Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Metabolic reprogramming of TAMs...
The regulation of tumor...
TAM-mediated...
Targeting metabolic interactions...
Conclusions and Outlooks
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(13):5109-5126. doi:10.7150/ijbs.99680 This issue Cite
Review
A new perspective on the therapeutic potential of tumor metastasis: targeting the metabolic interactions between TAMs and tumor cells
Xuan Zhao1,2,3*, Tong Ren1,2,3*, Sijin Li1,2,3, Xu Wang1,2,3, Rui Hou4, Zhangchun Guan1,2,3, Dan Liu1,2,3 ![]() , Junnian Zheng2,3
, Junnian Zheng2,3 ![]() , Ming Shi1,2,3
, Ming Shi1,2,3 ![]()
1. Cancer Institute, Xuzhou Medical University, China.
2. Center of Clinical Oncology, The Affiliated Hospital of Xuzhou Medical University, China.
3. Jiangsu Center for the Collaboration and Innovation of Cancer Biotherapy, Xuzhou Medical University, China.
4. College of Pharmacy, Xuzhou Medical University, Xuzhou, Jiangsu, China.
*These authors contributed equally to this work.
Received 2024-6-15; Accepted 2024-9-2; Published 2024-9-23
Abstract

Tumor-associated macrophages (TAMs) undergo metabolic reprogramming, encompassing glucose, amino acid, fatty acid metabolism, tricarboxylic acid (TCA) cycle, purine metabolism, and autophagy, within the tumor microenvironment (TME). The metabolic interdependencies between TAMs and tumor cells critically influence macrophage recruitment, differentiation, M2 polarization, and secretion of epithelial-mesenchymal transition (EMT)-related factors, thereby activating intratumoral EMT pathways and enhancing tumor cell invasion and metastasis. Tumor cell metabolic alterations, including hypoxia, metabolite secretion, aerobic metabolism, and autophagy, affect the TME's metabolic landscape, driving macrophage recruitment, differentiation, M2 polarization, and metabolic reprogramming, ultimately facilitating EMT, invasion, and metastasis. Additionally, macrophages can induce tumor cell EMT by reprogramming their aerobic glycolysis. Recent experimental and clinical studies have focused on the metabolic interactions between macrophages and tumor cells to control metastasis and inhibit tumor progression. This review highlights the regulatory role of TAM-tumor cell metabolic codependencies in EMT, offering valuable insights for TAM-targeted therapies in highly metastatic tumors. Modulating the metabolic interplay between tumors and TAMs represents a promising therapeutic strategy for treating patients with metastatic cancers.
Keywords: Tumor-associated macrophages, Tumor cells, Epithelial-mesenchymal transition, Metabolism, Tumor microenvironment
Introduction
The tumor microenvironment (TME) hosts a variety of immune cells, such as macrophages, lymphocytes, neutrophils, and myeloid suppressor cells. Macrophages, originating from bone marrow monocytes, dominate, constituting 30-50% of these immune cells [1]. These macrophages are classified into two main subtypes: M1 macrophages, which effectively eradicate infectious microorganisms and exhibit strong tumoricidal properties, and M2 macrophages, which facilitate tumor epithelial-mesenchymal transition (EMT) and progression [2]. The metabolic reprogramming of tumor-associated macrophages (TAMs) results in the secretion of various cytokines that advance cancer progression by inducing EMT, angiogenesis, metabolic reprogramming, multidrug resistance (MDR), and cancer stem cell (CSC) traits [3].
EMT is a complex, dynamic process where epithelial cells transform into a mesenchymal phenotype, significantly driving tumor metastasis. Several extracellular signaling factors are essential in inducing EMT in tumor cells. Metabolically reprogrammed M2-type TAMs secrete EMT-promoting factors, engage in various signaling pathways, and modulate marker expression—down-regulating epithelial markers and up-regulating mesenchymal markers—thus transforming tumor cells from an epithelial to a mesenchymal phenotype [4, 5].
The interaction of numerous metabolic pathways between TAMs and tumor cells underscores the pivotal role of EMT in cancer progression. The metabolic interaction between tumor cells and TAMs in the TME, along with their intrinsic changes, are critical drivers of tumor progression. The metabolic reprogramming of TAMs and tumor cells is a dynamic, reversible process in cancer progression, involving elements such as metabolic pathways and metabolites. Some of these elements have been utilized in the design and clinical application of targeted drugs.
This review summarizes the regulation of EMT through metabolic codependencies between TAMs and tumor cells, including: (1) the metabolic reprogramming of macrophages in the TME-promoting tumor EMT, (2) the metabolic microenvironment shaped by tumor cells regulating macrophage EMT-promoting activity, (3) macrophages inducing EMT in tumor cells via metabolic reprogramming, and (4) targeting metabolic interactions between TAMs and tumor cells to treat metastatic tumors. To date, various potential therapeutic strategies targeting the metabolic codependencies between TAMs and tumor cells have been confirmed, providing a basis for developing novel clinical anti-tumor combination therapies targeting TAMs and new anti-tumor drugs.
Metabolic reprogramming of TAMs promotes EMT in cancer cells
Metabolic reprogramming, a hallmark of cancer, endows macrophages with tumor-promoting properties. Tumors actively reprogram macrophage metabolism—including glucose, fatty acid, and amino acid metabolism, the TCA cycle, purine metabolism, aerobic metabolism, and autophagy—through metabolites, cytokines, and other signaling mediators. This metabolic reprogramming enhances the recruitment, M2 polarization, and secretion of EMT-related factors by macrophages, thereby promoting tumor EMT and progression (Figure 1).
Metabolic reprogramming of TAMs within the TME enhances their pro-EMT potential. TAMs in the TME undergo diverse metabolic transitions, including glucose utilization, fatty acid catabolism, amino acid metabolism, TCA cycle activities, purine metabolism, aerobic respiration, and autophagy. Such metabolic reconfiguration enables TAMs to shift towards a tumor-promoting M2 phenotype, thereby amplifying their capacity to facilitate EMT.
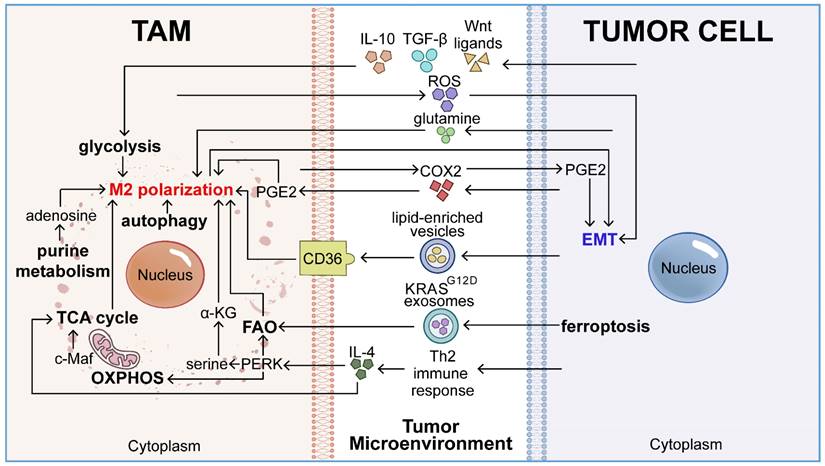
Glucose metabolism
Macrophages are the primary cell population responsible for glucose uptake and metabolism within the TME. Enhanced glucose metabolism in TAMs significantly contributes to the accumulation of tumor metabolites. For instance, lactate and succinate exhibit potent tumor-promoting effects and promote O-GlcNAcylation of lysosomal cathepsin B via O-GlcNAcylation transferase (OGT), thereby facilitating tumor metastasis [6, 7]. In the early stages of pancreatic tumor development, the inflammatory hyperglycemic microenvironment profoundly influences macrophage-mediated EMT in malignant tumors [8]. Under hyperglycemic conditions, benign and premalignant pancreatic ductal epithelial cells (PDEC) exhibit glucose- and macrophage-dependent EMT triggers in a coculture system. This induction is associated with the upregulation of EMT inducers such as interleukin (IL)-6 and tumor necrosis factor-alpha (TNF-α), along with EMT transcription factors, promoting the downregulation of the epithelial marker E-cadherin [8]. Pancreatic ductal adenocarcinoma (PDAC) cells specifically induce DNA methylation and downregulation of glucose metabolism and oxidative phosphorylation (OXPHOS) genes in M1 macrophages, reprogramming them into M2 macrophages to support tumor growth, EMT, and metastasis [9].
Glycolysis is a key pathway of glucose metabolism, and the glycolytic metabolism of macrophages has bidirectional effects on tumorigenesis, exhibiting both tumor-promoting and tumor-inhibiting properties. The aerobic glycolysis in M1 macrophages promotes phagocytosis through reactive oxygen species (ROS) production. Conversely, the glycolytic metabolism of M2 macrophages proceeds at a slower rate under aerobic conditions. M2 macrophages suppress ROS and nitric oxide (NO) production by blocking the glycolysis/pentose phosphate pathway (PPP)/ nicotinamide adenine dinucleotide phosphate (NADPH)/ROS and TCA cycle/L-arginine/ inducible nitric oxide synthase (iNOS)/NO pathways, thereby attenuating the tumoricidal functions of macrophages [10]. Tumor cells can trigger M2 polarization and pro-EMT activities in macrophages by secreting various factors that induce macrophage glycolysis. Tumor-derived soluble factors, such as hyaluronan fragments, promote glycolysis in TAMs by upregulating the glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) [11]. IL-10, transforming growth factor beta (TGF-β), and Wnt ligands from hepatocellular carcinoma (HCC) cells upregulate Wnt2b expression and promote M2 polarization in TAMs by activating the Wnt2b/β-catenin/c-Myc signaling axis and glycolysis [12]. Furthermore, the aerobic glycolysis exhibited by tumor-conditioned macrophages promotes angiogenesis, tumor cell extravasation, EMT, and metastasis in PDAC [13].
Fatty acid metabolism
Fatty acid metabolism is essential for macrophage M2 polarization, with tumor cells manipulating this process to induce a pro-EMT phenotype in macrophages.
Fatty acid intake and oxidation
The scavenger receptor CD36, upregulated in metastasis-associated macrophages (MAMs), plays a significant role in promoting liver metastasis. Deleting CD36 in MAMs markedly reduces liver metastasis in mice. In patients with liver metastases, high CD36 expression correlates with increased M2 MAM infiltration. These MAMs accumulate lipid droplets and uniquely phagocytose long-chain fatty acids from tumor cells, which are transported via extracellular vesicles. These lipid-enriched vesicles are selectively internalized by macrophages through the CD36 receptor, providing the energy necessary for macrophage tumor-promoting activities [14]. IL-4 in the TME induces both macrophage M2 polarization and fatty acid uptake and oxidation [10]. Oxidative stress prompts PDAC cells to secrete the mutant KRAS (KRASG12D) protein via autophagy-dependent ferroptosis. KRASG12D is then transferred to macrophages through exosomes, leading to M2 polarization via a STAT3-dependent FAO mechanism [15]. Recent research also highlights the role of fatty acid binding protein 4 in transferring saturated fatty acids to induce macrophage pyroptosis, mediating the NOD-like receptor thermal protein domain-associated protein 3 (NLRP3)/IL-1β axis, further regulating EMT, and enhancing pancreatic cancer cell metastasis in patients with obesity [16].
Arachidonic acid metabolism
Arachidonic acid, a polyunsaturated fatty acid, plays a pivotal role in the TME. Exosomes released from PDAC cells, such as AsPC-1, increase their fusion rate with THP-1-derived macrophages through the mediation of arachidonic acid, switching these macrophages to the M2 phenotype and triggering the secretion of pro-EMT factors, including VEGF, monocyte chemoattractant protein-1 (MCP-1), IL-6, IL-1β, matrix metalloproteinase (MMP)-9, and TNF-α [17].
