Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(1):143-159. doi:10.7150/ijbs.100212 This issue Cite
Research Paper
TBK1 inhibitor amlexanox exerts anti-cancer effects against endometrial cancer by regulating AKT/NF-κB signaling
Jiha Shin1†, Jihoon Lim1†, Daewon Han1, Solji Lee1, Nak Song Sung2, Jong-Seok Kim3, Do Kyung Kim4, Hoi Young Lee5, Sung Ki Lee3,6, Jongdae Shin1,3, Jeong Sig Kim7 ![]() , Hwan-Woo Park1,3
, Hwan-Woo Park1,3 ![]()
1. Department of Cell Biology, Konyang University College of Medicine, Daejeon 35365, Republic of Korea.
2. Department of Surgery, Konyang University Hospital, Daejeon 35365, Republic of Korea.
3. Myunggok Medical Research Institute, Konyang University College of Medicine, Daejeon 35365, Republic of Korea.
4. Department of Anatomy, Konyang University College of Medicine, Daejeon 35365, Republic of Korea.
5. Department of Pharmacology, Konyang University College of Medicine, Daejeon 35365, Republic of Korea.
6. Department of Obstetrics and Gynecology, Konyang University Hospital, Daejeon, Republic of Korea.
7. Department of Obstetrics and Gynecology, Soonchunhyang University Seoul Hospital, Seoul 04401, Republic of Korea.
†These authors contributed equally to this work.
Received 2024-6-27; Accepted 2024-11-15; Published 2025-1-1
Abstract

Endometrial cancer, a common gynecological malignancy, poses significant clinical challenges, particularly in advanced or recurrent cases. TANK-binding kinase 1 (TBK1), a serine/threonine kinase, plays crucial roles in inflammation and immunity by activating nuclear factor (NF)-κB and interferon regulatory factor 3. However, its specific roles in endometrial cancer remain unknown. In this study, we aimed to investigate the anti-cancer effects and underlying mechanisms of amlexanox, a TBK1 inhibitor, against endometrial cancer. The main genetic mutations in TBK1 were found to be mRNA downregulation and missense mutations. Kaplan-Meier plotter analysis revealed that low TBK1 expression was associated with a good prognosis in patients with uterine corpus endometrial carcinoma (UCEC). In vitro experiments demonstrated that TBK1 knockdown or amlexanox significantly inhibited the proliferation, cell cycle progression, and migration of endometrial cancer cells. Furthermore, the inhibitory effects of targeting TBK1 on cancer cell proliferation and migration were mediated by the protein kinase B (AKT)/NF-κB signaling pathway. Xenograft experiments revealed that both amlexanox treatment and TBK1 knockdown effectively suppressed the tumor growth. Overall, this study highlights the potent anti-cancer effects of amlexanox against endometrial cancer by modulating AKT/NF-κB signaling, thus providing a new avenue for the development of novel TBK1-targeting therapeutic strategies for UCEC.
Keywords: TBK1, endometrial cancer, xenograft, shRNA, prognosis, NF-κB
Introduction
Endometrial cancer, the most common gynecological malignancy, poses a significant global health burden, with increasing incidence worldwide [1, 2]. Despite advances in treatment, management of advanced or recurrent endometrial cancer remains a clinical challenge, necessitating the development of new therapeutic strategies [3, 4]. Targeting specific signaling pathways involved in endometrial cancer progression has emerged as a promising therapeutic approach.
TANK-binding kinase 1 (TBK1), a serine/threonine kinase, plays important roles in various cellular processes, including inflammation and innate immunity, via activation of nuclear factor (NF)-κB and interferon regulatory factor 3 pathways [5-7]. TBK1 is involved in malignant cell growth in different cancer types, including non-small cell lung cancer, breast cancer, and pancreatic ductal adenocarcinoma [8-11]. Increased TBK1 expression and aberrant TBK1 activity promote cancer cell survival and proliferation [9, 12, 13]. TBK1 is considered a potential therapeutic target owing to its crucial roles in various diseases, including autoimmune disorders, metabolic diseases, and cancer [6, 7, 14]. Inhibition of TBK1 reduces the viability and proliferation of lung and thyroid cancer cells [15, 16]. Although TBK1 is closely associated with cancer progression and cell viability, its specific roles in endometrial cancer remain unknown.
Recently, various TBK1-targeting inhibitors have been developed. These inhibitors have exhibited high efficacy in the treatment of TBK1-associated diseases [17]. Amlexanox, a TBK1 inhibitor approved for the treatment of aphthous ulcers and asthma, inhibits obesity and type 2 diabetes [18-21]. It has garnered interest owing to its potential anti-cancer effects [22, 23]. NF-κB signaling pathway plays vital roles in cancer development and progression by affecting cell survival, proliferation, and inflammation [24, 25]. In lung and thyroid cancer, TBK1 is a key regulator of the NF-κB and protein kinase B (AKT) signaling pathways [12, 16]. Amlexanox shows potential for endometrial cancer targeting by targeting TBK1. However, its specific anti-cancer effects and mechanisms against endometrial cancer remain unclear.
In this study, we aimed to investigate the effects of amlexanox and TBK1 short hairpin (sh) RNA (shRNA) on the proliferation and migration of endometrial cancer cells by inhibiting TBK1 expression. Moreover, we conducted bioinformatic analyses of TBK1, including genetic alteration, survival, DNA methylation, copy number variation, and somatic cell mutation analyses, in patients with uterine corpus endometrial carcinoma (UCEC). We also explored the molecular mechanisms underlying the regulation of AKT/NF-κB signaling by TBK1 inhibition in endometrial cancer cells. Our findings provide valuable insights for the development of targeted therapies and clinical management of patients with endometrial cancer.
Materials and Methods
Reagents
Amlexanox, BX-795, and MK-2206 were purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). Immunoblotting and immunostaining were performed using antibodies against phosphorylated (p)-TBK1, total TBK1, N-cadherin, E-cadherin, snail, vimentin, p-AKT, total AKT, p-NF-κB, total NF-κB, p-p70S6K, total p70S6K (Cell Signaling Technology, Danvers, MA, USA), α-tubulin, and β-actin (Developmental Studies Hybridoma Bank, Iowa City, IA, USA). Transforming growth factor (TGF)-β1 and insulin growth factor-1 were purchased from PeproTech Inc. (Rocky Hill, NJ, USA).
Cell culture and treatment
HEC-1A (ATCC, USA) and Ishikawa human endometrial adenocarcinoma cell lines were cultured in the Dulbecco's modified Eagle's medium (DMEM; Welgene, Korea) supplemented with 10% fetal bovine serum (FBS; Welgene), 4.5 g/L D-glucose with L-glutamine, and 100 U/mL penicillin-streptomycin. Human embryonic kidney (HEK)-293T cells were cultured in DMEM supplemented with 10% FBS, 4.5 g/L D-glucose with L-glutamine, 110 mg/L sodium pyruvate, and 100 U/mL penicillin-streptomycin. Both cultures were maintained in a humidified 5% CO2 atmosphere at 37 °C. For TBK1 or AKT inhibitor treatment, HEC-1A and Ishikawa cells were incubated with amlexanox (50, 100, and 200 μM) or MK-2206 (25, 50, and 100 nM) for the indicated time points. The same volume of dimethyl sulfoxide was used as vehicle control.
cBioPortal
cBioPortal (https://www.cbioportal.org/) is a comprehensive open resource for the exploration and visualization of large-scale cancer genomic databases, including The Cancer Genomic Atlas (TCGA), International Cancer Genome Consortium (ICGC), and Gene Expression Omnibus databases, which provide information on mutations, copy number alterations, and clinical correlations [26]. We used cBioPortal to explore the mutation data of TBK1 in UCEC, evaluate the prognostic value of TBK1 in patients with UCEC, and determine the correlation between TBK1 methylation and gene expression levels.
University of California Santa Cruz (UCSC) Xena
UCSC Xena (http://xena.ucsc.edu/) is a web-based resource for the visualization and analysis of cancer genomic databases, including TCGA, ICGC, TARGET, Genotype-Tissue Expression, and CCL [27]. The UCSC Xena database was used to generate heat maps of TBK1 mRNA expression, methylation, copy number, and somatic mutations in patients with UCEC and controls. Data were obtained from TCGA-UCEC dataset with 606 samples.
Tumor Immune Estimation Resource (TIMER) database analysis
TIMER2.0 (http://timer.cistrome.org/) is a web-based resource for the comprehensive analysis of gene expression and tumor-infiltrating immune cells in various cancer types. We used the Gene_Corr module of TIMER2.0 to analyze the correlation in expression levels between TBK1 and mechanistic target of rapamycin kinase and epithelial-mesenchymal transition (EMT) gene markers.
Plasmids and viral transduction
Using a polyethylenimine reagent, HEK293T cells were transfected with the sh-luciferase (sh-Luc), sh-TBK1 (The RNAi Consortium, Broad Institute, Cambridge, MA, USA), GFP, and TBK1 (Addgene, Cambridge, MA, USA) constructs with packaging plasmids. Briefly, lentiviral supernatants were collected, filtered at 48 and 72 h after transfection, and concentrated using the Lenti-X concentrator (Takara, Japan), according to the manufacturer's protocol. Then, HEC-1A and Ishikawa cells were transduced with the concentrated lentiviruses in the presence of 4 μg/mL polybrene.
