field concerning the development of small-molecule compounds and lncRNA that target these RCD subroutines. In the treatment of patients with EC, different RCD subroutines may have varying roles and significance among different patients, which could depend on multiple factors, including but not limited to the stage and classification of EC, genetic characteristics and treatment goals. The heterogeneity of patients with EC means that a one-size-fits-all RCD subroutine may not be effective for all patients, which underscores the need for tailored treatment strategies. In addition, interestingly, despite RCD mostly serving to promote cell death in cancer, it is still a double-edged sword in some cases since some studies have proven that some forms of RCD subroutines, such as pyroptosis, necroptosis and autophagy, can also promote tumor growth when certain conditions are met. For instance, in the context of autophagy, in the early stages of tumor progression, autophagy functions as a protective mechanism, preserving cellular and genomic integrity. However, as the tumor advances to a more advanced stage, autophagy can transition into a survival and defense mechanism for cancer cells, supplying vital nutrients to withstand environmental stresses. The conflicting roles of such RCD subroutines pose challenges for targeted therapy in this context. Regulating autophagy, apoptosis, and necrosis has demonstrated efficacy as therapeutic approaches in EC cell lines and animal models. However, determining whether to activate or inhibit these RCD subroutines requires further investigation and careful consideration. Hence, current research should focus on how to selectively manipulate RCD to treat patients with EC. To achieve this goal, it is essential to investigate the causes and mechanisms of different effects of RCD in tumor progression and suppression.
The decision to use RCD-targeted therapies as standalone treatments or in combination with other treatment modalities is typically based on both preclinical and clinical trials. In line with existing preclinical reports, RCD-targeted therapies can not only be used alone to directly induce cell death in EC cells or inhibit their survival and growth in some cases, but also improve treatment outcomes of treatment modalities including chemotherapy, radiotherapy and targeted therapies in some other cases. This means that RCD-targeted therapies may have potential to be used individually or in combination with other treatment modalities. It's critical to note that the clinical trials assessing the impact of RCD-targeted therapies in the treatment of EC are still lacking and ongoing. Therefore, further evidence is needed to fully understand the optimal role and effectiveness of RCD-targeted modulators and their potential combinations with other treatment modalities in EC.
It is crucial to understand the advantages and disadvantages of using small-molecule compounds in modulation of RCD subroutines to evaluate their potential clinical relevance and feasibility. The advantages of small-molecule compounds can easily penetrate cell membranes and target engagement due to their relatively small size, which can be designed to specifically target key molecules or pathways involved in RCD subroutines. In addition, they can not only be delivered through multiple routes, but also their pharmacokinetic properties can be optimized for efficacy and safety. However, there are still many challenges in applying these small-molecule compounds in clinical practice, especially ubiquitous bioavailability issues and off-target effects. Furthermore, the effectiveness of small-molecule compounds targeting a single target can be significantly limited by the activation of functional bypass or alternative compensatory pathways, as well as mutations in the drug target. In this scenario, dual-target or multi-target small molecules are being extensively studied as novel regimens to overcome drug resistance in cancer treatment, which have better pharmacokinetic and decreased toxic side effects and risk of drug-drug interaction. The resistance of cancer cells to a specific type of RCD may be avoided by simultaneously manipulating multiple RCD signaling pathways via dual-target or multi-target small molecules. There is a necessity to dedicate additional efforts towards the design of novel dual-target or multi-target compounds that exhibit enhanced anti-cancer activity and selectivity, aiming for more potent therapeutic effects. In addition to traditional approaches involving pharmacophore-based combination methods and structure-based modifications of existing inhibitors, the utilization of artificial intelligence (AI) in target identification and drug design has gained considerable attention. This presents a promising opportunity for enhancing the discovery of dual-target or multi-target drugs. In addition, further comprehensive investigations into the underlying mechanisms of individual RCD subroutines within cells are imperative. These studies will not only deepen our understanding of intracellular signaling molecules and the maintenance of cellular homeostasis but also facilitate the development of innovative precision-based dual-target or multi-target drugs capable of effectively eliminating EC cells. Notably, recent studies have demonstrated that nanoparticles play a crucial role in cancer therapy based on their good hydrophilicity and targeting ability and thus can be used to package and deliver the abovementioned small-molecule compounds. In addition, exosomes are also regarded as a suitable vehicle for small-molecule compounds delivery due to their low immunogenicity and high biocompatibility. To some extent, the combination of nanoparticles or exosomes is a promising future strategy for the application of small-molecule compounds and lncRNAs targeting RCD subroutines (Figure 7). Further research in this domain is warranted to explore its full potential in the future.
New strategies for treating EC. Using small-molecule compounds and lncRNAs to precisely target RCD subroutines is a promising approach to treat patients with EC, which is expected to improve the clinical outcomes of existing treatment modalities, including chemotherapy, radiotherapy and immunotherapy. And the combination of nanoparticles and exosomes is a promising future strategy for the application of small-molecule compounds and lncRNAs targeting RCD subroutines. (Created with BioRender.com)
Abbreviations
3-MA: 3-methyladenine; ACD: Accidental cell death; AKT: Protein kinase B; ART: artemisinin; ATG: Autophagy-associated protein; ASC: Adaptor protein apoptosis-associated speck-like protein; AML: Acute myeloid leukemia; BCL-2: B-cell lymphoma-2; BAX: BCL-2-associated X protein; BAK1: BCL-2 homologous antagonist/killer 1; BID: BAX-like BH3 protein; BA: Betulinic acid; BSO: L-buthionine-sulfoximine; BH4: Tetrahydrobiopterin; cTNM: Clinical tumor enodeemetastasis; CT: Computed tomography; Ch‑19: 2,4,6‑trimethoxy‑4'‑nitrochalcone; cIAP1: cellular inhibitor of apoptosis 1; cIAP2: Cellular inhibitor of apoptosis 2; CPP: Periplocin; CMSP: P-hydroxylcinnamaldehyde; CPS-C: Capilliposide C; CUR: Curcumin; CQ: Chloroquine; Cys2: Cystine; Cys: Cysteine; CIS: Cisplatin; CoQ10: Coenzyme Q10; CoQ10H2: Ubiquinol-10; CTL: Cytotoxic T lymphocyte; CAR: Chimeric antigen receptor; CUR: Curcumin; DISC: Death-inducing signaling complex; DR: Death receptor; DSC: 3-deoxysappanchalcone; DVDMS: Sinoporphyrin sodium; DEX: Dexmedetomidine; DHA: Dihydroartemisinin; DPI: Diverse pharmacological inhibitor; DPP8/9: Dipeptidases 8/9; DAMPs: Damage-associated molecular patterns; EC: Esophageal cancer; EAC: Esophageal adenocarcinoma; ESCC: Esophageal squamous cell carcinoma; EUS: Endoscopic ultrasound; ESD: Endoscopic submucosal dissection; EMRL: Ligation-assisted endoscopic mucosal resection; EMRC: Cap-assisted endoscopic mucosal resection; EMSO: European Society for Medical Oncology; Ech: Echinatin; FLOT: 5-fluorouracile-leucovorine-oxaliplatine-docetaxel; FADD: Fas-associated protein with death domain; FINs: Ferroptosis inducers; FDA: the U.S. Food and Drug Administration; FSP1: Ferroptosis suppressor protein 1; GLP: Ganoderma lucidum polysaccharide; GSR: Glutathione reductase; GPX4: Glutathione peroxidase 4; Glu: Glutamate; GSS: Glutathione synthetase; GCL: Glutamate-cysteine ligase; GSH: Glutathione; GSSH: Oxidized glutathione; GCH1: GTP cyclohydrolase I; GSDM: Gasdermin; GZMA: Granzyme A; GZMB: Granzyme B; HCQ: Hydroxyquinoline; IARC: International Agency for Research on Cancer; IAPs: Inhibitor of apoptosis proteins; IKE: Imidazole ketone erastin; LBP: Lobaplatin; LPS: Lipopolysaccharide; LBP: Lobaplatin; LncRNAs: Long non-coding ribonucleic acids; LOX-1: Lectin-like oxidized low-density lipoprotein receptor-1; MCBS: Magnitude of Clinical Benefifit Score; MBM: Multiband mucosectomy; MIO: Minimally invasive esophagectomy; MCL-1: Myeloid cell leukemia-1; MOMP: Mitochondria outer membrane permeability; MAPK: mitogen-activated protein kinase; mTOR: Mammalian target of rapamycin; MDP: Muramyl dipeptide; MLKL: Mixed lineage kinase domain-like protein; NK: Natural killer; NOD2: Nucleotide binding oligomerization domain containing 2; NBT: Neobractatin; ORI: Oridonin; OPD': Ophiopogonin D'; PET: Positron emission tomography; PARAs: Proapoptotic receptor agonists; PDT: Photodynamic therapy; PPT: Picropodophyllotoxin; PI3K: Phosphatidylinositol 3-kinase; PE: Piperazine erastin; PUFA: Polyunsaturated fatty acid; PUFA-PLs: Polyunsaturated fatty acid phospholipids; PRRs: Cytosolic pattern recognition receptors; PAMPs: Pathogen-associated molecular patterns; PD-L1: Programmed death-ligand 1; QoL: Quality of life; Quina: Quinalizarin; RCD: Regulated cell death; RhTRAIL: Recombinant human TNF-related apoptosis-inducing ligand; ROS: Reactive oxygen species; Ro: Ginsenoside Ro; REA: Realgar; RIPK: Receptor-interacting protein kinase; SEMSs: Self-expanding metal stents; SMAC: Second mitochondrial-derived activators of caspases; SLC7A11: Solute carrier family 7 member 11; SCD1: Stearoyl-CoA desaturase; TRAIL: TNF-related apoptosis-inducing ligand; tBID: Truncated Bid; TF: Transferrin; TFR: Transferrin receptor; TNFα: Tumor necrosis factor alpha; TNFR: Tumor necrosis factor receptor; TRADD: Tumor necrosis factor receptor 1-associated death domain protein; TLR: Toll-like receptors; TRAF: Tumour necrosis factor receptor-associated factor; TRIF: TIR domain-containing adapter-inducing IFN-β; UA: Ursolic acid; ULK1: UNC-51-like kinase 1; VES: Vitamin E succinate; XIAP: X-linked inhibitor of apoptosis protein; ZBP1: Z-DNA-binding protein 1.
