Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Anti-cancer potential and...
3. Celastrol and its target...
4. Drug combination and...
5. Side effects of celastrol
6. Anti-inflammatory effects of...
7. Discussion
8. Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(14):5510-5530. doi:10.7150/ijbs.99592 This issue Cite
Review
Recent Trends in anti-tumor mechanisms and molecular targets of celastrol
Yongping Zhu1#, Yuqing Meng1#, Junzhe Zhang1, Rui Liu2, Shengnan Shen1, Liwei Gu1, Yin-kwan Wong3, Ang Ma1, Xin Chai1, Ying Zhang1, Yanqing Liu1 ![]() , Jigang Wang1,4,5
, Jigang Wang1,4,5 ![]()
1. State Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, Artemisinin Research Center, and Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China.
2. College of Animal Science and Technology, Henan Agricultural University, Zhengzhou 450002, China.
3. Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117600, Singapore.
4. Department of Critical Care Medicine, Guangdong Provincial Clinical Research Center for Geriatrics, Shenzhen Clinical Research Center for Geriatric, Shenzhen People's Hospital (The Second Clinical Medical College, Jinan University; The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen 518020, China.
5. State Key Laboratory of Antiviral Drugs, School of Pharmacy, Henan University, Kaifeng, China.
# These authors contributed equally to this work.
Received 2024-6-13; Accepted 2024-9-24; Published 2024-10-7
Abstract

Celastrol, a compound derived from traditional Chinese medicine, has therapeutic effects and has been used to treat inflammation-related diseases, cancer, cardiovascular diseases, and neurodegenerative diseases. However, current reviews lack a comprehensive and systematic summary of the anti-tumor mechanisms and molecular targets of celastrol. For this reason, this paper reviews the anticancer properties of celastrol and the molecular mechanisms underlying its anticancer effects. This paper primarily focuses on the mechanism of action of celastrol in terms of inhibition of cell proliferation and regulation of the cell cycle, regulation of apoptosis and autophagy, inhibition of cell invasion and metastasis, anti-inflammation, regulation of immunotherapy, and angiogenesis. More importantly, the target proteins of celastrol identified by chemical proteomics or other methods are highlighted, providing detailed targets with novel therapeutic potential for anti-tumor treatment. In addition, we describe the side effects and strategies to improve the bioavailability of celastrol. In summary, this paper analyzes celastrol, a natural compound with therapeutic effects and clear targets, aiming to draw more attention from the scientific and pharmacological communities and accelerating its clinical application for the benefit of cancer patients.
Keywords: celastrol, pharmacology, mechanisms, targets
1. Introduction
Cancer remains one of the most lethal human diseases, with high annual incidence and mortality rates. The latest cancer statistics report indicates 19.3 million new cases and 10 million deaths worldwide in 2020 alone [1]. Surgery, chemotherapy, and radiotherapy are the preferred, recognized, and widely used cancer treatments globally [2-4], with immunotherapy also playing an increasingly important role in tumor treatment in recent years [5,6]. Despite the availability of therapeutic modalities and targeted drugs, cancer continues to be the leading cause of death worldwide. Therefore, further research into new strategies and the discovery of potential new drugs for cancer treatment is urgently needed.
Natural resources have become a significant part of the drug market, overshadowing synthetic compounds in drug development over the past half-century. Recently, natural medicinal plants and bioactive molecules have attracted immense interest for their potent efficacy, minimal side effects, lower cost, and in some cases, extraordinary anti-inflammatory, anti-obesity, anticancer, and neuroprotective properties [7]. It is widely believed that such substances can be used as a reasonable alternative to synthetic substances. As a prominent natural product molecule, celastrol has been identified as one of the five most promising natural pharmaceutical products. Also known as tripterygium, celastrol is a red acicular crystal and a quinone methyl pentacyclic triterpenoid compound isolated from Tripterygium wilfordii, a traditional Chinese medicine [8]. Recent studies have highlighted the diverse pharmacological activities of celastrol, including anti-inflammatory (9), anti-diabetes and obesity [10-12], immunomodulatory [13], and particularly, anti-tumor effects [14]. Celastrol displays broad-spectrum and efficient anti-tumor activity. It has been reported to inhibit various cancer cells, including lung, ovarian, stomach, liver, leukemia, colon, and breast cancers [15], and to enhance the sensitization effects of radiotherapy and chemotherapy [16].
This review comprehensively and systematically summarizes the anti-tumor effects of celastrol, including its molecular mechanisms, target proteins, drug combinations, bioavailability, and potential side effects. In particular, the mechanism of action and targets of celastrol are highlighted. Drugs generally prevent and treat diseases by acting on these drug targets and further modulating up- and downstream signaling pathways. Identifying drug targets is therefore essential for research and development of novel cancer drugs and therapies [17]. We hope that this review will provide novel and comprehensive design ideas and a theoretical basis for further research on celastrol, as well as theoretical guidance for its clinical prescription and acceleration of clinical trials.
2. Anti-cancer potential and molecular mechanisms of celastrol
2.1 Celastrol inhibits cell proliferation and regulates the cell cycle
The uncontrolled proliferation of tumor cells results from dysregulation of the cell cycle, and targeting this dysregulation is a key strategy in current cancer therapies. Studies have demonstrated that celastrol affects the cell cycle, including the modulation of cyclins and cyclin kinases, and inhibits the proliferation of tumor cells across various cancer types. Li et al. reported that treating ovarian cancer cells with celastrol led to the inhibition of Pin1 protein expression, resulting in the down-regulation of Cyclin D1, CDK2, and CDK4, and the arrest of the cell cycle in the G2/M phase [18]. Similarly, other studies have shown that celastrol upregulates Cyclin B1 and downregulates Cyclin E in a dose-dependent manner, effectively blocking the cell cycle in the G2/M phase [19]. Celastrol also enhances p21 and p27 expression while decreasing Cyclin D1 and Cyclin E levels, inhibiting cell proliferation and leading to G1 phase arrest in multiple myeloma cells [20]. Additionally, celastrol significantly increases the amount of reactive oxygen species (ROS) in colon cancer cells at both the cytoplasmic and mitochondrial levels, causing cell cycle arrest in the S phase [21]. Treatment of a human osteosarcoma cell line with celastrol resulted in cell cycle arrest at the G2/M phase, followed by apoptosis through the activation of the OS/JNK signaling pathway [22]. In gastric cancer cells, celastrol induced G2/M cell-cycle arrest by inhibiting the miR-21-mTOR signaling pathway and increasing the p27 protein level [23]. Furthermore, celastrol caused cell cycle arrest in the G2/M phase of nasopharyngeal carcinoma by upregulating the mitogen-activated protein kinase (p38 MAPK) and extracellular signal-regulated Kinase (ERK) pathways, triggering apoptosis via both endogenous and exogenous pathways, thus exerting an anti-tumor effect [24]. In essence, celastrol impedes the growth and proliferation of cancer cells by influencing cell cycle progression.
2.2 Celastrol regulates cell apoptosis
Apoptosis, a process of programmed cell death under physiological or pathological conditions, is influenced by the regulation of intracellular genes and some extracellular factors. It affects various processes including growth, metabolism, immunological defense, cell differentiation, nervous system damage, and tumor development [25]. The apoptosis of tumor cells is closely associated with abnormal expression of the endogenous apoptosis pathway and the exogenous pathway [26]. Studies have demonstrated that celastrol can induce apoptosis in various tumor cells through multiple mechanisms, such as the activation of caspases [27-30]. Celastrol inhibits tumor cell proliferation, reduces migration and invasion capabilities, and induces cell cycle arrest by increasing the expression of cleaved-PARP, cleaved-caspase-3, cleaved-caspase-8, and cleaved-caspase-9, ultimately leading to apoptosis [27-28].
The FAS gene belongs to the TNF receptor family. After the Fas protein binds with its ligand (Fas-L), target cells can be induced to undergo apoptosis via activating the caspase cascade. Abnormal expression of Fas and FasL has been shown to inhibit apoptosis in normal cells and promote the survival of poorly differentiated cells, leading to tumor formation [31]. The administration of celastrol has been reported to induce apoptosis by activating mitochondria and Fas/Fas related pathways in non-small cell lung cancer cells [32]. In glioma cells, celastrol activates the cell death receptor pathway by increasing the expression levels of death receptor 5 (DR5), caspase-8, caspase-3, and PARP proteins, thereby inducing apoptosis [33]. Additionally, celastrol increases the expression of TNF receptor superfamily members (TNRSF) 1A and 10B, and TNFRSF1A is associated with the death domain, Fas, and Fas-associated via the death domain, leading to cell cycle arrest in the G1 phase and G2/M phase, and inducing apoptosis [34].
Celastrol promotes apoptosis by inducing the mitochondrial apoptosis pathway [35-36]. Mitochondria, the direct energy suppliers of cellular activities, undergo structural remodeling and increased outer membrane permeability during apoptosis, leading to the release of cytochrome c (CytC) which activates downstream caspases involved in the apoptosis process [37]. Celastrol induces apoptosis in acute promyelocytic leukemia (HL-60 and NB-4) by inhibiting cysteine metabolism, increasing ROS levels, and activating the mitochondrial apoptosis pathway [35]. Furthermore, celastrol triggers the mitochondrial apoptosis pathway by upregulating the expression of CytC and the pro-apoptotic protein Bax, activating caspase-3 and caspase-9, and leading to the cleavage of PARP in human osteosarcoma cells [36]. Studies have also shown that celastrol directly binds to peroxiredoxin-2 (Prdx2) and inhibits its enzyme activity, increasing ROS levels and resulting in ROS-dependent endoplasmic reticulum (ER) stress, mitochondrial dysfunction, and apoptosis in gastric cancer cells [38].
Celastrol also induces apoptosis through the ER stress (ERS) emergency pathway [39-41]. The ER, a major cellular organelle that modifies and secretes proteins, activates ERS under constant and intense stimulation, leading to protein folding errors and excessive accumulation of unfolded proteins, triggering the unfolded protein response and initiating apoptosis. ERS regulates tumor cell proliferation, migration, and apoptosis [42]. Chen and colleagues revealed that celastrol inhibits the expression levels of ERS-related proteins (Bip, PERK, p-PERK, Ero1-Lα and calnexin), mitochondrial apoptosis-related proteins, and autophagy-related proteins in human osteosarcoma cells, inducing apoptosis [39]. Similarly, celastrol upregulates the expression of CHOP, Bip, XBP1s, and IRE1 proteins, increased the transcription of ERS target genes such as BIM, induced Bax translocation into mitochondria, and activated the ER and mitochondrial apoptosis pathways in cervical cancer HeLa cells [40]. Moreover, in non-small cell lung cancer (NSCLC), celastrol significantly increases intracellular ROS levels and triggered the activation of the ERS pathway, ultimately leading to apoptosis [41]. Although celastrol induces apoptosis in different ways, these pathways are not independent of each other but are closely linked. For example, both the death receptor pathway and the mitochondrial pathway can activate caspase proteins, which in turn leads to apoptosis. Celastrol also does not necessarily induce apoptosis through only a single signaling pathway. For example, it can induce apoptosis via both the ER pathway and the mitochondrial pathway [39-40]. In conclusion, celastrol exerts its anti-tumor effects partially through the induction of apoptosis by affecting one or more apoptotic pathways. However, how celastrol regulates multiple apoptotic pathways and the links between these pathways still need to be further studied.
Celastrol regulates cell autophagy
Autophagy is a form of programmed cell death in which cellular material is transported into the lysosome for degradation, allowing for the turnover of cellular components, and providing energy and macromolecular precursors [43]. In the context of cancer, autophagy serves a dual role, both by inhibiting tumorigenesis and by supporting tumor progression. It is generally accepted that autophagy suppresses tumor formation, yet evidence indicates that established tumors rely on the autophagic process for sustained cell growth and increased metabolic activity, thus depending on autophagy for maintenance [44]. Previous studies have demonstrated that celastrol can induce autophagy in cancer cells [45]. Treatment with celastrol has been shown to increase the expression of Atg5 and Atg7, facilitate the formation of autophagosomes, and promote the degradation of p62, thereby enhancing autophagy activity and reducing cell viability [46]. In human colorectal cancer cells, celastrol triggers apoptosis and autophagy by inhibiting Nur79, leading to increased expression of Atg7 and activation of the Nur77/Atg7 signaling pathway [47]. Furthermore, celastrol activates the LXRα signaling pathway, inducing autophagy and lipid droplet degradation, promoting ABCA1-mediated cholesterol efflux and impairing EMT progression, thereby playing an anti-invasive and anti-metastastic role in clear cell renal cell carcinoma [48]. Studies have also shown that celastrol upregulates the conversion of autophagy markers such as microtubule-associated protein 1 light chain 3I (LC3I) to LC3II, thereby inducing autophagy in cancer cell lines A549, PC-3, and HeLa [49]. Additionally, celastrol-treated gastric cancer cells exhibited enhanced levels of autophagy-related proteins Atg5, Atg7, Beclin1, and LC3I and II, which induce autophagy [50]. All these data indicate that celastrol can induce autophagy in cancer cells which inhibits tumorigenesis and suppresses tumor progression, and more specific molecular mechanisms and in vivo validation of the regulation of autophagy by celastrol are worthy of further study.
2.4 Celastrol inhibits cell invasion and metastasis
Metastasis occurs when tumor cells invade lymphatic vessels, blood vessels, or body cavities from their original site, and are transported via the bloodstream or lymph flow to another site or organ to continue growing, forming tumors identical to the primary tumor. Data indicate that over 90% of cancer-related deaths are caused by metastasis [51]. Chemokines, small cytokines or signaling proteins secreted by cells, constitute a family of more than 60 members with molecular weights ranging from 8 to 10 kDa [52]. Chemokine receptors (CXCRs) are G-protein-linked transmembrane proteins that interact with these cytokines to facilitate the spread of malignant cells, making drugs capable of blocking this interaction potential inhibitors of tumor cell metastasis [53]. CXCR4 is a crucial receptor involved in the interaction between tumor cells and their microenvironment. Studies have shown that celastrol decreases CXCR4 expression in a dose-dependent manner and downregulates related downstream pathways, including the PI3K/AKT pathway, significantly reducing tumor cell proliferation and migration capacity [54-55]. Additionally, celastrol indirectly influences the expression of miR-223-3p by downregulating circ_SLIT3 and affecting CXCR4, thereby restraining the migration and invasion of HCC cells [56].
