Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The structure and function of...
HBV infection and mTOR
Non-viral HCC and mTOR
mTOR inhibitor treatment in HCC
Summary and Outlook
Funding
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(11):4178-4189. doi:10.7150/ijbs.95894 This issue Cite
Review
mTOR Signaling: Roles in Hepatitis B Virus Infection and Hepatocellular Carcinoma
Ling Mei, MPH1,2,3, Huizhen Sun, PhD1,2, Ying Yan, PhD1,2, Huimin Ji, PhD1,2, Qian Su, PhD1,2,3, Le Chang, PhD1,2,3 ![]() , Lunan Wang, PhD1,2,3
, Lunan Wang, PhD1,2,3 ![]()
1. National Center for Clinical Laboratories, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing Hospital/ National Center of Gerontology, Beijing, 100730, P.R. China.
2. Beijing Engineering Research Center of Laboratory Medicine, Beijing Hospital, Beijing, 100730, P.R. China.
3. National Center for Clinical Laboratories, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, 100730, P.R. China.
Received 2024-3-3; Accepted 2024-7-24; Published 2024-8-1
Abstract

Currently, chronic hepatitis B virus infection is still one of the most serious public health problems in the world. Though current strategies are effective in controlling infection and slowing down the disease process, it remains a big challenge to achieve a functional cure for chronic hepatitis B in a majority of patients due to the inability to clear the cccDNA pool. The mammalian target of rapamycin (mTOR) integrates nutrition, energy, growth factors, and other extracellular signals, participating in gene transcription, protein translation, ribosome synthesis, and other biological processes. Additionally, mTOR plays an extremely important role in cell growth, apoptosis, autophagy, and metabolism. More and more evidence show that HBV infection can activate the mTOR pathway, suggesting that HBV uses or hijacks the mTOR pathway to facilitate its own replication. Therefore, mTOR signaling pathway may be a key target for controlling HBV infection. However, the role of the central cytokine mTOR in the pathogenesis of HBV infection has not yet been systematically addressed. Notably, mTOR is commonly activated in hepatocellular carcinoma, which can progress from chronic hepatitis B. This review systematically summarizes the role of mTOR in the life cycle of HBV and its impact on the clinical progression of HBV infection.
Keywords: mammalian target of rapamycin, hepatitis B virus, hepatocellular carcinoma, mTOR inhibitors
Introduction
Hepatitis B virus (HBV) infection remains a major global public health concern, with an estimated 257 million individuals worldwide suffering from chronic HBV infection[1]. Chronic HBV infection can lead to serious progressive liver diseases, such as cirrhosis, liver failure, and hepatocellular carcinoma (HCC)[2,3], which is the third leading cause of cancer-related deaths globally[4]. While effective vaccines have been instrumental in reducing HBV infection rates, particularly in infants, the efficacy of existing medications, including alpha interferon and nucleoside analogues that block viral DNA polymerase is hindered by low rates of sustained response, adverse effects, and the emergence of drug resistance[1,5-7]. The pathogenesis of HBV infection emphasizes that the specific immune response is not only responsible for viral clearance but also results in hepatocyte inflammatory and regenerative responses. It triggers mitogenic stimuli and mutagenic factors for the formation of DNA damage that can lead to the development of HCC[8-12].
It is worth noting that there is a contradiction between the theory of the pathogenesis of HBV infection and the clinical practice of medication. Currently, the pathogenesis emphasizes that the damage caused by HBV infection is mediated by immunity, while the clinical treatment focuses on inhibiting HBV replication to alleviate liver disease. This incongruity raises questions about the need for additional supplements to the pathogenesis of HBV infection, which could enhance our understanding of the disease and optimize the treatment plans. The mammalian target of rapamycin (mTOR) signaling pathway has been found to regulate the life cycle of many viruses[13,14], and understanding the role of mTOR in the life cycle of HBV is not only significant for clarifying the pathogenesis of HBV, but also for developing effective treatment strategies[15].
HCC is one of the most common tumors worldwide, and its treatment methods include surgical resection, percutaneous ethanol injection (PEI), radiofrequency ablation (RFA), transcatheter arterial chemoembolization (TACE), and liver transplantation. However, these methods are only suitable for a small number of patients and often lead to postoperative complications[16]. Fewer than 10% of HCC patients are cured, and most eventually progress to advanced HCC. At present, only systemic treatment can effectively delay the natural course of the disease[17]. The current frontline treatment for systemic therapy in clinical practice is the tyrosine kinase inhibitor (TKI) Sorafenib, both the RAF/MEK/ERK pathway and receptor tyrosine kinases to inhibit tumor growth and angiogenesis[18]. However, its median survival is only 10.7 months[19]. Given that mTOR is activated in 40% -50% HCC cases[20-23], it is essential to elaborate on the role of mTOR in the progression of HCC. This review aims to summarize the latest developments in the interaction between HBV and mTOR, as well as the impact of mTOR on HCC progression.
The structure and function of mTOR
The mTOR protein is a member of the phosphoinositide 3-kinase (PI3K)-related kinase (PIKK) family and is an evolutionarily conserved Ser/Thr kinase. it was discovered that mTOR and yeast TOR/DRR proteins, previously identified as rapamycin targets in genetic tests for rapamycin resistance, shared a similarity[24-26]. The mTOR protein consists of about 2,500 amino acids and contains functional domains including HEAT repeats, FAT, FRB, Kinase, and FATC (Figure 1). The FAT and FATC domains participate in the interaction between mTOR and its ligands, while the FRB (FKBP12-Rapamycin Binding) domain is involved in the binding of Rapamycin and FKBP12 to inhibit mTOR activity[27,28], and the Kinase domain is responsible for the phosphorylation of downstream targets of mTOR. Overall, the structure of mTOR is intricate, with different domains participating in different functions in signal transduction, protein synthesis, and cellular metabolism[29].
The composition of both mTORC1 and mTORC1. mTORC1 comprises a core of mTOR, mLST8, and Raptor that is suppressed by PRAS40 and DEPTOR. mTORC2 is composed of a basic complex of mTOR, mLST8, and Rictor, which is inhibited by DEPTOR and regulated by mSin1 and Protor1/2. A combination consisting of rapamycin and the cytoplasmic receptor FKBP12 binds to the FRB domain, allosterically inhibiting mTOR action.
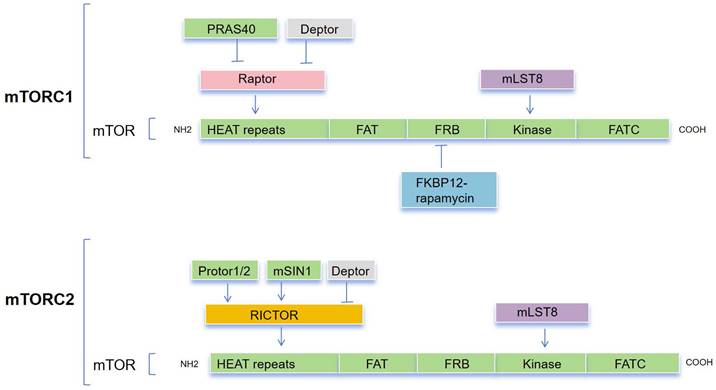
mTOR is the catalytic subunit of two complexes, mTORC1 and mTORC2[30] (Figure 1), and their activation and function depend on their subcellular localization [31]. These complexes can be distinguished by their specific substrates and activities, auxiliary proteins, and varying sensitivity to rapamycin. Three primary constituents comprise mTORC1: the catalytic subunit mTOR, the regulatory subunit Raptor, and mLST8[32,33]. Raptor guarantees appropriate subcellular localization and aids in the recruitment of substrates into the complex. By attaching itself to the catalytic domain, mLST8 keeps the kinase activation loop stable. In addition to these elements, mTORC1 contains two inhibitory subunits: DEPTOR and PRAS40 (Akt substrate). mTORC2 consists of mLST8, mTOR subunit and Rictor, which is insensitive to rapamycin and comparable with Raptor. Together with the other regulatory subunits, mSin1 and Protor1/2, mTORC2 also contains the inhibitory Deptor subunit[34-37].