Cell proliferation assay
HEC-1A and Ishikawa cells were seeded into 96-well plates at a density of 1 × 105 cells/well with 100 μL of the respective culture medium. After allowing the cells to adhere overnight, they were treated with amlexanox and vehicle or infected with sh-TBK1 or sh-Luc. Cell proliferation was assessed via water-soluble tetrazolium salt (WST)-8 assay (Biomax, Korea), according to the manufacturer's instructions. WST-8 reagent was added to each well and incubated for 30 min at 37 °C. Absorbance was measured at 450 nm using the Epoch 2 microplate reader (BioTek Instruments, Winooski, VT, USA).
BrdU incorporation assay
Cells were incubated with 10 μM 5-bromo-2'-deoxyuridine (BrdU) for 4 h. Following incubation, the cells were washed with PBS and fixed with 4% paraformaldehyde (pH 7.4, Biosesang, Seongnam, Korea). DNA denaturation was then achieved by treating the cells with 2N HCl. The incorporated BrdU was detected using an anti-BrdU antibody (Invitrogen, Carlsbad, CA, USA), followed by incubation with an Alexa Fluor 488-conjugated secondary antibody (Thermo Fisher Scientific). Nuclei were counterstained with 4',6-diamidino-2-phenylindole (Thermo Fisher Scientific). The labeling index was expressed as the proportion of BrdU-positive cells relative to the total cell count.
Colony formation assay
HEC-1A and Ishikawa cells were seeded into 6-well plates at a density of 1,000 cells/well. After allowing the cells to attach overnight, they were cultured for 2 weeks in complete medium. The formed colonies were then fixed with a pre-chilled methanol for 10 min and stained with 0.5% crystal violet for 15 min. The number of colonies was counted from three independent experiments.
Cell cycle assay
HEC-1A and Ishikawa cells were fixed with 70% cold ethanol for 30 min. For DNA staining, the fixed cells were washed and resuspended in phosphate-buffered saline (PBS) with propidium iodide (PI; 0.5 mg/mL) and RNase A (100 µg/mL) in the dark at 22-25 °C for 30 min. PI-stained cells were analyzed using the CytoFLEX S flow cytometer (Beckman-Coulter, USA).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from xenograft tumor tissues, HEC-1A cells, and Ishikawa cells using TRIzol reagent (Takara), according to the manufacturer's instructions. It was reverse transcribed to cDNA using the cDNA synthesis kit (BioFact, Seoul, Korea). qPCR was performed using the QuantStudio 3 real-time PCR system (Life Technologies, Carlsbad, CA, USA) with the SYBR Green qPCR Master Mix (BioFact). The qPCR reaction was set up as follows: 10 μL of SYBR Green Master Mix, 1 μL of each primer (final concentration of 500 nM), 2 μL of cDNA template, and 6 μL of nuclease-free water in a total volume of 20 μL. The qPCR thermal cycling conditions were as follows: 5 min at 95 °C, 40 cycles of 15 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C, followed by a 5 min final extension at 72 °C. The relative expression of each target gene was analyzed using the comparative threshold cycle (Ct) method with an internal control gene to normalize the target gene expression level. All primers used in this study are listed in Table S1.
Western blotting
Total protein was extracted from endometrial cancer tissues, HEC-1A cells, and Ishikawa cells using a radioimmunoprecipitation assay buffer with a protease inhibitor cocktail (Roche Diagnostics, Germany). Samples were homogenized and incubated on ice for 20 min, followed by centrifugation at 18,000 × g for 15 min at 4 °C. The total protein concentration was determined via bicinchoninic acid protein assay (Thermo Fisher Scientific, Waltham, MA, USA). Lysates were mixed with the sodium dodecyl sulfate Laemmli sample buffer, denatured by heating at 95 °C for 5 min, and separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were transferred onto a polyvinylidene fluoride membrane (Merck Millipore, Germany) using a semi-dry transfer system (Trans-Blot Turbo; Bio-Rad, Hercules, CA, USA). The transferred membranes were blocked with 5% non-fat milk in TBS-T for 1 h at room temperature. After blocking, the blots were incubated with primary antibodies against N-cadherin, E-cadherin, snail, vimentin, p-NF-κB, total NF-κB, p-AKT, total AKT, p-p70S6K, total p70S6K, β-actin, and α-tubulin overnight at 4 °C. After washing thrice with TBS-T for 10 min, the blots were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at 22-25 °C. After washing, the protein bands were visualized using the SuperSignal West Femto chemiluminescent substrate (Thermo Fisher Scientific) and detected using the Fusion Solo imaging system (Vilber Lourmat, France). ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used to calculate the protein band intensities.
Wound healing assay
HEC-1A and Ishikawa cells were seeded into 6-well plates and treated with amlexanox or TGF-β1 or transduced with sh-TBK1 or sh-Luc. When cells reached 90% confluency, a wound was created in the cell monolayer using a sterile 200-μL pipette tip to scratch a straight line across the well. Phase-contrast images of cell migration into the wound were captured at indicated time points using an inverted microscope (Ts2-FL; Nikon, Japan). The wound area at each time point was measured using the ImageJ software to determine the extent of wound closure.
Transwell migration assay
HEC-1A and Ishikawa cells were treated with amlexanox or vehicle or transduced with sh-TBK1 or sh-Luc. After reaching 80-90% confluency, the cells were detached using trypsin-EDTA. Transwell inserts with 8-μm pores (BD Falcon, Durham, NC, USA) were inserted into 24-well plates. The cells were resuspended in a medium containing 1% FBS at a density of 5 × 105 cells/mL. The cell suspension (200 μL) was added to the upper chamber of each transwell insert. The lower chamber was filled with a medium supplemented with 10% FBS to induce cell migration. After 24 h, non-migratory cells in the upper transwell chamber were removed with a cotton swab, and the migratory cells in the lower chamber were fixed with cold methanol and stained with crystal violet (Sigma-Aldrich, St. Louis, MO, USA). The average number of migrated cells was counted in five random fields at 100× magnification.
Immunocytochemistry
HEC-1A and Ishikawa cells were fixed with 4% paraformaldehyde (pH 7.4) for 15 min at room temperature, washed thrice with PBS, and permeabilized with methanol at -20 °C. The cells were incubated for 1 h in a blocking solution with an anti-NF-κB antibody at 4 °C in a humidified chamber. After washing thrice with PBS, the cells were incubated with the Alexa Fluor 488-conjugated secondary antibody for 1.5 h at 22-25 °C. Nuclei were counterstained with 4',6-diamidino-2-phenylindole, and coverslips were mounted on slides using the ProLong Gold antifade reagent (Thermo Fisher Scientific). Fluorescent images were obtained using a laser scanning confocal microscope (LSM 700; Carl Zeiss, Germany).
Establishment of a tumor xenograft model
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Konyang University (approval no. P-23-30-A-01). Five-week-old female nude BALB/c mice (14-19 g, Nara Biotech, Korea) were housed under pathogen-free conditions with appropriate care and monitoring throughout the study period. HEC-1A cells (4 × 106 cells/animal) were resuspended in 100 μL PBS and subcutaneously injected into the dorsal flank of the mice. When average tumor volume reached approximately 50 mm3 (day 0), tumor-bearing mice were randomly divided into two groups (n = 6) and intraperitoneally administered with amlexanox (5 mg/kg) or vehicle containing 5% Tween 80 daily for two weeks. To establish sh-Luc and sh-TBK1 tumor models, five-week-old female BALB/c nude mice were randomly divided into two groups (n = 6). Lentiviral sh-Luc or sh-TBK1-infected HEC-1A cells (4 × 106 cells/animal) were resuspended in 100 μL PBS and subcutaneously injected into the dorsal flank of the mice. After one week, tumor growth was monitored every three days using linear calipers. The tumor volume was calculated using the following formula: Volume = length × width2 × 0.52 (mm3). Mice were sacrificed via CO2 asphyxiation 4-5 weeks after tumor implantation. Then, their tissues were excised, weighed, and used for subsequent analysis.
Statistical analyses
Results are presented as the mean ± standard error of the mean. Unless otherwise indicated, data shown in the figures represent at least three independent experiments. Statistical analysis of data of the two groups was conducted using a two-tailed Student's t-test. Multiple groups were compared using one-way analysis of variance with Tukey's post-hoc test. Statistical significance was set at p < 0.05.