Acknowledgements
Funding
This work was supported in part by National Natural Science Foundation of China (Grant No. 81970481 and Grant No. 82173666), Sichuan Science and Technology Program (2022YFS0048), Shenzhen science and technology research and development funds (Grant No. JCYJ20210324094612035).
Author contributions
Bo Liu and Yong Yuan designed the review. Haowen Zhang and Siyuan Luan drafted the manuscript. Haowen Zhang and Jin Zhang prepared the figures. Zhiying Liu and Xiaokun Li summed up the literature and the compounds. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca-Cancer J Clin. 2021;71:209-49
2. Smyth EC, Lagergren J, Fitzgerald RC, Lordick F, Shah MA, Lagergren P. et al. Oesophageal cancer. Nat Rev Dis Primers. 2017;3:17048
3. Edgren G, Adami HO, Weiderpass E, Nyren O. A global assessment of the oesophageal adenocarcinoma epidemic. Gut. 2013;62:1406-14
4. di Pietro M, Canto MI, Fitzgerald RC. Endoscopic Management of Early Adenocarcinoma and Squamous Cell Carcinoma of the Esophagus: Screening, Diagnosis, and Therapy. Gastroenterology. 2018;154:421-36
5. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. Ca-Cancer J Clin. 2022;72:7-33
6. Lagergren J, Smyth E, Cunningham D, Lagergren P. Oesophageal cancer. Lancet. 2017;390:2383-96
7. Galluzzi L, Bravo-San PJ, Vitale I, Aaronson SA, Abrams JM, Adam D. et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58-73
8. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Brit J Cancer. 1972;26:239-57
9. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347-64
10. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486-541
11. Kahlson MA, Dixon SJ. Copper-induced cell death. Science. 2022;375:1231-2
12. Strasser A, Vaux DL. Cell Death in the Origin and Treatment of Cancer. Mol Cell. 2020;78:1045-54
13. Moujalled D, Strasser A, Liddell JR. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021;28:2029-44
14. Del RD, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol Rev. 2019;99:1765-817
15. Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L. et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Tar. 2022;7:286
16. Maimaitizunong R, Wang K, Li H. Ferroptosis and its emerging role in esophageal cancer. Front Mol Biosci. 2022;9:1027912
17. Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L. et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Tar. 2022;7:286
18. Wu J, Ye J, Xie Q, Liu B, Liu M. Targeting Regulated Cell Death with Pharmacological Small Molecules: An Update on Autophagy-Dependent Cell Death, Ferroptosis, and Necroptosis in Cancer. J Med Chem. 2022;65:2989-3001
19. Ye J, Wu J, Liu B. Therapeutic strategies of dual-target small molecules to overcome drug resistance in cancer therapy. Bba-Rev Cancer. 2023;1878:188866
20. Morita M, Kumashiro R, Kubo N, Nakashima Y, Yoshida R, Yoshinaga K. et al. Alcohol drinking, cigarette smoking, and the development of squamous cell carcinoma of the esophagus: epidemiology, clinical findings, and prevention. Int J Clin Oncol. 2010;15:126-34
21. Gao Y, Yuan F, Liu J, Li L, He Y, Gao R. et al. Identification of New Candidate Genes and Chemicals Related to Esophageal Cancer Using a Hybrid Interaction Network of Chemicals and Proteins. Plos One. 2015;10:e129474
22. Riley CA, Wu EL, Hsieh MC, Marino MJ, Wu XC, McCoul ED. Association of Gastroesophageal Reflux With Malignancy of the Upper Aerodigestive Tract in Elderly Patients. Jama Otolaryngol. 2018;144:140-8
23. Uhlenhopp DJ, Then EO, Sunkara T, Gaduputi V. Epidemiology of esophageal cancer: update in global trends, etiology and risk factors. Clin J Gastroenterol. 2020;13:1010-21
24. Zhu H, Ma X, Ye T, Wang H, Wang Z, Liu Q. et al. Esophageal cancer in China: Practice and research in the new era. Int J Cancer. 2022
25. Obermannova R, Alsina M, Cervantes A, Leong T, Lordick F, Nilsson M. et al. Oesophageal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33:992-1004
26. Malik S, Sharma G, Sanaka MR, Thota PN. Role of endoscopic therapy in early esophageal cancer. World J Gastroentero. 2018;24:3965-73
27. Spadaccini M, Bhandari P, Maselli R, Spaggiari P, Alkandari AA, Varytimiadis L. et al. Multi-band mucosectomy for neoplasia in patients with Barrett's esophagus: in vivo comparison between two different devices. Surg Endosc. 2020;34:3845-52
28. Deng J, Su Q, Ren Z, Wen J, Xue Z, Zhang L. et al. Comparison of short-term outcomes between minimally invasive McKeown and Ivor Lewis esophagectomy for esophageal or junctional cancer: a systematic review and meta-analysis. Oncotargets Ther. 2018;11:6057-69
29. Yang YS, Shang QX, Yuan Y, Wu XY, Hu WP, Chen LQ. Comparison of Long-term Quality of Life in Patients with Esophageal Cancer after Ivor-Lewis, Mckeown, or Sweet Esophagectomy. J Gastrointest Surg. 2019;23:225-31
30. Maas KW, Cuesta MA, van Berge HM, Roig J, Bonavina L, Rosman C. et al. Quality of Life and Late Complications After Minimally Invasive Compared to Open Esophagectomy: Results of a Randomized Trial. World J Surg. 2015;39:1986-93
31. van der Sluis PC, van der Horst S, May AM, Schippers C, Brosens L, Joore H. et al. Robot-assisted Minimally Invasive Thoracolaparoscopic Esophagectomy Versus Open Transthoracic Esophagectomy for Resectable Esophageal Cancer: A Randomized Controlled Trial. Ann Surg. 2019;269:621-30
32. Ychou M, Boige V, Pignon JP, Conroy T, Bouche O, Lebreton G. et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol. 2011;29:1715-21
33. Al-Batran SE, Homann N, Pauligk C, Goetze TO, Meiler J, Kasper S. et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, phase 2/3 trial. Lancet. 2019;393:1948-57
34. Crosby T, Hurt CN, Falk S, Gollins S, Staffurth J, Ray R. et al. Long-term results and recurrence patterns from SCOPE-1: a phase II/III randomised trial of definitive chemoradiotherapy +/- cetuximab in oesophageal cancer. Brit J Cancer. 2017;116:709-16
35. Guyer DL, Almhanna K, McKee KY. Palliative care for patients with esophageal cancer: a narrative review. Ann Transl Med. 2020;8:1103
36. Kim KY, Tsauo J, Song HY, Kim PH, Park JH. Self-Expandable Metallic Stent Placement for the Palliation of Esophageal Cancer. J Korean Med Sci. 2017;32:1062-71
37. Zhu HD, Guo JH, Mao AW, Lv WF, Ji JS, Wang WH. et al. Conventional stents versus stents loaded with (125)iodine seeds for the treatment of unresectable oesophageal cancer: a multicentre, randomised phase 3 trial. Lancet Oncol. 2014;15:612-9
38. Kawamoto T, Nakamura N, Saito T, Tonari A, Wada H, Harada H. et al. Palliative brachytherapy and external beam radiotherapy for dysphagia from esophageal cancer: a nationwide survey in Japan. Jpn J Clin Oncol. 2021;51:950-5
39. Pardo J, Wallich R, Martin P, Urban C, Rongvaux A, Flavell RA. et al. Granzyme B-induced cell death exerted by ex vivo CTL: discriminating requirements for cell death and some of its signs. Cell Death Differ. 2008;15:567-79
40. Dostert C, Grusdat M, Letellier E, Brenner D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol Rev. 2019;99:115-60
41. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65-80
42. Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G. et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477-82
43. S SR, Eastman A. BCL2 Inhibitors as Anticancer Drugs: A Plethora of Misleading BH3 Mimetics. Mol Cancer Ther. 2016;15:2011-7
44. Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, Strasser A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell. 2018;34:879-91
45. Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A, Kelly GL. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer. 2022;22:45-64
46. Surman DR, Xu Y, Lee MJ, Trepel J, Brown K, Ramineni M. et al. Therapeutic Synergy in Esophageal Cancer and Mesothelioma Is Predicted by Dynamic BH3 Profiling. Mol Cancer Ther. 2021;20:1469-80
47. Song S, Chen Q, Li Y, Lei G, Scott A, Huo L. et al. Targeting cancer stem cells with a pan-BCL-2 inhibitor in preclinical and clinical settings in patients with gastroesophageal carcinoma. Gut. 2021;70:2238-48
48. Stein MN, Goodin S, Gounder M, Gibbon D, Moss R, Portal D. et al. A phase I study of AT-101, a BH3 mimetic, in combination with paclitaxel and carboplatin in solid tumors. Invest New Drug. 2020;38:855-65
49. Lin QH, Que FC, Gu CP, Zhong DS, Zhou D, Kong Y. et al. ABT-263 induces G1/G0-phase arrest, apoptosis and autophagy in human esophageal cancer cells in vitro. Acta Pharmacol Sin. 2017;38:1632-41
50. Qiao Z, Cheng Y, Liu S, Ma Z, Li S, Zhang W. Casticin inhibits esophageal cancer cell proliferation and promotes apoptosis by regulating mitochondrial apoptotic and JNK signaling pathways. N-S Arch Pharmacol. 2019;392:177-87
51. Huang P, Sun LY, Zhang YQ. A Hopeful Natural Product, Pristimerin, Induces Apoptosis, Cell Cycle Arrest, and Autophagy in Esophageal Cancer Cells. Anal Cell Pathol. 2019;2019:6127169
52. Wu C, Huang H, Choi HY, Ma Y, Zhou T, Peng Y. et al. Anti-esophageal Cancer Effect of Corilagin Extracted from Phmllanthi Fructus via the Mitochondrial and Endoplasmic Reticulum Stress Pathways. J Ethnopharmacol. 2021;269:113700
53. Jiang JH, Pi J, Jin H, Yang F, Cai JY. Chinese herb medicine matrine induce apoptosis in human esophageal squamous cancer KYSE-150 cells through increasing reactive oxygen species and inhibiting mitochondrial function. Pathol Res Pract. 2018;214:691-9
54. Yang Y, Wu H, Zou X, Chen Y, He R, Jin Y. et al. A novel synthetic chalcone derivative, 2,4,6-trimethoxy-4'-nitrochalcone (Ch-19), exerted anti-tumor effects through stimulating ROS accumulation and inducing apoptosis in esophageal cancer cells. Cell Stress Chaperon. 2022;27:645-57
55. Zhang L, Lu Z, Zhao X. Targeting Bcl-2 for cancer therapy. Bba-Rev Cancer. 2021;1876:188569
56. Nir S, Sarit L. Inhibiting the inhibitors: Targeting anti-apoptotic proteins in cancer and therapy resistance. Drug Resist Update. 2020;52:100712
57. Zhou S, Ye W, Shao Q, Qi Y, Zhang M, Liang J. Prognostic significance of XIAP and NF-kappaB expression in esophageal carcinoma with postoperative radiotherapy. World J Surg Oncol. 2013;11:288
58. Hikami S, Shiozaki A, Kitagawa-Juge M, Ichikawa D, Kosuga T, Konishi H. et al. The Role of cIAP1 and XIAP in Apoptosis Induced by Tumor Necrosis Factor Alpha in Esophageal Squamous Cell Carcinoma Cells. Digest Dis Sci. 2017;62:652-9
59. Xu Y, Zhou L, Huang J, Liu F, Yu J, Zhan Q. et al. Role of Smac in determining the chemotherapeutic response of esophageal squamous cell carcinoma. Clin Cancer Res. 2011;17:5412-22
60. Fulda S. Smac Mimetics to Therapeutically Target IAP Proteins in Cancer. Int Rev Cel Mol Bio. 2017;330:157-69
61. Zhao XY, Wang XY, Wei QY, Xu YM, Lau A. Potency and Selectivity of SMAC/DIABLO Mimetics in Solid Tumor Therapy. Cells-Basel. 2020;9:1012-37
62. Qin Q, Zuo Y, Yang X, Lu J, Zhan L, Xu L. et al. Smac mimetic compound LCL161 sensitizes esophageal carcinoma cells to radiotherapy by inhibiting the expression of inhibitor of apoptosis protein. Tumour Biol. 2014;35:2565-74
63. Jiang N, Zhang WQ, Dong H, Hao YT, Zhang LM, Shan L. et al. SMAC exhibits anti-tumor effects in ECA109 cells by regulating expression of inhibitor of apoptosis protein family. World J Clin Cases. 2021;9:5019-27
64. Chen F, Xu C, Du L, Wang Y, Cao J, Fu Y. et al. Tat-SmacN7 induces radiosensitization in cancer cells through the activation of caspases and induction of apoptosis. Int J Oncol. 2013;42:985-92
65. Yuan X, Gajan A, Chu Q, Xiong H, Wu K, Wu GS. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metast Rev. 2018;37:733-48
66. Marini P, Junginger D, Stickl S, Budach W, Niyazi M, Belka C. Combined treatment with lexatumumab and irradiation leads to strongly increased long term tumour control under normoxic and hypoxic conditions. Radiat Oncol. 2009;4:49
67. Luster TA, Carrell JA, McCormick K, Sun D, Humphreys R. Mapatumumab and lexatumumab induce apoptosis in TRAIL-R1 and TRAIL-R2 antibody-resistant NSCLC cell lines when treated in combination with bortezomib. Mol Cancer Ther. 2009;8:292-302
68. Jin H, Yang R, Ross J, Fong S, Carano R, Totpal K. et al. Cooperation of the agonistic DR5 antibody apomab with chemotherapy to inhibit orthotopic lung tumor growth and improve survival. Clin Cancer Res. 2008;14:7733-40
69. von Pawel J, Harvey JH, Spigel DR, Dediu M, Reck M, Cebotaru CL. et al. Phase II trial of mapatumumab, a fully human agonist monoclonal antibody to tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1), in combination with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Clin Lung Cancer. 2014;15:188-96
70. Deng D, Shah K. TRAIL of Hope Meeting Resistance in Cancer. Trends Cancer. 2020;6:989-1001
71. Gibson SB, Oyer R, Spalding AC, Anderson SM, Johnson GL. Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL. Mol Cell Biol. 2000;20:205-12
72. Cheng H, Hong B, Zhou L, Allen JE, Tai G, Humphreys R. et al. Mitomycin C potentiates TRAIL-induced apoptosis through p53-independent upregulation of death receptors: evidence for the role of c-Jun N-terminal kinase activation. Cell Cycle. 2012;11:3312-23
73. Senbabaoglu F, Cingoz A, Kaya E, Kazancioglu S, Lack NA, Acilan C. et al. Identification of Mitoxantrone as a TRAIL-sensitizing agent for Glioblastoma Multiforme. Cancer Biol Ther. 2016;17:546-57
74. Chen L, Meng Y, Sun Q, Zhang Z, Guo X, Sheng X. et al. Ginsenoside compound K sensitizes human colon cancer cells to TRAIL-induced apoptosis via autophagy-dependent and -independent DR5 upregulation. Cell Death Dis. 2016;7:e2334
75. Liu Y, Tong Y, Yang X, Li F, Zheng L, Liu W. et al. Novel histone deacetylase inhibitors derived from Magnolia officinalis significantly enhance TRAIL-induced apoptosis in non-small cell lung cancer. Pharmacol Res. 2016;111:113-25
76. Yang J, Qian S, Cai X, Lu W, Hu C, Sun X. et al. Chikusetsusaponin IVa Butyl Ester (CS-IVa-Be), a Novel IL6R Antagonist, Inhibits IL6/STAT3 Signaling Pathway and Induces Cancer Cell Apoptosis. Mol Cancer Ther. 2016;15:1190-200
77. Hori T, Kondo T, Kanamori M, Tabuchi Y, Ogawa R, Zhao QL. et al. Nutlin-3 enhances tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through up-regulation of death receptor 5 (DR5) in human sarcoma HOS cells and human colon cancer HCT116 cells. Cancer Lett. 2010;287:98-108
78. Sung B, Prasad S, Ravindran J, Yadav VR, Aggarwal BB. Capsazepine, a TRPV1 antagonist, sensitizes colorectal cancer cells to apoptosis by TRAIL through ROS-JNK-CHOP-mediated upregulation of death receptors. Free Radical Bio Med. 2012;53:1977-87
79. Fassl A, Tagscherer KE, Richter J, De-Castro AJ, Savini C, Rosl F. et al. Inhibition of Notch1 signaling overcomes resistance to the death ligand Trail by specificity protein 1-dependent upregulation of death receptor 5. Cell Death Dis. 2015;6:e1921
