Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(11):4532-4550. doi:10.7150/ijbs.98856 This issue Cite
Research Paper
The Essential Role of Angiogenesis in Adenosine 2A Receptor Deficiency-mediated Impairment of Wound Healing Involving c-Ski via the ERK/CREB Pathways
Yan Peng, Renping Xiong, Bo Wang, Xing Chen, Yalei Ning, Yan Zhao, Nan Yang, Jing Zhang, Changhong Li, Yuanguo Zhou, Ping Li ![]()
Department of Army Occupational Disease, State Key Laboratory of Trauma, Burn and Combined Injury, Daping Hospital, Army Medical University (Third Military Medical University), 10 Changjiang Zhilu, Chongqing 400042, People's Republic of China.
Received 2024-5-24; Accepted 2024-8-7; Published 2024-8-19
Abstract

Adenosine receptor-mediated signaling, especially adenosine A2A receptor (A2AR) signaling, has been implicated in wound healing. However, the role of endothelial cells (ECs) in A2AR-mediated wound healing and the mechanism underlying this effect are still unclear. Here, we showed that the expression of A2AR substantially increased after wounding and was especially prominent in granulation tissue. The delaying effects of A2AR knockout (KO) on wound healing are due mainly to the effect of A2AR on endothelial cells, as shown with A2AR-KO and EC-A2AR-KO mice. Moreover, the expression of c-Ski, which is especially prominent in CD31-positive cells in granulation tissue, increased after wounding and was decreased by both EC-A2AR KO and A2AR KO. In human microvascular ECs (HMECs), A2AR activation induced EC proliferation, migration, tubule formation and c-Ski expression, whereas c-Ski depletion by RNAi abolished these effects. Mechanistically, A2AR activation promotes the expression of c-Ski through an ERK/CREB-dependent pathway. Thus, A2AR-mediated angiogenesis plays a critical role in wound healing, and c-Ski is involved mainly in the regulation of angiogenesis by A2AR via the ERK/CREB pathway. These findings identify A2AR as a therapeutic target in wound repair and other angiogenesis-dependent tissue repair processes.
Keywords: Wound healing, A2AR, Angiogenesis, c-Ski, Endothelial cell
Introduction
The enhancement of wound healing has been a goal of medical practitioners for thousands of years. Therapeutic interventions that activate the innate repair mechanism of native tissue and promote healing are of particular interest. Adenosine, a potent endogenous physiological mediator, and its receptors play key roles in tissue repair and wound healing[1-4]. Moreover, caffeine, a major component of many beverages, such as coffee and tea, is an adenosine receptor antagonist and has inhibitory effects on tissue repair and wound healing[5-7]. Therefore, exploration of the mechanisms of adenosine and its receptors in wound repair is very important.
The adenosine A2A receptor (A2AR), a subtype of G protein-coupled adenosine receptor, is broadly expressed on most cell types involved in wound healing, including macrophages, fibroblasts and microvascular endothelial cells (ECs)[8]. We and others have demonstrated that the topical application of A2AR agonists promotes healing of normal wounds[9-11] and diabetic wounds[8, 12, 13], and one such agonist, sonedenoson, is currently being evaluated as a therapy for diabetic foot ulcers[8, 11]. In addition, polydeoxyribonucleotides have been used to improve wound healing in clinical studies through A2AR activation[14]. Although A2AR activation reportedly accelerates the wound healing process by affecting ECs and stimulating angiogenesis[15], relieving inflammation[15, 16], and affecting fibroblasts and epithelial cells[13, 17, 18], the roles of these regulatory effects in A2AR-mediated healing have not been fully elucidated. Specifically, the regulation of ECs in A2AR-mediated wound healing and the mechanism underlying this effect are still unclear.
The cellular Sloan-Kettering Institute (c-Ski) is an intracellular homolog of the virus oncogene v-ski and is involved in various physiological and pathological processes, such as the proliferation of hematopoietic cells, muscle regeneration, bone and nervous system development, synaptic projection, and tumorigenesis[19-23]. Further research has shown that c-Ski is involved in the repair of injured skeletal muscle, liver and other tissues[24, 25]. In recent years, we demonstrated that c-Ski is a wound repair factor that can accelerate wound healing in skin tissue and the brain[26, 27]. These effects are related to c-Ski's role as a multifunctional transcription regulator[28, 29] that is involved in cell proliferation, transformation, secretion of the extracellular matrix and the inflammatory response. In particular, c-Ski is expressed on ECs[30], and its overexpression promotes the proliferation of ECs[31, 32]. However, whether A2AR regulates wound healing through c-Ski's effects on ECs is unclear.
The extracellular signal-regulated kinase (ERK)/cAMP response element binding protein (CREB) pathway, an upstream signal, can transform extracellular stimulation into intracellular responses to promote the proliferation and differentiation of cells[33]. Moreover, the ERK/CREB pathway has a regulatory effect on angiogenesis[34-36] and is also the canonical pathway of A2AR regulation[37-39]. Additionally, it has been reported that the expression of c-Ski can be regulated by the ERK/CREB pathway[40]. Thus, we speculated that A2AR may regulate c-Ski expression through the ERK/CREB pathway.
In this study, we explored the effect and mechanism of A2AR in regulating skin wound healing and its relationship with c-Ski. First, a full-thickness excisional wound model in mice was used to observe the expression of A2AR and explore its role in angiogenic regulation during wound healing via genetic [A2AR knockout (A2AR KO) and EC-specific A2AR knockout (EC-A2AR KO) mice] blockade of A2AR. Furthermore, we examined the effects of A2AR on the expression of c-Ski to explore its role in A2AR-mediated angiogenesis and wound healing by using A2AR KO and EC-A2AR KO mice. Next, we analyzed the effects of A2AR on the regulation of angiogenesis in human microvascular ECs (HMECs) in vitro and the role of c-Ski in this process by RNAi. Finally, the ERK1/2 antagonist PD98059 was used to confirm that A2AR regulates the expression of c-Ski through the ERK/CREB pathway in vitro.
Materials and methods
Animals
A2AR knockout (KO) mice (A2AR-/-) and their wild-type (WT) controls were bred on a C57BL/6 background. The A2AR+/- breeding pairs were backcrossed to C57BL/6 mice for 10 generations, resulting in a congenic C57BL/6 genetic background. Heterozygous interbreedings (A2AR+/- × A2AR+/-) were used so that the global A2AR homozygous KO mice (A2AR-/-) and their WT littermates were generated from the same breeding pairs [41]. Mice with a 'floxed' adenosine A2AR gene (A2Aflox/flox mice) have been described previously[42] and were provided by Dr. Chen. TeK-Cre transgenic mice (Tek, a strain that targets ECs)[43, 44] were obtained from Shanghai Biomodel Organism Science & Technology Development Co., Ltd. (No. NMX-TG-192000, Shanghai, China). Global A2AR knockout mice, A2Aflox/flox mice and TeK-Cre transgenic mice were backcrossed for 10 to 12 generations to C57BL/6 mice. TeK-Cre transgenic mice were then crossed with A2Aflox/flox mice to generate TeK-A2AR KO mice and TeK-A2AR WT littermates, which were injected with 120 mg/kg tamoxifen (10540-29-1, Sigma Aldrich, St. Louis, MO, USA) in oil via intraperitoneal injection once every 48 hours 5 times in total, followed by a 14-day waiting period between the final injection and full-thickness wounding. The mice were used at 90 days of age. The animals were treated in accordance with the guidelines of the Animal Ethical and Welfare Committee of the Army Medical University and according to the protocol approved by the Administration of Affairs Concerning Experimental Animals Guidelines of The Army Medical University (No. AMUWEC20172073).
Reagents
We purchased MCDB 131 medium (10372019), fetal bovine serum (FBS) (10099141C), TrypLE™ Express (12605028), penicillin‒streptomycin (15140122), and L-glutamine (25030081) from Fisher Scientific (Thermo Fisher, MA, USA). Hydrocortisone (M3451) was obtained from Abmole (Abmole, TX, USA), and recombinant mouse EGF protein (ab126695) was obtained from Abcam (Abcam, Cambridge, UK). The A2AR agonist 2-p-[2-carboxyethyl]phenethyl-amino-5'-N-ethylcarboxamido-adenosine (CGS21680) (1063) and the MEK inhibitor PD98059 (1213) were purchased from Tocris (Bio-Techne, MN, USA). Hydrocortisone, CGS21680, and PD98059 were prepared as stock solutions in dimethyl sulfoxide (DMSO), aliquoted, and stored at -80°C.
Model and observation of wound healing
A mouse model of wound healing was generated as previously described with some modifications[26]. Briefly, to minimize discomfort and pain, mice were anesthetized with intraperitoneal injection of 50 mg/kg sodium pentobarbital, and two 1 cm diameter, full-thickness circular skin flaps were cut from the buttocks of the mice (at least six animals in each group). The wounds were covered with a piece of sterile gauze. The mice were then returned to their cages and maintained separately to avoid any further wound damage under a 12:12 h light/dark cycle, and supplied with unrestricted food and water.
Wound healing was observed as described previously[26, 45]. At 0, 3, 6, 9 and 15 d after wounding, wound closure was traced in each group using transparent film. And, percentage wound closure at each time point was derived by the formula: [1- (current wound area/original wound area)]×100%. The scar areas were calculated every 2 weeks using image processing software.
Histological and quantitative image analysis
After perfusion, the mice were anesthetized with intraperitoneal injection of 50 mg/kg sodium pentobarbital and perfused with ice-cold normal saline and then with 4% paraformaldehyde solution. Next, the entire wound and its associated normal skin was excised and then was fixed overnight in 4% paraformaldehyde solution overnight for further fixation at 3, 6 and 9 d postwounding. After routine dehydration and paraffin embedding, the tissue sections (4 mm) were stained with hematoxylin and eosin (H&E) for morphological assessment. Re-epithelialization and granulation tissue formation were assessed as previously described[26, 45].
Immunofluorescence was performed as previously described[46, 47]. Briefly, after dewaxing and antigen repair, primary antibodies were incubated with the samples overnight at 4°C in 0.01% Triton X-100 and 10% goat serum. The primary antibodies included goat anti-A2AR (1:200, Frontier Institute, AB_2571655), rabbit anti-CD31 (1:100, Servicebio, GB113151), rabbit anti-Ski (1:100, Santa Cruz, sc-9140), rabbit anti-CD11b (1:2000, Abcam, ab133357), rabbit anti-vimentin (1:400, Abway, CY5134), and rabbit anti-F4/80 (1:400, Cell Signaling, 70076). The tissue sections were washed with phosphate-buffered saline (PBS) and then treated with an Alexa Fluor 488-conjugated goat anti-rabbit antibody (1:500, Abcam, ab150077) and/or a Cy3-conjugated donkey anti-goat (1:500, Abcam, ab6949) antibody for 50 min at room temperature. The nuclei were stained with DAPI (Solarbio, C0060), and an autofluorescence quenching kit (Vector Laboratories, SP-8400) was used to remove unwanted fluorescence.