mTORC1 plays a crucial role in regulating numerous essential cellular processes, including glucose homeostasis, lipid synthesis, and autophagy, by functioning as a sensor for growth factors, pressure, energy status, oxygen, and amino acids[38,39]. On the other hand, the mTORC2 complex regulates cell survival and cytoskeletal structure[34,35]. It also participates in the transcription factor forkhead box protein O3 (FOXO3) pathway, which controls autophagy[40]. Through a negative feedback loop between the mTORC1 and PI3K-AKT pathways, mTORC1 also controls mTORC2 signaling[41]. Tuberous sclerosis 1 (TSC1) and 2 (TSC2) are key upstream regulators of mTORC1. The GTP-bound form of Ras homolog enriched in the brain (Rheb) directly interacts with mTORC1 and strongly stimulates its kinase activity[42-44]. However, TSC1/2 negatively regulates mTORC1 activity by reversing Rheb into its inactive GDP-bound state [45,46]. Conversely, mTORC2 is a rapamycin-insensitive companion of mTOR[47] and can be directly activated by PI3K[48,49]. Compared with the mTORC1 pathway, the mTORC2 pathway is much less understood. Its downstream targets include several members of the AGC kinase subfamily, such as Akt, serum and glucocorticoid-induced protein kinase 1 (SGK1) and protein kinase C-a (PKC-a). mTORC2 directly activates Akt by phosphorylating the hydrophobic motif (Ser473) of Akt, which is required for its maximum activation[50]. Akt then regulates cellular metabolism, survival, apoptosis, growth and proliferation through the phosphorylation of several effectors. This involves the classic PI3K-AKT-mTOR pathway.
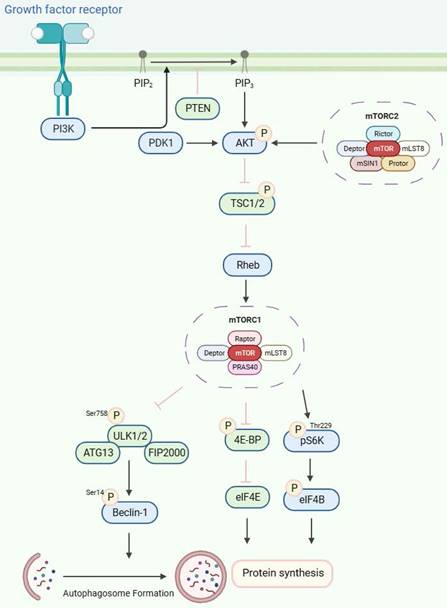
The growth factor-mediated receptor tyrosine kinases (RTKs)/PI3K/Akt signaling pathway is an important upstream signaling pathway for the mTOR protein molecule[51](Figure 2). Upon stimulation by growth factors, the RTKs initiate signaling cascades that activate PI3K. Generally, PI3K activity is tightly controlled to a basal level under normal conditions. Subsequently, PI3K catalyzes the synthesis of phosphatidylinositol 3,4,5-triphosphate (PIP3) by phosphorylating phosphatidylinositol 4,5-bisphosphate (PIP2). This process is antagonized by phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a tumor suppressor that converts PIP3 to PIP2[52]. PIP2 and PIP3 directly interact with the pleckstrin homology (pH) domain of AKT[53-55], resulting in its phosphorylation by PDK1 at Thr308[56-58]. Additionally, the phosphorylation of AKT on Ser473 by mTORC2 is also necessary for its activity[50,59]. TSC2 undergoes inactivation through Akt-dependent phosphorylation, which destabilizes TSC2 and disrupts its interaction with TSC1[60], and acts as a GTPase-activating protein (GAP) complex toward the GTPase RAS homolog enriched in the brain (Rheb)[61]. The mTORC1, a direct target of Rheb-GTP, activates the TOR kinase[62]. The mobilized mTORC1 then relays signaling by phosphorylating two key substrate protein molecules, p70S6K, and 4E-binding protein1 (4EBP1), resulting in activation of p70S6K at Thr229 and inactivation of 4EBP1[63]. Activated p70S6K subsequently phosphorylates eIF4B to initiate protein synthesis[64]. 4E-BP1 also releases inhibition on eIF4E to enhance protein synthesis [65,66]. Furthermore, mTORC1, similar to yeast TOR, phosphorylates mammalian ULK1, ATG13, and FIP200 complex[67,68], thereby inhibiting ULK1 and ULK2 kinase activity by phosphorylating ULK1 at Ser758. Then, the activated ULK1 phosphorylates Beclin-1 at Ser14, which is necessary to induce autophagy[49,67,69-72].
The mTORC1/2 signaling pathways. The receptor tyrosine kinases (RTKs)/PI3K/Akt signaling pathway, stimulated by growth factors, is pivotal to mTOR protein regulation. PI3K, typically maintained at a basal level, is activated to synthesize phosphatidylinositol 3,4,5-triphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate (PIP2). This process is counteracted by the tumor suppressor PTEN. PIP2 and PIP3 trigger AKT phosphorylation, leading to TSC2 inactivation and Rheb-GTP generation. Upon phosphorylation by mTORC1, ULK1/2 induces autophagy by phosphorylating Beclin-1.
HBV infection and mTOR
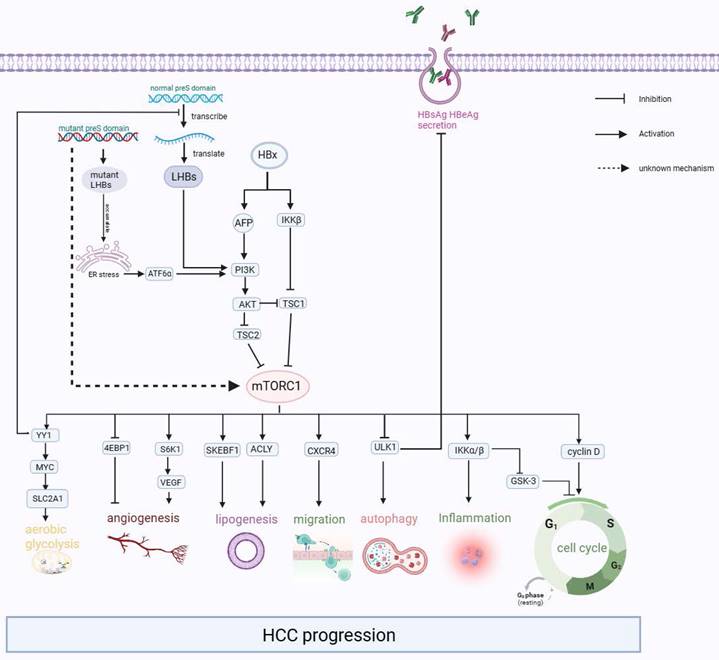
After the HBV particles enter liver cells through receptor sodium taurocholate cotransporting polypeptide (NTCP) and heparan sulfate proteoglycan (HSPG)[73,74], they undergo uncoating in the nucleus, and relaxed circular DNA (rcDNA) is released from the nucleocapsid into the liver nucleus. The rcDNA is converted into the template cccDNA[75,76], which is transcribed and then translated to L-HBsAg, M-HBsAg, S-HBsAg, HBx proteins, HBeAg, and core proteins[77]. In some cases, these viral proteins can affect the normal physiological activities of liver cells, resulting in ER stress, activating mTOR, and ultimately leading to the progression of liver-related diseases such as cirrhosis and liver cancer (Figure 3).
The Interaction between mTOR pathway and HCC. HBx activates the PI3K/AKT/mTOR signaling pathway by increasing the expression of AFP and activating IKKβ, which encourages malignant transformation. Besides, the accumulation of mutant L-HBsAg and excessive L-HBsAg could also activate the mTOR signaling pathway. Then the activated mTOR promotes carcinogenic-related life activities, such as aerobic glycolysis, angiogenesis, lipogenesis, migration, inflammation, and cell cycle.