80. Han L, Dai S, Li Z, Zhang C, Wei S, Zhao R. et al. Combination of the natural compound Periplocin and TRAIL induce esophageal squamous cell carcinoma apoptosis in vitro and in vivo: Implication in anticancer therapy. J Exp Clin Canc Res. 2019;38:501
81. Lu Z, Zhang G, Zhang Y, Hua P, Fang M, Wu M. et al. Isoalantolactone induces apoptosis through reactive oxygen species-dependent upregulation of death receptor 5 in human esophageal cancer cells. Toxicol Appl Pharm. 2018;352:46-58
82. Ma M, Zhang C, Xiang XH, Deng XQ, Dai SL, Wei SS. et al. p-Hydroxylcinnamaldehyde from cochinchinamomordica seed reverses resistance to TRAIL in human oesophageal squamous cell carcinoma via the activation of the p38 mitogen-activated protein kinase signalling pathway. Biomed Pharmacother. 2020;121:109611
83. Kong AN, Yu R, Chen C, Mandlekar S, Primiano T. Signal transduction events elicited by natural products: role of MAPK and caspase pathways in homeostatic response and induction of apoptosis. Arch Pharm Res. 2000;23:1-16
84. Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191-205
85. Yue J, Lopez JM. Understanding MAPK Signaling Pathways in Apoptosis. Int J Mol Sci. 2020 21
86. Kwak AW, Lee MJ, Lee MH, Yoon G, Cho SS, Chae JI. et al. The 3-deoxysappanchalcone induces ROS-mediated apoptosis and cell cycle arrest via JNK/p38 MAPKs signaling pathway in human esophageal cancer cells. Phytomedicine. 2021;86:153564
87. Shi Y, Zhang B, Feng X, Qu F, Wang S, Wu L. et al. Apoptosis and autophagy induced by DVDMs-PDT on human esophageal cancer Eca-109 cells. Photodiagn Photodyn. 2018;24:198-205
88. Zang YQ, Zhai YQ, Feng YY, Ju XY, Zuo F. Molecular mechanisms of quinalizarin induces apoptosis and G0/G1 cell cycle of human esophageal cancer HCE-4 cells depends on MAPK, STAT3, and NF-kappaB signaling pathways. Environ Toxicol. 2021;36:276-86
89. Kwak AW, Choi JS, Lee MH, Oh HN, Cho SS, Yoon G. et al. Retrochalcone Echinatin Triggers Apoptosis of Esophageal Squamous Cell Carcinoma via ROS- and ER Stress-Mediated Signaling Pathways. Molecules. 2019 24
90. Kwak AW, Yoon G, Lee MH, Cho SS, Shim JH, Chae JI. Picropodophyllotoxin, an Epimer of Podophyllotoxin, Causes Apoptosis of Human Esophageal Squamous Cell Carcinoma Cells Through ROS-Mediated JNK/P38 MAPK Pathways. Int J Mol Sci. 2020 21
91. Wani ZA, Guru SK, Rao AV, Sharma S, Mahajan G, Behl A. et al. A novel quinazolinone chalcone derivative induces mitochondrial dependent apoptosis and inhibits PI3K/Akt/mTOR signaling pathway in human colon cancer HCT-116 cells. Food Chem Toxicol. 2016;87:1-11
92. Pan S, Sun Y, Sui D, Yang T, Fu S, Wang J. et al. Lobaplatin promotes radiosensitivity, induces apoptosis, attenuates cancer stemness and inhibits proliferation through PI3K/AKT pathway in esophageal squamous cell carcinoma. Biomed Pharmacother. 2018;102:567-74
93. Javid H, Afshari AR, Zahedi AF, Asadi J, Hashemy SI. Aprepitant Promotes Caspase-Dependent Apoptotic Cell Death and G2/M Arrest through PI3K/Akt/NF-kappaB Axis in Cancer Stem-Like Esophageal Squamous Cell Carcinoma Spheres. Biomed Res Int. 2021;2021:8808214
94. Cao J, Lv W, Wang L, Xu J, Yuan P, Huang S. et al. Ricolinostat (ACY-1215) suppresses proliferation and promotes apoptosis in esophageal squamous cell carcinoma via miR-30d/PI3K/AKT/mTOR and ERK pathways. Cell Death Dis. 2018;9:817
95. Yang P, Zhao J, Hou L, Yang L, Wu K, Zhang L. Vitamin E succinate induces apoptosis via the PI3K/AKT signaling pathways in EC109 esophageal cancer cells. Mol Med Rep. 2016;14:1531-7
96. Jin Z, Yan W, Jin H, Ge C, Xu Y. Psoralidin inhibits proliferation and enhances apoptosis of human esophageal carcinoma cells via NF-kappaB and PI3K/Akt signaling pathways. Oncol Lett. 2016;12:971-6
97. Lv X, Li R, Li Z, Wang J. Purification of Gekko Small Peptide Fraction and Its Effect of Inducing Apoptosis of EC 9706 Esophageal Cancer Cells by Inhibiting PI3K/Akt/GLUT1 Signaling Pathway. Chem Biodivers. 2021;18:e2000720
98. Jiang JH, Pi J, Jin H, Cai JY. Oridonin-induced mitochondria-dependent apoptosis in esophageal cancer cells by inhibiting PI3K/AKT/mTOR and Ras/Raf pathways. J Cell Biochem. 2019;120:3736-46
99. Gao Y, Chen DL, Zhou M, Zheng ZS, He MF, Huang S. et al. Cordycepin enhances the chemosensitivity of esophageal cancer cells to cisplatin by inducing the activation of AMPK and suppressing the AKT signaling pathway. Cell Death Dis. 2020;11:866
100. Guo J, Zhang S, Wang J, Zhang P, Lu T, Zhang L. Hinokiflavone Inhibits Growth of Esophageal Squamous Cancer By Inducing Apoptosis via Regulation of the PI3K/AKT/mTOR Signaling Pathway. Front Oncol. 2022;12:833719
101. Zhu X, Li Z, Li T, Long F, Lv Y, Liu L. et al. Osthole inhibits the PI3K/AKT signaling pathway via activation of PTEN and induces cell cycle arrest and apoptosis in esophageal squamous cell carcinoma. Biomed Pharmacother. 2018;102:502-9
102. Shen Z, Xu L, Li J, Zhang N. Capilliposide C Sensitizes Esophageal Squamous Carcinoma Cells to Oxaliplatin by Inducing Apoptosis Through the PI3K/Akt/mTOR Pathway. Med Sci Monitor. 2017;23:2096-103
103. Hu Y, Qiu LL, Zhao ZF, Long YX, Yang T. Dexmedetomidine represses proliferation and promotes apoptosis of esophageal cancer cells by regulating C-Myc gene expression via the ERK signaling pathway. Eur Rev Med Pharmaco. 2021;25:950-6
104. Kobayashi T, Makino T, Yamashita K, Saito T, Tanaka K, Takahashi T. et al. APR-246 induces apoptosis and enhances chemo-sensitivity via activation of ROS and TAp73-Noxa signal in oesophageal squamous cell cancer with TP53 missense mutation. Brit J Cancer. 2021;125:1523-32
105. Qin T, Zhao J, Liu X, Li L, Zhang X, Shi X. et al. Luteolin combined with low-dose paclitaxel synergistically inhibits epithelial-mesenchymal transition and induces cell apoptosis on esophageal carcinoma in vitro and in vivo. Phytother Res. 2021;35:6228-40
106. Cao YY, Yu J, Liu TT, Yang KX, Yang LY, Chen Q. et al. Plumbagin inhibits the proliferation and survival of esophageal cancer cells by blocking STAT3-PLK1-AKT signaling. Cell Death Dis. 2018;9:17
107. Xie X, Liu K, Liu F, Chen H, Wang X, Zu X. et al. Gossypetin is a novel MKK3 and MKK6 inhibitor that suppresses esophageal cancer growth in vitro and in vivo. Cancer Lett. 2019;442:126-36
108. Wang T, Wang J, Ren W, Liu ZL, Cheng YF, Zhang XM. Combination treatment with artemisinin and oxaliplatin inhibits tumorigenesis in esophageal cancer EC109 cell through Wnt/beta-catenin signaling pathway. Thorac Cancer. 2020;11:2316-24
109. Xu N, Lu M, Wang J, Li Y, Yang X, Wei X. et al. Ivermectin induces apoptosis of esophageal squamous cell carcinoma via mitochondrial pathway. Bmc Cancer. 2021;21:1307
110. Yang G, Sheng B, Li R, Xu Q, Zhang L, Lu Z. Dehydrocostus Lactone Induces Apoptosis and Cell Cycle Arrest through Regulation of JAK2/STAT3/PLK1 Signaling Pathway in Human Esophageal Squamous Cell Carcinoma Cells. Anti-Cancer Agent Me. 2022;22:1742-52
111. Deng X, Sheng J, Liu H, Wang N, Dai C, Wang Z. et al. Cinobufagin Promotes Cell Cycle Arrest and Apoptosis to Block Human Esophageal Squamous Cell Carcinoma Cells Growth via the p73 Signalling Pathway. Biol Pharm Bull. 2019;42:1500-9
112. Sha B, Chen X, Wu H, Li M, Shi J, Wang L. et al. Deubiquitylatinase inhibitor b-AP15 induces c-Myc-Noxa-mediated apoptosis in esophageal squamous cell carcinoma. Apoptosis. 2019;24:826-36
113. Liu Q, He J, Zhou X, Han M, Li J, Liu C. et al. ACP-5862 suppresses esophageal squamous cell carcinoma growth through inducing apoptosis via activation of endoplasmic reticulum stress and ROS production. Biochem Bioph Res Co. 2021;534:995-1002