The results were captured by laser scanning confocal microscopy (TCS-SP8, Leica), and the quantitative analysis was performed independently by two observers who were blinded to the treatment conditions. The number of CD31-positive cells was counted and normalized to the total number of cells in each high-power field (HPF, 400×). The ratio of the integrated density (IntDen) to the total number of cells was determined with ImageJ software, and three fields were randomly chosen from each slice (three fields per section, three sections per mouse, five mice from each analyzed group).
RNA isolation and RT‒PCR
At 0, 6, 9, and 15 d postwounding, total RNA was extracted from the full-thickness skin of the mouse wounds using a TRIzol Reagent RNA Extraction Kit (Invitrogen, 15596026) according to the manufacturer's instructions and then reverse transcribed to cDNA using a kit (Promega, A2791). A qPCR master mix kit (Promega, A600A) was used for quantitative PCR. The methods used were previously reported[48]. The primers for c-Ski were as follows: 5′-TCAACTCGGTGTGCGATG-3′ and 5′-CGTCCGTCTTGGTGATGAG-3′. The primers for GAPDH were as follows: 5′-AGGTTGTCTCCTGCGACTT-3′ and 5′-TGGTCCAGGGTTTCTTACTCC-3′. The samples were denatured by heating at 95°C for 30 s, followed by 40 cycles of 95°C for 30 s, 58°C for 30 s and 72°C for 30 s, and the relative abundance of the target gene was calculated via normalization to GAPDH.
Western blotting
Western blotting was performed as previously described[46]. Briefly, at 0, 6, 9, and 15 d postwounding, the tissues were lysed in RIPA lysis buffer (Beyotime, P0013B) containing a protease and phosphatase inhibitor cocktail (Thermo Fisher, 78440). The total protein concentration was measured using a BCA kit (Aidlab, PP0102). The samples were resolved on 10% SDS‒PAGE and then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, IPVH00010). After the membranes were blocked with 5% bovine serum albumin (Solarbio, A8020) in Tris-buffered saline (pH 7.6) containing 0.1% Tween-20 (TBS-T) at room temperature for 1 hour, they were probed with the following primary antibodies overnight at 4°C: anti-Ski (1:100, Santa Cruz, sc-9140), anti-A2A adenosine receptor (1:1000, Abcam, ab3461), and anti-GAPDH (1:5000, Abcam, ab9485). After 3 washes with TBS-T incubation for 1 h at room temperature with HRP-conjugated goat anti-mouse IgG or goat anti-rabbit IgG secondary antibodies (1:8000, Abways, AB0035), the membranes were visualized using Clarity Western ECL Substrate (Beyotime, P0018). The band intensity was quantified by ImageJ software, and the relative quantity of the target protein was normalized to that of GAPDH or the nonphosphorylated protein for three independent experiments.
Cell culture and treatment
HMEC-1 cells were provided by Dr. Lan[49] and were characterized by Feiouer Biotechnology Co., Ltd. (Chengdu, China) using short tandem repeat (STR) markers (Fig. S9). The cells were maintained at 37°C at 5% CO2 in MCDB 131 medium supplemented with 10% FBS, 10 ng/mL recombinant mouse EGF protein, 1 μg/mL hydrocortisone, 1% penicillin‒streptomycin, and L-glutamine.
To determine the possible signaling mechanism, the cells were treated with 20 nM, 100 nM, or 500 nM of the A2AR agonist CGS21680, which was dissolved in DMSO, for 24 h or were pretreated with 100 µM of the MEK inhibitor PD98059 for 60 min before 100 nM CGS21680 treatment according to the protocol previously described. DMSO was used as the vehicle control.
Lentivirus infection
Short hairpin RNA (shRNA)-knockdown lentiviral particles for mouse c-Ski (sh c-Ski) and short hairpin negative control (sh Nc) lentiviruses were purchased from Shanghai Jikai Company (Shanghai, China). For c-Ski knockdown experiments, HMEC-1 cells were transfected with 5 nM, 10 nM, or 20 nM sh c-Ski for 24 h following the manufacturer's instructions before 100 nM CGS21680 treatment for 24 h.
Cell proliferation and scratch assays
A Cell Counting Kit (CCK)-8 assay (Beyotime, C0038) was used to assess cell proliferation[47]. Briefly, HMEC-1 cells were trypsinized using TrypLE™ Express and centrifuged at 1000g for 5 min at room temperature and then were seeded in 96-well plates with 100 μL of medium at a density of 8,000 cells/well. After various treatments, 10 µL of CCK-8 solution was added to each well and incubated for 2 h at 37°C and 5% CO2. The optical density (OD) values were measured at 450 nm using a microplate reader (Gene, ELX800ux). Wells without cells served as blank controls. Each experiment was performed in triplicate.
The scratch assay was performed as previously described[49]. Briefly, HMEC-1 cells were seeded in 6-well plates. The monolayer was scratched with a 200 μL pipette tip and rinsed with PBS after various treatments. Images were taken under an inverted phase contrast microscope (Zeiss, Primo Vert) with CCD cameras at 0, 24, 48, and 72 h after scratching. The experiment was repeated independently three times.
Tube formation
Matrigel (Corning, 356,234) was added to a precooled Millicell EZ SLIDE 8-well glass slide (200 μL/well) and polymerized for 40 min at 37°C. HMEC-1 cells were grown to 80% confluence and trypsinized using TrypLE™ Express. 1 × 104 cells in each treatment group were seeded on the Matrigel, incubated for 4 h in the corresponding medium at 37°C during tubule formation and then and washed with Hank's balanced salt solution (HBSS). For CGS21680 treatment, the cells were treated with calcein AM fluorescent dye (1:300, Corning, 354,216) for 4h. For CGS21680 and c-Ski RNAi treatment, the cells were treated with DiIC12(3) fluorescent dye (1:1000, Corning, 354,218). The results were captured by laser scanning confocal microscopy (TCS-SP8, Leica), and the number of junctions of the tubular-like structures was determined via Angiotool software [50]. Three independent experiments were performed.
In vitro immunofluorescence and western blotting
In vitro immunofluorescence and western blot assays were performed according to previously described methods. Briefly, for immunofluorescence analyses, HMEC-1 cells were fixed with 4% paraformaldehyde for 10 minutes and then incubated with the primary antibodies goat anti-A2AR (1:200, Frontier Institute, AB_2571655), rabbit anti-Ski (1:100, Santa Cruz, sc-9140), anti-p-PKA (1:500, Abcam, ab32390), anti-p-ERK1/2 (1:200, Cell Signaling, 9101), and anti-p-CREB (1:500, Cell Signaling, 9198) at 4°C overnight. The cells were then rinsed with phosphate-buffered saline (PBS) and treated with an Alexa Fluor 488-conjugated goat anti-rabbit antibody or a Cy3-conjugated donkey anti-goat antibody for 1 h at 37°C. The nuclei were stained with DAPI. The results were captured by laser scanning confocal microscopy (TCS-SP8, Leica), and the quantitative analysis was performed independently by two observers who were blinded to the treatment conditions. The ratio of the integrated density (IntDen) to the total number of cells was determined via ImageJ software, and three fields were randomly chosen from each well (five wells per group and 3 independent experiments).
For western blot assays, HMEC-1 cells were seeded into 25 cm2 vented cap flasks. After various treatments, the whole-cell lysates were collected and then separated on a 10% SDS‒polyacrylamide gel. After blocking with 5% BSA in Tris-buffered saline (pH 7.6) containing 0.1% Tween 20 (TBS-T) at room temperature for 1 hour, the PVDF membranes were probed with the following primary antibodies overnight at 4°C: anti-Ski (1:100, Santa Cruz, sc-9140), anti-A2A adenosine receptor (1:1000, Abcam, ab3461), anti-PKA (1:1000, Cell Signaling, 4782), anti-p-PKA (1:1000, Abcam, ab32390), anti-CREB (1:300, Cell Signaling, 9197), anti-p-CREB (1:1000, Cell Signaling, 9198), anti-ERK1/2 (1:1000, Cell Signaling, 9102), anti-p-ERK1/2 (1:1000, Cell Signaling, 9101), and anti-GAPDH (1:5000, Abcam, ab9485). After incubation with HRP-conjugated goat anti-mouse IgG or goat anti-rabbit IgG secondary antibodies (1:8000, Abways, AB0035), the membranes were washed with TBS-T 3 times and visualized using Clarity Western ECL Substrate (Beyotime, P0018). The band intensity was quantified by ImageJ software, and the relative quantity of the target protein was normalized to that of GAPDH or the nonphosphorylated protein for three independent experiments.
Statistical analysis
All procedures and analyses were performed by an experienced researcher who was blinded to all groups. All of the results are expressed as the means ± standard errors of the means. Statistical analyses were performed using GraphPad Prism 6 software (San Diego, CA, USA). Two-group comparisons were performed using Student's t test. One-way and two-way analysis of variance (ANOVA) was used for data with one variable and multiple conditions, followed by Tukey's multiple comparisons test. A value of P < 0.05 was considered statistically significant.
Results
A2AR expression in full-thickness excisional wounds
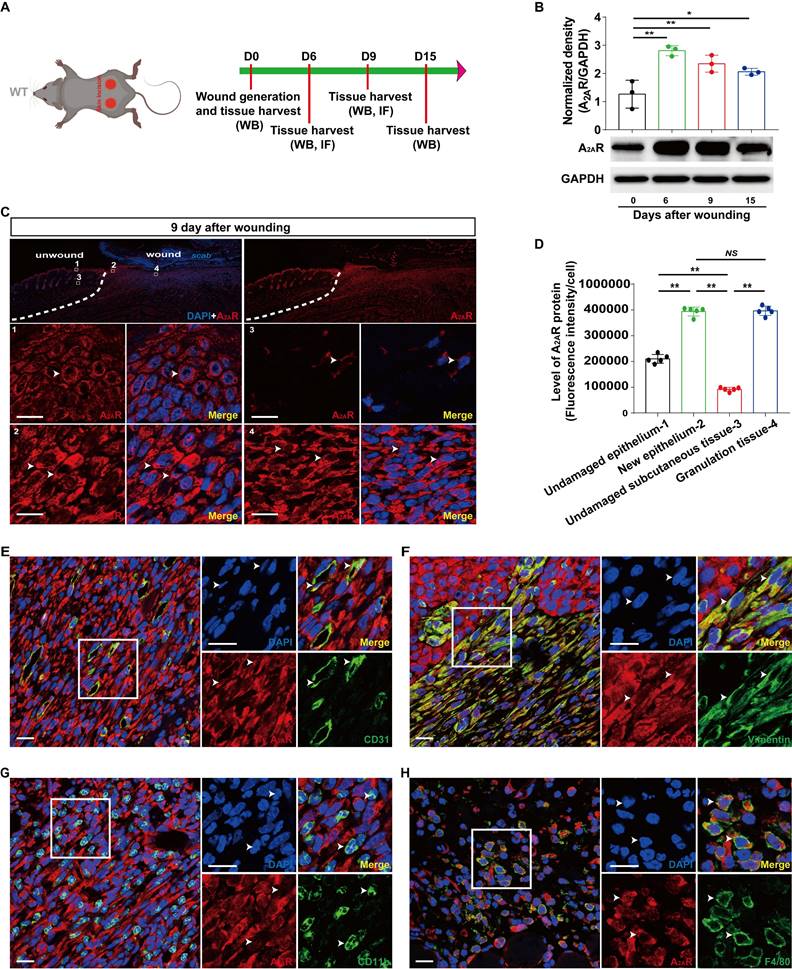
To study the role of A2AR in wound healing, we assessed the expression profile of A2AR in mouse dermal excisional wounds (Fig. 1A). Western blot analyses revealed that A2AR expression began to increase after the injury, peaked at 6 days, and was maintained at a high level during the late stages of wound healing (Fig. 1B). Next, immunofluorescence staining revealed that A2AR was widely distributed in the undamaged epidermis and subcutaneous tissue around the wound, resulting in a hole-like morphology (Fig. 1C), similar to our previous results[39, 42]. Moreover, A2AR expression increased in the epidermis and granulation tissue in the wound at 9 days postwounding and was especially prominent in granulation tissue (Fig. 1C-D). To characterize the expression of A2AR in granulation tissue further, we performed double immunofluorescence staining and revealed that A2AR was widely expressed in repair cells; was especially prominent in CD31+ ECs, vimentin+fibroblasts and F4/80+ macrophages; and was expressed at low levels in CD11b+ inflammatory cells (Fig. 3E-G). Thus, changes in the expression of A2AR during wound healing and its localization in repair cells suggest that this molecule is involved in the regulation of healing.