Mounting evidence suggests that extracellular proteases, such as matrix metalloproteinases (MMPs), which are capable of degrading various components of the extracellular matrix (ECM), play crucial roles in tumor invasion and metastasis by breaking down the histological barriers to tumor cell invasion [57]. Celastrol effectively inhibits cell migration and invasion in human osteosarcoma cells through down-regulating MMP2 and MMP9, by inhibiting the PI3K/AKT/NF-κB signaling pathway [58-60]. Furthermore, celastrol suppresses the expression of MMP3 and MMP7 in colorectal cancer cells by inhibiting the PI3K/AKT pathway, thereby affecting their proliferation and migration of [61]. Additionally, further evidence suggests that celastrol downregulates the phosphorylation of focal adhesion kinase (FAK) by activating the p38 MAPK signaling pathway, blocking the adhesion of the mouse melanoma cell line B16F10 and human lung cancer cell line 95-D to the extracellular matrix, and thereby inhibiting adhesion plaque-dependent cell migration and invasion [62].
The tumor microenvironment plays a vital role in tumor cell migration, invasion and angiogenesis [63]. It has been found that celastrol can reverse macrophage polarization from M2 to M1 in vivo and in vitro by affecting the colorectal tumor microenvironment, as well as mechanistically polarizing macrophages through the MAPK signaling pathway [64]. In addition, celastrol and siRNA have been delivered to retinoblastoma cells in the form of polymeric micelles to achieve synergistic anti-tumor and anti-angiogenic effects. The co-delivery system specifically and synergistically inhibited the expression of HIF-1α and VEGF in retinoblastoma cells, inhibited the HIF-1α /VEGF/VEGFR signaling pathway, ameliorated the hypoxic microenvironment of the tumors, and hindered the proliferation, migration, and invasion of vascular endothelial cells [65]. In summary, celastrol inhibits cell invasion and metastasis by influencing the tumor microenvironment and the expression of cytokines.
2.5 Celastrol regulates tumor immunotherapy
Immunotherapy has emerged as a powerful clinical strategy for the treatment of cancer. Advanced biomaterials and delivery systems, such as those utilizing T-cell delivery or nanoparticles, can be used to effectively utilize immunotherapies and increase their efficacy while reducing toxic side effects. Studies have shown that celastrol in combination with nanoparticle delivery systems has promising applications in melanoma immunotherapy [66-68]. It was found that celastrol not only induced immunogenic cell death (ICD), but also down-regulated PD-L1 expression in tumor cells, activated both dendritic cells and T cells, and interrupted the PD-1/PD-L1 pathway between T cells and tumor cells. In a bilateral tumor model, intratumoral injection of a high concentration of celastrol nanoemulsion effectively activated the immune system, thereby suppressing both treated and distant untreated tumors (i.e., the ascites effect) in mice [66]. Wang et al. constructed an ER-targeted celastrol nanoparticle, TSE-CEL/NP, which specificlly aggregated in the ER and induced ER stress. Excessive ER stress exposed the cells to calcreticulin, which released ATP and HMGB1 extracellularly. This enhanced the recruitment and maturation of dendritic cells, which in turn increased the proliferation of cytotoxic T lymphocytes (CD8+ T cells), thereby enhancing the effectiveness of immunotherapy against melanoma [67]. In addition, celastrol directly binds to IL-2, disrupts the binding of IL-2 and CD25, markedly inhibits the proliferation and signaling of IL-2-dependent mouse T cells, and increases the number of CD8+ T cells, thereby inhibiting tumor growth. The group also reported that celastrol failed to inhibit tumor growth in T cell-deficient BALB/c nude mice, suggesting that its activity was mediated by T cell responses. In addition, combination therapy with low-dose celastrol and a TNFR2 antagonist synergistically improved the efficacy of the single agent by increasing the ratio of intratumoral CD8+ T cells/Treg cells and inhibiting the expression of Foxp3. Celastrol thus exerts antitumor activity by targeting IL-2 to inhibit CD25 binding and mediating T cell responses, and has the potential to be a candidate for melanoma combination therapy in cancer immunotherapy [68]. In conclusion, while celastrol has shown great potential in tumor immunotherapy, few studies have been conducted in this area and more research is needed to elucidate the role of celastrol in immunotherapy.
2.6 Anti-inflammatory properties of celastrol
The immune system plays crucial roles in both tumor development and treatment. Adaptive immunity works to prevent or suppress tumors through immune surveillance, whereas innate immunity and inflammation often promote the onset and malignant progression of new cancers [69]. Chronic inflammation induced by certain infectious diseases has been reliably linked to cancer development. Thus, anti-infective agents play a significant role in reducing the burden of inflammation-related cancers. Nuclear factor-κB (NF-κB) and signal transducer and activator of transcription 3 (STAT3) are key regulators of inflammation, cell transformation, tumor cell survival, proliferation, and metastasis [70]. NF-κB is an important nuclear transcription factor involved in multiple complex biological processes, including immune regulation, inflammation, cell proliferation, and apoptosis, and is often overexpressed in various tumors, such as myeloma, breast, ovarian, and prostate cancers [71-72]. As an NF-κB inhibitor, celastrol suppresses the activity and expression of NF-κB and its associated genes, thereby inhibiting the proliferation, migration, and invasion of tumor cells. In prostate adenocarcinoma cells, celastrol modulated the protein expression of IκBα, IKKα (kappa B inhibitory kinase α subunit), p50, and NF-κB, promoted the degradation of the anti-apoptotic protein Mcl-1, and activated the pro-apoptotic protein Noxa. It also blocked the expression of IL-6, ultimately inhibiting tumor cell proliferation and invasion [73-74]. Celastrol additionally obstructed NF-κB and its downstream gene products, such as CXCR4 and MMP9, and reduced serum IL-6 and TNF-α levels to inhibit cell invasion and migration in vivo [75]. Studies have indicated that celastrol inhibits IκBα phosphorylation, preventing IκBα degradation and NF-κB accumulation. This in turn suppresses the expression and activity of the NF-κB target MMP9, thus blocking the NF-κB pathway and resulting in anti-ovarian cancer cell invasion activity [76]. Moreover, celastrol reduces the level of IL-6 by inhibiting the NF-κB signaling pathway in breast cancer cells, inhibiting cell proliferation, migration, and invasion, thereby playing an anti-tumor role [77].
As a nexus of numerous carcinogenic signaling pathways, STAT3 centrally regulates the anti-tumor immune response. It is widely over-activated in cancer cells, playing a crucial role in promoting the expression of immunosuppressive factors and inhibiting the production of key immune activation regulators [78]. Research has shown that celastrol inhibits STAT3 by suppressing the activation of upstream kinases c-Src and Janus-activated kinases -1 and -2 (JNK 1/2), impacting tumor cell proliferation [79]. By significantly increasing the expression levels of miR-181b and miR-24, celastrol reduces the phosphorylation of STAT3 and the ratio of B-cell lymphoma-2 (Bcl-2)/Bcl-2 associated X (Bax), thereby inhibiting cell proliferation and inducing apoptosis [80]. These results provide a theoretical basis for further exploration of the anti-tumor properties of celastrol through the regulation of the immune system.
2.7 Celastrol and angiogenesis
During tumor progression, angiogenesis is a complex process often triggered by hypoxia, which leads to increases in angiopoietin, vascular endothelial growth factor (VEGF), and hypoxia-inducing factor (HIF-1) levels. Angiopoietin and VEGF are considered pivotal in tumor angiogenesis [81]. The development of new antiangiogenic drugs represents a crucial strategy for treating multiple solid tumors by reducing or eliminating the blood supply to tumor microregions, causing extensive hypoxic necrosis of solid tumor tissues. These drugs are vital in treating certain cancers [82]. Previous studies have demonstrated that celastrol inhibits the growth of human glioma xenografts in nude mice by suppressing the expression and transcription of VEGF receptor (VEGFR) 1 and vascular dermal growth factor receptor (VDGFR) 2 [83]. Another study showed that celastrol inhibited the growth and migration of colorectal cancer cells, and this effect is associated with the inhibited expression of key genes and proteins associated with the angiogenesis pathway including PDGF, MMP-9, Serpin E1 and TIMP-4 [84]. Additionally, treatment with celastrol has been reported to inhibit tumor growth and angiogenesis in vitro and in vivo by targeting the mTOR/AKT/S6K kinase signaling pathway [14]. Thus, celastrol may partly exert its anti-tumor effects across various cancers by inhibiting angiogenesis. However, further comprehensive studies will be necessary to elucidate the mechanisms behind the anti-angiogenic properties of celastrol.
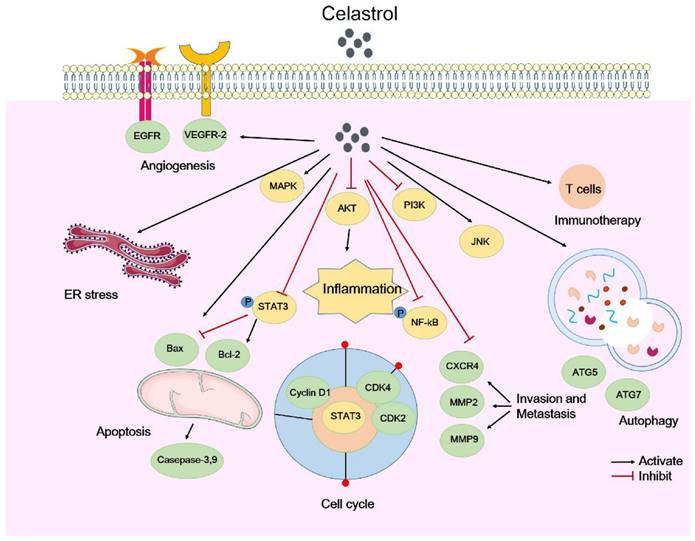
In summary, celastrol mainly attenuates tumor growth by inhibiting cell proliferation and regulating the cell cycle, regulating cell apoptosis and autophagy, inhibiting cell invasion and metastasis, and through its anti-inflammatory, antioxidant, and anti-angiogenic activities. Here, we briefly summarize the types of celastrol-induced anticancer activities and their mechanisms (Table 1). The molecular mechanisms by which celastrol inhibits tumors are depicted in Figure 1. Importantly, celastrol exerts these anti-tumor effects by binding to different cellular targets and affecting various upstream and downstream signaling pathways. In the next section, we will focus on these target proteins of celastrol.
Summary of detailed experimental study of celastrol against different tumor types in this article.