114. Xu LJ, Yu XJ, Wei B, Hui HX, Sun Y, Dai J. et al. LncRNA SNHG7 promotes the proliferation of esophageal cancer cells and inhibits its apoptosis. Eur Rev Med Pharmaco. 2018;22:2653-61
115. Wang B, Hua P, Zhang L, Li J, Zhang Y. LncRNA-IUR up-regulates PTEN by sponging miR-21 to regulate cancer cell proliferation and apoptosis in esophageal squamous cell carcinoma. Esophagus-Tokyo. 2020;17:298-304
116. Li Z, Qin X, Bian W, Li Y, Shan B, Yao Z. et al. Exosomal lncRNA ZFAS1 regulates esophageal squamous cell carcinoma cell proliferation, invasion, migration and apoptosis via microRNA-124/STAT3 axis. J Exp Clin Canc Res. 2019;38:477
117. Du W, Xu P, Yin H. LncRNA NEAT1 regulates the growth, migration, and invasion of the human esophageal cancer cells via the miR-377/E2F3 axis. Acta Biochim Pol. 2022;69:731-6
118. Ma W, Zhang CQ, Li HL, Gu J, Miao GY, Cai HY. et al. LncRNA FER1L4 suppressed cancer cell growth and invasion in esophageal squamous cell carcinoma. Eur Rev Med Pharmaco. 2018;22:2638-45
119. Fang Y, Zhang S, Yin J, Shen YX, Wang H, Chen XS. et al. LINC01535 promotes proliferation and inhibits apoptosis in esophageal squamous cell cancer by activating the JAK/STAT3 pathway. Eur Rev Med Pharmaco. 2020;24:3694-700
120. Zong M, Feng W, Wan L, Yu X, Yu W. LncRNA TUG1 promotes esophageal cancer development through regulating PLK1 expression by sponging miR-1294. Biotechnol Lett. 2020;42:2537-49
121. Zhao W, Huang Z, Liu H, Wang C. LncRNA GIHCG Promotes the Development of Esophageal Cancer by Modulating miR-29b-3p/ANO1 Axis. Oncotargets Ther. 2020;13:13387-400
122. Jia J, Li H, Chu J, Sheng J, Wang C, Jia Z. et al. LncRNA FAM83A-AS1 promotes ESCC progression by regulating miR-214/CDC25B axis. J Cancer. 2021;12:1200-11
123. Su W, Guo C, Wang L, Wang Z, Yang X, Niu F. et al. LncRNA MIR22HG abrogation inhibits proliferation and induces apoptosis in esophageal adenocarcinoma cells via activation of the STAT3/c-Myc/FAK signaling. Aging (Albany NY). 2019;11:4587-96
124. Ghaffar M, Khodahemmati S, Li J, Shahzad M, Wang M, Wang Y. et al. Long Non-coding RNA LINC01234 Regulates Proliferation, Invasion and Apoptosis in Esophageal Cancer Cells. J Cancer. 2018;9:4242-9
125. Li HM, Yu YK, Liu Q, Wei XF, Zhang J, Zhang RX. et al. LncRNA SNHG1 Regulates the Progression of Esophageal Squamous Cell Cancer by the miR-204/HOXC8 Axis. Oncotargets Ther. 2020;13:757-67
126. Gai L, Huang Y, Zhao L, Li F, Zhuang Z. Long non-coding RNA HAGLROS regulates the proliferation, migration, and apoptosis of esophageal cancer cells via the HAGLROS-miR-206-NOTCH3 axis. J Gastrointest Oncol. 2021;12:2093-108
127. Jin G, Yang Y, Tuo G, Wang W, Zhu Z. LncRNA TUG1 promotes tumor growth and metastasis of esophageal squamous cell carcinoma by regulating XBP1 via competitively binding to miR-498. Neoplasma. 2020;67:751-61
128. Wang J, Yang X, Li R, Zhang R, Hu D, Zhang Y. et al. LncRNA SNHG6 Inhibits Apoptosis by Regulating EZH2 Expression via the Sponging of MiR-101-3p in Esophageal Squamous-Cell Carcinoma. Oncotargets Ther. 2020;13:11411-20
129. Zhu JL, Xue WB, Jiang ZB, Feng W, Liu YC, Nie XY. et al. Long noncoding RNA CDKN2B-AS1 silencing protects against esophageal cancer cell invasion and migration by inactivating the TFAP2A/FSCN1 axis. Kaohsiung J Med Sci. 2022;38:1144-54
130. Liu Z, Yang S, Chen X, Dong S, Zhou S, Xu S. LncRNA LINC00467 acted as an oncogene in esophageal squamous cell carcinoma by accelerating cell proliferation and preventing cell apoptosis via the miR-485-5p/DPAGT1 axis. J Gastroen Hepatol. 2021;36:721-30
131. Ye H, Mulmi SS, Zhu J, Ding Y, Shi R. Long non-coding RNA LINC00491 promotes proliferation and inhibits apoptosis in esophageal squamous cell carcinoma. Int J Mol Med. 2021 47
132. Shen SN, Li K, Liu Y, Yang CL, He CY, Wang HR. Down-regulation of long noncoding RNA PVT1 inhibits esophageal carcinoma cell migration and invasion and promotes cell apoptosis via microRNA-145-mediated inhibition of FSCN1. Mol Oncol. 2019;13:2554-73
133. Li W, Zhao W, Lu Z, Zhang W, Yang X. Long Noncoding RNA GAS5 Promotes Proliferation, Migration, and Invasion by Regulation of miR-301a in Esophageal Cancer. Oncol Res. 2018;26:1285-94
134. Han D, Yuan RX, Su F. LINC00511 can promote the proliferation, migration and invasion of esophageal cancer cells through regulating microRNA-150-5p. Eur Rev Med Pharmaco. 2020;24:2462-9
135. Su W, Wang L, Niu F, Zou L, Guo C, Wang Z. et al. LINC00857 knockdown inhibits cell proliferation and induces apoptosis via involving STAT3 and MET oncogenic proteins in esophageal adenocarcinoma. Aging (Albany NY). 2019;11:2812-21
136. Yang X, Shen Z, Tian M, Lin Y, Li L, Chai T. et al. LncRNA C9orf139 can regulate the progression of esophageal squamous carcinoma by mediating the miR-661/HDAC11 axis. Transl Oncol. 2022;24:101487
137. Li H, Jia J, Yang L, Chu J, Sheng J, Wang C. et al. LncRNA MIR205HG Drives Esophageal Squamous Cell Carcinoma Progression by Regulating miR-214/SOX4 Axis. Oncotargets Ther. 2020;13:13097-109
138. Lu T, Wang R, Cai H, Cui Y. Long non-coding RNA DLEU2 promotes the progression of esophageal cancer through miR-30e-5p/E2F7 axis. Biomed Pharmacother. 2020;123:109650
139. Fleming A, Bourdenx M, Fujimaki M, Karabiyik C, Krause GJ, Lopez A. et al. The different autophagy degradation pathways and neurodegeneration. Neuron. 2022;110:935-66
140. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Bio. 2018;19:349-64
141. De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435-92
142. Levy J, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528-42
143. Kocaturk NM, Akkoc Y, Kig C, Bayraktar O, Gozuacik D, Kutlu O. Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci. 2019;134:116-37
144. Popova NV, Jucker M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int J Mol Sci. 2021 22
145. Jian Z, Han Y, Zhang W, Li C, Guo W, Feng X. et al. Anti-tumor effects of dual PI3K-HDAC inhibitor CUDC-907 on activation of ROS-IRE1alpha-JNK-mediated cytotoxic autophagy in esophageal cancer. Cell Biosci. 2022;12:135
146. Wu N, Zhu Y, Xu X, Zhu Y, Song Y, Pang L. et al. The anti-tumor effects of dual PI3K/mTOR inhibitor BEZ235 and histone deacetylase inhibitor Trichostatin A on inducing autophagy in esophageal squamous cell carcinoma. J Cancer. 2018;9:987-97
147. Liu KS, Zhang Y, Ding WC, Wang SX, Xiang YF, Yang P. et al. The selective Hsp90 inhibitor BJ-B11 exhibits potent antitumor activity via induction of cell cycle arrest, apoptosis and autophagy in Eca-109 human esophageal squamous carcinoma cells. Int J Oncol. 2012;41:2276-84
148. Qin WJ, Su YG, Ding XL, Zhao R, Zhao ZJ, Wang YY. CDK4/6 inhibitor enhances the radiosensitization of esophageal squamous cell carcinoma (ESCC) by activating autophagy signaling via the suppression of mTOR. Am J Transl Res. 2022;14:1616-27
149. Hong P, Liu QW, Xie Y, Zhang QH, Liao L, He QY. et al. Echinatin suppresses esophageal cancer tumor growth and invasion through inducing AKT/mTOR-dependent autophagy and apoptosis. Cell Death Dis. 2020;11:524
150. Liu H, Zhao J, Fu R, Zhu C, Fan D. The ginsenoside Rk3 exerts anti-esophageal cancer activity in vitro and in vivo by mediating apoptosis and autophagy through regulation of the PI3K/Akt/mTOR pathway. Plos One. 2019;14:e216759
