Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. The individual or potentially...
2. The cGAS-STING pathway in...
3. The PERK-eIF2α in CVDs
4. The Implication of the...
5. Inhibition of the...
6. The nanomedicine for the...
7. Conclusion and Limitations
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2024; 20(15):5868-5887. doi:10.7150/ijbs.101247 This issue Cite
Review
The cGAS-STING/PERK-eIF2α: Individual or Potentially Collaborative Signaling Transduction in Cardiovascular Diseases
Xueqi Wan1#, Huan Zhang1#, Jinfan Tian1#, Libo Liu1, Ziyu An1, Xin Zhao1, Lijun Zhang2, Xueyao Yang1, Changjiang Ge1 ![]() , Xiantao Song1
, Xiantao Song1 ![]()
1. Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart, Lung and Blood Vessel Disease, Beijing 100029, P.R. China.
2. Department of Radiology, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart, Lung and Blood Vessel Disease, Beijing 100029, P.R. China.
# These authors contributed equally to this work and shared the first authorship.
Received 2024-7-31; Accepted 2024-10-19; Published 2024-10-28
Abstract

Over the past several decades, a canonical pathway called the cyclic GMP-AMP (cGAMP) synthase (cGAS)-stimulator of interferon genes (STING) mediating type I interferon (IFN) release via TANK-binding kinase 1(TBK1) / IFN regulatory factor 3 (IRF3) pathway has been widely investigated and characterized. Unexpectedly, recent studies show that the cGAS-STING noncanonically activates the protein kinase RNA-like ER kinase (PERK)-eukaryotic initiation factor 2α (eIF2α), an essential branch of unfolded protein response (UPR), even before the activation of the TBK1/IRF3 signaling. Additionally, we found that the PERK could regulate the STING signaling besides being modulated by upstream cGAS-STING. However, earlier evidence solely focused on the unidirectional regulation of STING and PERK, lacking their functional crosstalk. Hence, we postulate that there is a complex relationship between the cGAS-STING and PERK-eIF2α pathways and that, through convergent downstream signaling, they may collaboratively contribute to the pathophysiology of cardiovascular diseases (CVDs) via the cGAS-STING/PERK-eIF2α signaling axis. This study provides a novel pathway for the development of CVDs and paves the foundation for potential therapeutic targets for CVDs.
Keywords: cGAS-STING, PERK-eIF2α, UPR, CVDs, therapeutics
Introduction
Cardiovascular diseases (CVDs), including atherosclerosis (AS), cardiomyopathy, and myocardial infarction (MI), are prevalent global health concerns. These diseases are associated with high morbidity and mortality [1-3]. The mechanisms underlying the pathogenesis and progression of CVDs are complicated and include biological processes such as cellular proliferation, migration, apoptosis, necrosis, ferroptosis, pyroptosis, macrophage phenotype switching, inflammation, and more [4-6]. Broadly, a constellation of signal transduction pathways determine cellular phenotypes, and aberrant signaling activation associated with CVDs leads to pathological changes and poor clinical prognosis [7].
Prior research has demonstrated that the cyclic GMP-AMP (cGAMP) synthase (cGAS)-stimulator of interferon genes (STING) signaling pathway, which is a conventional immunological and inflammatory system, is associated with numerous CVDs [8-10]. In the traditional signaling pathway, cGAS senses DNA misplaced in the cytosol and responds by synthesizing cyclic guanosine monophosphate adenosine monophosphate (cGAMP), which binds to STING, a small molecule located in the endoplasmic reticulum (ER). This interaction results in the conformation and movement of STING to the ER-Golgi membrane and the subsequent recruitment of TANK-binding kinase 1 (TBK1). TBK1 then phosphorylates IRF3. IRF3, which acts as a transcription factor, elevates the expression of type I interferon (IFN-Is)-stimulated genes (ISGs). The activation of TBK1 also recruits IκB kinase-ε (IKKε), followed by the nuclear factor kappa beta (NF-ΚB) activation. Next, type I interferons (IFN-Is) and other proinflammatory cytokines are promptly released and initiate a series of reactions to defend against the invasion of microbial or other pathogens [11, 12]. The cGAS-STING-TBK1-IRF3 axis has been thoroughly studied and verified as a primary and standard pathway that is involved in a series of diseases. As the connections between STING and ER stress are close, and the PERK is indispensable for the ER stress signaling transduction, there has been a growing focus on the regulation of cGAS-STING for the PERK-eIF2α pathway [13-15].
Protein kinase RNA-like ER kinase (PERK), which is localized to the ER, is a kinase capable of detecting ER distress and then halting translation to prevent protein misfolding. This process is known as the unfolded protein response (UPR). Under normal circumstances, PERK's lumen domain is tethered to glucose-regulated protein (GRP78) and silenced. However, in cases of ER disorder, PERK can detach from GRP78 and undergo phosphorylation, ensuring the initiation of downstream signaling pathways [16-19]. More recent research has also shown that PERK is closely associated with the cGAS-STING pathway [20]. The cGAMP-stimulated STING can also activate PERK, preceding TBK1-IRF3 initiation. This STING-activated PERK then interacts with eukaryotic initiation factor 2α (eIF2α) and participates in a variety of biological activities in the ER [20]. PERK-mediated phosphorylation of eIF2α induced by ER stress and other stimulation ultimately culminates in the attenuation of protein biogenesis and potential cell death [21]. It is reported that the PERK-eIF2α pathway plays a crucial role in CVD pathology [42, 113, 131]. The phosphorylation of eIF2α enables the translation of specific mRNAs that encode activating transcription factor 4 (ATF4), which is an essential transcriptional factor for regulating redox reactions and amino acid metabolism. Excessive levels of ATF4, in turn, dephosphorylate eIF2α by upregulating the protein phosphatase 1 regulatory subunit (e.g., the C terminus) of the growth arrest and DNA damage gene GADD34 to restore protein synthesis and maintain ER protein homeostasis [17, 22, 23]. Remarkably, the PERK-eIF2α complex also partially regulates cGAS-STING signaling, which is essentially involved in host-pathogen interactions. Previous evidence has shown that STING can also be activated via non-canonical pathways that are independent of cGAS conjunction [24, 25]. Additionally, PERK plays a crucial role in regulating STING alone, leading to its translocation and the activation of downstream cascades [26-28].
While several studies have highlighted potential regulatory relationships between the cGAS-STING and PERK-eIF2α pathways, they primarily illustrate a one-way modulation rather than a reciprocal interaction. Both pathways appear to significantly influence various CVDs. However, existing research has largely focused on the actions of individual pathways, neglecting a comprehensive understanding of the combined roles of the cGAS-STING and PERK-eIF2α pathways in CVDs.
We propose that a complex interplay exists between cGAS-STING and PERK-eIF2α signaling, and their potential mutual interactions may contribute to various pathological changes associated with CVDs. Investigating the connections between these pathways and their effects on different CVDs could reveal novel mechanisms and therapeutic targets, ultimately informing more effective clinical treatments.
1. The individual or potentially collaborative interplay of the cGAS-STING/PERK-eIF2α signaling
1.1 Overview of the cGAS-STING pathway
The cGAS-STING pathway is intimately involved in an array of immunological and inflammatory responses, which partially contribute to the pathogenesis and development of many diseases [29, 30]. The initial step of this pathway is the cGAS sensing misplaced DNA from exogenous (e.g., extracellular) microbe or endogenous (e.g., from the mitochondria) cell [31, 32]. The cGAS then binds to double-stranded DNA (dsDNA), allosterically increasing its catalytic activity and giving rise to the production of cGAMP, a second messenger molecule that activates STING [30, 33]. STING, which is a type of DNA adaptor that is located in the ER, consists of a transmembrane (TM) domain with four TM helices, a cytoplasmic ligand-binding domain, which is necessary for dimerization and cGAMP binding, and a C-terminal tail which hosts the TBK1 phosphorylation site that is needed to initiate its downstream cascade [34]. Under normal conditions, STING forms a domain-swapped homodimer and is oligomerized upon cGAMP stimulation. This activated STING then translocates from the ER through the ER-Golgi intermediate compartment (ERGIC) to the Golgi with the assistance of coatomer protein complex II (COPII) vesicles [35, 36]. Upon reaching the ERGIC and Golgi compartments, STING can recruit TBK1 and facilitate its autophosphorylation, which is followed by the phosphorylation and nuclear relocation of IRF3 and NF-κB [11, 31, 37]. IRF3 enables the activation of a series of target genes, including interferon-stimulated genes (ISGs), and NF-κB contributes to the expression of inflammation-related factors, including interleukin (IL)-6 and IL-12 [29]. The newly released IFNs, ILs, and other cytokines instigate immune and inflammatory responses. Although the activation of cGAS-STING is crucial in the defense of pathogens or viruses, the excessive activation of STING could lead to autoimmune diseases. Therefore, the activated cGAS-STING cascade needs to be restrained to maintain the immune homeostasis of the cell. Emerging evidence has elucidated that several elements of autophagy are involved in the suppression of STING signaling, such as autophagy-related (ATG) 9A and LC3 [38, 39]. CSNK1A1/CK1α, a serine/threonine protein kinase, is responsible for impeding the overactivation of STING-mediated type I IFN signaling via promoting autophagic degradation of STING [40]. In addition to being eliminated by the autophagic process, cGAS-STING also participates in the orchestration of autophagy, which contributes to the obliteration of foreign nucleic acids in the host cell cytosol. Earlier research demonstrated that autophagy could be induced by STING trafficking, independent of TBK1 and downstream signaling. An in-depth investigation has found that upon cGAMP stimulation, STING translocates to the ERGIC, which serves as a source for the LC3 lipidation, resulting in the formation of autophagosomes. Notably, sea anemone, a sort of extremely ancient species, was uncovered to be able to induce LC3 conversion in reaction to cGAMP, while its STING structure lacked the C-terminal TBK1 activation domain, indicating that the autophagy activation could be the primordial and intrinsic function of the cGAS-STING pathway [41]. Overall, this signaling axis allows the cGAS-STING complex to regulate a broad repertoire of cellular defense mechanisms.
1.2 Overview of the PERK-eIF2α pathway
PERK, which is localized in the ER, is the pivotal branch point that mediates the UPR. It is highly vulnerable and capable of being activated in response to persistent ER stress caused by disturbances in aberrant calcium levels, hypoxia, glucose deprivation, or the accumulation of unfolded and misfolded proteins. The UPR is a series of cellular responses that are triggered by certain stimuli- including ER stress or increases in misfolded proteins—and which stops translation and aids correct protein folding by up-regulating molecular chaperones and/or triggering cellular apoptosis [42]. There are three key branches orchestrated by membrane-related sensors in the UPR process: inositol-requiring enzyme 1(IRE1α), PERK, and ATF6. First, PERK can oligomerize and phosphorylate eIF2α, which reduces cellular protein synthesis and prompts the translation of the ATF4. Second, IRE1 RNase can splice X-box binding protein (XBP1) mRNA and produce the activated transcription factor XBP1. Thirdly, the ATF6 translocates from the ER to the Golgi apparatus, where it is cleaved by site-1 protease and site-2 protease to release an NH2-terminal domain that enters into the nucleus to promote the related gene expression [22, 43, 44]. Under normal states, PERK binds with chaperone proteins such as immunoglobulin heavy chain-binding protein (BiP) (also known as glucose-regulated protein 78 (GRP78) to form complexes to be silenced [45]. When faced with certain stressors, PERK dissociates from its complex and is activated. The specific downstream signaling pathway that PERK activates depends on the intensity of the stressors. Under low-intensity ER stress, the PERK branch mainly relieves the ER protein folding burden by phosphorylating eIF2α, impeding the translation of certain proteins, and mobilizing transcription factor 4 (ATF4) and other proteins. Under high-intensity ER stress, hyperactivated PERK can facilitate the higher expression of C/EBP-homologous protein (CHOP), which reduces the expression of the gene encoding antiapoptotic BCL-2 and dictates cellular demise [45-48].
eIF2 serves as the major hub for inducing translational repression. It is composed of three main subunits: α, β, and γ. This tripolymer constitutes a ternary complex with guanosine triphosphate (GTP) and initiator-methionyl-tRNA (Met)-tRNAi [49]. The tetramer can combine with the 40s subunit of ribosomes and be incorporated (along with other eukaryotic initiation factors) to create the 43S preinitiation complex. Afterward, the 43S complex recognizes the AUG initiation codon and forms the larger 48S complex. The process requires GTP as an energy source. Hydrolysis of GTP frees eIF2-GDP and other eIFs from the ribosome, allowing them to conduct their subsequent functions. The phosphorylation of eIF2α at ser 51 is a major checkpoint for the suspension of protein synthesis [45, 49, 50]. Generally, eIF2α phosphorylation is accomplished by the assembly of four kinases: PERK, protein kinase double-stranded RNA-dependent (PKR), general control non-derepressible-2 (GCN2), and heme-regulated inhibitor (HRI) [51]. In adaptative responses, the phosphorylation of eIF2α minimizes cellular toxicity by reducing protein burdens and boosting the transcription of selective genes, while its prolonged activation elicits cell death via CHOP [52-54]. It is reasonable to hypothesize that eIF2α phosphorylation can either lead to cell death or survival depending on the duration and degree of the subsequent signaling cascade. However, non-phosphorylated eIF2α is ubiquitinated and degraded by the E3 ubiquitin ligase carboxyl terminus of the HSC70-interaction protein (CHIP) in the absence of chaperone proteins, which upregulates cyclic AMP-dependent transcription factors. The eIF2α phosphorylation can also block ubiquitination [55]. Once ER homeostasis is restored, p-eIF2α levels return to their normal baseline (which is dominated by PP1) [54, 56].