Cyclooxygenase 2 (COX-2) converts arachidonic acid into the fatty acid derivative prostaglandin (PG). Primarily secreted by TAMs in the TME, elevated COX-2 expression is associated with poor prognosis in patients with breast cancer. TAM-derived COX-2 promotes its expression in breast cancer cells, thereby enhancing macrophage M2 polarization [18]. PDAC cell-derived exosomes can increase Prostaglandin E2 (PGE2) production, further activating M2 polarization in macrophages [19]. Macrophage-secreted TGF-β can elevate COX-2 expression by upregulating VEGF, connective tissue growth factor (CTGF), hepatocyte growth factor (HGF), fibroblast growth factor (FGF), and TNF-α. In colon cancer, COX-2 overexpression in TAMs can also upregulate TGF-β through a paracrine pathway, activating TGF-β-induced signaling pathways independent of Smads, such as the NF-kappaB (NF-κB) pathway. Furthermore, COX-2 secreted by TAMs induces PGE2 and IL-6 secretion, activating the extracellular signal-regulated kinase (ERK) 1/2, STAT3, β-catenin, and phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT/PKB) signaling pathways. These pathways are pivotal in mediating EMT in breast, colorectal, lung, and osteosarcoma cancer cells [18, 20].
Amino acid metabolism
Amino acid metabolism in macrophages, including the activation of glutamine catabolism and the hexosamine biosynthesis pathway, significantly influences M2 macrophage polarization.
Tumor cell-secreted glutamine induces M2 polarization in macrophages [10]. Upon lipopolysaccharide (LPS) activation, macrophages exhibit increased glutamine consumption and enhanced glycine production. Glycine inhibits LPS-induced NO production and macrophage activation, while glutaminolysis is essential for inducing M2 polarization. M2 macrophages heavily depend on glutamine entry into the TCA cycle and primarily rely on fatty acid β-oxidation and TCA cycling, facilitating the conversion of L-arginine into polyamines and L-proline through arginase 1 (Arg1) to support tumor growth [21].
Helper T cell 2 cytokine IL-4 and the TME potentiate the activation of the protein kinase RNA-like ER kinase (PERK)-signaling cascade in TAMs, enhancing immunosuppressive M2 polarization and proliferation. PERK signaling promotes mitochondrial respiration and FAO to meet cellular energy demands and regulates phosphoserine aminotransferase 1 (PSAT1) activity through ATF4 to mediate the serine biosynthesis pathway. Increased serine biosynthesis enhances mitochondrial function and α-ketoglutarate (α-KG) production. Mitochondrial OXPHOS, FAO, and α-KG drive tumor EMT, metastasis, and progression by inducing M2 polarization in macrophages [22].
TCA cycle
The TCA cycle, prevalent in TAMs, supports pro-tumorigenic bioenergetic functions and phenotypic plasticity within the macrophage population. Due to the slower rate of aerobic glycolysis, M2 macrophages sustain adenosine triphosphate (ATP) production through the TCA cycle, reducing ROS generation and compromising their tumoricidal capabilities [10]. High expression of the transcription factor cellular musculoaponeurotic fibrosarcoma (c-Maf) in TAMs supports their immunosuppressive function. Additionally, c-Maf acts as a metabolic checkpoint regulating the TCA cycle and uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) biosynthesis, promoting M2 macrophage polarization and activation, thereby fostering tumor cell EMT [6]. IL-4-induced polarization of M2 macrophages can interchangeably utilize glucose or tumor-derived lactate as a TCA cycling carbon source. This maximizes adenosine triphosphate-citrate lyase (ACLY)-dependent histone acetylation on M2 gene-specific promoters [23].
Purine metabolism
The lipid metabolism of TAMs can be reprogrammed to purine metabolism within the TME. Upregulation of purine metabolism characterizes TAMs with a pro-tumor and end-differentiation phenotype, correlating with poor responsiveness to immune checkpoint blockade. Tumor-reprogrammed macrophages with high purine metabolism exhibit decreased gene expression levels related to phagocytosis and antigen presentation while displaying elevated levels of immunosuppressive and angiogenic genes [24]. While granulocyte-macrophage colony-stimulating factor (GM-CSF)-dependent macrophages exhibit typical M1 type high stimulatory activity, macrophage colony-stimulating factor (M-CSF)-dependent macrophages adopt an M2 phenotype. This M2 phenotype can result from the purinergic pathway, which directs the release of extracellular ATP and its conversion to adenosine through co-expressed exonucleotidases CD39 and CD73. Adenosine drives these cells toward the M2 state and increases the secretion of EMT-related cytokines, such as IL-1β, IL-6, IL-10, and VEGF [21].
Aerobic metabolism
ROS, an aerobic metabolite of cells, is significantly elevated in macrophages and gastric adenocarcinoma cells compared to normal cells [25]. ROS secreted by macrophages can activate the TNF-α/NF-κB, hypoxia inducible factor (HIF)-1α, TGF-β/Smad, PI3K/AKT/glycogen synthase kinase 3 beta (GSK3β)/β-catenin, nuclear factor erythroid 2-related factor 2 (Nrf2)/Notch1, or ERK signaling pathways, leading to the upregulation of activator protein 1 (AP-1), MMPs, urokinase-type plasminogen activator (uPA), and urokinase-type plasminogen activator receptor (uPAR). Consequently, this triggers the EMT of tumor cells [26].
Autophagy
Autophagy, a key catabolic process, significantly impacts the generation, function, and polarization of macrophages [27]. In monocytes and TAMs, autophagy facilitates monocyte-macrophage differentiation, promotes M2 polarization, enhances tumor-promoting activities, and impairs phagocytosis and antigen presentation, thereby potentiating EMT and metastasis of tumor cells [28].
Tumor cells induce autophagy in monocytes and TAMs by secreting cytokines such as M-CSF, GM-CSF, and IL-4. Specifically, M-CSF alone or GM-CSF combined with IL-4 induces autophagy in monocytes by activating C-Jun NH2-terminal kinase and blocking ATG5 cleavage, promoting M2 differentiation and preventing monocyte apoptosis [29, 30]. Colon cancer-derived cathepsin S stimulates autophagy to induce M2 transformation of TAMs, promoting tumor development. Glioma-derived exosomes enriched in IL-6 and miR-155-3p initiate autophagy and promote M2 polarization in TAMs through the IL-6-p-STAT3-miR-155-3p-autophagy-p-STAT3 positive feedback loop, enhancing glioma progression [31]. Autophagic TAMs secrete TGF-β1 via the fucosyltransferase 4 (FUT4)/p-ezrin pathway, inducing EMT in lung adenocarcinoma cells [32]. In a mouse model of ovarian cancer with peritoneal metastasis, TIM4+ TAMs reduce mTORC1 activity through arginase-1-mediated arginine depletion, exhibiting higher mitochondrial OXPHOS and mitophagy activity, thus promoting ovarian cancer growth and peritoneal metastasis [33]. In breast cancer, lysosome associated membrane protein type 2A (LAMP2a) on the lysosomal membrane of TAMs degrades peroxiredoxin 1 (PRDX1) and CREB-regulated transcription coactivator 1 (CRTC1) via chaperone-mediated autophagy (CMA), reducing ROS generation and activating M2 polarization and tumor-promoting activities [34].
Interestingly, inhibiting macrophage autophagy can also induce tumor cell EMT. Reduced autophagy levels in TAMs within the HCC microenvironment are linked to poor prognosis and increased microvascular metastasis. HCC suppresses macrophage autophagy initiation by upregulating mechanistic target of rapamycin (mTOR) and unc-51-like kinase 1 (ULK1) phosphorylation. The accumulation of the NLRP3 inflammasome due to autophagy inhibition in TAMs promotes the cleavage, maturation, and release of IL-1β, accelerating HCC metastasis by enhancing EMT. Additionally, autophagy inhibition triggers macrophage self-recruitment via the C-C motif chemokine ligand (CCL) 20/C-C chemokine receptor (CCR) 6 signaling axis, further contributing to HCC progression [35]. This paradox highlights the complex role of autophagy in regulating TAMs.
The regulation of tumor metabolic microenvironment on TAM-mediated EMT
The elevated metabolic activities of tumor cells, including hypoxia [36], metabolite secretion [37], aerobic metabolism [26], and autophagy [38], define the unique metabolic landscape of the TME. TAMs, a resilient cell population, remain activated within this distinctive metabolic milieu shaped by tumor cells [39]. This tumor-altered metabolic microenvironment profoundly affects macrophage functions, thereby facilitating EMT and promoting the metastatic progression of tumor cells (Figure 2).
The distinct metabolic microenvironment of tumors profoundly influences TAM-mediated EMT. Elevated metabolic activities within this oncogenic setting—such as metabolite release (lactate, adenosine, α-ketoglutarate, glutamine, and reactive oxygen species), aerobic glycolysis, and autophagy—induce systemic biochemical shifts in the tumor. These changes in the TME promote M2 polarization of TAMs and trigger the secretion of EMT-related mediators (TGF-β1, IL-1β, IL-10, and VEGF). This cascade of events subsequently enhances the activation of EMT in tumor cells.
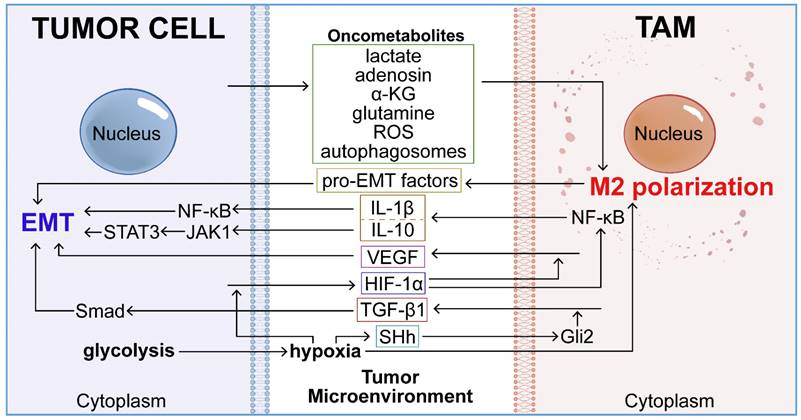
Tumor cell-induced hypoxia
Under aerobic conditions, normal cells produce ATP primarily through OXPHOS, while glycolysis predominantly generates ATP in the absence of oxygen. However, the rapid proliferation of tumor cells increases their energy demands, leading to the preferential use of glycolysis even in the presence of oxygen, a phenomenon known as the Warburg effect or aerobic glycolysis [13]. The combination of insufficient blood supply due to tumor growth and aerobic glycolysis can result in hypoxia [36]. M2 macrophages are more abundant in the hypoxic regions of solid tumors compared to M1 macrophages. The migration of macrophages to these hypoxic regions is driven by soluble factors secreted by the tumor into the circulation, including VEGF, endothelial monocyte-activating polypeptide II (EMAPII), endothelin-2, C-X-C chemokine receptor 4 (CXCR4), and semaphorin3A [40]. Hypoxia in the TME can stimulate the secretion of HIF-1α by tumors and the release of EMT-promoting factors by macrophages.