| Effects of celastrol on cancers | Cancer Type | Cell line | Dose of administration | Mechanisms of action of celatrol | Ref. |
|---|---|---|---|---|---|
| Inhibits cell proliferation and regulates cell cycle | ovarian cancer | A2780, SKOV3, OVCAR3 | 0, 0.25, 0.5, 1, 2 and 4 μM | inhibited Pin1 protein, CyclinD1↓, CDK2↓, CDK4 ↓genes | [18] |
| A2780 and SKOV3 | 0, 0.1, 0.3, 1, 3, and 10 µM | induced cell cycle arrest in G2/M phase with Cyclin B1↑ and Cyclin E↓ | [19] | ||
| Multiple myeloma | U266, RPMI 8226, RPMI-8226Dox-6, RPMI-8226-LR-5 | 0, 0.1, 0.25, 0.5, 1, 2.5 and 5 µM | inhibited cell proliferation, ub-G1 phase arrests cyclin D1↓ and cyclin E↓, p21↑ and p27↑ | [20] | |
| colon cancer | LOVO, LOVO/DX | 0, 0.1, 0.5, 1.5, 10 and 20 µM | increased ROS amount, arrest in the S and G2/M phase | [21] | |
| osteosarcoma | HOS, MG-63, U-2OS, Saos-2 | 0, 1, 2, 3, 4 µM | induced apoptosis via activation of the OSs/JNK signaling pathway, cell cycle arrest at the G2/M phase | [22] | |
| gastric cancer | BGC-823, MGC-803 | 2 μM | inhibited miR-21-mTOR signaling pathway, p27↑, induced G2/M cell-cycle arrest in gastric cancer cells | [23] | |
| nasopharyngeal carcinoma cancer | NPC-039, NPC-BM, Cis-039, Cis-BM | 0, 1, 2, 4 µM | induced cell cycle arrest in the 2G /M phase of nasopharyngeal carcinoma by up-regulating p38 MAPK and ERK pathway, and induces apoptosis mediated by endogenous and exogenous apoptotic pathways, thus playing an anti-tumor role | [24] | |
| Regulates cell apoptosis | hepatocellular carcinoma | HepG2 | 10, 20 and 40 μM | inhibited the phosphorylation of mTOR, cleaved-PARP↑, cleave-caspase-3↑, cleave-caspase-8↑, cleave-caspase-9↑ | [26] |
| Canine mammary tumor | CMT-7364 | 0, 0.1, 0.4, 0.6, 0.8, 1.2 and 1.6 µM | inhibited the proliferation, reduced the migration and invasion, cleaved caspase-3 ↑ and cleaved caspase-9 ↑, induced cell apoptosis | [27] | |
| ovarian cancer | OVCAR3 | 0, 0.25, 0.5, 1, 2, 4, 6 µM | inhibited PI3K/ Akt-NF-κB signaling pathway, induced apoptosis and inhibited cell proliferation caspase-3↑ and caspase-9↑ | [28] | |
| non-small-cell lung cancer | A549 | 0, 0.5, 1, 2, 4, 8 µM | activated mitochondria and Fas/Fas related pathway, Bax↑, Bcl-2↓, inhibited Akt phosphorylation | [32] | |
| glioblastoma | U87-MG, U251, and LN229 | 0, 0.5, 1, 2, 4 µM | activated cell death receptor pathway, DR5↑, caspase-8↑, caspase-3↑and PARP↑, inhibited tumor cell proliferation and inducing cell apoptosis | [33] | |
| Nasopharyngeal carcinoma | HONE-1 and NPC-039 | 0, 1, 2, 4 µM | TNRSF1A↑ and 10B↑, TNFRSF1A associated via death domain↑, Fas↑, Fas-associated via death domain↑, lead to cell cycle G1 phase and G2/M phase and induced cell apoptosis | [34] | |
| Acute leukemia | HL-60 cells and NB-4 | 0, 0.125, 0.25, 0.5 µM | induced cell apoptosis by inhibiting cysteine metabolism, increased reactive oxygen species (ROS) level, and activated mitochondrial apoptosis pathway | [35] | |
| gastric cancer | SGC-7901 and BGC-823 | 0, 1, 2, 3 µM | inhibited the activity of Prdx2 to increase ROS levels, leading to ROS-dependent ER stress, mitochondrial dysfunction and apoptosis | [38] | |
| osteosarcoma | MG- 63, U-2OS and HOS | 0, 0.5, 1, 2, 4, 6 µM | triggered mitochondrial apoptosis pathway, Bax↑, cytochrome c↑, procaspase-9↓, cleavaged PARP↓ | [36] | |
| Hepatocellular carcinoma | HepG2 and Bel7402 | 0, 0.625, 1.25, 2.5, 5.0 and 10 μM | inhibited the levels of ERS-related proteins, mitochondrial apoptosis-related proteins and autophagy related proteins, cause G2/M phase rest and inhibit proliferation | [39] | |
| Cervical cancer | HeLa | 0, 0.5, 1, 2, 4 µM | up-regulated the expression of Bip, CHOP, XBP1s and IRE1 proteins, increase the transcription of endoplasmic reticulum stress target genes such as BIM, induced Bax translocation into mitochondria, and finally activated the endoplasmic reticulum and mitochondrial apoptosis pathway | [40] | |
| Non-small cell lung cancer | H460, PC-9, H520 and BEAS-2B | 0, 1, 2, 4 µM | increased intracellular ROS levels, activated ERS pathway and inhibited P-STAT3 pathway, induced cell apoptosis | [41] | |
| Regulates cell autophagy | prostate cancer | LNCaP, 22Rv1, DU145 and PC-3 | 2 µM | suppressed cell viability and upregulated autophagic activity, Atg5↑ and Atg7↑, increased autophagosome formation and promoted p62 degradation | [46] |
| colorectal cancer | NCM460, HCT-116, SW480 | 0, 1.25, 2.5, 5 µM | induced apoptosis and autophagy by inhibiting Nur77, induced high expression of Atg 7 and Nur77/ Atg 7 signaling pathways | [47] | |
| cervix, lung and prostate cancer | HeLa cells, A549 cells and PC-3 | 0.5, 1.2, 4 µM | triggered autophagy and lipid droplet degradation, activated LXRα signaling | [49] | |
| gastric cancer | MKN1, MKN45, SNU216, SNU-668, and YCC-2 | 0, 0.25, 0.5, 1 µM | enhancement of autophagy-related proteins Atg 5↑, Atg 7↑, LC3 I↑, LC3 II and Beclin1↑ | [50] | |
| Inhibits cell invasion and metastasis | Hepatocellular Carcinoma | HCC, HepG2 and Hepa3B | 0, 0.1, 0.3, 0.625, 1 µM | attenuated the proliferation and migration capacity of cells, CXCR4↓, PI3K, Akt | [54] |
| colon and pancreatic cancer | MCF-7, KBM-5, SCC-4, Caco-2, HCT116, MDA-MB-231 | 0, 1, 2, 3 µM | regulated the expression of CXCR4, thereby inhibiting the invasion of tumor cells | [55] | |
| cervical cancer | HeLa | 1, 10, 100 μM | inhibited the proliferation, invasion and migration of HeLa, MMP- 2↓ and MMP- 9↓. | [58] | |
| colorectal cancer | CRC cells SW480 and HCT116 | 0, 0.1, 0.5, 1, 2 µM | affected the PI3K/AKT signaling pathway, inhibited the proliferation and migration of colorectal cancer cells, MMP3↓ and MMP7↓ | [61] | |
| osteosarcoma | U-2OS | 0, 2.5, 4 µM | suppress the cell invasion and migration, inhibited the PI3K/Akt/NF-κB signaling pathway, MMP-2↓and MMP -9↓ | [60] | |
| Malignant cancer, lung cancer | B16F10 and 95-D | 0, 1, 2, 4, 8 µM | down-regulated the phosphorylation of FAK, activated the p38 MAPK signaling pathway, inhibited the adhesion plaque dependent cell migration and invasion | [62] | |
| Colorectal cancer | HCT116 and SW480 | 0.75 µM | reversed macrophage polarization from M2 to M1 by affecting the colorectal tumor microenvironment, polarized the macrophages through the MAPK signaling pathway | [64] | |
| Retinoblastoma cancer | Y79 cells | 0, 0.5, 1, 2, 4 µg/mL | inhibited the HIF-1α /VEGF/VEGFR signaling pathway, ameliorated the hypoxic microenvironment of the tumors, and hindered the proliferation, migration, and invasion of vascular endothelial cells | [65] | |
| Cel regulates tumor immunotherapy | Melanoma | B16F10, A375 and M10 cell | 0, 0.1, 0.2, 0.5,1, 2, 4, 8 µM | induced immunogenic cell death, down-regulated PD-L1 expression, activated both dendritic cells and T cells, interrupted the PD-1/PD-L1 pathway | [66] |
| Melanoma | B16F10 | 0.5 μg/mL | induced ER stress, promoted the recruitment and maturation of dendritic cells, leading to an increase in the proliferation of cytotoxic T lymphocytes (CD8+ T cells) and ultimately improving the efficacy of immunotherapy against melanoma | [67] | |
| melanoma | B16F10 | 0, 1, 2, 4,6, 8, 10 µM | bond to IL-2, disrupted the binding of IL-2 and CD25, markedly inhibited the proliferation and signaling of IL-2-dependent mouse T cells, and increased the number of CD8+ T cells, thereby inhibiting tumor growth | [68] | |
| Anti-inflammatory properties of celastrol | Multiple Myeloma | U266, H929, and KMS11 | 0, 1, 2.5, 5 µM | inhibited the activation of NF-κB, CXCR4↓, MMP-9↓, reduced serum IL-6 and TNF-α levels | [75] |
| ovarian cancer | OVCAR-3 and SKOV-3 | 0, 0.125, 0.25, 0.5, 1. 2 µM | blocked the NF-κB pathway, inhibited i-κBα phosphorylation, prevented i-κBα degradation and NF-κB accumulation, MMP-9↓, exerted anti-invasion activity of cancer cells | [76] | |
| Breast Cancer | MDA-MB-468 and MDA-MB-231 | 0, 0.1, 0.5, 1, 2, 5 µM | repressed migration and invasion through decreasing IL-6 levels, inactivated NF-kB signaling | [77] | |
| Hepatocellular Carcinoma | C3A, HepG2, Hep3B, and PLC/ PRF5 | 0, 0.5, 1, 2.5, 5 µM | inhibited STAT3 activation, inhibited the activation of kinases c-Src, and janus activates kinases -1 and -2 (JNK 1/2), affected tumor cell proliferation | [79] | |
| lung adenocarcinoma | A549 | 0, 0.5, 3, 4.5, 6 µM | up-regulated the expression levels of miR-24 and miR-181b, decreased STAT3 phosphorylation and inactivation and Bcl-2/ Bax ratio, inhibited cell proliferation and induced cell apoptosis | [80] | |
| celastrol and angiogenesis | glioma | SHG44 | 0, 1, 2, 4 mg/kg/ day; | inhibited the growth of human glioma xenografts in nude mice, inhibited the expression and transcription of VEGFR1 and VEGFR2 | [83] |
| colorectal cancer | HT29, and HCT116 | 0, 2.5, 5, 10 µM | inhibited the growth and migration of cells inhibited the expression of key genes and proteins associated with the angiogenesis pathway | [84] | |
| prostate cancer | HUVECs or PC-3 cells | 0,0.1, 0.5, 1, 2 µM | inhibited tumor angiogenesis and tumor growth in vitro and in vivo, targeted the AKT/mTOR/S6K kinase signaling pathway | [14] |
The molecular mechanisms of tumor suppression by celastrol. Celastrol attenuates tumor growth through various mechanisms, including inhibiting cell proliferation and regulating the cell cycle, regulating cell apoptosis and autophagy, inhibiting cell invasion and metastasis, anti-inflammatory properties, regulating tumor immunotherapy and angiogenesis.
3. Celastrol and its target proteins
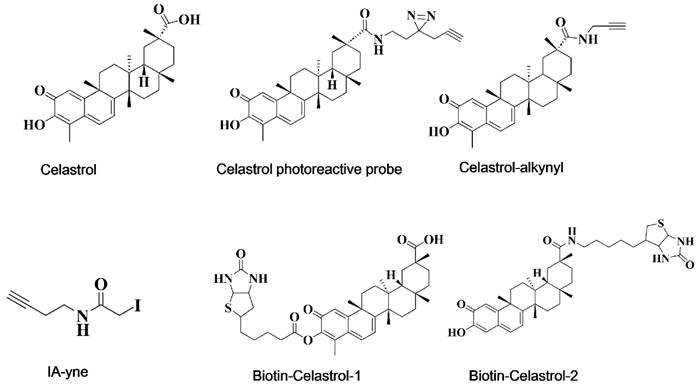
Approximately 123 out of the 154 anticancer drugs approved by the U.S. Food and Drug Administration (FDA) function through protein targets, underscoring the importance of drug target discovery and identification in oncology [85]. Drug targets are direct binding sites on biological macromolecules in vivo, encompassing proteases, receptors, ion channels, nucleic acids (DNA and RNA), and other biological macromolecules [86-88]. The identification of drug targets is crucial for researching and developing novel cancer therapies. Omics technology has become increasingly recognized by scientists for its role in unbiased target prediction and has been widely adopted in the field. Chemical proteomics, a multidisciplinary approach that combines chemical synthesis, mass spectrometry (MS) and cell biology, offers an unbiased platform for identifying small-molecule-protein interactions across a range of natural products and compounds, and has been successfully applied in prominent compounds such as artemisinin, celastrol, curcumin, aspirin, and eupalinolide B [89-93]. This summary presents the available probes (Figure 2) and the reported targets of celastrol, along with its mechanism of action (MOA), to enhance the understanding of celastrol targets and its pharmacological actions on cancer [93-96].
Chemical structures of celastrol and its available probes.
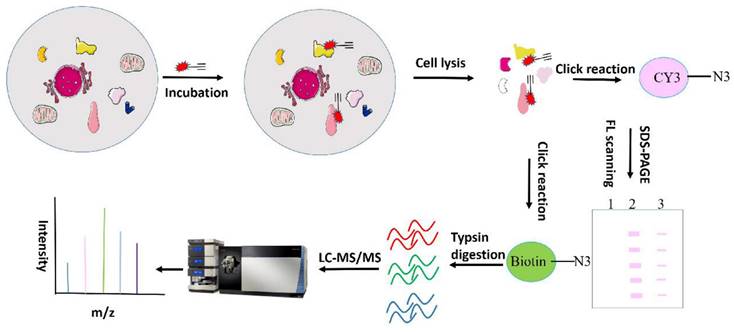
Zhou Y et al. have identified over 60 celastrol-binding proteins using iodoacetamide-derived cysteine reaction probes (iodoacetamide-alkyne, IA-yne) via competitive chemical proteomics in human cervical cancer HeLa cells [97]. Bioinformatics analysis revealed that celastrol exerts anti-tumor effects by promiscuously interacting with numerous proteins engaged in various cellular pathways and biological processes. Among the identified targets, only three (Hsp90 co-chaperone Cdc37, Annexin A2, and elongation factor 1-alpha 1 (eEF1A)) were consistent with previously-reported celastrol-binding proteins, likely due to celastrol's relatively low protein alkylation efficiency. Another study synthesized a clickable photoreactive probe for comprehensive analysis of targets and identified 700 celastrol-binding proteins, among which celastrol targeted various proteins associated with the biosynthesis, regulation and transport of cholesterol and its metabolites, and is involved in cholesterol metabolism and signaling [95]. Klaić and colleagues synthesized and developed an active biotinated celastrol probe and performed affinity pull-down experiments with PANC-1 cell lysates to identify cell targets of celastrol, in which Annexin II and eEF1A were identified. Notably, HSP90 was not determined to be a direct target in their study, but was indirectly targeted by triggering redox imbalances [93]. By utilizing an alkyne-linked Cel-p, our research group previously investigated the covalent binding targets and underlying mechanisms of celastrol in liver fibrosis, ischemia stroke, colon cancer and obesity depression comorbidity (79,98-100). We will next discuss the identified celastrol targets and their roles in tumors. A flowchart for chemical proteomics analysis of celastrol is shown in Figure 3.
Flow chart of chemical proteomics related to celastrol.