HBx protein activate mTOR
The HBV genome consists of four overlapping open reading frames (ORFs). One of these, ORFs X, encodes the small non-structural regulatory HBV X protein (HBx). HBx is composed of 154 amino acids and has a molecular mass of about 17.5 kDa[78]. The multifunctional HBx protein interacts with several host factors to affect cellular signal transduction pathways, transcriptional regulation, cell cycle progression, DNA repair, apoptosis, and genetic stability[79].
Early research found that in vitro cell experiments conducted on Chinese Hamster Lung cells showed that, compared to the control group, cells transfected with HBx exhibited significantly higher PI3K and Akt activities[80]. However, this study did not use the liver cell line. Subsequently, experiments on human liver cancer cell lines and HBx-transgenic mice have demonstrated that overexpression of HBx increases the level of phospho-S6K1, which is downstream of mTOR[81].
Further studies suggested that the expression of p-mTOR and its upstream p-AKT was significantly upregulated in liver-derived cells after transfection with HBx[82-84]. This upregulation leads to increased cell proliferation, which is also linked to inflammation and tumor angiogenesis. The occurrence of this result is based on the following clarified mechanisms: (1) HBx inhibits TSC1 and then activates mTOR through the IκB kinase (IKK) complex subunit β (IKK-β), ultimately promoting cell proliferation and new vessel formation by enhancing vascular endothelial growth factor A (VEGF-A), which promotes malignant transformation of liver cells[81,85]. (2) HBx prevented hepatocyte apoptosis and accelerated the cell cycle from the G1 phase to the S phase by increasing the expression of cyclinD1 through the Akt/mTOR signaling pathway[82,83,86-88]. (3) HBX induced Alpha-fetoprotein (AFP) expression to activate the PI3K/AKT/mTOR signaling pathway by binding PTEN with AFP, and then p-mTOR (Ser2448) enhanced HIF-1α binding to the promoters of Src, C-X-C chemokine receptor 4 (CXCR4), and Ras genes, which are oncogenes[89]. Or p-mTOR (Ser2448) promotes CXCR4 expression through binding to CXCR4 gene promoter elements directly[90]. Finally, overexpression of these oncogenes promotes invasion and metastasis in hepatocytes[91-95]. To summarize, it can be observed that mTOR signaling plays an important role as a molecular regulatory factor in connecting metabolic disorders and cancer in chronic HBV infection (Figure 3).
HBsAg activates mTOR
According to the research, accumulating wild-type and mutant HBsAg can cause ER stress and turn on the Akt/mTOR signaling pathway to induce cell transformation and inflammation[96-99]. Particularly, the overexpression of large hepatitis B surface antigen (L-HBsAg) may participate in HBV-related hepatocarcinogenesis by activating the PI3K/Akt/mTOR pathway[100]. Besides, overexpression of small hepatitis B surface antigen (S-HBsAg) cannot change the phosphorylation levels of mTOR[101]. Additionally, numerous studies have shown that mutations in the pre-S region are associated with the formation of liver cancer through the mediating mTOR signaling pathway, which is situated within the coding region of L-HBsAg [97,102-110].
Firstly, pre-S1/2 deletion mutants (pre-S1: nt 3040-3111, pre-S2 mutant: nt 4-57) induced the enhanced expression of p-Akt and p-mTOR in HuH-7 cells[97]. The pre-S2 deletion mutant-induced mTOR activation signal cascade can not only promote lipogenesis by activating key regulators of lipid metabolism, such as sterol regulatory element binding transcription factor 1 (SREBF1) and ATP citrate lyase (ACLY), but also stimulate cell proliferation, both of which may lead to the occurrence of HCC[108]. Besides, pre-S2 deletion mutants may activate mTOR/Yin Yang 1(YY1) /myelocytomatosis oncogene (MYC) signaling to upregulate SLC2A1, which would sustain high activation rates of aerobic glycolysis and lead to tumorigenesis[109,110]. Interestingly, Teng et al. indicated that some pre-S1 deletions and site mutants can activate mTOR in HuH-7 cells. In turn, the up-regulated mTOR inhibited L-HBsAg synthesis at the transcriptional stage through the transcription factor YY1, which binds to the preS1 promoter (nt 2812-2816)[102].
mTOR affects the transcription and replication of HBV
mTOR inhibits HBV transcription and replication. Related studies have shown that constitutively active Akt1 significantly suppressed HBV RNA transcription, which in turn decreased HBV DNA replication. Given that the mTOR inhibitor rapamycin reversed this decrease in HBV gene transcription, it appears that mTOR activation was the cause[111]. Besides, treatment with mTOR inhibitors (rapamycin) on HepG2.2.15 cells increased the transcription of 3.5-kb and 2.4-kb viral RNA, the replication of HBV DNA within the cell, and the secretion of HBeAg and HBsAg [111-113]. In addition, when using small interfering RNA (siRNA) specific to Akt and mTOR, similar to the use of chemical inhibitors, HBV replication and secretion of HBsAg and HBeAg were also significantly increased[112,113]. Another study found that these inhibitors enhance HBV replication and transcription in an HBx-dependent manner[114]. Furthermore, in HBV-infected dHepaRG cells, prolonged treatment with PI3K-AKT inhibitors also increased the extracellular HBV DNA[114].
The specific physiological mechanism impacting the replication of HBV following the activation of the Akt/mTOR signaling pathway is based on autophagy[112,113,115]. Studies have shown that inhibiting the mTOR/ULK1 signaling pathway promotes autophagy, thereby upregulating HBV replication[113,116]. Conversely, knocking down the downstream effector ULK1 results in a significant decrease in HBV replication as well as HBsAg and HBeAg secretion in HepG2.2.15 cells. ULK1, as a classic autophagy mediator, is essential for the initiation of autophagosome formation[117,118]. and silencing ULK1 significantly reduces the frequency of autophagic puncta[113]. The formation of autophagic cells is crucial for the effective replication of HBV in various cells and animal models during HBV infection[101,115,116,119-123]. If autophagy is suppressed, there is only a slight reduction in HBV RNA levels and pgRNA packaging. However, it significantly inhibits HBV DNA replication. This indicates autophagy primarily enhances HBV replication during the process of viral DNA replication[115]. In turn, HBV can induce the formation of autophagic lysosomes in liver cells, as supported by in vitro cell experiments, animal experiments using HBV transgenic mice, and clinical pathology. It is worth noting that HBV enhances autophagy flux without increasing the degradation rate of autophagic proteins, suggesting a positive role of autophagy in HBV DNA replication[115].
Non-viral HCC and mTOR
The mTOR signaling pathway is frequently dysregulated in cancer and metabolic diseases[124]. Constitutive activation of the mTOR pathway may cause diet-independent HCC[125]. A comprehensive microarray study on a large number of human hepatocellular carcinoma patients shows that activation of Akt1 is one of the most consistent characteristics of HBV-induced HCC[126]. Inhibition of the PI3K/Akt/mTOR pathway can induce apoptosis and autophagy in hepatocellular carcinoma cells[127]. The previous mentioned evidence has shown that HBV infection can regulate the activity of the mTOR signaling pathway through different mechanisms, thus affecting the occurrence and development of HCC. However, some non-HBV infection factors can also lead to the activation of mTOR, which in turn leads to the occurrence of HCC (Figure 3).