Hypoxia-induced HIF-1α
TME hypoxia induces tumor cells to secrete the hypoxia-responsive transcription factor HIF-1α. HIF-1α influences both tumor cells and TAMs, stimulating the expression of EMT-related factors such as VEGF [41], histone deacetylase 3 (HDAC3) [42], AXL [43], membrane-type 4 matrix metalloproteinase (MT4-MMP) [44], MMP-9 [36], lysyl oxidase (LOX) [5], COX-2 [18], Twist, Snail1/2, and Zeb1 [42], and activating the Notch [5] and Hedgehog (Hh) [42] pathways. These actions collectively promote EMT in tumor cells. Additionally, hypoxic exosomes derived from pancreatic cancer cells can induce macrophages to adopt an M2 phenotype in a manner dependent on HIF-1α or HIF-2α, further promoting EMT in tumor cells [19].
Hypoxia-induced EMT-related factor secretion of macrophages
Tumor cell-induced hypoxia enhances the secretion of EMT-promoting factors such as TGF-β1, IL-1β, IL-10, and VEGF by macrophages.
TGF-β1 is a vital regulator of EMT. Tumor-induced hypoxia elevates glioma-associated oncogene homolog (Gli2) expression in TAMs through the Sonic Hedgehog (SHh) signaling pathway, inducing M2 polarization of TAMs and increasing TGF-β1 secretion [45]. Hypoxia-induced necrotic fragments of cancer cells, like HCC cells, and HIF-1α secreted by hypoxic tumor cells recruit TLR4 to the macrophage cell membrane, activating the toll-like receptor (TLR) 4/TIR-domain-containing adaptor-inducing interferon-β (TRIF)/NF-κB pathway. This activation upregulates IL-1β and IL-10 expression in macrophages, inducing EMT in breast cancer, pancreatic cancer, HCC, and others [46, 47]. Hypoxic TAMs are a rich source of VEGF, significantly enhancing tumor angiogenesis, exacerbating hypoxia, and promoting EMT [40]. Under hypoxic conditions, HIF-1α/2α triggers the upregulation of VEGF-A expression in TAMs, thereby promoting TAM-induced angiogenesis and metastasis [41].
Tumor acidic metabolites
Aerobic glycolysis in tumor cells produces substantial hydrogen ions and acidic metabolites, creating an acidic microenvironment [37]. In human HCC, macrophages express high levels of carbonic anhydrase XII (CA12). A transient glycolytic activation triggers sustained CA12 expression on tumor-infiltrating monocytes and macrophages via autocrine cytokines and HIF-1α signaling pathways. CA12 enables macrophage survival in the acidic TME and stimulates the production of large amounts of CCL8, facilitating tumor EMT and metastasis [48]. High glycolytic activity in melanoma cells leads to pronounced acidification of the TME, which induces inducible cAMP early repressor (ICER) expression in TAMs, resulting in M2-type polarization and promoting tumor growth [49].
Lactate, an acidic metabolite from tumors, stimulates TAM polarization to the M2 phenotype, promoting tumor EMT and metastasis. In a lactate-rich TME, GM-CSF-activated macrophages undergo M2 polarization and produce large amounts of anti-inflammatory cytokines [37].
Lactate generated within tumor cells interacts with GPR132 [50], promotes TGF-β and VEGF expression, and induces M2 polarization in TAMs via HIF-1α activation, thereby promoting tumor growth [51, 52]. Additionally, lactate stimulates histone lysine lactylation in M1 macrophages, triggering the expression of M2-like homeostatic genes such as VEGF and Arg1, and promoting the transition to the M2 phenotype [53]. Tumor-derived lactic acid induces macrophage M2 polarization by activating the ERK/STAT3 pathway in breast cancer and the MCT/HIF-1α pathway in gastric cancer [54, 55]. Lactate also induces TAM-like phenotypes in macrophages and stimulates CCL5 secretion via Notch signaling. CCL5 promotes aerobic glycolysis in breast cancer cells by modulating AMP-activated protein kinase (AMPK) signaling, further inducing EMT and migration [56]. Lactate secreted by HCC and PDAC cells increases ROS levels in macrophages, inducing M2 polarization and upregulating VEGF expression through Nrf2 activation. In turn, M2 macrophages activate the Nrf2 signaling pathway in cancer cells via paracrine VEGF secretion, inducing tumor EMT [57].
Autophagy in tumor cells
Autophagy in tumor cells can reprogram monocytes and TAMs through various mechanisms, such as secreting autophagosomes, promoting monocyte-to-macrophage differentiation, and reducing the phagocytic and cytotoxic properties of TAMs [58], thereby driving tumor cell EMT. Tumor cell-released autophagosomes (TRAPs) can convert macrophages into an M2-like phenotype via the TLR4/MyD88/p38/STAT3 pathway, subsequently suppressing T cell activation [59]. Interferon-gamma (IFN-γ)-induced autophagy in tumor cells and CSCs leads to the production of arginine resynthesis precursor asymmetric dimethylarginine (ADMA). ADMA impacts macrophages by delaying phagocytosis, reducing proliferation, and NO production, and inducing M2 polarization, thus promoting tumor progression [1]. Inhibition of autophagy in triple-negative breast cancer (TNBC) has been shown to promote ROS-dependent macrophage migration inhibitory factor (MIF) secretion and stimulate M1 polarization [60]. Furthermore, M2 TAMs can promote EMT by suppressing autophagy in tumor cells. For instance, renal cell carcinoma (RCC) can induce M2-type macrophages to secrete CCL2, inhibiting muscleblind-like protein 2 (MBNL2)/B-cell lymphoma 2 (Bcl-2)/beclin 1-mediated autophagy in RCC cells, leading to cell growth and EMT [61].
Other oncometabolites
Beyond acidic metabolites secreted by tumor cells, macrophage reprogramming within the TME is influenced by various oncometabolites such as adenosine, glutamine, α-KG, and ROS. These oncometabolites drive monocyte recruitment, promote M2 macrophage polarization, and inhibit M1 activation, thereby facilitating EMT.
Adenosine promotes M2 macrophage polarization and monocyte migration towards tumor cells [10]. Activation of the Adenosine A2A receptor reduces TNF-α, IL-6, and IL-12 production in macrophages while increasing IL-10, VEGF, and IL-1β levels [21, 62, 63]. Similarly, the A2B receptor decreases TNF-α and IL-12 production and enhances IL-6 and IL-10 production in macrophages [21].
α-KG is a crucial regulator of M2 macrophage activation, enhancing FAO and inducing epigenetic reprogramming of gene expression in M2 macrophages [64]. Additionally, α-KG inhibits M1 activation by upregulating the NF-κB pathway, destabilizing HIF-1α, and accelerating glutamine decomposition, promoting M2 polarization [10].
Hypermetabolic cancer cells utilize ROS regulation, mediated by cell-cell interactions, to select macrophages that enhance their survival and malignancy [25]. Tumor-derived ROS facilitate macrophage recruitment and M2 polarization, with macrophages reciprocally increasing ROS secretion, ultimately promoting tumor metastasis [26]. ROS also activates the transcription factor aryl hydrocarbon receptor (AHR), further recruiting monocytes and activating macrophages [65]. Non-small cell lung carcinoma (NSCLC) activates the ROS/PI3K/AKT axis, releases cytokines such as VEGF-C, CCL7, and IL-8 to recruit macrophages, and upregulates M-CSF expression to drive M2 polarization, thereby facilitating NSCLC progression [66].
TAM-mediated glycolysis-dependent EMT in tumor cells
Aerobic glycolysis in tumor cells induces EMT through various intracellular pathways. TAMs further enhance this process by secreting cytokines that reprogram the aerobic glycolysis of tumor cells, thereby promoting EMT.
The mechanism of glycolysis-dependent EMT in tumor cells
Aerobic glycolysis is associated with various cellular processes, including EMT, angiogenesis, hypoxia, lactate production, macrophage polarization, and T-cell activation [53]. Pathway enrichment analysis has shown significant enrichment of EMT-related signaling pathways in patients with elevated glycolytic activity [67]. The metabolic shift from OXPHOS to glycolysis in tumor cells enhances EMT by upregulating EMT-inducing transcription factors (EMT-TFs), suppressing E-cadherin, promoting MMP secretion, and inducing cytoskeleton remodeling. Glycolytic enzymes such as glucose-6-phosphate isomerase/autocrine motility factor (PGI/AMF) and alpha-enolase (ENO1) play pivotal roles in activating EMT through the regulation of EMT-TFs [68].
TAMs induce glycolysis-dependent EMT in tumor cells by secreting cytokines
TAMs can reprogram the aerobic glycolysis of tumor cells by secreting cytokines like tumor necrosis factor TNF-α, chemokines CCL5 and CCL18, and growth factors VEGF and TGF-β1 [69], thereby inducing EMT (Figure 3).
The roles of TAMs in glycolytic-driven EMT. Activated macrophages secrete key cytokines, including CCL5, CCL18, VEGF, TNF-α, and TGF-β, which interact with various signaling pathways to stimulate glycolysis within tumor cells. This promotion of glycolysis leads to elevated transcription of EMT-TFs through glycolytic enzymes such as PGI/AMF and ENO1, thereby intensifying the EMT process and contributing to tumor metastasis.
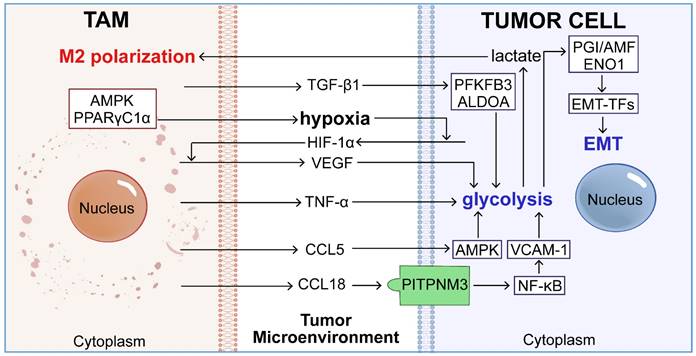
Tumor necrosis factor
Pancreatic cancer cells exhibit a high glycolytic activity, characterized by overexpression of glycolytic enzymes and increased lactate production, partially driven by TAMs [68]. TAMs enhance their own AMPK and peroxisome proliferator-activated receptor-gamma coactivator 1α (PPARγC1α) to promote tumor hypoxia while releasing TNF-α to boost glycolysis in tumor cells [19].
Chemokine
CCL5 secreted by TAMs activates AMPK signaling in breast cancer cells, promoting aerobic glycolysis, thereby inducing EMT and migration [56]. M2 TAMs highly express CCL18, which interacts with PITPNM3 in PDAC cells to activate the NF-kappaB (NF-κB) pathway, resulting in overexpression of vascular cell adhesion molecule-1 (VCAM-1). Elevated VCAM-1 further enhances aerobic glycolysis and lactate secretion in PDAC cells, facilitating macrophage M2 polarization and PDAC progression [70].
Growth factor
M2 TAM-derived exosomal metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) induces aerobic glycolysis in gastric cancer cells by activating the β-catenin and HIF-1α signaling pathways, enhancing their proliferation, metastasis, and chemoresistance in a glycolysis-dependent manner [71]. Glycolytic cancer cells can induce TAMs to express VEGF in a HIF-1α-dependent manner and promote M2 polarization via lactate secretion, further enhancing aerobic glycolysis in PDAC cells [68]. TGF-β1 secreted by TAMs stimulates glycolysis and enhances PANC-1 cell invasion by upregulating the expression of 6-phosphofructo 2-kinase/fructose 2, 6-bisphosphatase 3 (PFKFB3) and the glycolysis gene aldolase A (ALDOA) [68]. Additionally, TGF-β secreted by M2-type TAMs promotes glycolysis in bladder cancer cells via the Smad2/3 pathways, further driving EMT [52].
Targeting metabolic interactions between tumors and TAMs for metastatic tumor therapies
The inhibition of tumor metastasis by blocking EMT has consistently captured the research community's interest, with a growing focus on the intricate interplay between TAMs and tumor cells. Numerous research institutions are currently conducting clinical studies on the metabolic reprogramming of macrophages and tumor cells to treat aggressive tumors (Figure 4).