3.1 Celastrol targets HSP90 for anti-tumor effects
Heat shock proteins (HSPs) are considered to be involved in important molecular processes related to cancer development and spread, and are potential clinical targets for cancer treatment and biomarkers for cancer detection. HSP90, a key molecular chaperone, facilitates the folding of various proteins into their mature and stable forms. Given the vital role of HSP90 and its client proteins in tumors, celastrol as a HSP90-targeting compound could represent an important novel regulator of the HSP90 pathway [101]. In fact, celastrol treatment is known to lead to the accumulation of ubiquitinated proteins [102], and this accumulation may be due to the inhibition of HSP90 and the stress response, as well as the subsequent redirection of proteins through the proteasome pathway [103]. Sreeramulu et al. discovered that celastrol covalently binds to Cdc37 and forms disulfide bonds with it, inactivating Cdc37 and disrupting the HSP90-Cdc37 complex [104]. Moreover, researchers synthesized a celastrol analog based on its structure and found that the analog significantly disrupted HSP90-Cdc37 protein interaction and induced apoptosis in cancer cells by binding (hydrogen and/or covalent bonding) to Cdc37 [105]. Owing to their ATP-dependent nature, HSP90 inhibitors typically target ATP-binding pockets, leading to the non-selective degradation of HSP90 client proteins. Celastrol however is known to trigger oligomerization into amyloid fibrils by binding to the cochaperone protein p23, selectively destabilizing steroid receptors over kinase clients [106]. Our mass spectrometry research also showed that celastrol directly binds to HSP90 and HSP70, exerting antitumor and neuroprotective effects [99,107]. These findings suggest that celastrol may inhibit tumor growth by disrupting the interaction between HSP90 and its client proteins. Although celastrol has been reported to target HSP90 or affect the interaction between HSP90 and its service proteins, more in-depth studies are needed to elucidate how celastrol affects HSP90 and its client proteins, which in turn affect downstream signaling pathways to exert anti-tumor effects.
3.2 Celastrol exerts anti-tumor activity by inhibiting STAT3
We previously mentioned that STAT3 plays a central role in modulating anti-tumor immune responses as an intersection of many carcinogenic signaling pathways [78]. Through the integration of proteome microarrays, competitive binding, molecular simulation, and surface plasmon resonance (SPR) techniques, STAT3 was identified as a direct binding target of celastrol. Celastrol appears to bind to the coiled-coil domain (CCD, Leu-207) and SH2 domain (Gln-635/Val-637) of STAT3, reducing its tyrosine phosphorylation and nuclear translocation. Interestingly, the overexpression of STAT3 partially counteracted the inhibitory effect of celastrol on STAT3 activity. These findings suggest that celastrol suppresses angiopoietin II-induced hypertrophy and fibrosis in rat primary cardiomyocytes and H9C2 cells by attenuating STAT3 activity [108]. Radhamani et al. observed that celastrol downregulateed phosphorylated STAT3 levels in a dose- and time-dependent manner, significantly inhibiting the translocation of STAT3 from the cytoplasm to the nucleus. Since STAT3 activation is mediated by soluble tyrosine kinases from the Src kinase family [109], this study further explored celastrol's effect on the constitutive activation of Src kinase in U266 cells, and reported that celastrol inhibited Src kinase phosphorylation over time [109]. Given that the cytokine IL-6 is a primary stimulator and inflammatory mediator of STAT3, promoting tumor survival by blocking programmed cell death, treatment with celastrol was shown to inhibit IL-6-induced STAT3 phosphorylation in a time-dependent manner, reducing both constitutive and inducible STAT3 activation [20]. Similarly, another study also showed that celastrol affects the IL-6/STAT3 signaling pathway in NSCLC cells. After treatment of H460, H520 and PC-9 cells with IL-6 and celastrol, IL-6 stimulation of STAT3 was reversed in a dose-dependent manner by celastrol. Immunofluorescence staining analysis also showed that IL-6 treatment induces cytoplasmic-nuclear P-STAT3 transfer, whereas celastrol restrained this transfer [41]. Therefore, we hypothesize that celastrol may have potential for the treatment of various cancers through its interaction with STAT3.
3.3 Nur77 is a potential anti-tumor target of celastrol
Nuclear receptors (NRs) are a large superfamily of post-transcriptional factors involved in various biological processes, making them significant pharmacological targets. Various diseases, including diabetes, cardiovascular disease and cancer have been associated with compromised NR activity [110]. Nur77, also known as NR4A1/TR3/NGFIB, features a typical nuclear receptor structure, and its expression and localization are closely related to its roles in apoptosis and cell proliferation. Although Nur77 is considered an orphan receptor owing to the unidentified nature of its endogenous ligand, an increasing number of small molecules that target Nur77, such as celastrol, have demonstrated therapeutic potential in cancer and other diseases [110].
Research indicates that celastrol binds to Nur77 via a specific non-covalent interaction, positioning celastrol near the sulfhydryl group (C551) of the active cysteine to form a reversible covalent bond [111]. Further studies revealed that celastrol induces ubiquitination of Nur77's C-terminal LBD and its interaction with p62, resulting in the formation of Nur77-LBD/p62 condensates. This process also necessitated further interaction between Nur77's N-terminal IDR and p62 PB1, highlighting the importance of coordinated multivalent interactions between Nur77 and p62 [112]. Further investigations have shown that celastrol facilitates the transport of Nur77 from the nucleus to the mitochondria, where it engages with tumor necrosis factor receptor-associated factor 2 (TRAF2) to inhibit TNF-α-induced ubiquitination of TRAF2 and Lys63-linked ubiquitination of Nur77. This ubiquitinated Nur77 then interacts with p62/SQSTM1, inducing autophagy to remove damaged mitochondria. Additionally, the interaction of celastrol-induced Nur77 with TRAF2 obstructs the IKK-NF-κB pathway by curbing IKK-α/β phosphorylation and reducing the TNF-α-induced degradation of iκBα, thereby suppressing inflammation, and promoting autophagy [113]. In addition, researchers have shown that Nur77 is one of the targets of celastrol and is associated with anti-colorectal cancer activity. Celastrol may upregulate Atg7 by targeting Nur77, resulting in increased autophagy, thus playing an anti-tumor role, which may provide a new understanding of the anti-tumor effect of celastrol in CRC [47]. Overall, Nur77 is a vital target of celastrol, and more studies are needed to determine how celastrol targets Nur77 for its anti-tumor effects.
3.4 Celastrol and Nedd4
Nedd4 is an important E3 ubiquitin ligase protein, which is primarily involved in ubiquitination-dependent protein degradation in the lysosome, proteasome, and endoplasmic reticulum [114]. Recent research has highlighted the critical role of Nedd4 in tumorigenesis by regulating various downstream signaling pathways. Salah et al. reported that Nedd4 directly interacts with and ubiquitinates LATS1, reducing LATS1 levels which inhibits the activity of the Hippo signaling pathway and suppresses tumorigenicity [115]. Furthermore, Nedd4 is known to counteract the carcinogenic Notch signaling pathway by promoting the degradation of Notch and Deltex [116]. Our recent study demonstrated that celastrol mitigated oxidative damage in cerebral ischemia-reperfusion injury (CIRI) by boosting the expression of nuclear factor E2-associated factor 2 (Nrf2). Proteomic methods showed that Nedd4 was the direct target of celastrol, and that significantly blocks ubiquitination of the Nedd4-K48 link and inhibits the degradation of Nrf2, thereby reducing the production of reactive oxygen species by astrocytes in CIRI and exerting a protective effect on neurons [117].
3.5 Celastrol and peroxiredoxin proteins
Peroxiredoxins (Prdxs) are a ubiquitous and highly conserved family of redox regulatory proteins capable of eliminating various reactive oxygen species (ROS). This family, consisting of mercaptan-dependent peroxidase enzymes, degrades hydrogen peroxide and plays a crucial role in maintaining and reacting to altered cellular redox homeostasis [11]. The relationship between oxidative stress and cancer is complex, as normal cells rely on antioxidant systems to limit oxidant production, neutralize active substances, and repair oxidative damage. Disruption of these systems may lead to cancer [118].
Recent studies have demonstrated that celastrol targets Prdx proteins, which play critical roles in liver fibrosis and tumor management both in vitro and in vivo [38,98,119-120]. A previous study suggested that celastrol might play an anti-inflammatory role by activating the AMPK-SIRT3 pathway to prevent liver fibrosis, yet the direct targets and precise mechanisms of celastrol remain unclear [121]. Our research group developed an activity-based celastrol-probe (cel-p) to explore the direct targets of celastrol in liver fibrosis. We found that celastrol enhances ROS-mediated signaling pathways and induces ferroptosis in hepatic stellate cells (HSCs), effectively mitigating liver injury and fibrosis caused by carbon tetrachloride (CCl4). Through activity-based protein profiling (ABPP), we discovered that celastrol binds directly to Prdx1, Prdx2, Prdx4, and Prdx6 via active cysteine sites, inhibiting their antioxidant activity without affecting protein expression. Celastrol also targeted heme oxygenase-1 (HO-1), increasing its expression in activated hematopoietic stem cells. Knockdown of these target proteins in hematopoietic stem cells led to increased cell ROS levels and induced ferroptosis. Thus, our research identified specific protein targets and molecular mechanisms by which celastrol ameliorates liver fibrosis, laying a theoretical foundation for its potential as a therapeutic agent. Chen et al. combined biotin-labeled celastrol (bio-celastrol) with recombinant proteins on a HuProtTM human protein microarray to identify potential celastrol-binding proteins in gastric cancer cells. They reported that celastrol bound directly to Prdx2, inhibiting its enzyme activity, increasing cellular ROS levels and leading to ROS-dependent mitochondrial dysfunction, endoplasmic reticulum stress and apoptosis [38].
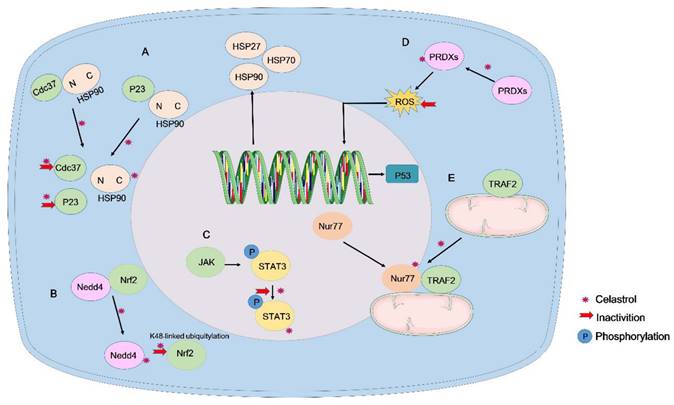
Chen et al. developed a new computational tool, OTTER, and combined it with experimental analyses to determine that Prdx1 is a target protein of celastrol that regulates ROS in colorectal cancer. They resolved the high-resolution crystal structure of Prdx1 bound to celastrol and synthesized a novel derivative, 19-048, with strong anti-Prdx1 activity and selectivity towards Prdx2~Prdx6. Treatment with celastrol or its analogs upregulated the expression of cell cycle arrest and apoptosis-related genes in the p53 signaling pathway, effectively inhibiting the proliferation and inducing apoptosis in colorectal cancer cells [120]. Consequently, the anti-tumor effect of celastrol may be attributed to the inhibition of peroxiredoxin activation. The multifaceted activity of celastrol could explain its therapeutic effects across various cancers (Figure 4).
Direct binding targets of celastrol. (A) Celastrol covalently binds to the HSP90 co-chaperones Cdc37 and p23 to disrupt the Cdc37-HSP90 or p23-HSP90 complex. (B). Celastrol directly binds to Nedd4 and inhibits the interaction between Nedd4 and Nrf2, reducing the K48-linked ubiquitylation of Nrf2. (C). Celastrol directly binds and inhibits STAT3 tyrosine phosphorylation and nuclear translocation. (D). Celastrol targets Prdx1 to inhibit ROS production and suppress the proliferation of colorectal cancer cells. (E). Celastrol covalently binds to Nur77 and induces Nur77 interaction with TRAF2 to inhibit the classical IKK/NF-κB pathway.
Celastrol exerts its antitumor effects by binding to different targets. However, the synergism or antagonism between different targets has seldom been investigated. It was found that luteolin reduced the binding of HSP90 to STAT3 and released the phosphorylated STAT3 from the HSP90 chaperone proteasome, which may lead to the simultaneous exposure of phosphorylated STAT3 to phosphatases and proteases. This leads to a subsequent decrease in the level of STAT3 phosphorylation, thus inhibiting the viability of gastric cancer cells with inactivation of STAT3 and exerting anti-tumor effects [122-123]. XL888, an HSP90 inhibitor, attenuates the formation of the HSP90 and STAT3 complex, leading to a decrease in the expression levels of STAT3 and p-STAT3. It also inhibits the expression levels of Mcl-1 and cleaved-caspase 3 in vivo and in vitro, and promotes heat-induced apoptosis in HCC cells [124]. While there are currently no studies on the interaction between the targets of celastrol, we have hypothesized that celastrol may also play an anti-tumor role by reducing the binding of HSP90 to STAT3 and reducing STAT3 phosphorylation, and we will conduct experiments to verify our conjecture in the future.
4. Drug combination and bioavailability
4.1 Drug combinations
As an active component of Chinese medicine with promising anti-tumor prospects, the clinical application of celastrol is nevertheless limited by its severe hepatic and renal toxicity and low bioavailability [15]. Currently, combination therapy has emerged as the preferred clinical treatment for various tumors [125,126], enhancing the efficacy of individual drugs and reducing their dosage and side effects, thereby improving patient tolerance. Additionally, the diverse mechanisms of action of different drugs minimize the risk of tumor cells developing resistance, making combination therapy an effective strategy to combat drug resistance [127]. Hence, combining celastrol with other agents offers a novel avenue for cancer treatment.