1.3 Overview of the cGAS-STING/PERK-eIF2α pathway
1.3.1 Crosstalk among STING, ER-stress, and UPR
As discussed earlier, the ER stress and UPR are closely associated with the PERK activation. Besides, as an essential DNA adapter located in the ER, STING is tightly linked to the ER stress response, thus suggesting an underlying crosstalk among STING, ER stress, and UPR [57]. It is widely accepted that mtDNA plays an indispensable role in the activation of the cGAS-STING pathway and connects with ER stress [10, 58]. Chronic ER stress and ISG expressions have been observed under autoimmune conditions. Furthermore, this gene expression was offset by the deficiency of STING or UPR branches, including PERK and ATF6, where the mtDNA acts as a mediator. Overall, the ER stress set in motion UPR chains, leading to the mtDNA release and ROS production, followed by the cGAS-STING signaling cascade and IFN release [59]. Existing evidence has unveiled that the function of STING is not completely dependent on the IFN-related pathways. To explore the mechanisms of how STING primes T-cell death, Wu et al. established the STINGN153S/+ mouse model and revealed that these mutants could trigger chronic activation of ER stress and UPR, leading to T-cell apoptosis. Moreover, the ER stress inhibitor impedes T-cell death. Further experiments verified that the motif-helix aa322-343, called the “UPR motif”, which is different from the domain that is necessary for setting off IFN, is required for inducing UPR. These findings suggest that certain stimulators can interact with the UPR motif of STING and instigate ER stress and UPR [57]. Additionally, due to the innate immune function of STING, its deficiency can lead to infection. It has been shown that Seneca Valley Virus infection leads to immune escape via the degradation of reticulophagy mediated by PERK and ATF6, however, the intrinsic mechanisms remain unclear [60]. Besides immunological effects, the STING-NOD-like receptor protein 3 (NLRP3) mediates inflammatory responses [29, 61]. Interestingly, during Brucella abortus infection, the STING promotes inflammasome formation via UPR point IRE1-α. The IRE1-α activation facilitates mitochondrial ROS (mROS) release, stabilizes hypoxia-inducible factor-1 (HIF-1), and enhances inflammation in macrophages [62]. These findings were consistent with the previous conclusion that ROS production could be an upstream trigger for the NLPR3 activation [63]. Additionally, a study by Lu et al. has shown that the inhibition of NLRP3 inflammasome decreases ROS production, suggesting a feedback loop between ROS and the NLRP3 inflammasome formation [64]. ROS is closely associated with ER stress. Previous studies have shown that ER stress activates NLRP3 inflammasome formation [65]. It is reported that contact sites between the ER and mitochondria-associated ER membranes (MAM) are crucial for various cellular activities. The calcium imbalance causes ER stress and NLRP3-mediated inflammation via MAM [66]. Therefore, it is reasonable to speculate that STING, ER stress, and UPR may coordinately orchestrate the inflammation activation. These consequences may indirectly suggest that there may be complicated crosstalk between STING, ER, and UPR during inflammation and immunity-related processes, where the ROS and MAM might be crucial mediators.
1.3.2 The cGAS-STING/PERK-eIF2α interaction
As mentioned above, the cGAS-STING pathway mediates canonical immunological and inflammatory responses via TBK1-IRF3 activation and IFN release, which is intimately associated with various diseases, including CVDs. Notably, recent studies have delineated that the cGAS-STING also mediates the activation of the PERK-eIF2α, even prior to the activation of the TBK1-IRF3. PERK-eIF2α is one of the cascade branches orchestrating ER stress-induced UPR. Its activation generally hinders the production of proteins and specifically enhances the transcription of particular mRNAs, which can contribute to the development of various illnesses. Additionally, previous evidence has shown a robust link between the downstream effects of the cGAS-STING pathway and ER stress-related signaling [14, 15, 57], which also indirectly indicates a certain relationship between cGAS-STING/PERK-eIF2α.
Existing evidence has elucidated the probable connections between STING and PERK. Notably, Zhang et al. [20] visualized the interaction between PERK and STING using proximity ligation assays and domain-mapping assays after co-immunoprecipitation and determined a direct domain interaction between STING and PERK. Mechanistically, the STING C-terminal domain fusion continuously elevated the PERK kinase domain's autoactivation capacity. Surprisingly, a previous study demonstrated that, during viral infections, STING promoted IFN release and inhibited protein synthesis in an IFN-independent manner. However, the intrinsic mechanism remains unknown. PERK was then confirmed to intertwine with STING and be activated by STING [67]. Another article demonstrated that PERK activation constrained viral replication by promoting STING-induced IFN release [68]. These experiments indicate that the STING-orchestrated and PERK-mediated pathways interact with each other during immune-mediated inhibition of viral replication. Additional data supports the notion that STING can be modulated by PERK, although not all studies come to this conclusion. As one example, eliminating PERK thwarted Nrf2 and promoted mitochondrial disfunction and mtDNA accumulation. Subsequently, STING and its downstream effectors were stimulated to enhance IFN release and increase anti-tumor effects [26]. Another paper showed that PERK phosphorylation inhibited the translocation of STING, resulting in the immunosuppression of myeloid-derived suppressor cells (MDSCs) [69]. By contrast, in another paper, inhibiting PERK quenched the STING-mediated classical route and IFN production in neurons after traumatic brain injury (TBI), thus alleviating brain injury and cell loss [27]. Likewise, in macrophages infected by M. bovis, PERK activation promoted both the translation of STING and its downstream signaling. Phosphorylated IRF3 is shown to initiate apoptosis in three ways: entering the nucleus to trigger the transcription of apoptotic genes, associating with Bax to induce the mitochondrial apoptotic pathway, and activating caspase 8 [70]. Therefore, the regulation between STING and PERK is mostly positive, and the controversies about the interactions between PERK and STING may have partially arisen from differences in the cell types or pathological contexts studied.
The sum of the evidence suggests that the cGAS-STING pathway and the PERK-eIF2α pathway may undergo reciprocal regulation and that a combined cGAS-STING/PERK-eIF2α pathway may arise to function in pathological contexts. Rather than unidirectional regulation, we surmise that STING and PERK collaborate to affect other cellular pathways and undergo mutual regulation. We also propose a probable mechanism underlying the cGAS-STING/PERK-eIF2α interaction (Figure 1): Under normal conditions, the cGAS-STING pathway and ER-stress-related PERK-eIF2α pathways are both inactive. However, upon stimulation from DNA leakage, ER-localized STING can activate PERK and thus inhibit overall protein synthesis. In turn, ER-stress-induced phosphorylated PERK simultaneously interacts with STING to promote its translocation to ERGIC, where it triggers the TBK1 and IRF3 cascades. Therefore, both activated STING and activated PERK can regulate each other. Studies have shown that inhibiting PERK does not affect the STING-mediated canonical pathways, which might be result from STING signaling initiated by other potent stimulators. In summary, cross-regulation between STING and PERK is intricate, and cGAS-STING/PERK-eIF2α positive crosstalk plays a vital role in various pathological conditions.
cGAS-STING/PERK-eIF2α interactions under different conditions (created with BioRender.com). Under normal conditions, both the cGAS-STING pathway and ER stress-related PERK-eIF2α pathways are in states of silence. Upon stimulation from leaked DNA, either from exogenous pathogens or intracellular mitochondria, STING that is localized to the ER can bind with cGAMP produced by cGAS to be activated and translocated to the ER-Golgi intermediate compartment (ERGIC). This translocation promotes downstream TBK1 and IRF3 phosphorylation to mediate IFN release. STING remains at the ER to interact with PERK and activate it, which inhibits overall protein synthesis. In turn, in times of ER stress, PERK is phosphorylated and facilitates the activation of eIF2α and ATF4, simultaneously interacting with STING. This interaction promotes STING's translocation to ERGIC to induce the TBK1 and IRF3 cascade. Under complex pathological conditions, both the cGAS-STING axis and the PERK-eIF2α axis are activated. Interactions between STING and PERK are also augmented, meaning that the activation of either molecule can promote the initiation of the other to mediate the pathological effects.
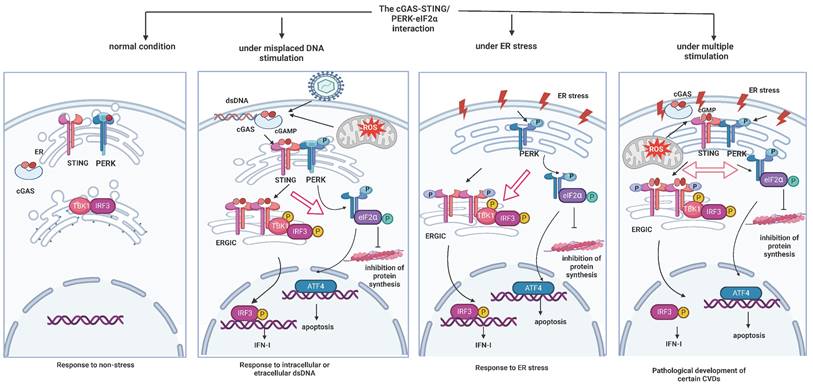
2. The cGAS-STING pathway in CVDs (Table 1)
2.1 The role of the cGAS-STING in atherosclerosis (AS)
An array of mechanisms, including inflammatory responses, endothelial disruption, lipid accretion, macrophage phenotype conversion and foam cell formation, aberrant smooth muscle cells (SMC) proliferation, and migration [4, 5, 71, 72], are involved in the pathogenesis and development of AS. Mounting evidence has shown that the cGAS-STING pathway is involved in each of these cellular activities and thus accelerates AS. DNA leakage is the primary stimulus for the cGAS-STING activation. Increases in cytosolic cell-free DNA and mtDNA have been observed in AS patients compared with the general population. Further analysis has shown that cigarette smoke leads to the deposition of plasma DNA, the initiation of the cGAS-STING pathway, and the release of inflammatory factors like IL-6, which may be a crucial mechanism underlying AS progression [9]. IQ motif-containing GTPase-activating protein 1 (IQGAP1) is thought to be an essential scaffolding protein that modulates mitochondrial function and affects endothelial cell behavior. IQGAP1 expression levels increase in high-fat diet (HFD)-triggered AS models in apolipoprotein E-deficient ApoE-/- and Ldlr-/- mice models. Mechanistically, the mitochondrial dysfunction indirectly activates the cGAS-STING and NLRP3-directed pyroptosis. Furthermore, IQGAP1 knockdown reverses the activation of this signaling pathway. To explore the underlying mechanism, human umbilical vein endothelial cells (HUVECs) were treated with palmitic acid (PA) to construct an in vitro AS model. Results show that silencing either IQGAP1 or cGAS-STING attenuated endothelial injury in AS by decreasing the lactate dehydrogenase (LDH) activity, cytokine factor secretion, and cellular pyroptosis [5]. Transactive response DNA-binding protein of 43 kDa (TDP43) is a protein that combines with DNA and is involved in nucleic acid metabolism, including the release of mitochondrial DNA. It also acts as an activator of the cGAS-STING [73, 74]. TDP43 induces cGAS-STING by promoting the mtDNA release and NF-κB signaling, exacerbating lipid absorption in macrophages, and eventually encouraging foam cell accumulation. As expected, the genetic knockout of TDP43 in macrophages decreased the expression of pro-inflammatory markers and lipid uptake. Eliminating TDP43 in AS mice also blunted lesion areas [75]. Even in the context of chronic kidney disease (CKD), the cGAS-STING pathway plays a significant role in the development of atherosclerotic (AS) plaques. In CKD models, heightened oxidative stress induces mitochondrial damage, which activates cGAS-STING signaling. This activation led to phenotypic switching, premature senescence, and increased plaque instability. Notably, the depletion of either cGAS or STING resulted in reduced plaque areas and diminished plaque instability [10]. These results indicate that the cGAS-STING pathway drives the progression of AS by affecting cell inflammation, pyroptosis, lipid deposition, and SMC phenotypic switching.