Due to the liver being an organ for fat metabolism, the impact of mTOR on the development of HCC through lipid metabolism cannot be ignored in the special microenvironment of hepatocytes. The process of aliphatic acid production by hepatocytes is mediated by insulin through the PI3K/AKT/mTOR signaling pathway. Imbalance in mTOR pathway can lead to the production of lipid synthesis intermediates, which can ultimately result in steatosis and cancer[128,129]. The enhancement of lipid synthesis leads to the pathological accumulation of fatty acids(FA), thereby promoting inflammation and contributing to tumor progression[130]. It is found that mTORC2 can regulate the expression of certain key lipid synthesis genes at the transcriptional level[128]. However, on the contrary, another study suggested that the imbalance in the mTOR pathway is associated with cancer, rather than the development of liver steatosis[125]. In addition to lipid synthesis, mTOR can also regulate lipid lipolysis and the mobilization of lipid storage in liver cells by regulating autophagy and lysosomes[131-133].
mTOR inhibitor treatment in HCC
Due to the abnormal activation of the mTOR pathway in 40% to 50% of HCC patients[134-136], there is growing interest in developing HCC treatment strategies that target mTOR. First generation mTOR inhibitors are Rapamycin (also known as Sirolimus) and its derivatives, targeting mTOR and FKBP12 to directly inhibit mTORC1. Derivatives include medications such as Temsirolimus (CCI-779), Deforolimus (AP23573), and Everolimus (RAD001)[137]. Some of them have been approved by the FDA for the treatment of advanced renal cell carcinoma or neuroendocrine tumors. For example, Sirolimus has been approved by the FDA for reducing organ rejection in patients undergoing renal transplantation in 1999[138-140], and temsirolimus has been approved by the FDA for patients with advanced renal cell carcinoma (RCC) in 2007[141]. Besides, Everolimus was approved by the FDA for the treatment of adult patients with specific pulmonary and gastrointestinal neuroendocrine tumors in 2016[142,143]. Although the second and third generations of mTOR inhibitors have been developed, the majority of current clinical trials related to HCC still choose the first generation for research. Recent years, mTOR inhibitors have shown effectiveness in inhibiting the growth of HCC cells in both cell and animal experiments[144-146]. However, clinical trials conducted to test the efficacy of Rapalogs in treating HCC showed mixed results (Table 1).
Summary of completed clinical trials with mTOR inhibitors in HCC
| Drug | Trail Phase | Number of enrolled patients | HCC Stage | Child-Pugh Score | Study design | Countries And Regions | Result | ID | |
|---|---|---|---|---|---|---|---|---|---|
| Adjuvant therapy after liver transplantation | Sirolimus vs mTOR inhibitor free | III | 510 | Milan criteria and extended | NA | Randomized | global multicenter | Completed[157,158,174]. Sirolimus prolonged OS and reduced the risk of death. | NCT00355862 |
| III | 397 | Exceeding the Milan criteria | NA | Non-randomized | China | Not yet completed[175]. | ChiCTR2100042869 | ||
| Sirolimus vs Tacrolimus | II | 45 | Exceeding the Milan criteria | NA | Randomized | Korea | Completed [159]. While sirolimus prolongs OS, it does not reduce HCC recurrence. | NCT01374750 | |
| III | 220 | Exceeding the Milan criteria | NA | Randomized | China | Recruiting. | NCT00554125 | ||
| Adjuvant therapy after TACE | TACE +/- Everolimus | I/II | 27 | Intermediate stage B | A, B (<8) | Randomized | Switzerland multicenter | The study was terminated due to low enrollment. | NCT01009801 |
| II | 65 | Intermediate stage B | A, early B | Randomized | Asia, multicenter | This study was terminated due to low enrollment. | NCT01379521 | ||
| Single agent for advanced HCC | Sirolimus | pilot | 21 | I to IV (TNM) | A/B/C | Non-randomized | Sweden | Completed[176]. A temporary disease-control rate (PR + SD) was observed. | NA |
| II | 25 | B,C (BCLC) | A/B | Non-randomized | France | Completed [177]. Sirolimus shows antitumoural efficacy. | Sirolimus | ||
| pilot | 18 | B,C,D (BCLC) | A/B/C | Non-randomized | Austria | Completed [150]. Sirolimus shows minimal effectiveness in patients with liver cirrhosis and advanced HCC. | NA | ||
| Temsirolimus | II | 45 | advanced | A | Non-randomized | Hong Kong, China | Completed [151]. The targeted PFS endpoint was not reached. | NCT00321594 | |
| II | 25 | advanced | A/B | NA | America | Completed [152]. Temsirolimus showed higher responses than previous report. | NCT01567930 | ||
| Everolimus | I/II | 28 | B,C (BCLC) | A/B | Non-randomized | America | Completed[178]. Everolimus was observed initial anticancer activity. | NA | |
| I/II | 39 | advanced | A, B(≤9) | Randomized | Taiwan, China | Completed[179]. This study recommends that future HCC studies on the dosage of everolimus should be conducted at a dose of 7.5mg/day. | NCT00390195 | ||
| III | 546 | advanced | A | Randomized | global multicenter | Completed [149]. The OS of patients with HCC was not improved by everolimus. | NCT01035229 | ||
| Adjuvant combination therapy for advanced HCC | Sorafenib and Temsirolimus | I | 25 | III, IV | A, B (≤7) | Non-randomized | America multicenter | Completed[180]. The maximum-tolerated dose (MTD) was sorafenib 200 mg twice daily plus temsirolimus 10mg/week. | NA |
| I | 25 | III, IV (AJCC) | A, B (≤7) | Non-randomized | America multicenter | Not yet published. | NCT01008917 | ||
| II | 29 | II,III,IV(AJCC) | A, B (≤7) | Non-randomized | America multicenter | Not yet published. | NCT01687673 | ||
| Sorafenib and Everolimus | II | 30 | advanced | A | Randomized | America | Completed [181]. The MTD was sorafenib 400 mg twice daily plus everolimus 2.5mg/day. | NA | |
| II | 106 | B,C (BCLC) | A, B (≤7) | Randomized | global multicenter | Completed[148]. Sorafenib and Everolimus combination failed to improve the efficiency compared to Sorafenib solely. | NCT01005199 | ||
| Temsirolimus + Bevacizumab | II | 28 | advanced | A | NA | Canada | Completed [155]. The overall response rate and median OS have both improved under Temsirolimus and Bevacizumab combination therapy. | NCT01010126 | |
| Everolimus+ Bevacizumab | II | 36 | B,C (BCLC) | A/B | randomized | Germany multicenter | Not yet published. | NCT00775073 | |
| Sirolimus + Bevacizumab | II | 27 | advanced | A/B | Non-randomized | Singapore | Completed[156]. The recommended dose was bevacizumab 5mg/kg /14 days and rapamycin 4 mg/day. | NCT00467194 | |
| Everolimus and Pasireotide | II | 24 | C (BCLC) | A(≤6) | randomized | America multicenter | Not yet published. | NCT01488487 |
For patients with advanced-stage HCC, the first-line treatment drug is the multi-kinase inhibitor Sorafenib[147]. However, when combined with Everolimus, there is no improved efficacy compared to Sorafenib alone[148]. Additionally, using single-agent Sirolimus was found to be modestly beneficial for patients. While Everolimus combined with the best supportive care did not improve survival compared to the placebo group[149,150]. Except for Everolimus and Sirolimus, Termosimox also fails to achieve the targeted progression free survival (PFS) endpoint of HCC[151]. However, intravenous injection of Termosimox showed a higher response in HCC compared to previously reported systemic Prescription[152]. Despite compelling preclinical evidence and scientific justification[20,22,23,144,146,153,154], mTOR inhibitors demonstrated only modest efficacy in advanced HCC patients.
Owing to the limited effectiveness of Rapalogs as a standalone therapy for HCC, numerous clinical trials have been conducted or are currently underway to evaluate their therapeutic efficacy in combination therapies. Temsirolimus in combination with Bevacizumab increased the objective response rate (ORR) and overall survival (OS) in this population with advanced HCC[155], which is promising but calls for more research at a more optimal dose and timing. The therapeutic efficacy of Sirolimus/ Bevacizumab doublet for advanced HCC also showed evidence of anti-vascular activity[156]. The differences in the effectiveness of mTOR inhibitors in single and combination therapies may be due to the complexity of the mTOR pathway and the heterogeneity of liver tumors.
mTOR inhibitors are not only effective in treating advanced HCC but also demonstrate impressive efficacy following liver transplantation for HCC. In the clinical trials of Sirolimus as adjuvant therapy after liver transplantation for HCC, it significantly prolonged OS and reduced the risk of death. Significantly, the benefits of Sirolimus in the initial 3-5 years of recurrence free survival (RFS) and OS are more pronounced in low-risk patients compared to high-risk groups[157]. Moreover, subgroups with AFP≥10ng/ml benefited mostly from sirolimus treatment, which improved OS, disease free survival (DFS) and HCC recurrence[158,159]. Therefore, focusing on the subgroups of HCC patients in clinical trials of mTOR inhibitors may be more promising.