Results
TBK1 genetic mutations and their associations with the overall survival (OS) and disease-free survival (DFS) of patients with UCEC
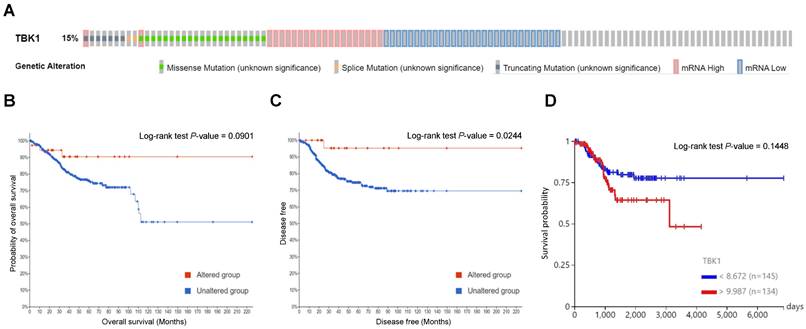
To determine the mutation characteristics of TBK1 and their correlation with patient survival in UCEC, we used the c-BioPortal database. TBK1 exhibited a high mutation frequency of 15% in UCEC on TCGA and PanCancer Atlas. The main genetic mutations in TBK1 were mRNA downregulation and missense mutations (Fig. 1A). Kaplan-Meier plots and log-rank tests revealed that aberrant expression of TBK1 mRNA was significantly associated with high OS (Fig. 1B) and DFS (Fig. 1C) in patients with UCEC. Furthermore, low TBK1 expression was associated with a better prognosis than those with high TBK1 expression in patients with UCEC (Fig. 1D). Subsequently, associations between TBK1 expression levels and copy number variance, somatic mutations, and DNA methylation were analyzed. As shown in Fig. S1A, no obvious alternations in the copy numbers, somatic mutations, and DNA methylation were observed that contributed to the TBK1 expression. Correlation analysis revealed that two TBK1 CpG sites (cg04466273 and cg21722680) were not significantly associated with TBK1 expression in patients with UCEC (Fig. S1B and C).
Genetic mutations in TANK-binding kinase 1 (TBK1) and their association with endometrial cancer prognosis. (A) Oncoprint analysis revealed the proportion and distribution of samples with different types of alterations in TBK1. (B and C) Kaplan-Meier curves of the overall and disease-free survival of patients with endometrial cancer in TBK1-altered and TBK1-unaltered groups. (D) Kaplan-Meier curves of overall survival according to TBK1 expression level in patients with endometrial cancer.
TBK1 deficiency suppressed proliferation and cell cycle progression in endometrial cancer cells
We examined the expression of phosphorylated and total TBK1 in the three endometrial cancer cell lines. The phosphorylated and total forms of endogenous TBK1 were expressed in all three cell lines. The phosphorylated form showed the highest expression in HEC-1A and SNU-1077 cells, whereas the total form showed the highest expression in HEC-1A and Ishikawa cells (Fig. 2A). Subsequently, TBK1 was knocked down in HEC-1A and Ishikawa cells, and the knockdown was confirmed via western blotting (Fig. 2B). The effect of TBK1 on endometrial cancer cell proliferation was investigated using the WST-8 assay. TBK1 knockdown significantly decreased the proliferation of HEC-1A and Ishikawa cells compared to that of the control cells (Fig. 2C). We further confirmed the inhibitory effect of TBK1 knockdown on the expression levels of the proliferation marker, MKI67, in HEC-1A and Ishikawa cells (Fig. 2D). Next, we investigated the effect of TBK1 on cell cycle progression. Flow cytometry revealed that TBK1 knockdown significantly increased the percentage of cells in G1 phase and decreased the percentage of cells in S phase of the cell cycle (Fig. 2E). Therefore, TBK1 deficiency induced G1 cell cycle arrest in endometrial cancer cells. Next, gene expression levels of the cyclin-dependent kinase (CDK) inhibitors, CDKN1A (encoding p21) and CDKN1B (encoding p27), were determined via RT-qPCR. TBK1 knockdown significantly increased the expression levels of CDKN1A and CDKN1B (Fig. S2). These results suggest that TBK1 knockdown regulates the cell cycle-related gene expression in endometrial cancer cells.
TBK1 knockdown suppresses proliferation and promotes cell cycle arrest in endometrial cancer cells. (A) Immunoblots of p-TBK1 and TBK1 in the cell lysates of three endometrial cancer cell lines. (B) Immunoblots of TBK1 in the cell lysates of lentiviral sh-TBK1- or sh-Luc-infected HEC-1A and Ishikawa cells. Band intensities were quantified and normalized to the control levels. (C) Water-soluble tetrazolium salt (WST)-8 assay of HEC-1A and Ishikawa cells infected with sh-TBK1 or sh-Luc lentiviruses at 24 h, 48 h, and 72 h. (D) Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis of MKI67 mRNA levels in HEC-1A and Ishikawa cells infected with sh-TBK1 or sh-Luc lentiviruses. (E) Cell cycle analysis of HEC-1A and Ishikawa cells infected with sh-TBK1 or sh-Luc lentiviruses via propidium iodide (PI) staining followed by flow cytometry. (F) Immunoblots of TBK1 in the cell lysates of TBK1 knockdown cells transfected with GFP or TBK1-Flag. (G) WST-8 assay of TBK1 knockdown cells transfected with GFP or TBK1-Flag. Data are represented as the mean ± standard error of the mean (SEM). *p < 0.05, **p < 0.01, and ***p < 0.001.
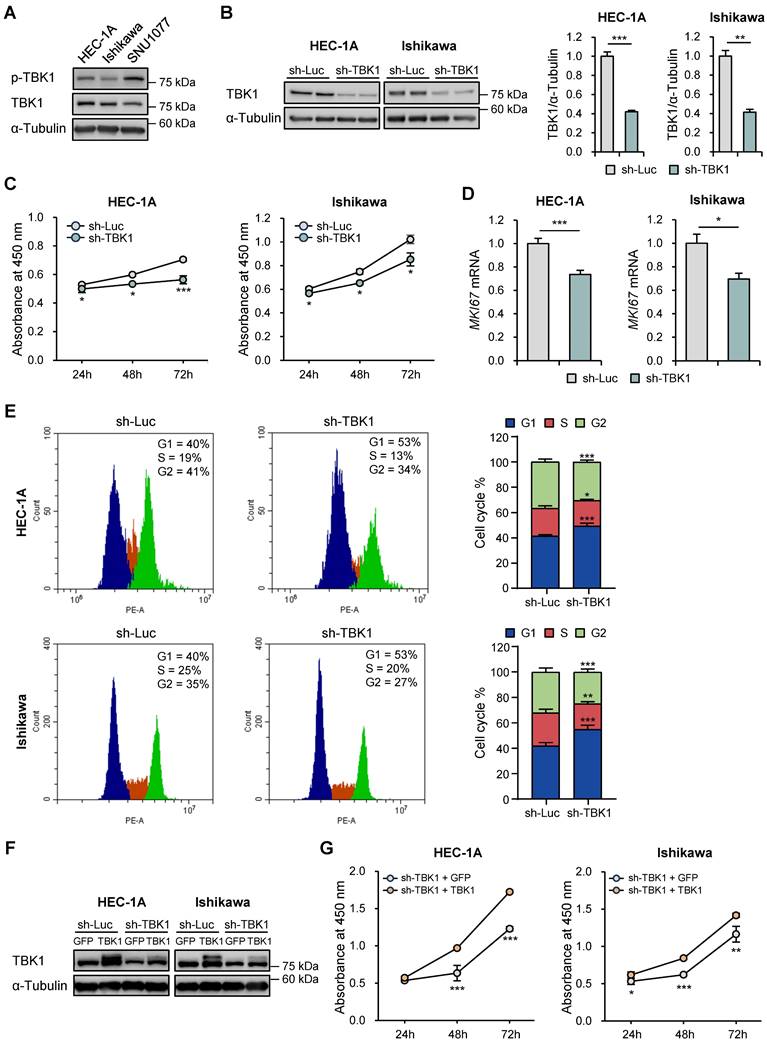
To determine whether restoring TBK1 expression could reverse the effects of TBK1 knockdown on endometrial cancer cell proliferation, we reintroduced TBK1 into the TBK1 knockdown cells using a TBK1 expression vector. Immunoblotting analysis confirmed that TBK1 expression was restored in HEC-1A and Ishikawa cells (Fig. 2F). The WST-8 assay showed that overexpression of TBK1 rescued the proliferation defect induced by TBK1 knockdown (Fig. 2G).