The anti-tumor effects of celastrol are potentiated when used in conjunction with other therapeutic agents. Research has indicated that celastrol, combined with 17-N-Allylamino-17-demethoxygeldanamycin (17-AAG), inhibits the toxic stress response of HSP90-targeted proteins, reduces the sensitization of human glioblastomas to celastrol treatment, and increases the accumulation of polyubiquitylated aggregates and p62, thereby enhancing protein stability and overcoming multidrug resistance (MDR) in glioblastoma [128]. Similarly, another study revealed that celastrol, when combined with EGFR tyrosine kinase inhibitors (EGFR-TKIs), effectively inhibits the growth and invasion of T790M mutant human lung cancer H1975 cells by suppressing the expression and phosphorylation of EGFR, STAT3, p-Akt, and p-ERK [129]. These findings suggest that blocking the p-EGFR pathway could inhibit cancer cell invasion. In vivo experiments further confirmed that celastrol, when used with EGFR-TKIs, exhibited a significantly greater inhibitory effect on tumor growth than either agent alone. Moreover, when celastrol is used in combination with histone deacetylase inhibitors (HDACis), it upregulates H4K16 acetylation (H4K16ac), modulates H3K4 trimethylation and H3S10 phosphorylation, synergistically inhibiting cancer cell proliferation. Celastrol also significantly boosts the efficacy of HDACis in suppressing mouse allogeneic lung cancer cell grafts by upregulating H4K16ac [130]. These findings suggest that the combination of celastrol and HDACis could represent a new therapeutic approach in cancer treatment. In summary, the results above hint to us that the synergistic sensitization of celastrol with other drugs may result in better anti-tumor outcomes.
4.2 Celastrol bioavailability
While celastrol demonstrates promising antitumor activity, its clinical application is hindered by several physicochemical and pharmacokinetic challenges, such as low water solubility, low bioavailability, and inadequate targeting [131]. The advent of nanotechnology in drug delivery has introduced innovative approaches to cancer treatment. Nano formulations are characterized by their robust drug loading capacity, prolonged circulation in vivo, and the ability to target tumor tissues either passively or actively through control over particle size, structural modifications, and surface alterations [132]. This emerging field offers potential solutions to enhance the therapeutic efficacy, oral bioavailability, and tissue targeting capabilities of celastrol [133].
Choi et al. formulated composite nanoparticles loaded with celastrol/aceitinib by loading celastrol into mesoporous silica nanoparticles and aceitinib into a pegylated lipid bilayer, which was approximately 120 nm in size and had a narrow polydispersity index (about 0.07). In vitro cytotoxicity experiments showed that the composite nanoparticles effectively inhibited angiogenesis and mitochondrial function, and effectively internalized rat squamous cell carcinoma SCC-7 cells, human breast cancer BT-474 cells and human neuroblastoma SH-SY5Y cells. Meanwhile, in the heterogeneous tumor model, the tumor inhibition rate of the composite nanoparticle group was significantly greater than that of the single agent group, reaching 64% [134]. Liu et al. developed an innovative chemo-immune strategy based on targeted delivery of mitoxantrone and celastrol, and found that when mitoxantrone and celastrol were effectively co-delivered to the tumor site at the optimal ratio (5:1), immunogenic tumor cell apoptosis was significantly triggered and tumor antigen recognition was restored. This approach stimulated overall anti-tumor immunity, reducing drug dosage and adverse reactions through synergistic effects. The nano-vector-mediated chemo-immunotherapy successfully altered the fibrotic and immunosuppressed tumor microenvironment, impeding tumor growth and further inhibiting metastasis to major organs [135]. Another investigation developed novel celastrol-loaded poly (ε-caprolactone) nanoparticles (cel-NPs) to assess their antitumor effects on prostate cancer cells. These cel-NPs dose-dependently inhibited cell proliferation across all the tested prostate cancer cell lines, particularly at low/medium doses (0.5 and 1.0 µM), and significantly enhanced the cytotoxicity against the DU145 and PC3 cell lines by modulating apoptosis and cell cycle regulatory proteins [136]. Moreover, a self-assembled nanomedicine was made by coupling celastrol with p-selectin targeting peptide (PSN) and low molecular heparin (LMWH). The results of in vivo and in vitro experiments confirmed that the prepared nanomedicine enhanced the anti-tumor and anti-metastatic ability of celastrol and significantly reduced organ toxicity [137].
In summary, nanotechnology-based drug delivery systems offer a versatile platform for the simultaneous transport of one or more drugs, enhancing drug solubility, pharmacokinetic and pharmacological properties, and facilitating targeted drug delivery while minimizing accumulation in non-targeted organs. Therefore, nano-delivery systems present a novel pathway for the development and clinical application of celastrol, potentially overcoming existing limitations and maximizing its therapeutic potential. Strategies to improve the bioavailability of celastrol are shown in Figure 5 and Table 2.
Strategies to improve the bioavailability of celastrol. We summarize the methods used to ameliorate the shortcomings of low bioavailability, low water solubility and inadequate targeting of celastrol, including drug combination, nano-delivery and target identification.
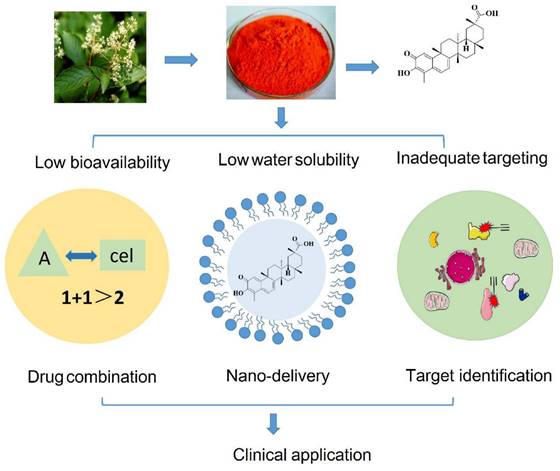
Strategies to improve the bioavailability and reduce the toxicity of celastrol.
| Strategy | Drugs/ Drug delivery | Cancer Types | Concentration/IC50 In Vitro/Drug Dose in Vivo | Mechanism of Action | Aim | Ref. |
|---|---|---|---|---|---|---|
| Drug combination | Celastrol and 17-AAG /direct | Glioblastoma | celastrol alone LD50: 1.03±0.12 μM without 17-AAG versus 0.69±0.11 μM with 17-AAG | increased the accumulation of polyubiquitylated aggregates and p62, thereby enhancing protein stability and overcoming multidrug resistance (MDR) in glioblastoma | enhanced the antitumor efficacy and reduce the toxicity | [128] |
| Celastrol and HDACi/direct | Lung cancer | Erlotinib (100 mg/kg/day/mouse), celastrol (1 mg/kg/day/ mouse), or erlotinib + celastrol | suppressed the expression and phosphorylation of EGFR, STAT3, p-Akt, and p-ERK | enhanced the antitumor effects of EGFR-TKI on NSCLC. | [129] | |
| Celastrol and HDACi/direct | Lung cancer | MS275 (5 mg/kg), celastrol (3 mg/kg), celastrol plus MS275 (3 mg/kg + 5 mg/kg, for 4 weeks | celastrol significantly boosts the efficacy of HDACi in suppressing mouse allogeneic lung cancer cell grafts by upregulating H4K16ac | enhanced the suppressive effects of HDACi on lung cancer | [130] | |
| Nano formulations | Celastrol and aceitinib/ nanoparticles | multi-targeted cancer | Injected with 1 mg/kg of aceitinib, celastrol, aceitinib+celastrol cocktail | inhibited angiogenesis and mitochondrial function, | the tumor inhibition rate of the composite nanoparticle group was significantly higher than that of the single agent, reaching 64%. | [134] |
| Celastrol and mitoxantrone/ nanocarrier | Desmoplastic Melanoma | celastrol alone IC50: 2.5 μM; mitoxantrone alone IC50:16 μM; celastrol + mitoxantrone (5:1) IC50:0.9/4.5 μM | triggered apoptosis of immunogenic tumor cells and restoration of tumor antigen recognition | reduced drug dosage and adverse reactions | [135] | |
| celastrol/ nanoparticles | prostate cancer | celastrol IC50: <2.5 μM; cel-NPs IC50: 0.5 or 1 μM | modulated apoptosis and cell cycle regulatory proteins | enhanced the cytotoxicity against DU145 and PC3 cell lines | [136] | |
| celastrol/ nanodrug | primary and metastatic cancer | celastrol formulations (Cel, LC-NP, and PLC-NP) at a dose of 2 mg/kg every day for a total of 5 times. | inhibited PI3K/Akt/mTOR signaling pathway and ROS-mediated mitochondrial dysfunction | enhanced the anti-tumor and anti-metastatic ability of and reduced organ toxicity | [137] |
5. Side effects of celastrol
Currently, celastrol is mainly used in the clinical treatment of rheumatoid arthritis, ankylosing spondylitis, glomerulonephritis, lupus erythematosus and various skin diseases in the form of celastrol tablets, celastrol polyglycoside tablets, and celastrol extract tablets. Despite its therapeutic applications, the use of celastrol in cancer treatment is impeded by several significant challenges, including its pronounced hepatic and renal toxicity and potential for causing immunosuppression [15]. Here, we summarize the toxicity of celastrol, which includes hepatotoxicity, cardiotoxicity, infertility toxicity, hematopoietic system toxicity and nephrotoxicity. The major toxicities of celastrol are shown in Figure 6.
The main toxicities of celastrol include hepatotoxicity, cardiotoxicity, infertility toxicity, hematopoietic system toxicity and nephrotoxicity.
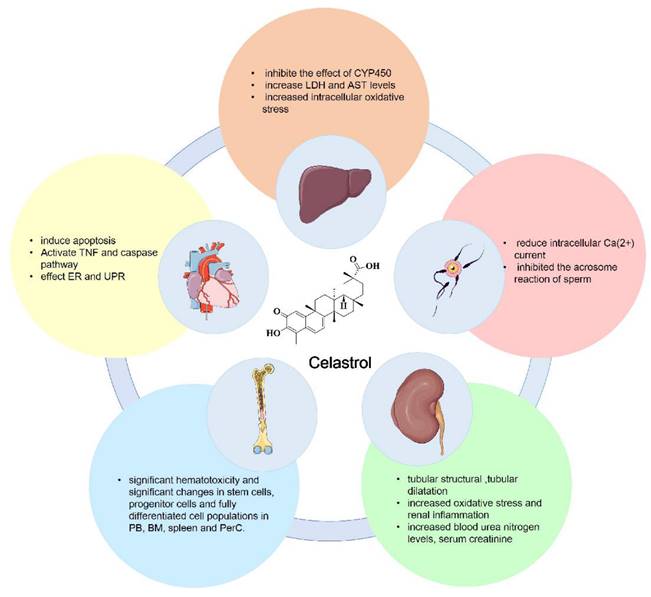
5.1 Hepatotoxicity
Cytochrome P450 (CYP450) is part of a membrane-bound hemoprotein family that functions as a metabolic enzyme crucial for the detoxification, metabolism, and homeostasis of exogenous substances. Its induction or inhibition can alter metabolic pathways in the body and even affect the effectiveness and safety of drugs [138]. The hepatotoxic effects of celastrol were examined by assessing the impact of CYP450 inhibition following 24 hours of administration in vitro [139]. Treatment with celastrol substantially reduced the viability of rat primary hepatocytes, leading to increased levels of AST and LDH, as well as heightened intracellular oxidative stress. Phenobarbital (a CYP450 enzyme attractant) reduces the hepatotoxicity of celastrol, whereas 1-aminobentriazole (a broad-spectrum inhibitor of cyp450) potentiates the hepatotoxicity of celastrol [140].
5.2 Cardiotoxicity
Utilizing mass spectrometry-based metabolomics, network toxicology strategies and molecular biology, the mechanisms underlying celastrol-induced cardiac damage have been elucidated. The findings revealed that celastrol treatment significantly increased plasma palmitic acid levels in rats, leading to an oxidative stress response in vivo. This activation further stimulated the TNF signaling pathway and the caspase family, which induces apoptosis. Celastrol dose-dependently induced cardiac dysfunction in mice, manifesting as cardiomyocyte hypertrophy, left ventricular dilation and myocardial interstitial fibrosis, and significantly reduced the activity and promoted apoptosis of neonatal ventricular myocytes (NRVMs) in rats. More importantly, celastrol was found to exert its pro-apoptotic effect through ER stress and the unfolded protein response [141]. Additionally, a study utilizing a zebrafish model to assess the toxicity of celastrol in vivo found that administering different concentrations of celastrol to embryos for 120 hours post-fertilization significantly reduced the incubation rate of embryos at concentrations of 1.0 μM or higher. This led to severe edema in the larval sac, tail bending or hook-like tail deformities, and pericardial edema [142]. The above studies indicate that celastrol has obvious cardiotoxicity, suggesting that the cardiovascular effects of celastrol should be carefully evaluated during clinical use.
5.3 Infertility toxicity
Bai and colleagues investigated the impact of celastrol on Ca2+ channels and the acrosome reaction in mouse spermatogenic cells via whole-cell patch-clamping and chlortetracycline staining. Their findings demonstrated that celastrol significantly reduced intracellular Ca2+ currents in a time-dependent and irreversible manner, additionally inhibiting the progesterone-induced acrosome reaction in sperm. These observations suggest that celastrol may exhibit anti-fertility effects by impeding Ca2+ currents [143].
5.4 Hematopoietic system toxicity
Researchers have explored the hematotoxic effects of celastrol by administering 5 mg/kg celastrol intraperitoneally to 8-10-week-old BALB/c mice daily. After four days, the mice in the administered group developed a hunched posture and wrinkled fur compared with those in the control group, and cells from the peripheral blood (PB), bone marrow (BM), spleen, and peritoneal cells (PerC) of mice were collected and analyzed. The results showed that celastrol treatment led to significant hematotoxicity and marked alterations in stem, progenitor, and fully differentiated cell populations in the PB, BM, spleen, and PerC of mice. These results underscore the importance of understanding the toxicity and side effects of celastrol to better inform its clinical use [144].