The above clinical experiments have shown that the success of raptalogs in the treatment of HCC is not significant, and the limited therapeutic effect may be due to the following reasons: (1) mTORC1 inhibition leads to AKT activation through a negative feedback loop stemming from S6K1 or upregulating the insulin-like growth factor-1 receptor (IGF-R1)[160,161]. Therefore, it may actually reduce the anticancer activity of rapalogs[162]. (2) Raptalogs mainly inhibits its substrate S6K, which is related to cell proliferation. But another key substrate, 4E-BP1, has not been fully blocked[163]. (3) Patients exhibit rapamycin resistance mutations in the FRB domain of mTOR after or before treatment [164,165]. Furthermore, elevated amounts of the antiapoptotic proteins Bcl-2, survivin, or Bcl-XL have resulted in rapalog resistance[166-169]. Therefore, genetic testing can be performed before medication to prevent rapalog resistance from delaying treatment.
In addition, mTORC1 can also be inhibited by acyclic nucleoside phosphonates (ANPs) in vitro, which are a kind of nucleoside analogue for clinical use of HBV infection. Studies have shown that ANPs reduce IL-10 production by inhibiting mTOR, and downregulation of IL-10 may promote HBV clearance in vivo by restoring the function of T cells and NK cells [170-172]. Can nucleoside analogues contribute to inhibit HCC development? However, in another article, activation of mTOR was found in the liver tissue of patients taking the same type of ANP (ADV or TDF)[173]. Considering the presence of two complexes of mTOR, further research is needed on the specific effects of nucleoside analogues on mTOR and HCC progression.
Summary and Outlook
This review has illustrated the pivotal role of mTOR in the nexus of nutrition, growth, aging, and disease, particularly in the replication of HBV particles at the level of DNA replication, RNA transcription, and antigen secretion. Simultaneously, activated mTOR, serving as an important carcinogenic pathway, also exhibits intricate interactions in HBV-related and non-viral HCC. Some studies suggest that the pathogenesis of HBV is attributed to the high-level replication of the virus, as the continuous accumulation of transcription templates will lead to the damage of infected cells. This elevated level of replication will lead to the consumption of cellular resources, such as synthetic cell membranes, lipoproteins, phospholipids, etc[182]. Additionally, mTOR is the central regulatory factor for nutrition and growth. Therefore, in the pathogenesis of HBV, mTOR is likely to have special regulation. The activation of mTOR signaling pathway during HBV-related tumorigenesis negatively regulates HBV replication and surface antigen synthesis. Therefore, the decrease of HBsAg and HBV DNA levels in serum or liver cells may not necessarily represent favorable disease improvement during the natural course of HCC, but on the contrary, it may suggest that the disease is developing toward a tumor, especially in the late stage of the disease. Considering the important role of the mTOR signaling pathway in various types of cancer[183-185], currently, many rapamycin analogs, such as Everolimus and Temsirolimus, have been approved by the Food and Drug Administration[186], Previous studies reported the importance of completely inhibiting mTORC1 effectors (RPS6 and eIF4E) in hepatocarcinogenesis[187]. In addition to treating HCC, mTOR inhibitors have also been found to be effective in reducing HCC recurrence after liver transplantation[188]. Moreover, the study of mTOR and its impact on the life cycle of HBV is valuable for the development of safe and effective therapies against viral effects. Although there have been many studies on mTOR signaling pathway in carcinogenesis, the unique characteristics of liver cells and the particularity of the HBV life cycle make the exploration of mTOR in HBV-related HCC both meaningful and worthy of further investigation.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (No. 82202612) and the Natural Science Foundation of Beijing Municipal (No.7232142).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liang TJ, Block TM, McMahon BJ. et al. Present and future therapies of hepatitis B: From discovery to cure. Hepatology. 2015;62:1893-908
2. Tang LSY, Covert E, Wilson E, Kottilil S. Chronic Hepatitis B Infection: A Review. JAMA. 2018;319:1802-13
3. McMahon BJ. Epidemiology and natural history of hepatitis B. Semin Liver Dis. 2005;25(Suppl 1):3-8
4. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245-55
5. Subic M, Zoulim F. How to improve access to therapy in hepatitis B patients. Liver Int. 2018;38(Suppl 1):115-21
6. Fanning GC, Zoulim F, Hou J, Bertoletti A. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat Rev Drug Discov. 2019;18:827-44
7. Locarnini S. Molecular virology and the development of resistant mutants: implications for therapy. Semin Liver Dis. 2005;25(Suppl 1):9-19
8. Chisari FV, Ferrari C. Hepatitis B virus immunopathology. Springer Semin Immunopathol. 1995;17:261-81
9. Shimizu Y. T cell immunopathogenesis and immunotherapeutic strategies for chronic hepatitis B virus infection. World J Gastroenterol. 2012;18:2443-51
10. Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825-9
11. Wieland SF, Spangenberg HC, Thimme R, Purcell RH, Chisari FV. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc Natl Acad Sci U S A. 2004;101:2129-34
12. Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A. 2004;101:6669-74
13. Bossler F, Hoppe-Seyler K, Hoppe-Seyler F. PI3K/AKT/mTOR Signaling Regulates the Virus/Host Cell Crosstalk in HPV-Positive Cervical Cancer Cells. Int J Mol Sci. 2019;20:2188
14. Ranadheera C, Coombs KM, Kobasa D. Comprehending a Killer: The Akt/mTOR Signaling Pathways Are Temporally High-Jacked by the Highly Pathogenic 1918 Influenza Virus. EBioMedicine. 2018;32:142-63
15. Wang X, Wei Z, Jiang Y, Meng Z, Lu M. mTOR Signaling: The Interface Linking Cellular Metabolism and Hepatitis B Virus Replication. Virol Sin. 2021;36:1303-14
16. Anwanwan D, Singh SK, Singh S, Saikam V, Singh R. Challenges in liver cancer and possible treatment approaches. Biochim Biophys Acta Rev Cancer. 2020;1873:188314
17. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15:599-616
18. Abdelgalil AA, Alkahtani HM, Al-Jenoobi FI. Sorafenib. Profiles Drug Subst Excip Relat Methodol. 2019;44:239-66
19. Llovet JM, Ricci S, Mazzaferro V. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
20. Sahin F, Kannangai R, Adegbola O, Wang J, Su G, Torbenson M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin Cancer Res. 2004;10:8421-5
21. Schumacher G, Oidtmann M, Rueggeberg A. et al. Sirolimus inhibits growth of human hepatoma cells alone or combined with tacrolimus, while tacrolimus promotes cell growth. World J Gastroenterol. 2005;11:1420-5
22. Sieghart W, Fuereder T, Schmid K. et al. Mammalian target of rapamycin pathway activity in hepatocellular carcinomas of patients undergoing liver transplantation. Transplantation. 2007;83:425-32
23. Semela D, Piguet A-C, Kolev M. et al. Vascular remodeling and antitumoral effects of mTOR inhibition in a rat model of hepatocellular carcinoma. J Hepatol. 2007;46:840-8
24. Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905-9
25. Cafferkey R, Young PR, McLaughlin MM. et al. Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol Cell Biol. 1993;13:6012-23
26. Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73:585-96
27. Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;273:239-42
28. Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol. 2003;4:117-26
29. Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation by the rapamycin-binding domain. Nature. 2013;497:217-23
30. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471-84
31. Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol. 2013;203:563-74
32. Kim D-H, Sarbassov DD, Ali SM. et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163-75
33. Hara K, Maruki Y, Long X. et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177-89
34. Sabatini DM. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc Natl Acad Sci U S A. 2017;114:11818-25
35. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;169:361-71
36. Samidurai A, Kukreja RC, Das A. Emerging Role of mTOR Signaling-Related miRNAs in Cardiovascular Diseases. Oxid Med Cell Longev. 2018;2018:6141902
37. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183-203
38. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926-45
39. Kim YC, Guan K-L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25-32
40. Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem. 2003;278:12361-6
41. Hsu PP, Kang SA, Rameseder J. et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317-22
42. Jewell JL, Russell RC, Guan K-L. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14:133-9
43. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274-93
44. Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013;15:555-64
45. Inoki K, Li Y, Xu T, Guan K-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829-34
46. Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control mTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr Biol. 2022;32:733-4
47. Jacinto E, Loewith R, Schmidt A. et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122-8
48. Sarbassov DD, Ali SM, Kim D-H. et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296-302
49. Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15:155-62
50. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098-101
51. Oldham S, Hafen E. Insulin/IGF and target of rapamycin signaling: a TOR de force in growth control. Trends Cell Biol. 2003;13:79-85
52. Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903-10
53. Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665-8
54. Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, Hemmings BA. High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/protein kinase B and their influence on kinase activity. J Biol Chem. 1997;272:8474-81
55. Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17:338-44
56. Stephens L, Anderson K, Stokoe D. et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710-4
57. Alessi DR, James SR, Downes CP. et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261-9
58. Stokoe D, Stephens LR, Copeland T. et al. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567-70
59. Alessi DR, Andjelkovic M, Caudwell B. et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541-51
60. Inoki K, Li Y, Zhu T, Wu J, Guan K-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648-57
61. Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259-68
62. Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702-13
63. Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci U S A. 1998;95:1432-7
64. Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569-80
65. Hara K, Yonezawa K, Kozlowski MT. et al. Regulation of eIF-4E BP1 phosphorylation by mTOR. J Biol Chem. 1997;272:26457-63
66. Gingras AC, Gygi SP, Raught B. et al. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 1999;13:1422-37
67. Jung CH, Jun CB, Ro S-H. et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992-2003
68. Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X. ULK1·ATG13·FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy. J Biol Chem. 2009;284:12297-305
69. Neufeld TP. TOR-dependent control of autophagy: biting the hand that feeds. Curr Opin Cell Biol. 2010;22:157-68
70. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2-11
71. Jung CH, Seo M, Otto NM, Kim D-H. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7:1212-21
72. Russell RC, Tian Y, Yuan H. et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741-50
73. Yan H, Zhong G, Xu G. et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049
74. Sureau C, Salisse J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus a-determinant. Hepatology. 2013;57:985-94
75. Rabe B, Glebe D, Kann M. Lipid-mediated introduction of hepatitis B virus capsids into nonsusceptible cells allows highly efficient replication and facilitates the study of early infection events. J Virol. 2006;80:5465-73
76. Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64:1972-84
77. Bruss V. Hepatitis B virus morphogenesis. World J Gastroenterol. 2007;13:65-73
78. Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004;78:12725-34
79. Motavaf M, Safari S, Saffari Jourshari M, Alavian SM. Hepatitis B virus-induced hepatocellular carcinoma: the role of the virus x protein. Acta Virol. 2013;57:389-96
80. Lee YI, Kang-Park S, Do SI, Lee YI. The hepatitis B virus-X protein activates a phosphatidylinositol 3-kinase-dependent survival signaling cascade. J Biol Chem. 2001;276:16969-77
81. Yen C-J, Lin Y-J, Yen C-S. et al. Hepatitis B virus X protein upregulates mTOR signaling through IKKβ to increase cell proliferation and VEGF production in hepatocellular carcinoma. PLoS One. 2012;7:e41931
82. Wang P, Guo Q-S, Wang Z-W, Qian H-X. HBx induces HepG-2 cells autophagy through PI3K/Akt-mTOR pathway. Mol Cell Biochem. 2013;372:161-8
83. Wang H-Y, Yang S-L, Liang H-F, Li C-H. HBx Protein Promotes Oval Cell Proliferation by Up-Regulation of Cyclin D1 via Activation of the MEK/ERK and PI3K/Akt Pathways. Int J Mol Sci. 2014;15:3507-18
84. Liu Y, Xu L, Lu B. et al. LncRNA H19/microRNA-675/PPARα axis regulates liver cell injury and energy metabolism remodelling induced by hepatitis B X protein via Akt/mTOR signalling. Mol Immunol. 2019;116:18-28
85. Lee D-F, Kuo H-P, Chen C-T. et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440-55
86. Kato JY, Sherr CJ. Inhibition of granulocyte differentiation by G1 cyclins D2 and D3 but not D1. Proc Natl Acad Sci U S A. 1993;90:11513-7
87. Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl). 2016;94:1313-26
88. Wang X, Huo B, Liu J, Huang X, Zhang S, Feng T. Hepatitis B virus X reduces hepatocyte apoptosis and promotes cell cycle progression through the Akt/mTOR pathway in vivo. Gene. 2019;691:87-95
89. Zhu M, Guo J, Li W. et al. Hepatitis B virus X protein induces expression of alpha-fetoprotein and activates PI3K/mTOR signaling pathway in liver cells. Oncotarget. 2015;6:12196-208
90. Zhu M, Guo J, Xia H. et al. Alpha-fetoprotein activates AKT/mTOR signaling to promote CXCR4 expression and migration of hepatoma cells. Oncoscience. 2015;2:59-70
91. Darash-Yahana M, Pikarsky E, Abramovitch R. et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004;18:1240-2
92. Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008;267:226-44
93. Zlotnik A. New insights on the role of CXCR4 in cancer metastasis. J Pathol. 2008;215:211-3
94. Xiang Z-L, Zeng Z-C, Tang Z-Y. et al. Chemokine receptor CXCR4 expression in hepatocellular carcinoma patients increases the risk of bone metastases and poor survival. BMC Cancer. 2009;9:176
95. Zeng H, Wei W, Xu X. Chemokine (C-X-C motif) receptor 4 RNA interference inhibits bone metastasis in breast cancer. Oncol Lett. 2014;8:77-81
96. Jing Z-T, Liu W, Wu S-X. et al. Hepatitis B Virus Surface Antigen Enhances the Sensitivity of Hepatocytes to Fas-Mediated Apoptosis via Suppression of AKT Phosphorylation. J Immunol. 2018;201:2303-14