Amlexanox inhibited proliferation and survival in endometrial cancer cells
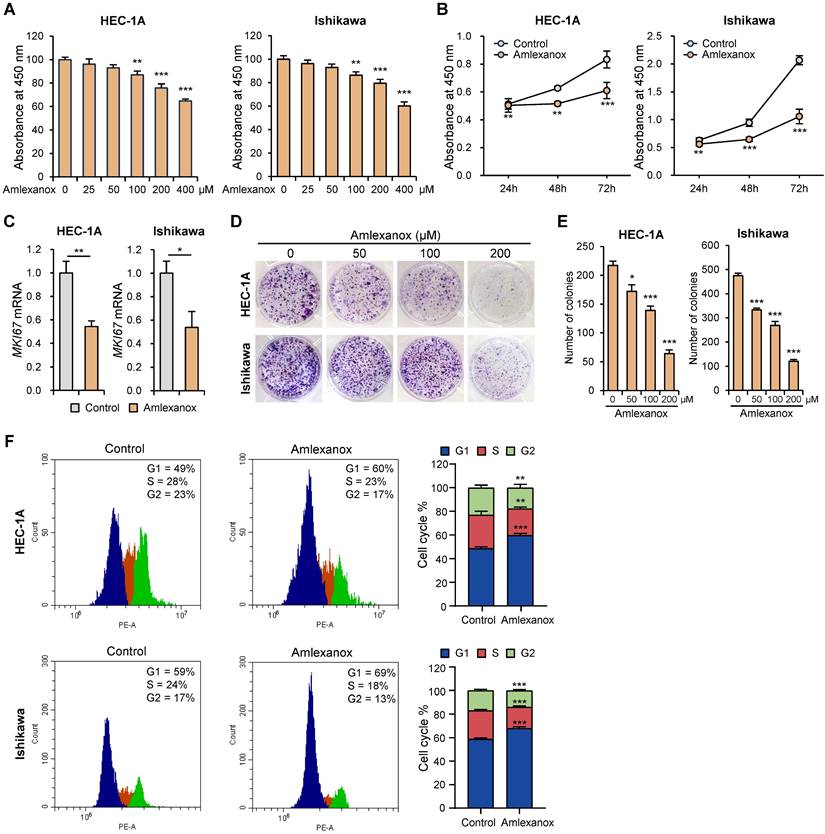
To determine the effect of amlexanox on endometrial cancer cell growth, we assessed cell proliferation using the WST-8 assay. Amlexanox suppressed the proliferation of HEC-1A and Ishikawa cells in a dose-dependent manner (Fig. 3A). It also significantly decreased the proliferation of HEC-1A and Ishikawa cells in a time-dependent manner (Fig. 3B). Next, we examined the effect of amlexanox on MKI67 expression. Amlexanox markedly decreased MKI67 mRNA levels in HEC-1A and Ishikawa cells (Fig. 3C). BrdU incorporation assay showed a significant reduction in DNA synthesis in amlexanox-treated cells compared to control cells (Fig. S3A and S3B). We also conducted a colony formation assay to evaluate the long-term effects of amlexanox on cell survival and proliferation. The results showed a marked decrease in both the number and size of colonies formed by cells treated with amlexanox (Fig. 3D and E), suggesting a substantial inhibition of cell proliferation and clonogenic potential. To determine whether amlexanox affects cell cycle progression, we investigated the cell cycle phases using flow cytometry. Consistent with the inhibitory effect of TBK1 knockdown on cell cycle, the G1 phase percentages of HEC-1A and Ishikawa cells were significantly higher in amlexanox-treated cells than in the control cells (Fig. 3F). Subsequently, we measured the expression levels of cell cycle-related genes. Amlexanox increased the expression of CDKN1A and CDKN1B in HEC-1A and Ishikawa cells (Fig. S3C). Immunoblotting analysis revealed that amlexanox increased the levels of cleaved PARP in a dose-dependent manner (Fig. S3D). Collectively, these data suggest that amlexanox suppresses proliferation and promotes cell cycle arrest and apoptosis in endometrial cancer cells.
Amlexanox suppresses proliferation and promotes cell cycle arrest in endometrial cancer cells. (A) WST-8 assay of HEC-1A and Ishikawa cells treated with the indicated concentrations of amlexanox (25-400 μM) for 24 h. (B) WST-8 assay of HEC-1A and Ishikawa cells treated with 100 μM amlexanox for 24 h, 48 h, and 72 h. (C) RT-qPCR analysis of MKI67 mRNA levels in HEC-1A and Ishikawa cells treated with 100 μM amlexanox for 24 h. (D) Colony formation assays of HEC-1A and Ishikawa cells treated with the indicated concentrations of amlexanox (50-200 μM). (E) Quantification of the colony formation assay shown in (D). (F) Cell cycle analysis of HEC-1A and Ishikawa cells treated with 100 μM amlexanox for 24 h via PI staining followed by flow cytometry. Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Downregulation of TBK1 expression was associated with EMT and migration in endometrial cancer cells
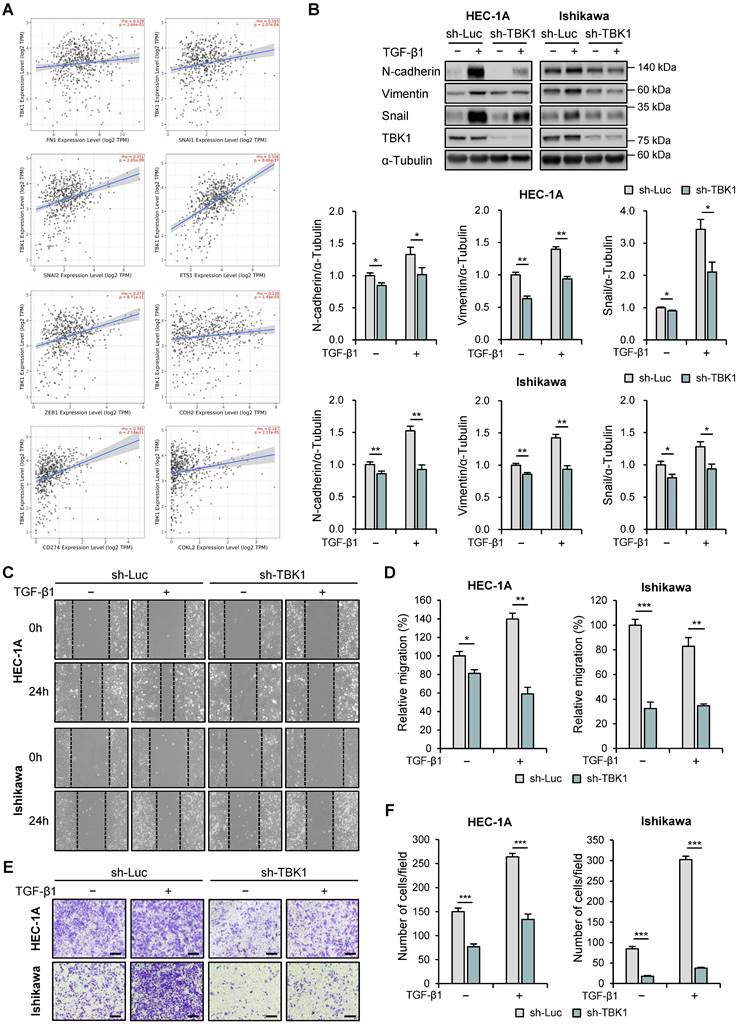
Next, we used the TIMER2.0 database to analyze the correlation between the expression levels of TBK1 and EMT-related genes using TCGA-UCEC data. TBK1 expression levels were positively correlated with the expression levels of fibronectin 1 (FN1; r = 0.129, p = 2.64e-3), snail (SNAI1; r = 0.165, p = 1.07e-4), slug (SNAI2; r = 0.251, p = 2.85e-9), E26 transformation-specific (ETS) transcription factors 1 (ETS1; r = 0.506, p = 8.46e-37), zinc finger E-box binding homeobox 1 (ZEB1; r = 0.273, p = 8.71e-11), E-cadherin (CDH1; r = 0.136, p = 1.49e-3), PD-L1 (CD274; r = 0.391, p = 2.14e-21), and cyclin-dependent protein kinase-like 2 (CDKL2; r = 0.187, p = 1.17e-5) (Fig. 4A). We then determined the effects of TBK1 knockdown on the expression levels of EMT-related proteins (N-cadherin, E-cadherin, vimentin, and snail) in HEC-1A and Ishikawa cells. Immunoblotting analysis revealed decreased levels of N-cadherin, vimentin, and snail in TBK1-knocked-down cells (Fig. 4B). However, TBK1 knockdown did not affect the E-cadherin levels in HEC-1A and Ishikawa cells (Fig. S4). These results suggest that TBK1 is involved in EMT. We further investigated the metastasis of endometrial cancer cells following TBK1 knockdown. Wound healing assay revealed that TBK1 knockdown significantly reduced the migration of HEC-1A and Ishikawa cells compared to that of control cells (Fig. 4C and D). Transwell migration assay revealed that TBK1 knockdown significantly inhibited the migration of endometrial cancer cells (Fig. 4E and F). To determine whether restoring TBK1 expression could reverse the effects of TBK1 knockdown on cell migration, we reintroduced TBK1 into TBK1 knockdown cells. Wound healing and transwell migration assays showed that overexpression of TBK1 significantly rescued the migration capability of the cells (Fig. S5A-D), indicating that TBK1 is crucial for maintaining the migratory properties associated with EMT.