The cGAS-STING pathway in CVDs
| Model | Targets | Inhibitors | Activators | Cell phenotype | Related molecules | Results | Reference |
|---|---|---|---|---|---|---|---|
| HUVECs | cGAS, STING | shRNA-cGAS | CSE | apoptosis, inflammation | mtDNA | CSE induced endothelial dysfunction and aggravated AS progression via activation of cGAS-STING signaling | [9] |
| ApoE-/-mice, oxLDL-treated macrophages and PBMCs | mitochondrial DNA | shRNA-cGAS | TDP43 | inflammation, lipid uptake of macrophages | mtDNA, NF-κB | TDP43 promoted inflammation and foam cell formation, augmenting AS progression through triggering mitochondrial DNA release to activate cGAS-STING signaling | [75] |
| HFD-fed ApoE-/-mice and Ldlr-/-mice, HUVECs treated with PA | cGAS, STING | RU.521 C-176 siRNA-cGAS siRNA-STING | IQGAP1 | pyroptosis, oxidative stress | NLRP3 | IQGAP1 induced mitochondrial damage and activation of cGAS-STING pathway during the development of AS | [5] |
| CKD/ApoE-/- mice, human VSMC and Mice VSMC | cGAS, STING | knockout of cGAS and STING, C-176 | premature, senescence and phenotypic switching of VSMC | mtDNA, IFN-1 | The activation of cGAS-STING decreased content of VSMC and fibrous cap, alleviated plaque area and increased plaque stability | [10] | |
| HFD-fed mice | STING | shRNA-STING | diABZI | inflammation, pyroptosis | NLRP3 | The inhibition of STING improved HFD-induced cardiac function and mitigated myocardial fibrosis | [76] |
| HFD-fed db/db mice, PA-induced H9C2 cells | cGAS, STING | siRNA-STING, C-176 | extracted mtDNA | inflammation, apoptosis | mtDNA, IRF3, NF-κB | The inhibition of STING improved cardiac function and mitigated myocardial fibrosis in DCM | [58] |
| HFD induced mice, HG and PA-induced H9c2 cells | STING | C-176 | diABZI | pyroptosis, apoptosis | MITOL, NLRP3, GSDMD | MITOL activation ameliorated DCM- induced myocardial injury and cardiac disorder by inhibiting cGAS/STING signaling | [78] |
| STZ-induced (T1D) mice and (db/db) (T2D) mice, HG-induced neonatal rat cardiomyocytes | STING | Metrnl | siRNA- Metrnl | Apoptosis, autophagy | LKB1, AMPK, ULK1 | Metrnl ameliorates DCM injury and cardiac function via inactivation of cGAS/STING signaling | [80] |
| LPS-induced mice and H9C2 cells | cGAS | siRNA-cGAS ALDH2 | inflammation, and apoptosis, oxidative stress | ALDH2 | ALDH2 alleviated LPS-induced cardiac dysfunction, and protected against SIC-induced injury via inhibition of cGAS/STING pathway | [84] | |
| LPS-induced mice, H9C2 cells and macrophages | STING | ICA69 | inflammation, ferroptosis, oxidative stress | ICA69 | ICA69 deficiency alleviated septic cardiac dysfunction and damage via upregulation of STING mediated pathway | [85] | |
| LPS-induced mice NRCMs, and H9C2 cells | STING | knockout of STING, siRNA-STING | inflammation, apoptosis, pyroptosis | NLRP3, IRF3 | Deficiency of STING alleviated SIC-triggered cardiac function and injury | [61] | |
| Dox-induced mice and HL-1 cardiomyocytes | STING | siRNA-STING | inflammation, apoptosis | NLRP3 | The knockdown of STING improved survival and cardiac function of DOX-treated mice and alleviated cell injury | [79] | |
| Mice model of LMNA-related cardiomyopathy | cGAS, STING | knockout of cGAS and STING | nuclear envelope rupture | cGAS or STING deletion does not rescue dilated cardiomyopathy in LMNA-CKO mice | [82] | ||
| Lmna F/F mice | cGAS | knockout of cGAS | inflammation, apoptosis | TBK1, IRF3, and NF-kB | The ablation of cGAS prolonged survival, improved cardiac function, and attenuated myocardial fibrosis in the LMNA-deficient mice | [87] | |
| MI mice, cardiac cardiomyocyte and fibroblasts after co-culturing them with the supernatant of stimulated BMDMs | cGAS, STING | H-151 | DMXAA | inflammation, apoptosis | TBK1, IRF3 | The inhibition of cGAS/STING mediated pathway alleviated cardiac function and myocardial fibrosis | [89] |
| MI model | STING | H-151 | inflammation | The STING blockage decreased infarction size and scarring, relieved cardiac function and reduced myocardial fibrosis and hypertrophy after reperfused MI | [92] | ||
| Mice challenged by TAC, neonatal rat ventricular myocytes | cGAS, STING | knockout of STING, shRNA-cGAS, shRNA-STING | iNOS, compound 3 | inflammation, macrophage polarization | mtDNA | The inhibition of cGAS/STING alleviated the myocardial fibrosis and cardiac function, and mitigated the pressure- overload induced myocardial injury | [93] |
| I/R mice model, H9c2 cells exposed to H/R | cGAS, STING | RU.521 H-151 | apoptosis | Bcl‑2/Bax/ Caspase‑3 | Scutellarin ameliorated the cardiac function and infarct area, decreasing the I/R injury | [94] | |
| Diabetic mice with I/R, H9C2 cells challenged by HG and PA | STING | H-151 | decreased mitofusins 2 | inflammation | mtDNA | The inhibition of cGAS/STING improved the cardiac function and alleviated the diabetic I/R injury | [95] |
| Mice chanllenged by TAC-induced HF | cGAS | sh-cGAS | Inflammation, apoptosis | The inhibition of cGAS relieved the cardiac function and remodeling, mitigating HF induced by pressure-overload | [97] | ||
| mice model with aPD-1-induced myocarditis | STING | STING mutant | Gasdermin-E | Pyroptosis, inflammation, T cell infiltration | mtDNA | The knockout of Gasdermin-E improved the myocardial morphology, and cardiac function, mitigating myocarditis | [98] |
HUVECs: Human umbilical vein endothelial cells; CSE: cigarette smoke; mtDNA: mitochondrial DNA; TDP43: transactive response DNA-binding protein∼43kDa; PBMCs: peripheral blood mononuclear cells; HFD: high-fat diet; IQGAP1: IQ motif-containing GTPase-activating protein; NLRP3: NOD-like receptor protein 3; IRF3: interferon regulatory factor 3; NF-κB: nuclear factor-kappa B; DCM: diabetic cardiomyopathy; PA: palmitic acid; HG: high-glucose; MITOL: mitochondrial ubiquitin ligase; GSDMD: gasdermin D; Dox: doxorubicin; STZ: streptozotocin; LKB1: liver kinase B1; AMPK: adenosine monophosphate activated protein kinase; ULK1: Unc-51 like autophagy activating kinase 1; ALDH2: mitochondrial aldehyde dehydrogenase 2; SIC: sepsis-induced cardiomyopathy; ICA69: Islet cell autoantigen 69; NRCMs: neonatal rat cardiomyocytes; MI: myocardial infraction; BMDMs: bone marrow-derived macrophages; DMXAA: 5, 6-dimethylxanthenone-4-acetic acid; TBK1: TANK-binding kinase 1; TAC: transverse aortic constriction; iNOS: inducible NO synthase; I/R: ischemia/reperfusion; H/R: hypoxia/reoxygenation; HF: heart failure.
2.2 The Role of cGAS-STING in Cardiomyopathy
The cGAS-STING signaling is involved in various kinds of cardiomyopathy. Many factors can contribute to the development of cardiomyopathy, including lipid toxicity, obesity, pathological states like diabetes mellitus (DM), infectious diseases, chemical drugs, and hereditary mutations. Each of these factors can cause disruptions in cardiac contractile or diastolic functioning [58, 76-79]. Obesity is a common risk factor for a constellation of CVDs, including but not limited to myocardial dysfunction. Recent work showed that a STING-mediated NLRP3 pathway was hyperactivated in the HFD-induced obese mice. Eight weeks of aerobic exercise could ameliorate STING-NLRP3-induced inflammation and pyroptosis and improve cardiac function, but these improvements were abolished after the injection of a STING agonist. These results demonstrate that suppressing STING can protect against obesity-related cardiopathy to some extent [76].
Diabetic cardiomyopathy (DCM) is a fairly common complication of DM [78]. The cardiac function is undermined by DM because the pathological changes augmented cardiac fibrosis and pyroptosis, which are partially driven by the cGAS-STING pathway. A specific polypeptide, Irisin, played a protective role in the DCM by blocking the cGAS-STING signaling. Additionally, initiation of the STING pathway partially offsets Irisin's favorable effects. Inactivation of cGAS-STING signaling, however, prevented cardiomyocyte disruption by raising sensitivity to autophagy in both high-glucose-stimulated H9C2 cells and DCM mice. Mechanistically, the liver kinase B1 (LKB1)/adenosine monophosphate-activated protein kinase (AMPK)/Unc-51-like autophagy activating kinase 1 (ULK1) axis is activated in the presence of protective agents, followed by STING dephosphorylation and translocation to the mitochondria. This translocation required tumor necrosis factor receptor-associated factor 2 (TRAF2), an E3 ubiquitin ligase that regulates inflammatory signaling pathways, to bind it and form the STING/TRAF2 complex, causing the ubiquitination and degradation of STING. This series of interactions promoted autophagy and protected against DCM injury [80, 81]. Another study examined the relationship between cGAS-STING and obesity-induced cardiomyopathy using HFD-fed db/db mice and H9C2 cells that were exposed to PA stimulation. Their results indicate that mitochondria-derived cytosolic DNA is an essential mediator of cGAS-STING signaling in DCM. In-depth investigations showed that genetic and pharmacologic blockage of STING ameliorated cardiomyocyte damage by decreasing NF-κB-related inflammatory signaling and apoptosis [58]. Taken together, these experiments suggest that cGAS-STING activation exacerbates DCM progression.
Sepsis is a life-threatening syndrome that can lead to multiple organ failure. The heart is the most susceptible organ to sepsis, and its damage is always serious and irreversible, which causes related cardiomyopathy and even heart failure (HF) [82-84]. Liu et al. [84] used lipopolysaccharide (LPS) stimulation to establish in vivo and in vitro models of sepsis-induced cardiomyopathy (SIC) and to study the mechanism by which mitochondrial aldehyde dehydrogenase 2 (ALDH2) alleviates SIC. They demonstrated that ALDH2's protective effects were mediated by the eradication of the cGAS-STING axis, which was followed by the mitigation of inflammation and apoptosis. These mechanisms decreased cardiac injury markers, including creatine kinase-MB (CK-MB) and LDH, and improved cardiac function. This study suggests that inhibiting cGAS-STING is capable of weakening the effects of SIC. In particular, the STING trafficking augments sepsis-induced cardiac dysfunction by aggravating ROS production, ferroptosis, and inflammation [85]. In addition, underlying cGAS-STING-mediated exacerbations of SIC, and the cGAS-STING activation could also fuel the phosphorylation and nuclear translocation of IRF3, resulting in NLRP3 inflammasome formation. Overall, activation of the pathological signaling axis aggravated inflammation, pyroptosis, and apoptosis, thus accelerating cardiac disorder [61]. The cGAS-STING activation also contributes to drug-induced cardiomyopathy [79]. Doxorubicin (DOX) administration significantly increases cGAS and STING expression levels and downstream effectors, including TBK1, IRF3, and NF-κB p65. Intriguingly, the NLRP3 level is also elevated under this condition. As expected, these changes in molecular expression after the DOX challenge could be partially reversed by silencing STING. This condition can improve myocardial injury and cardiac function dramatically, as shown by decreased activity of myocardial damage markers and increased ejection fractions (EF), fractional shortening (FS). In addition to these apparent changes, other mechanisms may include the inhibition of inflammation and apoptosis.
Hereditary dilated cardiomyopathy is caused by mutations in various genes and is a pervasive, genetically heterogeneous disease that leads to HF. Mutations in the Lamina A/C protein (LMNA) gene, which encodes the nuclear envelope protein Lamin-A/C, are one of the most common abnormalities that lead to dilated cardiomyopathy [86-88]. Sirisha M et al. [87] found that the expression of cGAS-related signaling molecules such as STING1, TBK1, IRF3, and NF-kB were elevated in LMNA deficient mice compared to control mice. Furthermore, genetic deletion of the cGAS cascade blocked inflammation and apoptosis, increased survival rates, and improved cardiac function. These results support targeting cGAS-STING to treat lamin-related cardiomyopathy.
2.3 The Role of cGAS-STING in the ischaemic cardiomyopathy
Ischemic cardiomyopathy is a severe type of CVD that is responsible for most cardiac-related deaths around the world. After myocardial ischemia, the inflammatory response is triggered to promote the repair of cardiomyocytes. However, excessive inflammation can lead to secondary damage to cardiomyocytes. The cells become irreversibly dysfunctional, ultimately culminating in cardiac remodeling and HF [89, 90]. For patients going through acute myocardial infarction (AMI), timely and efficient reperfusion treatment can save injured myocardial tissues, while it tends to result in dampening the cardiac damage upon the restoration of blood flow. This phenomenon is called myocardial ischemia-reperfusion (MI/R) injury, which drives a series of reactions such as inflammation, oxidative stress, and cell apoptosis [91]. The cGAS-STING pathway also contributes to the pathogenesis of MI and MI/R [6, 12, 91, 92]. The instigation of the cGAS-STING pathway contributes to the recruitment of inflammation-related cells like macrophages and the release of chemokines in the infarct area, leading to myocardial necrosis and fibrosis [91, 93]. Finally, the cardiac structure and function could be damaged, resulting in heart failure (HF). Since the essential role of cGAS-STING initiation in the MI, the ablation of STING-related molecules protects against MI-induced injuries [8]. H-151, a selective antagonist of STING, has been shown to abate inflammation and improve cardiac function after MI, reduce TNF-α, chemokine (C-X-C motif) ligands (CXCL)10, and IL-1β levels, and restore EF and FS. In vitro experiments showed that when co-cultured with bone marrow-derived macrophages stimulated by dsDNA, cardiomyocytes treated by H-151 show decreased apoptosis rates and inflammation levels, and their fibroblasts produce less collagen [89]. The H-151 is also used to inhibit STING to help repair the infarct areas, infarction-induced myocardial hypertrophy, fibrosis, and cytokine release after reperfusion. In addition, it is reported that cardiomyocyte induced by hypoxia/reoxygenation (H/R) possesses higher expression of cGAS-STING, and either the inhibition of cGAS with RU.521 or suppression of STING with H151 can mitigate the cell apoptosis and cardiac disruption after MI/R, probably via regulation for Bcl‑2/Bax/Caspase‑3 signaling pathways [94]. Type 2 diabetes is the risk factor for accelerating the damage of MI/R. It is established that during the development of diabetes, the morphological and functional properties alter, leading to the increased mtDNA leak into the cytosol and activation of the cGAS-STING signaling, exacerbating the MI impacts. The abrogation of cGAS-STING alleviates the inflammation and myocardial damage in diabetic MI/R rats. In vitro experiments demonstrated that the inhibition of STING improved the mitochondrial function and cell viability of H9C2 cardiomyocytes challenged by HG and H/R [95]. These data imply that obliterating STING can alleviate myocardial damage and improve cardiac function after MI [92].