5.5 Nephrotoxicity
Some research has demonstrated that treatment with celastrol exacerbates LPS-induced acute kidney injury in mice, which is mainly characterized by damage to renal tubular structure (loss of brush border) and tubular dilatation, increased oxidative stress and renal inflammation, as well as elevated blood urea nitrogen, serum creatinine, neutrophil gelatinase-associated lipocalcin, and kidney injury molecule-1 levels [145]. These findings highlight the nephrotoxic potential of celastrol, urging clinicians to carefully consider its nephrotoxic effects when it is prescribed. Despite the known toxic properties of celastrol, comprehensive data are still scarce. Consequently, further research is necessary to elucidate the mechanism of action of celastrol and its role in various disease models, aiming to gather more detailed and reliable data for a better understanding and application of celastrol in clinical settings.
6. Anti-inflammatory effects of celastrol in other diseases
Celastrol has anti-inflammatory and immunosuppressive effects that can be used to treat a variety of inflammatory and immune disorders, such as asthma, rheumatoid arthritis, inflammatory bowel disease and systemic lupus erythematosus. Studies have reported that celastrol inhibits TNF-α-induced proliferation of fibroblast-like synoviocytes (FLSs) decreases the secretion of pro-inflammatory cytokines while increasing the levels of autophagosomes and expression of LC3-II and Beclin-1 proteins in TNF-α-treated FLSs, and decreases the phosphorylation of mTOR and AKT thereby reducing the degree of rheumatoid arthritis [146]. In addition, celastrol inhibits the repolarization of macrophages to the pro-inflammatory M1 phenotype by modulating the NF-κB and Notch1 pathways, which significantly reduces the secretion of a variety of pro-inflammatory cytokines and inhibits the progression of RA [147]. Celastrol covalently binds to and dissociates the COMMD3/8 complex, a signaling adapter for chemoreceptors, inhibiting B-cell migration, decreasing antibody responses, and halting the progression of RA [148]. Based on the integration of systems pharmacology, proteomics, transcriptomics, and single-cell transcriptomics, it has been revealed that celastrol may exert immunomodulatory effects and thus treat RA by modulating the PI3K/AKT signaling pathway through the regulation of key targets such as TNF and IL6 [149]. Celastrol can also play a therapeutic role in a variety of inflammatory and autoimmune diseases by regulating various signaling pathways and inducing different responses in targets such as nuclear factor kappa B (NF-κB), mitogen-activated protein kinase (MAPK), PI3K Akt-Mtor, Ednrb/Kng1, NLRP3, AMPK-SIRT3, and Ca2+ [150].
7. Discussion
Compared with synthetic compounds, natural products constitute an important part of the pharmaceutical market because of their safety, efficacy, and low cost [7]. Celastrol, a standout natural compound, has emerged as one of the five most promising natural drugs due to its reported effectiveness against a broad spectrum of tumors, including those of the breast, colon, lung, adipose, liver, and pancreas, both in vivo and in vitro. In this review, we conclude that the effective amelioration and treatment of multiple tumors by celastrol is closely related to a variety of molecular mechanisms, such as the inhibition of cell proliferation, the regulation of cell cycle, apoptosis and autophagy, the inhibition of cell invasion and metastasis, and anti-inflammatory, regulation of immunotherapy and angiogenic mechanisms. However, the current research on the anti-tumor effects of celastrol remains lacking, and further in-depth studies are needed in the future to reveal the MOA of celastrol in tumor treatment.
Currently, celastrol is widely used to treat rheumatoid arthritis (RA), ankylosing spondylitis (AS), systemic lupus erythematosus (SLE) and Crohn's disease (CD). Meanwhile, celastrol has also been reported to have great potential for the treatment of tumors, but its poor water solubility, high toxicity, low bioavailability, and rapid metabolism limit its clinical application. In this review, we have summarized the side effects of celastrol, as well as methods to ameliorate these side effects and low bioavailability of celastrol, including combination drugs and nano-administration. In addition, in order to reduce the toxicity of celastrol and improve its bioavailability, Tan and colleagues prepared a mitochondria-targeting nanoparticle by modifying liposome carrier CSOSA with lipophilic cationic CTPP. It has been demonstrated that micelles enable rapid release of celastrol into mitochondria, reduce drug leakage into the cytoplasm and lysosomes, and improving the anti-tumor capacity of celastrol [151]. Additionally, researchers have summarized the major functional groups on celastrol that can be modified, including the C-2/3 oxygen, C-6 hydrogen, C-20 carboxyl, and quinone-methyl portions of the A- and B-rings, as well as their structure-activity relationships, which provide guidance for the development of celastrol analogs or derivatives [152]. In addition, a variety of delivery methods can be used to increase the bioavailability and reduce the toxicity of celastrol. These include glucolipid-celastrol conjugates, nucleic acid aptamer-celastrol conjugates, dendrimers, albumin, polymers, and vesicular carriers [153]. Therefore, celastrol can be used for clinical antitumor applications based on its optimized structural design, reduced toxicity, and improved bioavailability.
8. Conclusion
Alongside improvements in quality of life and increasing lifespans, the incidence of cancer has also increased in modern times, posing an enormous burden in terms of health and economic costs. Despite the proliferation of therapeutic approaches and targeted drugs, cancer remains the leading cause of death worldwide. Therefore, there is an urgent need for further research into new strategies for cancer treatment and the discovery of potential new drugs. This review comprehensively describes the current mechanisms of action and targets of celastrol in cancer, providing promising preclinical evidence for its treatment of cancer. Despite the positive therapeutic potential of celastrol, concerns about its clinical development remain due to its poor pharmacokinetic profile, potential toxicity and lack of long-term data. With the availability of various nano-formulations, combination therapies, analogs and other therapies, the development of celastrol formulations seems promising. However, more information on its dosage, formulation, efficacy, and toxicological profile needs to be obtained before the drug can enter human trials. In conclusion, the present review comprehensively reviews and summarizes the molecular mechanisms and targets of celastrol, providing new research ideas for improving therapeutic strategies, reducing side effects, expanding new fields and establishing a theoretical basis for the further development and clinical application of celastrol.
Abbreviations
TNRSF: TNF receptor superfamily members; CytC: cytochrome c; ROS: reactive oxygen species; MOA: mechanism of action; Prdx2: peroxoredotoxin-2; ER: Endoplasmic reticulum; NSCLC: non-small cell lung cancer; LC3I: Microtubule-associated protein 1 light chain 3I; CXCR: Chemokine receptors; NF-κB: nuclear factor-κB; STAT3: signal transducers and transcriptional activator 3; JNK 1/2: Janus activated kinases -1 and -2; Bcl-2: B cell proportional lymphomato-2; Bax: Bcl-2 associated X; VEGF: vascular endothelial growth factor; HIF-1: hypoxia-inducing factor; VEGFR: Vascular Endothelial Growth Factor Receptor; VDGFR: vascular dermal growth factor receptor; MAPK: Mitogen-activated protein kinase; ERK: Extraneous signal-regulated Kinase; EB: Eupalinolide B; SPR: surface plasmon resonance; CIRI: cerebral ischemia-reperfusion injury; Nrf2: nuclear factor E2-associated factor 2; Prdxs: Peroxredoxins; ABPP: Activity-Based Protein Profiling; HO-1: heme oxygenase-1; FLSs: fibroblast-like synoviocytes.
Acknowledgements
We thank the other members of Jigang Wang lab for their comments and discussions.
Funding
This work was supported by the Fundamental Research Funds for the National Key Research and Development Program of China (2020YFA0908000), National Natural Science Foundation (No. 82104480 and 82204672), CACMS Innovation Fund (CI2023E002), the Central Public Welfare Research Institutes (ZZ13-YQ-105, ZZ13-YQ-103, ZZ14-YQ-058, ZZ15-YQ-065), the Science and Technology Foundation of Shenzhen (Shenzhen Clinical Medical Research Center for Geriatric Diseases), the Shenzhen Medical Research Fund (B2302051), and the National Natural Science Foundation of Shenzhen (JCYJ20230807111911024).
Author contributions
Yongping Zhu: Writing-review & editing, Writing-original draft, Conceptualization. Yanqing Liu: Writing-review & editing, Conceptualization. Yuqing Meng: Writing-original draft. Jigang Wang: Writing-review & editing, Writing-original draft, Funding acquisition, Conceptualization, Supervision. Junzhe Zhang: Writing-review. Rui Liu: Writing-original draft. Shengnan Shen: Writing-review & editing. Liwei Gu: Writing- review & editing. Yin-kwan Wong: Writing-review. Ang Ma: Writing-review & editing. Xin Chai: Writing-review & editing. Ying Zhang: Writing-review & editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209-49
2. Schaue D, McBride WH. Opportunities and challenges of radiotherapy for treating cancer. Nat Rev Clin Oncol. 2015;12:527-40
3. Wyld L, Audisio RA, Poston GJ. The evolution of cancer surgery and future perspectives. Nat Rev Clin Oncol. 2015;12:115-24
4. Galmarini D, Galmarini CM, Galmarini FC. Cancer chemotherapy: a critical analysis of its 60 years of history. Crit Rev Oncol Hematol. 2012;84:181-99
5. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18:175-96
6. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17:807-21
7. Cascão R, Fonseca JE, Moita LF. Celastrol: A Spectrum of Treatment Opportunities in Chronic Diseases. Front Med (Lausanne). 2017;4:69
8. Corson TW, Crews CM. Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell. 2007;130:769-74
9. Luo P, Zhang Q, Zhong TY, Chen JY, Zhang JZ, Tian Y. et al. Celastrol mitigates inflammation in sepsis by inhibiting the PKM2-dependent Warburg effect. Mil Med Res. 2022;9:22
10. Xu S, Feng Y, He W, Xu W, Xu W, Yang H. et al. Celastrol in metabolic diseases: Progress and application prospects. Pharmacol Res. 2021;167:105572
11. Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999-1011
12. Ma X, Xu L, Alberobello AT, Gavrilova O, Bagattin A, Skarulis M. et al. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1-PGC1α transcriptional axis. Cell Metab. 2015;22:695-708
13. Enkatesha SH, Dudics S, Astry B, Moudgil KD. Control of autoimmune inflammation by celastrol, a natural triterpenoid. Pathog Dis. 2016;74:ftw059
14. Pang X, Yi Z, Zhang J, Lu B, Sung B, Qu W. et al. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010;70:1951-9
15. Wang C, Dai S, Zhao X, Zhang Y, Gong L, Fu K. et al. Celastrol as an emerging anticancer agent: Current status, challenges and therapeutic strategies. Biomed Pharmacother. 2023;163:114882
16. Dai Y, DeSano JT, Meng Y, Ji Q, Ljungman M, Lawrence TS. et al. Celastrol potentiates radiotherapy by impairment of DNA damage processing in human prostate cancer. Int J Radiat Oncol Biol Phys. 2009;74:1217-25
17. Thompson N, Lyons J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr Opin Pharmacol. 2005;5:350-6
18. Li X, Wang H, Ding J, Nie S, Wang L, Zhang L. et al. Celastrol strongly inhibits proliferation, migration and cancer stem cell properties through suppression of Pin1 in ovarian cancer cells. Eur J Pharmacol. 2019;842:146-56
19. Xu LN, Zhao N, Chen JY, Ye PP, Nan XW, Zhou HH. et al. Celastrol Inhibits the Growth of Ovarian Cancer Cells in vitro and in vivo. Front Oncol. 2019;9:2
20. Kannaiyan R, Hay HS, Rajendran P, Li F, Shanmugam MK, Vali S. et al. Celastrol inhibits proliferation and induces chemosensitization through down-regulation of NF-κB and STAT3 regulated gene products in multiple myeloma cells. Br J Pharmacol. 2011;164:1506-21
21. Moreira H, Szyjka A, Paliszkiewicz K, Barg E. Prooxidative activity ofcelastrol induces apoptosis, DNA damage, and cell cycle arrest in drug-resistant human colon cancer cells. Oxid Med Cell Longev. 2019;2019:6793957
22. Li HY, Zhang J, Sun LL, Li BH, Gao HL, Xie T. et al. Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: an in vitro and in vivo study. Cell Death Dis. 2015;6:e1604