97. Yang J-C, Teng C-F, Wu H-C. et al. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology. 2009;49:1962-71
98. Choi Y-M, Lee S-Y, Kim B-J. Naturally Occurring Hepatitis B Virus Mutations Leading to Endoplasmic Reticulum Stress and Their Contribution to the Progression of Hepatocellular Carcinoma. Int J Mol Sci. 2019;20:597
99. Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012;22:274-82
100. Liu H, Xu J, Zhou L. et al. Hepatitis B virus large surface antigen promotes liver carcinogenesis by activating the Src/PI3K/Akt pathway. Cancer Res. 2011;71:7547-57
101. Li J, Liu Y, Wang Z. et al. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol. 2011;85:6319-33
102. Teng C-F, Wu H-C, Tsai H-W, Shiah H-S, Huang W, Su I-J. Novel feedback inhibition of surface antigen synthesis by mammalian target of rapamycin (mTOR) signal and its implication for hepatitis B virus tumorigenesis and therapy. Hepatology. 2011;54:1199-207
103. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64:S84-101
104. Yang Y, Sun J-W, Zhao L-G, Bray F, Xiang Y-B. Quantitative evaluation of hepatitis B virus mutations and hepatocellular carcinoma risk: a meta-analysis of prospective studies. Chin J Cancer Res. 2015;27:497-508
105. Chen C-H, Changchien C-S, Lee C-M. et al. Combined mutations in pre-s/surface and core promoter/precore regions of hepatitis B virus increase the risk of hepatocellular carcinoma: a case-control study. J Infect Dis. 2008;198:1634-42
106. Huang H-P, Hsu H-Y, Chen C-L. et al. Pre-S2 deletions of hepatitis B virus and hepatocellular carcinoma in children. Pediatr Res. 2010;67:90-4
107. Yeung P, Wong DK-H, Lai C-L, Fung J, Seto W-K, Yuen M-F. Association of hepatitis B virus pre-S deletions with the development of hepatocellular carcinoma in chronic hepatitis B. J Infect Dis. 2011;203:646-54
108. Teng C-F, Wu H-C, Hsieh W-C, Tsai H-W, Su I-J. Activation of ATP citrate lyase by mTOR signal induces disturbed lipid metabolism in hepatitis B virus pre-S2 mutant tumorigenesis. J Virol. 2015;89:605-14
109. Teng C-F, Hsieh W-C, Wu H-C. et al. Hepatitis B Virus Pre-S2 Mutant Induces Aerobic Glycolysis through Mammalian Target of Rapamycin Signal Cascade. PLoS One. 2015;10:e0122373
110. Amann T, Maegdefrau U, Hartmann A. et al. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am J Pathol. 2009;174:1544-52
111. Guo H, Zhou T, Jiang D. et al. Regulation of Hepatitis B Virus Replication by the Phosphatidylinositol 3-Kinase-Akt Signal Transduction Pathway. J Virol. 2007;81:10072-80
112. Huang W, Zhao F, Huang Y. et al. Rapamycin Enhances HBV Production by Inducing Cellular Autophagy. Hepat Mon. 2014;14:e20719
113. Lin Y, Deng W, Pang J. et al. The microRNA-99 family modulates hepatitis B virus replication by promoting IGF-1R/PI3K/Akt/mTOR/ULK1 signaling-induced autophagy. Cell Microbiol. 2017 19
114. Xiang K, Wang B. Role of the PI3K-AKT-mTOR pathway in hepatitis B virus infection and replication. Mol Med Rep. 2018;17:4713-9
115. Sir D, Tian Y, Chen W, Ann DK, Yen T-SB, Ou J-HJ. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U S A. 2010;107:4383-8
116. Wang X, Lin Y, Kemper T. et al. AMPK and Akt/mTOR signalling pathways participate in glucose-mediated regulation of hepatitis B virus replication and cellular autophagy. Cell Microbiol. 2020;22:e13131
117. Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017;61:585-96
118. Ro S-H, Jung CH, Hahn WS. et al. Distinct functions of Ulk1 and Ulk2 in the regulation of lipid metabolism in adipocytes. Autophagy. 2013;9:2103-14
119. Lin Y, Wu C, Wang X. et al. Hepatitis B virus is degraded by autophagosome-lysosome fusion mediated by Rab7 and related components. Protein Cell. 2019;10:60-6
120. Tian Y, Sir D, Kuo C-F, Ann DK, Ou J-HJ. Autophagy required for hepatitis B virus replication in transgenic mice. J Virol. 2011;85:13453-6
121. Lin Y, Wu C, Wang X. et al. Synaptosomal-associated protein 29 is required for the autophagic degradation of hepatitis B virus. FASEB J. 2019;33:6023-34
122. Lin Y, Wu C, Wang X. et al. Glucosamine promotes hepatitis B virus replication through its dual effects in suppressing autophagic degradation and inhibiting MTORC1 signaling. Autophagy. 2020;16:548-61
123. Wang J, Chen J, Liu Y. et al. Hepatitis B Virus Induces Autophagy to Promote its Replication by the Axis of miR-192-3p-XIAP Through NF kappa B Signaling. Hepatology. 2019;69:974-92
124. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424-30
125. Menon S, Yecies JL, Zhang HH. et al. Chronic Activation of mTOR Complex 1 is Sufficient to Cause Hepatocellular Carcinoma. Sci Signal. 2012;5:ra24
126. Boyault S, Rickman DS, de Reyniès A. et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42-52
127. Yang J, Pi C, Wang G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed Pharmacother. 2018;103:699-707
128. Guri Y, Colombi M, Dazert E. et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell. 2017;32:807-823.e12
129. Lee G, Zheng Y, Cho S. et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell. 2017;171:1545-1558.e18
130. Park EJ, Lee JH, Yu G-Y. et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197-208
131. Lin C-W, Zhang H, Li M. et al. Pharmacological Promotion of Autophagy Alleviates Steatosis and Injury in Alcoholic and Non-alcoholic Fatty Liver Conditions in Mice. J Hepatol. 2013;58:993-9
132. Zhang H, Yan S, Khambu B. et al. Dynamic MTORC1-TFEB feedback signaling regulates hepatic autophagy, steatosis and liver injury in long-term nutrient oversupply. Autophagy. 2018;14:1779-95
133. Singh R, Kaushik S, Wang Y. et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131-5
134. Villanueva A, Chiang DY, Newell P. et al. Pivotal Role of mTOR Signaling in Hepatocellular Carcinoma. Gastroenterology. 2008;135:1972-198411
135. Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60:855-65
136. Wang C, Wang X, Su Z, Fei H, Liu X, Pan Q. The novel mTOR inhibitor Torin-2 induces autophagy and downregulates the expression of UHRF1 to suppress hepatocarcinoma cell growth. Oncol Rep. 2015;34:1708-16
137. Chen Y, Zhou X. Research progress of mTOR inhibitors. Eur J Med Chem. 2020;208:112820
138. Toniato de Rezende Freschi J, Cristelli MP, Viana LA. et al. A Head-to-head Comparison of De Novo Sirolimus or Everolimus Plus Reduced-dose Tacrolimus in Kidney Transplant Recipients: A Prospective and Randomized Trial. Transplantation. 2024;108:261-75
139. Tomita Y, Uehara S, Takiguchi S, Nakamura M. Effect of Mammalian Target of Rapamycin Inhibition on Activated Regulatory T-Cell Expansion in Kidney Transplantation. Transplant Proc. 2023;55:792-6
140. Nunes Ficher K, Dreige Y, Gessolo Lins PR. et al. Long-term Efficacy and Safety of Everolimus Versus Mycophenolate in Kidney Transplant Recipients Receiving Tacrolimus. Transplantation. 2022;106:381-90
141. Kwitkowski VE, Prowell TM, Ibrahim A. et al. FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist. 2010;15:428-35
142. Motzer RJ, Escudier B, Oudard S. et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449-56
143. Yao JC, Fazio N, Singh S. et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387:968-77
144. Huynh H, Chow KHP, Soo KC. et al. RAD001 (everolimus) inhibits tumour growth in xenograft models of human hepatocellular carcinoma. J Cell Mol Med. 2009;13:1371-80
145. Hui IC-F, Tung EK-K, Sze KM-F, Ching Y-P, Ng IO-L. Rapamycin and CCI-779 inhibit the mammalian target of rapamycin signalling in hepatocellular carcinoma. Liver Int. 2010;30:65-75
146. Buitrago-Molina LE, Pothiraju D, Lamlé J. et al. Rapamycin delays tumor development in murine livers by inhibiting proliferation of hepatocytes with DNA damage. Hepatology. 2009;50:500-9