Localization and expression of A2AR in full-thickness excisional wounds of WT mice. (A) Experimental procedure. (B) A2AR levels were detected by western blotting at 0, 6, 9 and 15 days postwounding. **p < 0.01, *p <0.05 (n=3); NS, not significant. Immunohistochemistry for A2AR (red, white arrow) in the wound and surrounding area at 9 days postwounding (C) and its quantitative analysis (D) in WT mice. **p < 0.01 (n=5); NS, not significant. The lower panel shows higher magnification sections of the white squares in the upper panel. Scale bar, 50 µm. Immunohistochemistry for CD31 (E), vimentin (F), CD11b (G), F4/80 (H) (green) and A2AR (red) in wound granulation tissue at 9 days postwounding in WT mice. The right panel shows higher magnification sections of the white squares on the left, and the white arrow indicates that the green fluorescent-labeled cells colocalized with the red fluorescent-labeled cells. Short scale bar, 50 µm; long scale bar, 50 µm.
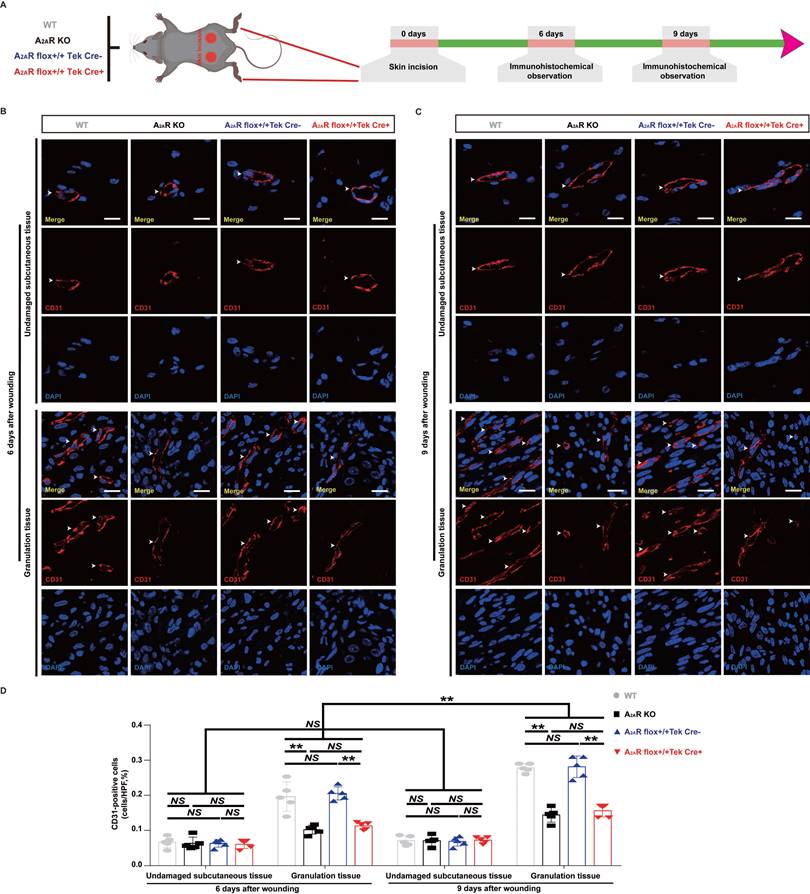
Changes in angiogenesis in A2AR KO and EC-A2AR KO mice after full-thickness wounding. (A) Experimental procedure. (B) Immunohistochemistry for CD31 in A2AR KO and EC-A2AR KO mice at 6 and 9 days postwounding. Scale bar, 50 µm. (C) Quantitative analysis of CD31-positive cells in each group. **p < 0.01 (n = 3); NS, not significant.
Both A2AR KO and EC-A2AR KO delay full-thickness excisional wound healing and inhibit granulation tissue formation
To directly confirm the role of A2AR in angiogenic regulation during wound healing through ECs, we used A2AR-KO mice and A2AR flox+/+ Tek-Cre+ mice (EC-A2AR KO and Tek-Cre+ mice were bred with A2ARflox/flox mice for a generation). Western blot analysis revealed that A2AR protein was not detected in the A2AR-KO mice (Fig. S1A), and its level was slightly lower in the EC-A2AR-KO mice compared with the A2AR flox+/+ Tek-Cre- control (EC-A2AR control) mice (Fig. S1B). Furthermore, immunofluorescence staining revealed that the A2AR protein level in CD31-positive cells was not significantly different between the wild-type (WT) and EC-A2AR control mice in undamaged subcutaneous tissue or in the granulation tissue of the wound at 9 days post-wounding (Fig. S1C and S2). In contrast, A2AR protein was not detected in any cells from the A2AR-KO mice and was found only in CD31-positive cells from the EC-A2AR-KO mice (Fig. S1A-C and S2). Collectively, these results suggest that the A2AR-KO and EC-A2AR-KO mice were successfully established.
Next, we assessed the effects of A2AR-KO and EC-A2AR KO on wound healing using full-thickness excisional wounds as described in a previous experiment[26, 45] (Fig. 2A). Comparison of the wound size revealed a clear delay in wound healing in the A2AR-KO and EC-A2AR-KO mice compared with the WT and EC-A2AR control mice, particularly at 9 days postwounding (Fig. 2B). Moreover, there were no significant differences in the rate of wound closure or healing time between the WT and EC-A2AR control mice, while both the A2AR-KO and EC-A2AR-KO mice presented a decreased rate of wound closure and delayed healing time (Fig. 2C-D). Furthermore, there was no significant difference in the rate of wound closure or healing time between the A2AR-KO and EC-A2AR-KO mice (Fig. 2C-D). Collectively, these results demonstrate that the delayed wound healing induced by A2AR KO is due mainly to the endothelial A2AR effect and suggest that A2AR-mediated angiogenesis plays a critical role in wound healing.
Wound healing and histopathological characteristics of A2AR KO and EC-A2AR KO mice after full-thickness wounding. (A) Experimental procedure. (B) Photographs of representative wounds of mice at 0, 3 and 9 days postwounding. (C) Wound healing time for each experimental and control group (n = 7 for each group), **p < 0.01. (D) Time course of wound closure for each experimental and control group (n = 9 for each group). **p <0.01, compared with the appropriate controls; ##p <0.01, compared with the previous adjacent time point. (E) Histopathological observation of wound healing at 6 and 9 days postwounding. Short scale bar, 200 µm; long scale bar, 50 µm. (F) Quantitative analysis of granulation tissue thickness at 6 and 9 days postwounding. **p < 0.01 (n = 5); NS, not significant. (G) Quantitative analysis of the crawling distance at 6 and 9 days postwounding; **p < 0.01 (n = 5); NS, not significant.
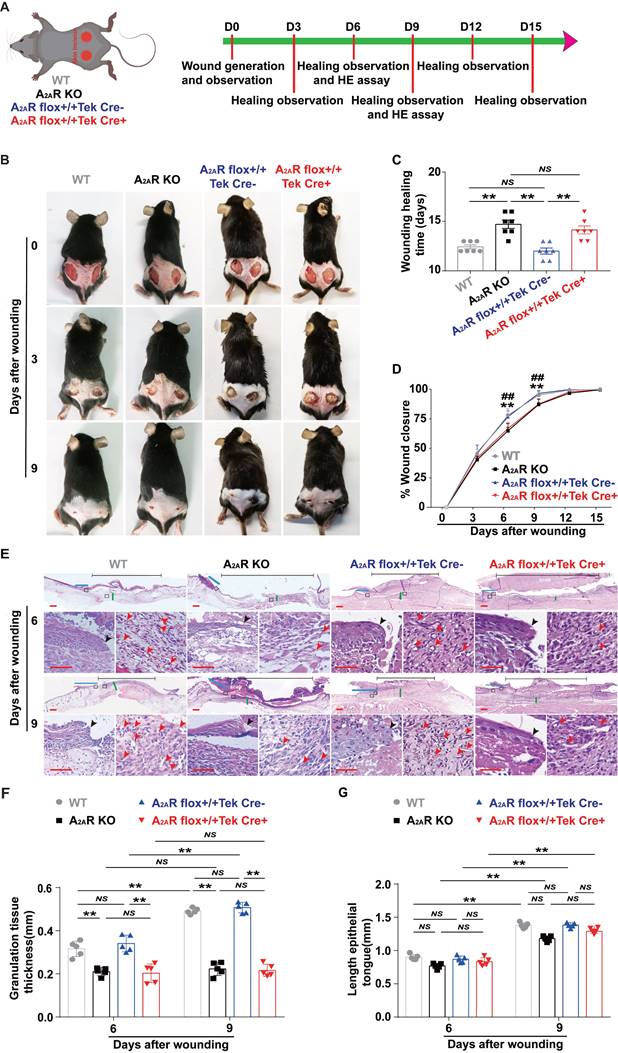
Additionally, H&E-stained sections revealed that granulation tissue grew significantly slower in both the A2AR-KO and EC-A2AR-KO mice than in the WT and EC-A2AR control mice at 6 and 9 days post wounding (Fig. 2E-F). Moreover, there was no significant difference in granulation tissue formation between the A2AR-KO and EC-A2AR-KO mice or the WT and EC-A2AR control mice (Fig. 2E-F). Conversely, there was no significant difference in re-epithelialization at 6 and 9 days postwounding in any group, including the WT, A2AR-KO, EC-A2AR control and EC-A2AR-KO mice (Fig. 2E and G). However, there was an obvious decrease in tube-like structures in both A2AR-KO and EC-A2AR-KO mice (Fig. 2E). Taken together, these results suggest that A2AR plays an important role in granulation tissue formation, but not epithelialization, and it is possible that A2AR affects granulation tissue formation mainly by regulating angiogenesis.