151. Lee NR, Meng RY, Rah SY, Jin H, Ray N, Kim SH. et al. Reactive Oxygen Species-Mediated Autophagy by Ursolic Acid Inhibits Growth and Metastasis of Esophageal Cancer Cells. Int J Mol Sci. 2020 21
152. Chen X, He LY, Lai S, He Y. Dihydroartemisinin inhibits the migration of esophageal cancer cells by inducing autophagy. Oncol Lett. 2020;20:94
153. Deng L, Wu X, Zhu X, Yu Z, Liu Z, Wang J. et al. Combination effect of curcumin with docetaxel on the PI3K/AKT/mTOR pathway to induce autophagy and apoptosis in esophageal squamous cell carcinoma. Am J Transl Res. 2021;13:57-72
154. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19:12
155. Lu J, Zhu L, Zheng LP, Cui Q, Zhu HH, Zhao H. et al. Overexpression of ULK1 Represents a Potential Diagnostic Marker for Clear Cell Renal Carcinoma and the Antitumor Effects of SBI-0206965. Ebiomedicine. 2018;34:85-93
156. Yun M, Bai H, Zhang J, Rong J, Weng H, Zheng Z. et al. ULK1: A Promising Biomarker in Predicting Poor Prognosis and Therapeutic Response in Human Nasopharygeal Carcinoma. Plos One. 2015;10:e117375
157. Zou Y, Chen Z, He X, He X, Wu X, Chen Y. et al. High expression levels of unc-51-like kinase 1 as a predictor of poor prognosis in colorectal cancer. Oncol Lett. 2015;10:1583-8
158. Sun D, Yang Z, Zhen Y, Yang Y, Chen Y, Yuan Y. et al. Discovery of 5-bromo-4-phenoxy-N-phenylpyrimidin-2-amine derivatives as novel ULK1 inhibitors that block autophagy and induce apoptosis in non-small cell lung cancer. Eur J Med Chem. 2020;208:112782
159. Zou L, Liao M, Zhen Y, Zhu S, Chen X, Zhang J. et al. Autophagy and beyond: Unraveling the complexity of UNC-51-like kinase 1 (ULK1) from biological functions to therapeutic implications. Acta Pharm Sin B. 2022;12:3743-82
160. Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ. et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015;59:285-97
161. Driessen S, Berleth N, Friesen O, Loffler AS, Bohler P, Hieke N. et al. Deubiquitinase inhibition by WP1130 leads to ULK1 aggregation and blockade of autophagy. Autophagy. 2015;11:1458-70
162. Martin KR, Celano SL, Solitro AR, Gunaydin H, Scott M, O'Hagan RC. et al. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. Iscience. 2018;8:74-84
163. Petherick KJ, Conway OJ, Mpamhanga C, Osborne SA, Kamal A, Saxty B. et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015;290:11376-83
164. Lazarus MB, Shokat KM. Discovery and structure of a new inhibitor scaffold of the autophagy initiating kinase ULK1. Bioorgan Med Chem. 2015;23:5483-8
165. Lazarus MB, Novotny CJ, Shokat KM. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. Acs Chem Biol. 2015;10:257-61
166. Jiang S, Li Y, Zhu YH, Wu XQ, Tang J, Li Z. et al. Intensive expression of UNC-51-like kinase 1 is a novel biomarker of poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Sci. 2011;102:1568-75
167. Perez-Hernandez M, Arias A, Martinez-Garcia D, Perez-Tomas R, Quesada R, Soto-Cerrato V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers. 2019 11
168. Li X, Liu S, Jin L, Ma Y, Liu T. NOD2 inhibits the proliferation of esophageal adenocarcinoma cells through autophagy. J Cancer Res Clin. 2022
169. Liu PF, Tsai KL, Hsu CJ, Tsai WL, Cheng JS, Chang HW. et al. Drug Repurposing Screening Identifies Tioconazole as an ATG4 Inhibitor that Suppresses Autophagy and Sensitizes Cancer Cells to Chemotherapy. Theranostics. 2018;8:830-45
170. Fu Y, Hong L, Xu J, Zhong G, Gu Q, Gu Q. et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy. 2019;15:295-311
171. Pengo N, Prak K, Costa JR, Luft C, Agrotis A, Freeman J. et al. Identification of Kinases and Phosphatases That Regulate ATG4B Activity by siRNA and Small Molecule Screening in Cells. Front Cell Dev Biol. 2018;6:148
172. Zhang L, Guo M, Li J, Zheng Y, Zhang S, Xie T. et al. Systems biology-based discovery of a potential Atg4B agonist (Flubendazole) that induces autophagy in breast cancer. Mol Biosyst. 2015;11:2860-6
173. Khan T, Relitti N, Brindisi M, Magnano S, Zisterer D, Gemma S. et al. Autophagy modulators for the treatment of oral and esophageal squamous cell carcinomas. Med Res Rev. 2020;40:1002-60
174. McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S. et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. P Natl Acad Sci Usa. 2012;109:8253-8
175. Baquero P, Dawson A, Mukhopadhyay A, Kuntz EM, Mitchell R, Olivares O. et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia. 2019;33:981-94
176. Lam YH, Than H, Sng C, Cheong MA, Chuah C, Xiang W. Lysosome Inhibition by Mefloquine Preferentially Enhances the Cytotoxic Effects of Tyrosine Kinase Inhibitors in Blast Phase Chronic Myeloid Leukemia. Transl Oncol. 2019;12:1221-8
177. Sharma N, Thomas S, Golden EB, Hofman FM, Chen TC, Petasis NA. et al. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012;326:143-54
178. Goodall ML, Wang T, Martin KR, Kortus MG, Kauffman AL, Trent JM. et al. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy. 2014;10:1120-36
179. Rebecca VW, Nicastri MC, McLaughlin N, Fennelly C, McAfee Q, Ronghe A. et al. A Unified Approach to Targeting the Lysosome's Degradative and Growth Signaling Roles. Cancer Discov. 2017;7:1266-83
180. Greene LM, Nolan DP, Regan-Komito D, Campiani G, Williams DC, Zisterer DM. Inhibition of late-stage autophagy synergistically enhances pyrrolo-1,5-benzoxazepine-6-induced apoptotic cell death in human colon cancer cells. Int J Oncol. 2013;43:927-35
181. Xie Z, Xie Y, Xu Y, Zhou H, Xu W, Dong Q. Bafilomycin A1 inhibits autophagy and induces apoptosis in MG63 osteosarcoma cells. Mol Med Rep. 2014;10:1103-7
182. Wu K, Na K, Chen D, Wang Y, Pan H, Wang X. Effects of non-steroidal anti-inflammatory drug-activated gene-1 on Ganoderma lucidum polysaccharides-induced apoptosis of human prostate cancer PC-3 cells. Int J Oncol. 2018;53:2356-68
183. Goard CA, Schimmer AD. An evidence-based review of obatoclax mesylate in the treatment of hematological malignancies. Core Evid. 2013;8:15-26
184. Zheng K, Li Y, Wang S, Wang X, Liao C, Hu X. et al. Inhibition of autophagosome-lysosome fusion by ginsenoside Ro via the ESR2-NCF1-ROS pathway sensitizes esophageal cancer cells to 5-fluorouracil-induced cell death via the CHEK1-mediated DNA damage checkpoint. Autophagy. 2016;12:1593-613
185. Shan C, Hui W, Li H, Wang Z, Guo C, Peng R. et al. Discovery of Novel Autophagy Inhibitors and Their Sensitization Abilities for Vincristine-Resistant Esophageal Cancer Cell Line Eca109/VCR. Chemmedchem. 2020;15:970-81
186. Li C, Liu F, Yang X, Guo B, Li G, Yin J. et al. Targeting lectin-like oxidized low-density lipoprotein receptor-1 triggers autophagic program in esophageal cancer. Cell Death Differ. 2022;29:697-708
187. Chen YS, Song HX, Lu Y, Li X, Chen T, Zhang Y. et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis Esophagus. 2011;24:437-43
188. Chen Y, Li X, Guo L, Wu X, He C, Zhang S. et al. Combining radiation with autophagy inhibition enhances suppression of tumor growth and angiogenesis in esophageal cancer. Mol Med Rep. 2015;12:1645-52
189. Li F, Zhou X, Chen M, Fan W. Regulatory effect of LncRNA DRAIC/miR-149-5p/NFIB molecular network on autophagy of esophageal cancer cells and its biological behavior. Exp Mol Pathol. 2020;116:104491
190. Yang C, Shen S, Zheng X, Ye K, Ge H, Sun Y. et al. Long non-coding RNA LINC00337 induces autophagy and chemoresistance to cisplatin in esophageal squamous cell carcinoma cells via upregulation of TPX2 by recruiting E2F4. Faseb J. 2020;34:6055-69