2.4 The role of cGAS-STING in other CVDs
HF is the end-stage of cardiac dysfunction, which is mainly caused by various kinds of cardiogenic factors. The progression of HF is reported to be associated with chronic inflammation [96], and further investigation has identified the crucial role of the cGAS-STING pathway in HF [12, 97]. Under the stimulation of pressure overload, the cardiomyocytes may undergo mitochondrial injury, followed by the mtDNA leakage to the cytosol and the activation of the cGAS-STING axis. Then, the undue inflammation cascade and cell apoptosis occurs, driving the hypertrophy and fibrosis of cardiomyocytes. However, the genetic ablation of cGAS in mice challenged by transverse aortic constriction blocked the early inflammatory response and apoptosis, alleviating cardiac remodeling and restoring cardiac functions [97]. Additionally, the cGAS-STING signaling pathway is involved in the development of myocarditis. In mice treated with aPD-1 therapy to induce immune checkpoint inhibitor-related myocarditis, activated GSDME mediates pyroptosis, leading to mitochondrial injury and subsequent release of mtDNA. This release activates the cGAS-STING pathway, which exacerbates myocardial inflammation. In contrast, genetically blocking STING in mice results in lower levels of markers associated with myocardial damage, reduced T-cell infiltration, and improved cardiac function. These beneficial effects are comparable to those achieved by the ablation of gasdermin E (GSDME) [98]. Similarly, the administration of X6 in a mouse model of autoimmune myocarditis effectively inhibited the release of cGAMP and the subsequent cGAS-STING-induced production of IFN-β and expression of ISGs. This intervention ultimately reduced endocardial inflammation and fibrosis. These findings suggest that targeting the cGAS-STING pathway could alleviate the progression of HF and myocarditis.
3. The PERK-eIF2α in CVDs
3.1 The PERK-eIF2α pathway in different cellular phenotypes
The PERK-eIF2α mediated pathway plays a major role in several cellular disorders, including inflammation, apoptosis, autophagy, and pyroptosis (Figure 2) [99]. Inhibiting PERK-eIF2α has been shown to reduce ER stress-related inflammation by preventing the translocation of NF-κB and decreasing the release of tumor necrosis factor-α (TNF-α), IL-6, and monocyte chemoattractant protein-1 (MCP-1) [100]. Notably, PERK-eIF2α downregulation alleviated the inflammation and apoptosis of cardiomyocytes that occurs during the development of ischemic stroke and thus protected against resultant injury [99, 101]. Apoptosis was also blunted by the deacetylation of PERK-regulated eIF2α, which is responsible for the amelioration of cardiomyocyte death and cardiac function [102]. Additionally, the levels of several ER-stress-related molecules, including p-PERK, p-eIF2α, ATF4, and CHOP, were up-regulated in alveolar type II epithelial cells after perfluorooctane sulfonate treatment. However, the use of a PERK inhibitor partially attenuated GSDME-triggered pyroptosis and inflammatory factor secretion, thereby mitigating lung injury [103]. There is a significant relationship between ER stress and autophagy. It is established that activating PERK-eIF2α increased rates of autophagy by up-regulating ATG5 and ATG7 [104]. Additionally, ATG12 and p62 were confirmed to be involved in this process [105]. Notably, autophagy negatively regulates the PERK-eIF2α pathway, which also participates in the interplay between apoptosis and autophagy. By targeting PERK-eIF2α mediated pathways, the neurotoxins were shown to instigate both autophagy and apoptosis in neurons [106]. Further studies using a related inhibitor verified that activating autophagy was protective against cellular injury due to the alleviation of apoptosis. Activation of PERK-eIF2α can lead to favorable effects, such as regulating mitochondrial functions. Previous studies have shown that PERK located in MAMs physically interacts with Mitofusin‑2 (Mfn2) and regulates mitochondrial morphology, calcium (Ca2+) homeostasis, and oxidative stress [16, 107-109]. The PERK-eIF2α pathway also stabilizes mitochondrial function in a multitude of ways, including promoting mitochondrial hyperfusion via the PERK-eIF2α-ATF4 pathway, increasing the production of mitochondrial cristae via the PERK-O-linked N-acetyl-glucosamine transferase pathway [110], facilitating mitophagy through the PERK-transcription factor EB (TFEB) pathway [16, 111, 112], and increasing oxidative phosphorylation via the PERK-nuclear factor erythroid 2-related factor 2 (Nrf2) pathway [43]. Thus, PERK-eIF2α is closely involved in diverse signaling transduction and plays a key role in numerous normal and aberrant mitochondrial functions.
The PERK-eIF2α pathway connects to different cellular activities (created with BioRender.com). The PERK-eIF2α pathway plays a master role in several critical cellular functions. Its fundamental effects are the selective transcription of genes such as ATF4 and the arrest of protein translation. Excessive ATF4 activation can promote cell apoptosis. It can also induce ER stress-related inflammation by increasing the translocation of NF-κB, thus increasing the release of tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and monocyte chemoattractant protein-1 (MCP-1). Additionally, it triggers pyroptosis via gasdermin E (GSDME). PERK-eIF2α activation can positively regulate autophagy by up-regulating autophagy related 5 (ATG5) and ATG7, as well as involving ATG12 and p62. Finally, its activation stabilizes mitochondrial functions in several ways: it promotes mitochondrial hyperfusion via the PERK-eIF2α-ATF4 pathway, it increases the formation of mitochondrial cristae via the PERK-O-linked N-acetyl-glucosamine transferase pathway, it facilitates mitophagy through the PERK-transcription factor EB (TFEB) pathway, and it favors oxidative phosphorylation via the PERK-nuclear factor erythroid 2-related factor 2 (Nrf2) pathway.
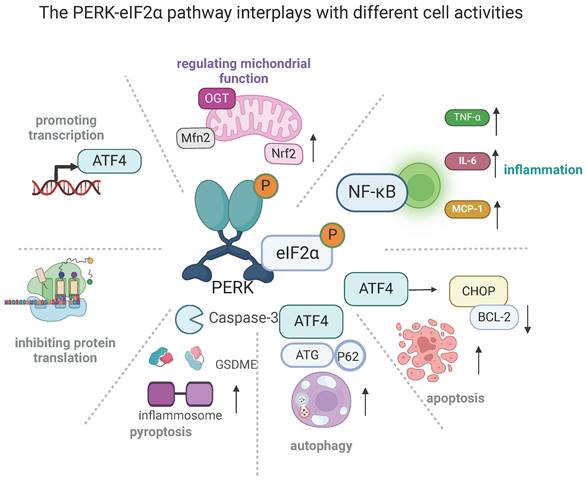
3.2 The role of PERK-eIF2α in AS
Oxidized low-density lipoprotein (ox-LDL) is a crucial component in the development of AS. As cholesterol continues to accumulate within a vascular lesion, smooth muscle cells (SMCs) switch phenotype in a process that is characterized by the downregulation of SMC differentiation genes and the upregulation of macrophage or fibroblast-related genes. This process accelerates the formation of atherosclerotic plaques. Previous studies have shown that the p-PERK and p-eIF2α levels are higher in the ApoE-/- mice [113]. In addition, the influx of cholesterol into cardiomyocytes facilitates the UPR in the ER, thus activating PERK, IRE 1α, and ATF. This cascade can also promote the modulation of SMC-related phenotypic conversion, leading to an increase in plaque loads [42]. Pericentrin is a key component of the microtubule organizing center, which enables the accurate completion of cellular division. Pericentrin deficiency in SMCs can actuate heat shock factor 1 (HSF1), and consequently augment the viability of HMG-CoA reductase (HMGCR), thus increasing cholesterol biosynthesis. The increases in cholesterol biosynthesis induced both PERK signaling and the phenotypic modulation of SMCs, which in turn increased atherosclerotic plaque burdens [13, 114]. However, blocking PERK decreased the expression of several macrophage- and UPR-related molecules, such as phosphorylated eIF2α and ATF4, thereby alleviating AS progression [115]. Vascular calcification (VC), particularly intimal VC, is a risk factor for the development of vulnerable atherosclerotic plaques [116, 117]. Intriguingly, the expression of ER-related molecules such as PERK, eIF2α, ATF4, GRP78, and CHOP was found to be significantly higher in VC rat models compared to wild-type rats. Allicin administration, however, suppressed PERK activation and relieved aortic VC. Further study identified that the pharmacological inhibition of eIF2α phosphorylation helped ameliorate VC in the aorta [116, 118]. The above findings indicate that the activation of the PERK-eIF2α plays an essential role in the process of AS, and the blockage of PERK may dampen the advancement of AS burden.
3.3 The role of PERK-eIF2α in cardiomyopathy and HF
Accumulating evidence suggests that PERK-eIF2α plays a major role in the development of cardiomyopathy, although some disagreement persists [17, 119-121]. The PERK's involvement in dilated cardiomyopathy has been well-documented [122]. For instance, melatonin alleviated dilated cardiomyopathy by targeting the PERK-eIF2α pathway. To be exact, melatonin mitigated the tether between PERK and GRP78, thereby promoting PERK's dissociation and phosphorylation. The activated PERK then mitigated myocardial injury and improved cardiac function by up-regulating autophagy pathways. In-depth investigations demonstrated that inhibiting PERK weakened the protective effects of melatonin in dilated cardiomyopathy [17]. Paradoxically, it is documented that the lncRNA H19 downregulated the expression of p-PERK and CHOP, which then ameliorated cardiomyocyte apoptosis and fibrosis, and remarkably improved cardiac function in DCM mice [123]. Likewise, LCZ696 was found to protect against DCM-induced inflammation, ER Stress, and apoptosis by inhibiting advanced glycation end-products (AGEs)/NF-κB and PERK-eIF2α directed cascades, thus improving glucose metabolism, lipid metabolism, and myocardial injury [119]. In vitro experiments, inhibiting PERK and its downstream molecules decreased apoptosis and ER stress. [120].
GCN2 is an enzyme that phosphorylates PERK, and its deficiency is observed to be protective in DCM [124]. Loss of GCN2 could mitigate cardiac contractile disruption, myocardial fibrosis, apoptosis, and oxidative stress in DOX-induced cardiomyopathy by attenuating eIF2α-CHOP, and knockdown of eIF2α in the DOX-challenged H9C2 cells resulted in the higher survival rates and decreased oxidative stress [125]. Aiming to understand the ER stress effects in Lamin-A/C-variant-induced dilated cardiomyopathy, Pietrafesa et al. [126] established an in vitro model and discovered that levels of PERK and its downstream signal molecules were elevated in the context of this mutation. Additionally, inhibiting PERK promoted myocardial survival, probably by increasing levels of phosphorylated A serine/threonine-specific protein kinase B (AKT). Phospholamban is a crucial modulator of cardiac calcium balance and contractility, and arginine deficiency at position 14 is associated with both dilated cardiomyopathy and ventricular arrhythmias. Surprisingly, the protective effects of PERK have been validated in cardiomyopathy by utilizing PERK knockdown in human induced pluripotent stem cells (hiPSC-CMs) to establish an R14del model [127]. Concerning other types of cardiomyopathies, PERK-eIF2α mediated ER stress is also found to be involved in the protective effects of hydrogen sulfide against SIC [128]. These findings may imply that the PERK-eIF2α participated in the pathophysiology of cardiomyopathy via regulation of cell death, inflammation, fibrosis, and metabolism. HF is the final stage of various cardiomyopathies [129, 130]. To investigate the mechanism underlying dapagliflozin's protective effects in HF, Ren et al. [131] created a transverse aortic constriction (TAC) mouse model, as well as angiotensin (Ang) II-induced cardiomyocytes. They determined that the expression of p-PERK-p-eIF2α-ATF4-CHOP was increased in model groups. However, dapagliflozin administration improved cardiac function and reduced myocardial hypertrophy and fibrosis. Cardiomyocyte apoptosis also decreased, as exhibited by increases in pro-apoptotic markers such as caspase 3 and Bax-1 and decreases in TUNEL-stained cells. Several protective effects were also decreased after the use of ER stress inducers. These results indicate that inhibiting PERK-eIF2α-directed apoptosis helped alleviate myocardial remodeling during the development of HF. Additionally, this inhibition was achieved in a Sirtuin 1 (SIRT1)-dependent way, which also suppressed cardiomyocyte apoptosis in DCM via the PERK-eIF2α pathway [132]. Activating PERK, however, was also verified to help alleviate hypertrophic remodeling [133]. It is therefore feasible that PERK may have divergent effects on cardiac disease depending on the context and signaling involved.
3.4 PERK-eIF2α's role in MI injury
Myocardial reperfusion injury primarily derives from excessive oxidative stress, increased ER stress, and/or cardiomyocyte apoptosis [134-137]. Consistently, in both cellular H/R models and animal myocardial I/R models, Perk/eIF2α/ATF4 signaling pathways were highly activated compared with control groups [134]. Anaerobic exercise exerts protective effects in I/R mice, which were in part mediated by the inactivation of the PERK/eIF2α/ATF4 pathway. Mechanistically, anaerobic exercise attenuated apoptosis and mitophagy by downregulating the PERK/eIF2α/ATF4 pathway, which consequently reduced myocardial damage and improved cardiac function. Furthermore, the PERK takes action in the pathological development of certain arrhythmias. Liu et al. [138] found that both genetic and pharmacological inhibition of PERK shortened Q and T waves on ECGs (thus shortening QTc intervals), decreased ventricular tachycardia episodes, and heightened survival rates after MI, suggesting that inhibiting PERK may reduce arrhythmia risk after MI.