23. Sha M, Ye J, Luan ZY, Guo T, Wang B, Huang JX. Celastrol induces cell cycle arrest by MicroRNA-21-mTOR-mediated inhibition p27 protein degradation in gastric cancer. Cancer Cell Int. 2015;15:101
24. Hsieh MJ, Wang CW, Lin JT, Chuang YC, Hsi YT, Lo YS. et al. Celastrol, a plant-derived triterpene, induces cisplatin-resistance nasopharyngeal carcinoma cancer cell apoptosis though ERK1/2 and p38 MAPK signaling pathway. Phytomedicine. 2019;58:152805
25. Shin SA, Moon SY, Park D, Park JB, Lee CS. Apoptotic cell clearance in the tumor microenvironment: a potential cancer therapeutic target. Arch Pharm Res. 2019;42:658-71
26. Abraha AM, Ketema EB. Apoptotic pathways as a therapeutic target for colorectal cancer treatment. World J Gastrointest Oncol. 2016;8:583-91
27. Shen B, Chen HB, Zhou HG, Wu MH. Celastrol induces caspase-dependent apoptosis of hepatocellular carcinoma cells by suppression of mammalian target of rapamycin. J Tradit Chin Med. 2021;41:381-9
28. Ou G, Jiang X, Gao A, Li X, Lin Z, Pei S. Celastrol Inhibits Canine Mammary Tumor Cells by Inducing Apoptosis via the Caspase Pathway. Front Vet Sci. 2022;8:801407
29. Zhang H, Li J, Li G, Wang S. Effects of celastrol on enhancing apoptosis of ovarian cancer cells via the downregulation of microRNA-21 and the suppression of the PI3K/Akt-NF-κB signaling pathway in an in vitro model of ovarian carcinoma. Mol Med Rep. 2016;14:5363-8
30. Ye Y, Aihui Z, Anmao LI. Celastrol promotes apoptotic cell death in children neuroblastoma cells through caspases dependent pathway. J Tradit Chin Med. 2022;42:877-84
31. Sharma S, Carmona A, Skowronek A, Yu F, Collins MO, Naik S. et al. Apoptotic signalling targets the post-endocytic sorting machinery of the death receptor Fas/CD95. Nat Commun. 2019;10:3105
32. Mou H, Zheng Y, Zhao P, Bao H, Fang W, Xu N. Celastrol induces apoptosis in non-small-cell lung cancer A549 cells through activation of mitochondria- and Fas/FasL-mediated pathways. Toxicol In Vitro. 2011;25:1027-32
33. Cha Z, Cheng J, Xiang H, Qin J, He Y, Peng Z. et al. Celastrol enhances TRAIL-induced apoptosis in human glioblastoma via the death receptor pathway. Cancer Chemother Pharmacol. 2019;84:719-28
34. Lin HF, Hsieh MJ, Hsi YT, Lo YS, Chuang YC, Chen MK. et al. Celastrol-induced apoptosis in human nasopharyngeal carcinoma is associated with the activation of the death receptor and the mitochondrial pathway. Oncol Lett. 2017;14:1683-90
35. Chen M, Yang J, Li L, Hu Y, Lu X, Sun R. et al. Metabolomics Reveals that Cysteine Metabolism Plays a Role in Celastrol-Induced Mitochondrial Apoptosis in HL-60 and NB-4 Cells. Sci Rep. 2020;10:471
36. Yu X, Zhou X, Fu C, Wang Q, Nie T, Zou F. et al. Celastrol induces apoptosis of human osteosarcoma cells via the mitochondrial apoptotic pathway. Oncol Rep. 2015;34:1129-36
37. Burke PJ. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer. 2017;3:857-870
38. Chen X, Zhao Y, Luo W, Chen S, Lin F, Zhang X. et al. Celastrol induces ROS-mediated apoptosis via directly targeting peroxiredoxin-2 in gastric cancer cells. Theranostics. 2020;10:10290-308
39. Ren B, Liu H, Gao H, Liu S, Zhang Z, Fribley AM. et al. Celastrol induces apoptosis in hepatocellular carcinoma cells via targeting ER-stress/UPR. Oncotarget. 2017;8:93039-50
40. Feng L, Zhang D, Fan C, Ma C, Yang W, Meng Y. et al. ER stress-mediated apoptosis induced by celastrol in cancer cells and important role of glycogen synthase kinase-3β in the signal network. Cell Death Dis. 2013;4:e715
41. Zhao Z, Wang Y, Gong Y, Wang X, Zhang L, Zhao H. et al. Celastrol elicits antitumor effects by inhibiting the STAT3 pathway through ROS accumulation in non-small cell lung cancer. J Transl Med. 2022;20:525
42. Zhu P, Li T, Li Q, Gu Y, Shu Y, Hu K. et al. Mechanism and Role of Endoplasmic Reticulum Stress in Osteosarcoma. Biomolecules. 2022;12:1882
43. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528-42
44. Debnath J, Gammoh N, Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. 2023;24:560-75
45. Zhang C, Wang W, Du C, Li H, Zhou K, Luan Z. et al. Autophagy in the pharmacological activities of celastrol (Review). Exp Ther Med. 2023;25:268
46. Liu M, Fan Y, Li D, Han B, Meng Y, Chen F. et al. Ferroptosis inducer erastin sensitizes NSCLC cells to celastrol through activation of the ROS-mitochondrial fission-mitophagy axis. Mol Oncol. 2021;15:2084-105
47. Zhang W, Wu Z, Qi H, Chen L, Wang T, Mao X. et al. Celastrol upregulated ATG7 triggers autophagy via targeting Nur77 in colorectal cancer. Phytomedicine. 2022;104:154280
48. Zhang CJ, Zhu N, Long J, Wu HT, Wang YX, Liu BY. et al. Celastrol induces lipophagy via the LXRα/ABCA1 pathway in clear cell renal cell carcinoma. Acta Pharmacol Sin. 2021;42:1472-85
49. Wang WB, Feng LX, Yue QX, Wu WY, Guan SH, Jiang BH. et al. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J Cell Physiol. 2012;227:2196-206
50. Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH, Chun KH. Celastrol inhibits gastric cancer growth by induction of apoptosis and autophagy. BMB Rep. 2014;47:697-702
51. Li X, Wang J. Mechanical tumor microenvironment and transduction: cytoskeleton mediates cancer cell invasion and metastasis. Int J Biol Sci. 2020;16:2014-28
52. Zhou W, Guo S, Liu M, Burow ME, Wang G. Targeting CXCL12/CXCR4 Axis in Tumor Immunotherapy. Curr Med Chem. 2019;26:3026-41
53. Itatani Y, Kawada K, Inamoto S, Yamamoto T, Ogawa R, Taketo MM. et al. The Role of Chemokines in Promoting Colorectal Cancer Invasion/Metastasis. Int J Mol Sci. 2016;17:643
54. Kun-Ming C, Chih-Hsien C, Chen-Fang L, Ting-Jung W, Hong-Shiue C, Wei-Chen L. Potential Anticancer Effect of Celastrol on Hepatocellular Carcinoma by Suppressing CXCR4-related Signal and Impeding Tumor Growth in Vivo. Arch Med Res. 2020;51:297-302
55. Yadav VR, Sung B, Prasad S, Kannappan R, Cho SG, Liu M. et al. Celastrol suppresses invasion of colon and pancreatic cancer cells through the downregulation of expression of CXCR4 chemokine receptor. J Mol Med (Berl). 2010;88:1243-53
56. Si H, Wang H, Xiao H, Fang Y, Wu Z. Anti-Tumor Effect of Celastrol on Hepatocellular Carcinoma by the circ_SLIT3/miR-223-3p/CXCR4 Axis. Cancer Manag Res. 2021;13:1099-111
57. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
58. Zhang J, Wang R, Cheng L, Xu H. Celastrol inhibit the proliferation, invasion and migration of human cervical HeLa cancer cells through down-regulation of MMP-2 and MMP-9. J Cell Mol Med. 2021;25:5335-38
59. Kim Y, Kang H, Jang SW, Ko J. Celastrol inhibits breast cancer cell invasion via suppression of NF-ĸB-mediated matrix metalloproteinase-9 expression. Cell Physiol Biochem. 2011;28:175-84
60. Yu X, Wang Q, Zhou X, Fu C, Cheng M, Guo R. et al. Celastrol negatively regulates cell invasion and migration ability of human osteosarcoma via downregulation of the PI3K/Akt/NF-κB signaling pathway in vitro. Oncol Lett. 2016;12:3423-8
61. Bufu T, Di X, Yilin Z, Gege L, Xi C, Ling W. Celastrol inhibits colorectal cancer cell proliferation and migration through suppression of MMP3 and MMP7 by the PI3K/AKT signaling pathway. Anticancer Drugs. 2018;29:530-8
62. Zhu H, Liu XW, Cai TY, Cao J, Tu CX, Lu W. et al. Celastrol acts as a potent antimetastatic agent targeting beta1 integrin and inhibiting cell-extracellular matrix adhesion, in part via the p38 mitogen-activated protein kinase pathway. J Pharmacol Exp Ther. 2010;334:489-99
63. Lee HE, Lee JY, Yang G, Kang HC, Cho YY, Lee HS. et al. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci Rep. 2019;9:12277
64. Wang S, Hu G, Chen L, Ma K, Hu C, Zhu H. et al. Celastrol acts as a new histone deacetylase inhibitor to inhibit colorectal cancer cell growth via regulating macrophage polarity. Cell Biol Int. 2023;47:492-501
65. Yang F, Guo Z, Shi L, Li Z, Zhang J, Chai C. et al. Antiangiogenic and Antitumor Therapy for Retinoblastoma with Hypoxia-Inducible Factor-1α siRNA and Celastrol Co-Delivery Nanomicelles. J Biomed Nanotechnol. 2020;16:1471-81
66. Qiu N, Liu Y, Liu Q, Chen Y, Shen L, Hu M. et al. Celastrol nanoemulsion induces immunogenicity and downregulates PD-L1 to boost abscopal effect in melanoma therapy. Biomaterials. 2021;269:120604
67. Wang F, Lai W, Xie D, Zhou M, Wang J, Xu R. et al. Nanoparticle-mediated celastrol ER targeting delivery amplify immunogenic cell death in melanoma. J Adv Res. 2024 S2090-1232(24)00248-0
68. Cho O, Lee JW, Jeong YJ, Kim LK, Jung BK, Heo TH. Celastrol, which targets IL-2/CD25 binding inhibition, induces T cell-mediated antitumor activity in melanoma. Eur J Pharmacol. 2024;962:176239
69. Hou J, Karin M, Sun B. Targeting cancer-promoting inflammation - have anti-inflammatory therapies come of age? Nat Rev Clin Oncol. 2021;18:261-79
70. Yadav VR, Prasad S, Sung B, Kannappan R, Aggarwal BB. Targeting inflammatory pathways by triterpenoids for prevention and treatment of cancer. Toxins (Basel). 2010;2:2428-66
71. Kaltschmidt C, Greiner JFW, Kaltschmidt B. The Transcription Factor NF-κB in Stem Cells and Development. Cells. 2021;10:2042
72. Rasmi RR, Sakthivel KM, Guruvayoorappan C. NF-κB inhibitors in treatment and prevention of lung cancer. Biomed Pharmacother. 2020;130:110569
73. Chiang KC, Tsui KH, Chung LC, Yeh CN, Chen WT, Chang PL. et al. Celastrol blocks interleukin-6 gene expression via downregulation of NF-κB in prostate carcinoma cells. PLoS One. 2014;9:e93151
74. Dai Y, Desano J, Tang W, Meng X, Meng Y, Burstein E. et al. Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS One. 2010;5:e14153
75. Shanmugam MK, Ahn KS, Lee JH, Kannaiyan R, Mustafa N, Manu KA. et al. Celastrol Attenuates the Invasion and Migration and Augments the Anticancer Effects of Bortezomib in a Xenograft Mouse Model of Multiple Myeloma. Front Pharmacol. 2018;9:365
76. Wang Z, Zhai Z, Du X. Celastrol inhibits migration and invasion through blocking the NF-κB pathway in ovarian cancer cells. Exp Ther Med. 2017;14:819-24
77. Yan F, Wu Z, Li Z, Liu L. Celastrol Inhibits Migration and Invasion of Triple-Negative Breast Cancer Cells by Suppressing Interleukin-6 via Downregulating Nuclear Factor-κB (NF-κB). Med Sci Monit. 2020;26:e922814
78. Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in Cancer Immunotherapy. Mol Cancer. 2020;19:145
79. Rajendran P, Li F, Shanmugam MK, Kannaiyan R, Goh JN, Wong KF. et al. Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev Res (Phila). 2012;5:631-43
80. Yan YF, Zhang HH, Lv Q, Liu YM, Li YJ, Li BS. et al. Celastrol suppresses the proliferation of lung adenocarcinoma cells by regulating microRNA-24 and microRNA-181b. Oncol Lett. 2018;15:2515-21
81. Qi S, Deng S, Lian Z, Yu K. Novel Drugs with High Efficacy against Tumor Angiogenesis. Int J Mol Sci. 2022;23:6934
82. Al-Ostoot FH, Salah S, Khamees HA, Khanum SA. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat Res Commun. 2021;28:100422
83. Huang Y, Zhou Y, Fan Y, Zhou D. Celastrol inhibits the growth of human glioma xenografts in nude mice through suppressing VEGFR expression. Cancer Lett. 2008;264:101-6
84. Gao Y, Zhou S, Pang L, Yang J, Li HJ, Huo X. et al. Celastrol suppresses nitric oxide synthases and the angiogenesis pathway in colorectal cancer. Free Radic Res. 2019;53:324-34
85. Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG. et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16:19-34
86. Zhang J, Zhang F, Li H, Liu C, Xia J, Ma L. et al. Recent progress and future potential for metal complexes as anticancer drugs targeting G-quadruplex DNA. Curr Med Chem. 2012;19:2957-75
87. Bagal SK, Brown AD, Cox PJ, Omoto K, Owen RM, Pryde DC. et al. Ion channels as therapeutic targets: a drug discovery perspective. J Med Chem. 2013;56:593-624
88. Sharma A, Shah SR, Illum H, Dowell J. Vemurafenib: targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs. 2012;72:2207-22
89. Zhang XW, Feng N, Liu YC, Guo Q, Wang JK, Bai YZ. et al. Neuroinflammation inhibition by small-molecule targeting USP7 noncatalytic domain for neurodegenerative disease therapy. Sci Adv. 2022;8:eabo0789
90. Wang J, Zhang CJ, Chia WN, Loh CC, Li Z, Lee YM. et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun. 2015;6:10111
91. Wang J, Zhang J, Zhang CJ, Wong YK, Lim TK, Hua ZC. et al. In situ Proteomic Profiling of Curcumin Targets in HCT116 Colon Cancer Cell Line. Sci Rep. 2016;6:22146
92. Wang J, Zhang CJ, Zhang J, He Y, Lee YM, Chen S. et al. Mapping sites of aspirin-induced acetylations in live cells by quantitative acid-cleavable activity-based protein profiling (QA-ABPP). Sci Rep. 2015;5:7896