Both A2AR KO and EC-A2AR KO decrease angiogenesis in the granulation tissue of full-thickness excisional wounds and the expression of c-Ski
Previous studies have indicated that adenosine receptor-mediated stimulation of angiogenesis promotes wound closure in mice treated with the adenosine receptor agonist CGS21680 and A2AR-KO mice[15, 51]. We first used EC-A2AR KO mice to examine the vascularity of wounds in total KO, EC-specific A2AR KO and control mice (Fig.3A and S4A). We observed a nearly twofold decrease in CD31-positive cells in both the A2AR-KO and EC-A2AR-KO mice compared with the WT and EC-A2AR control mice at 6 and 9 days postwounding (Fig. 3B-D, S2, S3A-B, S4B), but the differences between the A2AR-KO and EC-A2AR-KO mice or the WT and EC-A2AR control mice were no longer significant (Fig. 3B-D, S2, S4B). Furthermore, the number of CD31-positive cells in the A2AR-KO or EC-A2AR-KO mice decreased to a certain extent between 6 and 9 days postwounding but did not significantly differ (Fig. 3B-D). Additionally, the number of CD31-positive cells in the undamaged subcutaneous tissue of these four groups was not significantly different (Fig. 3B-D). These results suggest that A2AR affects granulation tissue formation mainly by regulating angiogenesis.
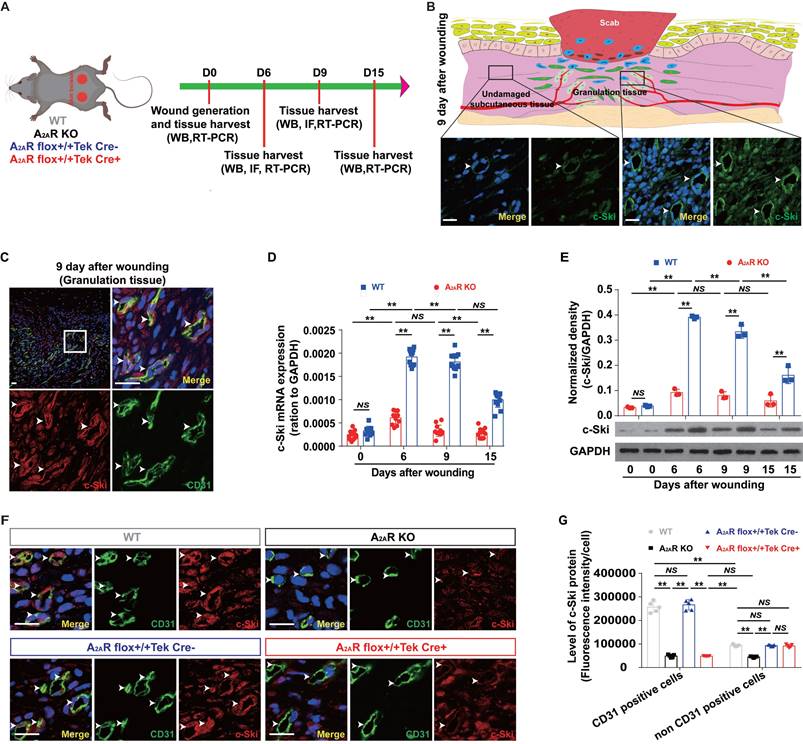
To determine whether the deletion of A2AR affects angiogenesis during wound healing through c-Ski, we analyzed the effects of A2AR on the expression of c-Ski in A2AR-KO and EC-A2AR-KO mice (Fig.4A and S5A). Consistent with our previous results[39, 42], the level of c-Ski in the wound area was greater than that in the surrounding area (Fig. 4B and S5B), and c-Ski expression significantly increased after injury and was maintained at a high level during the late stages of wound healing (Fig. 4D-E). Furthermore, immunofluorescence revealed that the c-Ski level in CD31-positive cells was greater than that in non-CD31-positive cells at 9 days post wounding (Fig. 4C and S5B). Moreover, c-Ski was colocalized with A2AR in both undamaged subcutaneous tissue and the granulation tissue of the wound (Fig. S5C). In contrast to WT mice, the expression and level of c-Ski were significantly reduced in A2AR-KO mice after wounding (Fig. 4D-E). Immunofluorescence staining further confirmed that the c-Ski protein level in CD31-positive cells was not significantly different between the WT and EC-A2AR control mice (Fig. 4F-G), whereas c-Ski protein expression was significantly decreased in all cells of the A2AR-KO mice and was found only in CD3-positive cells of the EC-A2AR-KO mice (Fig. 4F-G and S6). Collectively, these results suggest that A2AR regulates angiogenesis through c-Ski.
The expression of c-Ski in A2AR KO and EC-A2AR KO mice after full-thickness wounding. (A) Experimental procedure. (B) Immunohistochemistry for c-Ski (green, white arrow) in the granulation tissue of the wound and subcutaneous tissue of the surrounding wound at 9 days postwounding. The lower panel shows higher magnification sections of the black squares in the upper panel. Scale bar, 50 µm. (C) Double-label immunofluorescence for c-Ski (red) and CD31 (green) in the granulation tissue of the wound at 9 days postwounding revealed the colocalization of c-Ski and CD31 (white arrow). The surrounding area is a higher magnification section of the white square. Short scale bar, 50 µm; long scale bar, 50 µm. Expression of c-Ski detected by (D) real-time PCR (n = 9) and (E) western blot analysis (n = 3) at 0, 6, 9 and 15 days postwounding. **p < 0.01; NS, not significant. (F) Double-label immunofluorescence for c-Ski (red) and CD31 (green) in the granulation tissue of the wound at 9 days postwounding in each group. The white arrow indicates the colocalization of c-Ski and CD31. Scale bar, 50 µm. (G) Quantitative analysis of (F). **p < 0.01 (n=5); NS, not significant.
A2AR activation induces a proangiogenic effect, as shown by the application of an A2AR agonist in vitro
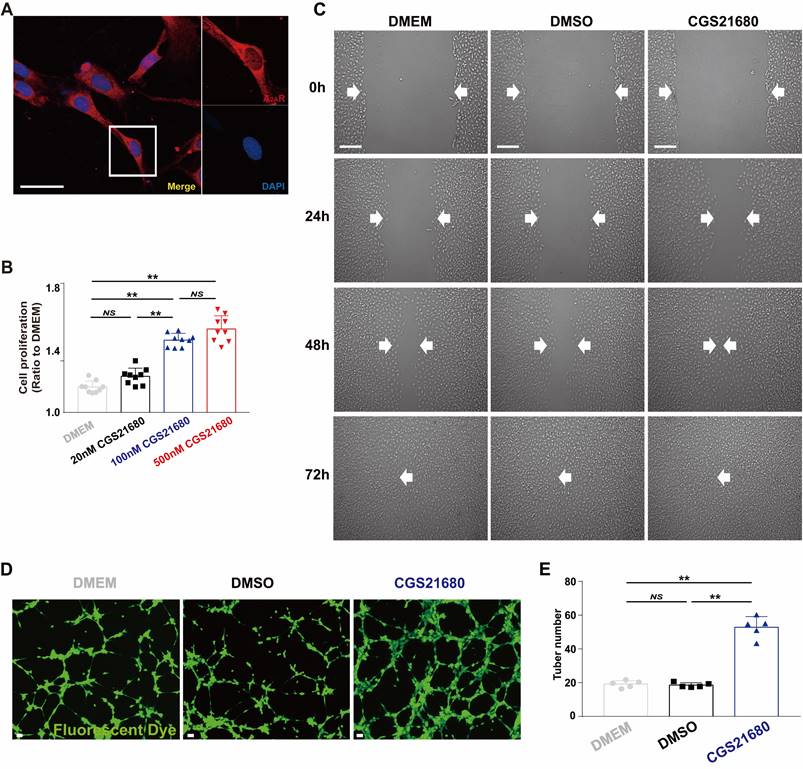
After confirming the essential role of angiogenesis in A2A R deficiency-mediated impairment of wound healing involving c-Ski, we further verified the proangiogenic effect of A2A R in vitro, laying the foundation for future exploration of the role of c-Ski in this effect. Previous studies have indicated that adenosine, which acts through A2AR, promotes angiogenesis in pulmonary ECs[52-54], vascular ECs[55, 56], retinal ECs[57], umbilical vein ECs[58], lymphatic ECs[59], and human EA.hy926 ECs[60]. However, the activation of A2AR does not affect angiogenesis in bone fracture medium[55] or human lung microvascular ECs[61]. We used HMECs, a critical component of granulation tissue involved in wound healing, to verify the proangiogenic effect induced by A2AR activation via cell proliferation, migration and tube formation assays, which are often used to evaluate angiogenesis in vitro[53, 62, 63]. Immunofluorescence staining revealed that A2AR was expressed in HMECs and exhibited a hole-like morphology (Fig. 5A). Moreover, application of the A2AR agonist CGS21680 promoted cell proliferation in a dose-dependent manner (Fig. 5B). In the scratch assay, cell migration was obviously greater in the CGS21680 group than in the DMEM and DMSO control groups (Fig. 5C). Moreover, CGS21680 treatment resulted in a significant increase in the tube formation of HMECs compared with that of the DMEM and DMSO control groups in angiogenesis assays in vitro (Fig. 5D-F). Consistent with other studies[53, 54, 56, 64], the activation of A2AR by CGS21680 significantly promoted the proliferation, migration and angiogenesis of HMECs.
Changes in the proangiogenic effect in HMECs by the application of an A2AR agonist in vitro. (A) Immunohistochemistry of A2AR (red) in HMECs. Scale bar, 50 µm. (B) Cell proliferation was measured by the CCK-8 assay 24 h after treatment with different concentrations of CGS21680. **p < 0.01 (n=9); NS, not significant. (C) Representative images of cell migration evaluated by scratch wound healing assays at 0, 24, 48 and 72 h after CGS21680 treatment. Scale bar, 50 µm. (D) Representative images of tube formation 24 h after treatment with CGS21680. Scale bar, 50 µm. (E) Quantitative analysis of (D). **p < 0.01 (n=5); NS, not significant.