191. Kakeji Y, Oshikiri T, Takiguchi G, Kanaji S, Matsuda T, Nakamura T. et al. Multimodality approaches to control esophageal cancer: development of chemoradiotherapy, chemotherapy, and immunotherapy. Esophagus-Tokyo. 2021;18:25-32
192. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
193. Sun WY, Tyurin VA, Mikulska-Ruminska K, Shrivastava IH, Anthonymuthu TS, Zhai YJ. et al. Phospholipase iPLA2beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol. 2021;17:465-76
194. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S. et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639-43
195. Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018;38:12
196. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317-31
197. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247-50
198. Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell. 2021;12:836-57
199. Hassannia B, Vandenabeele P, Vanden BT. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell. 2019;35:830-49
200. Liu CC, Li HH, Lin JH, Chiang MC, Hsu TW, Li AF. et al. Esophageal Cancer Stem-like Cells Resist Ferroptosis-Induced Cell Death by Active Hsp27-GPX4 Pathway. Biomolecules. 2021 12
201. Shishido Y, Amisaki M, Matsumi Y, Yakura H, Nakayama Y, Miyauchi W. et al. Antitumor Effect of 5-Aminolevulinic Acid Through Ferroptosis in Esophageal Squamous Cell Carcinoma. Ann Surg Oncol. 2021;28:3996-4006
202. Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ. et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol. 2020;16:497-506
203. Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG. et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radical Bio Med. 2003;34:496-502
204. Friedmann AJ, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180-91
205. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radical Bio Med. 2020;152:175-85
206. Chio I, Tuveson DA. ROS in Cancer: The Burning Question. Trends Mol Med. 2017;23:411-29
207. Feng L, Zhao K, Sun L, Yin X, Zhang J, Liu C. et al. SLC7A11 regulated by NRF2 modulates esophageal squamous cell carcinoma radiosensitivity by inhibiting ferroptosis. J Transl Med. 2021;19:367
208. Shi ZZ, Tao H, Fan ZW, Song SJ, Bai J. Prognostic and Immunological Role of Key Genes of Ferroptosis in Pan-Cancer. Front Cell Dev Biol. 2021;9:748925
209. Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B. et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133:1732-42
210. Jyotsana N, Ta KT, DelGiorno KE. The Role of Cystine/Glutamate Antiporter SLC7A11/xCT in the Pathophysiology of Cancer. Front Oncol. 2022;12:858462
211. Liang C, Zhang X, Yang M, Dong X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv Mater. 2019;31:e1904197
212. Raimondi V, Ciccarese F, Ciminale V. Oncogenic pathways and the electron transport chain: a dangeROS liaison. Brit J Cancer. 2020;122:168-81
213. Kong H, Chandel NS. Regulation of redox balance in cancer and T cells. J Biol Chem. 2018;293:7499-507
214. Gamcsik MP, Kasibhatla MS, Teeter SD, Colvin OM. Glutathione levels in human tumors. Biomarkers. 2012;17:671-91
215. Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC. et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211-22
216. Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol. 2018;217:2291-8
217. Circu ML, Stringer S, Rhoads CA, Moyer MP, Aw TY. The role of GSH efflux in staurosporine-induced apoptosis in colonic epithelial cells. Biochem Pharmacol. 2009;77:76-85
218. Trompier D, Chang X, Barattin R, D Hardemare ADM, Di Pietro A, Baubichon-Cortay H. Verapamil and Its Derivative Trigger Apoptosis through Glutathione Extrusion by Multidrug Resistance Protein MRP1. Cancer Res. 2004;64:4950-6
219. Dury L, Nasr R, Lorendeau D, Comsa E, Wong I, Zhu X. et al. Flavonoid dimers are highly potent killers of multidrug resistant cancer cells overexpressing MRP1. Biochem Pharmacol. 2017;124:10-8
220. Dacevic M, Isakovic A, Podolski-Renic A, Isakovic AM, Stankovic T, Milosevic Z. et al. Purine nucleoside analog-sulfinosine modulates diverse mechanisms of cancer progression in multi-drug resistant cancer cell lines. Plos One. 2013;8:e54044
221. Liao W, Xiang W, Wang FF, Wang R, Ding Y. Curcumin inhibited growth of human melanoma A375 cells via inciting oxidative stress. Biomed Pharmacother. 2017;95:1177-86
222. Cruz A, Mota P, Ramos C, Pires RF, Mendes C, Silva JP. et al. Polyurea Dendrimer Folate-Targeted Nanodelivery of l-Buthionine sulfoximine as a Tool to Tackle Ovarian Cancer Chemoresistance. Antioxidants-Basel. 2020 9
223. Peng L, Linghu R, Chen D, Yang J, Kou X, Wang XZ. et al. Inhibition of glutathione metabolism attenuates esophageal cancer progression. Exp Mol Med. 2017;49:e318
224. Ku CM, Lin JY. Anti-inflammatory effects of 27 selected terpenoid compounds tested through modulating Th1/Th2 cytokine secretion profiles using murine primary splenocytes. Food Chem. 2013;141:1104-13
225. Li Y, Li N, Shi J, Ahmed T, Liu H, Guo J. et al. Involvement of Glutathione Depletion in Selective Cytotoxicity of Oridonin to p53-Mutant Esophageal Squamous Carcinoma Cells. Front Oncol. 2019;9:1525
226. Yang H, Wang J, Khan S, Zhang Y, Zhu K, Zhou E. et al. Selective synergistic anticancer effects of cisplatin and oridonin against human p53-mutant esophageal squamous carcinoma cells. Anti-Cancer Drug. 2022;33:e444-52
227. Zhang J, Wang N, Zhou Y, Wang K, Sun Y, Yan H. et al. Oridonin induces ferroptosis by inhibiting gamma-glutamyl cycle in TE1 cells. Phytother Res. 2021;35:494-503
228. Godwin AK, Meister A, O'Dwyer PJ, Huang CS, Hamilton TC, Anderson ME. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. P Natl Acad Sci Usa. 1992;89:3070-4
229. Xiong Y, Xiao C, Li Z, Yang X. Engineering nanomedicine for glutathione depletion-augmented cancer therapy. Chem Soc Rev. 2021;50:6013-41
230. Doll S, Freitas FP, Shah R, Aldrovandi M, Da SM, Ingold I. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693-8
231. Yoshioka H, Kawamura T, Muroi M, Kondoh Y, Honda K, Kawatani M. et al. Identification of a Small Molecule That Enhances Ferroptosis via Inhibition of Ferroptosis Suppressor Protein 1 (FSP1). Acs Chem Biol. 2022;17:483-91
232. Zhang S, Guo J, Zhang H, Tong L, Zhang L. Gliotoxin Induced Ferroptosis by Downregulating SUV39H1 Expression in Esophageal Cancer Cells. Recent Pat Anti-Canc. 2022
233. Yang R, Chen F, Xu H, Guo Z, Cao C, Zhang H. et al. Exploring the Mechanism of Realgar against Esophageal Cancer Based on Ferroptosis Induced by ROS-ASK1-p38 MAPK Signaling Pathway. Evid-Based Compl Alt. 2022;2022:3698772
234. Ballout F, Lu H, Chen Z, Hu T, Chen L, Washington MK. et al. Targeting NRF2 Sensitizes Esophageal Adenocarcinoma Cells to Cisplatin through Induction of Ferroptosis and Apoptosis. Antioxidants-Basel. 2022 11
235. Luo H, Wang X, Song S, Wang Y, Dan Q, Ge H. Targeting stearoyl-coa desaturase enhances radiation induced ferroptosis and immunogenic cell death in esophageal squamous cell carcinoma. Oncoimmunology. 2022;11:2101769
236. Hou J, Huang Q, Fan Z, Sang H, Wu S, Cheng S. et al. LncRNA OIP5-AS1 Knockdown Facilitated the Ferroptosis and Immune Evasion by Modulating the GPX4 in Oesophageal Carcinoma. Comput Math Method M. 2022;2022:8103198
237. Pan C, Chen G, Zhao X, Xu X, Liu J. lncRNA BBOX1-AS1 silencing inhibits esophageal squamous cell cancer progression by promoting ferroptosis via miR-513a-3p/SLC7A11 axis. Eur J Pharmacol. 2022;934:175317
238. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167-9
239. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660-5
240. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Tar. 2021;6:128
241. Du T, Gao J, Li P, Wang Y, Qi Q, Liu X. et al. Pyroptosis, metabolism, and tumor immune microenvironment. Clin Transl Med. 2021;11:e492
242. Qiao L, Wu X, Zhang J, Liu L, Sui X, Zhang R. et al. alpha-NETA induces pyroptosis of epithelial ovarian cancer cells through the GSDMD/caspase-4 pathway. Faseb J. 2019;33:12760-7
243. Pizato N, Luzete BC, Kiffer L, Correa LH, de Oliveira SI, Assumpcao J. et al. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triple-negative breast cancer cells. Sci Rep-Uk. 2018;8:1952
244. Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J. et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10:193
245. Zhang CC, Li CG, Wang YF, Xu LH, He XH, Zeng QZ. et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24:312-25
246. Chen L, Weng B, Li H, Wang H, Li Q, Wei X. et al. A thiopyran derivative with low murine toxicity with therapeutic potential on lung cancer acting through a NF-kappaB mediated apoptosis-to-pyroptosis switch. Apoptosis. 2019;24:74-82
247. Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K. et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171-85
248. Wu M, Wang Y, Yang D, Gong Y, Rao F, Liu R. et al. A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. Ebiomedicine. 2019;41:244-55
249. Wang L, Li K, Lin X, Yao Z, Wang S, Xiong X. et al. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR-497/PELP1 axis. Cancer Lett. 2019;450:22-31
250. Li L, Song D, Qi L, Jiang M, Wu Y, Gan J. et al. Photodynamic therapy induces human esophageal carcinoma cell pyroptosis by targeting the PKM2/caspase-8/caspase-3/GSDME axis. Cancer Lett. 2021;520:143-59
251. Jiang M, Wu Y, Qi L, Li L, Song D, Gan J. et al. Dihydroartemisinin mediating PKM2-caspase-8/3-GSDME axis for pyroptosis in esophageal squamous cell carcinoma. Chem-Biol Interact. 2021;350:109704
252. Li RY, Zheng ZY, Li ZM, Heng JH, Zheng YQ, Deng DX. et al. Cisplatin-induced pyroptosis is mediated via the CAPN1/CAPN2-BAK/BAX-caspase-9-caspase-3-GSDME axis in esophageal cancer. Chem-Biol Interact. 2022;361:109967
253. Tan JQ, Li Z, Chen G, Wu M, Feng JL, Kong SY. et al. The natural compound from Garcinia bracteata mainly induces GSDME-mediated pyroptosis in esophageal cancer cells. Phytomedicine. 2022;102:154142
254. Chen JH, Guo QF, Liu QG, He BX, Song WP, Yin ZH. et al. Chaetoglobosin E inhibits tumor growth and promotes the anti-tumor efficacy of cytotoxic drugs in esophageal squamous cell carcinoma by targeting PLK1. Ann Transl Med. 2022;10:1236
255. Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB. et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045-56
256. Chai D, Liu N, Li H, Wang G, Song J, Fang L. et al. H1/pAIM2 nanoparticles exert anti-tumour effects that is associated with the inflammasome activation in renal carcinoma. J Cell Mol Med. 2018;22:5670-81
257. Yue E, Tuguzbaeva G, Chen X, Qin Y, Li A, Sun X. et al. Anthocyanin is involved in the activation of pyroptosis in oral squamous cell carcinoma. Phytomedicine. 2019;56:286-94
258. Li Y, Wang W, Li A, Huang W, Chen S, Han F. et al. Dihydroartemisinin induces pyroptosis by promoting the AIM2/caspase-3/DFNA5 axis in breast cancer cells. Chem-Biol Interact. 2021;340:109434
259. Wang F, Liu W, Ning J, Wang J, Lang Y, Jin X. et al. Simvastatin Suppresses Proliferation and Migration in Non-small Cell Lung Cancer via Pyroptosis. Int J Biol Sci. 2018;14:406-17
260. Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC. et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med. 2018;24:1151-6
261. Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep-Uk. 2016;6:36107
262. Chen J, Peng R, Niu Z, Zhou H, Kang C. Betulinic acid enhanced the chemical sensitivity of esophageal cancer cells to cisplatin by inducing cell pyroptosis and reducing cell stemness. Ann Palliat Med. 2020;9:1912-20
263. Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263-8
264. Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K. et al. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019;18:100
265. Hockendorf U, Yabal M, Herold T, Munkhbaatar E, Rott S, Jilg S. et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell. 2016;30:75-91
266. Strilic B, Yang L, Albarran-Juarez J, Wachsmuth L, Han K, Muller UC. et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536:215-8
267. McCormick KD, Ghosh A, Trivedi S, Wang L, Coyne CB, Ferris RL. et al. Innate immune signaling through differential RIPK1 expression promote tumor progression in head and neck squamous cell carcinoma. Carcinogenesis. 2016;37:522-9
268. Seifert L, Werba G, Tiwari S, Giao LN, Alothman S, Alqunaibit D. et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532:245-9
269. Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS. et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25:707-25
270. Feng X, Song Q, Yu A, Tang H, Peng Z, Wang X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma. 2015;62:592-601
271. Nugues AL, El BH, Hetuin D, Berthon C, Loyens A, Bertrand E. et al. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis. 2014;5:e1384
272. Sun Y, Zhai L, Ma S, Zhang C, Zhao L, Li N. et al. Down-regulation of RIP3 potentiates cisplatin chemoresistance by triggering HSP90-ERK pathway mediated DNA repair in esophageal squamous cell carcinoma. Cancer Lett. 2018;418:97-108
273. He L, Peng K, Liu Y, Xiong J, Zhu FF. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Oncotargets Ther. 2013;6:1539-43
274. Colbert LE, Fisher SB, Hardy CW, Hall WA, Saka B, Shelton JW. et al. Pronecrotic mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer-Am Cancer Soc. 2013;119:3148-55
275. Ertao Z, Jianhui C, Kang W, Zhijun Y, Hui W, Chuangqi C. et al. Prognostic value of mixed lineage kinase domain-like protein expression in the survival of patients with gastric caner. Tumour Biol. 2016;37:13679-85
276. Li X, Guo J, Ding AP, Qi WW, Zhang PH, Lv J. et al. Association of Mixed Lineage Kinase Domain-Like Protein Expression With Prognosis in Patients With Colon Cancer. Technol Cancer Res T. 2017;16:428-34
277. Zhou J, Li G, Han G, Feng S, Liu Y, Chen J. et al. Emodin induced necroptosis in the glioma cell line U251 via the TNF-alpha/RIP1/RIP3 pathway. Invest New Drug. 2020;38:50-9
278. Lu Z, Wu C, Zhu M, Song W, Wang H, Wang J. et al. Ophiopogonin D' induces RIPK1dependent necroptosis in androgendependent LNCaP prostate cancer cells. Int J Oncol. 2020;56:439-47
279. Li Y, Tian X, Liu X, Gong P. Bufalin inhibits human breast cancer tumorigenesis by inducing cell death through the ROS-mediated RIP1/RIP3/PARP-1 pathways. Carcinogenesis. 2018;39:700-7
280. Xie Y, Zhu S, Zhong M, Yang M, Sun X, Liu J. et al. Inhibition of Aurora Kinase A Induces Necroptosis in Pancreatic Carcinoma. Gastroenterology. 2017;153:1429-43
281. Xu H, Zeng X, Wei X, Xue Z, Chen N, Zhu W. et al. Rosin Derivative IDOAMP Inhibits Prostate Cancer Growth via Activating RIPK1/RIPK3/MLKL Signaling Pathway. Oxid Med Cell Longev. 2022;2022:9325973
Author contact
![]() Corresponding authors: Bo Liu (liubo2400com), Yong Yuan (yongyuanedu.cn); Tel./Fax.: (+86)-28-85164063.
Corresponding authors: Bo Liu (liubo2400com), Yong Yuan (yongyuanedu.cn); Tel./Fax.: (+86)-28-85164063.