93. Klaić L, Morimoto RI, Silverman RB. Celastrol analogues as inducers of the heat shock response. Design and synthesis of affinity probes for the identification of protein targets. ACS Chem Biol. 2012;7:928-37
94. Wang J, Gao L, Lee YM, Kalesh KA, Ong YS, Lim J. et al. Target identification of natural and traditional medicines with quantitative chemical proteomics approaches. Pharmacol Ther. 2016;162:10-22
95. Geng Y, Xu J, Li W, Li Q, Shen C, Deng Z. et al. Chemoproteomic profiling reveals celastrol as a potential modulator of cholesterol signaling. Chem Commun (Camb). 2022;58:1914-17
96. Zhou Y, Li M, Shen T, Yang T, Shi G, Wei Y. et al. Celastrol Targets Cullin-Associated and Neddylation-Dissociated 1 to Prevent Fibroblast-Myofibroblast Transformation against Pulmonary Fibrosis. ACS Chem Biol. 2022;17:2734-43
97. Zhou Y, Li W, Wang M, Zhang X, Zhang H, Tong X. et al. Competitive profiling of celastrol targets in human cervical cancer HeLa cells via quantitative chemical proteomics. Mol Biosyst. 2016;13:83-91
98. Luo P, Liu D, Zhang Q, Yang F, Wong YK, Xia F. et al. Celastrol induces ferroptosis in activated HSCs to ameliorate hepatic fibrosis via targeting peroxiredoxins and HO-1. Acta Pharm Sin B. 2022;12:2300-14
99. Liu DD, Luo P, Gu L, Zhang Q, Gao P, Zhu Y. et al. Celastrol exerts a neuroprotective effect by directly binding to HMGB1 protein in cerebral ischemia-reperfusion. J Neuroinflammation. 2021;18:174
100. Zhu C, Yang J, Zhu Y, Li J, Chi H, Tian C. et al. Celastrol alleviates comorbid obesity and depression by directly binding amygdala HnRNPA1 in a mouse model. Clin Transl Med. 2021;11:e394
101. Klaić L, Trippier PC, Mishra RK, Morimoto RI, Silverman RB. Remarkable stereospecific conjugate additions to the Hsp90 inhibitor celastrol. J Am Chem Soc. 2011;133:19634-7
102. Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese "Thunder of God Vine," is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758-65
103. Mimnaugh EG, Xu W, Vos M, Yuan X, Isaacs JS, Bisht KS. et al. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol Cancer Ther. 2004;3:551-66
104. Sreeramulu S, Gande SL, Göbel M, Schwalbe H. Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew Chem Int Ed Engl. 2009;48:5853-5
105. Li N, Xu M, Wang B, Shi Z, Zhao Z, Tang Y. et al. Discovery of Novel Celastrol Derivatives as Hsp90-Cdc37 Interaction Disruptors with Antitumor Activity. J Med Chem. 2019;62:10798-815
106. Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A. et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321-30
107. Zhang X, Zhou J, Zhu Y, Wong YK, Liu D, Gao P. et al. Quantitative chemical proteomics reveals anti-cancer targets of Celastrol in HCT116 human colon cancer cells. Phytomedicine. 2022;101:154096
108. Ye S, Luo W, Khan ZA, Wu G, Xuan L, Shan P. et al. Celastrol Attenuates Angiotensin II-Induced Cardiac Remodeling by Targeting STAT3. Circ Res. 2020;126:1007-23
109. Schreiner SJ, Schiavone AP, Smithgall TE. Activation of STAT3 by the Src family kinase Hck requires a functional SH3 domain. J Biol Chem. 2002;277(47):45680-7
110. Wu L, Chen L. Characteristics of Nur77 and its ligands as potential anticancer compounds (Review). Mol Med Rep. 2018;18:4793-801
111. Zhang D, Chen Z, Hu C, Yan S, Li Z, Lian B. et al. Celastrol binds to its target protein via specific noncovalent interactions and reversible covalent bonds. Chem Commun (Camb). 2018;54:12871-4
112. Hu M, Luo Q, Alitongbieke G, Chong S, Xu C, Xie L. et al. Celastrol-Induced Nur77 Interaction with TRAF2 Alleviates Inflammation by Promoting Mitochondrial Ubiquitination and Autophagy. Mol Cell. 2017;66:141-53
113. Peng SZ, Chen XH, Chen SJ, Zhang J, Wang CY, Liu WR. et al. Phase separation of Nur77 mediates celastrol-induced mitophagy by promoting the liquidity of p62/SQSTM1 condensates. Nat Commun. 2021;12:5989
114. Shearwin-Whyatt L, Dalton HE, Foot N, Kumar S. Regulation of functional diversity within the Nedd4 family by accessory and adaptor proteins. Bioessays. 2006;28:617-28
115. Salah Z, Cohen S, Itzhaki E, Aqeilan RI. NEDD4 E3 ligase inhibits the activity of the Hippo pathway by targeting LATS1 for degradation. Cell Cycle. 2013;12:3817-23
116. Sakata T, Sakaguchi H, Tsuda L, Higashitani A, Aigaki T, Matsuno K. et al. Drosophila Nedd4 regulates endocytosis of notch and suppresses its ligand-independent activation. Curr Biol. 2004;14:2228-36
117. Hong Z, Cao J, Liu D, Liu M, Chen M, Zeng F. et al. Celastrol targeting Nedd4 reduces Nrf2-mediated oxidative stress in astrocytes after ischemic stroke. J Pharm Anal. 2023;13:156-69
118. Hampton MB, Vick KA, Skoko JJ, Neumann CA. Peroxiredoxin Involvement in the Initiation and Progression of Human Cancer. Antioxid Redox Signal. 2018;28:591-608
119. Zhao Q, Ding Y, Deng Z, Lee OY, Gao P, Chen P. et al. Natural products triptolide, celastrol, and withaferin A inhibit the chaperone activity of peroxiredoxin I. Chem Sci. 2015;6:4124-30
120. Xu H, Zhao H, Ding C, Jiang D, Zhao Z, Li Y. et al. Celastrol suppresses colorectal cancer via covalent targeting peroxiredoxin 1. Signal Transduct Target Ther. 2023;8:51
121. Wang Y, Li C, Gu J, Chen C, Duanmu J, Miao J. et al. Celastrol exerts anti-inflammatory effect in liver fibrosis via activation of AMPK-SIRT3 signalling. J Cell Mol Med. 2020;24:941-53
122. Song S, Su Z, Xu H, Niu M, Chen X, Min H. et al. Luteolin selectively kills STAT3 highly activated gastric cancer cells through enhancing the binding of STAT3 to SHP-1. Cell Death Dis. 2017;8:e2612
123. Fu J, Chen D, Zhao B, Zhao Z, Zhou J, Xu Y. et al. Luteolin induces carcinoma cell apoptosis through binding Hsp90 to suppress constitutive activation of STAT3. PLoS One. 2012;7:e49194
124. Sun C, Bai M, Ke W, Wang X, Zhao X, Lu Z. The HSP90 inhibitor, XL888, enhanced cell apoptosis via downregulating STAT3 after insufficient radiofrequency ablation in hepatocellular carcinoma. Life Sci. 2021;282:119762
125. Aumeeruddy MZ, Mahomoodally MF. Combating breast cancer using combination therapy with 3 phytochemicals: Piperine, sulforaphane, and thymoquinone. Cancer. 2019;125:1600-11
126. Wang X, Li J, Chen R, Li T, Chen M. Active Ingredients from Chinese Medicine for Combination Cancer Therapy. Int J Biol Sci. 2023;19:3499-525
127. Jaaks P, Coker EA, Vis DJ, Edwards O, Carpenter EF, Leto SM. et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature. 2022;603:166-73
128. Boridy S, Le PU, Petrecca K, Maysinger D. Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis. 2014;5:e1216
129. Wang Y, Liu Q, Chen H, You J, Peng B, Cao F. et al. Celastrol improves the therapeutic efficacy of EGFR-TKIs for non-small-cell lung cancer by overcoming EGFR T790M drug resistance. Anticancer Drugs. 2018;29:748-55
130. Chen G, Zhu X, Li J, Zhang Y, Wang X, Zhang R. et al. Celastrol inhibits lung cancer growth by triggering histone acetylation and acting synergically with HDAC inhibitors. Pharmacol Res. 2022;185:106487
131. Shi J, Li J, Xu Z, Chen L, Luo R, Zhang C. et al. Celastrol: A Review of Useful Strategies Overcoming its Limitation in Anticancer Application. Front Pharmacol. 2020;11:558741
132. Sharma M, Pandey C, Sharma N, Kamal MA, Sayeed U, Akhtar S. Cancer Nanotechnology-An Excursion on Drug Delivery Systems. Anticancer Agents Med Chem. 2018;18:2078-92
133. Guo L, Zhang Y, Al-Jamal KT. Recent progress in nanotechnology-based drug carriers for celastrol delivery. Biomater Sci. 2021;9:6355-80
134. Choi JY, Ramasamy T, Kim SY, Kim J, Ku SK, Youn YS. et al. PEGylated lipid bilayer-supported mesoporous silica nanoparticle composite for synergistic co-delivery of axitinib and celastrol in multi-targeted cancer therapy. Acta Biomater. 2016;39:94-105
135. Liu Q, Chen F, Hou L, Shen L, Zhang X, Wang D. et al. Nanocarrier-Mediated Chemo-Immunotherapy Arrested Cancer Progression and Induced Tumor Dormancy in Desmoplastic Melanoma. ACS Nano. 2018;12:7812-25
136. Sanna V, Chamcheu JC, Pala N, Mukhtar H, Sechi M, Siddiqui IA. Nanoencapsulation of natural triterpenoid celastrol for prostate cancer treatment. Int J Nanomedicine. 2015;10:6835-46
137. Zhou M, Liao J, Lai W, Xu R, Liu W, Xie D. et al. A celastrol-based nanodrug with reduced hepatotoxicity for primary and metastatic cancer treatment. EBioMedicine. 2023;94:104724
138. Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389-430
139. Jin C, Wu Z, Wang L, Kanai Y, He X. CYP450s-Activity Relations of Celastrol to Interact with Triptolide Reveal the Reasons of Hepatotoxicity of Tripterygium wilfordii. Molecules. 2019;24:2162
140. Qiu D, Chu X, Hua L, Yang Y, Li K, Han Y. et al. Gpr174-deficient regulatory T cells decrease cytokine storm in septic mice. Cell Death Dis. 2019;10:233
141. Chen Z, Zhuang Z, Meng C, Zhu Z, Zhang Y, Zhang Z. Induction of the ER stress response in NRVMs is linked to cardiotoxicity caused by celastrol. Acta Biochim Biophys Sin (Shanghai). 2022;54:1180-92
142. Wang S, Liu K, Wang X, He Q, Chen X. Toxic effects of celastrol on embryonic development of zebrafish (Danio rerio). Drug Chem Toxicol. 2011;34:61-5
143. Bai JP, Shi YL, Fang X, Shi QX. Effects of demethylzeylasteral and celastrol on spermatogenic cell Ca2+ channels and progesterone-induced sperm acrosome reaction. Eur J Pharmacol. 2003;464:9-15
144. Kusy S, Ghosn EE, Herzenberg LA, Contag CH. Development of B cells and erythrocytes is specifically impaired by the drug celastrol in mice. PLoS One. 2012;7:e35733
145. Wu M, Chen W, Yu X, Ding D, Zhang W, Hua H. et al. Celastrol aggravates LPS-induced inflammation and injuries of liver and kidney in mice. Am J Transl Res. 2018;10:2078-86
146. Yang J, Liu J, Li J, Jing M, Zhang L, Sun M. et al. Celastrol inhibits rheumatoid arthritis by inducing autophagy via inhibition of the PI3K/AKT/mTOR signaling pathway. Int Immunopharmacol. 2022;112:109241
147. An L, Li Z, Shi L, Wang L, Wang Y, Jin L. et al. Inflammation-Targeted Celastrol Nanodrug Attenuates Collagen-Induced Arthritis through NF-κB and Notch1 Pathways. Nano Lett. 2020;20:7728-36
148. Shirai T, Nakai A, Ando E, Fujimoto J, Leach S, Arimori T. et al. Celastrol suppresses humoral immune responses and autoimmunity by targeting the COMMD3/8 complex. Sci Immunol. 2023;8:eadc9324
149. Zeng L, Yu G, Yang K, He Q, Hao W, Xiang W. et al. Exploring the mechanism of Celastrol in the treatment of rheumatoid arthritis based on systems pharmacology and multi-omics. Sci Rep. 2024;14:1604
150. Fang G, Tang B. Current advances in the nano-delivery of celastrol for treating inflammation-associated diseases. J Mater Chem B. 2020;8:10954-65
151. Tan Y, Zhu Y, Zhao Y, Wen L, Meng T, Liu X. et al. Mitochondrial alkaline pH-responsive drug release mediated by Celastrol loaded glycolipid-like micelles for cancer therapy. Biomaterials. 2018;154:169-81
152. Hou W, Liu B, Xu H. Celastrol: Progresses in structure-modifications, structure-activity relationships, pharmacology and toxicology. Eur J Med Chem. 2020;189:112081
153. Sun Y, Wang C, Li X, Lu J, Wang M. Recent advances in drug delivery of celastrol for enhancing efficiency and reducing the toxicity. Front Pharmacol. 2024;15:1137289
Author contact
![]() Corresponding authors: Yanqing Liu, State Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, Artemisinin Research Center, and Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, E-mail: yqliuac.cn. Jigang Wang, State Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, Artemisinin Research Center, and Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China. Department of Critical Care Medicine, Shenzhen Clinical Research Centre for Geriatrics, Shenzhen People's Hospital, The First Affiliated Hospital, Southern University of Science and Technology, Shenzhen 518020, Guangdong, China. State Key Laboratory of Antiviral Drugs, School of Pharmacy, Henan University, Kaifeng, China. E-mail: jgwangac.cn.
Corresponding authors: Yanqing Liu, State Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, Artemisinin Research Center, and Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, E-mail: yqliuac.cn. Jigang Wang, State Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, Artemisinin Research Center, and Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China. Department of Critical Care Medicine, Shenzhen Clinical Research Centre for Geriatrics, Shenzhen People's Hospital, The First Affiliated Hospital, Southern University of Science and Technology, Shenzhen 518020, Guangdong, China. State Key Laboratory of Antiviral Drugs, School of Pharmacy, Henan University, Kaifeng, China. E-mail: jgwangac.cn.