A2AR regulates angiogenesis by regulating c-Ski, as shown by RNAi of c-Ski expression in HMECs
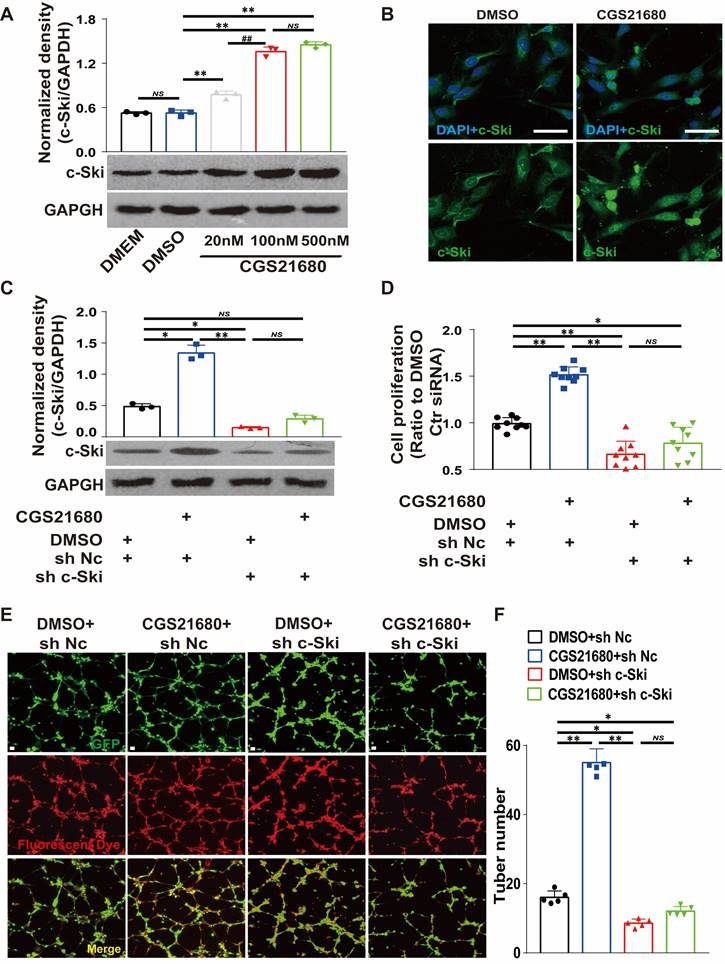
To determine whether c-Ski is required for A2AR-mediated angiogenesis, we used CGS21680 to activate A2AR and RNAi to inhibit the expression of c-Ski in HMECs. Transfection with sh c-Ski successfully reduced the protein level of c-Ski in a dose-dependent manner (Fig. S8A-B), and CGS21680 treatment increased the c-Ski protein level in a dose-dependent manner (Fig. 6A). Immunofluorescence staining revealed that the c-Ski protein level increased after CGS21680 treatment, and c-Ski clearly aggregated in the nucleus (Figs. 6B and S7A). Interestingly, the c-Ski protein level was greater in dividing cells (Fig. S7B), suggesting an important role of c-Ski in cell proliferation. RNAi not only significantly reduced the level of the c-Ski protein compared with that of the control but also significantly reduced the increase in the c-Ski protein induced by CGS21680 treatment (Fig. 6C). Similarly, c-Ski depletion reduced the proliferation and tube formation of HMECs and abolished the effects of CGS21680 treatment on promoting cell proliferation and tube formation (Fig. 6D-F). Overall, these cell proliferation and tube formation assays strongly indicate that c-Ski plays an essential role in A2AR-mediated angiogenesis in HMECs.
Effects of c-Ski on the proangiogenic effect of A2AR activation in HMECs. (A) Levels of c-Ski detected by western blots 24 h after treatment with different concentrations of CGS21680. ** p < 0.01, ##p <0.01, compared with the previous adjacent group (n = 3); NS, not significant. (B) Immunohistochemistry for c-Ski (green) in HMECs 24 h after CGS21680 treatment. Scale bar, 50 µm. (C) Changes in the CGS21680-induced increase in the intracellular protein level of c-Ski in HMECs after 24 h of c-Ski RNAi. ** p < 0.01, *p <0.05 (n = 3); NS, not significant. (D) Changes in CGS21680-induced cell proliferation for 24 h in HMECs after c-Ski RNAi. ** p < 0.01 (n = 9); NS, not significant. (E) Representative images of CGS21680-induced tube formation in HMECs after c-Ski RNAi treatment for 24 h. Scale bar, 50 µm. (F) Quantitative analysis of (E). **p < 0.01, *p <0.05 (n=5); NS, not significant.
A2AR regulates the expression of the c-Ski protein through the ERK/CREB pathway in HMECs
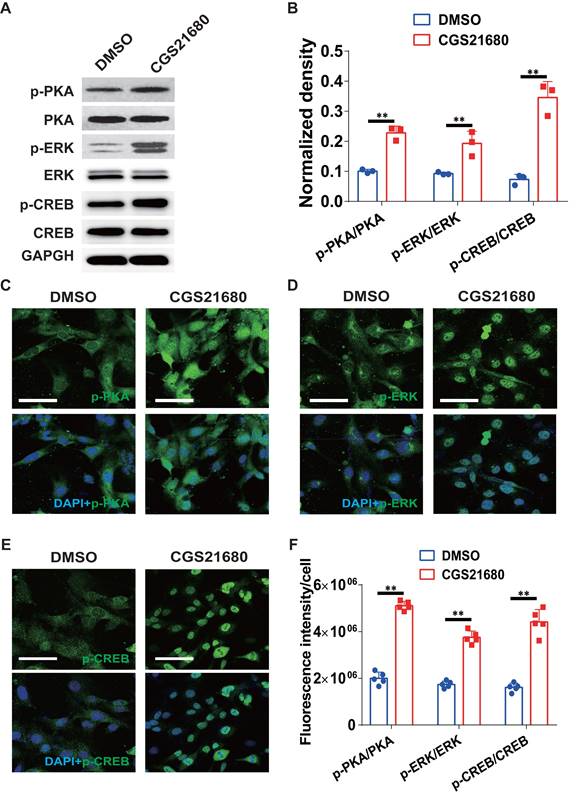
To characterize the underlying mechanisms by which A2AR regulates the expression of the c-Ski protein, we surveyed the ERK/CREB signaling pathway. As shown in Fig. 7A-B, CGS21680 treatment significantly increased the PKA, ERK and CREB phosphorylation levels in HMECs. Similar results were obtained by immunofluorescence staining at 24 h after CGS21680 treatment, indicating that activation of A2AR by CGS21680 can enhance the PKA/ERK/CREB signaling pathways in HMECs (Fig. 7C-F).
Changes in the ERK/CREB signaling pathways after A2AR activation in HMECs. (A) Representative immunoblot images of the levels of PKA, p-PKA, ERK, p-ERK, CREB and p-CREB in HMECs 24 h after treatment with CGS21680. (B) Quantitative analysis of (A). **p < 0.01 (n=3); Immunohistochemistry for p-PKA (C), p-ERK (D) and p-CREB (E) in HMECs 24 h after treatment with CGS21680. Scale bar, 50 µm. (F) Quantitative analysis of (C-E). **p < 0.01, *p <0.05 (n=5).
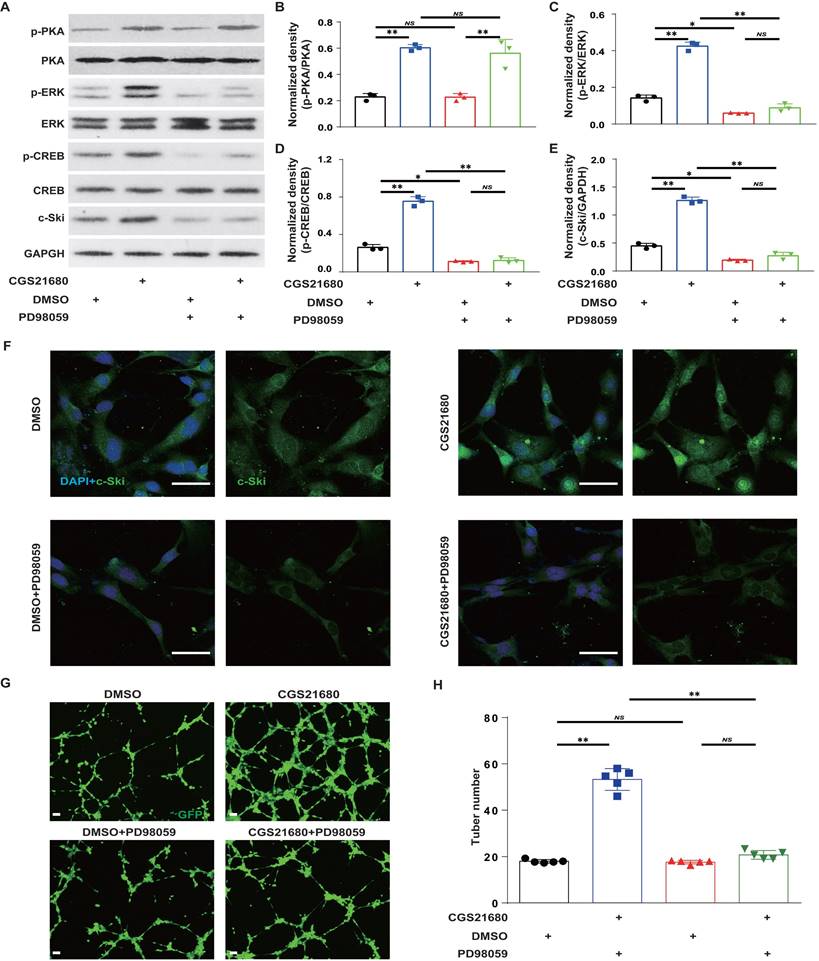
To further verify that A2AR regulates c-Ski protein expression through the ERK/CREB pathway in HMECs, we used CGS21680 to activate A2AR and the MEK1/2 inhibitor PD98059[40, 65] to inhibit ERK activity in HMECs. Compared with the lack of effect on p-PKA (Fig. 8A-B), PD98059 alone significantly reduced p-ERK, p-CREB and c-Ski levels and abrogated the CGS21680 treatment-induced increases in p-ERK, p-CREB and c-Ski levels (Fig. 8A and C-E). Similarly, immunofluorescence staining revealed that PD98059 not only decreased the fluorescence intensity of c-Ski but also abrogated the CGS21680 treatment-induced increase in the fluorescence intensity of c-Ski (Fig. 8F). Moreover, compared with DMSO, PD98059 not only decreased the tube formation of HMECs but also abolished the CGS21680 treatment-induced increase in the tube formation of HMECs in angiogenesis assays in vitro (Fig.8G-H). Taken together, these results suggest that A2AR regulates c-Ski protein expression in HMECs through the ERK/CREB pathway.
Effects of the ERK/CREB signaling pathways on A2AR-induced expression of c-Ski in HMECs. Representative immunoblot images of the CGS21680-induced changes in the intracellular protein levels of PKA, p-PKA, ERK, p-ERK, CREB and p-CREB in HMECs after ERK/CREB inhibition. (B-E) Quantitative analysis of (A). **p < 0.01 (n=3); NS, not significant. (F) Immunohistochemistry for c-Ski (green) induced by treatment with CGS21680 in HMECs after ERK/CREB inhibition. Scale bar, 50 µm. (G) Representative images of tube formation 24 h after treatment with CGS21680. Scale bar, 50 µm. (H) Quantitative analysis of (G). **p < 0.01 (n=5); NS, not significant.
Discussion
Some studies have shown that A2AR is expressed on most cell types involved in wound healing, including macrophages, fibroblasts and microvascular ECs, in vitro[8, 18, 61, 66]. We first explored the types of expression in these repair-associated cells in vivo. According to the histochemical results of full-thickness excisional wounds, A2A R was expressed on most repair cells, including epidermal cells, ECs, macrophages and fibroblasts (Fig. 1), suggesting that it regulates wound healing. Interestingly, the increase was greater in the granulation tissue of the wound than in the epidermis relative to the undamaged side, suggesting that regulating granulation tissue formation is more important than re-epithelialization in A2AR regulation of wound healing, which is consistent with the findings of some studies[15, 54]. Importantly, the peak expression level of A2AR during wound healing occurred at 6 days postwounding (Fig. 1), which is also the fast period of wound healing (Fig. 2)[67-69]. In contrast, A2AR protein levels are lower at 7 days postwounding than at 3 days during the healing of refractory wounds in diabetic mice[70]. Taken together, these results suggest that A2AR may have a wound-promoting effect. A2AR agonists can promote skin wound healing and radiation-impaired wound healing[8-13], whereas A2AR gene knockout delays the course of skin wound healing[10, 15, 71], which also indicates the promotion of healing by A2AR.