A variety of antitumor therapeutic modalities targeting metabolic processes in both TAMs and tumor cells have emerged to combat invasive and metastatic malignancies. These treatments reprogram TAMs from a tumorigenic state to an anti-tumor state. Simultaneously, metabolic regulators aimed at cancer cells and their bioproducts can significantly modify the TME, indirectly reducing the EMT-inducing potential of TAMs. These therapeutic strategies offer pathways to inhibit EMT, thereby decelerating tumor progression.
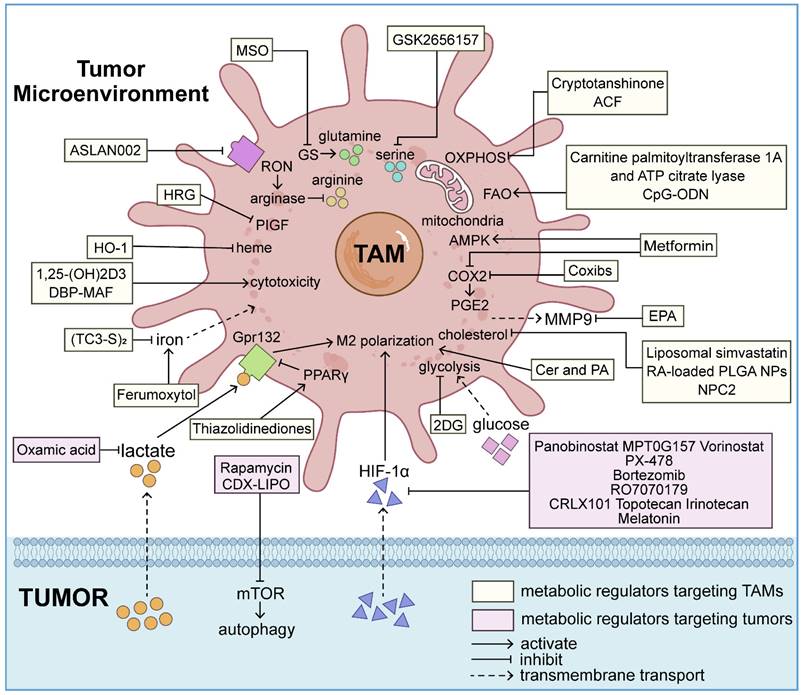
Targeting metabolism of TAMs
Tumor-induced metabolic reprogramming of TAMs enhances their pro-tumorigenic capabilities, including promoting EMT and metastasis. Targeting glucose [13], lipid [72], amino acid [73], mitochondrial [38], iron [74], vitamin [74], heme metabolism [75], and the AMPK metabolic pathway [76] in macrophages presents promising therapeutic strategies for augmenting antitumor activities and inducing TAM M1 polarization (Table 1).
Targeting metabolism of TAMs
| Drug | Research phase of the drug | Types of cancer | Drug-specific properties & Mechanism of inhibiting tumor cell EMT | References |
|---|---|---|---|---|
| 2-deoxy-D-glucose (2DG) | Pre-clinical | Ehrlich ascites cancer, PDAC | HK2 competitive inhibitor; Target: glucose metabolism | PMID: 26597503 PMID: 27622062 |
| Carnitine palmitoyltransferase 1A and ATP citrate lyase | Pre-clinical | PDAC | Enzymes; Target: lipid metabolism | PMID: 31813459 PMID: 30664738 |
| Cytidine-phosphate-guanosine oligodeoxynucleotide (CpG-ODN) | Pre-clinical | PDAC | TLR9 agonist; Target: lipid metabolism | PMID: 30664738 PMID: 33831324 |
| Ceramide (Cer) and palmitic acid (PA) | Pre-clinical | Colorectal cancer, breast cancer | Natural lipids; Target: lipid metabolism | PMID: 32222879 PMID: 35457057 |
| Eicosapentaenoic acid (EPA) | Pre-clinical | Colon cancer | Omega-3 polyunsaturated fatty acid; Target: lipid metabolism | PMID: 26864323 |
| Liposomal simvastatin | Pre-clinical | NSCLC | Liposome and statin based lipid-lowering drug; Target: cholesterol metabolism | PMID: 30662566 |
| Retinoic acid-loaded PLGA nanocarriers | Pre-clinical | Colorectal cancer | Drug delivery system; Target: cholesterol metabolism | PMID: 36857852 |
| Niemann-pick type C2 (NPC2) | Pre-clinical | Lung cancer | Cholesterol-binding protein; Target: cholesterol metabolism | PMID: 26183450 |
| Celecoxib | FDA approval | Bladder cancer, colon cancer, Glioblastoma multiforme | NSAIDs and selective COX-2 inhibitors; Target: fatty acid metabolism (COX-2/PGE2 signal pathway) | PMID: 28096371 PMID: 21730361 PMID: 27693715 |
| Rofecoxib | FDA approval | Glioblastoma multiforme | PMID: 27693715 | |
| Valdecoxib | FDA approval | Glioblastoma multiforme | PMID: 27693715 | |
| Etodolac | FDA approval | Gastrointestinal cancer | PMID: 12483244 | |
| NS-398 | Pre-clinical | Glioblastoma multiforme | PMID: 27693715 | |
| Metformin | FDA approval | Pancreatic cancer, colorectal cancer, breast cancer, prostate cancer | Hypoglycemic drugs; Target: COX-2/PGE2 (fatty acid metabolism) and AMPK signal pathways | PMID: 26641266 PMID: 25552600 PMID: 35740547 PMID: 28157701 PMID: 30012567 |
| ASLAN002/BMS777607 | Phase I clinical trial (NCT01721148) | Breast cancer | RON/MET receptor tyrosine kinase inhibitor; Target: arginine (amino acid metabolism) | PMID: 23612011 |
| Methionine sulfoximine (MSO) | Pre-clinical | Lung carcinoma | GS inhibitor; Target: GS (amino acid metabolism) | PMID: 28813676 |
| GSK2656157 | Pre-clinical | Melanoma | PERK inhibitor; Target: serine (amino acid metabolism) | PMID:35228694 |
| Histidine-rich glycoprotein (HRG) | Pre-clinical | Fibrosarcoma, pancreatic adenocarcinoma | Plasma glycoprotein; Target: histidine (amino acid metabolism) | PMID: 21215706 |
| Cryptotanshinone | Pre-clinical | TNBC | Immunomodulator; Target: mitochondrial metabolism | PMID: 35860009 |
| Acriflavine (ACF) | Pre-clinical | PDAC | Antibacterial drug and metabolic inhibitor of OXPHOS; Target: mitochondrial metabolism | PMID: 32764982 |
| (TC3-S)2 | Pre-clinical | Breast cancer | Intracellular iron chelator; Target: iron metabolism | PMID: 27806101 |
| Ferumoxytol | FDA approval | Early mammary cancer | Iron supplement; Target: iron metabolism | PMID: 28449873 PMID: 27668795 |
| 1,25-dihydroxyvitamin D3 [1,25-(OH)2D3] | FDA approval | Burkitt's lymphoma, breast cancer | Bioactive form of vitamin D; Target: vitamin metabolism | PMID: 25855493 PMID: 24821711 |
| Vitamin D-binding protein-derived macrophage-activating factor (DBP-MAF) | Pre-clinical | Breast cancer | Macrophage-activating factor; Target: vitamin metabolism | PMID: 22213287 |
| Heme oxygenase-1 (HO-1) | Pre-clinical | Prostate cancer | Metabolic enzyme; Target: heme metabolism | PMID: 26418896 |
Glucose metabolism
Inhibition of macrophage glycolysis by the competitive inhibitor hexokinase II (HK2), 2-deoxy-D-glucose (2DG), reduces the production of pro-EMT factors, such as IL-10, M-CSF, and MMP-9, facilitating M1 polarization of TAMs and reversing TAM-mediated angiogenesis, PDAC cell extravasation, and EMT [13, 77]. The 18F-fluoro-2DG (18F-FDG) radiopharmaceutical is widely employed in Positron Emission Tomography/Computed Tomography (PET/CT) examinations [78].
Lipid metabolism
Drugs targeting lipid metabolism are demonstrating efficacy in anti-tumor therapy. Targeting FAO, lipids, cholesterol, and COX2 can reverse macrophage M2 polarization by modulating lipid metabolism in macrophages, thereby inhibiting tumor progression.
Targeting FAO
Targeting peroxisome proliferator-activated receptor-gamma (PPARγ), PPARγ-coactivator-1β (PGC-1β), or STAT6 effectively reverses macrophage M2 polarization by inhibiting the FAO pathway. Combining FAO with fatty acid synthesis enhances macrophage antitumor activity [10]. Carnitine palmitoyltransferase 1A and ATP citrate lyase increase macrophage antitumor activity by promoting the integration of FAO and lipid biosynthesis [79].
Intratumoral injection of TLR agonists increases monocyte recruitment and infiltration, inducing M1 polarization of M2 TAMs [74, 80]. The TLR9 agonist cytidine-phosphate-guanosine oligodeoxynucleotide (CpG-ODN) effectively activates de novo lipid biosynthesis by promoting the shunting of macrophage FAO and TCA cycle intermediates, thereby inducing macrophage phagocytosis of CD47+ cancer cells [79]. CpG-ODN is now recognized as the most potent immunostimulant among known vaccine adjuvants [81].
Targeting lipids
Exogenously applied lipids and lipid analogs have demonstrated anti-tumor activity in several cancers. The combination of natural lipids ceramide (Cer) and palmitic acid (PA) induces macrophage M1 polarization and inhibits M2 macrophage-driven EMT in breast cancer and colorectal cancer [82, 83]. Additionally, an extra-pure formulation of eicosapentaenoic acid (EPA), an omega-3 polyunsaturated fatty acid, as free fatty acid (EPA-FFA), prevents colon cancer EMT and progression in colitis-associated cancer (CAC) by counteracting macrophage-derived MMP-9 upregulation of Notch1 signaling [84].
Targeting cholesterol
Targeting cholesterol metabolism in TAMs can promote M1 polarization and reverse tumor cell EMT. High cholesterol levels in the TME promote greater infiltration of M2 macrophages in breast cancer [85].
Simvastatin, delivered via liposomes, restores sensitivity to paclitaxel in EMT-associated drug-resistant NSCLC cells by inhibiting the cholesterol/lipid raft/integrin β3/FAK pathway. Simvastatin also promotes TAM M1 polarization and suppresses EMT through its negative regulation of the cholesterol-related liver X receptor (LXR)/ATP-binding cassette protein A1 (ABCA1) pathway [72].
Cancer cells' high metabolic activity can amplify IL-4 receptor activity by upregulating macrophage cholesterol efflux transporters, shifting TAMs towards an M2 phenotype. A poly(lactic-co-glycolic acid) (PLGA) nanoparticle (NP)-based drug delivery system loaded with retinoic acid (RA) and coated with cholesterol (CHO) can impede colorectal cancer cell EMT and M2 polarization. The CHO coating enhances membrane fusion capability and directs NPs towards M1 polarizing signals by suppressing cholesterol efflux and retinoid X receptors in TAMs [86].
Premalignant lung adenomas recruit immature macrophage-lineage cells (IMCs) to their stroma through CCR1-mediated pathways, promoting EMT. The recruitment of IMCs is impeded by the cholesterol-binding protein Niemann-Pick type C2 (NPC2), which regulates cholesterol trafficking from the late endosomes/lysosomes to the cytosol. Exogenous NPC2 decreases cholesterol levels in IMCs, suppressing CCR1 ligand CCL6 secretion [87].