TBK1 expression is correlated with the epithelial-mesenchymal transition (EMT)-related marker expression and cell migration in endometrial cancer. (A) Spearman correlation analysis between TBK1 and EMT-related gene expression in The Cancer Genomic Atlas (TCGA) endometrial cancer database. (B) Immunoblots of N-cadherin, vimentin, snail, and TBK1 in the cell lysates of lentiviral sh-TBK1- or sh-Luc-infected HEC-1A and Ishikawa cells treated with 10 ng/mL transforming growth factor (TGF)-β1 or vehicle. Band intensities were quantified and normalized to the control levels. (C) Wound-healing assay of lentiviral sh-TBK1- or sh-Luc-infected HEC-1A and Ishikawa cells treated with 10 ng/mL TGF-β1 or vehicle. Images were taken 24 h after the scratch wound. (D) Quantification of cell migration expressed as a percentage of control values. (E) Transwell migration assay of lentiviral sh-TBK1- or sh-Luc-infected HEC-1A and Ishikawa cells treated with 10 ng/mL TGF-β1 or vehicle. Images were taken after 24 h under a light microscope. Scale bars, 200 μm. (F) Quantification of cell migration expressed as a percentage of control values. Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Amlexanox suppressed the expression of EMT-related markers and migration in endometrial cancer cells
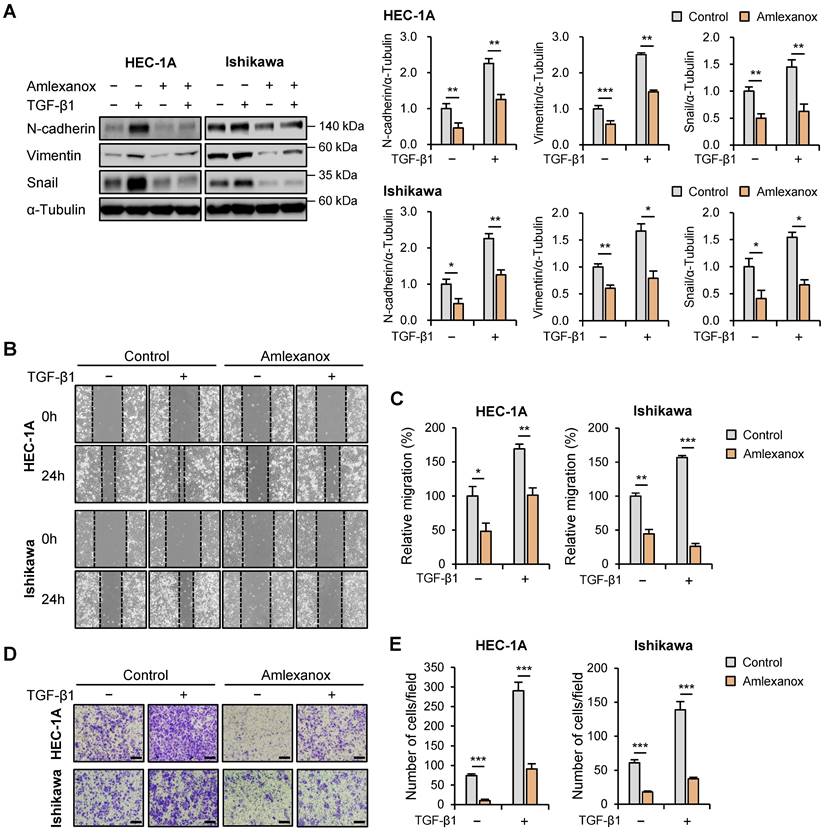
Subsequently, we investigated the effects of amlexanox on the expression levels of EMT-related proteins in HEC-1A and Ishikawa cells. Protein levels of N-cadherin, vimentin, and snail were significantly reduced in amlexanox-treated cells compared to those in control cells (Fig. 5A); however, E-cadherin expression levels were not affected (Fig. S6). We also examined the effects of BX795, another well-known TBK1 inhibitor, on the expression levels of EMT-related proteins. Similar to amlexanox, treatment with BX795 resulted in a significant decrease in the levels of N-cadherin and snail proteins (Fig. S7), suggesting that BX795 can effectively suppress EMT-related processes in these cells as well. We further evaluated the potential regulatory effect of amlexanox on endometrial cancer cell migration. Wound healing assay revealed that amlexanox markedly suppressed cell migration in both HEC-1A and Ishikawa cells (Fig. 5B and C). Transwell migration assay showed that the migration of HEC-1A and Ishikawa cells was significantly reduced after amlexanox treatment (Fig. 5D and E). Taken together, these data suggest that amlexanox suppresses EMT and migration of endometrial cancer cells.
Amlexanox inhibits the migration of endometrial cancer cells. (A) Immunoblots of N-cadherin, vimentin, and snail in the cell lysates of HEC-1A and Ishikawa cells treated with 10 ng/mL TGF-β1 or 100 μM amlexanox for 36 h. Band intensities were quantified and normalized to the control levels. (B) Wound-healing assay of HEC-1A and Ishikawa cells treated with 10 ng/mL TGF-β1 or 100 μM amlexanox. Images were taken 24 h after the scratch wound. (C) Quantification of cell migration expressed as a percentage of control values. (D) Transwell migration assay of HEC-1A and Ishikawa cells treated with 10 ng/mL TGF-β1 or 100 μM amlexanox. Images were taken after 24 h under a light microscope. Scale bars, 200 μm. (E) Quantification of cell migration expressed as a percentage of control values. Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
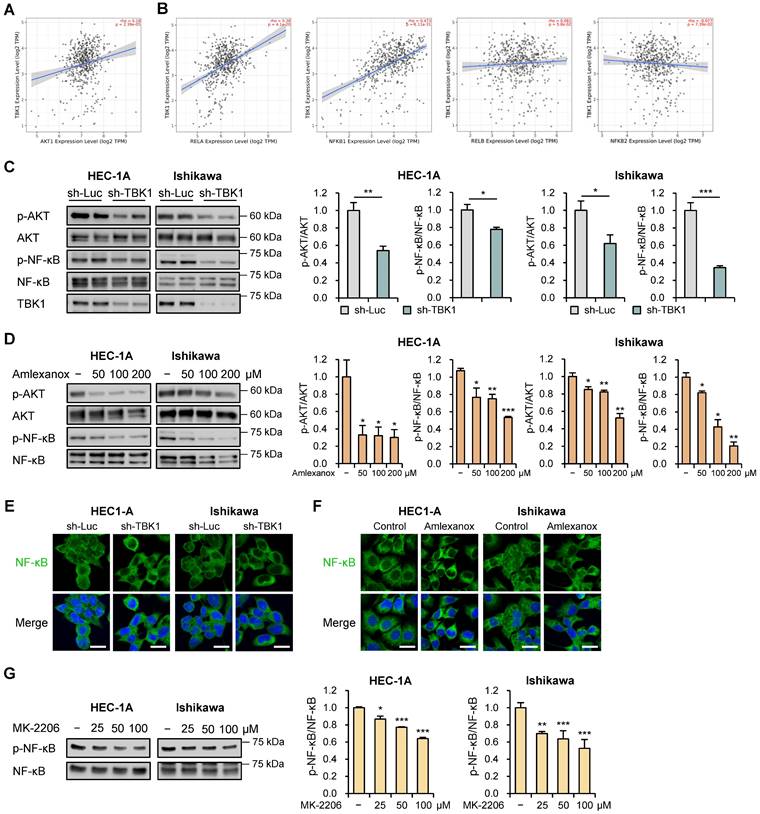
Targeting TBK1 inhibited the proliferation and migration of endometrial cancer cells via the AKT/NF-κB pathway
AKT/NF-κB signaling pathway contributes to tumor progression and invasion [28, 29]. TBK1 directly interacts with and activates this pathway [30]. To investigate their association, we evaluated the correlations between TBK1 and AKT/NF-κB pathway genes using the TIMER2.0 database. We observed strong correlations between TBK1 and AKT1 (r = 0.18, p = 2.39e-5), RELA (r = 0.38, p = 4.1e-20), and NFKB1 (r = 0.473, p = 1.11e-31) levels in TCGA-UCEC data (Fig. 6A and B). However, no associations were observed between TBK1 levels and levels of non-canonical NF-κB subunit genes, RELB and NFKB2 (Fig. 6B). Next, we explored whether AKT/NF-κB pathway is necessary for the anti-metastatic effects of targeting TBK1 against endometrial cancer cells. Immunoblotting analysis showed that the levels of p-AKT and p-NF-κB were significantly reduced by TBK1 knockdown in HEC-1A and Ishikawa cells (Fig. 6C). Consistent with the inhibitory effect of TBK1 knockdown on AKT/NF-κB pathway, amlexanox reduced the levels of p-AKT and p-NF-κB in a dose-dependent manner in cells (Fig. 6D). Moreover, TBK1 knockdown and amlexanox treatment decreased the levels of p-NF-κB and NF-κB levels in the nuclei of both HEC-1A and Ishikawa cells, compared with those in the nuclei of control cells (Fig. 6E, F, S8A and S8B). We further assessed whether AKT inhibition affects NF-κB activity in endometrial cancer cells. AKT inhibitor MK-2206 significantly inhibited the phosphorylation of NF-κB in HEC-1A and Ishikawa cells (Fig. 6G). These findings suggest that targeting TBK1 decreases AKT phosphorylation and suppresses NF-κB pathway activation.