Similar to the findings of a previous experiment[15, 54], our study revealed that A2AR KO delayed wound healing compared with WT mice. Importantly, there was no significant difference in the rate of wound closure or healing time between the A2AR-KO and EC-A2AR-KO mice, suggesting that the effects of A2AR on wound healing are mainly mediated through endothelial cells. However, the mechanism by which A2AR regulates wound healing has not been established and is likely multifactorial, as it affects inflammation[15, 16], fibroblasts and epithelial cells [13, 17, 18]. On the one hand, these findings regarding the effects on fibroblasts and epithelial cells are based on the use of agonists and in vitro experiments[2, 17, 72, 73], and there are specific concerns about the effects of agonists and their widespread regulation compared with those of exogenous substances. Moreover, regarding the effect of A2AR on fibroblasts, researchers have focused mainly on its role in fibrotic diseases[71, 74]. On the other hand, for the regulation of inflammatory cells, an induced deficiency in A2AR is not associated with a defect in the acute inflammatory response or with a difference in the number or functional capacity of A2AR-deficient leukocytes[15], and convincing in vivo experimental evidence for a role of A2AR in wound healing through inflammatory reactions is lacking[75]. In addition, some research has shown that A2AR regulates inflammation through ECs[76]. Thus, A2AR's effects on ECs play a critical role in the regulation of wound healing. However, further studies are needed to determine the role of A2AR in other cells during wound healing.
Interestingly, we also found that the deficit in granulation tissue was obvious at 6 and 9 days postwounding, whereas the extent of re-epithelialization did not differ between the WT controls and the A2AR KO mice, which is consistent with the findings of a previous experiment[15, 54]. Moreover, EC-A2AR KO mice presented similar results and were not significantly different from A2AR KO mice, strongly suggesting that A2AR regulates granulation tissue formation, but not epithelialization, during wound healing. Previous studies have indirectly demonstrated that A2A R is involved in the angiogenic effects of ECs by total A2AR KO or the application of agonists or antagonists[13, 15, 51], which critically affects the formation of granulation tissue, and these results are consistent with the observed defects in granulation tissue formation. Importantly, our study directly demonstrated that A2AR affects granulation tissue formation mainly by regulating angiogenesis by using EC-A2AR KO mice, further indicating that the angiogenic effects of A2AR on ECs play a critical role in the regulation of wound healing. Similarly, Liu et al. demonstrated that A2AR is crucial for pathological angiogenesis in proliferative retinopathies using EC-A2AR-KO mice[57].
Angiogenesis promotes growth, development, and wound healing through the formation of granulation tissue[77]. Among the many cells involved in the angiogenic process, ECs play an essential role in new vessel formation via an increase in cell proliferation, migration, and tube formation capacity[78]. Although cell proliferation, migration, and tube formation capacity were assessed in vitro in this study, only the number of CD31-positive cells was determined in vivo. In future experiments, we should use other parameters to clarify this process in vivo. In addition to the above effects, further exploration is needed to determine whether A2AR regulates angiogenesis through other mechanisms or can regulate angiogenesis by influencing other cellular effects (for example, inflammatory cells).
However, previous reports have shown that the angiogenic effect of A2AR activation is likely mediated by increased expression of VEGF in ECs[79-81]. Other studies have also reported that A2AR stimulation increases angiogenesis through the antiangiogenic matrix proteins thrombospondin 1[64], annexin A2[56], and MyD88[16] and macrophage exosomes[55].
We demonstrated that A2AR regulates angiogenesis through c-Ski. c-Ski was significantly expressed on ECs during wound healing and was significantly decreased after A2AR KO and EC-A2AR KO. A2AR activation induced a proangiogenic effect and increased the expression of c-Ski, whereas c-Ski depletion abolished the proangiogenic effects of A2AR activation. In addition, similar to our previous research[26, 82], the expression of c-Ski increased after wounding, peaked at 6 days and then significantly decreased, which is consistent with the changes in A2AR expression after wounding. Furthermore, c-Ski is expressed on ECs[30], and reduced c-Ski expression in the wound results in significantly slower wound healing[26], similar to the results of A2A R KO. Taken together, these results strongly indicate that c-Ski plays an essential role in A2AR-mediated angiogenesis. More importantly, c-Ski depletion reduced the proliferation and tube formation of HMECs and abolished the promotive effects on cell proliferation and tube formation caused by A2AR activation in vitro, further demonstrating the important role of c-Ski in A2AR-mediated angiogenesis.
Although there is little research on the role of c-Ski in angiogenesis or EC regulation, as a multieffector factor, this molecule can regulate the proliferation and migration of fibroblasts[83], vascular smooth muscle cells[84], cardiomyocytes[85], astrocytes[86], and tumor cells[87, 88]. These results indicate that c-Ski may regulate angiogenesis through these effects. Moreover, the overexpression of c-Ski was shown to promote the proliferation of ECs[31, 32]. In addition, an analysis of the regulated signal transduction pathway revealed that c-Ski not only synergizes with factors such as activator protein-1 (AP-1)[89] and signal transducer and activator of transcription 3 (STAT-3)[90], which upregulate the expression of the essential angiogenic factor VEGF[91, 92], but can also reduce the expression of the antiangiogenic factor thrombospondin-1 (TSP-1)[93], suggesting that c-Ski plays an important role in angiogenesis. Interestingly, A2AR can regulate angiogenesis not only through VEGF[18,19,20] but also through TSP1[64], further indicating that A2AR can regulate angiogenesis through c-Ski. Nevertheless, the mechanism of the proangiogenic effect of c-Ski remains to be further explored.
As reported in previous studies, the ERK/CREB pathway is an important pathway for regulating c-Ski expression[40]. Our study revealed that MEK inhibition markedly reduced c-Ski expression and abrogated A2AR-mediated promotion of c-Ski expression with CGS21680 in HMECs, indicating that A2AR regulates c-Ski protein expression in HMECs through the ERK1/2 pathway. Our results demonstrated that c-Ski expression is regulated by A2AR in ECs through this pathway, and the results of other studies support this finding. Several studies have demonstrated that the ERK pathway is important in angiogenesis[94, 95] and A2AR activation can activate the ERK pathway in ECs[52, 96-98]. In particular, Liu et al. reported that A2AR activation promotes pathological angiogenesis via ERK-dependent translational activation[57]. In addition, our study revealed that A2AR activation by CGS21680 enhanced the ERK1/2 pathway through PKA, which is consistent with the findings of several previous reports[99, 100]. However, further in vivo experimental verification of A2AR activation inducing the expression of c-Ski through the ERK/CREB pathway is needed.
Conclusions
Taken together, our results demonstrate that the delayed effects of A2AR KO on wound healing are due mainly to the effects of A2AR on endothelial cells, indicating that A2AR-mediated angiogenesis plays a critical role in wound healing. Moreover, our results demonstrate a novel cellular and molecular mechanism whereby endothelial A2AR deficiency delays wound healing and decreases angiogenesis through c-Ski deficiency. A2AR regulates the expression of the c-Ski protein through the ERK/CREB pathway in HMECs. Thus, these findings provide new insights into a previously unrecognized effect of A2AR on angiogenesis by ECs in wounds and highlight the mechanism of A2AR in wound repair, which not only identifies A2AR as a therapeutic target in wound repair but also highlights other angiogenesis-dependent tissue repair processes.
Abbreviations
A2AR: adenosine A2A receptor; ECs: endothelial cells; c-Ski: Cellular Sloan-Kettering Institute; A2AR KO: A2AR knockout; EC-A2AR KO: EC-specific A2AR knockout; HMECs: human microvascular ECs; ERK1/2: signal-regulated kinase 1/2; WT: wild-type; H&E: hematoxylin and eosin; CGS21680: 2-p-[2-carboxyethyl]phenethyl-amino-5'-N-ethylcarboxamido-adenosine; DMSO: dimethyl sulfoxide; AP-1: activator protein-1; STAT-3: signal transducer and activator of transcription 3; TSP-1: thrombospondin-1; FBS: fetal bovine serum; PBS: phosphate-buffered saline; IntDen: integrated density; STR: short tandem repeat; HBSS: Hank's balanced salt solution; OD: optical density.
Supplementary Material
Supplementary figures.
Acknowledgements
We would like to thank AJE (www.aje.com) for English language editing.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No: 81701915) and the projects of Chongqing (cstc2021yszx-jcyjX0001).
Author contributions
PL, YP, and YZ designed the experiments; YP, RX, XC, and BW performed the experiments and acquired the data; YZ, BW, YN, YZ, NY, JZ, and CL reviewed and interpreted the data; PL and YP wrote the first draft of the paper; and all of the authors contributed to the final manuscript before submission.