Targeting PGE2
TAMs facilitate tumor metastasis by responding to lipid metabolites secreted by cancer cells or TAMs, particularly PGE2. Nonsteroidal anti-inflammatory drugs (NSAIDs) like coxibs (celecoxib, rofecoxib, valdecoxib) [88], etodolac [89], and NS-398 [88], are notable cancer prevention agents that inhibit COX-2 by elevating ROS concentrations, thereby reducing cancer recurrence. Celecoxib effectively reduces PGE2 expression in bladder cancer and promotes M2 polarization of TAMs in colon cancer [90, 91]. Additionally, metformin inhibits TAM infiltration by suppressing the COX2/PGE2 axis, thus slowing prostate cancer progression [92].
Amino acid metabolism
TAMs possess a high catabolic capacity for arginine and tryptophan, which are essential for T cell anti-tumor activity. Replenishing these nutrients by suppressing arginase, indoleamine 2, 3-dioxygenase (IDO), and nitric oxide synthase can enhance T cell survival and function [74]. Recepteur d'Origine Nantais (RON) signaling in macrophages promotes macrophage spreading, phagocytosis, and M2 polarization by stimulating arginase expression and reducing responses to pro-inflammatory stimuli like IFN-γ and LPS [74]. In breast cancer, the RON/MET receptor tyrosine kinase inhibitor BMS-777607/ASLAN002 increases the abundance of pro-inflammatory macrophages and reduces lung metastasis [93].
Glutamine catabolism in macrophages stimulates M2 polarization. Methionine sulfoximine (MSO) inhibits glutamine synthase (GS), diverting glucose rather than glutamine into the TCA cycle, inducing M1 polarization and preventing lung carcinoma metastasis [94]. The PERK inhibitor GSK2656157 suppresses macrophage serine biosynthesis and immunosuppressive activity, delaying melanoma growth [22]. Histidine-rich glycoprotein (HRG) induces M1 polarization of TAMs and promotes vascular normalization by downregulating placental growth factor (PLGF) [73].
Mitochondrial metabolism
Treatment of M2 TAMs with the immunomodulator cryptotanshinone activates apoptosis signal-regulating kinase 1 (ASK1), subsequently stimulating autophagy and promoting M1 polarization. This process impairs mitochondrial OXPHOS and fusion in M2 macrophages, reduces their oxygen consumption rate, and stimulates NO and ROS production, thereby inhibiting the proliferation and motility of TNBC cells [38]. In vitro studies have shown that the antibacterial agent and OXPHOS metabolic inhibitor acriflavine (ACF) reduces pancreatic cancer EMT and invasion, and shifts macrophages to a M1-like phenotype [95].
Iron metabolism
The divergent iron metabolism in M1 and M2 macrophages is notable. M1 macrophages exhibit heightened ferritin expression, promoting intracellular iron retention. In contrast, M2 macrophages show increased ferroportin expression, promoting iron efflux and providing tumor cells with the iron necessary for proliferation [74]. The intracellular iron chelator (TC3-S)2 has been shown to reverse the iron-processing function of M2 macrophages from iron release to sequestration, blocking their tumor-promoting effects [96]. Additionally, external iron supplementation with ferumoxytol has demonstrated therapeutic efficacy in slowing the metastasis of early mammary cancers by stimulating M1 macrophage polarization and ROS production [97].
Vitamin metabolism
Vitamin D influences the tumor-promoting and anti-tumor activities of macrophages through various mechanisms. Vitamin D-1-hydroxylase CYP27B1, expressed at lower levels in TAMs, catalyzes the conversion of 25-hydroxyvitamin D (25D) to 1, 25-dihydroxyvitamin D3 [1, 25-(OH)2D3]. Macrophages secrete the host-defense peptide cathelicidin, which effectively lyses proliferating B cell lymphoma cells. Stimulation of M2 macrophages with 1, 25-(OH)2D3 restores cathelicidin production and cytotoxicity against B-cell lymphoma [98]. Overexpression of the Vitamin D receptor (VDR) can prevent EMT in breast cancer cells co-cultured with macrophages. In breast cancer cells, macrophage-secreted TNF-α inhibits VDR expression. Administration of calcitriol [1, 25-(OH)2D3] mitigates macrophage-induced EMT and metastasis of breast cancer cells by preserving VDR [99]. Vitamin D-binding protein-derived macrophage-activating factor (DBP-MAF) stimulates macrophages, downregulates vimentin expression, and reverses EMT in breast cancer cells [100].
Heme metabolism
Heme oxygenase-1 (HO-1), a key metabolic enzyme for heme degradation, restricts the differentiation and polarization of TAMs. The HO-1-derived catalytic product, carbon monoxide (CO), from TAMs, enhances mitochondrial activity in cancer cells, leading to increased E-cadherin levels and inhibiting EMT and prostate cancer progression [75]. Therefore, therapeutic strategies targeting TAM modulation via HO-1 could yield significant anti-tumor effects.
AMPK metabolic pathway
AMPK, a serine/threonine protein kinase, functions as a central metabolic sensor in cellular energy homeostasis [101]. Metformin enhances AMPK activity in macrophages, reduces STAT3 phosphorylation, and promotes mTOR suppression, thereby slowing monocyte transformation into macrophages, reducing TAM infiltration, and inhibiting M2 polarization [76, 102, 103]. Metformin's influence on the AMPK pathway in macrophages decreases the expression of several genes in PDAC involved in ECM remodeling (including MMPs) and EMT [76]. However, the specific regulatory mechanisms of metformin on macrophage metabolism via the AMPK pathway remain unclear.
Targeting metabolism of tumor cells
Metabolic alterations within tumor cells can induce macrophage recruitment, M2 polarization, and secretion of EMT-related factors. Targeting metabolic pathways such as hypoxia, metabolite secretion, and autophagy in tumor cells can reverse macrophage manipulation by the metabolic microenvironment, effectively preventing cancer EMT and progression (Table 2).
Targeting metabolism of tumor cells
| Drug | Research phase of the drug | Types of cancer | Drug-specific properties & Mechanism of inhibiting tumor cell EMT | References |
|---|---|---|---|---|
| Panobinostat | FDA approval | Hodgkin lymphoma | HDAC inhibitors; Target: HIF-1α (tumor hypoxia) | PMID: 22408261 |
| MPT0G157 | Pre-clinical | Colorectal cancer | PMID: 26087180 | |
| Vorinostat | FDA approval | T-cell lymphoma | PMID: 17438089 | |
| PX-478 | Phase I clinical trial (NCT00522652) | Prostate cancer, breast cancer, colorectal adenocarcinoma, pancreatic cancer | HIF-1α inhibitor; Target: HIF-1α (tumor hypoxia) | PMID: 18202012 |
| Bortezomib | FDA approval | Multiple myeloma | Proteasome inhibitor; Target: HIF-1α (tumor hypoxia) | PMID: 14657528 |
| RO7070179 (EZN-2968) | Phase I clinical trial (NCT02564614) | Advanced HCC | ASO; Target: HIF-1α (tumor hypoxia) | PMID: 30949444 |
| CRLX101 | Phase II clinical trial (NCT03531827) | Rectal cancer | Topoisomerase I inhibitor camptothecin and its analogs; Target: HIF-1α (tumor hypoxia) | PMID: 27784746 |
| Topotecan | FDA approval | TNBC | PMID: 26623560 | |
| Irinotecan | FDA approval | Malignant glioma | PMID: 20066473 | |
| Melatonin | FDA approval | Prostate cancer | Hormonal drug; Target: HIF-1α (tumor hypoxia) | PMID: 21392092 |
| Oxamic acid | Pre-clinical | Breast cancer | Lactate dehydrogenase inhibitor; Target: lactate (tumor metabolite) | PMID: 24823638 |
| Thiazolidinediones | FDA approval | Breast cancer | PPARγ agonists Target: lactate (tumor metabolite) | PMID: 27692066 |
| Rapamycin | FDA approval | GBM, RCC | Autophagy activator and mtor inhibitor; Target: autophagy (catabolism) | PMID: 32020472 PMID: 36244911 |
| Liposomal honokiol and disulfiram/copper codelivery system (CDX-LIPO) | Pre-clinical | GBM | Liposome; Target: autophagy (catabolism) | PMID: 32817393 |
Targeting tumor hypoxia
TME hypoxia induces HIF-1α secretion in tumor cells, regulating EMT by enhancing TAM functions. HDAC inhibitors (including panobinostat, MPT0G157, vorinostat), the HIF-1α inhibitor PX-478, proteasome inhibitor bortezomib [104], antisense oligonucleotide (ASO) RO7070179 (EZN-2968), camptothecin and its analogs (including CRLX101, topotecan, irinotecan), and melatonin [105] inhibit HIF-1α expression through various strategies. Additionally, exosomes derived from M1 macrophages can be engineered to express catalase in the membrane or carry DNA damage repair inhibitors, effectively alleviating tumor hypoxia and enhancing tumor DNA damage [106].
Targeting tumor metabolites
Specific targeting of the tumor metabolite lactate is currently being explored in preclinical cancer models. Lactate recognition by the G-protein coupled receptor Gpr132 on TAMs promotes M2 polarization, while PPARγ transcription factor suppresses Gpr132 expression [74]. Inhibition of lactate production by pretreating MDA-MB-231 cells with the lactate dehydrogenase inhibitor oxamic acid can shift the polarization of tumor-derived GM-CSF-stimulated macrophages towards a pro-inflammatory phenotype [37].
PPARγ agonists, such as thiazolidinediones, or siRNA silencing of Gpr132 have shown efficacy in rendering TAMs insensitive to lactate stimulation, significantly impeding breast cancer progression [107].
Targeting tumor cell autophagy
Combining the autophagy activator rapamycin with hydroxychloroquine (HCQ) reduces macrophage M2 polarization and lowers CD47 and SIRPα expression levels in tumor cells and macrophages. This enhances macrophage phagocytic ability and increases glioblastoma (GBM) sensitivity to immune checkpoint inhibitors (ICIs). Rapamycin also reverses M2 macrophage-induced autophagy inhibition and EMT in RCC cells [61, 108]. Rapamycin has been approved by the FDA for managing advanced RCC [109]. Liposomal honokiol and the disulfiram/copper codelivery system (CDX-LIPO) significantly stimulate autophagy in GBM cells by interfering with the mTOR pathway, inhibiting aerobic glycolysis and lactic acid production, thereby converting M2 TAMs to the M1 type [110].
Conclusions and Outlooks
Understanding and studying the effects of metabolic pathways that connect and diversify TAMs and tumor cells on EMT is essential for identifying potential TAM-associated immunotherapy targets and enhancing therapeutic efficacy of metastatic tumors. This review elucidates the metabolic crosstalk between TAMs and tumor cells within the TME, presenting a novel approach for immunotherapy of metastatic tumors.
Despite these advancements, several obstacles and challenges remain in developing and clinically implementing anti-tumor drugs targeting TAMs or tumor cell metabolism. Innovative research methodologies and strategies may provide solutions to these issues. This prospective analysis summarizes the existing challenges and potential strategies to overcome them.
Identification and selection of metabolic therapeutic targets for TAMs or tumor cells
Tumors and TAMs exhibit a vast and diverse array of metabolic pathways and gene expression profiles, with significant variations in cellular metabolic states observed among different patients. One of the primary challenges in the clinical application of metabolic-related treatments is identifying metabolic targets that can be broadly applied across patients with various cancers, while also developing personalized treatment strategies based on individual metabolic profiles.