147. Kudo M, Finn RS, Qin S. et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163-73
148. Koeberle D, Dufour J-F, Demeter G. et al. Sorafenib with or without everolimus in patients with advanced hepatocellular carcinoma (HCC): a randomized multicenter, multinational phase II trial (SAKK 77/08 and SASL 29). Ann Oncol. 2016;27:856-61
149. Zhu AX, Kudo M, Assenat E. et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. JAMA. 2014;312:57-67
150. Schöniger-Hekele M, Müller C. Pilot study: rapamycin in advanced hepatocellular carcinoma. Aliment Pharmacol Ther. 2010;32:763-8
151. Yeo W, Chan SL, Mo FKF. et al. Phase I/II study of temsirolimus for patients with unresectable Hepatocellular Carcinoma (HCC)- a correlative study to explore potential biomarkers for response. BMC Cancer. 2015;15:395
152. Sachdev JC, Javed AY, Weir AB. et al. A phase II study of temsirolimus in previously treated advanced hepatocellular carcinoma (HCC). JCO. 2014;32:4098-4098
153. Baba HA, Wohlschlaeger J, Cicinnati VR. et al. Phosphorylation of p70S6 kinase predicts overall survival in patients with clear margin-resected hepatocellular carcinoma. Liver Int. 2009;29:399-405
154. Fornari F, Milazzo M, Chieco P. et al. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2010;70:5184-93
155. Knox JJ, Qin R, Strosberg JR. et al. A phase II trial of bevacizumab plus temsirolimus in patients with advanced hepatocellular carcinoma. Invest New Drugs. 2015;33:241-6
156. Choo SP, Chowbay B, Ng QS. et al. A Phase 1 dose-finding and pharmacodynamic study of rapamycin in combination with bevacizumab in patients with unresectable hepatocellular carcinoma. Eur J Cancer. 2013;49:999-1008
157. Geissler EK, Schnitzbauer AA, Zülke C. et al. Sirolimus Use in Liver Transplant Recipients With Hepatocellular Carcinoma: A Randomized, Multicenter, Open-Label Phase 3 Trial. Transplantation. 2016;100:116-25
158. Schnitzbauer AA, Filmann N, Adam R. et al. mTOR Inhibition Is Most Beneficial After Liver Transplantation for Hepatocellular Carcinoma in Patients With Active Tumors. Ann Surg. 2020;272:855-62
159. Lee K-W, Kim SH, Yoon KC. et al. Sirolimus Prolongs Survival after Living Donor Liver Transplantation for Hepatocellular Carcinoma Beyond Milan Criteria: A Prospective, Randomised, Open-Label, Multicentre Phase 2 Trial. J Clin Med. 2020;9:3264
160. Carracedo A, Ma L, Teruya-Feldstein J. et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065-74
161. Tamburini J, Chapuis N, Bardet V. et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood. 2008;111:379-82
162. Gupta M, Ansell SM, Novak AJ, Kumar S, Kaufmann SH, Witzig TE. Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood. 2009;114:2926-35
163. Choo AY, Yoon S-O, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414-9
164. Wagle N, Grabiner BC, Van Allen EM. et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med. 2014;371:1426-33
165. Rodrik-Outmezguine VS, Okaniwa M, Yao Z. et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534:272-6
166. Mahalingam D, Medina EC, Esquivel JA. et al. Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin Cancer Res. 2010;16:141-53
167. Marinov M, Ziogas A, Pardo OE. et al. AKT/mTOR pathway activation and BCL-2 family proteins modulate the sensitivity of human small cell lung cancer cells to RAD001. Clin Cancer Res. 2009;15:1277-87
168. Wendel H-G, Malina A, Zhao Z. et al. Determinants of sensitivity and resistance to rapamycin-chemotherapy drug combinations in vivo. Cancer Res. 2006;66:7639-46
169. Majumder PK, Febbo PG, Bikoff R. et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594-601
170. Murata K, Tsukuda S, Suizu F. et al. Immunomodulatory Mechanism of Acyclic Nucleoside Phosphates in Treatment of Hepatitis B Virus Infection. Hepatology. 2020;71:1533-45
171. Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MBA. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301-9
172. Peppa D, Micco L, Javaid A. et al. Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic hepatitis B virus infection. PLoS Pathog. 2010;6:e1001227
173. Wang Z, Kawaguchi K, Honda M. et al. Distinct notch signaling expression patterns between nucleoside and nucleotide analogues treatment for hepatitis B virus infection. Biochem Biophys Res Commun. 2018;501:682-7
174. Schnitzbauer AA, Zuelke C, Graeb C. et al. A prospective randomised, open-labeled, trial comparing sirolimus-containing versus mTOR-inhibitor-free immunosuppression in patients undergoing liver transplantation for hepatocellular carcinoma. BMC Cancer. 2010;10:190
175. Su R-Y, Ling S-B, Shan Q-N. et al. Efficacy and safety of sirolimus early conversion protocol in liver transplant patients with hepatocellular carcinoma: A single-arm, multicenter, prospective study. Hepatobiliary Pancreat Dis Int. 2022;21:106-12
176. Rizell M, Andersson M, Cahlin C, Hafström L, Olausson M, Lindnér P. Effects of the mTOR inhibitor sirolimus in patients with hepatocellular and cholangiocellular cancer. Int J Clin Oncol. 2008;13:66-70
177. Decaens T, Luciani A, Itti E. et al. Phase II study of sirolimus in treatment-naive patients with advanced hepatocellular carcinoma. Dig Liver Dis. 2012;44:610-6
178. Zhu AX, Abrams TA, Miksad R. et al. Phase 1/2 study of everolimus in advanced hepatocellular carcinoma. Cancer. 2011;117:5094-102
179. Shiah H-S, Chen C-Y, Dai C-Y. et al. Randomised clinical trial: comparison of two everolimus dosing schedules in patients with advanced hepatocellular carcinoma. Aliment Pharmacol Ther. 2013;37:62-73
180. Kelley RK, Nimeiri HS, Munster PN. et al. Temsirolimus combined with sorafenib in hepatocellular carcinoma: a phase I dose-finding trial with pharmacokinetic and biomarker correlates. Ann Oncol. 2013;24:1900-7
181. Finn RS, Poon RTP, Yau T. et al. Phase I study investigating everolimus combined with sorafenib in patients with advanced hepatocellular carcinoma. J Hepatol. 2013;59:1271-7
182. Zhang Y-Y, Hu K-Q. Rethinking the pathogenesis of hepatitis B virus (HBV) infection. J Med Virol. 2015;87:1989-99
183. Li J, Cheng D, Zhu M. et al. OTUB2 stabilizes U2AF2 to promote the Warburg effect and tumorigenesis via the AKT/mTOR signaling pathway in non-small cell lung cancer. Theranostics. 2019;9:179-95
184. Xiong D, Jin C, Ye X. et al. TRIM44 promotes human esophageal cancer progression via the AKT/mTOR pathway. Cancer Sci. 2018;109:3080-92
185. Zheng Y-L, Li L, Jia Y-X. et al. LINC01554-Mediated Glucose Metabolism Reprogramming Suppresses Tumorigenicity in Hepatocellular Carcinoma via Downregulating PKM2 Expression and Inhibiting Akt/mTOR Signaling Pathway. Theranostics. 2019;9:796-810
186. Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. The Journal of Clinical Investigation. 2011;121:1231
187. Wang C, Cigliano A, Jiang L. et al. 4EBP1/eIF4E and p70S6K/RPS6 axes play critical and distinct roles in hepatocarcinogenesis driven by AKT and N-Ras proto-oncogenes in mice. Hepatology. 2015;61:200-13
188. Duvoux C, Toso C. mTOR inhibitor therapy: Does it prevent HCC recurrence after liver transplantation? Transplant Rev (Orlando). 2015;29:168-74
Author contact
![]() Corresponding authors: Le Chang; Email: changle4652cn; Tel.: +86 10 85133609; Add.: No.1 Dahua Road, Dongcheng District, Beijing 100730, P.R. China. Lunan Wang; Email: lunan99com; Tel.: +86 10 85133609; Add.: No.1 Dahua Road, Dongcheng District, Beijing 100730, P.R. China.
Corresponding authors: Le Chang; Email: changle4652cn; Tel.: +86 10 85133609; Add.: No.1 Dahua Road, Dongcheng District, Beijing 100730, P.R. China. Lunan Wang; Email: lunan99com; Tel.: +86 10 85133609; Add.: No.1 Dahua Road, Dongcheng District, Beijing 100730, P.R. China.