Availability of data and materials
Raw data supporting the conclusions of this article are available from the corresponding author upon reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hoque J, Zeng Y, Newman H, Gonzales G, Lee C, Varghese S. Microgel-Assisted Delivery of Adenosine to Accelerate Fracture Healing. ACS Biomater Sci Eng. 2022;8:4863-72
2. Kim J, Shin JY, Choi YH, Lee SY, Jin MH, Kim CD. et al. Adenosine and Cordycepin Accelerate Tissue Remodeling Process through Adenosine Receptor Mediated Wnt/beta-Catenin Pathway Stimulation by Regulating GSK3b Activity. Int J Mol Sci. 2021;22:5571
3. Cheng X, Yin C, Deng Y, Li Z. Exogenous adenosine activates A2A adenosine receptor to inhibit RANKL-induced osteoclastogenesis via AP-1 pathway to facilitate bone repair. Mol Biol Rep. 2022;49:2003-14
4. Shaikh G, Cronstein B. Signaling pathways involving adenosine A2A and A2B receptors in wound healing and fibrosis. Purinergic Signal. 2016;12:191-7
5. Ojeh N, Stojadinovic O, Pastar I, Sawaya A, Yin N, Tomic-Canic M. The effects of caffeine on wound healing. Int Wound J. 2016;13:605-13
6. Bezerra JP, de Siqueira A, Pires AG, Marques MR, Duarte PM, Bastos MF. Effects of estrogen deficiency and/or caffeine intake on alveolar bone loss, density, and healing: a study in rats. J Periodontol. 2013;84:839-49
7. Supit T, Susilaningsih N, Prasetyo A, Najatullah. Effects of Caffeine Consumption on Autologous Full-Thickness Skin Graft Healing in an Animal Model. Indian J Plast Surg. 2021;54:314-20
8. Valls MD, Cronstein BN, Montesinos MC. Adenosine receptor agonists for promotion of dermal wound healing. Biochem Pharmacol. 2009;77:1117-24
9. Montesinos MC, Desai-Merchant A, Cronstein BN. Promotion of Wound Healing by an Agonist of Adenosine A2A Receptor Is Dependent on Tissue Plasminogen Activator. Inflammation. 2015;38:2036-41
10. Dai SS, Xiong RP, Yang N, Li W, Zhu PF, Zhou YG. [Different effects of adenosine A2A receptors in the models of traumatic brain injury and peripheral tissue injury]. Sheng Li Xue Bao. 2008;60:254-8
11. Victor-Vega C, Desai A, Montesinos MC, Cronstein BN. Adenosine A2A receptor agonists promote more rapid wound healing than recombinant human platelet-derived growth factor (Becaplermin gel). Inflammation. 2002;26:19-24
12. Altavilla D, Squadrito F, Polito F, Irrera N, Calo M, Lo Cascio P. et al. Activation of adenosine A2A receptors restores the altered cell-cycle machinery during impaired wound healing in genetically diabetic mice. Surgery. 2011;149:253-61
13. Montesinos MC, Gadangi P, Longaker M, Sung J, Levine J, Nilsen D. et al. Wound healing is accelerated by agonists of adenosine A2 (G alpha s-linked) receptors. J Exp Med. 1997;186:1615-20
14. Galeano M, Pallio G, Irrera N, Mannino F, Bitto A, Altavilla D. et al. Polydeoxyribonucleotide: A Promising Biological Platform to Accelerate Impaired Skin Wound Healing. Pharmaceuticals (Basel). 2021;14:1103
15. Montesinos MC, Desai A, Chen JF, Yee H, Schwarzschild MA, Fink JS. et al. Adenosine promotes wound healing and mediates angiogenesis in response to tissue injury via occupancy of A(2A) receptors. Am J Pathol. 2002;160:2009-18
16. Macedo L, Pinhal-Enfield G, Alshits V, Elson G, Cronstein BN, Leibovich SJ. Wound healing is impaired in MyD88-deficient mice: a role for MyD88 in the regulation of wound healing by adenosine A2A receptors. Am J Pathol. 2007;171:1774-88
17. Allen-Gipson DS, Wong J, Spurzem JR, Sisson JH, Wyatt TA. Adenosine A2A receptors promote adenosine-stimulated wound healing in bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L849-55
18. Perez-Aso M, Fernandez P, Mediero A, Chan ES, Cronstein BN. Adenosine 2A receptor promotes collagen production by human fibroblasts via pathways involving cyclic AMP and AKT but independent of Smad2/3. FASEB J. 2014;28:802-12
19. Pearson-White S, Deacon D, Crittenden R, Brady G, Iscove N, Quesenberry PJ. The ski/sno protooncogene family in hematopoietic development. Blood. 1995;86:2146-55
20. Berk M, Desai SY, Heyman HC, Colmenares C. Mice lacking the ski proto-oncogene have defects in neurulation, craniofacial, patterning, and skeletal muscle development. Genes Dev. 1997;11:2029-39
21. Atanasoski S, Notterpek L, Lee HY, Castagner F, Young P, Ehrengruber MU. et al. The protooncogene Ski controls Schwann cell proliferation and myelination. Neuron. 2004;43:499-511
22. Fumagalli S, Doneda L, Nomura N, Larizza L. Expression of the c-ski proto-oncogene in human melanoma cell lines. Melanoma Res. 1993;3:23-7
23. Rashidian J, Le Scolan E, Ji X, Zhu Q, Mulvihill MM, Nomura D. et al. Ski regulates Hippo and TAZ signaling to suppress breast cancer progression. Sci Signal. 2015;8:ra14
24. Soeta C, Suzuki M, Suzuki S, Naito K, Tachi C, Tojo H. Possible role for the c-ski gene in the proliferation of myogenic cells in regenerating skeletal muscles of rats. Dev Growth Differ. 2001;43:155-64
25. Macias-Silva M, Li W, Leu JI, Crissey MA, Taub R. Up-regulated transcriptional repressors SnoN and Ski bind Smad proteins to antagonize transforming growth factor-beta signals during liver regeneration. J Biol Chem. 2002;277:28483-90
26. Li P, Liu P, Xiong RP, Chen XY, Zhao Y, Lu WP. et al. Ski, a modulator of wound healing and scar formation in the rat skin and rabbit ear. J Pathol. 2011;223:659-71
27. Zhai Y, Ye SY, Wang QS, Xiong RP, Fu SY, Du H. et al. Overexpressed ski efficiently promotes neurorestoration, increases neuronal regeneration, and reduces astrogliosis after traumatic brain injury. Gene Ther. 2022;30:75-87
28. Liao HY, Da CM, Wu ZL, Zhang HH. Ski: Double roles in cancers. Clin Biochem. 2021;87:1-12
29. Bonnon C, Atanasoski S. c-Ski in health and disease. Cell Tissue Res. 2012;347:51-64
30. Wang J, Li H, Lv Z, Luo X, Deng W, Zou T. et al. The miR-214-3p/c-Ski axis modulates endothelial-mesenchymal transition in human coronary artery endothelial cells in vitro and in mice model in vivo. Hum Cell. 2022;35:486-97
31. Sun B, Huang Z, Yang H, Zhao X. MicroRNA-195-5p inhibits the progression of hemangioma via targeting SKI. Exp Ther Med. 2022;23:165
32. O TM, Tan M, Tarango M, Fink L, Mihm M, Ma Y. et al. Differential expression of SKI oncogene protein in hemangiomas. Otolaryngol Head Neck Surg. 2009;141:213-8
33. Li X, Sun C, Chen J, Ma JF, Pan YH. ERK-CREB pathway is involved in HSPB8-mediated glioma cell growth and metastatic properties. Exp Mol Pathol. 2021: 104653.
34. Rao J, Li H, Zhang H, Xiang X, Ding X, Li L. et al. Periplaneta Americana (L.) extract activates the ERK/CREB/BDNF pathway to promote post-stroke neuroregeneration and recovery of neurological functions in rats. J Ethnopharmacol. 2024;321:117400
35. Xin Y, Roh K, Cho E, Park D, Whang W, Jung E. Isookanin Inhibits PGE(2)-Mediated Angiogenesis by Inducing Cell Arrest through Inhibiting the Phosphorylation of ERK1/2 and CREB in HMEC-1 Cells. Int J Mol Sci. 2021;22:6466
36. Zhang Y, Zheng D, Zhou T, Song H, Hulsurkar M, Su N. et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat Commun. 2018;9:4080
37. Trinh PNH, Baltos JA, Hellyer SD, May LT, Gregory KJ. Adenosine receptor signalling in Alzheimer's disease. Purinergic Signal. 2022;18:359-81
38. Fredholm BB, Chern Y, Franco R, Sitkovsky M. Aspects of the general biology of adenosine A2A signaling. Prog Neurobiol. 2007;83:263-76
39. Li P, Rial D, Canas PM, Yoo JH, Li W, Zhou X. et al. Optogenetic activation of intracellular adenosine A2A receptor signaling in the hippocampus is sufficient to trigger CREB phosphorylation and impair memory. Mol Psychiatry. 2015;20:1339-49
40. Li P, Liu P, Peng Y, Zhang ZH, Li XM, Xiong RP. et al. The ERK/CREB pathway is involved in the c-Ski expression induced by low TGF-beta1 concentrations during primary fibroblast proliferation. Cell Cycle. 2018;17:1319-28
41. Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D. et al. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19:9192-200
42. Xu Y, Ning Y, Zhao Y, Peng Y, Luo F, Zhou Y. et al. Caffeine Functions by Inhibiting Dorsal and Ventral Hippocampal Adenosine 2A Receptors to Modulate Memory and Anxiety, Respectively. Front Pharmacol. 2022;13:807330
43. Zeng Q, Wu Z, Duan H, Jiang X, Tu T, Lu D. et al. Impaired tumor angiogenesis and VEGF-induced pathway in endothelial CD146 knockout mice. Protein Cell. 2014;5:445-56
44. Wang Z, Chai Q, Zhu M. Differential Roles of LTbetaR in Endothelial Cell Subsets for Lymph Node Organogenesis and Maturation. J Immunol. 2018;201:69-76
45. Peng Y, Li P, Zhao ZA, Chen L, Zhao XG, Chen X. et al. Comparative evaluation of the wound-healing potency of recombinant bFGF and ski gene therapy in rats. Growth Factors. 2016;34:119-27
46. Zhang ZH, Xu YW, Peng Y, Chen X, Li P, Zhou YG. Expression of a short antibody heavy chain peptide effectively antagonizes adenosine 2A receptor in vitro and in vivo. Expert Opin Ther Targets. 2020;24:707-17
47. Peng Y, Xiong RP, Zhang ZH, Ning YL, Zhao Y, Tan SW. et al. Ski promotes proliferation and inhibits apoptosis in fibroblasts under high-glucose conditions via the FoxO1 pathway. Cell Prolif. 2021;54:e12971
48. Li P, Wang QS, Zhai Y, Xiong RP, Chen X, Liu P. et al. Ski mediates TGF-beta1-induced fibrosarcoma cell proliferation and promotes tumor growth. J Cancer. 2020;11:5929-40
49. Jian L, Mei Y, Xing C, Rongdi Y. Haem relieves hyperoxia-mediated inhibition of HMEC-1 cell proliferation, migration and angiogenesis by inhibiting BACH1 expression. BMC Ophthalmol. 2021;21:104
50. Zudaire E, Gambardella L, Kurcz C, Vermeren S. A computational tool for quantitative analysis of vascular networks. PLoS One. 2011;6:e27385
51. Montesinos MC, Shaw JP, Yee H, Shamamian P, Cronstein BN. Adenosine A(2A) receptor activation promotes wound neovascularization by stimulating angiogenesis and vasculogenesis. Am J Pathol. 2004;164:1887-92
52. Ahmad A, Schaack JB, White CW, Ahmad S. Adenosine A2A receptor-dependent proliferation of pulmonary endothelial cells is mediated through calcium mobilization, PI3-kinase and ERK1/2 pathways. Biochem Biophys Res Commun. 2013;434:566-71
53. Ahmad A, Ahmad S, Glover L, Miller SM, Shannon JM, Guo X. et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc Natl Acad Sci U S A. 2009;106:10684-9
54. Troncoso F, Herlitz K, Acurio J, Aguayo C, Guevara K, Castro FO. et al. Advantages in Wound Healing Process in Female Mice Require Upregulation A(2A)-Mediated Angiogenesis under the Stimulation of 17beta-Estradiol. Int J Mol Sci. 2020;21:7145