Investigating the metabolic reprogramming of TAMs across different cancer types and disease stages is a prevalent research strategy, utilizing techniques such as single-cell sequencing, transcriptome sequencing, gene enrichment analysis, qPCR, and metabolomics [111-113]. The integration of these emerging research technologies allows for a comprehensive understanding of the alterations in TAMs' metabolic genes and their role in tumor metastasis on a patient-by-patient basis. This approach can identify key metabolic regulatory genes and metabolites that are applicable to populations with distinct cancer types. Additionally, it supports the development of personalized treatment strategies targeting the metabolism of patients with diverse cancers.
The safety considerations for TAMs or targeted metabolic therapy in tumor cells
Despite normal cells from different tissues exhibiting varied metabolic profiles, cancer cells also show significant metabolic diversity and plasticity. However, few metabolic characteristics consistently distinguish cancer cells from normal cells [114]. Both normal and tumor tissues produce numerous metabolites, and the same metabolite can play diverse roles in different tissues or organs [115]. Therefore, precisely targeting TAMs or tumor cells with metabolic regulators and minimizing adverse effects on normal tissue metabolism presents a substantial challenge for the clinical application of this anti-tumor strategy.
Developing meticulously targeted metabolic regulators that specifically engage TAMs or tumor cells and circumvent the crucial metabolic products in normal cells is essential. This issue might be addressed by utilizing exosomes as targeted drug carriers to TAMs or tumor cells. Exosomes, natural drug delivery vehicles, can be genetically engineered and chemically modified to enhance their targeting capacity, thereby mitigating medication side effects [116]. Other strategies based on small molecules, ligands, peptides, antibodies, and cells for targeted drug delivery also exist and can help mitigate the off-target effects of drugs [117].
The dynamic fluctuations of TAMs or tumor cell metabolites
The abundance of cellular metabolites fluctuates dynamically, heavily influenced by the nutritional environment and cellular context of cancer cells [115]. This variation spans both temporal and spatial dimensions. Therefore, a comprehensive understanding of the changing patterns of tumor metabolites across different cancer types and stages is essential. Additionally, metabolic modulators should be assessed through experimental pharmacokinetic trials to determine the optimal dosage, timing, and interval for administration that would best disrupt the pro-tumor metabolic interplay between TAMs and tumor cells.
Currently, various detection techniques enable the dynamic monitoring of metabolite levels in TAMs and tumor cells. Metabolomics, for example, involves the comprehensive quantification of diverse metabolites, including nutrients, drugs, signaling intermediates, and metabolic byproducts in blood, urine, tissue extracts, or other body fluids. Commonly employed metabolomics techniques include LC-MS, gas chromatography-mass spectrometry (GC-MS), and nuclear magnetic resonance (NMR) [118, 119]. These techniques facilitate the real-time monitoring of changes in metabolites, aiding in the determination of optimal administration timing.
Given the spatial heterogeneity of tumor metabolites, it is necessary to study the distribution of therapeutic drugs and their corresponding metabolites in both tumor and normal tissues to ensure accurate targeting. Molecular spectral imaging (MSI) is an innovative technique for constructing molecular images, precisely depicting the distribution of drugs and metabolites within tissues and their substructures. The most extensively utilized ionization technique in MSI research is matrix-assisted laser desorption/ionization mass spectrometry (MALDI), which provides information about the penetration of drugs and metabolites into target organs [120].
In conclusion, this review systematically describes the metabolic symbiosis between TAMs and tumor cells in regulating tumor EMT, offering a novel perspective for the development of potential TAM- or tumor-targeted metabolic drugs. It lays the theoretical groundwork for formulating clinical treatment strategies for metastatic tumors and aids subsequent researchers in deciphering novel molecular mechanisms involved.
Abbreviations
TAMs: Tumor-associated macrophages; TCA: Tricarboxylic acid; TME: Tumor microenvironment; EMT: Epithelial mesenchymal transition; MDR: Multidrug resistance; CSC: Cancer stem cell; OGT: O-GlcNAcylation transferase; PDEC: Pancreatic ductal epithelial cells; IL: Interleukin; TNF-α: Tumor necrosis factor alpha; PDAC: Pancreatic ductal adenocarcinoma; DNA: Deoxyribonucleic acid; OXPHOS: Oxidative phosphorylation; ROS: Reactive oxygen species; NO: Nitric oxide; PPP: Pentose phosphate pathway; NADPH: Nicotinamide adenine dinucleotide phosphate; iNOS: Inducible nitric oxide synthase; PFKFB3: 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; TGF-β: Transforming growth factor beta; HCC: Hepatocellular carcinoma; MAMs: Metastasis-associated macrophages; STAT: Signal transducer and activator of transcription; FAO: Fatty acid oxidation; NLRP3: NOD-like receptor thermal protein domain associated protein 3; VEGF: Vascular endothelial growth factor; MCP-1: Monocyte chemoattractant protein-1; MMP: Matrix metalloproteinase; COX-2: Cyclooxygenase 2; PG: Prostaglandins; PGE2: Prostaglandin E2; CTGF: Connective tissue growth factor; HGF: Hepatocyte growth factor; FGF: Fibroblast growth factor; NF-κB: NF-kappaB; ERK: Extracellular signal-regulated kinase; PI3K: Phosphatidylinositol-3-kinase; AKT/PKB: Protein kinase B; LPS: Lipopolysaccharide; Arg1: Arginase 1; PERK: Protein kinase RNA-like ER kinase; PSAT1: Phosphoserine aminotransferase 1; α-KG: Alpha-ketoglutarate; ATP: Adenosine triphosphate; c-Maf: Cellular musculoaponeurotic fibrosarcoma; UDP-GlcNAc: Uridine diphosphate N-acetylglucosamine; ACLY: Adenosine triphosphate-citrate lyase; GM-CSF: Granulocyte-macrophage colony-stimulating factor; M-CSF: Macrophage colony-stimulating factor; HIF: Hypoxia inducible factor; GSK3β: Glycogen synthase kinase 3 beta; Nrf2: Nuclear factor erythroid 2-related factor 2; AP-1: Activator protein 1; uPA: Urokinase-type plasminogen activator; uPAR: Urokinase-type plasminogen activator receptor; ATG5: Autophagy related 5; FUT4: Fucosyltransferase 4; mTORC1: Mechanistic target of rapamycin complex 1; LAMP2a: Lysosome associated membrane protein type 2A; PRDX1: Peroxiredoxin 1; CRTC1: CREB-regulated transcription coactivator 1; CMA: Chaperone-mediated autophagy; mTOR: Mechanistic target of rapamycin; ULK1: Unc-51-like kinase 1; CCL: C-C motif chemokine ligand; CCR: C-C chemokine receptor; EMAPII: Endothelial monocyte-activating polypeptide II; CXCR4: C-X-C chemokine receptor 4; HDAC: Histone deacetylase; MT4-MMP: Membrane-type 4 matrix metalloproteinase; LOX: Lysyl oxidase; Hh: Hedgehog; Gli: Glioma-associated oncogene homolog; SHh: Sonic Hedgehog; TLR: Toll-like receptor; TRIF: TIR domain containing adaptor molecule 1; CA12: Carbonic anhydrase XII; ICER: Inducible camp early repressor; MCT: Monocarboxylate channel transporter; AMPK: AMP-activated protein kinase; TRAPs: Tumor cell-released autophagosomes; IFN-γ: Interferon gamma; ADMA: Arginine resynthesis precursor asymmetric dimethylarginine; TNBC: Triple-negative breast cancer; MIF: Macrophage migration inhibitory factor; RCC: Renal cell carcinoma; MBNL2: Muscleblind-like protein 2; Bcl-2: B-cell lymphoma 2; AHR: Aryl hydrocarbon receptor; NSCLC: Non-small cell lung carcinoma; EMT-TFs: EMT-inducing transcription factors; PGI/AMF: Phosphoglucose isomerase/autocrine motility factor; ENO1: Enolase 1; PPARγC1α: Peroxisome proliferator-activated receptor-gamma coactivator 1α; VCAM-1: Vascular cellular adhesion molecule-1; MALAT1: Metastasis-associated lung adenocarcinoma transcript 1; ALDOA: Aldolase, fructose-bisphosphate A; HK2: Hexokinase II; 2DG: 2-deoxy-D-glucose; 18F-FDG: 18F-fluoro-2DG; PET/CT: Positron emission tomography/computed tomography; PPARγ: Peroxisome proliferator-activated receptor-gamma; PGC-1β: PPARγ-coactivator-1β; CpG-ODN: Cytidine-phosphate-guanosine oligodeoxynucleotide; Cer: Ceramide; PA: Palmitic acid; EPA: Eicosapentaenoic acid; FFA: Free fatty acid; CAC: Colitis-associated cancer; LXR: Liver X receptor; ABCA1: ATB-binding cassette protein A1; PLGA: Poly(lactic-co-glycolic acid) ; NP: Nanoparticle; RA: Retinoic acid; CHO: Cholesterol; IMCs: Immature macrophage-lineage cells; NPC2: Niemann-pick type C2; NSAIDs: Nonsteroidal anti-inflammatory drugs; IDO: Indoleamine 2, 3-dioxygenase; RON: Recepteur d'origine nantais; MSO: Methionine sulfoximine; GS: Glutamine synthase; HRG: Histidine-rich glycoprotein; PLGF: Placental growth factor; ASK1: Apoptosis signal-regulating kinase 1; ACF: Acriflavine; 1,25-(OH)2D3: 1,25-dihydroxyvitamin D3; VDR: Vitamin D receptor; DBP-MAF: Vitamin D-binding protein-derived macrophage-activating factor; HO-1: Heme oxygenase-1; CO: Carbon monoxide; ASO: Antisense oligonucleotide; HCQ: Hydroxychloroquine; GBM: Glioblastoma; ICIs: Immune checkpoint inhibitors; CDX-LIPO: Liposomal honokiol and disulfiram/copper codelivery system; qPCR: Quantitative real-time PCR; GC-MS: Gas chromatography-mass spectrometry; NMR: Nuclear magnetic resonance; MSI: Molecular spectral imaging; MALDI: Matrix-assisted laser desorption/ionization mass spectrometry.
Acknowledgements
Funding
This work was supported by National Natural Science Foundation of China (No.: 82003164, 82471378, 82273207, 82304535, 82204541 and 81972719), Jiangsu Province Natural Science Foundation (No.: BK20210910, BK20210913, and BK20220671), National science research in Universities of Jiangsu Province (No.: 21KJA320008 and 22KJB320009).