4. The Implication of the cGAS-STING/PERK-eIF2α pathway in CVDs
As discussed, there is a significant connection between the cGAS-STING and PERK-eIF2α pathways. Evidence suggests that inhibiting either pathway can alleviate the pathological progression of various CVDs, including atherosclerosis, myocardial ischemia-reperfusion injury, and cardiomyopathy, by reducing inflammation, apoptosis, macrophage accumulation, and smooth muscle cell phenotype switching. While both pathways have been extensively studied individually, few investigations have explored their combined effects. Therefore, research targeting the cGAS-STING/PERK-eIF2α pathway may uncover novel therapeutic targets for CVDs. Notably, this combined pathway has been shown to mitigate senescence and organ fibrosis, which are critical mechanisms in the development of CVDs [139, 140]. The STING-p-PERK-p-eIF2α-ATF4 pathway has also consistently been verified to be involved in inflammation and collagen deposition and synthesis while attenuating the cGAS-STING/PERK pathway reduces fibroblast activation and extracellular matrix production [141, 142]. Additionally, aiming to explore the association between ER stress and cGAS-STING in cardiac pathophysiology, Zhang et al. [143] first demonstrated greater p-PERK and p-eIF2α expression in patients with DCM and hypertrophic cardiomyopathy (HCM). Then they showed that knockout of STING resulted in the elimination of p-PERK and p-eIF2α in mice with aortic binding. Likewise, the suppression of PERK reduced STING expression. Blocking the cGAS-STING/PERK-eIF2α pathway alleviated cardiomyocyte hypertrophy, inflammation, and fibrosis, and thus attenuated myocardial damage. With regards to AS, p-STING and p-PERK expression were increased in patients with AS as well as in ox-LDL-treated endothelium. The knockdown of STING alleviated endothelial injury, inflammatory responses, and atherosclerotic plaque areas. The intrinsic mechanism was explored using chromatin immunoprecipitation sequencing, and results confirmed that, during the development of AS, activated STING recruits PERK, leading to the recruitment of transcription factors such as IRF3 and NF-kB. Simultaneously, the activated STING/PERK complex promotes interactions between IRF3, NF-kB, and bromodomain protein 4 (BRD4), which form a super-enhancer and further direct the transcription of inflammatory genes [7]. Taken together, it is conceivable to extrapolate that the cGAS-STING/PERK-eIF2α interaction plays a vital role in an array of CVDs. Targeting this pathway may be an exciting new approach to the treatment of CVDs.
The PERK-eIF2α pathway in CVDs
| Model | Targets | Inhibitors | Activators | Cell phenotype | Related molecules | Results | Reference |
|---|---|---|---|---|---|---|---|
| ApoE-/-mice, oxLDL- induced HUVECs | PERK, eIF2α | atorvastatin | AMPK | Atorvastatin reduced the level of p-PERK and p-eIF2α, alleviating AS plaque formation and increased plaque stability | [113] | ||
| Cholesterol-loaded VSMCs | PERK, eIF2α | 4-PBA, ISRIB, shRNA-PERK | phenotypic switch of SMCs | ATF4 | The inhibition of PERK decreased plaque burden and AS progression | [42] | |
| Hyperlipidemic mice, PcntSMC-/-SMCs | PERK, eIF2α | ISRIB | pericentrin deficiency | phenotypic modulation of SMCs | HSF-1 | Pericentrin deficiency augmented cholesterol biosynthesis and AS plaque burden through PERK activation | [13] |
| Hyperlipidemic mice, VSMCs induced by free cholesterol | PERK | knockout of PERK | phenotypic modulation of SMCs | none | The inhibiton of PERK blocked AS plaque formation | [115] | |
| Rat administrated by vitamin D3 | PERK, eIF2α | ISRIB | calcium deposition and ALP activity | ATF-4 | The inhibition of eIF2α ameliorated the pathogenesis of vascular calcification | [116] | |
| Rat administrated by vitamin D3 | PERK, eIF2α | allicin | calcium deposition and ALP activity osteoblastic differentiation of VSMCs | ATF-4 | Allicin decreased the level of p-PERK and p-eIF2α, ameliorating the vascular calcification and stiffness | [118] | |
| T1DM induced cardiomyopathy mice, high glucose-treated neonatal rat ventricular myocytes | PERK, eIF2α | GSK2656157 | Melatonin | autophagy | ATF-4 GRP78 | The activation of PERK improved cardiac function and ameliorated myocardial injury in DCM | [17] |
| DCM mice induced by STZ | PERK, eIF2α | LCZ696 | inflammation, apoptosis | CHOP | LCZ696 alleviated lipid and glucose metabolism, protected against DCM-Induced myocardial injury through inhibiting AGEs/NF-κB and PERK/CHOP | [119] | |
| DCM mice induced by STZ, HL-1 cardiomyocyte stimulated by high-glucose | PERK | LncRNA H19 | thapsigargin | apoptosis | H19 improved left ventricular dysfunction and decreased fibrosis in DCM | [123] | |
| DCM mice induced by STZ and HFD, H9C2 cardiomyocyte induced by high glucose or palmitic acid | eIF2α | GCN2 deficiency | inflammation, apoptosis | CHOP, ATF4 | GCN2 deficiency ameliorated cardiac dysfunction and remodeling in DCM by reducing fibrosis, lipo-toxicity and oxidative stress | [124] | |
| Mice induced by DOX | eIF2α | GCN2 deficiency | apoptosis | CHOP, ATF4 | GCN2 deficiency improved myocardial contractile function and fibrosis in DOX-induced cardiomyopathy | [125] | |
| Rats administrated by CLP | PERK, eIF2α | Hydrogen Sulfide | apoptosis | CHOP | Hydrogen sulfide ameliorated myocardial injury caused by sepsis via suppressing PERK-eIF2α related ER stress | [128] | |
| LMNA R321X-cardiomyocytes | PERK, eIF2α | empaglifozin | apoptosis | CHOP | Empaglifozin attenuated LMNA cardiomyopathy via inhibition of PERK | [126] | |
| TAC induced mice, Ang II-induced cardiomyocyte | PERK, eIF2α | Dapagliflozin | migration and transformation of fibroblast, apoptosis | STRT1 | Dapagliflozin attenuated pressure overload-induced myocardial hyper trophy and remodeling via inhibition of PERK- eIF2α | [131] | |
| I/R rats, H/R-indeced H9c2 cardiomyocyte | PERK, eIF2α | M2AChR | mitophagy and apoptosis | ATF-4 | M2AChR activation resisted I/R-induced myocardial injury and improving cardiac function via inhibition of PERK- eIF2α | [134] | |
| MIRI rats, H/R-indeced H9c2 cardiomyocyte | PERK, eIF2α | cFLIPL | apoptosis | p38 MAPK | cFLIPL attenuated activation of PERK, and reduced myocardial infarction and increased the viability of H9c2 cells | [135] | |
| DM+I/R rat, H9C2 cardiomyocyte treated with HG and H/R | Ferrostatin-1, Salubrinal, | ferroptosis | ATF4 | Inhibition of ERS inhibited ferroptosis and ameliorated diabetes MIRI | [136] | ||
| MI mice, isolated cardiomyocytes from remote zone of the left ventricle scar | PERK | knockout of PERK, GSK | ion channel activation | Inhibition of PERK reduces arrhythmia risk after MI | [138] |
AS: atherosclerosis; HUVECs: human umbilical vein endothelial cells; AMPK: adenosine monophosphate-activated protein kinase; VSMCs: vascular smooth cells; 4-PBA: 4-phenylbutyric acid; ISRIB: integrated stress response inhibitor; HSF-1: heat shock factor 1; Pcnt: Pericentrin; ATF4: activating transcription factor 4; ALP: alkaline phosphatase; T1DM: type 1 diabetes mellitus; GRP78: Glucose-regulated protein 78; STZ: streptozotocin; AGEs: advanced glycation end-products; CHOP: C/EBP homologous protein; GCN2: general control nonderepressible 2; DOX: doxorubicin; Lamina A/C protein; CHOP: CCAAT-enhancer-binding protein homologous protein; TAC: transverse aortic constriction; M2AChR:M2 Acetylcholine receptor; I/R:ischemia-reperfusion; H/R: hypoxia/reoxygenation; MIRI: myocardial ischemia-reperfusion injury; CLP: cecal ligation and puncture; ERS: endoplasmic reticulum stress.
5. Inhibition of the cGAS-STING/PERK-eIF2α in CVDs and potential clinical value
With a growing body of research highlighting the significance of the STING/PERK pathway in numerous disorders, our understanding of the inhibitors of this pathway has also improved. The common inhibitors in the application of pre-clinical experiments about CVDs are listed in Table 3. For STING, mature antagonists include H-151, C-176, and C-178, which block palmitoylation at cysteines 88 and 91 (C88/91) and thus block STING's activation [144]. C-176 and C-178 are nitrofuran derivatives that covalently bind with one of STING's nucleophilic side chains via their 4-position furan rings, followed by rearrangement of STING and blocking of STING palmitoylation. C-176 and C-178 are only able to react with mice STING, but H-151 can block human STING activation with high specificity [145]. Common inhibitors of cGAS include RU.521, G108, G150, and PF-06928215. These inhibitors bind with cGAS' active site with high affinity and inhibit the production of cGAMP [146]. Although RU.521 only reacts with murine cGAS, G108, and G150 can also combine with human cGAS [147]. PF-06928215 has a high affinity with cGAS residue to impede its activation, although it showed no activity in cellular cGAS assays assessing dsDNA-induced IFN expression. The alkyl chain of PF-06928215 interplay with a small hydrophobic pocket of cGAS formed between Tyr436 and His437 [148]. In addition, several natural products have been discovered to prohibit cGAS-STING-induced pathways, including Astin C, flavonoids, and Scutellarin [91, 94]. GSK2656157, GSK2606414, and AMG44 are known to negate PERK by occupying its ATP-binding site, which can inhibit PERK in the nanomolar range [149]. However, the potent inhibition of phospho-PERK and histologic changes showed no effects on the PERK-mediated classical signaling, since the improper concentration of it may induce other related enzymes that promote this cascade. Integrated stress response inhibitor (ISRIB) can be used to inhibit eIF-2α's activation [150, 151]. However, GSK2656157 specifically has no effects on eIF2α [152]. Considering the significant crosstalk between the cGAS-STING and PERK-eIF2α pathways, a combination of drugs targeting both STING and PERK or certain compounds that inhibit both STING and PERK may have better effects in certain CVDs.
The inhibitors of cGAS-STING/PERK-eIF2α in CVDs
| Inhibitor | Effects in CVDs | Reference |
|---|---|---|
| RU.521 C-176 | Alleviated mitochondrial damage and activation of cGAS-STING pathway during the development of AS | [5] |
| C-176 | Increased content of VSMC and fibrous cap, alleviated plaque area and increased plaque stability | [10] |
| C-176 | Improved cardiac function and mitigated myocardial fibrosis in DCM | [49] |
| H-151 | Alleviated cardiac function and myocardial fibrosis in MI mice | [62] |
| H-151 | Decreased infarction size and scarring, relieved cardiac function and reduced myocardial fibrosis and hypertrophy in reperfused MI mice | [60] |
| RU.521 H-151 | Decreased cell apoptosis and alleviated cardiac function in MI/R mice | [94] |
| H-151 | Alleviated the LDH release and infarction area, increased cell viability and cardiac function, mitigated the MI/R injury in mice | [95] |
| ISRIB | Decreased plaque burden and AS progression | [65] |
| ISRIB | Reduced cholesterol biosynthesis and AS plaque burden | [13] |
| ISRIB | Ameliorated the calcium deposition and pathogenesis of vascular calcification | [104] |
| GSK2656157 | Promoted cardiac disfunction and myocardial injury in DCM | [17] |
| GSK2606414 | Reduces arrhythmia risk after MI | [126] |
AS: atherosclerosis; VSMC: vascular smooth muscle cell; DCM: diabetic cardiomyopathy; MI: myocardial infarction; MIR: myocardial ischemia/reperfusion
As discussed in this article, the individual and collaborative roles of the cGAS-STING and PERK-eIF2α pathways are crucial in the progression of CVDs. Understanding these pathways opens new avenues for clinical strategies in the diagnosis, treatment, and prognosis of patients.
Firstly, in addition to traditional diagnostic methods such as measuring myocardial enzymes and conducting echocardiograms, advanced technologies can be employed to assess the concentrations of cGAS-STING, PERK-eIF2α, and related downstream molecules, including IFN, IL, MCP-1, ROS, and ATF. This assessment can help gauge disease severity and facilitate risk stratification among patients. A more comprehensive understanding of a patient's condition before reperfusion surgery could enable healthcare providers to develop more precise therapeutic approaches.
Secondly, many current clinical treatments are insufficient due to myocardial remodeling and fibrosis, and they may cause adverse effects such as severe hypotension, edema, liver and kidney dysfunction, or hemorrhage, leading to patient intolerance. With an increasing number of preclinical studies investigating inhibitors of the cGAS-STING and PERK-eIF2α pathways, there is a promising opportunity to introduce these therapies into clinical practice. Additionally, considering the interaction between STING and PERK, combining inhibitors of both pathways may enhance efficacy while maintaining safety.
Previously, STING inhibitors primarily targeted the classical cGAS-STING pathway, neglecting the STING-PERK axis. Furthermore, research on PERK antagonists remains scarce, highlighting the urgent need to explore novel compounds or natural products that can simultaneously target both STING and PERK, such as compounds centered on the motif-helix aa322-343 of STING.
Lastly, the application of these inhibitors could provide alternative therapies for patients who do not achieve optimal results with existing treatments or experience severe side effects. A more thorough evaluation of disease status coupled with personalized treatment plans could lead to better prognoses for patients. From a clinical perspective, targeting the cGAS-STING/PERK-eIF2α pathway may represent a valuable therapeutic strategy for alleviating CVDs.
6. The nanomedicine for the treatment of CVDs by targeting cGAS-STING/PERK-eIF2α pathway
Investigating compounds within the cGAS-STING/PERK-eIF2α pathway holds significant clinical promise. However, many pharmacological experiments have struggled due to poor local drug activity and systemic toxicity arising from a lack of selectivity for target cells. This challenge has fostered advancements in biomaterials to address these issues, particularly through the use of nanomaterials, which have gained increasing attention.