Targeting TBK1 attenuates nuclear factor (NF)-κB activation by inhibiting the protein kinase B (AKT) pathway in endometrial cancer cells. (A, B) Spearman correlation analysis between TBK1 and AKT- or NF-κB-related gene expression in TCGA endometrial cancer database. (C) Immunoblots of p-AKT, AKT, p-NF-κB, and NF-κB in the cell lysates of lentiviral sh-TBK1- or sh-Luc-infected HEC-1A and Ishikawa cells. (D) Immunoblots of p-AKT, AKT, p-NF-κB, and NF-κB in the cell lysates of HEC-1A and Ishikawa cells treated with amlexanox (50-200 μM) for 18 h. (E) Immunofluorescence staining for NF-κB in lentiviral sh-TBK1- or sh-Luc-infected HEC-1A and Ishikawa cells. (F) Immunofluorescence staining for NF-κB in HEC-1A and Ishikawa cells treated with 100 μM amlexanox for 24 h. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI; blue). Scale bars, 20 μm. (G) Immunoblots of p-NF-κB and NF-κB in the cell lysates of HEC-1A and Ishikawa cells treated with MK-2206 (25-100 μM) for 4 h. Band intensities were quantified and normalized to the control levels. Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
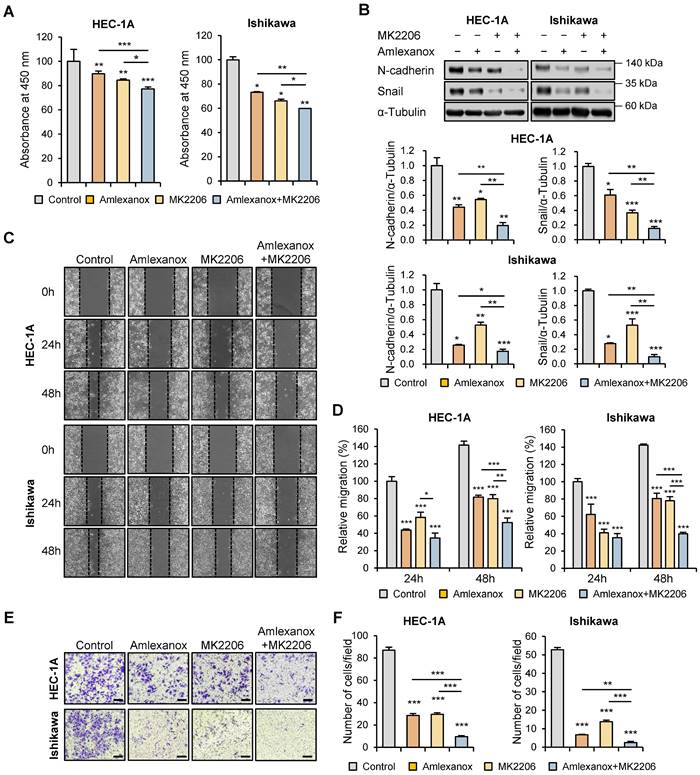
To further evaluate the regulatory effect of AKT inhibitor MK-2206 on endometrial cancer cell proliferation, WST-8 cell viability assay was performed using HEC-1A and Ishikawa cells after MK-2206 treatment. MK-2206 significantly suppressed cell proliferation, and this effect was enhanced by the combination of MK-2206 and amlexanox (Fig. 7A). Following this, we investigated whether AKT inhibition regulated the expression levels of EMT-related proteins in HEC-1A and Ishikawa cells. Immunoblotting analysis revealed that the protein levels of N-cadherin and snail were significantly reduced by MK-2206 and the combination of MK-2206 and amlexanox (Fig. 7B). Consistent with the inhibitory effects of the AKT inhibitor on the expression levels of EMT-related proteins, wound healing and transwell migration assays showed that MK-2206 suppressed the migration of both HEC-1A and Ishikawa cells (Fig. 7C-F). MK-2206 and amlexanox combination significantly reduced cell migration. These data suggest that the inhibitory effects of targeting TBK1 on cancer cell proliferation and migration are mediated by the AKT/NF-κB signaling pathway.
Targeting TBK1 inhibits the proliferation and migration of endometrial cancer cells via the AKT/NF-κB pathway. (A) WST-8 assay of HEC-1A and Ishikawa cells treated with 100 μM MK-2206 or 100 μM amlexanox for 24 h. (B) Immunoblots of N-cadherin and snail in the cell lysates of HEC-1A and Ishikawa cells pretreated with 100 μM MK-2206 or 100 μM amlexanox for 36 h in the presence of 10 ng/mL TGF-β1. Band intensities were quantified and normalized to the control levels. (C) Wound-healing assay of HEC-1A and Ishikawa cells pretreated with 100 μM MK-2206 or 100 μM amlexanox in the presence of 10 ng/mL TGF-β1. Images were taken 24 and 48 h after the scratch wound. (D) Quantification of cell migration expressed as a percentage of control values. (E) Transwell migration assay of HEC-1A and Ishikawa cells pretreated with 100 μM MK-2206 or 100 μM amlexanox in the presence of 10 ng/mL TGF-β1. Images were taken after 24 h under a light microscope. Scale bars, 200 μm. (F) Quantification of cell migration expressed as a percentage of control values. Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
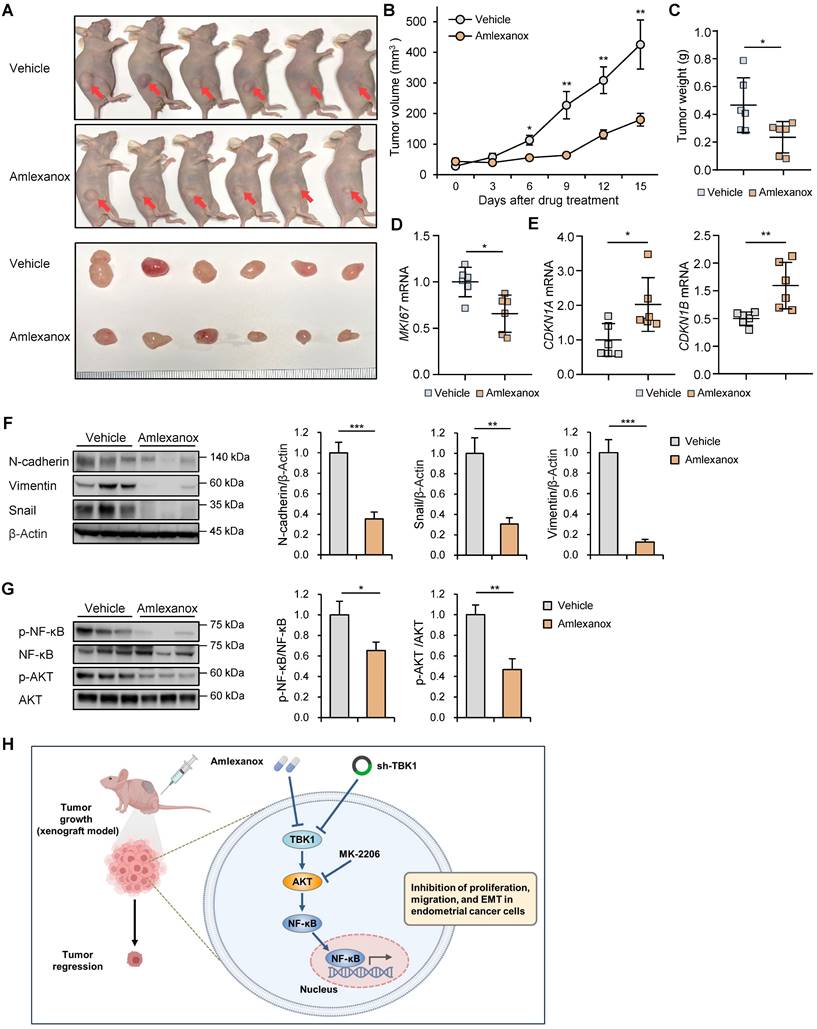
Targeting TBK1 inhibited tumor growth in vivo
To explore the roles of TBK1 in endometrial cancer-bearing mice, BALB/c nude mice were subcutaneously injected with lentiviral sh-TBK1- or sh-Luc-infected HEC-1A cells. Growth rate of xenograft tumors derived from the sh-TBK1 group was significantly lower than that of tumors derived from the sh-Luc group (Fig. S9A-C). To further verify the anti-tumor activity of amlexanox in xenograft models, HEC-1A cells were subcutaneously injected into BALB/c nude mice. The mice were intraperitoneally injected with amlexanox or vehicle for 15 d. Amlexanox significantly reduced the xenograft tumor growth in nude mice (Fig. 8A-C). Additionally, amlexanox inhibited MKI67 expression in xenograft tumors (Fig. 8D). Consistent with our results observed in HEC-1A and Ishikawa cells, RT-qPCR analysis showed that amlexanox treatment increased the expression of CDKN1A and CDKN1B in xenograft tumors (Fig. 8E). Immunoblotting analysis revealed that the expression levels of N-cadherin, vimentin, and snail were significantly decreased in the amlexanox-treated group compared with those in the control group (Fig. 8F). We further determined whether amlexanox treatment influenced AKT/NF-κB signaling in xenografts. Immunoblotting analysis revealed that phosphorylation of AKT and NF-κB was significantly lower in the amlexanox-treated group than in the control group (Fig. 8G). Collectively, these data suggest that targeting TBK1 inhibits the growth of endometrial cancer cells in vivo.