Author contributions
Xuan Zhao and Tong Ren wrote the main manuscript text, prepared figures and edited the manuscript. Sijin Li, Xu Wang, Rui Hou and Zhangchun Guan organized and draw tables. Dan Liu, Junnian Zheng and Ming Shi conceptualized, supervised, obtained funding, reviewed and edited the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen YL, Lowery AT, Lin S, Walker AM, Chen KE. Tumor cell-derived asymmetric dimethylarginine regulates macrophage functions and polarization. Cancer Cell Int. 2022;22:351
2. Italiani P, Boraschi D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Frontiers in immunology. 2014;5:514
3. Pan Y, Yu Y, Wang X, Zhang T. Corrigendum: Tumor-Associated Macrophages in Tumor Immunity. Frontiers in immunology. 2021;12:775758
4. Zhang J, Yao H, Song G, Liao X, Xian Y, Li W. Regulation of epithelial-mesenchymal transition by tumor-associated macrophages in cancer. Am J Transl Res. 2015;7:1699-711
5. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7:re8
6. Liu J, Cao X. Glucose metabolism of TAMs in tumor chemoresistance and metastasis. Trends Cell Biol. 2023;33:967-78
7. Shi Q, Shen Q, Liu Y, Shi Y, Huang W, Wang X. et al. Increased glucose metabolism in TAMs fuels O-GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer Cell. 2022;40:1207-22 e10
8. Otto L, Rahn S, Daunke T, Walter F, Winter E, Moller JL. et al. Initiation of Pancreatic Cancer: The Interplay of Hyperglycemia and Macrophages Promotes the Acquisition of Malignancy-Associated Properties in Pancreatic Ductal Epithelial Cells. International journal of molecular sciences. 2021;22:5086
9. Zhang M, Pan X, Fujiwara K, Jurcak N, Muth S, Zhou J. et al. Pancreatic cancer cells render tumor-associated macrophages metabolically reprogrammed by a GARP and DNA methylation-mediated mechanism. Signal Transduct Target Ther. 2021;6:366
10. Mehla K, Singh PK. Metabolic Regulation of Macrophage Polarization in Cancer. Trends in cancer. 2019;5:822-34
11. Chen DP, Ning WR, Jiang ZZ, Peng ZP, Zhu LY, Zhuang SM. et al. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3-PD-L1 axis in human hepatocellular carcinoma. J Hepatol. 2019;71:333-43
12. Jiang Y, Han Q, Zhao H, Zhang J. Promotion of epithelial-mesenchymal transformation by hepatocellular carcinoma-educated macrophages through Wnt2b/beta-catenin/c-Myc signaling and reprogramming glycolysis. J Exp Clin Cancer Res. 2021;40:13
13. Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi Ee P, San Luis B. et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology. 2016;5:e1191731
14. Yang P, Qin H, Li Y, Xiao A, Zheng E, Zeng H. et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat Commun. 2022;13:5782
15. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ. et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16:2069-83
16. Yang J, Liu S, Li Y, Fan Z, Meng Y, Zhou B. et al. FABP4 in macrophages facilitates obesity-associated pancreatic cancer progression via the NLRP3/IL-1beta axis. Cancer letters. 2023;575:216403
17. Closa D. Pancreatic cancer, stroma, and exosomes. J Physiol Biochem. 2023;79:205-11
18. Gomez-Valenzuela F, Escobar E, Perez-Tomas R, Montecinos VP. The Inflammatory Profile of the Tumor Microenvironment, Orchestrated by Cyclooxygenase-2, Promotes Epithelial-Mesenchymal Transition. Front Oncol. 2021;11:686792
19. Shao X, Hua S, Feng T, Ocansey DKW, Yin L. Hypoxia-Regulated Tumor-Derived Exosomes and Tumor Progression: A Focus on Immune Evasion. Int J Mol Sci. 2022;23:11789
20. Che D, Zhang S, Jing Z, Shang L, Jin S, Liu F. et al. Corrigendum to "Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE(2)/beta-catenin signalling pathway" [Mol. Immunol. 90 (2017) 197-210]. Mol Immunol. 2020;126:165-6
21. Ohradanova-Repic A, Machacek C, Charvet C, Lager F, Le Roux D, Platzer R. et al. Extracellular Purine Metabolism Is the Switchboard of Immunosuppressive Macrophages and a Novel Target to Treat Diseases With Macrophage Imbalances. Frontiers in immunology. 2018;9:852
22. Raines LN, Zhao H, Wang Y, Chen HY, Gallart-Ayala H, Hsueh PC. et al. PERK is a critical metabolic hub for immunosuppressive function in macrophages. Nat Immunol. 2022;23:431-45
23. Noe JT, Rendon BE, Geller AE, Conroy LR, Morrissey SM, Young LEA. et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci Adv. 2021;7:eabi8602
24. Li S, Yu J, Huber A, Kryczek I, Wang Z, Jiang L. et al. Metabolism drives macrophage heterogeneity in the tumor microenvironment. Cell Rep. 2022;39:110609
25. Sung JY, Cheong JH. Single Cell Analysis Reveals Reciprocal Tumor-Macrophage Intercellular Communications Related with Metabolic Reprogramming in Stem-like Gastric Cancer. Cells. 2022;11:2373
26. Huang R, Chen H, Liang J, Li Y, Yang J, Luo C. et al. Dual Role of Reactive Oxygen Species and their Application in Cancer Therapy. J Cancer. 2021;12:5543-61
27. Santoni M, Massari F, Amantini C, Nabissi M, Maines F, Burattini L. et al. Emerging role of tumor-associated macrophages as therapeutic targets in patients with metastatic renal cell carcinoma. Cancer Immunol Immunother. 2013;62:1757-68
28. Duan Y, Tian X, Liu Q, Jin J, Shi J, Hou Y. Role of autophagy on cancer immune escape. Cell Commun Signal. 2021;19:91
29. Jacquel A, Obba S, Boyer L, Dufies M, Robert G, Gounon P. et al. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood. 2012;119:4527-31
30. Zhang Y, Morgan MJ, Chen K, Choksi S, Liu ZG. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood. 2012;119:2895-905
31. Fan Y, Wang Y, Zhang J, Dong X, Gao P, Liu K. et al. Breaking Bad: Autophagy Tweaks the Interplay Between Glioma and the Tumor Immune Microenvironment. Front Immunol. 2021;12:746621
32. Wang K, Chen X. Autophagic tumor-associated macrophages promote the endothelial mesenchymal transition in lung adenocarcinomas through the FUT4/p-ezrin pathway. J Thorac Dis. 2021;13:5973-85
33. Xia H, Li S, Li X, Wang W, Bian Y, Wei S. et al. Autophagic adaptation to oxidative stress alters peritoneal residential macrophage survival and ovarian cancer metastasis. JCI Insight. 2020;5:e141115
34. Wang R, Liu Y, Liu L, Chen M, Wang X, Yang J. et al. Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. EBioMedicine. 2019;40:118-34
35. Gao Z, Feng SR, Chen JF, Li XG, Shi YH, Tang Z. et al. Inhibition of autophagy in macrophage promotes IL-1beta-mediated hepatocellular carcinoma progression via inflammasome accumulation and self-recruitment. Biomed Pharmacother. 2023;161:114560
36. Dehne N, Mora J, Namgaladze D, Weigert A, Brune B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr Opin Pharmacol. 2017;35:12-9
37. Su S, Liu Q, Chen J, Chen J, Chen F, He C. et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. 2014;25:605-20
38. Yen JH, Huang WC, Lin SC, Huang YW, Chio WT, Tsay GJ. et al. Metabolic remodeling in tumor-associated macrophages contributing to antitumor activity of cryptotanshinone by regulating TRAF6-ASK1 axis. Mol Ther Oncolytics. 2022;26:158-74
39. Dubey S, Ghosh S, Goswami D, Ghatak D, De R. Immunometabolic attributes and mitochondria-associated signaling of Tumor-Associated Macrophages in tumor microenvironment modulate cancer progression. Biochem Pharmacol. 2023;208:115369
40. McDonald PC, Chafe SC, Dedhar S. Overcoming Hypoxia-Mediated Tumor Progression: Combinatorial Approaches Targeting pH Regulation, Angiogenesis and Immune Dysfunction. Front Cell Dev Biol. 2016;4:27
41. Song W, Mazzieri R, Yang T, Gobe GC. Translational Significance for Tumor Metastasis of Tumor-Associated Macrophages and Epithelial-Mesenchymal Transition. Front Immunol. 2017;8:1106
42. Tam SY, Wu VWC, Law HKW. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1alpha and Beyond. Front Oncol. 2020;10:486
43. Ireland L, Luckett T, Schmid MC, Mielgo A. Blockade of Stromal Gas6 Alters Cancer Cell Plasticity, Activates NK Cells, and Inhibits Pancreatic Cancer Metastasis. Front Immunol. 2020;11:297
44. Huang CH, Yang WH, Chang SY, Tai SK, Tzeng CH, Kao JY. et al. Regulation of membrane-type 4 matrix metalloproteinase by SLUG contributes to hypoxia-mediated metastasis. Neoplasia. 2009;11:1371-82
45. Chen Z, Li H, Li Z, Chen S, Huang X, Zheng Z. et al. SHH/GLI2-TGF-beta1 feedback loop between cancer cells and tumor-associated macrophages maintains epithelial-mesenchymal transition and endoplasmic reticulum homeostasis in cholangiocarcinoma. Pharmacol Res. 2023;187:106564
46. Li X, Chen L, Peng X, Zhan X. Progress of tumor-associated macrophages in the epithelial-mesenchymal transition of tumor. Front Oncol. 2022;12:911410
47. Liu CY, Xu JY, Shi XY, Huang W, Ruan TY, Xie P. et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest. 2013;93:844-54
48. Ning WR, Jiang D, Liu XC, Huang YF, Peng ZP, Jiang ZZ. et al. Carbonic anhydrase XII mediates the survival and prometastatic functions of macrophages in human hepatocellular carcinoma. J Clin Invest. 2022;132:e153110
49. Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N. et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. 2018;19:1319-29
50. Liu Y, Jiang C, Xu C, Gu L. Systematic analysis of integrated bioinformatics to identify upregulated THBS2 expression in colorectal cancer cells inhibiting tumour immunity through the HIF1A/Lactic Acid/GPR132 pathway. Cancer Cell Int. 2023;23:253
51. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559-63
52. Shen C, Liu J, Jiao W, Zhang X, Zhao X, Yang X. et al. A feed-forward loop based on aerobic glycolysis and TGF-beta between tumor-associated macrophages and bladder cancer cells promoted malignant progression and immune escape. J Cancer Res Clin Oncol. 2023;149:12867-80
53. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y. et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575-80
54. Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B. et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. 2018;17:428-38
55. Zhang L, Li S. Lactic acid promotes macrophage polarization through MCT-HIF1alpha signaling in gastric cancer. Exp Cell Res. 2020;388:111846
56. Lin S, Sun L, Lyu X, Ai X, Du D, Su N. et al. Lactate-activated macrophages induced aerobic glycolysis and epithelial-mesenchymal transition in breast cancer by regulation of CCL5-CCR5 axis: a positive metabolic feedback loop. Oncotarget. 2017;8:110426-43
57. Feng R, Morine Y, Ikemoto T, Imura S, Iwahashi S, Saito Y. et al. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun Signal. 2018;16:54
58. Luo X, Qiu Y, Dinesh P, Gong W, Jiang L, Feng X. et al. The functions of autophagy at the tumour-immune interface. J Cell Mol Med. 2021;25:2333-41
59. Wen ZF, Liu H, Gao R, Zhou M, Ma J, Zhang Y. et al. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J Immunother Cancer. 2018;6:151
60. Cotzomi-Ortega I, Nieto-Yanez O, Juarez-Avelar I, Rojas-Sanchez G, Montes-Alvarado JB, Reyes-Leyva J. et al. Autophagy inhibition in breast cancer cells induces ROS-mediated MIF expression and M1 macrophage polarization. Cellular signalling. 2021;86:110075