The application of nanomaterial technology in inhibiting the cGAS-STING pathway is on the rise. Unlike conventional biomedical materials, nanomaterials exhibit enhanced properties, such as safety and biocompatibility, making them effective carriers for delivering cGAS or STING inhibitors to modulate the cGAS-STING signaling pathway [153, 154]. Research has shown that nanomedicine can have beneficial effects in treating various CVDs by primarily suppressing inflammation and immune responses [155-157]. For example, a biomimetic hydrogel system, Rb1/PDA-hydrogel, composed of ginsenoside Rb1 and polydopamine nanoparticles loaded into carboxylated chitosan, has been reported to improve outcomes in myocardial infarction treatment. This system reduces myocardial fibrosis, increases ventricular wall thickness, and enhances cardiac function by disrupting the crosstalk between mtDNA and cGAS, thereby inhibiting inflammation and macrophage polarization. Notably, this biomaterial demonstrates excellent adhesion to myocardial surfaces, making it suitable for deep myocardial injection due to its wet adhesion properties and conductivity [154]. While research on nanomedicine targeting PERK inhibition is limited, one study indicated that delivering a PERK inhibitor via biomimetic nanoclusters significantly improved cardiac function and inhibited aneurysmal dilatation. Remarkably, weekly injections of this targeted PERK inhibitor prevented the progression of pre-existing aneurysmal lesions [158]. Additionally, cysteine-based nanoparticles have been utilized to encapsulate the antifibrotic drug PF543, effectively delivering it to activated cardiac fibroblasts within the infarct area. This approach not only ensures prolonged drug release but also markedly reduces kidney and liver toxicity compared to traditional systemic therapies [155]. Overall, nanomaterials have shown great potential in treating CVDs by efficiently inhibiting inflammation and immune responses. Given the crucial role of macrophages in these processes, recent research has focused on macrophage-targeted nanomedicine, although challenges remain [159]. Future biomedical materials, especially nanomaterials, could be designed to specifically target cells involved in CVDs, such as macrophages and fibroblasts, to effectively inhibit macrophage polarization, inflammation, and fibrosis, ultimately alleviating conditions like atherosclerosis and myocardial infarction. In summary, the promising field of nanomedicine presents new possibilities for targeted therapies in the treatment of CVDs.
7. Conclusion and Limitations
In our review, we explored the critical roles of the cGAS-STING and PERK-eIF2α pathways in CVDs and hypothesized that these pathways may interact synergistically to accelerate disease progression. These insights could provide a new perspective on the cGAS-STING/PERK-eIF2α pathway, potentially enhancing the precision of targeted therapies for CVDs. With the rise of nanomedicine, the prospects for effectively inhibiting the cGAS-STING/PERK-eIF2α pathways appear increasingly promising. However, the more specific mechanisms underlying the association between STING and PERK remain unclear, and there is ongoing debate about how they regulate each other. Comprehensive investigations into these exact mechanisms are needed and could help transform the present basic research results into clinical practice. Additionally, due to the complexity of the crosstalk, activating this pathway can occasionally exert opposite effects depending on the context of CVDs, posing a challenge in determining whether to administer antagonists to patients with similar conditions. Therefore, we still need to determine the precise cardiac disorders within which PERK is activated and causes ensuing damage, as well as the cardiac disorders where PERK activation does not cause damage. Additionally, despite the evidence demonstrating the suppression of the cGAS-STING/PERK-eIF2α pathway in vivo and in vitro settings, there has been a lack of adequate implementation in clinical practice. The potential side effects of combining inhibitors targeting both pathways have yet to be thoroughly investigated. Additionally, compounds capable of simultaneously inhibiting both the cGAS-STING and PERK-eIF2α pathways remain largely unexplored. While promising advancements in nanomedicine have facilitated the inhibition of STING and PERK, several factors may hinder the widespread adoption of nanomaterials. For instance, their small size and enhanced penetration can increase the risk of toxicity to vital organs, such as the heart and brain. Furthermore, the release of nanomaterials into the environment could pose ecological risks [160, 161]. Therefore, further research is needed to establish appropriate dose ranges and to explore additional methods for assessing the toxicity of nanomaterials, including the use of various 3D models and experimental approaches. Additionally, the metabolic pathways of most nanoparticles in the body remain largely unexplored [162]. In other words, we need to develop advanced tracking modalities, such as radioactive tracing and fluorescence bioimaging, to accurately locate nanoparticles. Additionally, achieving quality control for nanoformulations requires sophisticated technology and a quality-by-design approach, along with multidisciplinary collaboration. Given both the potential benefits and uncertainties, the inhibition of the cGAS-STING/PERK-eIF2α pathway for treatment warrants ongoing and in-depth exploration in the future.
Abbreviations
cGAS: the cyclic 2′3′ GMP-AMP (cGAMP) synthase; cGAMP: cyclic guanosine monophosphate adenosine monophosphate; STING: stimulator of interferon genes; IFN: type I interferon; TBK1: TANK-binding kinase 1; IRF3: IFN regulatory factor 3; IL: interleukin; PERK: protein kinase RNA-like ER kinase; eIF2α: eukaryotic initiation factor 2α; UPR: unfolded protein response; CVDs: cardiovascular diseases; GRP78: glucose-regulated protein; ATF4: activating transcription factor 4; ISGs: type I interferon (IFN-I)-stimulated genes; IRE1α: inositol-requiring enzyme 1; CHOP: C/EBP-homologous protein; NLRP3: STING-NOD-like receptor protein 3; MAM: mitochondria-associated ER membranes; TRAF2: tumor necrosis factor receptor-associated factor 2; Nrf2: nuclear factor erythroid 2-related factor 2; MI/R: myocardial ischemia/reperfusion; TDP43: transactive response DNA-binding protein of 43 kDa; TNF-α: tumor necrosis factor-α; CKD: chronic kidney disease; DCM: diabetic cardiomyopathy.
Acknowledgements
Funding
This work was supported by Beijing Municipal Natural Science Foundation (7232043), Beijing Anzhen Hospital High Level Research Funding (2024AZB3003), the National Natural Science Foundation of China (82100486 and 82270341), High-level public technical talent construction project of Beijing Municipal Health Commission (Leading Talent-02-01), Beijing Hospitals Authority's Ascent Plan (DFL20220603), and Beijing Hospitals Authority Youth Programme (QMS20210603).
Author contributions
All authors made a significant contribution to the review article, whether that is in the conception, analysis, interpretation, or all these areas; participated in the drafting, revising, and reviewing of this article. All authors gave final approval of the version to be published and be accountable for all aspects of the work. All authors have reviewed the final version of the manuscript and approved it for publication.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Virani SS, Newby LK, Arnold SV, Bittner V, Brewer LC, Demeter SH. et al. 2023 AHA/ACC/ACCP/ASPC/NLA/PCNA Guideline for the Management of Patients With Chronic Coronary Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2023;82:833-955
2. Lejeune T, Derwael C, Bernard C, Letesson G, Daenen F, Hustinx R. Myocardial perfusion imaging and coronary artery disease: recent technological innovations. Rev Med Liege. 2023;78:580-5
3. Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circ Res. 2019;124:315-27
4. Wan X, Tian J, Hao P, Zhou K, Zhang J, Zhou Y. et al. cGAS-STING Pathway Performance in the Vulnerable Atherosclerotic Plaque. Aging Dis. 2022;13:1606-14
5. An C, Sun F, Liu C, Huang S, Xu T, Zhang C. et al. IQGAP1 promotes mitochondrial damage and activation of the mtDNA sensor cGAS-STING pathway to induce endothelial cell pyroptosis leading to atherosclerosis. Int Immunopharmacol. 2023;123:110795
6. Lv J, Zhu X, Xing C, Chen Y, Bian H, Yin H. et al. Stimulator of interferon genes (STING): Key therapeutic targets in ischemia/reperfusion injury. Biomed Pharmacother. 2023;167:115458
7. Li X, Chen X, Zheng L, Chen M, Zhang Y, Zhu R. et al. Non-canonical STING-PERK pathway dependent epigenetic regulation of vascular endothelial dysfunction via integrating IRF3 and NF-κB in inflammatory response. Acta Pharm Sin B. 2023;13:4765-84
8. King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP Jr. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23:1481-7
9. Ueda K, Sakai C, Ishida T, Morita K, Kobayashi Y, Horikoshi Y. et al. Cigarette smoke induces mitochondrial DNA damage and activates cGAS-STING pathway: application to a biomarker for atherosclerosis. Clin Sci (Lond). 2023;137:163-80
10. Bi X, Du C, Wang X, Wang XY, Han W, Wang Y. et al. Mitochondrial Damage-Induced Innate Immune Activation in Vascular Smooth Muscle Cells Promotes Chronic Kidney Disease-Associated Plaque Vulnerability. Adv Sci (Weinh). 2021;8:2002738
11. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. 2020;21:501-21
12. Oduro PK, Zheng X, Wei J, Yang Y, Wang Y, Zhang H. et al. The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm Sin B. 2022;12:50-75
13. Majumder S, Chattopadhyay A, Wright JM, Guan P, Buja LM, Kwartler CS. et al. Pericentrin deficiency in smooth muscle cells augments atherosclerosis through HSF1-driven cholesterol biosynthesis and PERK activation. JCI Insight. 2023;8:e173247
14. Sprooten J, Garg AD. Type I interferons and endoplasmic reticulum stress in health and disease. Int Rev Cell Mol Biol. 2020;350:63-118
15. Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B. et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell. 2017;171:809-23.e13
16. Almeida LM, Pinho BR, Duchen MR, Oliveira JMA. The PERKs of mitochondria protection during stress: insights for PERK modulation in neurodegenerative and metabolic diseases. Biol Rev Camb Philos Soc. 2022;97:1737-48
17. Zhang S, Tian W, Duan X, Zhang Q, Cao L, Liu C. et al. Melatonin attenuates diabetic cardiomyopathy by increasing autophagy of cardiomyocytes via regulation of VEGF-B/GRP78/PERK signaling pathway. Cardiovasc Diabetol. 2024;23:19
18. Zhao J, Xia C, Tang Y, Wan H. Role of PERK-mediated pathway in the effect of mild hypothermia after cerebral ischaemia/reperfusion. Eur J Clin Invest. 2023;53:e14040
19. Yu JE, Yeo IJ, Yoo SS, Kim SH, Son DJ, Yun J. et al. Induction of ER stress-mediated apoptosis through SOD1 upregulation by deficiency of CHI3L1 inhibits lung metastasis. Theranostics. 2023;13:2693-709
20. Zhang D, Liu Y, Zhu Y, Zhang Q, Guan H, Liu S. et al. A non-canonical cGAS-STING-PERK pathway facilitates the translational program critical for senescence and organ fibrosis. Nat Cell Biol. 2022;24:766-82
21. Vanhoutte D, Schips TG, Vo A, Grimes KM, Baldwin TA, Brody MJ. et al. Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated autophagy. Nat Commun. 2021;12:3928
22. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421-38
23. Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011-22
24. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL. et al. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol Cell. 2018;71:745-60.e5
25. Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB. et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol. 2012;13:737-43
26. Mohamed E, Sierra RA, Trillo-Tinoco J, Cao Y, Innamarato P, Payne KK. et al. The Unfolded Protein Response Mediator PERK Governs Myeloid Cell-Driven Immunosuppression in Tumors through Inhibition of STING Signaling. Immunity. 2020;52:668-82.e7
27. Sen T, Saha P, Gupta R, Foley LM, Jiang T, Abakumova OS. et al. Aberrant ER Stress Induced Neuronal-IFNβ Elicits White Matter Injury Due to Microglial Activation and T-Cell Infiltration after TBI. J Neurosci. 2020;40:424-46
28. Chin AC. PERK-STING Signaling Drives Neuroinflammation in Traumatic Brain Injury. J Neurosci. 2020;40:2384-6
29. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21:548-69
30. Zhou J, Zhuang Z, Li J, Feng Z. Significance of the cGAS-STING Pathway in Health and Disease. Int J Mol Sci. 2023;24:13316
31. Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020;10:26-39
32. Zhang W, Li G, Luo R, Lei J, Song Y, Wang B. et al. Cytosolic escape of mitochondrial DNA triggers cGAS-STING-NLRP3 axis-dependent nucleus pulposus cell pyroptosis. Exp Mol Med. 2022;54:129-42
33. Sun Z, Hornung V. cGAS-STING signaling. Curr Biol. 2022;32:R730-r4
34. Pan J, Fei CJ, Hu Y, Wu XY, Nie L, Chen J. Current understanding of the cGAS-STING signaling pathway: Structure, regulatory mechanisms, and related diseases. Zool Res. 2023;44:183-218
35. Fang R, Jiang Q, Yu X, Zhao Z, Jiang Z. Recent advances in the activation and regulation of the cGAS-STING pathway. Adv Immunol. 2022;156:55-102
36. Chen C, Xu P. Cellular functions of cGAS-STING signaling. Trends Cell Biol. 2023;33:630-48
37. Xu Y, Chen C, Liao Z, Xu P. cGAS-STING signaling in cell death: Mechanisms of action and implications in pathologies. Eur J Immunol. 2023;53:e2350386
38. Zhang K, Wang S, Gou H, Zhang J, Li C. Crosstalk Between Autophagy and the cGAS-STING Signaling Pathway in Type I Interferon Production. Front Cell Dev Biol. 2021;9:748485
39. Mitzel DN, Lowry V, Shirali AC, Liu Y, Stout-Delgado HW. Age-enhanced endoplasmic reticulum stress contributes to increased Atg9A inhibition of STING-mediated IFN-β production during Streptococcus pneumoniae infection. J Immunol. 2014;192:4273-83
40. Pan M, Hu T, Lyu J, Yin Y, Sun J, Wang Q. et al. CSNK1A1/CK1α suppresses autoimmunity by restraining the CGAS-STING1 signaling. Autophagy. 2024;20:311-28
41. Gui X, Yang H, Li T, Tan X, Shi P, Li M. et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567:262-6
42. Chattopadhyay A, Kwartler CS, Kaw K, Li Y, Kaw A, Chen J. et al. Cholesterol-Induced Phenotypic Modulation of Smooth Muscle Cells to Macrophage/Fibroblast-like Cells Is Driven by an Unfolded Protein Response. Arterioscler Thromb Vasc Biol. 2021;41:302-16
43. Read A, Schröder M. The Unfolded Protein Response: An Overview. Biology (Basel). 2021;10:384
44. Li T, Zhao H, Guo G, Xia S, Wang L. VMP1 affects endoplasmic reticulum stress sensitivity via differential modulation of the three unfolded protein response arms. Cell Rep. 2023;42:112209
45. Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr Mol Med. 2016;16:533-44
46. Zhou D, Yin M, Kang B, Yu X, Zeng H, Chen B. et al. CCT020312 exerts anti-prostate cancer effect by inducing G1 cell cycle arrest, apoptosis and autophagy through activation of PERK/eIF2α/ATF4/CHOP signaling. Biochem Pharmacol. 2024;221:116038
47. Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21:71-88
48. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173-94
49. Asano K, Clayton J, Shalev A, Hinnebusch AG. A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5, and initiator tRNA(Met) is an important translation initiation intermediate in vivo. Genes Dev. 2000;14:2534-46
50. Rajesh K, Krishnamoorthy J, Kazimierczak U, Tenkerian C, Papadakis AI, Wang S. et al. Phosphorylation of the translation initiation factor eIF2α at serine 51 determines the cell fate decisions of Akt in response to oxidative stress. Cell Death Dis. 2015;6:e1591
51. Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2α kinases: their structures and functions. Cell Mol Life Sci. 2013;70:3493-511
52. Perkins DJ, Barber GN. Defects in translational regulation mediated by the alpha subunit of eukaryotic initiation factor 2 inhibit antiviral activity and facilitate the malignant transformation of human fibroblasts. Mol Cell Biol. 2004;24:2025-40
53. Bond S, Lopez-Lloreda C, Gannon PJ, Akay-Espinoza C, Jordan-Sciutto KL. The Integrated Stress Response and Phosphorylated Eukaryotic Initiation Factor 2α in Neurodegeneration. J Neuropathol Exp Neurol. 2020;79:123-43
54. Kepp O, Semeraro M, Bravo-San Pedro JM, Bloy N, Buqué A, Huang X. et al. eIF2α phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol. 2015;33:86-92
55. Jin B, Wang M, Sun Y, Lee PAH, Zhang X, Lu Y. et al. CHIP suppresses the proliferation and migration of A549 cells by mediating the ubiquitination of eIF2α and upregulation of tumor suppressor RBM5. J Biol Chem. 2024;300:105673
56. Hicks D, Giresh K, Wrischnik LA, Weiser DC. The PPP1R15 Family of eIF2-alpha Phosphatase Targeting Subunits (GADD34 and CReP). Int J Mol Sci. 2023;24:17321
57. Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ. et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med. 2019;216:867-83
58. Ma XM, Geng K, Law BY, Wang P, Pu YL, Chen Q. et al. Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell Biol Toxicol. 2023;39:277-99
59. Rösing S, Ullrich F, Meisterfeld S, Schmidt F, Mlitzko L, Croon M. et al. Chronic endoplasmic reticulum stress in myotonic dystrophy type 2 promotes autoimmunity via mitochondrial DNA release. Nat Commun. 2024;15:1534
60. Bai L, Zhang R, Zheng H, Zhang Z, Zhang Z, Li Y. Seneca Valley Virus Degrades STING via PERK and ATF6-Mediated Reticulophagy. Viruses. 2023;15:2209
61. Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W. et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019;24:101215
62. Guimarães ES, Gomes MTR, Sanches RCO, Matteucci KC, Marinho FV, Oliveira SC. The endoplasmic reticulum stress sensor IRE1α modulates macrophage metabolic function during Brucella abortus infection. Front Immunol. 2022;13:1063221
63. Zhen Y, Zhang H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front Immunol. 2019;10:276
64. Jiang L, Yang F, Liao H, Chen W, Dai X, Peng C. et al. Molybdenum and cadmium cause blood-testis barrier dysfunction through ROS-mediated NLRP3 inflammasome activation in sheep. Sci Total Environ. 2024;906:167267
65. Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K. et al. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012;3:e261
66. Zhang Y, Li G, Zhao Y, Dai X, Hu M, Cao H. et al. Inhibition of calcium imbalance protects hepatocytes from vanadium exposure-induced inflammation by mediating mitochondrial-associated endoplasmic reticulum membranes in ducks. Poult Sci. 2023;102:103013
67. Franz KM, Neidermyer WJ, Tan YJ, Whelan SPJ, Kagan JC. STING-dependent translation inhibition restricts RNA virus replication. Proc Natl Acad Sci U S A. 2018;115:E2058-e67
68. Xue M, Fu F, Ma Y, Zhang X, Li L, Feng L. et al. The PERK Arm of the Unfolded Protein Response Negatively Regulates Transmissible Gastroenteritis Virus Replication by Suppressing Protein Translation and Promoting Type I Interferon Production. J Virol. 2018;92:e00431-18
69. Charbonneau ME, O'Riordan MXD. Reducing Stress PERKs up Anti-tumor Immunity. Immunity. 2020;52:575-7
70. Cui Y, Zhao D, Sreevatsan S, Liu C, Yang W, Song Z. et al. Mycobacterium bovis Induces Endoplasmic Reticulum Stress Mediated-Apoptosis by Activating IRF3 in a Murine Macrophage Cell Line. Front Cell Infect Microbiol. 2016;6:182
71. Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones. 2007;39:86-93
72. Liang SJ, Zeng DY, Mai XY, Shang JY, Wu QQ, Yuan JN. et al. Inhibition of Orai1 Store-Operated Calcium Channel Prevents Foam Cell Formation and Atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:618-28
73. Coyne AN, Zaepfel BL, Zarnescu DC. Failure to Deliver and Translate-New Insights into RNA Dysregulation in ALS. Front Cell Neurosci. 2017;11:243
74. Kumar S, Julien JP. TDP-43 triggers immune response via mitochondrial DNA release. Cell Res. 2021;31:379-80
75. Huangfu N, Wang Y, Xu Z, Zheng W, Tao C, Li Z. et al. TDP43 Exacerbates Atherosclerosis Progression by Promoting Inflammation and Lipid Uptake of Macrophages. Front Cell Dev Biol. 2021;9:687169
76. Xu Z, Ma Z, Zhao X, Zhang B. Aerobic exercise mitigates high-fat diet-induced cardiac dysfunction, pyroptosis, and inflammation by inhibiting STING-NLRP3 signaling pathway. Mol Cell Biochem. 2024;479:3459-3470
77. Wan X, Tian J, Hao P, Zhang J, Zhou Y, Ge C. et al. The cGAS-STING Pathway: A Ubiquitous Checkpoint Perturbing Myocardial Attributes. Curr Vasc Pharmacol. 2023;21:152-62
78. Lu L, Shao Y, Xiong X, Ma J, Zhai M, Lu G. et al. Irisin improves diabetic cardiomyopathy-induced cardiac remodeling by regulating GSDMD-mediated pyroptosis through MITOL/STING signaling. Biomed Pharmacother. 2024;171:116007
79. Xiao Z, Yu Z, Chen C, Chen R, Su Y. GAS-STING signaling plays an essential pathogenetic role in Doxorubicin-Induced Cardiotoxicity. BMC Pharmacol Toxicol. 2023;24:19
80. Lu QB, Ding Y, Liu Y, Wang ZC, Wu YJ, Niu KM. et al. Metrnl ameliorates diabetic cardiomyopathy via inactivation of cGAS/STING signaling dependent on LKB1/AMPK/ULK1-mediated autophagy. J Adv Res. 2023;51:161-79
81. Borghi A, Verstrepen L, Beyaert R. TRAF2 multitasking in TNF receptor-induced signaling to NF-κB, MAP kinases and cell death. Biochem Pharmacol. 2016;116:1-10
82. En A, Bogireddi H, Thomas B, Stutzman A, Ikegami S, LaForest B. et al. The cGAS-STING pathway is dispensable in a mouse model of LMNA-cardiomyopathy despite nuclear envelope rupture. bioRxiv. 2023
83. Beesley SJ, Weber G, Sarge T, Nikravan S, Grissom CK, Lanspa MJ. et al. Septic Cardiomyopathy. Crit Care Med. 2018;46:625-34
84. Liu H, Hu Q, Ren K, Wu P, Wang Y, Lv C. ALDH2 mitigates LPS-induced cardiac dysfunction, inflammation, and apoptosis through the cGAS/STING pathway. Mol Med. 2023;29:171
85. Kong C, Ni X, Wang Y, Zhang A, Zhang Y, Lin F. et al. ICA69 aggravates ferroptosis causing septic cardiac dysfunction via STING trafficking. Cell Death Discov. 2022;8:187
86. Chang L, Huang R, Chen J, Li G, Shi G, Xu B. et al. An alpha-helix variant p.Arg156Pro in LMNA as a cause of hereditary dilated cardiomyopathy: genetics and bioinfomatics exploration. BMC Med Genomics. 2023;16:229
87. Cheedipudi SM, Asghar S, Marian AJ. Genetic Ablation of the DNA Damage Response Pathway Attenuates Lamin-Associated Dilated Cardiomyopathy in Mice. JACC Basic Transl Sci. 2022;7:1232-45
88. Park HY. Hereditary Dilated Cardiomyopathy: Recent Advances in Genetic Diagnostics. Korean Circ J. 2017;47:291-8
89. Hu S, Gao Y, Gao R, Wang Y, Qu Y, Yang J. et al. The selective STING inhibitor H-151 preserves myocardial function and ameliorates cardiac fibrosis in murine myocardial infarction. Int Immunopharmacol. 2022;107:108658
90. Mauro AG, Bonaventura A, Mezzaroma E, Quader M, Toldo S. NLRP3 Inflammasome in Acute Myocardial Infarction. J Cardiovasc Pharmacol. 2019;74:175-87
91. Tian M, Li F, Pei H. The cGAS-STING Pathway: A New Therapeutic Target for Ischemia-Reperfusion Injury in Acute Myocardial Infarction? Biomedicines. 2024;12:1728
92. Rech L, Abdellatif M, Pöttler M, Stangl V, Mabotuwana N, Hardy S. et al. Small molecule STING inhibition improves myocardial infarction remodeling. Life Sci. 2022;291:120263
93. Guo Y, You Y, Shang FF, Wang X, Huang B, Zhao B. et al. iNOS aggravates pressure overload-induced cardiac dysfunction via activation of the cytosolic-mtDNA-mediated cGAS-STING pathway. Theranostics. 2023;13:4229-46
94. Li JK, Song ZP, Hou XZ. Scutellarin ameliorates ischemia/reperfusion injury-induced cardiomyocyte apoptosis and cardiac dysfunction via inhibition of the cGAS-STING pathway. Exp Ther Med. 2023;25:155
95. Xiong Y, Leng Y, Tian H, Deng X, Li W, Li W. et al. Decreased MFN2 activates the cGAS-STING pathway in diabetic myocardial ischaemia-reperfusion by triggering the release of mitochondrial DNA. Cell Commun Signal. 2023;21:192
96. Halade GV, Lee DH. Inflammation and resolution signaling in cardiac repair and heart failure. EBioMedicine. 2022;79:103992
97. Hu D, Cui YX, Wu MY, Li L, Su LN, Lian Z. et al. Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure. Am J Physiol Heart Circ Physiol. 2020;318:H1525-h37
98. Sun SJ, Jiao XD, Chen ZG, Cao Q, Zhu JH, Shen QR. et al. Gasdermin-E-mediated pyroptosis drives immune checkpoint inhibitor-associated myocarditis via cGAS-STING activation. Nat Commun. 2024;15:6640
99. Liu C, Chen H, Tao X, Li C, Li A, Wu W. ALKBH5 protects against stroke by reducing endoplasmic reticulum stress-dependent inflammation injury via the STAT5/PERK/EIF2α/CHOP signaling pathway in an m(6)A-YTHDF1-dependent manner. Exp Neurol. 2024;372:114629
100. Wang JL, Liu H, Jing ZC, Zhao F, Zhou R. 18β-Glycyrrhetinic acid ameliorates endoplasmic reticulum stress-induced inflammation in pulmonary arterial hypertension through PERK/eIF2α/NF-κB signaling. Chin J Physiol. 2022;65:187-98
101. Zuo MY, Tang TJ, Wang X, Gu JF, Wang L, Chen J. et al. Atractylenolide III Attenuates Apoptosis in H9c2 Cells by Inhibiting Endoplasmic Reticulum Stress through the GRP78/PERK/CHOP Signaling Pathway. Evid Based Complement Alternat Med. 2022;2022:1149231