Targeting TBK1 inhibits tumor growth in vivo. (A) Representative images of subcutaneous xenograft tumors in nude mice treated with 5 mg/kg amlexanox or vehicle. Red arrows indicate the xenograft tumor. (B) Tumor growth curve in each group. (C) Tumor weights in each group. (D, E) RT-qPCR analysis of MKI67, CDKN1A, and CDKN1B mRNA levels in the xenograft tissues of each group. (F, G) Immunoblots of N-cadherin, vimentin, snail, p-AKT, AKT, p-NF-κB, and NF-κB in the xenograft tumor lysates of nude mice of each group. Band intensities were quantified and normalized to the control levels. Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. (H) Schematic diagram showing the anti-cancer mechanism of TBK1 inhibition, leading to decreased tumor growth, migration, and EMT in endometrial cancer cells.
Discussion
Endometrial cancer is a significant health concern worldwide, necessitating the exploration of new therapeutic strategies to improve patient outcomes [1]. TBK1 is involved in various diseases, including metabolic diseases and cancer. [6, 7, 9, 12, 14] However, its specific roles in endometrial cancer remain unknown. In this study, we investigated the potential anti-cancer effects of amlexanox, a TBK1 inhibitor, against endometrial cancer and the mechanisms by which TBK1 regulates the proliferation and migration of endometrial cancer cells. We found that patients with UCEC with low TBK1 expression exhibited better prognosis than those with high TBK1 expression. Moreover, TBK1 expression was positively correlated with increased EMT-related marker expression in patients with UCEC. Treatment with a TBK1 inhibitor attenuated the growth of the endometrial cancer cells by inhibiting the AKT/NF-κB pathway (Fig. 8H). These findings are consistent with those of previous studies showing the beneficial effects of TBK inhibition on inflammatory and metabolic diseases and cancers [19, 22, 23, 31]. The present study supports these findings by demonstrating the potential of TBK1 inhibition to suppress the growth of endometrial cancer cells.
Amlexanox, initially developed as an anti-inflammatory agent, has garnered increasing attention due to its potential therapeutic applications [6, 32]. Our findings demonstrated that amlexanox effectively inhibited endometrial cancer cell proliferation and migration in vitro. These results are consistent with those of previous studies demonstrating the anti-cancer properties of amlexanox in non-small cell lung cancers, melanoma, and glioblastoma cells [22, 23, 33, 34]. TBK1 is involved in the development and progression of multiple solid tumors. In lung and breast cancer cell lines, TBK1 phosphorylates Cdc20 at Ser134 and Cdh1 at multiple sites, including Thr20, Ser39, Ser42, Ser58, Ser131, and Ser151 [12]. This phosphorylation suggests that TBK1 modulates the activity of the anaphase-promoting complex/cyclosome, thereby influencing cell cycle dynamics and promoting cancer cell proliferation. Importantly, the finding that low TBK1 expression predicts a better prognosis in patients with UCEC highlights its potential as a therapeutic target and prognostic factor. Moreover, we found that inhibition of TBK1 through knockdown resulted in a marked reduction in proliferation and cell cycle progression of endometrial cancer cells. These findings are consistent with a growing body of evidence supporting the anti-cancer effects of TBK1 inhibition [8, 9]. By disrupting critical signaling pathways, TBK1 inhibitors can effectively impair cancer cell survival and proliferation, thereby presenting a viable therapeutic strategy.
Our study demonstrated that amlexanox and TBK1 shRNA effectively suppressed the expression of EMT-related markers and reduced the migration capacity of endometrial cancer cells. This is particularly significant in the context of TGF-β signaling, which is known to play a dual role in cancer progression [35, 36]. In early-stage cancer, TGF-β has been shown to exert tumor-suppressive effects by inducing apoptosis and cell cycle arrest. However, as the cancer progresses, TGF-β can switch roles and promote tumor progression and metastasis. In the present study, TGF-β treatment increased the expression of EMT-related markers and promoted the migration of endometrial cancer cells. However, when amlexanox or TBK1 knockdown were applied, we observed a significant reduction in these TGF-β-induced effects. These results suggest that TBK1 may be crucial in mediating the pro-tumorigenic effects of TGF-β, particularly in the context of EMT and cell migration.
AKT pathway, a key regulator of cell growth, survival, and metabolism, is commonly activated in various cancers and exhibits somatic mutations, copy number alterations, and aberrant epigenetic regulation in different cancer types [37, 38]. Activated TBK1 induces AKT phosphorylation, thereby influencing the anti-apoptotic pathways and promoting cell survival [13, 30, 39]. Herein, we observed a significant decrease in AKT phosphorylation after TBK1 inhibition, indicating its involvement in mediating the anti-cancer effects. Moreover, blocking AKT activation using MK-2206 suppressed the endometrial cancer cell proliferation and migration. This is consistent with previous reports that activation, mutation, and amplification of the AKT pathway contribute to cancer cell proliferation and survival [40, 41]. AKT-mediated phosphorylation of downstream targets, including mTOR, GSK, and NF-κB, underscores its vital roles in driving the malignant phenotypes of cancer cells by promoting cell cycle progression, inhibiting apoptosis, and enhancing metabolic reprogramming [42-44]. In this study, we found that amlexanox and TBK1 shRNA suppressed NF-κB phosphorylation in endometrial cancer cells. NF-κB is a transcription factor implicated in cancer-related inflammation, immune response, and progression. NF-κB activation leads to the transcription of various genes involved in inflammation, apoptosis, and metastasis, such as cytokines, Bcl-2 family proteins, and matrix metalloproteinases [45, 46]. Dysregulated NF-κB activation promotes cancer cell survival, proliferation, and invasion, making it an attractive therapeutic target [45]. By suppressing NF-κB signaling, TBK1 inhibition disrupts the pro-tumorigenic microenvironment and impedes cancer progression. Here, correlation between TBK1 inhibition by amlexanox and AKT/NF-κB axis suggests that the anti-cancer effects of amlexanox are associated with its ability to suppress the proliferation and migration of endometrial cancer cells via the AKT/NF-κB pathway.
While studies in other cancers have shown that TBK1 is a key regulator of these oncogenic pathways, there is a gap in the literature directly addressing the role of TBK1 in endometrial cancer. This study contributes to filling this gap by providing initial insights into how TBK1 might influence endometrial cancer progression through its interaction with the AKT/NF-κB pathway [13, 47, 48]. The limited existing research underscores the need for further studies to explore the clinical relevance of TBK1 in endometrial cancer more comprehensively. This study highlights the potential of TBK1 inhibition as a therapeutic strategy for endometrial cancer. The ability of TBK1 inhibition to modulate multiple signals, including AKT and NF-κB, underscores its potential as a multi-target anti-cancer approach. Combination therapies concurrently targeting AKT and NF-κB signaling pathways may exhibit synergistic effects, amplifying the therapeutic efficacy of endometrial cancer treatment. However, further preclinical and clinical investigations are needed to validate the efficacy and safety of amlexanox as a therapeutic intervention for endometrial cancer. Moreover, mechanisms underlying the anti-cancer effects of TBK1 inhibition should be explored to facilitate its clinical translation.
In conclusion, our study revealed that TBK1 inhibition exerts anti-cancer effects against endometrial cancer by regulating the AKT/NF-κB pathway. Our findings suggest TBK1 inhibition as a promising therapeutic strategy for endometrial cancer, warranting further investigation for future translation into clinical practice.
Abbreviations
TBK1: TANK-Binding Kinase 1; AKT: Protein Kinase B; NF-κB: Nuclear Factor Kappa B; UCEC: Uterine Corpus Endometrial Carcinoma; shRNA: Short Hairpin RNA; DMEM: Dulbecco's Modified Eagle Medium; FBS: Fetal Bovine Serum; HEK: Human Embryonic Kidney; TGF: Transforming Growth Factor; TCGA: The Cancer Genomic Atlas; ICGC: International Cancer Genome Consortium; UCSC: University of California, Santa Cruz; TIMER: Tumor Immune Estimation Resource; EMT: Epithelial-Mesenchymal Transition; sh-Luc: sh-luciferase; WST: Water-Soluble Tetrazolium Salt; PBS: Phosphate-Buffered Saline; PI: Propidium Iodide; RT-qPCR: Reverse Transcription-Quantitative Polymerase Chain Reaction; Ct: Comparative Threshold; DAPI: 4',6-diamidino-2-phenylindole; p: Phosphorylated; OS: Overall Survival; DFS: Disease-Free Survival; IRB: Institutional Review Board; SEM: Standard Error of the Mean.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This study was supported by grants from the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2020R1A2C1102244 and NRF- 2023R1A2C1005469) and Ministry of Education (NRF-2017R1A6A1A03015713). This manuscript was edited for proper English language, grammar, punctuation, spelling, and overall style by the highly qualified native English-speaking editors at Editage.
Author contributions
Jiha Shin and Jihoon Lim contributed to the study design, data acquisition, analysis, interpretation, and drafting of the manuscript; Daewon Han and Solji Lee conducted analysis and in vitro studies; Nak Song Sung, Jong-Seok Kim, Do Kyung Kim, and Hoi Young Lee contributed to the analysis, interpretation, and critical revision of the manuscript; Sung Ki Lee and Jongdae Shin contributed to the analysis and interpretation of the study and acquired funding for the study; Jeong Sig Kim and Hwan-Woo Park contributed to the conception and interpretation of the study, acquired funding, supervised the work, and wrote the final version of the manuscript. All authors have read and approved the final version of this manuscript.