61. He C, Li Y, Chen ZY, Huang CK. Crosstalk of renal cell carcinoma cells and tumor-associated macrophages aggravates tumor progression by modulating muscleblind-like protein 2/B-cell lymphoma 2/beclin 1-mediated autophagy. Cytotherapy. 2023;25:298-309
62. Hasko G, Szabo C, Nemeth ZH, Kvetan V, Pastores SM, Vizi ES. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. Journal of immunology. 1996;157:4634-40
63. Nemeth ZH, Lutz CS, Csoka B, Deitch EA, Leibovich SJ, Gause WC. et al. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. Journal of immunology. 2005;175:8260-70
64. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S. et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18:985-94
65. Dey S, Murmu N, Mondal T, Saha I, Chatterjee S, Manna R. et al. Multifaceted entrancing role of glucose and its analogue, 2-deoxy-D-glucose in cancer cell proliferation, inflammation, and virus infection. Biomed Pharmacother. 2022;156:113801
66. Peng C, Li L, Luo G, Tan S, Xia R, Zeng L. Integrated analysis of the M2 macrophage-related signature associated with prognosis in ovarian cancer. Front Oncol. 2022;12:986885
67. Zeng L, Liang L, Fang X, Xiang S, Dai C, Zheng T. et al. Glycolysis induces Th2 cell infiltration and significantly affects prognosis and immunotherapy response to lung adenocarcinoma. Funct Integr Genomics. 2023;23:221
68. Yang J, Ren B, Yang G, Wang H, Chen G, You L. et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. 2020;77:305-21
69. Yi M, Li T, Niu M, Zhang H, Wu Y, Wu K. et al. Targeting cytokine and chemokine signaling pathways for cancer therapy. Signal transduction and targeted therapy. 2024;9:176
70. Ye H, Zhou Q, Zheng S, Li G, Lin Q, Wei L. et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;9:453
71. Wang Y, Zhang J, Shi H, Wang M, Yu D, Fu M. et al. M2 Tumor-Associated Macrophages-Derived Exosomal MALAT1 Promotes Glycolysis and Gastric Cancer Progression. Advanced science. 2024;11:e2309298
72. Jin H, He Y, Zhao P, Hu Y, Tao J, Chen J. et al. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin beta3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics. 2019;9:265-78
73. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C. et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19:31-44
74. Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv Drug Deliv Rev. 2017;114:206-21
75. Nemeth Z, Li M, Csizmadia E, Dome B, Johansson M, Persson JL. et al. Heme oxygenase-1 in macrophages controls prostate cancer progression. Oncotarget. 2015;6:33675-88
76. Incio J, Suboj P, Chin SM, Vardam-Kaur T, Liu H, Hato T. et al. Metformin Reduces Desmoplasia in Pancreatic Cancer by Reprogramming Stellate Cells and Tumor-Associated Macrophages. PLoS One. 2015;10:e0141392
77. Farooque A, Afrin F, Adhikari JS, Dwarakanath BS. Polarization of macrophages towards M1 phenotype by a combination of 2-deoxy-d-glucose and radiation: Implications for tumor therapy. Immunobiology. 2016;221:269-81
78. Hadad ZSH, Afzelius P, Sorensen SM, Jurik AG. Clinical relevance of 18F-FDG-PET/CT incidental findings. Dan Med J. 2020;67:A10190553
79. Liu M, O'Connor RS, Trefely S, Graham K, Snyder NW, Beatty GL. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated 'don't-eat-me' signal. Nat Immunol. 2019;20:265-75
80. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res. 2021;81:1201-8
81. Jin Y, Zhuang Y, Dong X, Liu M. Development of CpG oligodeoxynucleotide TLR9 agonists in anti-cancer therapy. Expert Rev Anticancer Ther. 2021;21:841-51
82. de Araujo Junior RF, Eich C, Jorquera C, Schomann T, Baldazzi F, Chan AB. et al. Ceramide and palmitic acid inhibit macrophage-mediated epithelial-mesenchymal transition in colorectal cancer. Mol Cell Biochem. 2020;468:153-68
83. He Y, Rezaei S, Junior RFA, Cruz LJ, Eich C. Multifunctional Role of Lipids in Modulating the Tumorigenic Properties of 4T1 Breast Cancer Cells. Int J Mol Sci. 2022;23:4240
84. Fazio C, Piazzi G, Vitaglione P, Fogliano V, Munarini A, Prossomariti A. et al. Inflammation increases NOTCH1 activity via MMP9 and is counteracted by Eicosapentaenoic Acid-free fatty acid in colon cancer cells. Sci Rep. 2016;6:20670
85. Brindisi M, Frattaruolo L, Fiorillo M, Dolce V, Sotgia F, Lisanti MP. et al. New insights into cholesterol-mediated ERRalpha activation in breast cancer progression and pro-tumoral microenvironment orchestration. FEBS J. 2023;290:1481-501
86. Junior RFA, Lira GA, Schomann T, Cavalcante RS, Vilar NF, de Paula RCM. et al. Retinoic acid-loaded PLGA nanocarriers targeting cell cholesterol potentialize the antitumour effect of PD-L1 antibody by preventing epithelial-mesenchymal transition mediated by M2-TAM in colorectal cancer. Transl Oncol. 2023;31:101647
87. Kamata T, Jin H, Giblett S, Patel B, Patel F, Foster C. et al. The cholesterol-binding protein NPC2 restrains recruitment of stromal macrophage-lineage cells to early-stage lung tumours. EMBO Mol Med. 2015;7:1119-37
88. Qiu J, Shi Z, Jiang J. Cyclooxygenase-2 in glioblastoma multiforme. Drug Discov Today. 2017;22:148-56
89. Noda M, Tatsumi Y, Tomizawa M, Takama T, Mitsufuji S, Sugihara H. et al. Effects of etodolac, a selective cyclooxygenase-2 inhibitor, on the expression of E-cadherin-catenin complexes in gastrointestinal cell lines. J Gastroenterol. 2002;37:896-904
90. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci U S A. 2017;114:1117-22
91. Nakanishi Y, Nakatsuji M, Seno H, Ishizu S, Akitake-Kawano R, Kanda K. et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis. 2011;32:1333-9
92. Liu Q, Tong D, Liu G, Gao J, Wang LA, Xu J. et al. Metformin Inhibits Prostate Cancer Progression by Targeting Tumor-Associated Inflammatory Infiltration. Clin Cancer Res. 2018;24:5622-34
93. Eyob H, Ekiz HA, Derose YS, Waltz SE, Williams MA, Welm AL. Inhibition of ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov. 2013;3:751-60
94. Palmieri EM, Menga A, Martin-Perez R, Quinto A, Riera-Domingo C, De Tullio G. et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017;20:1654-66
95. Bulle A, Dekervel J, Deschuttere L, Nittner D, Van Cutsem E, Verslype C. et al. Anti-Cancer Activity of Acriflavine as Metabolic Inhibitor of OXPHOS in Pancreas Cancer Xenografts. Onco Targets Ther. 2020;13:6907-16
96. Mertens C, Akam EA, Rehwald C, Brune B, Tomat E, Jung M. Intracellular Iron Chelation Modulates the Macrophage Iron Phenotype with Consequences on Tumor Progression. PLoS One. 2016;11:e0166164
97. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A. et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11:986-94
98. Bruns H, Buttner M, Fabri M, Mougiakakos D, Bittenbring JT, Hoffmann MH. et al. Vitamin D-dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci Transl Med. 2015;7:282ra47
99. Zhang Y, Guo Q, Zhang Z, Bai N, Liu Z, Xiong M. et al. VDR status arbitrates the prometastatic effects of tumor-associated macrophages. Mol Cancer Res. 2014;12:1181-91
100. Pacini S, Punzi T, Morucci G, Gulisano M, Ruggiero M. Effects of vitamin D-binding protein-derived macrophage-activating factor on human breast cancer cells. Anticancer Res. 2012;32:45-52
101. Chiang CF, Chao TT, Su YF, Hsu CC, Chien CY, Chiu KC. et al. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-kappaB signaling. Oncotarget. 2017;8:20706-18
102. Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015;64:2028-41
103. Kang J, Lee D, Lee KJ, Yoon JE, Kwon JH, Seo Y. et al. Tumor-Suppressive Effect of Metformin via the Regulation of M2 Macrophages and Myeloid-Derived Suppressor Cells in the Tumor Microenvironment of Colorectal Cancer. Cancers (Basel). 2022;14:2881
104. Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int J Mol Sci. 2021;22:5703
105. Bai R, Li Y, Jian L, Yang Y, Zhao L, Wei M. The hypoxia-driven crosstalk between tumor and tumor-associated macrophages: mechanisms and clinical treatment strategies. Mol Cancer. 2022;21:177
106. Ma X, Yao M, Gao Y, Yue Y, Li Y, Zhang T. et al. Functional Immune Cell-Derived Exosomes Engineered for the Trilogy of Radiotherapy Sensitization. Adv Sci (Weinh). 2022;9:e2106031
107. Cheng WY, Huynh H, Chen P, Pena-Llopis S, Wan Y. Macrophage PPARgamma inhibits Gpr132 to mediate the anti-tumor effects of rosiglitazone. Elife. 2016;5:e18501
108. Hsu SPC, Chen YC, Chiang HC, Huang YC, Huang CC, Wang HE. et al. Rapamycin and hydroxychloroquine combination alters macrophage polarization and sensitizes glioblastoma to immune checkpoint inhibitors. J Neurooncol. 2020;146:417-26
109. Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin Cancer Biol. 2019;59:125-32
110. Zheng Z, Zhang J, Jiang J, He Y, Zhang W, Mo X. et al. Remodeling tumor immune microenvironment (TIME) for glioma therapy using multi-targeting liposomal codelivery. J Immunother Cancer. 2020;8:e000207
111. Zhang S, Peng W, Wang H, Xiang X, Ye L, Wei X. et al. C1q(+) tumor-associated macrophages contribute to immunosuppression through fatty acid metabolic reprogramming in malignant pleural effusion. Journal for immunotherapy of cancer. 2023;11:e007441
112. Chen YJ, Li GN, Li XJ, Wei LX, Fu MJ, Cheng ZL. et al. Targeting IRG1 reverses the immunosuppressive function of tumor-associated macrophages and enhances cancer immunotherapy. Science advances. 2023;9:eadg0654
113. Cai Z, Li W, Hager S, Wilson JL, Afjehi-Sadat L, Heiss EH. et al. Targeting PHGDH reverses the immunosuppressive phenotype of tumor-associated macrophages through alpha-ketoglutarate and mTORC1 signaling. Cellular & molecular immunology. 2024;21:448-65
114. Finley LWS. What is cancer metabolism? Cell. 2023;186:1670-88
115. Wang YP, Li JT, Qu J, Yin M, Lei QY. Metabolite sensing and signaling in cancer. The Journal of biological chemistry. 2020;295:11938-46
116. Butreddy A, Kommineni N, Dudhipala N. Exosomes as Naturally Occurring Vehicles for Delivery of Biopharmaceuticals: Insights from Drug Delivery to Clinical Perspectives. Nanomaterials. 2021;11:1481
117. Zhao Z, Ukidve A, Kim J, Mitragotri S. Targeting Strategies for Tissue-Specific Drug Delivery. Cell. 2020;181:151-67
118. Schmidt DR, Patel R, Kirsch DG, Lewis CA, Vander Heiden MG, Locasale JW. Metabolomics in cancer research and emerging applications in clinical oncology. CA: a cancer journal for clinicians. 2021;71:333-58
119. Xiao J, Wang S, Chen L, Ding X, Dang Y, Han M. et al. 25-Hydroxycholesterol regulates lysosome AMP kinase activation and metabolic reprogramming to educate immunosuppressive macrophages. Immunity. 2024;57:1087-104 e7
120. Davoli E, Zucchetti M, Matteo C, Ubezio P, D'Incalci M, Morosi L. The Space Dimension at the Micro Level: Mass Spectrometry Imaging of Drugs in Tissues. Mass spectrometry reviews. 2021;40:201-14
Author contact
![]() Corresponding authors: E-mail addresses: liudan_bdcom (Dan Liu), jnzhengedu.cn (Junnian Zheng), mingshiedu.cn (Ming Shi). Cancer Institute, Xuzhou Medical University, 209 Tongshan Road, Xuzhou, Jiangsu 221004, China.
Corresponding authors: E-mail addresses: liudan_bdcom (Dan Liu), jnzhengedu.cn (Junnian Zheng), mingshiedu.cn (Ming Shi). Cancer Institute, Xuzhou Medical University, 209 Tongshan Road, Xuzhou, Jiangsu 221004, China.