102. Monceaux K, Gressette M, Karoui A, Pires Da Silva J, Piquereau J, Ventura-Clapier R. et al. Ferulic Acid, Pterostilbene, and Tyrosol Protect the Heart from ER-Stress-Induced Injury by Activating SIRT1-Dependent Deacetylation of eIF2α. Int J Mol Sci. 2022;23:6628
103. Li C, Zhang H, Mo J, Zuo J, Ye L. Caspase-3/GSDME dependent pyroptosis contributes to offspring lung injury induced by gestational PFOS exposure via PERK/ATF4 signaling. Arch Toxicol. 2024;98:207-21
104. Wen B, Yang L, Guo J, Chang W, Wei S, Yu S. et al. Peste des petits ruminants virus induces ERS-mediated autophagy to promote virus replication. Vet Microbiol. 2022;270:109451
105. Wang L, Pan Y, Yang F, Guo X, Peng J, Wang X. et al. New sight into interaction between endoplasmic reticulum stress and autophagy induced by vanadium in duck renal tubule epithelial cells. Chem Biol Interact. 2022;362:109981
106. Li Y, Hu P, Zhang Z, Yuan Z, Yang K, Sun Z. Protective autophagy alleviates neurotoxin-gelsenicine induced apoptosis through PERK signaling pathway in Neuro-2a cells. Toxicology. 2022;474:153210
107. Balsa E, Soustek MS, Thomas A, Cogliati S, García-Poyatos C, Martín-García E. et al. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2α Axis. Mol Cell. 2019;74:877-90.e6
108. Lebeau J, Saunders JM, Moraes VWR, Madhavan A, Madrazo N, Anthony MC. et al. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018;22:2827-36
109. Zhong Y, Liu J, Sun D, Guo T, Yao Y, Xia X. et al. Dioscin relieves diabetic nephropathy via suppressing oxidative stress and apoptosis, and improving mitochondrial quality and quantity control. Food Funct. 2022;13:3660-73
110. Latorre-Muro P, O'Malley KE, Bennett CF, Perry EA, Balsa E, Tavares CDJ. et al. A cold-stress-inducible PERK/OGT axis controls TOM70-assisted mitochondrial protein import and cristae formation. Cell Metab. 2021;33:598-614.e7
111. Li D, Shao R, Wang N, Zhou N, Du K, Shi J. et al. Sulforaphane Activates a lysosome-dependent transcriptional program to mitigate oxidative stress. Autophagy. 2021;17:872-87
112. Schuster EM, Epple MW, Glaser KM, Mihlan M, Lucht K, Zimmermann JA. et al. TFEB induces mitochondrial itaconate synthesis to suppress bacterial growth in macrophages. Nat Metab. 2022;4:856-66
113. Xiong W, Fei M, Wu C, Wang W, Luo R, Shen L. et al. Atorvastatin inhibits endoplasmic reticulum stress through AMPK signaling pathway in atherosclerosis in mice. Exp Ther Med. 2020;19:2266-72
114. Ma W, Viveiros MM. Depletion of pericentrin in mouse oocytes disrupts microtubule organizing center function and meiotic spindle organization. Mol Reprod Dev. 2014;81:1019-29
115. Chattopadhyay A, Guan P, Majumder S, Kaw K, Zhou Z, Zhang C. et al. Preventing Cholesterol-Induced Perk (Protein Kinase RNA-Like Endoplasmic Reticulum Kinase) Signaling in Smooth Muscle Cells Blocks Atherosclerotic Plaque Formation. Arterioscler Thromb Vasc Biol. 2022;42:1005-22
116. Dong J, Jin S, Guo J, Yang R, Tian D, Xue H. et al. Pharmacological inhibition of eIF2alpha phosphorylation by integrated stress response inhibitor (ISRIB) ameliorates vascular calcification in rats. Physiol Res. 2022;71:379-88
117. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. 2018;114:590-600
118. Wang Q, Lin P, Feng L, Ren Q, Xie X, Zhang B. Ameliorative effect of allicin on vascular calcification via inhibiting endoplasmic reticulum stress. Vascular. 2022;30:999-1007
119. Belali OM, Ahmed MM, Mohany M, Belali TM, Alotaibi MM, Al-Hoshani A. et al. LCZ696 Protects against Diabetic Cardiomyopathy-Induced Myocardial Inflammation, ER Stress, and Apoptosis through Inhibiting AGEs/NF-κB and PERK/CHOP Signaling Pathways. Int J Mol Sci. 2022;23:1288
120. Hou H, Zhang Q, Dong H, Ge Z. Matrine improves diabetic cardiomyopathy through TGF-β-induced protein kinase RNA-like endoplasmic reticulum kinase signaling pathway. J Cell Biochem. 2019;120:13573-82
121. Liu M, Kang GJ, Dudley SC Jr. Targeting PERK to treat arrhythmias. Trends Mol Med. 2023;29:583
122. Yang L, Zhao D, Ren J, Yang J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:209-18
123. Wang S, Duan J, Liao J, Wang Y, Xiao X, Li L. et al. LncRNA H19 inhibits ER stress induced apoptosis and improves diabetic cardiomyopathy by regulating PI3K/AKT/mTOR axis. Aging (Albany NY). 2022;14:6809-28
124. Feng W, Lei T, Wang Y, Feng R, Yuan J, Shen X. et al. GCN2 deficiency ameliorates cardiac dysfunction in diabetic mice by reducing lipotoxicity and oxidative stress. Free Radic Biol Med. 2019;130:128-39
125. Wang Y, Lei T, Yuan J, Wu Y, Shen X, Gao J. et al. GCN2 deficiency ameliorates doxorubicin-induced cardiotoxicity by decreasing cardiomyocyte apoptosis and myocardial oxidative stress. Redox Biol. 2018;17:25-34
126. Pietrafesa G, De Zio R, Scorza SI, Armentano MF, Pepe M, Forleo C. et al. Targeting unfolded protein response reverts ER stress and ER Ca(2+) homeostasis in cardiomyocytes expressing the pathogenic variant of Lamin A/C R321X. J Transl Med. 2023;21:340
127. Feyen DAM, Perea-Gil I, Maas RGC, Harakalova M, Gavidia AA, Arthur Ataam J. et al. Unfolded Protein Response as a Compensatory Mechanism and Potential Therapeutic Target in PLN R14del Cardiomyopathy. Circulation. 2021;144:382-92
128. Zhao YH, Cao GD, Guo LC, Cheng QH. Hydrogen Sulfide Ameliorates Myocardial Injury Caused by Sepsis Through Suppressing ROS-Mediated Endoplasmic Reticulum Stress. Sichuan Da Xue Xue Bao Yi Xue Ban. 2022;53:798-804
129. Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y. et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 2022;12:708-22
130. Brieler J, Breeden MA, Tucker J. Cardiomyopathy: An Overview. Am Fam Physician. 2017;96:640-6
131. Liu Z, Huang J, Wang X, Deng S, Zhou J, Gong Z. et al. Dapagliflozin suppress endoplasmic reticulum stress mediated apoptosis of chondrocytes by activating Sirt1. Chem Biol Interact. 2023;384:110724
132. Guo R, Liu W, Liu B, Zhang B, Li W, Xu Y. SIRT1 suppresses cardiomyocyte apoptosis in diabetic cardiomyopathy: An insight into endoplasmic reticulum stress response mechanism. Int J Cardiol. 2015;191:36-45
133. Ghadge SK, Messner M, Seiringer H, Maurer T, Staggl S, Zeller T. et al. Smooth Muscle Specific Ablation of CXCL12 in Mice Downregulates CXCR7 Associated with Defective Coronary Arteries and Cardiac Hypertrophy. Int J Mol Sci. 2021;22:5908
134. Chen W, Ma M, Song Y, Hua Y, Jia H, Liu J. et al. Exercise Attenuates Myocardial Ischemia-Reperfusion Injury by Regulating Endoplasmic Reticulum Stress and Mitophagy Through M(2) Acetylcholine Receptor. Antioxid Redox Signal. 2024;40:209-21
135. Li YZ, Wu H, Liu D, Yang J, Yang J, Ding JW. et al. cFLIP(L) Alleviates Myocardial Ischemia-Reperfusion Injury by Inhibiting Endoplasmic Reticulum Stress. Cardiovasc Drugs Ther. 2023;37:225-38
136. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis Is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury Through Endoplasmic Reticulum Stress. DNA Cell Biol. 2020;39:210-25
137. Guo W, Liu X, Li J, Shen Y, Zhou Z, Wang M. et al. Prdx1 alleviates cardiomyocyte apoptosis through ROS-activated MAPK pathway during myocardial ischemia/reperfusion injury. Int J Biol Macromol. 2018;112:608-15
138. Liu M, Liu H, Parthiban P, Kang GJ, Shi G, Feng F. et al. Inhibition of the unfolded protein response reduces arrhythmia risk after myocardial infarction. J Clin Invest. 2021;131:e147836
139. Hu C, Zhang X, Teng T, Ma ZG, Tang QZ. Cellular Senescence in Cardiovascular Diseases: A Systematic Review. Aging Dis. 2022;13:103-28
140. Evangelou K, Vasileiou PVS, Papaspyropoulos A, Hazapis O, Petty R, Demaria M. et al. Cellular senescence and cardiovascular diseases: moving to the "heart" of the problem. Physiol Rev. 2023;103:609-47
141. Xie X, Wu X, Zhao D, Liu Y, Du Q, Li Y. et al. Fluvoxamine alleviates bleomycin-induced lung fibrosis via regulating the cGAS-STING pathway. Pharmacol Res. 2023;187:106577
142. Deng J, He Y, Sun G, Yang H, Wang L, Tao X. et al. Tanreqing injection protects against bleomycin-induced pulmonary fibrosis via inhibiting STING-mediated endoplasmic reticulum stress signaling pathway. J Ethnopharmacol. 2023;305:116071
143. Zhang Y, Chen W, Wang Y. STING is an essential regulator of heart inflammation and fibrosis in mice with pathological cardiac hypertrophy via endoplasmic reticulum (ER) stress. Biomed Pharmacother. 2020;125:110022
144. Zhang S, Zheng R, Pan Y, Sun H. Potential Therapeutic Value of the STING Inhibitors. Molecules. 2023;28:3127
145. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A. et al. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559:269-73
146. Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y. et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. 2017;8:750
147. Lama L, Adura C, Xie W, Tomita D, Kamei T, Kuryavyi V. et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat Commun. 2019;10:2261
148. Hall J, Brault A, Vincent F, Weng S, Wang H, Dumlao D. et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS One. 2017;12:e0184843
149. Szaruga M, Janssen DA, de Miguel C, Hodgson G, Fatalska A, Pitera AP. et al. Activation of the integrated stress response by inhibitors of its kinases. Nat Commun. 2023;14:5535
150. Zyryanova AF, Kashiwagi K, Rato C, Harding HP, Crespillo-Casado A, Perera LA. et al. ISRIB Blunts the Integrated Stress Response by Allosterically Antagonising the Inhibitory Effect of Phosphorylated eIF2 on eIF2B. Mol Cell. 2021;81:88-103.e6
151. Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife. 2015;4:e05033
152. Krishnamoorthy J, Rajesh K, Mirzajani F, Kesoglidou P, Papadakis AI, Koromilas AE. Evidence for eIF2α phosphorylation-independent effects of GSK2656157, a novel catalytic inhibitor of PERK with clinical implications. Cell Cycle. 2014;13:801-6
153. Gao X, Wang J, Wang Y, Liu S, Dong K, Wu J. et al. Fucoidan-ferulic acid nanoparticles alleviate cisplatin-induced acute kidney injury by inhibiting the cGAS-STING pathway. Int J Biol Macromol. 2022;223:1083-93
154. Zheng Z, Sun J, Wang J, He S, Liu Z, Xie J. et al. Enhancing myocardial infarction treatment through bionic hydrogel-mediated spatial combination therapy via mtDNA-STING crosstalk modulation. J Control Release. 2024;371:570-87
155. Ji X, Meng Y, Wang Q, Tong T, Liu Z, Lin J. et al. Cysteine-Based Redox-Responsive Nanoparticles for Fibroblast-Targeted Drug Delivery in the Treatment of Myocardial Infarction. ACS Nano. 2023;17:5421-34
156. Li Y, Chen X, Jin R, Chen L, Dang M, Cao H. et al. Injectable hydrogel with MSNs/microRNA-21-5p delivery enables both immunomodification and enhanced angiogenesis for myocardial infarction therapy in pigs. Sci Adv. 2021;7:eabd6740
157. Zhang J, Guo Y, Bai Y, Wei Y. Application of biomedical materials in the diagnosis and treatment of myocardial infarction. J Nanobiotechnology. 2023;21:298
158. Yodsanit N, Shirasu T, Huang Y, Yin L, Islam ZH, Gregg AC. et al. Targeted PERK inhibition with biomimetic nanoclusters confers preventative and interventional benefits to elastase-induced abdominal aortic aneurysms. Bioact Mater. 2023;26:52-63
159. Chen W, Schilperoort M, Cao Y, Shi J, Tabas I, Tao W. Macrophage-targeted nanomedicine for the diagnosis and treatment of atherosclerosis. Nat Rev Cardiol. 2022;19:228-49
160. Cheng Y, Chen Z, Yang S, Liu T, Yin L, Pu Y. et al. Nanomaterials-induced toxicity on cardiac myocytes and tissues, and emerging toxicity assessment techniques. Sci Total Environ. 2021;800:149584
161. Juárez-Maldonado A, Tortella G, Rubilar O, Fincheira P, Benavides-Mendoza A. Biostimulation and toxicity: The magnitude of the impact of nanomaterials in microorganisms and plants. J Adv Res. 2021;31:113-26
162. Jia Y, Jiang Y, He Y, Zhang W, Zou J, Magar KT. et al. Approved Nanomedicine against Diseases. Pharmaceutics. 2023;15:774
Author contact
![]() Corresponding authors: Xiantao Song, MD, PhD, Professor of Medicine/Cardiology, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart, Lung and Blood Vessel Disease, 2 Anzhen Road, Beijing 100029, P.R. China; Email: song0929ccmu.edu.cn. Changjiang Ge, MD, PhD, Professor of Medicine/Cardiology, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart, Lung and Blood Vessel Disease, 2 Anzhen Road, Beijing 100029, P.R. China; Email: cjge1116ccmu.edu.cn.
Corresponding authors: Xiantao Song, MD, PhD, Professor of Medicine/Cardiology, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart, Lung and Blood Vessel Disease, 2 Anzhen Road, Beijing 100029, P.R. China; Email: song0929ccmu.edu.cn. Changjiang Ge, MD, PhD, Professor of Medicine/Cardiology, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart, Lung and Blood Vessel Disease, 2 Anzhen Road, Beijing 100029, P.R. China; Email: cjge1116ccmu.edu.cn.