Data availability
Data generated/analyzed in this study are available from the corresponding author upon request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Crosbie EJ, Kitson SJ, McAlpine JN, Mukhopadhyay A, Powell ME, Singh N. Endometrial cancer. Lancet. 2022;399:1412-28
2. Group SGOCPECW, Burke WM, Orr J, Leitao M, Salom E, Gehrig P. et al. Endometrial cancer: a review and current management strategies: part I. Gynecol Oncol. 2014;134:385-92
3. Bouwman F, Smits A, Lopes A, Das N, Pollard A, Massuger L. et al. The impact of BMI on surgical complications and outcomes in endometrial cancer surgery-an institutional study and systematic review of the literature. Gynecol Oncol. 2015;139:369-76
4. Suidan RS, He W, Sun CC, Zhao H, Fleming ND, Ramirez PT. et al. Impact of body mass index and operative approach on surgical morbidity and costs in women with endometrial carcinoma and hyperplasia. Gynecol Oncol. 2017;145:55-60
5. Helgason E, Phung QT, Dueber EC. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 2013;587:1230-7
6. Lee S, Shin J, Kim JS, Shin J, Lee SK, Park HW. Targeting TBK1 Attenuates LPS-Induced NLRP3 Inflammasome Activation by Regulating of mTORC1 Pathways in Trophoblasts. Front Immunol. 2021;12:743700
7. Cho CS, Park HW, Ho A, Semple IA, Kim B, Jang I. et al. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1-mediated p62/sequestosome 1 phosphorylation. Hepatology. 2018;68:1331-46
8. Runde AP, Mack R, S JP, Zhang J. The role of TBK1 in cancer pathogenesis and anticancer immunity. J Exp Clin Cancer Res. 2022;41:135
9. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108-12
10. Kim JY, Welsh EA, Oguz U, Fang B, Bai Y, Kinose F. et al. Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc Natl Acad Sci U S A. 2013;110:12414-9
11. Wei C, Cao Y, Yang X, Zheng Z, Guan K, Wang Q. et al. Elevated expression of TANK-binding kinase 1 enhances tamoxifen resistance in breast cancer. Proc Natl Acad Sci U S A. 2014;111:E601-10
12. Cooper JM, Ou YH, McMillan EA, Vaden RM, Zaman A, Bodemann BO. et al. TBK1 Provides Context-Selective Support of the Activated AKT/mTOR Pathway in Lung Cancer. Cancer Res. 2017;77:5077-94
13. Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T. et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41:458-70
14. Louis C, Burns C, Wicks I. TANK-Binding Kinase 1-Dependent Responses in Health and Autoimmunity. Front Immunol. 2018;9:434
15. Maan M, Agrawal NJ, Padmanabhan J, Leitzinger CC, Rivera-Rivera Y, Saavedra HI. et al. Tank Binding Kinase 1 modulates spindle assembly checkpoint components to regulate mitosis in breast and lung cancer cells. Biochim Biophys Acta Mol Cell Res. 2021;1868:118929
16. Jiang Q, Guan Y, Zheng J, Lu H. TBK1 promotes thyroid cancer progress by activating the PI3K/Akt/mTOR signaling pathway. Immun Inflamm Dis. 2023;11:e796
17. Xiang S, Song S, Tang H, Smaill JB, Wang A, Xie H. et al. TANK-binding kinase 1 (TBK1): An emerging therapeutic target for drug discovery. Drug Discov Today. 2021;26:2445-55
18. Beyett TS, Gan X, Reilly SM, Chang L, Gomez AV, Saltiel AR. et al. Carboxylic Acid Derivatives of Amlexanox Display Enhanced Potency toward TBK1 and IKKepsilon and Reveal Mechanisms for Selective Inhibition. Mol Pharmacol. 2018;94:1210-9
19. Oral EA, Reilly SM, Gomez AV, Meral R, Butz L, Ajluni N. et al. Inhibition of IKKvarepsilon and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 2017;26:157-70 e7
20. Bell J. Amlexanox for the treatment of recurrent aphthous ulcers. Clin Drug Investig. 2005;25:555-66
21. Makino H, Saijo T, Ashida Y, Kuriki H, Maki Y. Mechanism of action of an antiallergic agent, amlexanox (AA-673), in inhibiting histamine release from mast cells. Acceleration of cAMP generation and inhibition of phosphodiesterase. Int Arch Allergy Appl Immunol. 1987;82:66-71
22. Xiong J, Guo G, Guo L, Wang Z, Chen Z, Nan Y. et al. Amlexanox Enhances Temozolomide-Induced Antitumor Effects in Human Glioblastoma Cells by Inhibiting IKBKE and the Akt-mTOR Signaling Pathway. ACS Omega. 2021;6:4289-99
23. Liu Y, Lu J, Zhang Z, Zhu L, Dong S, Guo G. et al. Amlexanox, a selective inhibitor of IKBKE, generates anti-tumoral effects by disrupting the Hippo pathway in human glioblastoma cell lines. Cell Death Dis. 2017;8:e3022
24. Su C, Han Y, Zhang H, Li Y, Yi L, Wang X. et al. CiRS-7 targeting miR-7 modulates the progression of non-small cell lung cancer in a manner dependent on NF-kappaB signalling. Journal of cellular and molecular medicine. 2018;22:3097-107
25. Chen W, Li Z, Bai L, Lin Y. NF-kappaB in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front Biosci (Landmark Ed). 2011;16:1172-85
26. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
27. Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38:675-8
28. Xia L, Tan S, Zhou Y, Lin J, Wang H, Oyang L. et al. Role of the NFkappaB-signaling pathway in cancer. Onco Targets Ther. 2018;11:2063-73
29. Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13:738-47
30. Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G. et al. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci U S A. 2011;108:6474-9
31. Bailly C. The potential value of amlexanox in the treatment of cancer: Molecular targets and therapeutic perspectives. Biochem Pharmacol. 2022;197:114895
32. Reilly SM, Ahmadian M, Zamarron BF, Chang L, Uhm M, Poirier B. et al. A subcutaneous adipose tissue-liver signalling axis controls hepatic gluconeogenesis. Nature communications. 2015;6:6047
33. Challa S, Guo JP, Ding X, Xu CX, Li Y, Kim D. et al. IKBKE Is a Substrate of EGFR and a Therapeutic Target in Non-Small Cell Lung Cancer with Activating Mutations of EGFR. Cancer Res. 2016;76:4418-29
34. Moller M, Wasel J, Schmetzer J, Weiss U, Meissner M, Schiffmann S. et al. The Specific IKKepsilon/TBK1 Inhibitor Amlexanox Suppresses Human Melanoma by the Inhibition of Autophagy, NF-kappaB and MAP Kinase Pathways. Int J Mol Sci. 2020;21:4721
35. Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415-24
36. Moustakas A, Heldin P. TGFbeta and matrix-regulated epithelial to mesenchymal transition. Biochim Biophys Acta. 2014;1840:2621-34
37. Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol. 2016;82:943-56
38. Slomovitz BM, Coleman RL. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin Cancer Res. 2012;18:5856-64
39. Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL. et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157-70
40. Song M, Bode AM, Dong Z, Lee MH. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019;79:1019-31
41. Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74-88
42. He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW. et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther. 2021;6:425
43. Weng MC, Li MH, Chung JG, Liu YC, Wu JY, Hsu FT. et al. Apoptosis induction and AKT/NF-kappaB inactivation are associated with regroafenib-inhibited tumor progression in non-small cell lung cancer in vitro and in vivo. Biomed Pharmacother. 2019;116:109032
44. Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-kappaB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490-500
45. Xia Y, Shen S, Verma IM. NF-kappaB, an active player in human cancers. Cancer Immunol Res. 2014;2:823-30
46. Zhang T, Ma C, Zhang Z, Zhang H, Hu H. NF-kappaB signaling in inflammation and cancer. MedComm (2020). 2021;2:618-53
47. Shen RR, Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2011;30:631-41
48. Durand JK, Zhang Q, Baldwin AS. Roles for the IKK-Related Kinases TBK1 and IKKepsilon in Cancer. Cells. 2018;7:139
Author contact
![]() Corresponding author: Prof. Hwan-Woo Park, Department of Cell Biology, Myunggok Medical Research Institute, Konyang University College of Medicine, Daejeon 35365, Republic of Korea, E-mail: hwanwooparkac.kr; Prof. Jeong Sig Kim, Department of Obstetrics and Gynecology, Soonchunhyang University Seoul Hospital, Seoul 04401, Republic of Korea, E-mail: jskimac.kr.
Corresponding author: Prof. Hwan-Woo Park, Department of Cell Biology, Myunggok Medical Research Institute, Konyang University College of Medicine, Daejeon 35365, Republic of Korea, E-mail: hwanwooparkac.kr; Prof. Jeong Sig Kim, Department of Obstetrics and Gynecology, Soonchunhyang University Seoul Hospital, Seoul 04401, Republic of Korea, E-mail: jskimac.kr.