55. Wang D, Wang J, Zhou J, Zheng X. The Role of Adenosine Receptor A2A in the Regulation of Macrophage Exosomes and Vascular Endothelial Cells During Bone Healing. J Inflamm Res. 2021;14:4001-17
56. Valls MD, Soldado M, Arasa J, Perez-Aso M, Williams AJ, Cronstein BN. et al. Annexin A2-Mediated Plasminogen Activation in Endothelial Cells Contributes to the Proangiogenic Effect of Adenosine A(2A) Receptors. Front Pharmacol. 2021;12:654104
57. Liu Z, Yan S, Wang J, Xu Y, Wang Y, Zhang S. et al. Endothelial adenosine A2a receptor-mediated glycolysis is essential for pathological retinal angiogenesis. Nat Commun. 2017;8:584
58. Escudero C, Roberts JM, Myatt L, Feoktistov I. Impaired adenosine-mediated angiogenesis in preeclampsia: potential implications for fetal programming. Front Pharmacol. 2014;5:134
59. Allard B, Cousineau I, Allard D, Buisseret L, Pommey S, Chrobak P. et al. Adenosine A2a receptor promotes lymphangiogenesis and lymph node metastasis. Oncoimmunology. 2019;8:1601481
60. Bonyanian Z, Walker M, Du Toit E, Rose'Meyer RB. Multiple adenosine receptor subtypes stimulate wound healing in human EA.hy926 endothelial cells. Purinergic Signal. 2019;15:357-66
61. Feoktistov I, Goldstein AE, Ryzhov S, Zeng D, Belardinelli L, Voyno-Yasenetskaya T. et al. Differential expression of adenosine receptors in human endothelial cells: role of A2B receptors in angiogenic factor regulation. Circ Res. 2002;90:531-8
62. Ling X, Jiang X, Li Y, Han W, Rodriguez M, Xu Z. et al. Sequential Treatment of Bioresponsive Nanoparticles Elicits Antiangiogenesis and Apoptosis and Synergizes with a CD40 Agonist for Antitumor Immunity. ACS Nano. 2021;15:765-80
63. Wilhelm EN, Gonzalez-Alonso J, Parris C, Rakobowchuk M. Exercise intensity modulates the appearance of circulating microvesicles with proangiogenic potential upon endothelial cells. Am J Physiol Heart Circ Physiol. 2016;311:H1297-H310
64. Desai A, Victor-Vega C, Gadangi S, Montesinos MC, Chu CC, Cronstein BN. Adenosine A2A receptor stimulation increases angiogenesis by down-regulating production of the antiangiogenic matrix protein thrombospondin 1. Mol Pharmacol. 2005;67:1406-13
65. Huang K, Chen Y, Zhang R, Wu Y, Ma Y, Fang X. et al. Honokiol induces apoptosis and autophagy via the ROS/ERK1/2 signaling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2018;9:157
66. Khoa ND, Montesinos MC, Reiss AB, Delano D, Awadallah N, Cronstein BN. Inflammatory cytokines regulate function and expression of adenosine A(2A) receptors in human monocytic THP-1 cells. J Immunol. 2001;167:4026-32
67. Scrima M, Melito C, Merola F, Iorio A, Vito N, Giori AM. et al. Evaluation of Wound Healing Activity of Salvia haenkei Hydroalcoholic Aerial Part Extract on in vitro and in vivo Experimental Models. Clin Cosmet Investig Dermatol. 2020;13:627-37
68. Brett E, Zielins ER, Chin M, Januszyk M, Blackshear CP, Findlay M. et al. Isolation of CD248-expressing stromal vascular fraction for targeted improvement of wound healing. Wound Repair Regen. 2017;25:414-22
69. Milan PB, Lotfibakhshaiesh N, Joghataie MT, Ai J, Pazouki A, Kaplan DL. et al. Accelerated wound healing in a diabetic rat model using decellularized dermal matrix and human umbilical cord perivascular cells. Acta Biomater. 2016;45:234-46
70. Zhu W, Dong Y, Xu P, Pan Q, Jia K, Jin P. et al. A composite hydrogel containing resveratrol-laden nanoparticles and platelet-derived extracellular vesicles promotes wound healing in diabetic mice. Acta Biomater. 2022;154:212-30
71. Chan ES, Fernandez P, Merchant AA, Montesinos MC, Trzaska S, Desai A. et al. Adenosine A2A receptors in diffuse dermal fibrosis: pathogenic role in human dermal fibroblasts and in a murine model of scleroderma. Arthritis Rheum. 2006;54:2632-42
72. Tian Z, Zhang H, Dixon J, Traphagen N, Wyatt TA, Kharbanda K. et al. Cigarette Smoke Impairs A(2A) Adenosine Receptor Mediated Wound Repair through Up-regulation of Duox-1 Expression. Sci Rep. 2017;7:44405
73. Dignass AU, Becker A, Spiegler S, Goebell H. Adenine nucleotides modulate epithelial wound healing in vitro. Eur J Clin Invest. 1998;28:554-61
74. Zhang J, Corciulo C, Liu H, Wilder T, Ito M, Cronstein B. Adenosine A(2a) Receptor Blockade Diminishes Wnt/beta-Catenin Signaling in a Murine Model of Bleomycin-Induced Dermal Fibrosis. Am J Pathol. 2017;187:1935-44
75. Pasquini S, Contri C, Borea PA, Vincenzi F, Varani K. Adenosine and Inflammation: Here, There and Everywhere. Int J Mol Sci. 2021;22:7685
76. Sands WA, Martin AF, Strong EW, Palmer TM. Specific inhibition of nuclear factor-kappaB-dependent inflammatory responses by cell type-specific mechanisms upon A2A adenosine receptor gene transfer. Mol Pharmacol. 2004;66:1147-59
77. Mendez-Valdes G, Gomez-Hevia F, Lillo-Moya J, Gonzalez-Fernandez T, Abelli J, Cereceda-Cornejo A. et al. Endostatin and Cancer Therapy: A Novel Potential Alternative to Anti-VEGF Monoclonal Antibodies. Biomedicines. 2023;11:718
78. Stryker ZI, Rajabi M, Davis PJ, Mousa SA. Evaluation of Angiogenesis Assays. Biomedicines. 2019;7:37
79. Takagi H, King GL, Robinson GS, Ferrara N, Aiello LP. Adenosine mediates hypoxic induction of vascular endothelial growth factor in retinal pericytes and endothelial cells. Invest Ophthalmol Vis Sci. 1996;37:2165-76
80. Leibovich SJ, Chen JF, Pinhal-Enfield G, Belem PC, Elson G, Rosania A. et al. Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A(2A) receptor agonists and endotoxin. Am J Pathol. 2002;160:2231-44
81. Acurio J, Herlitz K, Troncoso F, Aguayo C, Bertoglia P, Escudero C. Adenosine A(2A) receptor regulates expression of vascular endothelial growth factor in feto-placental endothelium from normal and late-onset pre-eclamptic pregnancies. Purinergic Signal. 2017;13:51-60
82. Liu X, Zhang E, Li P, Liu J, Zhou P, Gu DY. et al. Expression and possible mechanism of c-ski, a novel tissue repair-related gene during normal and radiation-impaired wound healing. Wound Repair Regen. 2006;14:162-71
83. Zhang H, Wang JS, Chen XG, Kang L, Lin MB. Overexpression of c-Ski promotes cell proliferation, invasion and migration of gastric cancer associated fibroblasts. Kaohsiung J Med Sci. 2019;35:214-21
84. Feng M, Zhou Q, Tu W, Wang Y, Du Y, Xu K. ATF4 promotes brain vascular smooth muscle cells proliferation, invasion and migration by targeting miR-552-SKI axis. PLoS One. 2022;17:e0270880
85. Ling J, Cai Z, Jin W, Zhuang X, Kan L, Wang F. et al. Silencing of c-Ski augments TGF-b1-induced epithelial-mesenchymal transition in cardiomyocyte H9C2 cells. Cardiol J. 2019;26:66-76
86. Liao HY, Wang ZQ, Da CM, Zhou KS, Zhang HH. Ski regulates proliferation and migration of reactive astrocytes induced by lipopolysaccharide (LPS) through PI3K/Akt pathway. J Neuroimmunol. 2022;364:577807
87. Chen D, Lin Q, Box N, Roop D, Ishii S, Matsuzaki K. et al. SKI knockdown inhibits human melanoma tumor growth in vivo. Pigment Cell Melanoma Res. 2009;22:761-72
88. Zhao X, Fang Y, Wang X, Yang Z, Li D, Tian M. et al. Knockdown of Ski decreases osteosarcoma cell proliferation and migration by suppressing the PI3K/Akt signaling pathway. Int J Oncol. 2020;56:206-18
89. Xu W, Angelis K, Danielpour D, Haddad MM, Bischof O, Campisi J. et al. Ski acts as a co-repressor with Smad2 and Smad3 to regulate the response to type beta transforming growth factor. Proc Natl Acad Sci U S A. 2000;97:5924-9
90. Makino Y, Yoon JH, Bae E, Kato M, Miyazawa K, Ohira T. et al. Repression of Smad3 by Stat3 and c-Ski/SnoN induces gefitinib resistance in lung adenocarcinoma. Biochem Biophys Res Commun. 2017;484:269-77
91. Shih SC, Claffey KP. Role of AP-1 and HIF-1 transcription factors in TGF-beta activation of VEGF expression. Growth Factors. 2001;19:19-34
92. Hwang S, Seong H, Ryu J, Jeong JY, Kang TS, Nam KY. et al. Phosphorylation of STAT3 and ERBB2 mediates hypoxia-induced VEGF release in ARPE-19 cells. Mol Med Rep. 2020;22:2733-40
93. Kiyono K, Suzuki HI, Morishita Y, Komuro A, Iwata C, Yashiro M. et al. c-Ski overexpression promotes tumor growth and angiogenesis through inhibition of transforming growth factor-beta signaling in diffuse-type gastric carcinoma. Cancer Sci. 2009;100:1809-16
94. Pages G, Milanini J, Richard DE, Berra E, Gothie E, Vinals F. et al. Signaling angiogenesis via p42/p44 MAP kinase cascade. Ann N Y Acad Sci. 2000;902:187-200
95. Song YY, Liang D, Liu DK, Lin L, Zhang L, Yang WQ. The role of the ERK signaling pathway in promoting angiogenesis for treating ischemic diseases. Front Cell Dev Biol. 2023;11:1164166
96. Wyatt AW, Steinert JR, Wheeler-Jones CP, Morgan AJ, Sugden D, Pearson JD. et al. Early activation of the p42/p44MAPK pathway mediates adenosine-induced nitric oxide production in human endothelial cells: a novel calcium-insensitive mechanism. FASEB J. 2002;16:1584-94
97. Ribe D, Sawbridge D, Thakur S, Hussey M, Ledent C, Kitchen I. et al. Adenosine A2A receptor signaling regulation of cardiac NADPH oxidase activity. Free Radic Biol Med. 2008;44:1433-42
98. Thakur S, Du J, Hourani S, Ledent C, Li JM. Inactivation of adenosine A2A receptor attenuates basal and angiotensin II-induced ROS production by Nox2 in endothelial cells. J Biol Chem. 2010;285:40104-13
99. Ouyang X, Ghani A, Malik A, Wilder T, Colegio OR, Flavell RA. et al. Adenosine is required for sustained inflammasome activation via the A(2)A receptor and the HIF-1alpha pathway. Nat Commun. 2013;4:2909
100. Germack R, Dickenson JM. Characterization of ERK1/2 signalling pathways induced by adenosine receptor subtypes in newborn rat cardiomyocytes. Br J Pharmacol. 2004;141:329-39
Author contact
![]() Corresponding author: Ping Li, Department of Army Occupational Disease, State Key Laboratory of Trauma, Burn and Combined Injury, Daping Hospital, Army Medical University (Third Military Medical University), 10 Changjiang Zhilu, Chongqing 400042, People's Republic of China. E-mail addresses: ping__ping0074com; liping123123edu.cn.
Corresponding author: Ping Li, Department of Army Occupational Disease, State Key Laboratory of Trauma, Burn and Combined Injury, Daping Hospital, Army Medical University (Third Military Medical University), 10 Changjiang Zhilu, Chongqing 400042, People's Republic of China. E-mail addresses: ping__ping0074com; liping123123edu.cn.